
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomineralization-inspired synthesis of functional organic/inorganic hybrid materials: organic molecular control of self-organization of hybrids
Atsushi
Arakaki
*a,
Katsuhiko
Shimizu
b,
Mayumi
Oda
a,
Takeshi
Sakamoto
c,
Tatsuya
Nishimura
c and
Takashi
Kato
*c
aDivision of Biotechnology and Life Science, Institute of Engineering, Tokyo University of Agriculture and Technology, 2-24-16, Naka-cho, Koganei, Tokyo 184-8588, Japan. E-mail: arakakia@cc.tuat.ac.jp
bOrganization for Regional Industrial Academic Cooperation, Tottori University, 4-101, Minami, Koyama-cho, Tottori 680-8550, Japan
cDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kato@chiral.t.u-tokyo.ac.jp
First published on 14th October 2014
Abstract
Organisms produce various organic/inorganic hybrid materials, which are called biominerals. They form through the self-organization of organic molecules and inorganic elements under ambient conditions. Biominerals often have highly organized and hierarchical structures from nanometer to macroscopic length scales, resulting in their remarkable physical and chemical properties that cannot be obtained by simple accumulation of their organic and inorganic constituents. These observations motivate us to create novel functional materials exhibiting properties superior to conventional materials—both synthetic and natural. Herein, we introduce recent progress in understanding biomineralization processes at the molecular level and the development of organic/inorganic hybrid materials by these processes. We specifically outline fundamental molecular studies on silica, iron oxide, and calcium carbonate biomineralization and describe material synthesis based on these mechanisms. These approaches allow us to design a variety of advanced hybrid materials with desired morphologies, sizes, compositions, and structures through environmentally friendly synthetic routes using functions of organic molecules.
 Atsushi Arakaki | Atsushi Arakaki received his PhD from the Tokyo University of Agriculture and Technology in 2003 under the supervision of Prof. Tadashi Matsunaga. After his postdoctoral research at Waseda University, Japan, he joined the Tokyo University of Agriculture and Technology and became an associate professor in 2011. His current research focuses on understanding the mechanism controlling the magnetite morphology in magnetotactic bacteria and utilization of this mechanism for developing functional magnetic materials. He is a principal investigator of the “Fusion Materials” project. |
 Katsuhiko Shimizu | Katsuhiko Shimizu received his PhD in Biological Science at Tokyo Metropolitan University in 1992. He conducted his postdoctoral research at several institutions, including the Fusetani Biofouling Project, Exploratory Research for Advanced Technology (ERATO), Research Development Corporation of Japan (JRDC), and the University of California Santa Barbara under the guidance of Prof. Daniel E. Morse. He joined Tottori University, Japan, as an associate professor in 2010. His current research focuses on understanding the molecular mechanisms controlling silicon biomineralization in sponges for the development of novel, environmentally benign routes for manufacturing silicon-based inorganic–organic composite materials. He is a principal investigator of the “Fusion Materials” project. |
 Takashi Kato | Takashi Kato received his PhD from the University of Tokyo in 1988. After doing postdoctoral research at Cornell University under the guidance of Prof. Jean M. J. Fréchet, he joined the University of Tokyo where he is currently a professor. His research highlights the development of self-assembled materials, including supramolecular materials, liquid crystals, gels, ion-, electro-, and photo-functional materials, functional polymers, and bio-inspired organic/inorganic hybrids. He has published about 400 papers including original manuscripts, reviews, and chapters of books. He is an editorial and advisory board member of several scientific journals including RSC journals: Chem. Sci., J. Mater. Chem. C and New J. Chem. He is the project leader of a project of Grant-in-Aid for Scientific Research (KAKENHI) on Innovative Areas funded by the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT): “Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control”. |
Introduction
Biominerals are organic/inorganic hybrid materials that are essential components of living organisms and support important functions. Typical examples are hydroxyapatite (Ca10(PO4)6(OH)2) in bones and teeth of mammals,1 calcium carbonate (CaCO3) in molluscan shells,2 amorphous silica (SiO2) in diatoms3 and marine sponges,4 and magnetite (Fe3O4) in chiton teeth.5 These biominerals have elaborate hierarchical structures giving them conspicuously high mechanical hardness and flexibility, which are not provided by conventional synthetic materials.6,7 Single calcite crystals found in the skeletal construction of brittle stars are a component of specialized photosensory organs, possibly functioning as a compound eye.8 Magnetite nanocrystals in some organisms such as birds and bacteria function as biological geomagnetic sensors that help them find and survive in their habitats on the earth.9 The optical and mechanical properties of biominerals are derived from their complex structures consisting of organic and inorganic constituents.7,10,11 Most biominerals are formed using abundant elements in earth's crust. Moreover, they are synthesized under mild conditions at a near-neutral pH and ambient temperature. Owing to these interesting features, the formation processes of biominerals have attracted much attention with the aim of understanding the mechanisms and applying them to material synthesis in industry.Over the last three decades, a number of proteins that control biomineralization processes, including promotion of crystal formation,3,4,12 matrix-assisted orientation of crystals,13,14 growth inhibition by face-selective surface adsorption,15,16 and control of the crystal phase,16,17 have been isolated and analyzed (Table 1). Although it has been believed that proteins play important roles in biomineralization processes, most studies are still limited to fundamental analyses of proteins, such as amino acid sequence comparisons and biochemical characterization. The functions of proteins at the molecular level have remained largely unknown so far. A significant breakthrough in material synthesis using a biomineralization protein was achieved by a study involving sponges.18 The protein—known as silicatein—was isolated from silica spicules that form the skeletal structures of marine sponges.4 Surprisingly, this protein shows a direct catalytic function of silica biomineralization and it allows us to synthesize new materials with a non-natural composition and a wide spectrum of properties.19,20 After the identification of silicatein, many proteins were isolated from various biominerals in diverse organisms, which encouraged researchers to use them for material synthesis.3,14,17 Silaffins isolated from diatoms can induce and regulate silica precipitation.3 A small iron-binding protein, Mms6, isolated from magnetotactic bacteria regulates the surface structure of magnetite nanoparticles.17 A calcium-binding protein, Pif, regulates the nacre formation in the pearl oyster Pinctada fucata.14 These proteins, isolated from different biominerals, opened the door for bio-inspired synthesis of functional organic/inorganic hybrid materials with controlled micro- to nanoscale properties. On the basis of these fundamental molecular studies, the functional regions of organic molecules have been adopted for biomimetic nanofabrication processes and the development of bio-inspired materials.21–25
| Protein | Biomineral | Function | References |
|---|---|---|---|
| Calcium carbonate | |||
| Perlucin | The nacreous layer of the shell | Calcite precipitation | 12 |
| MSI31, 60 | The nacreous layer of the shell | Framework of the prismatic layer | 13 |
| Pif | The nacreous layer of the shell | Aragonite crystal formation | 14 |
| Ansocalcin | Goose egg shell matrix | Template for calcite nucleation | 15 |
| CAP-1 | The exoskeleton of crayfish | Crystal growth regulation | 16 |
| Silica | |||
| Silicatein | Sponge spicules | Silica polymerization | 4 |
| Silaffin | Diatom shells | Silica precipitation | 3 |
| Iron oxide | |||
| Mms6 | Bacterial magnetites | Crystal size and shape control of magnetites | 17 |
In this review, we introduce the recent advances in the fundamental molecular analyses of biomineralization in several biological systems. Here, we focus on the studies on silica, iron oxide, and calcium carbonate (Fig. 1). We also emphasize the application of the proteins associated with biomineralization toward the controlled synthesis of organic/inorganic functional materials.
 | ||
| Fig. 1 Schematic illustration of molecularly controlled processes of biomineralization. | ||
Silica biomineralization and application
Learning the essence of silica biomineralization in sponges
Silicon (Si) is the second most abundant element in the earth's crust, existing naturally as silicon dioxide or silicates. This common element is incorporated into a wide range of living organisms, including diatoms, sponges, and higher plants, which produce structures composed of amorphous hydrated silica.26 Biological siliceous structures (biosilica) are synthesized by biological processes and possess exquisite species-specific morphology with sizes ranging from the nanometer to, occasionally, the meter scale.26 Organic molecules, whose expression is under the control of genetic information, determine the uniqueness of biosilica. Scientists have taken advantage of recent advances in molecular biology and analytical technology to explore the organic molecules involved in biosilicification.3,4,27–29 Understanding its mechanism will lead to the development of new routes to synthesize functional materials under environmentally benign conditions.Biosilica occurs in sponges in the form of structures called spicules that are needle shaped, with dimensions ranging from micrometer to millimeter scales. However, some exceptions also exist; e.g., in the case of the hexactinellid sponge Monorhaphis chuni, the basal spicules can be as long as 3 m (diameter, 1 cm) and are the largest known biosilica structures on the earth (Fig. 2).30,31 Spicules of the Venus’ flower basket sponge Euplectella aspergillum are exquisitely weaved to construct skeletons that are hierarchically arranged with nano- to meter-scale structures. In sponges, the biosilica shows remarkable mechanical toughness and optical properties10 and provides support for the body, protection against predators, and light guides.
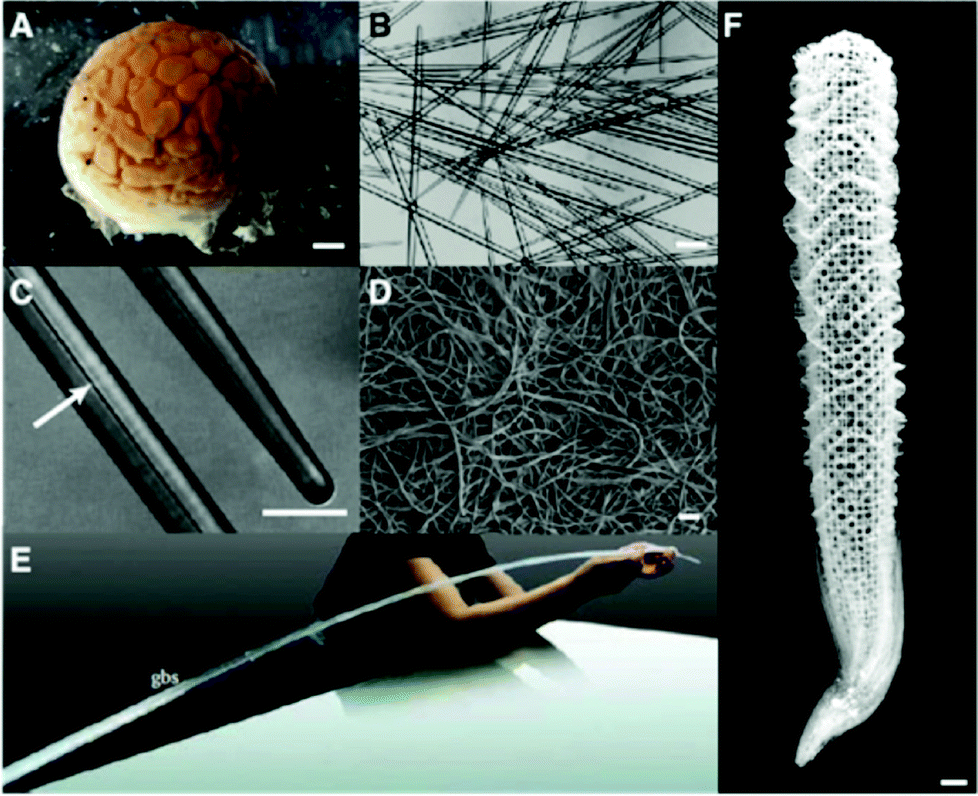 | ||
| Fig. 2 Sponge biosilica. (A) Demosponge Tethya aurantia. (B) Spicules of T. aurantia. (C) Enlarged image of (B) with an arrow indicating an axial filament that is composed of silicateins. (D) Axial filaments isolated by dissolving silica spicules. (B–D) Reproduced with permission from ref. 30. Copyright Wiley 2003 Wiley Periodicals Inc. (E) Giant basal spicules (gbs) from Monorhaphis chuni, with a length of 270 cm and a diameter of 10 mm. Reproduced from ref. 31. (F) Silica skeleton of the Venus flower basket sponge Euplectella aspergillum. | ||
Each silica spicule contains a proteinous central core along the long axis, called the axial filament.4 The protein filaments were isolated from the demosponge Tethya aurantia by dissolving the surrounding silica with HF and dissociated to yield three similar proteins: silicatein α, β, and γ (Table 1).4 Their molecular weights (27–29 kDa) and amino acid compositions are similar, suggesting that they are members of a single protein family. The amino acid sequence of silicatein α reveals that the protein is similar to cathepsin L and members of the papain-like cysteine protease family. The cysteine protease is named after the cysteine (Cys) at the active site; however, the amino acid at the corresponding site in silicateins is occupied by serine (Ser) (Fig. 3A). The Cys residues that form intramolecular disulfide bridges in the proteases are conserved, suggesting that the tertiary conformations of these proteins are also similar.18
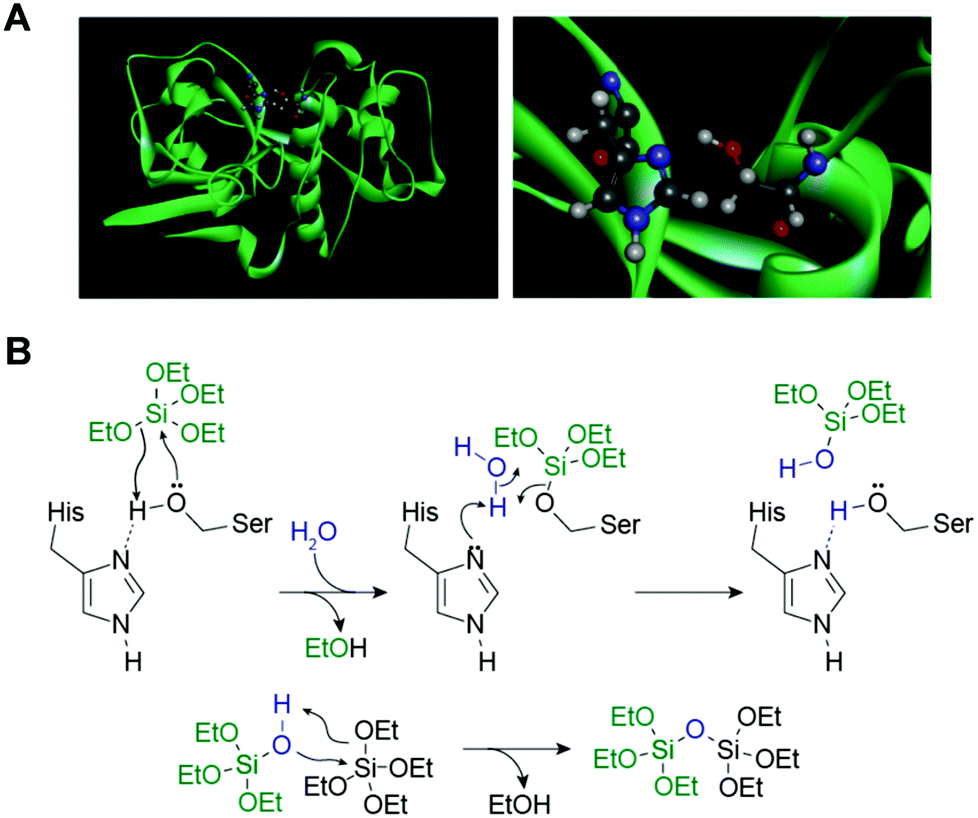 | ||
| Fig. 3 (A) Ribbon model of silicatein α (in green) (left) and enlarged serine-hydrolase catalytic site (right). At the site, a nucleophilic serine residue is presented for hydrogen-bonding with imidazole that enhances the hydrolytic activity of the enzyme.20 (B) Proposed reaction mechanism of hydrolysis of tetraethoxysilane and polycondensation of silica, as catalyzed by silicatein α. Proposed mechanism is based on a well-characterized mechanism of catalytic hydrolysis of peptide bonds by serine proteases.18 | ||
Since the first identification in T. aurantia, silicateins have been isolated from a wide variety of demosponge species.32–35 The demosponges Geodia cydonium and Ephydatia fluviatilis possess two morphologically different spicules.34,35 The characteristic morphology of spicules is considered to be due to the specific properties and combinatory functions of silicatein isoforms. Silicatein has also been identified in the hexactinellid sponges, Monorhaphis chuni,36Crateromorpha meyeri,37 and Aulosaccus schulzei, suggesting that silicatein is a common protein in sponges.38
Silicateins probably play two roles in sponge spicule formation. First, a catalytic role in sponge silica biomineralization is suggested. Cha et al.18 demonstrated that the axial filaments isolated from T. aurantia catalyzed the hydrolysis of various silicon alkoxides such as tetraethoxysilane (TEOS) and methyl- and phenyl-triethoxysilanes to yield silica and the corresponding silsesquioxanes in vitro at ambient temperature, pressure, and neutral pH. This activity was retained in silicatein monomers dissociated from the axial filaments and also reproduced with a recombinant form synthesized in genetically engineered Escherichia coli.39 The mutant silicateins, constructed by replacing the putative active site Ser (with a hydroxyl side-chain) at position 26 and/or His (with an imidizole side-chain) at position 165 of silcatein α with alanine (with a methyl side-chain), significantly decreased silica formation from the alkoxide substrate.39 Based on the mechanism for catalytic hydrolysis of the peptide bonds by the serine proteases (e.g., chymotrypsin, trypsin), in conjunction with the results of the site-directed mutagenesis experiments, the hydrolysis of silicon alkoxides and polymerization of silicic acids by silicatein are explained as follows (Fig. 3B).18 The Ser 26–His 165 side-chain couple in silcatein α to hydrolyze the silicon alkoxides acts as the active-site Ser–His pair for the serine proteases to catalyze peptide bond hydrolysis.20
The second role of silicateins is the templating and construction of the axial filaments within the structure. Self-assembly of the silicatein to form a filamentous structure appears to be mediated by the interaction of hydrophobic patches on the surfaces of the silicatein isoforms.40 The filaments guide the deposition of the polycondensed silica product along the entire filament length.18 This activity is abolished by treatment with heat or a detergent (sodium dodecylsulfate).18
Application of silicateins and sponge-biosilicification
Since the dual functions of silicatein were characterized, it has been widely used directly, as well as in a biologically motivated manner, for the synthesis of silica and other industrially important inorganic materials under mild conditions.41–44The discovery that silicatein filaments prepared from T. aurantia can biocatalytically hydrolyze silicon alkoxides and template the products led us to apply this technology to the synthesis and templating of other metal(loid) oxide materials. For example, nanocrystalline anatase TiO2 were prepared from titanium(IV) bis(ammonium lactato)dihydroxide (Ti(BALDH)), a water-stable alkoxide-like conjugate of titanium.45 Oriented aggregates of Ga2O3 nanocrystals were obtained from Ga(NO3)3,46 and nanostructured BaTiOF4,19 which is an intermediate for production of BaTiO3, was formed from BaTiF6 under ambient conditions. Silicateins are arranged to form a perfect lattice which templates a 3D mesoporous silica structure in a Monorhaphis chuni giant spicule.47 This capability was used to synthesize cristobalite and remarkable flexible rods of aligned calcite nanocrystals.48,49
Production of recombinant silicateins by genetic engineering broadens opportunities for application of these catalysts for manufacturing inorganic materials through environmentally benign routes. Recombinant silicatein catalyzes condensation of alkoxysilanes at neutral pH and ambient temperature to yield silica as well as silicones such as straight-chained poly(dimethylsiloxane) (PDMS).50 The formation of uniform silica films with controlled thickness, roughness, and hydrophilicity was achieved using immobilized silicateins on gold-coated surfaces, polystyrene, and silicon wafers.51 Recombinant silicateins were immobilized on silicon substrates by a microcontact printing-based physisorption method. Incubation of the immobilized recombinant silicatein with TEOS produced biosilica microstructures and/or silica films by simply varying the reaction time in precursor solution.52 Electrical insulation properties were demonstrated for potential device applications. Recombinant silicateins can be modified with the fusion of additional peptide sequences, including polyHis (His-tag), bearing a strong affinity to Ni2+ or Co2+ chelated with nitrilotriacetic acid (NTA), and polyglutamate (Glu-tag), with the affinity to hydroxyapatite, making them remarkably useful tools for immobilization. Tahir et al.53,54 showed that the recombinant silicatein with His-tag bound to the gold surface with Ni-NTA retained its biocatalytic activity to produce layered arrangements of titania and zirconia from Ti(BALDH) and hexafluorozirconate, respectively, in addition to silica from TEOS. Titania coating from Ti(BALDH) on the surface of the WS2 nanotubes was also mediated by the immobilized silicatein (Fig. 4). To prepare silicatein-immobilized WS2 nanotubes, the nanotubes were covered with the polymers carrying NTA side-chains, and then a His-tagged silicatein was bound to the surface of the polymer-coated WS2 nanotubes through NTA.55 Nanostructured cassiterite SnO2 was synthesized from sodium hexafluorostannate by the catalytic activity of His-tagged silicatein immobilized on glass surfaces using an NTA anchor.56 The immobilized His-tagged silicatein on magnetite nanoparticles also retains its native hydrolytic activity to catalyze the formation of silica by copolymerization of TEOS.57 His-tagged silicatein acts as a nanoreactor to synthesize and immobilize gold nanoparticles obtained from auric acid onto the core–shell polymer colloids, although the mechanism of gold synthesis in the presence of silicatein is not uncovered.58
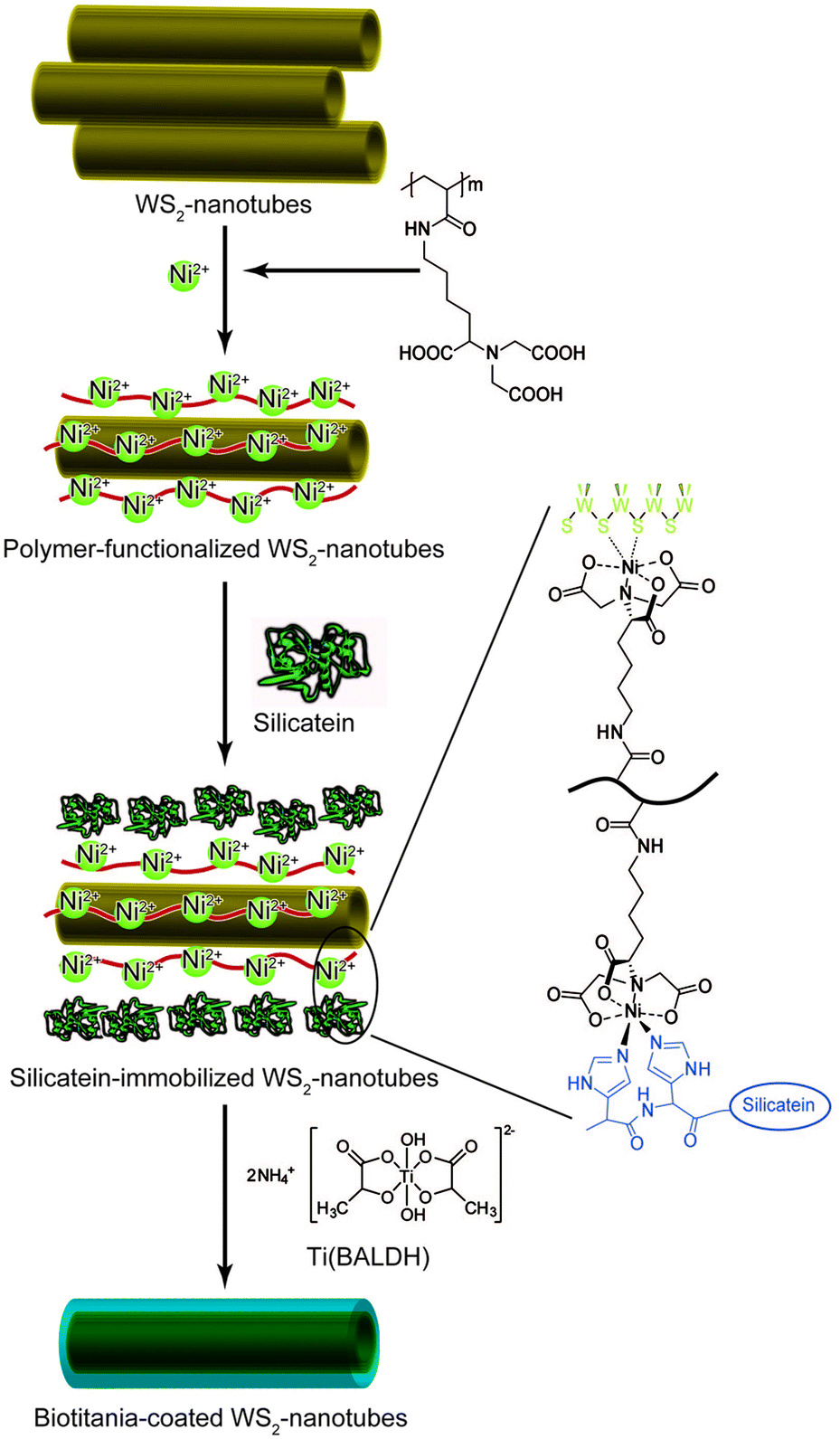 | ||
| Fig. 4 Schematic illustration for preparation of biotitania-coated inorganic nanotubes using silicatein.55 | ||
The development and availability of recombinant silicatein opens new possibilities for biomaterial and medical applications. Tissue culture plates modified with silicatein-mediated deposition of silica enhanced the activity of bone-mineralizing cells.59 Glu-tag-immobilized recombinant silicateins catalyzed the synthesis of biosilica coatings from sodium metasilicate on both synthetic hydroxyapatite nanofibrils and dental hydroxyapatite.60 This technology indicates that Glu-tagged silicateins have considerable biomedical potential with regenerative and prophylactic implementations.
Based on the essential participation of specific Ser and His residues in silicatein's catalytic active site, a synthetic mimic that provides both catalysis and the surface determinants necessary to structurally direct heterogeneous nucleation by condensation was developed. Kisailus et al.61 prepared gold nanoparticles separately functionalized with hydroxy (nucleophilic) and imidazole (hydrogen bonding) groups. The catalytic activity of silicatein was mimicked by a mixture of two gold nanoparticle types. When the two gold nanoparticles were mixed with TEOS at room temperature and neutral pH, silica production was observed. Subsequently, using lithographically patterned PDMS stamps, bifunctional self-assembled monolayer surfaces were fabricated with hydroxy- and imidazole-functionalized alkane thiols on gold substrates.20 The interface between chemically distinct self-assembled monolayer domains provided the necessary juxtaposition of nucleophilic (hydroxyl) and hydrogen-bonding (imidazole) agents to catalyze the hydrolysis of a Ga2O3 precursor and template the condensed product to form GaOOH and the defect spinel γ-Ga2O3 (Fig. 5). Using this approach, the production of patterned substrates for catalytic synthesis and templating of semiconductors for device applications can be envisioned. Silicateins inspired Adamson et al.62 to synthesize a catalytically active block copolymer poly(2-vinylpyridine-b-1,2-butadiene) functionalized by the introduction of hydroxyl groups at butadiene, which showed the catalytic action of the polymer on TEOS under neutral pH and ambient temperature conditions. Lee et al.63 demonstrated that the dipeptide His–Ser, mimicking the catalytic structure of silicatein, was used as an additive in the Ga(NO3)3 solution to form spindle-shaped GaOOH crystals, while only irregularly shaped aggregates of amorphous Ga(OH)3 were obtained in the absence of the dipeptide. This study suggests that a designed peptide with an active functionality can be further exploited to produce inorganic compounds with controlled nucleation and growth.
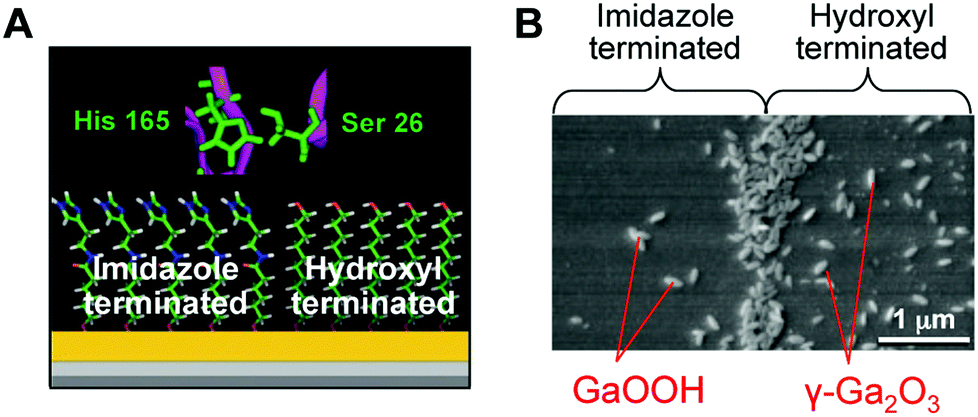 | ||
| Fig. 5 (A) Schematic representation of self-assembled monolayer domains providing the necessary juxtaposition of hydroxyl and imidazole moieties to catalyze the hydrolysis of the gallium oxide precursor and template the condensed product to form GaOOH and γ-Ga2O3. (B) SEM image of GaOOH and γ-Ga2O3 formed on a self-assembled monolayer surface. Reproduced with permission from ref. 20. Copyright 2005 National Academy of Sciences. | ||
Iron oxide biomineralization and applications
Learning the essence of magnetite biomineralization in bacteria
Magnetic particles are currently important materials in bio-industries.64–66 They are widely used in medical and diagnostic applications such as magnetic resonance imaging, cell separation, environmental inspections, drug delivery, and hyperthermia.64,67 The major advantage of magnetic particles in such applications is that they can be easily manipulated by a magnetic force. Magnetic particles enable rapid and easy separation of target molecules bound to the magnetic particles from reaction mixtures. This technology is beneficial for use in fully automated systems, providing minimal manual labor and superior reproducibility.64The presence of magnetic iron oxide in an organism was first discovered in the teeth of chitons, which are marine invertebrates.5 Their teeth are coated with magnetite and are used to scrape microalgae off the substratum. After this finding, iron oxide minerals were further identified as small crystals in a wide range of organisms, including honey bees, fish, and birds.9 In these organisms, the function of the iron oxide crystals is still under investigation, but they are suggested to function as biological compass needles to sense their locations through the earth's geomagnetic field. Magnetotactic bacteria have the ability to synthesize nano-sized single domain magnetite crystals (Fe3O4) that are aligned in a chain, enabling the cells to swim or to migrate along magnetic field lines (Fig. 6A and 6B). For the synthesis of magnetite crystals, the cells use a specialized intracellular membranous compartment, also referred to as a magnetosome.68 The diameter of magnetosomes typically varies from 20 to 100 nm. The biomineralization process in magnetosomes is strictly controlled, leading to a highly regular and specific crystal size, shape, number, and assembly for a given bacterial strain. Recent studies on the molecular biology of magnetotactic bacteria have progressed our understanding of the formation mechanisms. Moreover, this knowledge provides insights into the potential use of magnetosomes, their proteins, and the bacterial cells as magnetic materials for biotechnological applications.64
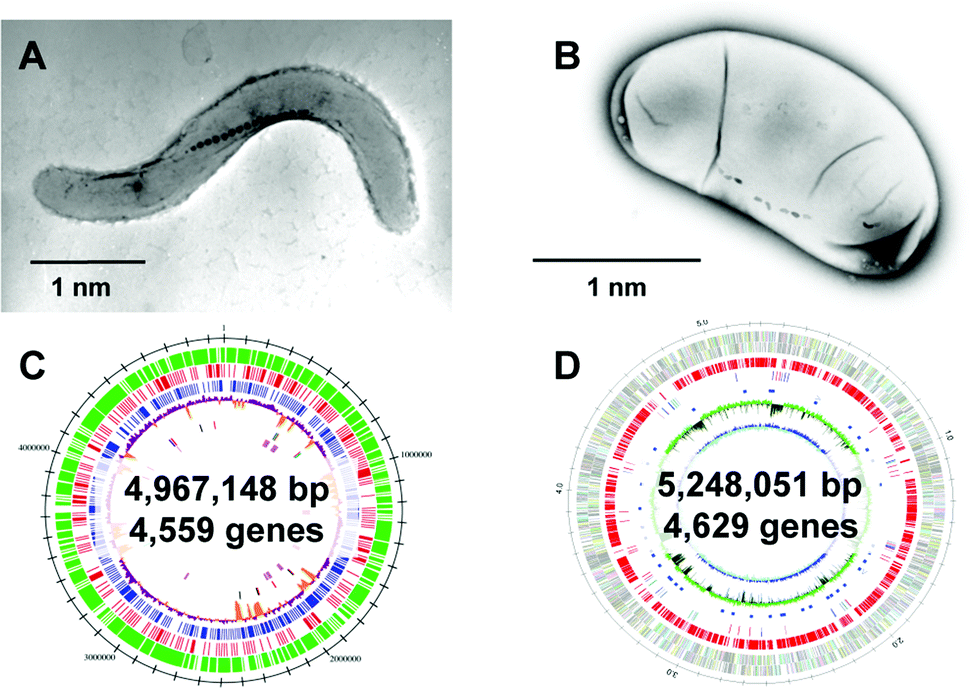 | ||
| Fig. 6 Two representative strains of magnetotactic bacteria. Transmission electron microscopy (TEM) images of (A) Magnetospirillum magneticum strain AMB-1 and (B) Desulfovibrio magneticus strain RS-1. Reproduced with permission from ref. 64. Copyright 2008 Royal Society of Chemistry. (C) Genome feature of Magnetospirillum magneticum AMB-1. Reproduced with permission from ref. 69. Copyright 2005 Oxford University Press. (D) Genomic feature of D. magneticus RS-1. Reproduced with permission from ref. 70. Copyright 2009 Cold Spring Harbor Laboratory Press. | ||
After clarification of the complete genome sequence of magnetotactic bacteria (Fig. 6C and 6D),69,70 tremendous improvements have been made in elucidating the molecular, biochemical, chemical and genetic bases of magnetosome formation and understanding how these unique intracellular organelles function. The genome information also allowed us to conduct comprehensive analyses of magnetotactic bacteria.64,71,72 The experimental data obtained from the molecular studies with Magnetospirillum sp. suggested that magnetosomes are assembled in a step-wise manner in which membrane biogenesis, magnetosome protein localization, and biomineralization are placed under discrete genetic control (Fig. 7).64,73,74 The first stage entails the invagination of the inner cell membrane to form a vesicle which serves as the precursor for magnetite crystals (Fig. 7A). The vesicles formed are assembled into a linear chain along with cytoskeletal filaments (Fig. 7B). Then, ferrous ions are accumulated into the vesicles through the transmembrane iron transporters (Fig. 7C). Internal iron is strictly controlled by an oxidation–reduction system. In the final stage, accumulated iron ions are crystallized within the vesicle (Fig. 7D). The formation and maintenance of membrane vesicles involves several proteins that might be functionally associated or spatially co-localized together. The MamY protein was isolated from small magnetite crystals.75 The MamY protein binds directly to the liposomes, causing formation of long tubules in vitro.75 The function of the MamY protein is considered to involve the constriction of the magnetosome membrane during magnetosome vesicle formation. The MamA protein, which has a tetratricopepetide repeat (TPR) domain—a protein–protein interaction module—was identified as a dominant magnetosome membrane protein in Magnetospirillum sp.76 Analysis of the crystal structure revealed that MamA folds as a sequential TPR protein with a unique hook-like shape.77 MamA may act as a scaffold protein for magnetosome formation by interacting with other magnetosome-forming proteins. Mms16 and MpsA proteins are speculated to prime the invagination site of the cytoplasmic membrane for vesicle formation.78 MagA is a proton-driving H+/Fe2+ antiporter protein.79 MamM, a cation diffusion facilitator family protein, is a multifunctional protein involved in the crystallization, initiation, and regulation of proper localization of other magnetosome proteins.80 MamB, which also exhibits similarity to the cation diffusion facilitator family, was observed to interact with several other proteins, including the PDZ1 domain of MamE.81 MamP is an iron oxidase that contributes to the magnetite crystal growth.82 MamK is a homolog of the bacterial actin-like protein, and has been shown to form filaments to establish the chain-like structure of magnetosomes through appropriate subcellular targeting.83 MamJ is an acidic protein associated with the MamK filament, and it directs the assembly of the magnetosome chain.84
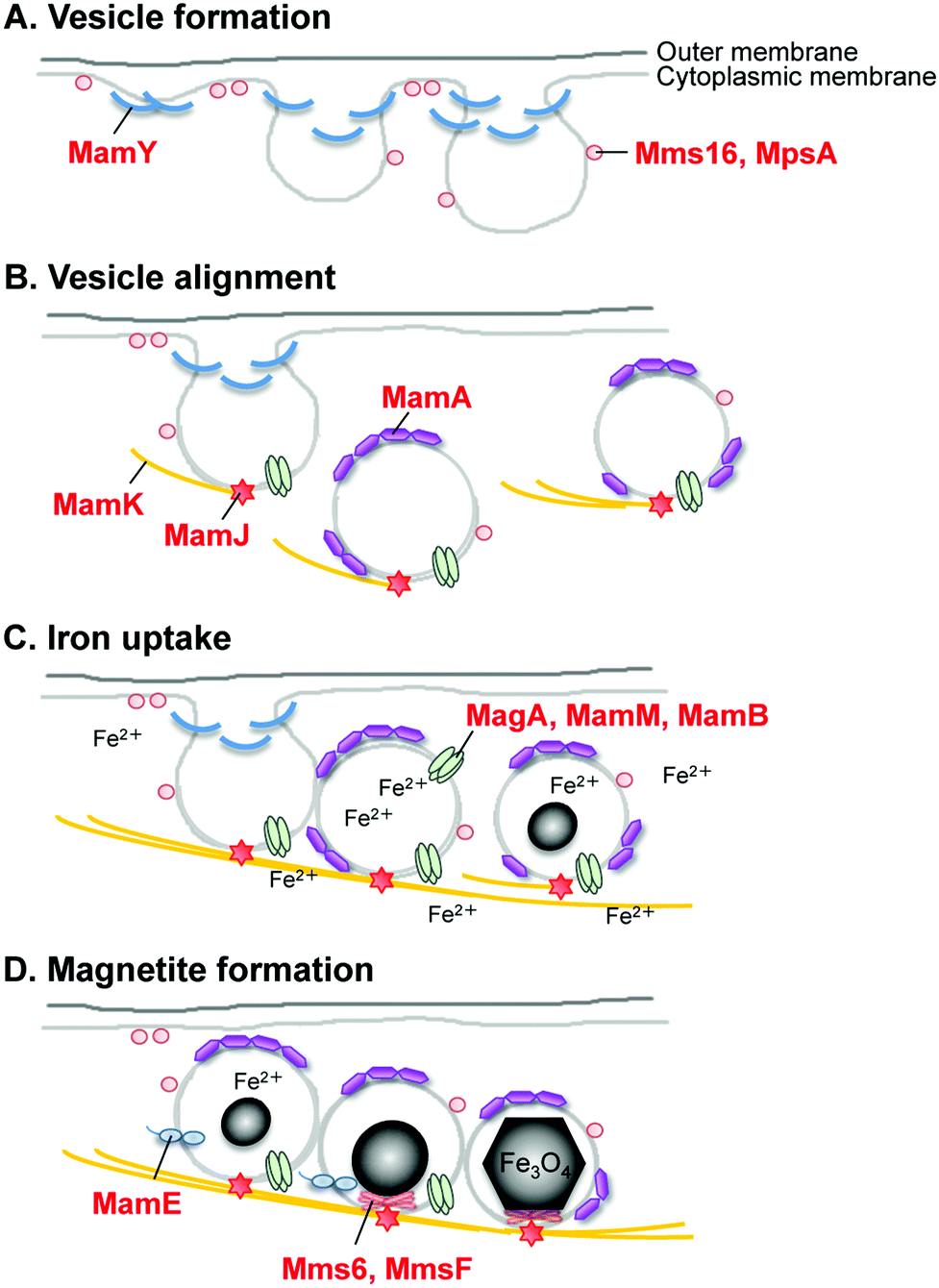 | ||
| Fig. 7 Schematic for the hypothesized mechanism of magnetite biomineralization in magnetotactic bacteria. | ||
A key protein that regulates the morphology of cubo-octahedral magnetite crystals was isolated in a study on the M. magneticum strain AMB-1 (Table 1).17 The Mms6 protein (Fig. 7D) represents a class of proteins that are tightly associated with the magnetite crystals. Three other proteins, designated as Mms5 (MamG), Mms7 (MamD), and Mms13 (MamC), were also isolated along with the Mms6 protein. Mms5, Mms6, and Mms7 showed a common amphiphilic characteristic containing hydrophobic N-terminal and hydrophilic C-terminal regions and could be categorized into a family of proteins. The N-terminal regions in Mms5, Mms6, and Mms7 possess a common leucine and glycine repetitive sequence. These amino acid sequences did not show any similarity to known proteins found in other organisms, suggesting that these proteins are specific in magnetotactic bacteria. Following competitive iron-binding analysis with other inorganic cations, it has been suggested that the C-terminal region is an iron-binding site.17 The N-terminal region is responsible for the self-aggregation of this protein.85,86 These characteristics are considered to contribute to magnetite biomineralization in bacteria.
The function of the Mms6 protein in bacterial cells was elucidated by the analysis of the gene deletion mutant mms6 (Fig. 8). Surprisingly, the mms6 deletion mutant synthesized smaller and irregular-shaped magnetite crystals (Fig. 8B), while the wild-type and complementation strains produced highly ordered cubo-octahedral crystals (Fig. 8A).87 Uncommon crystal faces, such as {210}, {211}, and {311}, were detected in magnetite crystals from the mms6 deletion mutant, in contrast to the cubo-octahedral magnetite crystals in wild-type and complementation strains, which had {111} and {100} crystal faces. These results indicate that the Mms6 protein regulates crystal surfaces to control the magnetite crystal morphology during crystal growth in magnetotactic bacterial cells. In addition, Mms5, Mms7, and Mms13 have similar but different functions from the Mms6 protein, and they cooperate in the formation of magnetites of consistent crystal size and morphology.88 This is the first example of a protein being involved in the regulation of a nano-sized crystallographic structure in in vivo biomineralization.
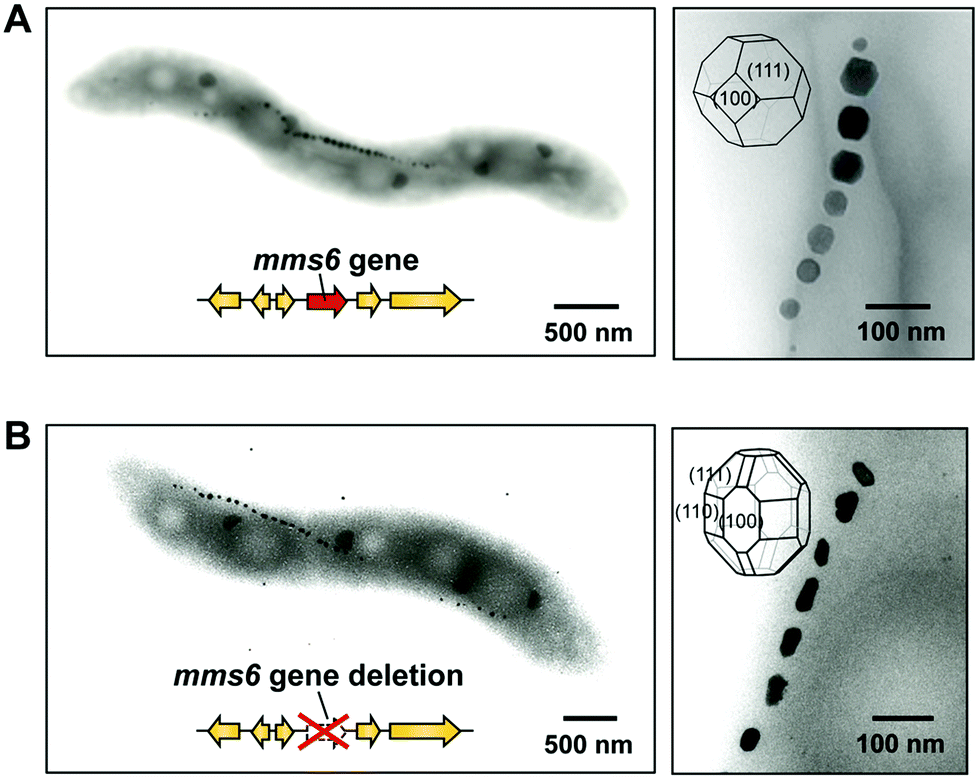 | ||
| Fig. 8 Elucidation of the function of the Mms6 protein in Magnetospirillum magneticum AMB-1 using gene deletion. (A) The wild-type strain synthesized highly ordered cubo-octahedral crystals. (B) The mms6 gene deletion mutant synthesized smaller magnetite crystals with uncommon crystal faces. Reproduced with permission from ref. 87. Copyright 2011 American Society for Biochemistry and Molecular Biology. | ||
Application of magnetite biomineralization proteins for magnetite synthesis
Particle size and morphology are important factors for the use of magnetites in practical applications because they strongly affect magnetic properties.67 The development of reliable and reproducible methods that enable synthesis of magnetic particles with controlled size and morphology has therefore been a major challenge in this field over the years.89 Given the increasing demand for magnetic nano-materials, alternative processes for the simple formation of size- and shape-controlled magnetite crystals in the aqueous environment are required.Biomimetic approaches that use the function of the Mms6 protein (Table 1) for preparing magnetite crystals have been investigated using two synthetic methods.17,85,90 Co-precipitation of ferrous and ferric ions in the presence of Mms6 produced uniform magnetite crystals with sizes ranging from 20 to 30 nm, while the absence of the protein resulted in the formation of crystals with irregular morphologies and sizes.17 The result suggests that Mms6 mediates crystal formation by binding precursors and/or magnetite crystals to regulate morphology. Prozorov et al. applied this synthetic method for the production of cobalt ferrite (CoFe2O3).91
Alternatively, a method involving partial oxidation of ferrous hydroxide in the presence of Mms6 was employed (Fig. 9).85 Mms6-mediated synthesis of magnetites by this method produced crystals of uniform size with narrow size distribution (approximately 20 nm in diameter) and a cubo-octahedral morphology consisting of {111} and {100} crystal faces (Fig. 9A and 9C). The crystal surfaces were similar to those of magnetosomes observed in the M. magneticum strain AMB-1. In contrast, the crystals formed in the absence of Mms6 were octahedral, consisting of {111} crystal faces, which were larger and had increased size distribution (approximately 30 nm in diameter) (Fig. 9B and 9D). Protein quantification analysis of Mms6 in the synthesized crystals indicated tight association of this protein with the crystal.85
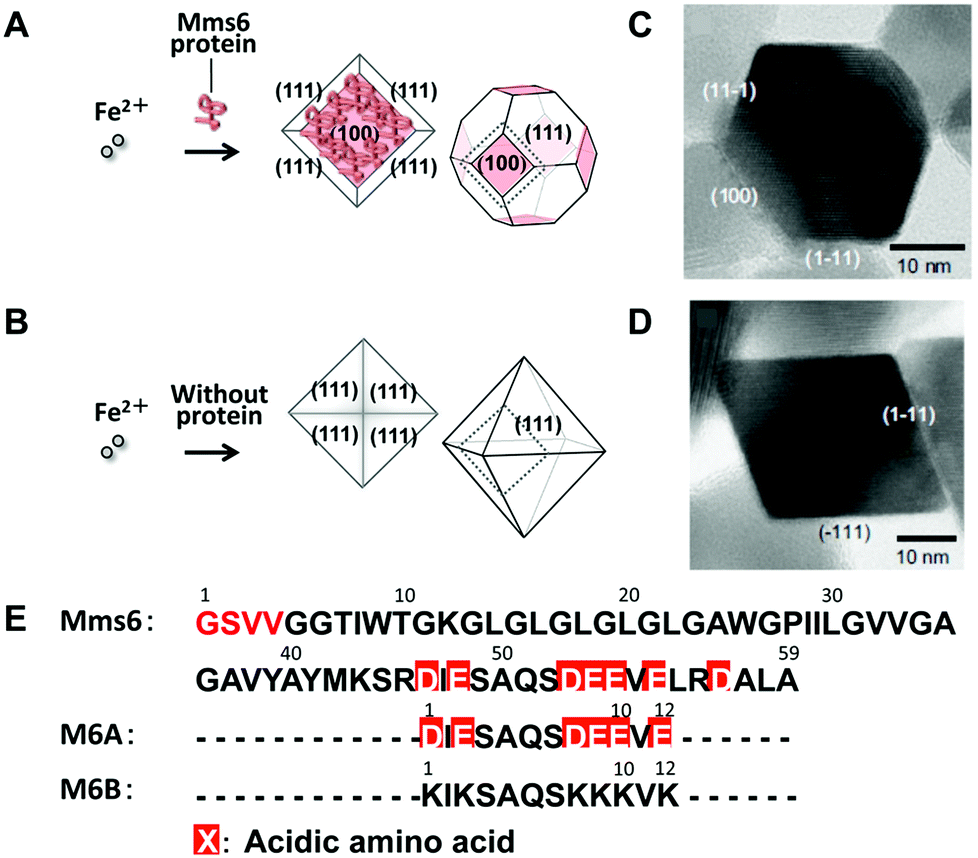 | ||
| Fig. 9 Schematic illustrations of magnetic particles synthesized (A) with or (B) without Mms6. TEM images of magnetic particles synthesized (C) in the presence of Mms6 and (D) in the absence of the Mms6 protein. Reproduced with permission from ref. 85. Copyright 2007 Elsevier. (E) Amino acid sequences of Mms6, M6A, and M6B. D: aspartic acid and E: glutamic acid. | ||
Short synthetic peptides that mimicked the characteristic amino acid sequences of Mms6 were further investigated for the synthesis of magnetite crystals (Fig. 9E).90 Magnetite synthesis using M6A peptides containing the C-terminal acidic region of Mms6 resulted in the formation of particles with a cubo-octahedral morphology similar to those of magnetosomes and particles formed in the presence of the Mms6 protein. Shape-regulated cobalt-doped magnetite nanoparticles were also synthesized through this method.92 High magnetic saturation, high coercivity magnetic particles with a narrow size distribution in the single-domain size can be produced by optimizing the conditions of the protein-mediated synthetic method.
Magnetite formation using Mms6 was also exploited to synthesize magnetic particles on planar substrates by bottom-up approaches.93,94 A silicon substrate was modified with a self-assembled monolayer of octadecyltrimethoxysilane and the Mms6 protein through hydrophobic interactions between the protein molecules and the monolayer. Active carboxylic groups of the Mms6 protein are arranged on the surface of substrates. The crystals were selectively formed in the specific regions modified with the Mms6 protein.93 Galloway et al. applied a soft-lithographic technique to fabricate surface patterning of the Mms6 protein and used it for the formation of biomimetic magnetite nanoparticle arrays.94 Nanocrystals of magnetites were successfully formed on the patterned surface. These methods offer an alternative strategy for magnetite formation under mild conditions and could be used for construction of high-density data storage.
Calcium carbonate biomineralization and applications
Learning the essence of calcium carbonate biomineralization
Calcium carbonate (CaCO3) is one of the most abundant inorganic compounds in biominerals. Mollusk shells, exoskeletons of crustaceans, coccolith of coccolithophores, and egg shells consist of CaCO3.2 Three different polymorphs, calcite, aragonite, and vaterite, are well-characterized in crystalline CaCO3. Amorphous calcium carbonate,95–98 which is observed in the cuticle of the exoskeleton of crustaceans, is also recognized. A typical example of the controlled CaCO3 hybrid structure is the nacre of the abalone shell (Fig. 10A, left).2 Plate-like aggregates of nanocrystalline aragonite submicrometer in thickness showing uniaxial crystallographic orientation are formed between the pre-organized macromolecular sheets (Fig. 10B). The stacks of the CaCO3 tablets, having uniform thickness, provide structural colors on the pearl's surface. The exoskeleton of crayfish is also an example of a CaCO3-based biomineral (Fig. 10A, right). The exoskeleton mainly consists of α-chitin/protein microfibril frameworks, and both calcite and amorphous calcium carbonate are closely associated with the fibrils. Mollusk shells and the exoskeletons of crustaceans are fascinating biominerals from a materials science perspective because they are tough and lightweight.6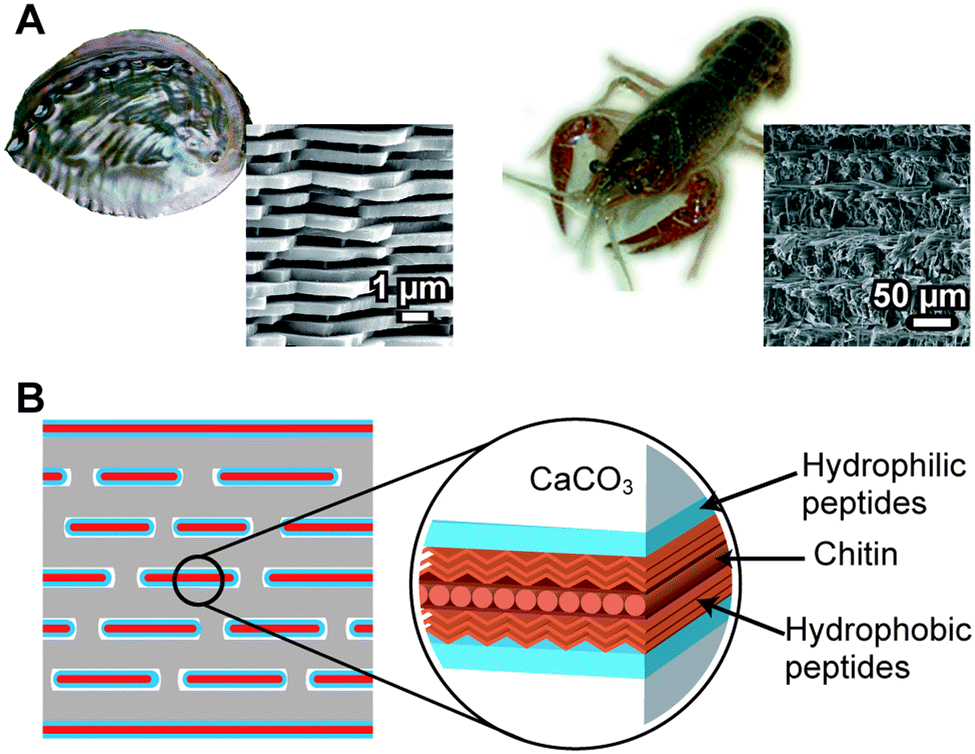 | ||
| Fig. 10 (A) Calcium carbonate (CaCO3)-based biominerals; left: abalone shell; right: the exoskeleton of crayfish. Insets show SEM images of biominerals. (B) Schematic of the nacre of the abalone shell. | ||
Similar to other biominerals described in previous sections, organic biopolymers in the form of proteins, glycoproteins, and polysaccharides are proposed to play main roles in the CaCO3 biomineralization processes.2,12–16 The biopolymers control the formation of minerals with uniform crystal size, nanostructural regularity, specific crystallographic phase, morphologies, and orientations. The highly organized and hierarchical structures with the organic/inorganic composition of the minerals achieve fracture resistance. To date, many proteins have been identified from different organisms or species forming CaCO3 biominerals.12–16 Materials scientists have been inspired by these molecularly controlled processes,21–25,99–103 although some of the formation mechanisms are yet unclear. In this section, we describe synthetic approaches for the formation of CaCO3/polymer hybrids as templates.
Inspired by the roles of biomacromolecules in biomineralization, synthetic systems for the formation of CaCO3/polymer hybrids have been developed.104–111 Thin-film hybrids have been obtained by the cooperative effects of polymer film matrices and soluble additives of acidic polymers. Fig. 11 shows a schematic representation of the formation of these hybrids, and their polarized optical microscopy and scanning electron microscopy (SEM) images. Spin-coated thin films of polysaccharides, such as chitin104,105 and chitosan106–108 on glass substrates, were used as insoluble templates. Thin-film CaCO3 with flat surfaces developed on the insoluble organic templates in the presence of acidic polymers such as poly(acrylic acid) (PAA), poly(aspartic acid), and poly(glutamic acid) until the polymer film was fully covered with CaCO3. The functional groups forming H-bonds, such as –OH and –NH groups on the surface of the organic matrix, interact with the acidic group of the soluble polymer additive.109 Then, acidic macromolecules induce a locally high concentration of calcium ions, resulting in the nucleation of crystalline thin films. Their polymorphs are calcite, which is the thermodynamically most stable state, and vaterite, which is the kinetically preferred state. These films are composed of assembled nanocrystals with radially oriented c axes, as revealed from the polarized optical microscopy image showing crossed extinction between the polarizers (Fig. 11). The supramacromolecules also act as soluble polymer additives, which have polyrotaxane structures composed of acidic cyclodextrins and polyethylene oxide.112
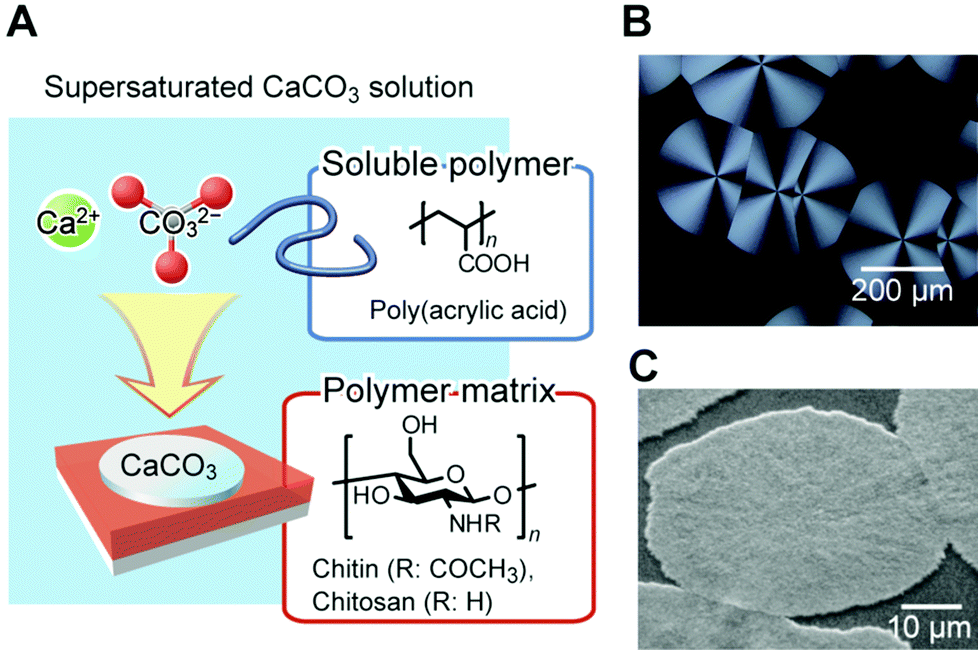 | ||
| Fig. 11 (A) Schematic illustration of the formation of thin-film CaCO3/organic hybrids. Reproduced with permission from ref. 100. Copyright 2010 Cambridge University Press. (B) Polarized optical microscopy image of a thin-film hybrid developed on a chitosan matrix in the presence of poly(acrylic acid). (C) Scanning electron microscopy image of CaCO3 hybrids formed on the chitin matrix. Reproduced with permission from ref. 104. Copyright 2000 The Chemical Society of Japan. | ||
Cooperative effects between soluble bio-macromolecules and the polysaccharide film matrices enable highly controlled crystallization of CaCO3 in synthetic systems. One of the peptides (CAP-1) involved in CaCO3 biomineralization was isolated from the exoskeleton of crayfish (Table 1) and used in soluble additives in a crystallization experiment in order to understand the structure–function relationships of the peptide.113 CAP-1 is a highly acidic peptide, which has interesting structural features (Fig. 12A). In particular, it has a six consecutive aspartate sequence at the C terminal. The chitin-binding motif, called the Rebers–Riddiford consensus sequence, is seen at the central part of CAP-1. This consensus sequence is found in many cuticle proteins in crustaceans and considered to be involved in binding to the crystalline structure of chitin.114 The serine at the 70th position of CAP-1 was phosphorylated, which may bind to calcium ions and stabilize the CaCO3 amorphous precursors.99,115
 | ||
| Fig. 12 (A) Amino acid sequence of the CAP-1 peptide isolated from the exoskeleton of crayfish. Amino acid sequence; D: aspartic acid; E: glutamic acid; pS: phosphoserine. (B) Scanning electron microscopy (SEM) images of CaCO3 crystals formed on chitin matrices in the presence of CAP-1. (C) SEM images of CaCO3 crystals formed on glass substrates in the presence of CAP-1. B, (C) Concentrations of CAP-1: 3.0 × 10−4 wt% (left): 1.0 × 10−3 wt% (center); 3.0 × 10−3 wt% (right). Reproduced with permission from ref. 113. Copyright 2006 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
In the presence of CAP-1, unidirectionally oriented thin-films of CaCO3/chitin hybrids, approximately on the ten-micrometer scale, were obtained on the matrix (Fig. 12B, right).113 These thin films are assemblies of nanocrystals of about 100 nm in size with single-crystal orientation. No oriented crystals are obtained on a glass substrate or on an amorphous chitosan matrix in the presence of CAP-1 (Fig. 12C). The cooperative interaction of each CAP-1 functional part induces the formation of the unidirectionally oriented CaCO3 thin film crystals on chitin fibrils. The CaCO3 amorphous precursors stabilized with CAP-1 are initially formed on the chitin fibrous matrix. The alignment of the functional groups of CAP-1 binding on the crystalline chitin fibrils induces the nucleation of CaCO3 in the deposited amorphous precursors.113 Analogous recombinant peptides of CAP-1 have been synthesized and used as additives for crystallization of CaCO3 to examine the effects of each functional part on CaCO3 crystal growth.116–118 The crystallographic orientation depends on the chitin-binding consensus sequence (the Ribers–Riddiford consensus sequence), and the functional group of the 70th residue is critical to the surface morphology of thin-film crystals.
The matrix proteins Pif80 and Pif97 were isolated from the pearl oyster P. fucata by Suzuki, Nagasawa, and coworkers14 (Table 1). Pif80 and Pif97 form a Pif complex in vivo via disulfide bonds (Fig. 13A). The Pif complex plays key roles in controlling CaCO3 crystallization for the formation of the nacreous layers. It was found that the Pif complex works on the interface between the organic framework and aragonite through an aragonite-binding domain in Pif80 and a chitin-binding domain in Pif97. These domains allow the organic framework and calcium carbonate crystals to interact. When the Pif complex is used as a soluble additive for calcium carbonate crystallization on the chitin matrix in vitro, the Pif complex induces the aragonite thin film with the c axis unidirectionally aligned perpendicular to the surface, as observed for natural nacre (Fig. 13B and 13C).
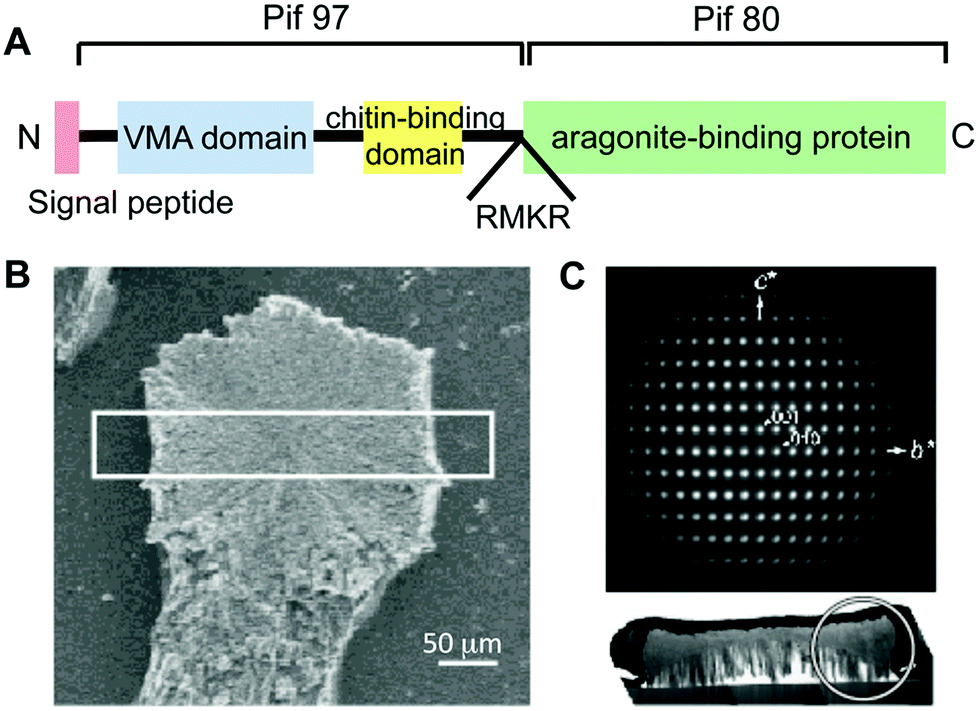 | ||
| Fig. 13 (A) Schematic illustration of the overall protein structure of the Pif complex. Red, blue, and yellow boxes represent the signal peptide, VWA domain, and peritrophin A-type chitin-binding domain, respectively. RMKR is a Kex2-like proteinase cleavage site. Green box is the aragonite-binding protein (Pif 80). (B) A fragment of the CaCO3 crystal observed by SEM. (C) Electron diffraction pattern of the white circled area in the cross-section of the white box in (B), indicating a single aragonite crystal and its c axis perpendicular to the glass plate. Reproduced with permission from ref. 14. Copyright 2009 American Association for the Advancement of Science. | ||
Application of calcium carbonate biomineralization mechanisms toward the formation of functional hybrids
In the biomimetic synthesis of CaCO3, designed templates and/or additives with specific functional groups have been used for the preparation of organic/inorganic hybrids.21–25,99–103 CaCO3 crystals with controlled polymorphs and crystallographic orientations are formed on insoluble templates such as self-assembled monolayers (SAMs)21,119 and polymer brushes.120–123 The use of soluble polymers, containing acidic functional groups such as carboxyl groups and phosphate groups that interact with inorganic substances, leads to the formation of a variety of complex structures.99–103,124–127 The experimental and simulation studies on the CaCO3 precursor stabilized by magnesium ions was also performed to reveal the formation mechanism of the hybrids.128,129 In this section, we focus on the hybrids of CaCO3, and synthetic and semi-synthetic polymers obtained by cooperative effects between film matrices and soluble additives.A significant aspect of biomineralization is that crystallization is precisely controlled by templates of aligned polymers. As mentioned above, well-controlled oriented aragonite nanocrystals are formed in the nacre of the abalone shell.2,6 Preparation of such controlled crystal structures is still challenging for the present technology, and both synthetic and biological approaches are needed. As a synthetic approach, the use of a crystalline poly(vinyl alcohol) (PVA) matrix leads to the induction of aragonite crystallization in the presence of PAA.110 The polycrystalline PVA matrix is prepared by annealing a spin-coated PVA film. The lattice of the PVA (100) face matches the aragonite lattice. It is considered that the PAA interacting with the hydroxy groups in the micro-domains of the crystalline PVA forms the local arrangement of carboxylic acid groups. These aligned carboxylates locally arrange the calcium ions, leading to the nucleation of aragonite. The distance between the functional groups on the PAA polymer backbone is the same as that for PVA, resulting in the selective formation of aragonite. In contrast, the use of poly(glutamic acid) (PGA) as a soluble additive leads to the formation of thin-film vaterite on the PVA matrix because of different distances between the functional groups.110
Unidirectionally oriented polymer/CaCO3 hybrids were obtained using macroscopically oriented polymer matrices through macromolecular templating in the presence of simple acidic polymers.130Fig. 14 shows polarized optical microscopy images of the oriented chitin matrix, made from chitin phenylcarbamate, forming lyotropic liquid-crystalline (LC) structures and the calcite crystals grown on the oriented matrix. The LC chitin derivatives were synthesized by the carbamation of chitin (Fig. 14A). The condensed solution of LC chitin derivatives was converted to the gel state by soaking in methanol. The gel was oriented by stretching. After cleavage by hydrolysis of the carbamate groups, the oriented chitin matrix was obtained (Fig. 14B) and immersed in a crystallization solution for the controlled crystal growth of CaCO3. Rod-shaped crystals were grown for 15 h to about 80 μm in the ordered chitin matrix and aligned parallel to the direction of the polymer main chains of the matrix (Fig. 14C). When polarized optical microscopy is used, the visibility of the rod crystals oscillates between bright and dark with every 45° rotation, as shown in Fig. 14D. This indicates that the crystallographic orientation of the rod crystals corresponds to the longitudinal axis of the rod crystals. The SEM images of the isolated CaCO3 rods (Fig. 14E) indicate that the rod crystals formed on the oriented matrix are assemblies of rhombohedral calcite crystals with fine facets of approximately 500 nm in size. Moreover, TEM images and the corresponding electron diffraction patterns of the selected area show that the rod calcite has a single crystalline orientation (Fig. 14F). The crystals exhibiting such features are categorized as mesocrystals.126,127,131 LC polymers are useful for control of oriented structures from the macroscopic to molecular levels. In nature, the silkworm cocoon and the exoskeleton of the jewel beetle also have a precisely ordered hierarchical structure. It is considered that these hierarchical structures are formed via liquid crystalline structures.132,133 This LC methodology may provide novel ways towards the development of hybrids with oriented structures based on functional inorganic crystals.134
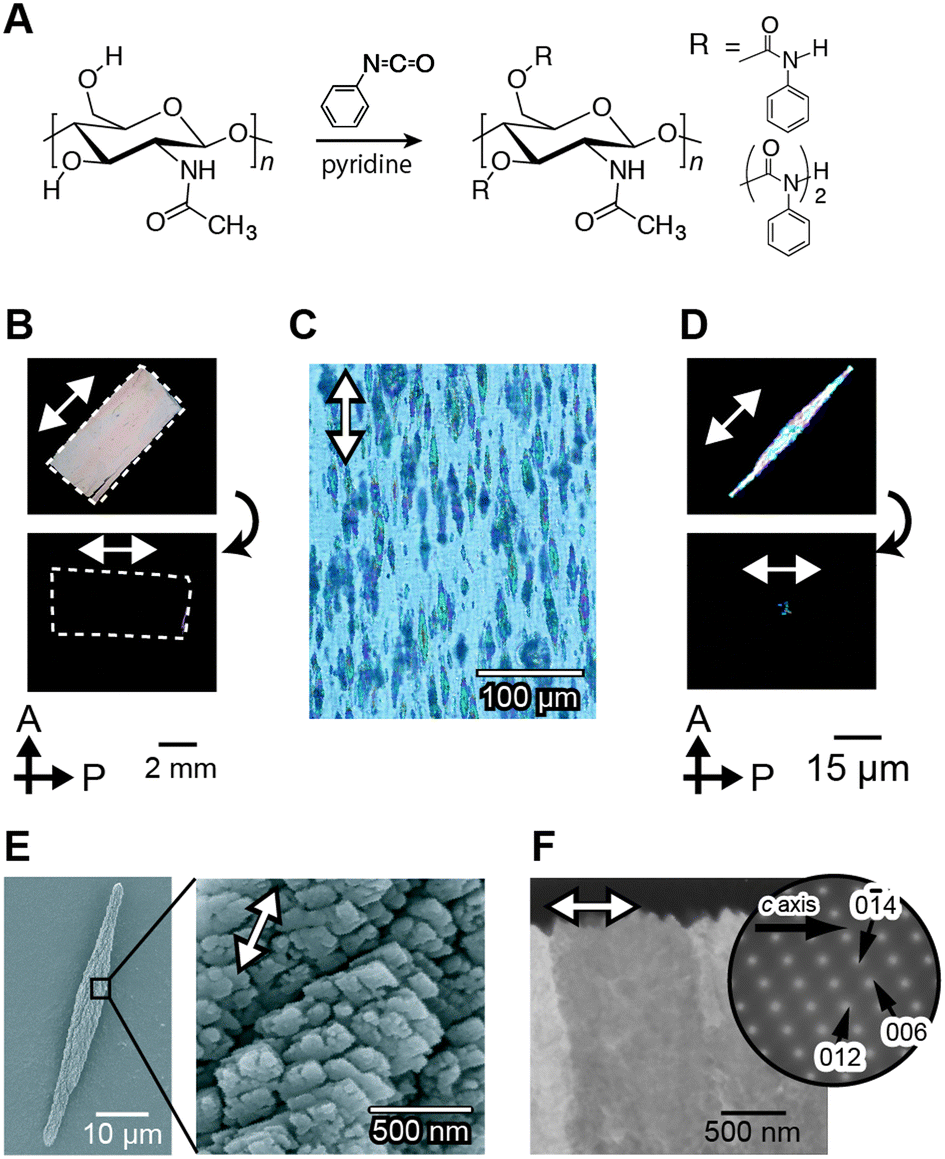 | ||
| Fig. 14 Macroscopically ordered CaCO3/organic hybrids grown on ordered macromolecular templates. (A) Synthetic scheme of chitin phenylcarbamate. Polarized optical microscopy images of (B) an oriented template, (C) the hybrids obtained, and (D) isolated CaCO3 crystals. (E) SEM images of the CaCO3 crystals. (F) TEM image of cross-sections of CaCO3 crystals. Reproduced with permission from ref. 130. Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Stiff, flexible, elastic, and transparent bulk materials are designed and synthesized by mimicking the structure of crustacean exoskeletons (Fig. 15).135 These elastic nanocomposite materials are prepared from a bulk matrix consisting of amorphous calcium carbonate and poly(acrylic acid), and reinforcing cellulose nanofibers. These unique characteristics cannot be obtained by simple mixing of cellulose and calcium carbonate. The strong interactions at the interface between the surface of the cellulose nanofibers and calcium carbonate are keys for these properties. The cellulose nanofiber matrix oxidized by the TEMPO-mediated system on the surface is used because carboxyl groups form on the surface of the nanofibers. Moreover the carboxylate group of the poly(acrylic acid) and the hydroxyl groups of cellulose interact at the interface of the bulk amorphous calcium carbonate and cellulose.
 | ||
| Fig. 15 (A) Elastic flexibility of the nanocellulose/ACC composites and (B) schematic illustration of the composites. Reproduced with permission from ref. 135. Copyright 2014 Royal Society of Chemistry. | ||
Not only the polymorphs of the CaCO3 films, but also the morphologies of the hybrids are affected by the polymer matrices. Film hybrids with regular surface-relief structures self-organize when “soft” hydrogel film matrices are used.111 A pullulan derivative, with the main chain of a water-soluble polysaccharide and hydrophobic cholesteryl groups as non-covalent crosslinking points, has been reported to form a physical hydrogel in aqueous solution. In this hydrogel matrix, CaCO3 crystals develop inside the gel in the presence of PAA as a thin-film hybrid with regular surface-relief structures. The period of the patterned relief structures ranges from sub-micrometers to a few micrometers. It is considered that the formation of patterned thin films is induced by spontaneous oscillation of the ion concentrations because of slow ion diffusion in the gels.
CaCO3 crystals with complex structures are self-organized by the cooperative effects between matrices of PVA derivatives in the presence of soluble additives (Fig. 16).136–140 PVA derivatives can also form “soft” and “hard” hydrogels as thin-film matrices depending on the degree of crosslinking. The hydrogel matrix is obtained by thermal treatment of a PVA-based copolymer and the properties of the gel matrices are altered by the thermal procedures. On the “soft” hydrogel matrix of PVA, thin-film hybrids with patterned surface structures are obtained.136
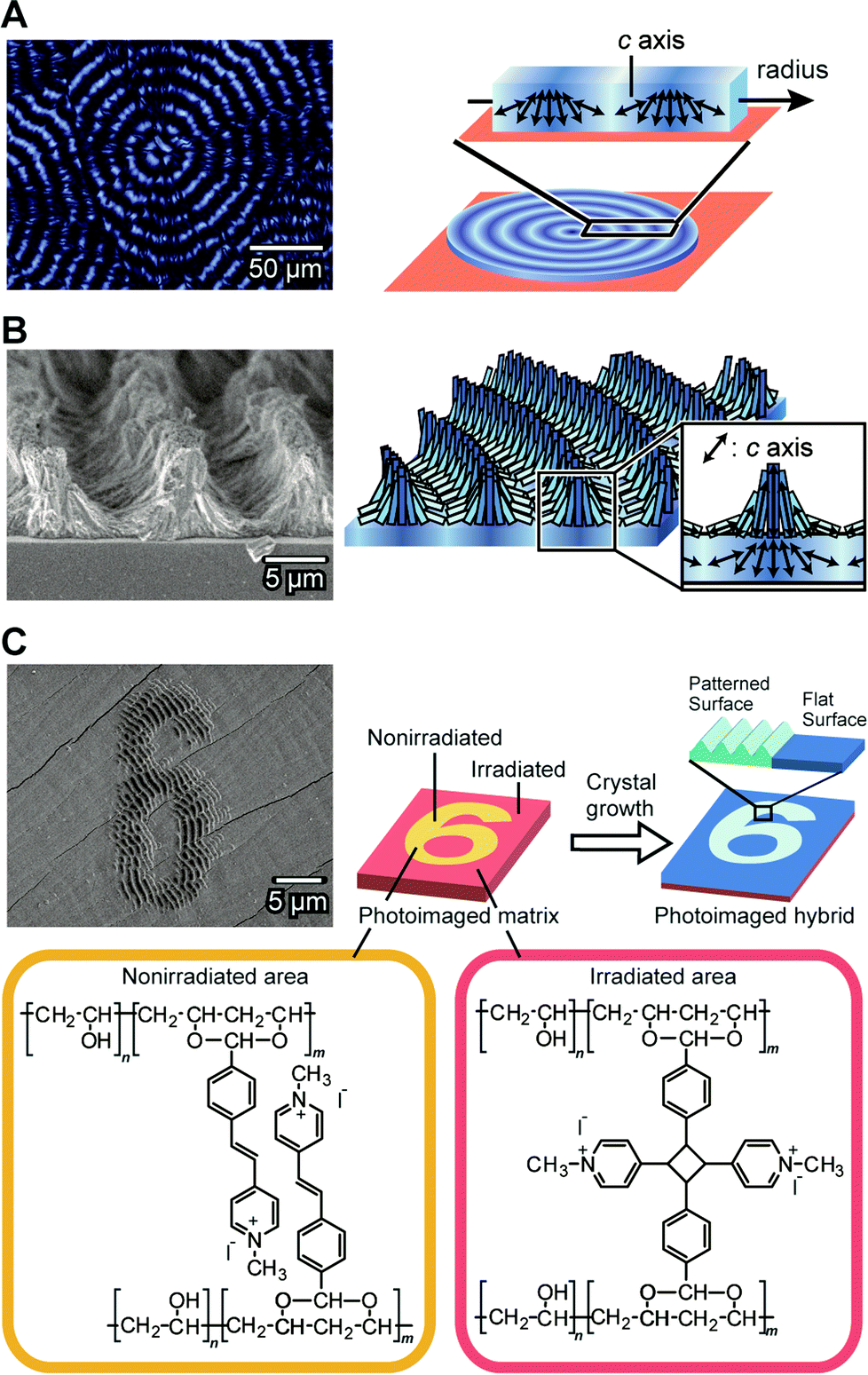 | ||
| Fig. 16 Self-organization of CaCO3 crystals with complex structures. (A) Thin-film showing periodic crystallographical orientations. Left: polarized optical microscopy image; right: schematic illustration of the structure. (B) Three-dimensional structures formed on the thin film shown in (A). Left: SEM image; right: schematic illustration of the structure. Reproduced with permission from ref. 137. Copyright 2009 American Chemical Society. (C) Photoimaged CaCO3/polymer hybrid films formed on the matrix of the photoreactive PVA-derivative. Upper left: SEM image; upper right: schematic illustration of the formation process; bottom: chemical structures of the photoreactive PVA-derivative. Reproduced with permission from ref. 138. Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
When we use a more cross-linked “hard” PVA hydrogel as a matrix for the crystal growth, three dimensional (3D) patterned relief objects, on the scale of about tens of micrometers, are formed in the presence of PAA (Fig. 16A and 16B).137 The 3D structures are formed in two steps and are different from the relief structures of the thin-film crystals. The first step is the formation of thin-film CaCO3 hybrids with a flat surface (Fig. 16A). These film crystals show concentric dark rings under crossed polarizers. In this flat film of CaCO3, the directions of the c axes for the CaCO3 crystals change periodically between the perpendicular orientation and the parallel orientation to the direction of the crystal growth. The self-organized periodic orientation in this first step results from diffusion-controlled crystal growth. The second step is the formation of the 3D relief objects composed of needle-like CaCO3 crystals on the flat surface of the template (Fig. 16B). The height of the relief objects is about 5 μm and the period is about 10 μm. The needle-like CaCO3 crystals are self-assembled into the micrometer-scale relief objects. These 3D-structured CaCO3 crystals can be used as substrates for cultivation of pre-osteoblastic cells.140 The ordered microgrooves enhanced adhesion, spreading, proliferation, and differentiation of the cells. X-ray diffraction studies of the relief objects show that in the ridge regions, the c axes of the needle-like crystals are aligned perpendicularly to the substrate. The crystals formed in the second step have the same orientation as those in the underlying thin film.137 These results indicate that the flat thin-film crystals formed in the first step serve as templates in the second step. In addition, the flat-film template also induces regular architectures for different kinds of carbonate crystals such as SrCO3.
Tuning the degree of cross-linking in the polymer matrix using photoreactions allows for photoimaging of CaCO3 hybrid films with self-organized structures (Fig. 16C).138 Figures written on a photoreactive polymer film were transferred into CaCO3 crystal morphologies. The CaCO3 crystal films with microscopic regular relief structures and flat surfaces are self-organized on the non-irradiated and UV-irradiated areas of the matrix, respectively. As a result, the hierarchically ordered polymer/CaCO3 hybrid films are formed through the combination of the top-down photoimaging method and the bottom-up self-organization method.
Conclusions
The results obtained from fundamental studies on biomineralization have allowed us to develop various organic/inorganic hybrid materials using proteins isolated from organisms and/or their mimicking macromolecules. Although biomineralization mechanisms in most biological systems are extremely complicated, current biomimetic and bio-inspired syntheses are based on only certain aspects of these mechanisms. The application of various synthetic routes using multiple molecules will enable tuning of the crystal size, morphology, surface structures, composition, crystallinity, and even hierarchical organization of materials. These principles will also facilitate the development of functional organic/inorganic hybrid materials with multiple combinations of outstanding properties including lightweight, high flexibility, mechanical strength, dynamic function and structural hierarchy through environmentally benign routes. More advanced materials will be created in the future for a wide array of tailor-made applications via collaborative work between researchers in a wide variety of scientific fields, including inorganic chemistry, organic chemistry, biology, computer science and engineering.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research (KAKENHI) on Innovative Areas of “Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control” (no. 2206) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT).Notes and references
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689 RSC.
- N. Kroeger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129 CrossRef.
- K. Shimizu, J. Cha, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6234 CrossRef CAS.
- H. A. Lowenstam, Geol. Soc. Am. Bull., 1962, 73, 435 CrossRef CAS.
- M. A. Meyers, J. McKittrick and P. Y. Chen, Science, 2013, 339, 773 CrossRef CAS PubMed.
- J. C. Weaver, G. W. Milliron, A. Miserez, K. Evans-Lutterodt, S. Herrera, I. Gallana, W. J. Mershon, B. Swanson, P. Zavattieri, E. DiMasi and D. Kisailus, Science, 2012, 336, 1275 CrossRef CAS PubMed.
- J. Aizenberg, A. Tkachenko, S. Weiner, L. Addadi and G. Hendler, Nature, 2001, 412, 819 CrossRef CAS PubMed.
- C. E. Diebel, R. Proksch, C. R. Green, P. Neilson and M. M. Walker, Nature, 2000, 406, 299 CrossRef CAS PubMed.
- J. Aizenberg, V. C. Sundar, A. D. Yablon, J. C. Weaver and G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3358 CrossRef CAS PubMed.
- F. Natalio, T. P. Corrales, M. Panthoefer, D. Schollmeyer, I. Lieberwirth, W. E. G. Mueller, M. Kapp, H. J. Butt and W. Tremel, Science, 2013, 339, 1298 CrossRef CAS PubMed.
- I. M. Weiss, S. Kaufmann, K. Mann and M. Fritz, Biochem. Biophys. Res. Commun., 2000, 267, 17 CrossRef CAS PubMed.
- S. Sudo, T. Fujikawa, T. Nagakura, T. Ohkubo, K. Sakaguchi, M. Tanaka, K. Nakashima and T. Takahashi, Nature, 1997, 387, 563 CrossRef CAS PubMed.
- M. Suzuki, K. Saruwatari, T. Kogure, Y. Yamamoto, T. Nishimura, T. Kato and H. Nagasawa, Science, 2009, 325, 1388 CrossRef CAS PubMed.
- R. Lakshminarayanan, R. Kini and S. Valiyaveettil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5155 CrossRef CAS PubMed.
- H. Inoue, N. Ozaki and H. Nagasawa, Biosci., Biotechnol., Biochem., 2001, 65, 1840 CrossRef CAS PubMed.
- A. Arakaki, J. Webb and T. Matsunaga, J. Biol. Chem., 2003, 278, 8745 CrossRef CAS PubMed.
- J. N. Cha, K. Shimizu, Y. Zhou, S. C. Christiansen, B. F. Chmelka, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 361 CrossRef CAS.
- R. L. Brutchey, E. S. Yoo and D. E. Morse, J. Am. Chem. Soc., 2006, 128, 10288 CrossRef CAS PubMed.
- D. Kisailus, Q. Truong, Y. Amemiya, J. C. Weaver and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5652 CrossRef CAS PubMed.
- J. Aizenberg, A. J. Black and G. M. Whitesides, Nature, 1999, 398, 495 CrossRef CAS PubMed.
- H. Imai, Y. Oaki and A. Kotachi, Bull. Chem. Soc. Jpn., 2006, 79, 1834 CrossRef CAS.
- T. Kato, A. Sugawara and N. Hosoda, Adv. Mater., 2002, 14, 869 CrossRef CAS.
- F. C. Meldrum and H. Coelfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS PubMed.
- F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582 CrossRef CAS PubMed.
- T. L. Simpson and B. E. Volcani, Silicon and Siliceous Structures in Biological Systems, Springer-Verlag, New York, USA, 1981 Search PubMed.
- N. Kroger, R. Deutzmann, C. Bergsdorf and M. Sumper, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14133 CrossRef CAS PubMed.
- S. Matsunaga, R. Sakai, M. Jimbo and H. Kamiya, ChemBioChem, 2007, 8, 1729 CrossRef CAS PubMed.
- H. Ehrlich, R. Deutzmann, E. Brunner, E. Cappellini, H. Koon, C. Solazzo, Y. Yang, D. Ashford, J. Thomas-Oates, M. Lubeck, C. Baessmann, T. Langrock, R. Hoffmann, G. Worheide, J. Reitner, P. Simon, M. Tsurkan, A. V. Ereskovsky, D. Kurek, V. V. Bazhenov, S. Hunoldt, M. Mertig, D. V. Vyalikh, S. L. Molodtsov, K. Kummer, H. Worch, V. Smetacek and M. J. Collins, Nat. Chem., 2010, 2, 1084 CrossRef CAS PubMed.
- J. C. Weaver and D. E. Morse, Microsc. Res. Tech., 2003, 62, 356 CrossRef CAS PubMed.
- X. Wang, L. Gan, K. P. Jochum, H. C. Schroeder and W. E. G. Mueller, Evid. Based Complementary Altern. Med., 2011, 2011, 1 Search PubMed.
- V. B. Kozhemyako, G. N. Veremeichik, Y. N. Shkryl, S. N. Kovalchuk, V. B. Krasokhin, V. A. Rasskazov, Y. N. Zhuravlev, V. P. Bulgakov and Y. N. Kulchin, Mar. Biotechnol., 2010, 12, 403 CrossRef CAS PubMed.
- A. Krasko, B. Lorenz, R. Batel, H. C. Schroeder, I. M. Mueller and W. E. G. Mueller, Eur. J. Biochem., 2000, 267, 4878 CrossRef CAS.
- W. E. G. Mueller, U. Schlossmacher, C. Eckert, A. Krasko, A. Boreiko, H. Ushijima, S. E. Wolf, W. Trernel, I. M. Mueller and H. C. Schröder, Eur. J. Cell Biol., 2007, 86, 473 CrossRef CAS PubMed.
- K. Mohri, M. Nakatsukasa, Y. Masuda, K. Agata and N. Funayama, Dev. Dyn., 2008, 237, 3024 CrossRef CAS PubMed.
- W. E. G. Mueller, C. Eckert, K. Kropf, X. H. Wang, U. Schlossmacher, C. Seckert, S. E. Wolf, W. Tremel and H. C. Schroeder, Cell Tissue Res., 2007, 329, 363 CrossRef CAS PubMed.
- W. E. G. Mueller, X. H. Wang, K. Kropf, A. Boreiko, U. Schlssmacher, D. Brandt, H. C. Schroeder and M. Wiens, Cell Tissue Res., 2008, 333, 339 CrossRef CAS PubMed.
- G. N. Veremeichik, Y. N. Shkryl, V. P. Bulgakov, S. V. Shedko, V. B. Kozhemyako, S. N. Kovalchuk, V. B. Krasokhin, Y. N. Zhuravlev and Y. N. Kulchin, Mar. Biotechnol., 2011, 13, 810 CrossRef CAS PubMed.
- Y. Zhou, K. Shimizu, J. N. Cha, G. D. Stucky and D. E. Morse, Angew. Chem., Int. Ed., 1999, 38, 780 CrossRef CAS.
- M. M. Murr and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11657 CrossRef CAS PubMed.
- D. E. Morse, Trends Biotechnol., 1999, 17, 230 CrossRef CAS.
- R. L. Brutchey and D. E. Morse, Chem. Rev., 2008, 108, 4915 CrossRef CAS PubMed.
- W. E. G. Mueller, X. Wang, F.-Z. Cui, K. P. Jochum, W. Tremel, J. Bill, H. C. Schroeder, F. Natalio, U. Schloßmacher and M. Wiens, Appl. Microbiol. Biotechnol., 2009, 83, 397 CrossRef CAS PubMed.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935 CrossRef CAS PubMed.
- J. L. Sumerel, W. J. Yang, D. Kisailus, J. C. Weaver, J. H. Choi and D. E. Morse, Chem. Mater., 2003, 15, 4804 CrossRef CAS.
- D. Kisailus, J. H. Choi, J. C. Weaver, W. J. Yang and D. E. Morse, Adv. Mater., 2005, 17, 314 CrossRef CAS.
- I. Zlotnikov, P. Werner, H. Blumtritt, A. Graff, Y. Dauphin, E. Zolotoyabko and P. Fratzl, Adv. Mater., 2014, 26, 1682 CrossRef CAS PubMed.
- I. Fuchs, Y. Aluma, M. Ilan and Y. Mastai, J. Phys. Chem. B, 2014, 118, 2104 CAS.
- F. Natalio, T. P. Corrales, M. Panthöfer, D. Schollmeyer, I. Lieberwirth, W. E. G. Muller, M. Kappl, H.-J. Butt and W. Tremel, Science, 2013, 339, 1298 CrossRef CAS PubMed.
- S. E. Wolf, U. Schlossmacher, A. Pietuch, B. Mathiasch, H. C. Schroeder, W. E. G. Mueller and W. Tremel, Dalton Trans., 2010, 39, 9245 RSC.
- A. Rai and C. C. Perry, J. Mater. Chem., 2012, 22, 4790 RSC.
- A. Polini, S. Pagliara, A. Camposeo, A. Biasco, H. C. Schroeder, W. E. G. Mueller and D. Pisignano, Adv. Mater., 2011, 23, 4674 CrossRef CAS PubMed.
- M. N. Tahir, P. Theato, W. E. G. Mueller, H. C. Schroeder, A. Borejko, S. Faiss, A. Janshoff, J. Huth and W. Tremel, Chem. Commun., 2005, 5533 RSC.
- M. N. Tahir, P. Theato, W. E. G. Mueller, H. C. Schroeder, A. Janshoff, J. Zhang, J. Huth and W. Tremel, Chem. Commun., 2004, 2848 RSC.
- M. N. Tahir, F. Natalio, H. A. Therese, A. Yella, N. Metz, M. R. Shah, E. Mugnaioli, R. Berger, P. Theato, H. C. Schroeder, W. E. G. Mueller and W. Tremel, Adv. Funct. Mater., 2009, 19, 285 CrossRef CAS.
- R. Andre, M. N. Tahir, H. C. C. Schroeder, W. E. G. Mueller and W. Tremel, Chem. Mater., 2011, 23, 5358 CrossRef CAS.
- M. I. Shukoor, F. Natalio, H. A. Therese, M. N. Tahir, V. Ksenofontov, M. Panthofer, M. Eberhardt, P. Theato, H. C. Schröder, W. E. G. Mueller and W. Tremel, Chem. Mater., 2008, 20, 3567 CrossRef CAS.
- B. Lange, N. Metz, M. N. Tahir, F. Fleischhaker, P. Theato, H. C. Schröder, W. E. G. Mueller, W. Tremel and R. Zentel, Macromol. Rapid Commun., 2007, 28, 1987 CrossRef CAS.
- O. V. Kaluzhnaya, S. I. Belikov, H. C. Schroeder, M. Wiens, M. Giovine, A. Krasko, I. M. Mueller and W. E. G. Mueller, Naturwissenschaften, 2005, 92, 134 CrossRef CAS PubMed.
- F. Natalio, T. Link, W. E. G. Mueller, H. C. Schroeder, F. Z. Cui, X. H. Wang and M. Wiens, Acta Biomater., 2010, 6, 3720 CrossRef CAS PubMed.
- D. Kisailus, M. Najarian, J. C. Weaver and D. E. Morse, Adv. Mater., 2005, 17, 1234 CrossRef CAS.
- D. H. Adamson, D. M. Dabbs, C. R. Pacheco, M. V. Giotto, D. E. Morse and I. A. Aksay, Macromolecules, 2007, 40, 5710 CrossRef CAS.
- I. Lee, J. Kwak, S. Haam and S. Y. Lee, J. Cryst. Growth, 2010, 312, 2107 CrossRef CAS PubMed.
- A. Arakaki, H. Nakazawa, M. Nemoto, T. Mori and T. Matsunaga, J. R. Soc. Interface, 2008, 5, 977 CrossRef CAS PubMed.
- K. Katagiri, Y. Imai, K. Koumoto, T. Kaiden, K. Kono and S. Aoshima, Small, 2011, 7, 1683 CrossRef CAS PubMed.
- A. Kanazawa, S. Kanaoka, N. Yagita, Y. Oaki, H. Imai, M. Oda, A. Arakaki, T. Matsunaga and S. Aoshima, Chem. Commun., 2012, 48, 10904 RSC.
- T. Osaka, T. Matsunaga, T. Nakanishi, A. Arakaki, D. Niwa and H. Iida, Anal. Bioanal. Chem., 2006, 384, 593 CrossRef CAS PubMed.
- R. P. Blakemore, Nature, 1975, 190, 377 CAS.
- T. Matsunaga, Y. Okamura, Y. Fukuda, A. T. Wahyudi, Y. Murase and H. Takeyama, DNA Res., 2005, 12, 157 CrossRef CAS PubMed.
- H. Nakazawa, A. Arakaki, S. Narita-Yamada, I. Yashiro, K. Jinno, N. Aoki, A. Tsuruyama, Y. Okamura, S. Tanikawa, N. Fujita, H. Takeyama and T. Matsunaga, Genome Res., 2009, 19, 1801 CrossRef CAS PubMed.
- T. Suzuki, Y. Okamura, R. J. Calugay, H. Takeyama and T. Matsunaga, J. Bacteriol., 2006, 188, 2275 CrossRef CAS PubMed.
- M. Tanaka, Y. Okamura, A. Arakaki, T. Tanaka, H. Takeyama and T. Matsunaga, Proteomics, 2006, 6, 5234 CrossRef CAS PubMed.
- A. Komeili, FEMS Microbiol. Rev., 2012, 36, 232 CrossRef CAS PubMed.
- D. Schüler, FEMS Microbiol. Rev., 2008, 32, 654 CrossRef PubMed.
- M. Tanaka, A. Arakaki and T. Matsunaga, Mol. Microbiol., 2010, 76, 480 CrossRef CAS PubMed.
- A. Komeili, H. Vali, T. J. Beveridge and D. K. Newman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3839 CrossRef CAS PubMed.
- N. Zeytuni, E. Ozyamak, K. Ben-Harush, G. Davidov, M. Levin, Y. Gat, T. Moyal, A. Brik, A. Komeili and R. Zarivach, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E480 CrossRef CAS PubMed.
- Y. Okamura, H. Takeyama and T. Matsunaga, J. Biol. Chem., 2001, 276, 48183 CrossRef CAS PubMed.
- C. Nakamura, J. G. Burgess, K. Sode and T. Matsunaga, J. Biol. Chem., 1995, 270, 28392 CrossRef CAS PubMed.
- D. Murat, A. Quinlan, H. Vali and A. Komeili, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5593 CrossRef CAS PubMed.
- R. Uebe, K. Junge, V. Henn, G. Poxleitner, E. Katzmann, J. M. Plitzko, R. Zarivach, T. Kasama, G. Wanner, M. Posfai, L. Bottger, B. Matzanke and D. Schüler, Mol. Microbiol., 2011, 82, 818 CrossRef CAS PubMed.
- M. I. Siponen, P. Legrand, M. Widdra, S. R. Jones, W. J. Zhang, M. C. Chang, D. Faivre, P. Arnoux and D. Pignol, Nature, 2013, 502, 681 CrossRef CAS PubMed.
- A. Komeili, Z. Li, D. K. Newman and G. J. Jensen, Science, 2006, 311, 242 CrossRef CAS PubMed.
- A. Scheffel, M. Gruska, D. Faivre, A. Linaroudis, J. M. Plitzko and D. Schüler, Nature, 2006, 440, 110 CrossRef CAS PubMed.
- Y. Amemiya, A. Arakaki, S. S. Staniland, T. Tanaka and T. Matsunaga, Biomaterials, 2007, 28, 5381 CrossRef CAS PubMed.
- L. Wang, T. Prozorov, P. E. Palo, X. Liu, D. Vaknin, R. Prozorov, S. Mallapragada and M. Nilsen-Hamilton, Biomacromolecules, 2012, 13, 98 CrossRef PubMed.
- M. Tanaka, E. Mazuyama, A. Arakaki and T. Matsunaga, J. Biol. Chem., 2011, 286, 6386 CrossRef CAS PubMed.
- A. Arakaki, A. Yamagishi, A. Fukuyo, M. Tanaka and T. Matsunaga, Mol. Microbiol., 2014, 93, 554 CrossRef CAS PubMed.
- F. Vereda, J. de Vicente and R. Hidalgo-Alvarez, J. Colloid Interface Sci., 2013, 392, 50 CrossRef CAS PubMed.
- A. Arakaki, F. Masuda, Y. Amemiya, T. Tanaka and T. Matsunaga, J. Colloid Interface Sci., 2010, 343, 65 CrossRef CAS PubMed.
- T. Prozorov, P. Palo, L. Wang, M. Nilsen-Hamilton, D. Jones, D. Orr, S. K. Mallapragada, B. Narasimhan, P. C. Canfield and R. Prozorov, ACS Nano, 2007, 1, 228 CrossRef CAS PubMed.
- J. M. Galloway, A. Arakaki, F. Masuda, T. Tanaka, T. Matsunaga and S. S. Staniland, J. Mater. Chem., 2011, 21, 15244 RSC.
- A. Arakaki, F. Masuda and T. Matsunaga, Mater. Res. Soc. Symp. Proc., 2009, 1187, KK03 CrossRef.
- J. Galloway, J. Bramble, A. Rawlings, G. Burnell, S. Evans and S. Staniland, Small, 2012, 8, 204 CrossRef CAS PubMed.
- J. H. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Diaz, Angew. Chem., Int. Ed., 2012, 51, 11960 CrossRef CAS PubMed.
- J. Ihli, W. C. Wong, E. H. Noel, Y.-Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169, DOI:10.1038/ncomms4169.
- E. H. Noel, Y.-Y. Kim, J. M. Charnock and F. C. Meldrum, CrystEngComm, 2013, 15, 697 RSC.
- S.-F. Chen, H. Coelfen, M. Antonietti and S.-H. Yu, Chem. Commun., 2013, 49, 9564 RSC.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS PubMed.
- T. Kato, T. Sakamoto and T. Nishimura, MRS Bull., 2010, 35, 127 CrossRef CAS.
- F. C. Meldrum, Int. Mater. Rev., 2003, 48, 187 CrossRef CAS PubMed.
- N. A. Sommerdijk and G. de With, Chem. Rev., 2008, 108, 4499 CrossRef CAS PubMed.
- A. Sugawara-Narutaki, Polym. J., 2013, 45, 269 CrossRef CAS.
- T. Kato, T. Suzuki and T. Irie, Chem. Lett., 2000, 29, 186 CrossRef.
- T. Kato, Adv. Mater., 2000, 12, 1543 CrossRef CAS.
- T. Kato, T. Suzuki, T. Amamiya, T. Irie, M. Komiyama and H. Yui, Supramol. Sci., 1998, 5, 411 CrossRef CAS.
- A. Sugawara and T. Kato, Chem. Commun., 2000, 487 RSC.
- A. Sugawara and T. Kato, Compos. Interfaces, 2004, 11, 287 CrossRef CAS PubMed.
- N. Hosoda and T. Kato, Chem. Mater., 2001, 13, 688 CrossRef CAS.
- N. Hosoda, A. Sugawara and T. Kato, Macromolecules, 2003, 36, 6449 CrossRef CAS.
- A. Sugawara, T. Ishii and T. Kato, Angew. Chem., Int. Ed., 2003, 42, 5299 CrossRef CAS PubMed.
- F. Zhu, T. Nishimura, H. Eimura and T. Kato, CrystEngComm, 2014, 16, 1496–1501 RSC.
- A. Sugawara, T. Nishimura, Y. Yamamoto, H. Inoue, H. Nagasawa and T. Kato, Angew. Chem., Int. Ed., 2006, 45, 2876 CrossRef CAS PubMed.
- J. E. Rebers and L. M. Riddiford, J. Mol. Biol., 1988, 203, 411 CrossRef CAS.
- A. Hecker, O. Testeniere, F. Marin and G. Luquet, FEBS Lett., 2003, 535, 49 CrossRef CAS.
- H. Inoue, T. Ohira and H. Nagasawa, Peptides, 2007, 28, 566 CrossRef CAS PubMed.
- Y. Yamamoto, T. Nishimura, A. Sugawara, H. Inoue, H. Nagasawa and T. Kato, Cryst. Growth Des., 2008, 8, 4062 Search PubMed.
- H. Kumagai, R. Matsunaga, T. Nishimura, Y. Yamamoto, S. Kajiyama, Y. Oaki, K. Akaiwa, H. Inoue, H. Nagasawa, K. Tsumoto and T. Kato, Faraday Discuss., 2012, 159, 483 RSC.
- J. Aizenberg, A. J. Black and G. M. Whitesides, J. Am. Chem. Soc., 1999, 121, 4500 CrossRef CAS.
- S. Tugulu, M. Harms, M. Fricke, D. Volkmer and H. A. Klok, Angew. Chem., Int. Ed., 2006, 45, 7458 CrossRef CAS PubMed.
- S. Kumar, T. Ito, Y. Yanagihara, Y. Oaki, T. Nishimura and T. Kato, CrystEngComm, 2010, 12, 2021 RSC.
- Y. Han, T. Nishimura and T. Kato, CrystEngComm, 2014, 16, 3540 RSC.
- Y. Han, T. Nishimura and T. Kato, Polym. J., 2014, 46, 499 CrossRef CAS.
- Y.-Y. Kim, A. S. Schenk, D. Walsh, A. N. Kulak, O. Cespedesc and F. C. Meldrum, Nanoscale, 2014, 6, 852 RSC.
- S.-S. Wang, A. Picker, H. Coelfen and A.-W. Xu, Angew. Chem., Int. Ed., 2013, 52, 6317 CrossRef CAS PubMed.
- Y.-Y. Kim, A. S. Schenk, J. Ihli, A. N. Kulak, N. B. J. Hetherington, C. C. Tang, W. W. Schmahl, E. Griesshaber, G. Hyett and F. C. Meldrum, Nat. Commun., 2014, 5, 4341, DOI:10.1038/ncomms5341.
- Y. Jiang, H. Gong, M. Grzywa, D. Volkmer, L. Gower and H. Coelfen, Adv. Funct. Mater., 2013, 23, 1547 CrossRef CAS.
- H. Tomono, H. Nada, F. Zhu, T. Sakamoto, T. Nishimura and T. Kato, J. Phys. Chem. B, 2013, 117, 14849 CrossRef CAS PubMed.
- F. Zhu, T. Nishimura, T. Sakamoto, H. Tomono, H. Nada, Y. Okumura, H. Kikuchi and T. Kato, Chem. – Asian J., 2013, 8, 3002 CrossRef CAS PubMed.
- T. Nishimura, T. Ito, Y. Yamamoto, M. Yoshio and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 2800 CrossRef CAS PubMed.
- H. Coelfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576 CrossRef CAS PubMed.
- Y. Bouligand, Tissue Cell, 1972, 4, 189 CrossRef CAS.
- J. E. Lydon, Liq. Cryst. Today, 2004, 13, 1 CrossRef CAS.
- Y. Yamamoto, T. Nishimura, T. Saito and T. Kato, Polym. J., 2010, 42, 583 CrossRef CAS.
- T. Saito, Y. Oaki, T. Nishimura, A. Isogai and T. Kato, Mater. Horiz., 2014, 1, 321 RSC.
- T. Sakamoto, A. Oichi, T. Nishimura, A. Sugawara and T. Kato, Polym. J., 2009, 41, 522 CrossRef CAS.
- T. Sakamoto, A. Oichi, Y. Oaki, T. Nishimura, A. Sugawara and T. Kato, Cryst. Growth Des., 2009, 9, 622 CAS.
- T. Sakamoto, Y. Nishimura, T. Nishimura and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 5856 CrossRef CAS PubMed.
- S. Kajiyama, T. Nishimura, T. Sakamoto and T. Kato, Small, 2014, 10, 1634 CrossRef CAS PubMed.
- X. Wu and S. Wang, Adv. Healthcare Mater., 2013, 2, 326 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |