
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding modes of a core-extended metalloporphyrin to human telomeric DNA G-quadruplexes†
Jenifer
Rubio-Magnieto‡
a,
Florent
Di Meo‡
b,
Mamadou
Lo
c,
Cécile
Delcourt
a,
Sébastien
Clément
c,
Patrick
Norman
b,
Sébastien
Richeter
*c,
Mathieu
Linares
*ab and
Mathieu
Surin
*a
aLaboratory for Chemistry of Novel Materials, Center for Innovation in Materials and Polymers, University of Mons – UMONS, 20 Place du Parc, B-7000 Mons, Belgium. E-mail: mathieu.surin@umons.ac.be; Tel: +32(0)65373868
bDepartment of Physics, Chemistry and Biology (IFM) Linköping University, SE-581 83 Linköping, Sweden. E-mail: mathieu@ifm.liu.se
cInstitut Charles Gerhardt – UMR 5253, Université de Montpellier 2 – CC1701, Place Eugène Bataillon, F-34095 Montpellier Cedex 05, France. E-mail: sebastien.richeter@univ-montp2.fr
First published on 17th December 2014
Abstract
The molecular recognition of human telomeric G-quadruplexes by a novel cationic π-extended NiII-porphyrin (NiII-TImidP4) is studied in aqueous solutions via (chir)optical spectroscopy, Fluorescence Resonance Energy Transfer (FRET) melting assay, and computational molecular modeling. The results are systematically compared with the recognition by a conventional meso-substituted NiII-porphyrin (NiII-TMPyP4), which allows us to pinpoint the differences in binding modes depending on the G-quadruplex topology. Importantly, FRET melting assays show the higher selectivity of NiII-TImidP4 towards human telomeric G4 than that of NiII-TMPyP4.
Introduction
Specific guanine-rich oligonucleotides can fold or assemble into quadruplex structures, i.e., four guanines (G) form a square-planar network via Hoogsteen hydrogen-bonds, a G-quartet, and G-quartets can stack on top of each other, stabilized by monovalent alkali cations such as potassium (K+) or sodium (Na+).1–4 There is solid evidence that the human telomeric DNA sequence d(TTAGGG)n forms intramolecular G-quadruplexes at the end of chromosomes, and that G-quadruplexes are over-represented in promoter regions such as oncogenes.5–9 Therefore, G-quadruplexes have been identified as therapeutic targets, and the search for new molecules (ligands) that stabilize G-quadruplex topologies has become an active field of research in anti-cancer drug design.10–13 This includes organometallic complexes, planar aromatic structures, and macrocyclic ligands such as porphyrin derivatives.14–17 Indeed, the structural and photophysical properties of porphyrins make these compounds interesting for DNA recognition and sensing.18–22 For instance, the tetrakis(N-methylpyridinium-4-yl)porphyrin (TMPyP4, see Chart 1 left, without a metal center) can show different types of DNA binding modes, which depend on different factors such as the sequence and structure of DNA, the peripheral substituents on the porphyrin ring, and the composition of the solution.23–26 If the porphyrin possesses a central metal (metalloporphyrin), the binding to G-quadruplexes depends on the nature of the metal: octahedral complexes (e.g. CoII porphyrins, possessing axial ligands) bind externally to the G-quadruplex due to their 3D structures hindering intercalation in between G-quartets, whereas square-planar complexes (CuII or NiII porphyrins) can possibly intercalate in between G-quartets.14,23,27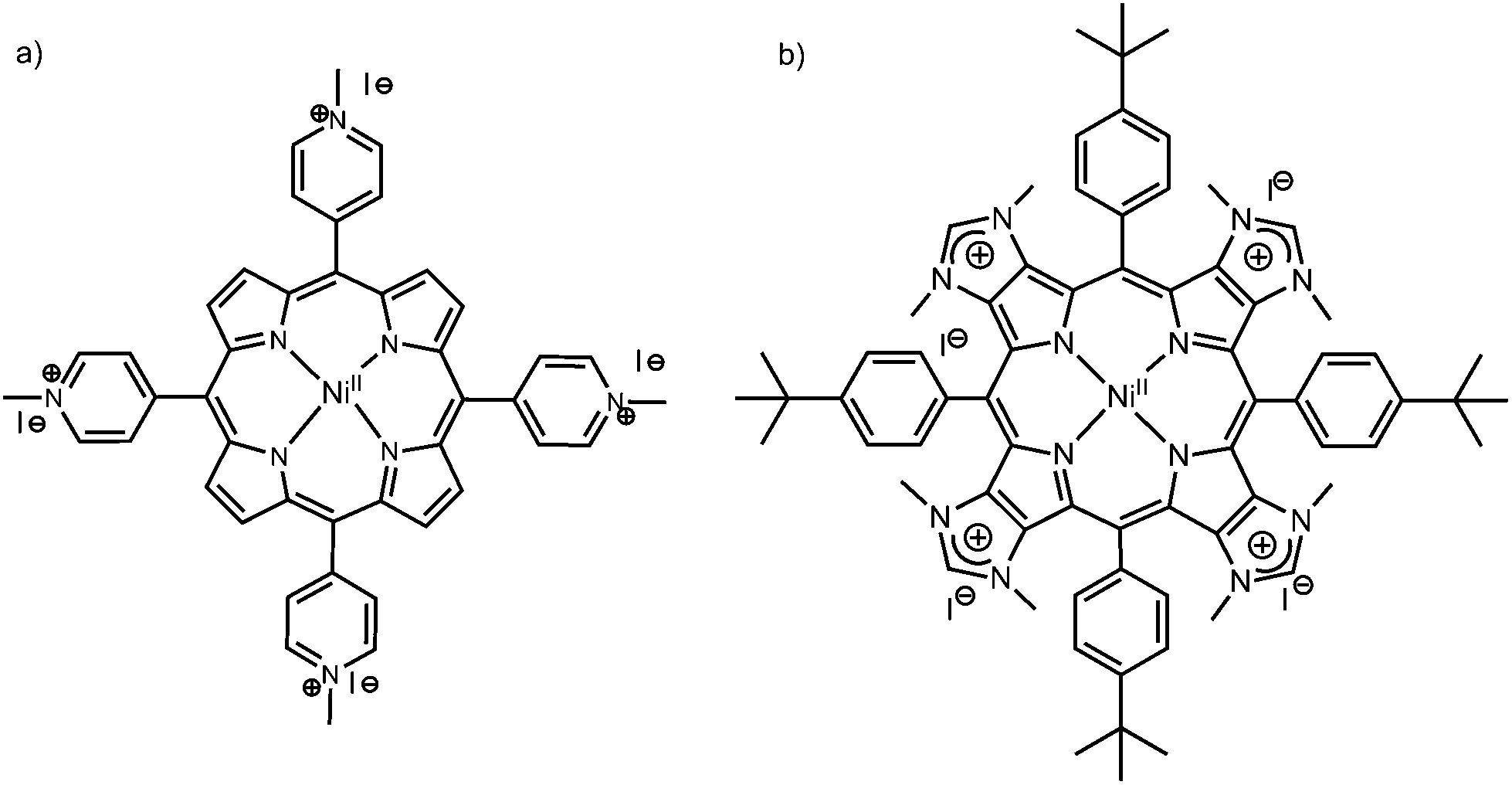 | ||
| Chart 1 Chemical structures of the metalloporphyrins under study: (a) NiII-TMPyP4 and (b) NiII-TImidP4. | ||
Herein, we describe the molecular recognition of intra-molecular G-quadruplexes (G4) by a metalloporphyrin NiII-TImidP4 (Chart 1, right). In particular, we focus on human telomeric DNA sequences that form G4, here 22-mer and 30-mer oligonucleotides. These G4 sequences were identified as therapeutic targets, for which binding to a ligand, such as a porphyrin derivative, may inhibit telomerase, an over-expressed enzyme in 85–90% cancers.13,17,28–30 In contrast with other metalloporphyrins studied so far for their interactions with DNA, the four positive charges of NiII-TImidP4 are located in the aromatic core of the porphyrin, i.e. on the fused imidazolium rings. The meso 4-tert-butylphenyl groups were used as solubilizing groups in the course of the synthesis of the corresponding tetraimidazole derivatives in organic solvents.31 However, these hydrophobic groups do not prevent the solubility of NiII-TImidP4 in water. Keeping nickel(II) in the porphyrin was also a deliberate choice as we had previously observed that free base porphyrins fused to imidazole rings may decompose before the quaternization reaction leading to the formation of NiII-TImidP4.32 This expands the family of porphyrins to more π-extended cationic structures, for which the DNA G-quadruplex recognition should be quite different due to the particular charge distribution and an extended π-conjugated plane of this porphyrin. Our results are systematically compared to the recognition of human telomeric G4 to NiII-TMPyP4 (Chart 1, left), for which the cationic substituents are placed in the four meso positions. Indeed, the porphyrin TMPyP4 has been well-studied (with or without a metal center) for its DNA-binding abilities, noticeably with G-quadruplexes.10,19,23,33 We carried out UV-Vis spectroscopy and Circular Dichroism (CD) in aqueous solutions, which allowed us to probe the G4/metalloporphyrin interactions as a function of the solution composition. Fluorescence Resonance Energy Transfer (FRET) melting assays provide valuable information on the stabilization and selectivity of the studied ligands towards G4. Molecular dynamics simulations are used to propose reliable supramolecular structures and to estimate binding free energies of the G4–porphyrin complexes. Finally, a comparison between CD experiments and computed chirality parameters support the identification of relevant binding modes.
Results and discussion
Synthesis of NiII-TImidP4
We have recently reported on the synthesis of a π-extended porphyrin fused to imidazole rings across their β,β′-pyrrolic positions and showed that up to four imidazole rings could be fused to the aromatic core of the porphyrin (one on each pyrrole unit).31 The tetrakis(imidazolium) salt NiII-TImidP4 (see Chart 1, right) was obtained by alkylating the tetrakis(imidazole) derivative with an excess of iodomethane. The molecular mass peak of NiII-TImidP4 was observed by using ESI-TOF mass spectrometry at m/z = 292.6498, as expected for this tetracationic species (calculated m/z = 292.6496, see ESI Fig. S1 and S2†). NiII-TImidP4 is well soluble in protic polar solvents such as water (green solutions) and is very poorly soluble in organic solvents such as CHCl3 (brown turbid solutions). The UV-visible spectrum of NiII-TImidP4 in water showed that the Soret absorption band is broad and split with a main absorption band centred at λ = 436 nm and a shoulder on the right (Fig. S3 in ESI†). This suggests that additional ligands (i.e. water molecules) are coordinated at the nickel center.34 Moreover, the four imidazolium rings acting as electron deficient units also favour the coordination of axial ligands on the central nickel(II). Knowing that square pyramidal and distorted octahedral nickel(II) porphyrin complexes are paramagnetic (S = 1, high-spin), axial coordination of one and/or two molecule(s) of solvent at the nickel(II) center may explain the observed broad and split signals for NiII-TImidP4 by 1H NMR spectroscopy in d6-methanol (Fig. S4 in ESI†). This is confirmed by the fact that the same phenomenon was observed with the porphyrin fused to three imidazolium rings in d6-methanol, while sharp and well-defined signals were observed for this compound in a non-coordinating solvent like CDCl3 (Fig. S5 in ESI†).Binding to human telomeric sequences
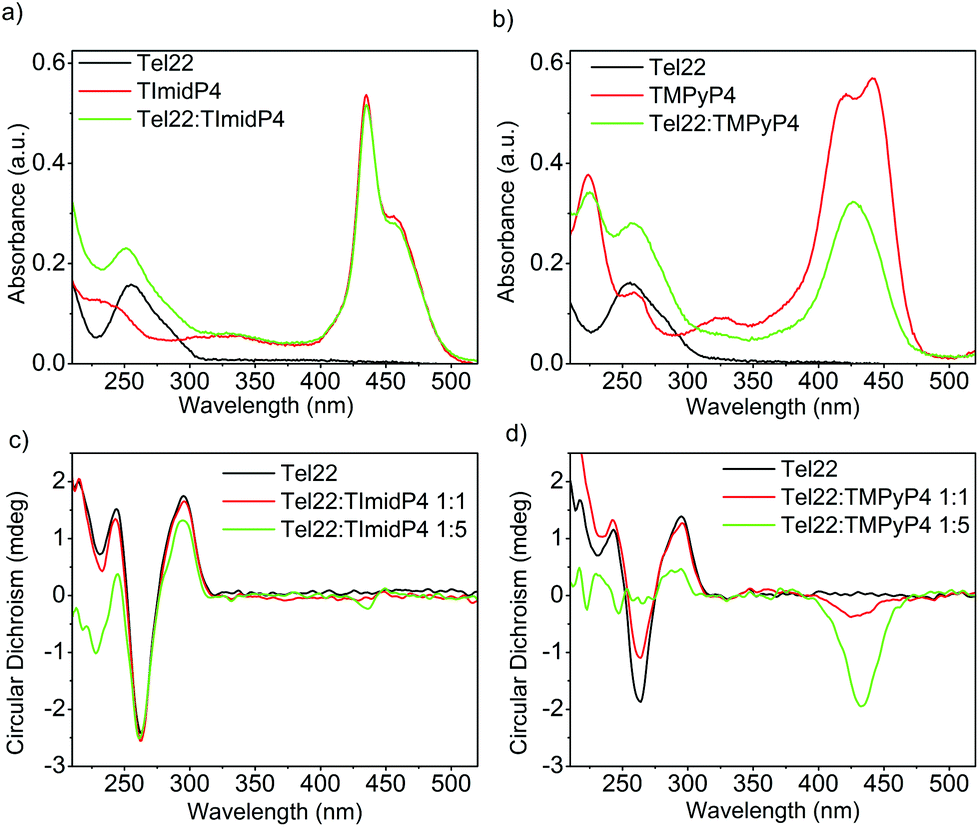
The human telomeric sequence d(TTAGGG)n can adopt different intramolecular G-quadruplex structures with backbone loop-shapes depending on the aqueous solution conditions, especially on the nature of the salt.35–37 Human telomeric G-quadruplexes possess large structural diversity, as recently reviewed.38 The main human telomeric sequence studied here d[AG3(T2AG3)3], hereafter referred to as Tel22, presents a parallel and/or anti-parallel G4 loop structure depending on the alkali cation in aqueous solution. In solutions of potassium ions (K+), this sequence forms a mixture of parallel and anti-parallel G-quadruplex conformations, in a dynamic equilibrium between hybrid structures.35,39–41 This is observed using circular dichroism (CD) experiments, showing a CD spectrum characterized by a positive maximum at 290 nm, a plateau at 265 nm, and a negative peak at 240 nm (Fig. 1 and S7 in ESI†). | ||
Fig. 1 UV-Vis (top) and CD spectra (bottom) of (a) and (c) Tel22: NiII-TImidP4 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N and (b) and (d) Tel22:NiII-TMPyP4 1:N in TE buffer + 100 mM KCl. The molar ratio for (a) and (b) is 1:5. :N and (b) and (d) Tel22:NiII-TMPyP4 1:N in TE buffer + 100 mM KCl. The molar ratio for (a) and (b) is 1:5. | ||
In the presence of sodium ions (Na+), Tel22 presents an anti-parallel “basket-type” structure, with a characteristic positive peak at 295 nm and an intense negative peak at 260 nm (Fig. 2 and S7 in ESI).37 The molecular recognition of Tel22 by each metalloporphyrin was studied under different solution conditions using CD and UV-Vis absorption. Fig. 1c shows the CD spectra of Tel22:NiII-TImidP4 mixtures in an aqueous solution containing 100 mM KCl. The typical CD signal of this telomeric hybrid G4 structure in K+ solution, i.e. a positive peak at 290 nm and a plateau at 265 nm, is maintained whatever the molar ratio. Moreover, we observe a weak negative induced CD signal (ICD) in the Soret band of the metalloporphyrin, which increases at a 1:5 molar ratio. This indeed suggests that NiII-TImidP4 interacts with Tel22. In contrast, for mixtures of Tel22:NiII-TMPyP4 (Fig. 1d), no ICD is observed at a 1:1 molar ratio, and at a molar ratio of 1:5 a weak negative ICD is observed but the CD signal of Tel22 (200–320 nm) completely vanishes. This is in agreement with other studies showing that the porphyrin TMPyP4 can unfold G-quadruplex structures.42,43 The CD spectra indicate that the core-extended metalloporphyrin NiII-TImidP4 stabilizes the initial G4 conformations of Tel22 in the presence of K+, while the binding to (an excess of) NiII-TMPyP4 may lead to unfolding of the G-quadruplex conformations. This is likely due to the stronger intermolecular interactions of NiII-TMPyP4 with DNA bases (see below).
 | ||
| Fig. 2 UV-Vis (top) and CD spectra (bottom) of (a) and (c) Tel22: NiII-TImidP4 1:N and (b) and (d) Tel22:NiII-TMPyP4 1:N in TE buffer + 100 mM NaCl. The molar ratio for (a) and (b) is 1:5. | ||
This is supported by the UV-Vis absorption spectra shown in Fig. 1a and b. Pure NiII-TMPyP4 shows two overlapping bands in the Soret region: one at 420 nm due to the diamagnetic four-coordinate form and one at 441 nm due to the paramagnetic diaquo complex (Fig. 1b). Upon interaction with Tel22, the UV-Vis spectrum shows a red-shifted absorption maximum (Δλmax ∼ 8 nm) and a large hypochromicity in the Soret band. This is different for NiII-TImidP4, for which UV-Vis spectra show no change in the shape and a smaller hypochromicity in the Soret band upon binding to Tel22 (Fig. 1a). As indicated above, a shoulder is noticed on the right which may indicate that one or two ligands (i.e. water molecules) are coordinated at the nickel center.34
For each metalloporphyrin, the same types of changes in the spectra were observed upon binding to the longer human telomeric sequence Tel30 (d[GT2A(GGGTTA)4GG]), a sequence also presenting a mixture of anti-parallel and parallel G4 conformations in KCl solutions (Fig. S8 in ESI†). This indicates that larger G-quadruplexes structures have rather identical recognition modes by these metalloporphyrins.
In aqueous solutions containing 100 mM NaCl, the Tel22 adopts an anti-parallel G-quadruplex conformation. As observed in the CD spectra shown in Fig. 2c, this anti-parallel G4 conformation is maintained upon addition of NiII-TImidP4 metalloporphyrin. No ICD in the Soret band is observed at a 1:1 molar ratio, and only a weak ICD signal is observed with 5 equivalents of NiII-TImidP4. In contrast, upon addition of NiII-TMPyP4 metalloporphyrin, at a 1:1 molar ratio, an ICD is observed in the Soret band, and at 1:5 the CD signals characteristic of the G4 structure Tel22 start to vanish (Fig. 2d and S9†). The negative ICD signal is much more pronounced for the Tel22:NiII-TMPyP4 mixture. This signal notably differs from what happens with the ZnII-TMPyP4 octahedral complex, yielding a bisignated ICD peak, which was ascribed to an end-stacking binding mode.44 The difference in binding modes to Tel22 between NiII-TMPyP4 and NiII-TImidP4 can be ascribed to the extended π-conjugated system and the saddle conformation of NiII-TImidP4, as discussed here below. Again, the UV-Vis experiments are consistent with ICD signals in that they show that, upon interaction with Tel22, the NiII-TMPyP4 shows larger perturbations in the Soret band than for NiII-TImidP4 (Fig. 2a and b).
In order to assess the binding properties of NiII-TImidP4 towards Tel22, a FRET melting assay was performed, as this is a valuable method to assess the stabilization and selectivity of G4 ligands.45,46 This method is based on the measurement of melting properties of a double-dye labelled oligonucleotide, as followed by fluorescence spectra showing FRET between the two dyes when the oligonucleotide is folded in G4 conformation. Here we used a modified human telomeric sequence end-capped with a fluorescein amidite dye (FAM) at the 5′-end and a tetramethylrhodamine (TAMRA) at the 3′-end. This labelled oligonucleotide FAM-5′-GGG(T2AG3)3-TAMRA-3′ named F21T was widely studied in the literature for G4-ligand studies. After a preliminary heating/cooling cycle of F21T, we added the studied porphyrin and followed the emission of the donor dye (i.e. FAM) as a function of temperature. This method has been shown to give more reproducible results than the sensitized emission of the acceptor (i.e. TAMRA).45 The G4 structure unfolds when the temperature increases and thereby the fluorescence emission of FAM occurs. This denaturation can be followed by plotting melting curves, which show the increase in FAM fluorescence as a function of temperature for pure F21T and for the mixtures F21T/NiII-TImidP4 and F21T/NiII-TMPyP4 at a 1:5 molar ratio under K+ buffer conditions (Fig. 3a). The determination of the half-melting temperature difference (ΔT1/2) between the pure oligonucleotide and the same oligonucleotide bound to a ligand is a quantitative analysis of the stabilization effect induced by the ligand (Fig. 3b). The results show that the ΔT1/2 is around 25 °C and around 23 °C for F21T/NiII-TImidP4 and F21T/NiII-TMPyP4, respectively (Fig. 3). The latter value is in agreement with the one obtained by Romera et al., who studied the influence of the incorporation of a metal ion on the G4-binding properties of TMPyP4 porphyrin.27 Thus, the π-extended metalloporphyrin NiII-TImidP4 could be considered as a good G4-ligand, as it induces an increase in the melting temperature of F21T close to, or slightly higher than, in the case of other well-considered G-quadruplex ligands.47
 | ||
| Fig. 3 (a) Melting curves of 200 nM pure F21T and in the presence of a 1 μM NiII-TImidP4 ligand or a 1 μM NiII-TMPyP4 ligand without or with the presence of 15 equiv. of competitor double-stranded DNA (ds20). The curves shown correspond to normalized FAM fluorescence. (b) Melting temperature differences ΔT1/2 from FRET melting assays for each metalloporphyrin without and with the presence of the competitor ds20. All measurements were performed in a 10 mM lithium cacodylate buffer (pH = 7.2) with 10 mM KCl and 90 mM LiCl. | ||
In order to evaluate the selectivity of this ligand, a competitor double-stranded DNA ds20 (oligonucleotide 5′-CGTCACGTAAATCGGTTAAC-3′ hybridized with its complementary sequence), was added to the F21T/NiII-TImidP4 and F21T/NiII-TMPyP4 complexes in an excess of 15 equivalents as compared to F21T. The decrease in ΔT1/2 shows that the NiII-TImidP4 ligand is affected by the presence of the ds20 competitor; however this decrease is more pronounced for the NiII-TMPyP4 (Fig. 3b). Therefore, the core-extended metalloporphyrin NiII-TImidP4 showed a higher selectivity for G-quadruplex DNA than the conventional NiII-TMPyP4.
Supramolecular structures of NiII-porphyrin–Tel22 complexes
Molecular dynamics (MD) simulations were carried out to give insights into the structures and binding modes of the studied porphyrins to model intramolecular G4: anti-parallel “basket-type” G4 (with Na+, PDB ID: 143D), parallel G4 (with K+, PDB ID: 1KF1) and hybrid G4 (mixed parallel/anti-parallel strands, PDB ID: 2HY9) structures of Tel22.37,41,48 Only 1:1 complexes were considered for the sake of computational cost, taking into account the water explicitly (TIP3P model). MD simulations were carried out on a 100 ns timescale, allowing us to explore the movements of a single porphyrin around each G4 type and the possible interaction sites. Five starting conformations were considered, as depicted in Fig. 4.
 | ||
| Fig. 4 View of the initial conformations of 1:1 NiII-TImidP4–parallel Tel22 complexes for MD simulations. The guanosines are shown in yellow (T and A bases are omitted for clarity), the phosphate backbone is shown as a grey tube. The porphyrin is shown in black, red, blue, green or violet depending on the considered conformation. | ||
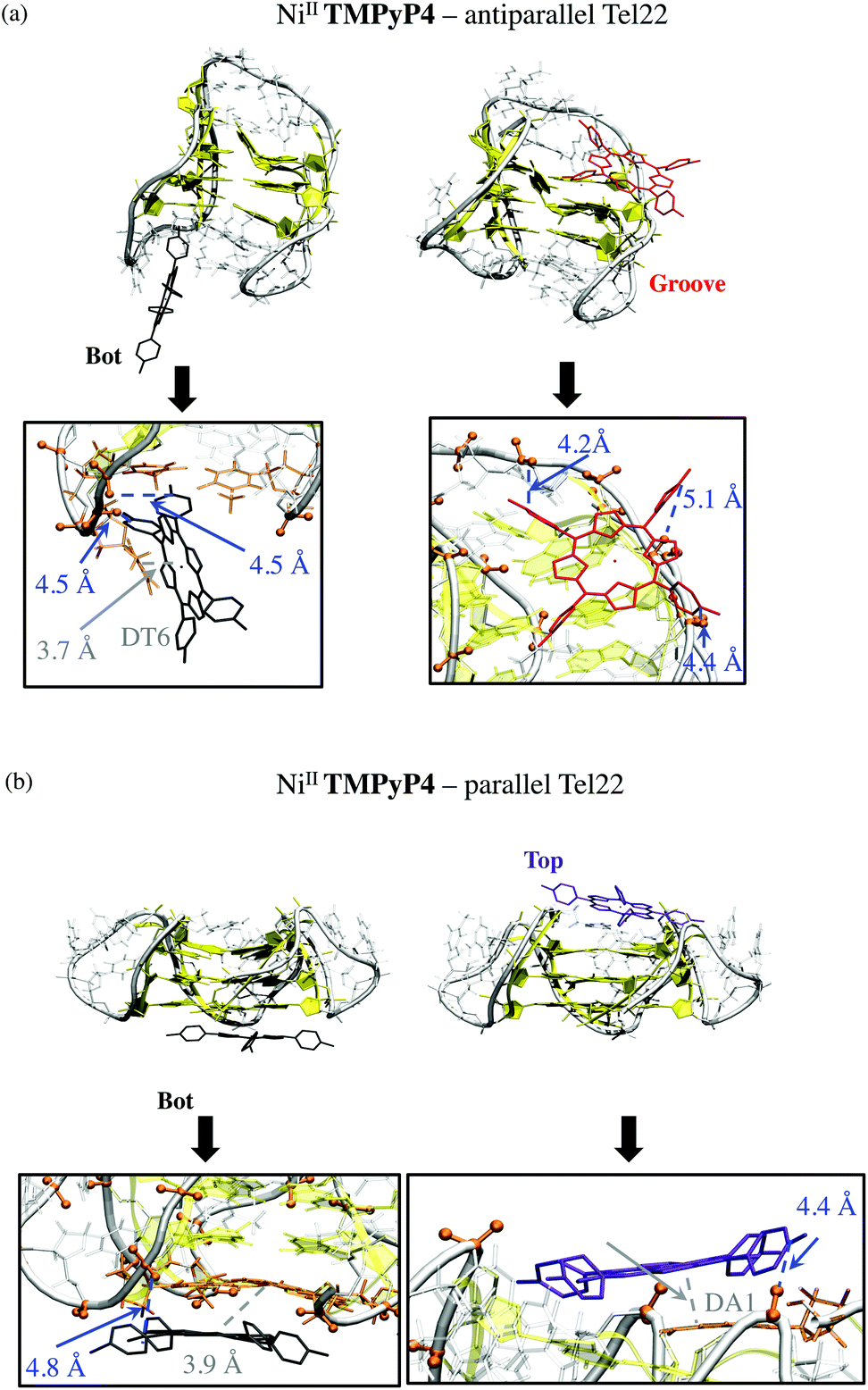
Of the five starting conformations of this porphyrin in interaction with the anti-parallel G-quadruplex, the outcome of the five starting conformations, MD runs reveal two preferential binding modes, namely Bot and Groove conformations, see Fig. 5 and S10 and Table S1 in ESI.† In the Bot conformation, the saddle shape of NiII-TImidP4 perfectly accommodates the d(TTA) loop structure (Fig. 5a). The imidazolium groups strongly interact with negatively-charged phosphate groups due to electrostatic interactions. Interestingly, the binding occurs on both facing imidazolium moieties with the two bottom d(TTA) loops. The π-stacking interactions between the G-quartet and NiII-TImidP4 are expected to be weak since the saddle shape of the porphyrin, together with the specific d(TTA) loop structure of this anti-parallel G4, covers the G-quartets. Note that different side-groove binding modes are observed (Fig. S10 in ESI†), for which NiII-TImidP4 is nested in the wide grooves of the anti-parallel G4, as for other organometallic complexes.50
 | ||
| Fig. 5 Conformations and zooms of representative snapshots at the end of MD simulations of 1:1 NiII-TImidP4 complex with (a) anti-parallel and (b) parallel G-quadruplexes. Porphyrins are coloured by binding modes (i.e., bot, top, and groove in black, violet and red/green, respectively). Relevant phosphates, residues are shown in orange. Relevant distances involving electrostatic and van der Waals interactions are shown in blue and grey, respectively. | ||
Three preferential binding modes are also obtained for NiII-TImidP4–parallel G-quadruplex complexes, namely Bot, Top and Groove, see Fig. 5b and S10.† In contrast to interactions with anti-parallel G4, Top conformations are accessible since the loops do not cover the G-quartets in this structure. The Groove binding mode is comparable to the one with anti-parallel G4, the imidazolium interacting here with dT12 (Fig. 4b). Note that MD simulations starting from hybrid parallel/anti-parallel G4 folding (PDB ID: 2HY9) yield the same preferential binding modes (Bot, Top, Groove). In this case the Bot conformation shows a π-stacking between NiII-TImidP4 and 3′-terminal adenine (Fig. S11†).
 | ||
| Fig. 6 Conformations and zooms of representative final snapshots from 100 ns MD simulations of 1:1 NiII-TMPyP4 with (a) anti-parallel and (b) parallel G-quadruplexes. Porphyrins are coloured by binding modes (i.e., bot, top, and groove in black, violet and red, respectively). Relevant phosphates groups are coloured in orange. Relevant distances involving predicted electrostatic and van der Waals interactions are shown in blue and grey, respectively. | ||
For complexes with the parallel G4 structure (Fig. 6b), the G-quartets are uncovered and solvent-accessible, which allows the planar structure of NiII-TMPyP4 to interact via π-stacking interactions. Such interactions allow both stable Bot and Top supramolecular complexes in which this porphyrin is also electrostatically anchored to phosphate groups (Fig. 6b). Note that side-groove supramolecular complexes are also observed, in which NiII-TMPyP4 is anchored to backbone phosphates through electrostatic interactions (Fig. S12†).
Binding energies in NiII-porphyrin–Tel22 complexes
To assess the preferential binding modes over the conformations obtained by MD, binding free energies (ΔΔGMM-GBSA, Table 1) for each porphyrin–G4 complex are estimated and are rationalized by calculating electrostatic and van der Waals energy contributions along MD runs, see Table 1. Although electrostatic contributions play a major role, the van der Waals interactions cannot be neglected since they significantly contribute to the total non-covalent interaction energy Enc (Enc = Eelec + EvdW). For example, in the Bot conformation of NiII-TImidP4 with anti-parallel G-quadruplex, it must be stressed that strong electrostatic contributions are in line with strong van der Waals contributions (Table 1a), because this porphyrin is anchored to G4 by both electrostatic interactions and π-stacking interactions with loop nucleobases (Fig. 5a).:1 complexes of NiII-TImidP4 (a) or NiII-TMPyP4 (b) – anti-parallel or parallel G-quadruplexes estimated from 100 ns MD simulations. Non-covalent binding energies are decomposed in electrostatic and van der Waals contributions (Eelec, EvdW, in kcal mol−1) and the relative binding free energies are computed in the MM-GBSA approach (ΔΔGMM-GBSA, kcal mol−1)
| (a) NiII-TImidP4 conformation | |||||||
|---|---|---|---|---|---|---|---|
| Anti-parallel G-quadruplex | Parallel G-quadruplex | ||||||
| E elec | E vdW | ΔΔGMM-GBSA | E elec | E vdW | ΔΔGMM-GBSA | ||
| Bot | −166.1 | −46.9 | 0.0 | Bot | −115.7 | −28.5 | +11.5 |
| Groove A | −112.7 | −32.4 | 0.0 | Top | −135.7 | −37.2 | 0.0 |
| Groove B | −87.6 | −19.6 | +9.0 | Groove | −124.8 | −24.6 | +9.4 |
| (b) NiII-TMPyP4 conformation | |||||||
|---|---|---|---|---|---|---|---|
| Anti-parallel G-quadruplex | Parallel G-quadruplex | ||||||
| E elec | E vdW | ΔΔGMM-GBSA | E elec | E vdW | ΔΔGMM-GBSA | ||
| Bot | −172.1 | −30.5 | +10.0 | Bot | −196.3 | −42.6 | 0.0 |
| Top | −124.1 | −24.1 | +13.1 | Top | −178.8 | −43.0 | +5.4 |
| Groove | −194.0 | −32.0 | 0.0 | Groove | −155.7 | −28.3 | +15.6 |
By analysing the estimates of the relative binding free energies (ΔΔGMM-GBSA), both NiII-porphyrins preferentially bind to anti-parallel G-quadruplexes in Bot or Groove conformations, respectively (ΔΔGMM-GBSA = 0 being by definition the most stable structure calculated for each type of complex), which are therefore the most likely conformations in Na+ solutions. Note that the binding free energy for Bot or Groove A conformations of NiII-TImidP4 with anti-parallel G4 is in the same range, while electrostatic and van der Waals contributions are significantly lower in Groove A. This can be rationalized by a lower destabilization of the G4 internal structure.
However, the preferential binding modes of the two studied porphyrins with parallel G4 are different. For NiII-TImidP4, the Top conformation is the most stable one, with a partial overlap of the aromatic core to the top G-quartet. The same Top preferential binding mode is obtained for conformations with the hybrid parallel/anti-parallel G4 (see Fig. S11†), which altogether indicates that this is likely the preferential binding mode for NiII-TImidP4–Tel22 in K+ solutions. In contrast, the Bot is the preferential conformation for NiII-TMPyP4. In this case, the total non-covalent interactions are the strongest, because the core of the porphyrin is fully adsorbed, π-stacked to a G-quartet, while the four pyridinium groups accommodate close to DNA phosphate groups. This is in line with experimental CD spectra, showing the highest ICD signals for this mixture at a 1:1 molar ratio, whereas almost no ICD is observed for NiII-TImidP4 under the same conditions. Note that the Bot and Top binding modes allowed an intercalative binding mode for 1:2 porphyrin–G4 complexes, in which a porphyrin was located in between two G-quartets structures, as suggested in the literature.51
Binding modes and (chir)optical properties
Both NiII-TMPyP4 and NiII-TImidP4 inherently exhibit C2v symmetry. These molecules are achiral and are CD-silent under the aqueous solution conditions used here (see Fig. S13 in ESI†). However, the electrostatic and π-stacking interactions with G-quadruplexes induce geometrical deformations that can break the inherent central symmetry of porphyrins. This is what is observed by the induced CD (ICD) signals reported here. Indeed, for specific mixtures our experiments exhibit a weak negative ICD band around 435 nm, i.e. in the Soret band of the metalloporphyrin assigned to a π–π* transition on the tetrapyrrole moiety. In view of correlating the ICD signals with binding modes, the geometrical deformations were assessed from MD simulations, and the chirality parameters were estimated, see Fig. 7. | ||
| Fig. 7 (a) Geometrical parameters assessing induced chirality of porphyrins. “Up” and “Down” annotations refer to side in contact or not with G-quadruplex, respectively. Only one example of pair angle is shown in the side view for the sake of clarity. (b) Averaged chirality parameters Δθdown, Δθup for the last 10 ns MD simulations of (a) NiII-TImidP4 with anti-parallel G4 and (b) parallel G4 and (c) NiII-TMPyP4 with anti-parallel G4 and (d) parallel G4. | ||
We focus on specific geometrical parameters of NiII-TImidP4: the angles θ1,2,3,4 formed by the Ni-atom, the N-atom of the pyrrole units and the corresponding CH-imidazolium moieties (Fig. 7a). For NiII-TMPyP4, the geometrical parameters are defined as the angles formed by the Ni-atoms, the N-atom of the pyrrole units and the centres of mass of the corresponding pyrrole C–C bonds. Δθup and Δθdown are defined as the angle differences between each facing angle (for example, θ1 − θ2 and θ3 − θ4). The “Up” annotation refers to the side in close contact with the G-quadruplex along MD runs. An ICD signal in the Soret region is observed if and only if each parameter is non-zero. The chirality parameters are shown in Fig. 7b, as estimated for the various binding modes obtained at the end of MD simulations depicted in Fig. 5 and 6.
The chirality parameters of NiII-TImidP4 metalloporphyrin upon interaction with Tel22 are higher when it interacts with the parallel or hybrid G4 (i.e. structures relevant to K+ solutions) than with the anti-parallel G4 (i.e. structures relevant to Na+ solutions), see Fig. 7b and S11.† Indeed, in the anti-parallel G4, the d(TTA) loops surround the Top and Bottom faces of the G-quartets, yielding a globular shape, which makes it easier for the curved-aromatic porphyrin NiII-TImidP4 to be nested in a wide Groove, leading to a minute conformational change in this metalloporphyrin. This is in fair agreement with the experimental CD shown in Fig. 1 and 2: the ICD signals of this metalloporphyrin are very weak in the presence of NaCl (i.e., anti-parallel conformation) and only appear at a 1:5 molar ratio.
Besides, the averaged chirality parameters Δθ for NiII-TMPyP4:Tel22 are slightly lower than for NiII-TImidP4:Tel22 (Fig. 6b), i.e. the planar tetrapyrrole is less deformed upon interaction with G4. However, the deformation of the core is not the only parameter influencing the chirality of the molecule. Indeed the rotation of the rings in the meso position can also affect the CD response. For TImidP4, the rotation is locked and only the deformation of the core is possible, while for TMPyP4 the relative orientation of the pyridinium groups can also induce chirality.
Conclusions
The recognition modes of a new cationic π-extended metalloporphyrin NiII-TImidP4 towards human telomeric G-quadruplexes have been studied. By comparing (chir)optical spectroscopy and MD simulations, we have revealed the differences in preferential binding modes with respect to a conventional meso-substituted NiII-TMPyP4. We show that NiII-TImidP4 maintains both anti-parallel and parallel G4 conformations up to 5 equivalents of Tel22. Importantly, FRET melting assays show the higher stabilization and selectivity of NiII-TImidP4 towards human telomeric G4 than with the conventional NiII-TMPyP4. These results can have significant implications in the context of G4 as therapeutic targets.Experimental
General experimental
Reagents and solvents were purchased from commercial suppliers (Aldrich, Fluka, or Merck) and were used without further purification. The oligonucleotides (ODNs) were purchased from Eurogentec (Belgium) with the highest purity grade (UltraPure Gold™, >95% pure in sequence) in a dried state, and the purity of the ODN sequences was checked with MALDI-ToF.Synthetic procedures
500), 458 sh (70300), 548 (5400), 592 (9700), 639 nm (10100 L mol−1 cm−1).
Preparation of the porphyrin–G4 mixtures
The buffer was prepared using tris(hydroxymethyl) aminomethane ((HOCH2)3CNH2), EDTA and Milli-Q water. The oligonucleotides were dissolved in a volume of TE buffer (pH 7.4, 10 mM Tris buffer and 1 mM EDTA) at a concentration of 100 μM. All ODN solutions obtained were centrifuged for 2 minutes at 2000 rpm. Small volumes of this solution were used in order to prepare different aliquots, to which were added TE buffer, or TE buffer + 3 M KCl or + 3 M NaCl in order to obtain a final volume of 300 μL and solutions in pure TE buffer or TE buffer + 100 mM K+ ions or TE buffer + 100 mM Na+ ions, respectively. The final solution was mixed using a vortex. The concentration of the aliquot of DNA in the buffer solution was determined by UV-Vis at 25 °C using the specific extinction coefficients at 260 nm (ε260) of each DNA, which are 228500 L mol−1 cm−1 and 306900 L mol−1 cm−1, for Tel22 (d[AG3(T2AG3)3]) and Tel30 (d[GT2A(GGGTTA)4GG]), respectively. The NiII-porphyrin samples were also dissolved in TE buffer (pH 7.4, 10 mM Tris buffer and 1 mM EDTA) or in TE buffer + 100 mM KCl or NaCl and the molar ratio between metalloporphyrins and DNA was adjusted using the calculated molar concentrations of DNA (around 3.5 μM). The molar extinction coefficient (ε) for NiII-TMPyP4 at 418 nm is 149000 L mol−1 cm−1 and for NiII-TImidP4 is 134500 L mol−1 cm−1 at 436 nm. The solution of metalloporphyrin was added to the DNA solution and was stirred using a vortex at vigorous speed for 2 min and allowed to equilibrate for 30 min.
UV-Vis absorption and circular dichroism spectroscopy
The UV-Vis absorption and circular dichroism (CD) measurements were recorded using a Chirascan™ Plus CD Spectrometer from Applied Photophysics. The measurements were carried out using 1 mm suprasil quartz cells from Hellma Analytics. The spectra were recorded between 200 and 650 nm, with a bandwidth of 1 nm, time per point 1 s and 2 repetitions. The buffered water solvent reference spectra were recorded as baselines and were automatically subtracted from the CD spectra of the samples.FRET melting assays
Fluorescence resonance energy transfer (FRET) melting assays were performed according to Mergny et al.45 using a synthetic double-dye labelled oligonucleotide F21 T 5′-FAM-GGG(T2AG3)3-TAMRA-3′ (from Eurogentec, Belgium). The solutions were prepared at a concentration of around 200 nM (base concentration) in 10 mM lithium cacodylate buffer (pH = 7.2) in the presence of 10 mM KCl + 90 mM LiCl (K+ medium). The oligonucleotide was first heated to 90 °C for 3 min under the corresponding buffer conditions and then put on ice to support the formation of G4 secondary structure. The metalloporphyrin was then added at a concentration of ∼1 μM, and the mixture was equilibrated at 25 °C for 5 minutes. The FRET spectra were recorded using a Chirascan™ Plus instrument equipped for fluorescence measurements. The samples were excited at 492 nm and the fluorescence emission spectra were recorded between 500 and 700 nm. The temperature was varied from 25 °C to 96 °C at a rate of 1 °C min−1. The melting of the F21 T was monitored by measuring the fluorescence of FAM (at 516 nm), as described in ref. 45. The FAM emission intensity was normalized and ΔT1/2 was defined as the temperature for which the normalized emission equals 0.5. For the selectivity studies, a solution of 15 equivalents of a dsDNA competitor (∼3 μM) was added into the F21 T/metalloporphyrin solution and the final solution was equilibrated at 25 °C for 5 minutes. The dsDNA competitor is 5′-CGTCACGTAAATCGGTTAAC-3′ hybridized with its complementary sequence.Molecular modeling simulations
:1 metalloporphyrin–G-quadruplexes MD simulations. Five different binding modes were used as initial structures for 1:1 metalloporphyrin–anti-parallel/parallel G-quadruplex structures (Fig. 3). The same protocol was used to equilibrate 1:1 complex structures (i.e., minimization, heating, and density equilibration). 100 ns MD simulations were then achieved in the (N,V,T) ensemble.
:1 complex.
Acknowledgements
This work was supported by the Fonds de la Recherche Scientifique-FNRS (Belgium) under the grant no. 2.4615.11-BINDER. J. R.-M. is a FNRS post-doctoral researcher and M.S. is a FNRS research associate. M. L. acknowledges SeRC (Swedish e-Science Research Center) for funding and FNRS for visiting professor position. P. N. and F. D. M. acknowledge financial support from the Swedish Research Council (grant no. 621-2010-5014). The authors acknowledge SNIC (Swedish National Infrastructure for Computing) for providing computer resources. S. R., S. C. and M. L. are grateful to the CNRS and the Agence Nationale de la Recherche (ANR) for financial support (research project ANR-09-JCJC-0089-01).References
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- O. Doluca, J. M. Withers and V. V. Filichev, Chem. Rev., 2013, 113, 3044–3083 CrossRef CAS PubMed.
- M. L. Bochman, K. Paeschke and V. A. Zakian, Nat. Rev. Genet., 2012, 13, 770–780 CrossRef CAS PubMed.
- Guanine Quartets: Structure and Applications, ed. W. Fritzsche and L. Spindler, Royal Society of Chemistry, 2013 Search PubMed.
- S. Rankin, A. P. Reszka, J. Huppert, M. Zloh, G. N. Parkinson, A. K. Todd, S. Ladame, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2005, 127, 10584–10589 CrossRef CAS PubMed.
- N. Maizels and L. T. Gray, PLoS Genet., 2013, 9, e1003468 CAS.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- B. Maji and S. Bhattacharya, Chem. Commun., 2014, 50, 6422–6438 RSC.
- Y. Wu and R. M. Brosh Jr., FEBS J., 2010, 277, 3470–3488 CrossRef CAS PubMed.
- D. Monchaud and M. P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 CAS.
- S. Balasubramanian and S. Neidle, Curr. Opin. Chem. Biol., 2009, 13, 345–353 CrossRef CAS PubMed.
- W. O. Tucker, K. T. Shum and J. A. Tanner, Curr. Pharm. Des., 2012, 18, 2014–2026 CrossRef CAS.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 RSC.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- A. De Cian, E. DeLemos, J.-L. Mergny, M.-P. Teulade-Fichou and D. Monchaud, J. Am. Chem. Soc., 2007, 129, 1856–1857 CrossRef CAS PubMed.
- D. Monchaud, A. Granzhan, N. Saettel, A. Guédin, J. L. Mergny and M. P. Teulade-Fichou, J. Nucleic Acids, 2010, 2010, 525862 Search PubMed.
- L. Wang, H. Wen, J. Liu, J. Zhou, C. Li and C. Wei, Org. Biomol. Chem., 2011, 9, 2648–2653 CAS.
- J. K. Choi, G. Sargsyan, M. Shabbir-Hussain, A. E. Holmes and M. Balaz, J. Phys. Chem. B, 2011, 115, 10182–10188 CrossRef CAS PubMed.
- M. Balaz, M. De Napoli, A. E. Holmes, A. Mammana, K. Nakanishi, N. Berova and R. Purrello, Angew. Chem., Int. Ed., 2005, 44, 4006–4009 CrossRef CAS PubMed.
- M. Balaz, K. Bitsch-Jensen, A. Mammana, G. A. Ellestad, K. Nakanishi and N. Berova, Pure Appl. Chem., 2007, 79, 801–809 CrossRef CAS.
- S. Ishihara, J. Labuta, W. Van Rossom, D. Ishikawa, K. Minami, J. P. Hill and K. Ariga, Phys. Chem. Chem. Phys., 2014, 16, 9713–9746 RSC.
- G. Sargsyan, B. M. Leonard, J. Kubelka and M. Balaz, Chem. – Eur. J., 2014, 20, 1878–1892 CrossRef CAS PubMed.
- R. F. Pasternack, Chirality, 2003, 15, 329–332 CrossRef CAS PubMed.
- D. R. McMillin, A. H. Shelton, S. A. Bejune, P. E. Fanwick and R. K. Wall, Coord. Chem. Rev., 2005, 249, 1451–1459 CrossRef CAS PubMed.
- J. K. Choi, A. D'Urso and M. Balaz, J. Inorg. Biochem., 2013, 127, 1–6 CrossRef CAS PubMed.
- Y. Ikawa, S. Touden and H. Furuta, Org. Biomol. Chem., 2011, 9, 8068–8078 CAS.
- C. Romera, O. Bombarde, R. Bonnet, D. Gomez, P. Dumy, P. Calsou, J. F. Gwan, J. H. Lin, E. Defrancq and G. Pratviel, Biochimie, 2011, 93, 1310–1317 CrossRef CAS PubMed.
- T.-m. Ou, Y.-j. Lu, J.-h. Tan, Z.-s. Huang, K.-Y. Wong and L.-q. Gu, ChemMedChem, 2008, 3, 690–713 CrossRef CAS PubMed.
- Q. Li, J. F. Xiang, H. Zhang and Y. L. Tang, Curr. Pharm. Des., 2012, 18, 1973–1983 CrossRef CAS.
- D. P. N. Goncalves, S. Ladame, S. Balasubramanian and J. K. M. Sanders, Org. Biomol. Chem., 2006, 4, 3337–3342 CAS.
- M. Lo, J.-F. Lefebvre, D. Leclercq, A. van der Lee and S. Richeter, Org. Lett., 2011, 13, 3110–3113 CrossRef CAS PubMed.
- M. Lo, J.-F. Lefebvre, N. Marcotte, C. Tonnelé, D. Beljonne, R. Lazzaroni, S. Clément and S. Richeter, Chem. Commun., 2012, 48, 3460–3462 RSC.
- J. Pan and S. Zhang, JBIC, J. Biol. Inorg. Chem., 2009, 14, 401–407 CrossRef CAS PubMed.
- R. F. Pasternack, E. J. Gibbs and J. J. Villafranca, Biochemistry, 1983, 22, 5409–5417 CrossRef CAS.
- J. Dai, M. Carver and D. Yang, Biochimie, 2008, 90, 1172–1183 CrossRef CAS PubMed.
- K. W. Lim, S. Amrane, S. Bouaziz, W. Xu, Y. Mu, D. J. Patel, K. N. Luu and A. T. Phan, J. Am. Chem. Soc., 2009, 131, 4301–4309 CrossRef CAS PubMed.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef CAS.
- A. T. Phan, FEBS J., 2010, 277, 1107–1117 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef CAS PubMed.
- M. Bončina, J. Lah, I. Prislan and G. Vesnaver, J. Am. Chem. Soc., 2012, 134, 9657–9663 CrossRef PubMed.
- J. Dai, C. Punchihewa, A. Ambrus, D. Chen, R. A. Jones and D. Yang, Nucleic Acids Res., 2007, 35, 2440–2450 CrossRef CAS PubMed.
- M. J. Morris, K. L. Wingate, J. Silwal, T. C. Leeper and S. Basu, Nucleic Acids Res., 2012, 40, 4137–4145 CrossRef CAS PubMed.
- M. Wieland and J. S. Hartig, Angew. Chem., Int. Ed., 2006, 45, 5875–5878 CrossRef CAS PubMed.
- P. Zhao, J. Z. Lu, F. Y. Hong, B. H. Ou, F. D. Zhang, L. N. Ma and H. M. Guo, Spectrochim. Acta, Part A, 2013, 108, 1–7 CrossRef CAS PubMed.
- A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M.-P. Teulade-Fichou, L. Lacroix and J.-L. Mergny, Methods, 2007, 42, 183–195 CrossRef CAS PubMed.
- D. Renčiuk, J. Zhou, L. Beaurepaire, A. Guédin, A. Bourdoncle and J.-L. Mergny, Methods, 2012, 57, 122–128 CrossRef PubMed.
- D. Monchaud, C. Allain, H. Bertrand, N. Smargiasso, F. Rosu, V. Gabelica, A. De Cian, J. L. Mergny and M. P. Teulade-Fichou, Biochimie, 2008, 90, 1207–1223 CrossRef CAS PubMed.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS PubMed.
- C. J. Medforth, M. O. Senge, K. M. Smith, L. D. Sparks and J. A. Shelnutt, J. Am. Chem. Soc., 1992, 114, 9859–9869 CrossRef CAS.
- S. Rickling, L. Ghisdavu, F. Pierard, P. Gerbaux, M. Surin, P. Murat, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Chem. – Eur. J., 2010, 16, 3951–3961 CrossRef CAS PubMed.
- X.-X. Huang, L.-N. Zhu, B. Wu, Y.-F. Huo, N.-N. Duan and D.-M. Kong, Nucleic Acids Res., 2014, 42, 8719–8731 CrossRef CAS PubMed.
- K. M. Kadish, D. Sazou, Y. M. Liu, A. Saoiabi, M. Ferhat and R. Guilard, Inorg. Chem., 1988, 27, 1198–1204 CrossRef CAS.
- M. C. Gomes, S. M. Woranovicz-Barreira, M. A. F. Faustino, R. Fernandes, M. G. P. M. S. Neves, A. C. Tome, N. C. M. Gomes, A. Almeida, J. A. S. Cavaleiro, A. Cunha and J. P. C. Tome, Photochem. Photobiol. Sci., 2011, 10, 1735–1743 CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Revision D.01, Wallingford, CT, 2009 Search PubMed.
- M. B. Peters, Y. Yang, B. Wang, L. s. Füsti-Molnár, M. N. Weaver and K. M. Merz, J. Chem. Theory Comput., 2010, 6, 2935–2947 CrossRef CAS PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- A. Onufriev, D. Bashford and D. A. Case, J. Phys. Chem. B, 2000, 104, 3712–3720 CrossRef CAS.
- A. Onufriev, D. Bashford and D. A. Case, Proteins: Struct., Funct., Bioinf., 2004, 55, 383–394 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2010, 51, 69–82 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization of the compounds, supplementary UV-Vis and CD spectra, and computational data are available. See DOI: 10.1039/c4ob02097a |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |