
Design and synthesis of potent hydroxamate inhibitors with increased selectivity within the gelatinase family†
José María
Zapico
a,
Anna
Puckowska
ab,
Kamila
Filipiak
ac,
Claire
Coderch
a,
Beatriz
de Pascual-Teresa
*a and
Ana
Ramos
*a
aDepartamento de Química y Bioquímica, Facultad de Farmacia, Universidad CEU San Pablo, Urbanización Monteprincipe, 28668 Madrid, Spain. E-mail: bpaster@ceu.es; aramgon@ceu.es; Fax: (+34) 913510496; Tel: (+34) 913724724, (+34) 913724796
bDepartment of Organic Chemistry, Faculty of Pharmacy, Medical University of Bialystok, Poland
cDepartment of Molecular Biology, Faculty of Biotechnology and Natural Sciences, The John Paul II Catholic University of Lublin, 20-718 Lublin, Poland
First published on 1st October 2014
Abstract
MMP-2 is a validated target for the development of anticancer agents. Herein we describe the synthesis of a new series of potent phenylalanine derived hydroxamates, with increased MMP-2/MMP-9 selectivity compared to analogous hydroxamates described previously. Docking and molecular dynamics experiments have been used to account for this selectivity, and to clarify the role of the triazole ring in the binding process.
Introduction
Proteases are classified into serine proteases, cysteine proteases, aspartate proteases and metalloproteinases, based on differences in the hydrolysis mechanism of peptide bonds.1 The human degradome (proteases produced by cells) consists of at least 569 proteases and homologues, with metalloproteinases representing the largest class (194 described in humans).2The Metzincin superfamily of metalloproteinases can, in turn, be classified into the following four subfamilies, according to the slight differences in their catalytic site and the presence of additional domains: matrix metalloproteinases (MMPs), adamalysins (ADAM, ADAMTS and class III snake venom proteins), astacins (BMP1/TLL proteins and meprins) and bacterial serralysins.3 Metzincins are distinguished by a highly conserved HEXXHXXGXXH domain that bears three histidine residues that bind a zinc atom at the active site.4
The 26 MMPs described so far in the vertebrate family can be classified into several subfamilies according to their degree of sequence homology and substrate specificity: collagenases, gelatinases, stromelysins, matrilysines and membrane-type MMPs (MT-MMPs).5
Basically, all MMPs have been linked to disease development,6,7 mainly in cardiovascular disorders,8–10 arthritis,11 acute lung injury,12 and cancer.13–15
Gelatinase A (MMP-2) and gelatinase B (MMP-9) play an important role in cancer. They are over-expressed or deregulated in a variety of malignant tumors.16 It has been shown, in an experiment in MMP-2 knockout mice, that angiogenesis and tumour progression can be controlled by inhibiting the activity of this enzyme, suggesting the use of MMP-2 inhibitors for chemotherapy of cancer and other diseases.17 However, while MMP-2 is a validated target in cancer therapy, MMP-9 has both pro- and anti-tumorigenic effects, and is an anti-target protein in advanced stages of the disease.18
A large number of MMP inhibitors (MMPIs) have been reported so far. MMPIs containing a Zinc Binding Group (ZBG) and a lipophilic chain (called the P1′-segment) which interacts with the hydrophobic S1′ pocket are very effective. The latter is considered the “selectivity pocket”, as it is the region where MMPs exhibit the largest differences.19–22
In our research group we are interested in the design of MMP-2 inhibitors with selectivity over other metalloproteinases, especially over MMP-9. Because the active sites of both enzymes are very similar, it is necessary to carefully select the fragment that has to interact with the S1′ pocket. Our approach is based on the use of Cu(I)-catalyzed azide–alkyne cycloaddition, one of the most effective click reactions, to easily connect the selected ZBG with different non-polar P1′ substituents in order to explore the S1′ pocket. Following this strategy we have obtained inhibitors with high potency and MMP-2/MMP-9 selectivity (Fig. 1A).23–25 The potent but not selective MMPI (2R)-[(4-biphenylylsulfonyl)amino]-N-hydroxy-3-phenylpropionamide (BiPS)26 (Fig. 1B) has been the starting point for the design of the new series of inhibitors described herein. With the aim of increasing the selectivity between both gelatinases we have followed the same approach based on click chemistry.
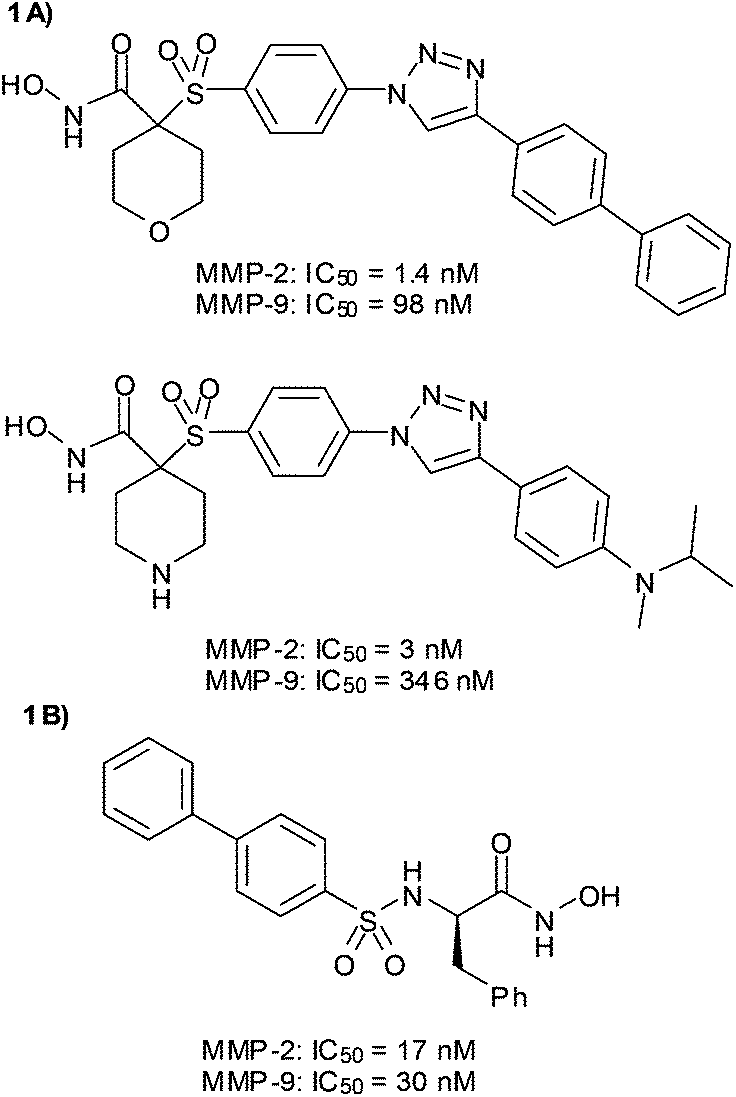 | ||
| Fig. 1 Structure and inhibitory activity of (A) clicked MMP-2 selective inhibitors and (B) BiPS. | ||
Results and discussion
Chemistry
Hydroxamates 1a–g were synthesized following the pathway outlined in Scheme 1. Compound 2 was obtained as described previously,27 starting from phenylalanine and 4-nitrobenzenesulfonyl chloride under basic conditions. The nitro group was reduced by catalytic hydrogenation to give amino acid 3, which was transformed into azide 4 by reaction with tert-butyl nitrite and azidotrimethylsilane. Reaction of the carboxylic acid with O-THP protected hydroxylamine, using EDCI as an amide coupling agent, gave hydroxamate 5. Copper(I) catalyzed Huisgen cycloaddition (CuAAC) between 5 and different alkynes gave compounds 6a–g, which were deprotected to obtain the final hydroxamates 1a–g.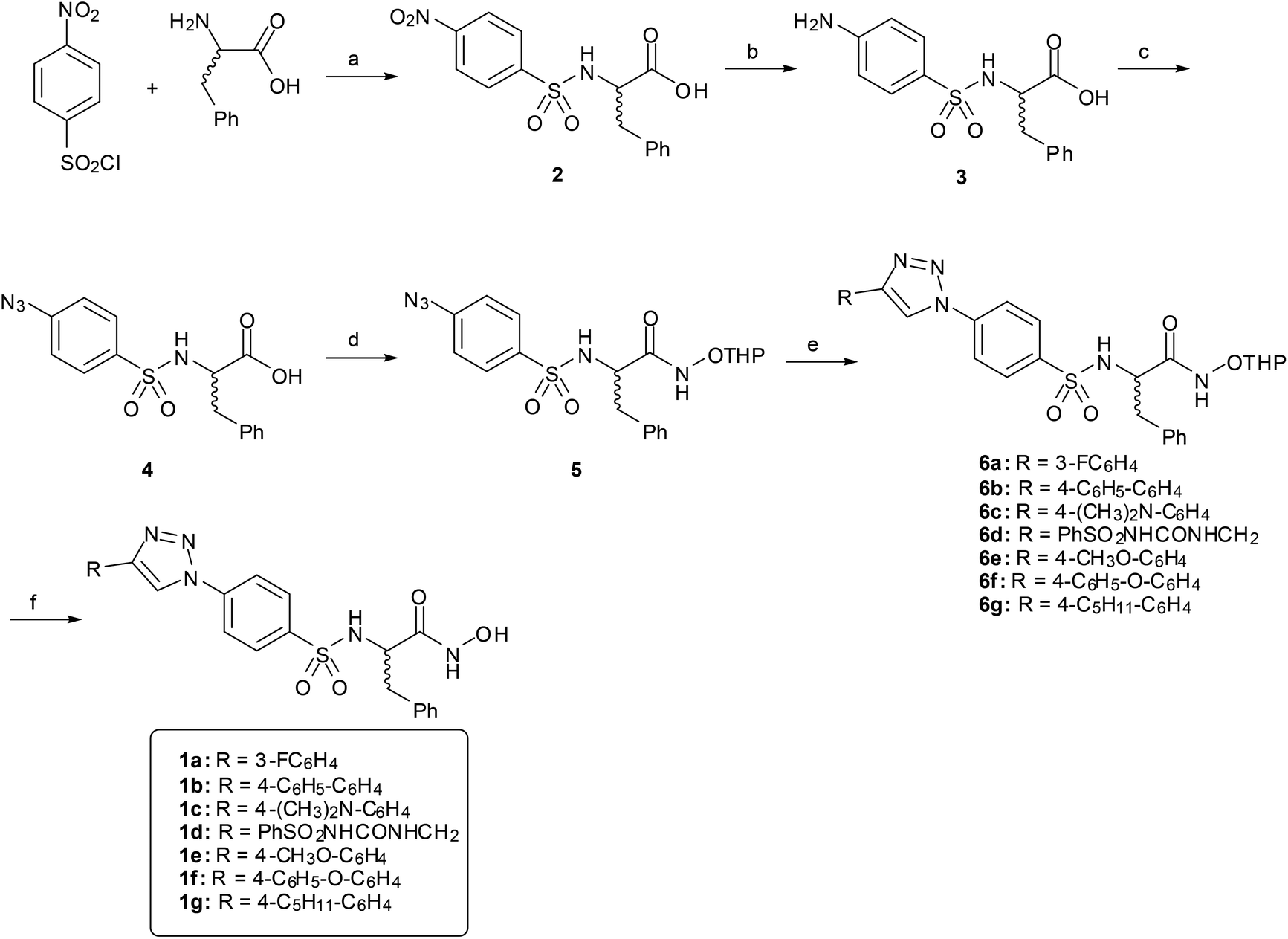 | ||
| Scheme 1 Synthesis of triazoles 1a–g. (a) NaOH–H2O; (b) H2, Pd/C, MeOH; (c) t-BuONO, TMSiN3, acetonitrile, 0 °C; (d) O-(tetrahydro-2H-pyran-2-yl)hydroxylamine, EDCI, HOBt, NMM, DMF; (e) alkyne, CuSO4, sodium ascorbate, DMF or t-BuOH–H2O; (f) 4 M HCl–dioxane, MeOH. | ||
(R) and (S) enantiomers of hydroxamates 1b, 1c and 1f were prepared separately following the same pathway starting from D or L phenylalanine.
In order to study the effect of the triazole ring on the activity, hydroxamate 7 was synthesized. This compound has a different substitution pattern in the triazole ring, with the biphenyl group attached to N1, instead of the C-4 position that is occupied in 1a–g. The synthetic pathway for 7 (Scheme 2) started from (±) phenylalanine methyl ester hydrochloride, which was transformed into sulphonamide 8 by reaction with pipsyl chloride. Sonogashira cross-coupling reaction with ethynyltrimethylsilane gave 9, which was deprotected under basic conditions to give 10, and was subsequently transformed into 11 by coupling with NH2OTHP. The terminal alkyne was clicked with 4-azidobiphenyl28 to give triazole 12, which was deprotected to give the desired compound 7.
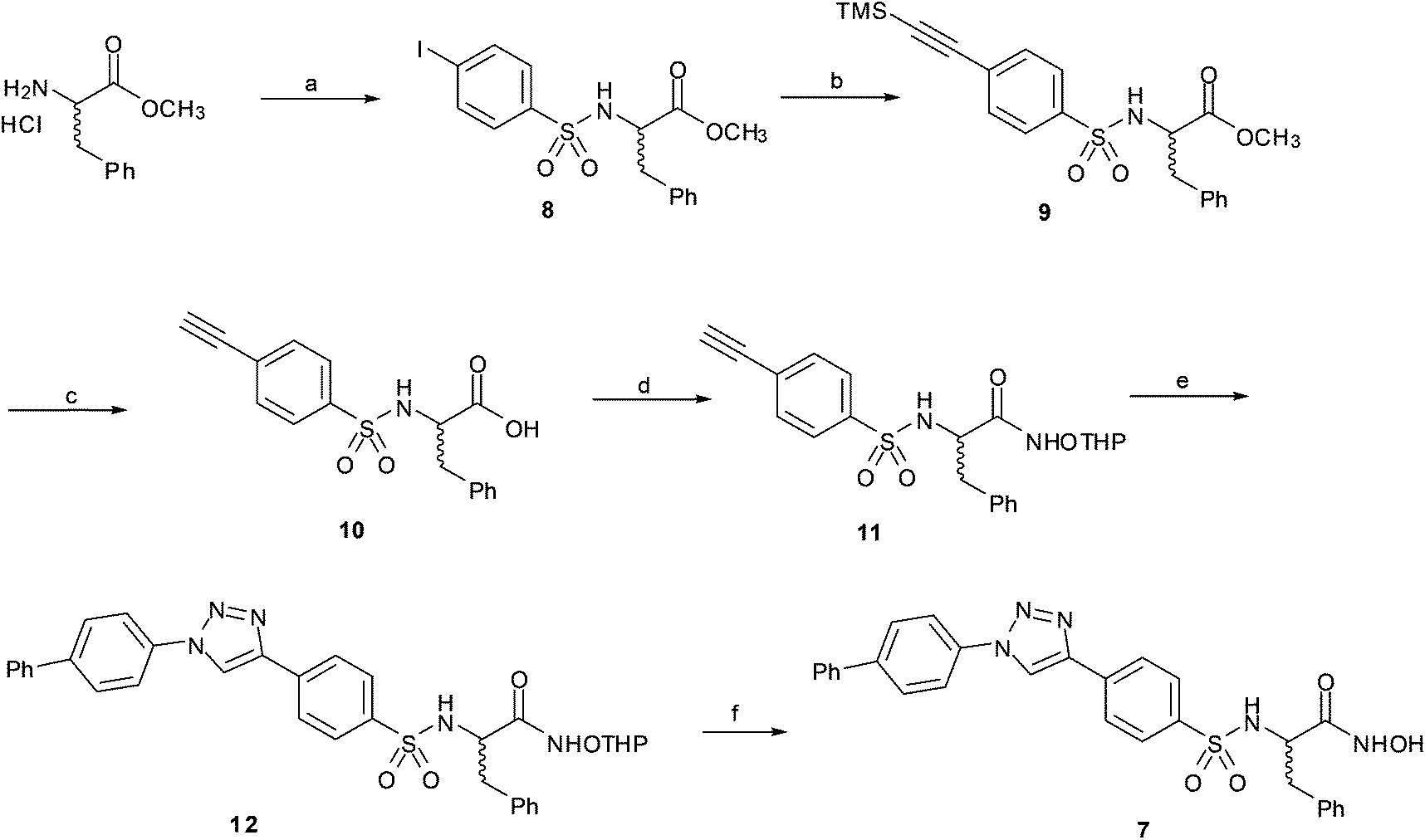 | ||
| Scheme 2 Synthesis of triazole 7: (a) pipsyl chloride, NMM, CH2Cl2; (b) TMSCCH, CuI, Et3N, Ph3P, Pd/C, reflux; (c) 1: KOH, dioxane, rt, 2: citric acid 20%; (d) NH2OTHP, EDCI, HOBt, NMM, DMF; (e) biphenylazide, CuSO4·5H2O, sodium ascorbate, DMF, (f) 4 M HCl–dioxane, MeOH. | ||
With the aim of improving the solubility of this class of compounds, a basic side chain was introduced into the sulphonamide nitrogen atom. We chose a morpholinoethyl moiety, as this fragment was used before to improve the solubility of valine-derived hydroxamates.29
Thus, compounds 13 and 14 were synthesized following the synthetic pathway shown in Scheme 3. Carboxylic acid 4 was protected as tert-butyl ester 15, and after alkylation with 4-(2-chloroethyl)morpholine in the presence of K2CO3, 16 was obtained. A sequence of tert-butyl deprotection to give 17 and coupling of the carboxylic acid with O-THP hydroxylamine gave azide 18. The CuAAC reaction with two different alkynes gave the corresponding triazoles 19–20 which were converted into hydroxamates 13 and 14 by acidic O-THP deprotection.
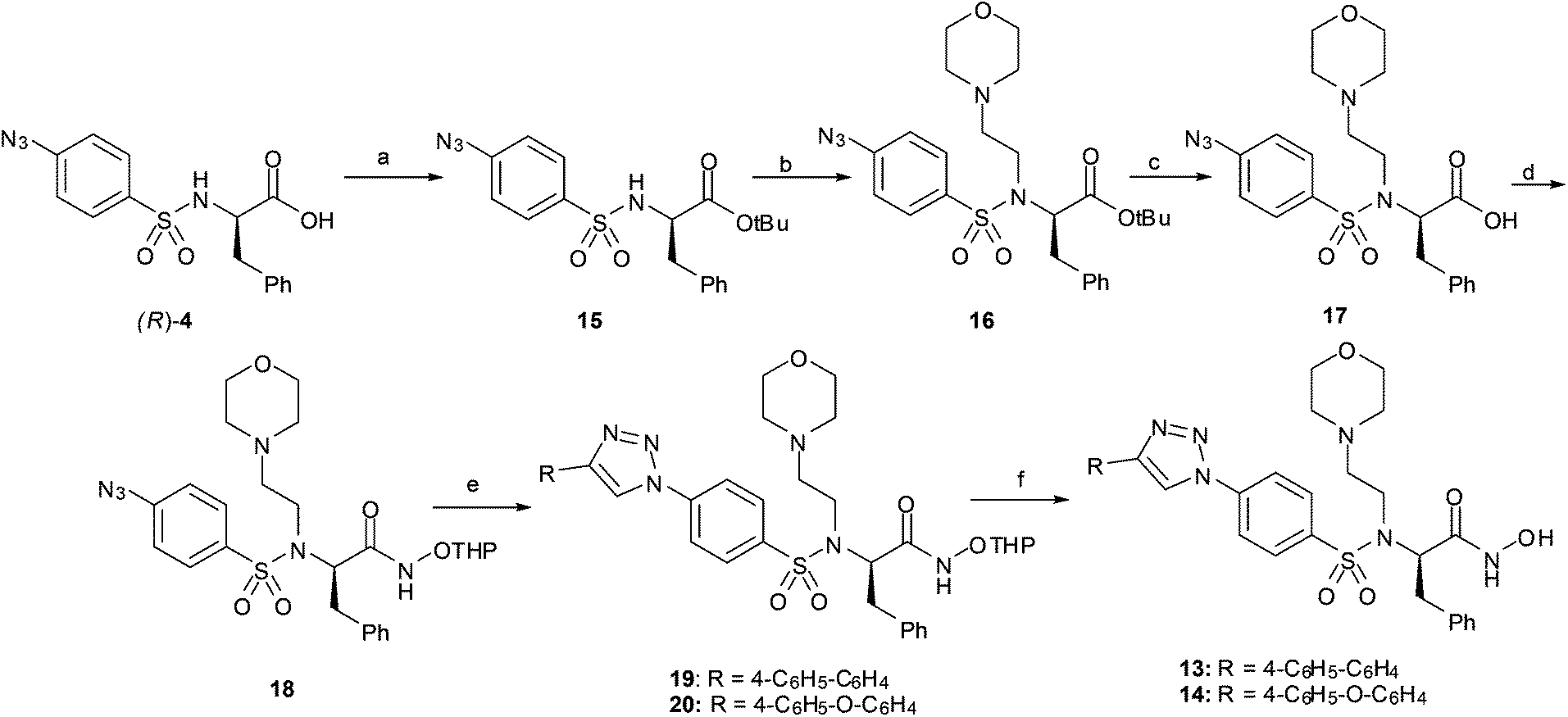 | ||
| Scheme 3 Synthesis of triazoles 13 and 14. (a) 2-Methylpropene, H2SO4, DCM, 86%; (b) 4-(2-chloroethyl)morpholine, K2CO3, DMF, 85%; (c) TFA, thioanisole, DCM, 80%; (d) NH2OTHP, EDC, HOBt, DMAP, DMF, 91%; (e) alkyne, CuSO4, sodium ascorbate, DMF; (f) HCl, dioxane, 60–75%. | ||
Biological evaluation
The inhibitory activities of hydroxamates 1a–g, 7, 13 and 14 against MMP-2 and MMP-9 were determined by the colorimetric method using a thiopeptide as a chromogenic substrate (Enzo Life Science Inc.). The assay conditions were similar to those described before [Fabre, 2013 #34].24 The results are collected as IC50 in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | MMP-2 IC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (nM) a (nM) |
MMP-9 IC50a (nM) |
Selectivity for MMP-9/MMP-2 |
| a IC50 values on the nM scale were determined using a colorimetric assay. Enzymatic data are the main values from three independent experiments. SD are within ±10%. | |||||
| Rac.-1a | 3-FC6H4 | H | 0.27 | 3.6 | 13 |
| Rac.-1b | 4-C6H5-C6H4 | H | 1.3 | 124 | 95 |
| (R)-1b | 4-C6H5-C6H4 | H | 0.75 | 58.5 | 78 |
| (S)-1b | 4-C6H5-C6H4 | H | 220 | >1000 | — |
| Rac.-1c | (CH3)2N-C6H4 | H | 0.88 | 2.11 | 2.4 |
| (R)-1c | (CH3)2N-C6H4 | H | 0.32 | 1.06 | 3.4 |
| (S)-1c | (CH3)2N-C6H4 | H | 49 | >1000 | — |
| Rac.-1d | PhSO2NHCONHCH2 | H | >500 | >1000 | — |
| Rac.-1e | 4-CH3O-C6H4 | H | 0.42 | 1.16 | 2.8 |
| Rac.-1f | 4-C6H5-O-C6H4 | H | 2.2 | 88 | 41 |
| (R)-1f | 4-C6H5-O-C6H4 | H | 1.08 | 36.8 | 34 |
| (S)-1f | 4-C6H5-O-C6H4 | H | >500 | >1000 | — |
| Rac.-1g | 4-C5H11-C6H4 | H | 8.18 | 372 | 46 |
| Rac.-7 | 1.32 | 5.54 | 4 | ||
| (R)-13 | 4-C6H5-C6H4 | 2-Morpholinoethyl | 2.66 | 6.34 | 2.4 |
| (R)-14 | 4-C6H5-O-C6H4 | 2-Morpholinoethyl | 1.99 | 29.9 | 15 |
All racemic compounds showed potent inhibitory activity against MMP-2, which was higher than the one reported for BiPS (IC50 = 17 nM).26
An exception was 1d, where the P1′ side chain is too polar to interact with the hydrophobic S1′ pocket of the enzyme. This result was expected, as we also found the lack of activity with this P1′ group in our previous studies.23
The best result in potency and selectivity was found for 1b, with an IC50 of 1.3 nM for MMP-2 and almost 100-fold less activity against MMP-9. This result shows that our click based strategy is a useful approach to improve the profile of this type of inhibitor, especially in the search for ligands capable of differentiating between the two gelatinases.
In the previously reported activity against MMP-3 of phenylalanine-derived hydroxamates, it was found that the preferred stereochemistry at the stereogenic centre corresponded to the R-isomer, which was more potent than the racemate.30 A similar result was reported for BiPS, which was 28-fold more active in MMP-9 than the S-isomer.26
In order to verify if the configuration of the stereogenic centre has an effect also on the inhibitory activity of 1b, 1c and 1f, both enantiomers of these compounds were synthesized and tested against both gelatinases. The results show that (R)-1b is 293-fold more potent against MMP-2 than (S)-1b, and 1.7-fold more potent than the racemic form, showing that the racemate activity is mainly due to the R-isomer. However, the MMP-2/MMP-9 selectivity of (R)-1b is slightly diminished with respect to the racemic mixture because the S-isomer was completely inactive in MMP-9 (IC50 > 1000). A similar effect was observed for 1c and 1f.
Compounds 13 and 14, where a basic chain was introduced into the sulphonamide nitrogen atom, presented better solubility during sample preparation. However, although the inhibition against MMP-2 was kept at the same level, the inhibitory activities against MMP-9 were increased giving a worse selectivity profile.
Interestingly, the same effect was observed for compound 7. This compound was obtained through a click reaction between 4-azidobiphenyl and an alkyne bearing the hydroxamate ZBG. The structure is closely related to 1b, with the only difference being in the substitution pattern of the triazole ring: the biphenyl group in 7 is attached to the N1 of the triazole, instead of the C-4 position that is occupied in 1b. While the activity of 7 against MMP-2 was maintained (IC50 = 1.32 nM), a high increase in the activity against MMP-9 was observed (IC50 = 5.54 nM) compared to the activity of 1b (Rac.-1b: IC50 = 124 nM; (R)-1b: 58.5 nM), with the corresponding loss of selectivity.
Compounds 1b, 1f and 1g, showing a promising selectivity profile within gelatinases, were chosen for the analysis of their activity towards other metalloproteinases (Table 2). The obtained profile showed that these compounds are devoid of activity towards MMP-1, whose inhibition is hypothesized to be connected with the musculoskeletal syndrome,31,32 and have lower activity towards MMP-8, an enzyme whose inhibition could enhance tumourigenesis and metastasis.33 Moreover, 1b and 1f displayed good activity towards MMP-13, an enzyme whose inhibition is targeted in the treatment of osteoarthritis, while having much lower potency towards MMP-14, proposed recently as an anti-target in the treatment of this disease.34
| 1b (nM) | 1f (nM) | 1g (nM) | |
|---|---|---|---|
| a Enzymatic data are the mean values from two independent experiments. SD are within ±10%. | |||
| MMP-1 | >10000 |
>10000 |
>1000 |
| MMP-2 | 1.3 | 2.2 | 8.2 |
| MMP-3 | 50 | 33.4 | 170 |
| MMP-7 | >500 | >1000 | >1000 |
| MMP-8 | >500 | 128.8 | 450 |
| MMP-9 | 124 | 88 | 372.5 |
| MMP-10 | 181 | 103.4 | 336 |
| MMP-12 | 3.8 | 2 | 14.2 |
| MMP-13 | <5 | 1.5 | 36.8 |
| MMP-14 | 555.7 | 256.6 | >200 |
In order to explain the difference in selectivity between compounds 1b and 7 we first studied both separately by means of ab initio quantum mechanics (QM) methods. Despite being structurally very similar, the difference in the substitution pattern of the triazole determines the relative orientation of the P1′-segment with the rest of the compound. The main difference arises from the energy profile of the dihedral angle that describes the torsion around Csp2–Csp2 and Csp2–Nsp2. The first dihedral angle presents energy minima at ±15° and global maxima of 3.3 kcal mol−1 at ±90°, whereas the dihedral angle between Csp2–Nsp2 presents energy minima between ±30–45°, a local maximum of 0.7 kcal mol−1 at 0°, and a global maximum of 1.6 kcal mol−1 at ±90° (ESI† Fig. 1). This means that compound 1b will be almost flat from the triazole to the end of the P1′-segment, whereas compound 7 will form an angle of around 45° between the triazole and the P1′-segment.
The docking experiments for compound 1b were carried out with a deprotonated hydroxamic acid moiety on MMP-2 and MMP-9, and with a protonated side chain of Glu404. The obtained poses were overall similar to those obtained in our previous work,35 so these complexes were used to build the complexes of 7 with MMP-2 and MMP-9 by changing the atom type in the triazole ring and allowing the geometries to relax in the molecular mechanics force field. During the 10 ns molecular dynamics (MD) simulations we monitored the per-residue protein–ligand interactions, especially those established between the P1′-segment and the S1′ pocket. Compounds 1b and 7 establish mainly van der Waals interactions in the complexes with both MMP-2 and MMP-9 (ESI† Fig. 2). Both compounds establish strong van der Waals interactions with the amino acids of the first segment of the Ω-loop within both MMPs, especially with Ile424(MMP-2)/Met422(MMP-9) that interacts with the triazole through the backbone; and Tyr425(MMP-2)/Tyr423(MMP-9) and Thr426(MMP-2)/Arg424(MMP-9) that stack their side chains with the triazole and biphenyl moieties. The slight difference in the interaction energy between both compounds and the side chain of Arg424 in the case of MMP-9 can be explained by the difference in flexibility at the end of the P1′-segment mentioned before. The Csp2–Csp2 bond present in the P1′ fragment of 1b is more rigid than the Csp2–Nsp2 bond present in 7, which induces fewer fluctuations in the interaction with the highly flexible side chain of Arg424, resulting in a better interaction energy for 1b compared to 7. The interactions established between compounds 1b and 7 and the last segment of the Ω-loop can account for the lack of selectivity of compound 7. In our previous work we proposed that the selectivity of this type of molecules, especially the biphenyl derivatives, arises from the presence of the Phe431-Arg432 motif in MMP-2, which is mutated to Pro429-Pro430 in MMP-9. This difference makes the Ω-loop more rigid in MMP-9 than in MMP-2.25 In the case of MMP-2, both compounds interact with the side chain of Phe431 (Fig. 2). Interestingly, the study of the MD trajectories and the minimized cooled structure obtained after the 10 ns simulations showed that compound 7, having a torsion of around 45° between the triazole and the P1′-segment, due to the Csp2–Nsp2 bond, is able to adopt more easily the shape of the S1′ pocket, establishing a van der Waals interaction with Pro429 (Fig. 2). This interaction is absent in compound 1b which is flatter and has less flexibility. This difference could explain the different in vitro activity observed for both compounds. Although we have explained the difference in selectivity between compounds 1b and 7, we were not able to give the rationale of the difference in binding affinity between both enantiomers of compound 1b as they did not show any significant differences in the dynamic behaviour and interactions with the two proteins in the time span and conditions of our simulations.
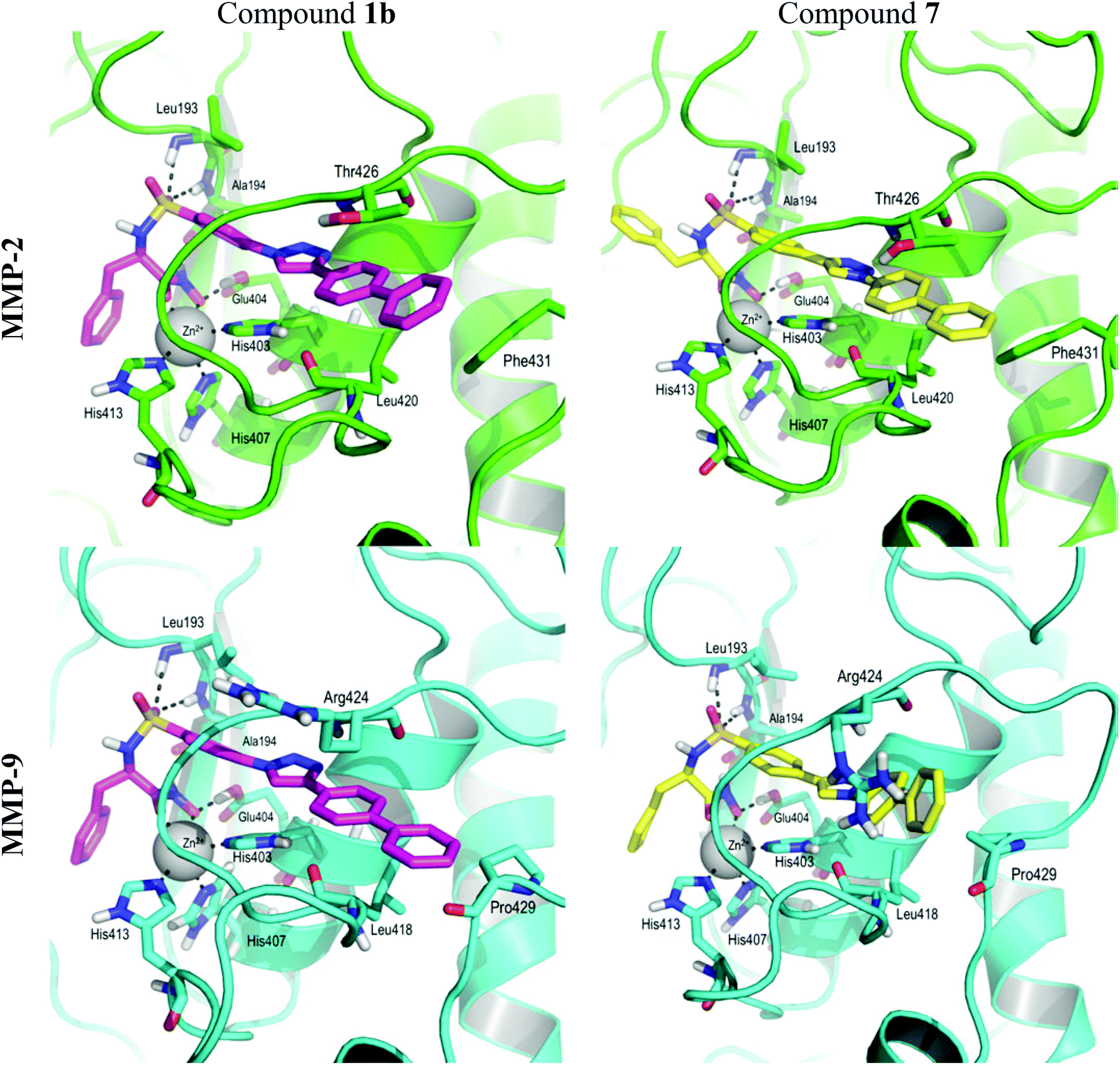 | ||
| Fig. 2 PyMOL cartoon representation of the complexes between MMP2 (green) and MMP9 (cyan) and compounds 1b (magenta) and 7 (yellow) respectively. For the sake of clarity, only polar hydrogens are displayed; and only the side chains of the amino acids that stabilize the ZBG-end and those that bring about the most different interactions in the Ω-loop are shown as sticks. | ||
Experimental section
Molecular modeling
The proteins were prepared and the docking experiments were carried out as in the previous work.25 The ligands (compounds 1b and 7) were built using the Maestro LigPrep module (http://www.Schrodinger.com). pKa of titratable groups were calculated using the Sparc pKa online calculator,36 and the non-protonated form of the hydroxamate was used for the docking calculations making use of the Glide module.37–39 The center of the box was positioned on the catalytic zinc ion present in the active site. The box size was set up to enclose the ligand-binding domain to ensure an adequate exploration of the binding poses. The docking procedure was performed with XP (extra precision) mode, and a van der Waals radii scale factor of 1.0/0.8 for the receptor and the ligand, respectively. The best-obtained result for each ligand in each complex was considered for analysis of the ligand–receptor interactions and subsequent molecular modeling simulations.For the MD simulations the charge distribution for the ligands was obtained by fitting the quantum mechanically calculated (RHF/6-31G**//RHF/3-21G*) molecular electrostatic potential (MEP) of the geometry-optimized molecules to a point charge model, as implemented in Gaussian 03 (Gaussian, Inc., Wallingford, CT). Consistent bonded and non-bonded AMBER parameters for both molecules were assigned by analogy or through interpolation from those already present in the AMBER database (ff03). Each MMP molecular system was immersed in a truncated octahedron containing ∼10000 TIP3P water molecules40 and four Na+ ions41 to achieve system electroneutrality. The sander and pmemd modules of the AMBER12 suite (http://ambermd.org/) were used for the restrained and unrestrained MD simulations, respectively. Periodic boundary conditions were applied and electrostatic interactions were treated using the smooth particle mesh Ewald method42 with a grid spacing of 1 Å. The cutoff distance for the non-bonded interactions was 9 Å, the SHAKE43 algorithm was applied to all bonds, and an integration step of 2.0 fs was used throughout. After an initial energy minimization of the water molecules and counterions, the system was heated to 300 K in 25 ps, after which the solvent was allowed to redistribute around the positionally restrained solute for 220 ps. After this time, the system was allowed to move freely so as to explore the mutual adaptation between the ligand and the protein. Snapshots from each 10 ns MD trajectory were collected every 20 ps for further analysis carried out with the ptraj module of AMBER to monitor the hydrogen bonding distances between the ligands and the protein. The per-residue energy decomposition of the 10 ns MD trajectory was carried out using the ISM program.44,45
Chemistry
:4) to afford 3 (1.57 g, 78%) as a white solid, mp 205–206 °C (EtOH). (Found: C, 56.04; H, 5.00; N, 8.82; S, 9.97. C15H16N2O4S requires C, 56.24; H, 5.03; N, 8.74; S, 10.01%); νmax (KBr)/cm−1 3372, 3306, 3284, 1719; δH (300 MHz, DMSO-d6) 2.68 (1H, dd, J 13.4, 7.9, ½CH2), 2.91 (1H, dd, J 13.4, 6.1, ½CH2), 3.69–3.76 (1H, m, CH), 5.90 (2H, bs, NH2), 6.48 (2H, d, J 8.5, ArH), 7.09–7.12 (2H, m, ArH), 7.17–7.27 (5H, m, ArH), 7.69 (1H, d, J 8.5, SO2NH), 12.64 (1H, bs, COOH); δC (75.4 MHz, DMSO-d6) 37.9, 57.1, 112.4, 125.8, 126.4, 128.1, 128.4, 129.2, 136.9, 152.4, 172.4. EM (ESI+) m/z 319.00 [M − H]+.
:3) to afford 5 (1.14 g, 68%) as a yellow solid. This compound is present as a diastereoisomeric mixture (55:45), mp 142–144 °C. (Found: C, 53.81; H, 5.14; N, 15.37; S, 7.18. C20H23N5O5S requires C, 53.92; H, 5.20; N, 15.72; S, 7.20%); νmax (KBr)/cm−1 3213, 2130, 2097, 1673; δH (300 MHz, CDCl3) 1.45–1.65 (4H, m, CH2), 1.68–1.82 (2H, m, CH2), 2.87–3.05 (2H, m, CH2Ph), 3.54–3.58 (1H, m, CHN), 3.81–3.92 (2H, m, OCH2), 4.72 (0.45H, m, O–CH–O, isomer b), 4.77 (0.55H, m, O–CH–O, isomer a), 5.47, (0.55H, d, J 7.8, ½SO2NH, isomer a), 5.52 (0.45H, d, J 7.8, ½SO2NH, isomer b), 6.96–7.02 (5H, m, ArH), 7.18–7.21 (2H, m, ArH), 7.59 (2H, d, J 8.4, ArH), 9.05 (1H, s, CONH, isomer a), 9.17 (1H, s, CONH, isomer b); δC (75.4 MHz, CDCl3): 18.4, 18.6, 24.9, 27.9, 38.6, 39.0, 56.6, 62.5, 62.7, 102.5, 119.5, 127.4, 128.9, 129.0, 129.3, 134.9, 135.2, 145.0, 167.5. EM (ESI+) m/z 468.00 [M + Na]+.
:2) to afford (R)-5 (594 mg, 92%) as a yellow solid. This compound is present as a diastereoisomeric mixture (55:45), mp 137–138 °C.
:2) to afford (S)-5 (589 mg, 92%) as a yellow solid. This compound is present as a diastereoisomeric mixture (55:45), mp 137–138 °C.
Preparation of triazoles: general procedure 1
To a suspension of azide 5 (1 equiv.) and the corresponding alkyne (1–1.5 equiv.) in t-BuOH–H2O (1:1) or DMF were added sodium ascorbate (2 equiv. of freshly prepared 1 M solution in water) and copper(II) sulfate pentahydrate (0.5 equiv. of a 0.25 M solution in water) under argon. The mixture was stirred vigorously overnight, and then diluted with water (20 mL) and extracted with AcOEt. The organic layer was washed successively with saturated aqueous NH4Cl and brine. The extract was dried (MgSO4), filtered and evaporated to dryness, and the residue was chromatographed on silica gel.
:1 + 2% MeOH), mp 146–147 °C. (Found: C, 59.06; H, 5.08; N, 12.27; S 5.70. C28H28FN5O5S requires C, 59.46; H, 4.99; N, 12.38; S, 5.67%); νmax (KBr)/cm−1 3203, 1665, 1620; δH (300 MHz, DMSO-d6) 1.43–1.59 (6H, m, 3CH2), 2.66–2.73 (1H, m, ½CH2Ph), 2.79–2.89 (1H, m, ½CH2Ph), 3.38–3.49 (1H, m, CHN), 3.76–3.85 (1H, m, OCH2, isomer a), 3.93–3.98 (1H, m, OCH2, isomer b), 4.39 (0.5H, s, O–CH–O, isomer a), 4.57 (0.5H, s, O–CH–O, isomer b), 7.11–7.29 (6H, m, ArH), 7.55–7.62 (1H, m, ArH), 7.76–7.84 (4H, m, ArH), 8.02 (2H, dd, J 8.7, 1.4, ArH), 8.54 (1H, bs, SO2NH), 9.51 (1H, s, triazole), 11.25 (0.5H, bs, CONH, isomer a) 11.32 (0.5H, bs, CONH, isomer b); δC (75.4 MHz, DMSO-d6): 18.2, 24.5, 27.7, 38.5, 55.2, 61.3 (61.4 b), 101.0, 112.0 (d, 2JCF 22.5), 115.17 (d, 2JCF 21.0), 119.9, 120.9 (d, 2JCF 74.2), 126.4, 128.0, 128.1, 129.2 (129.3 b), 131.3 (d, 3JCF 9.0), 132.31 (d, 3JCF 9.0), 136.5, 136.8, 138.6, 140.9, 146.4, 162.6 (d, 1JCF 241.5), 166.6 (166.7 b). EM (ESI+): m/z 588.26 [M + Na]+.
:1, 5 mL), (R)-6b (68 mg, 32%) was produced as a white solid after chromatography purification (DCM–MeOH 0.6% MeOH), mp 199–200 °C (hexane–EtOAc).
:1, 5 mL), (S)-6b (105 mg, 50%) was produced as a white solid after chromatography purification (DCM–MeOH 0.6% MeOH), mp 199–200 °C (hexane–EtOAc).
:1, 5 mL), (R)-6c (130 mg, 65%) was produced as a yellow solid after chromatography purification (DCM–MeOH 0.75% MeOH), mp 203–204 °C (hexane–EtOAc).
:1, 5 mL), (S)-6c (130 mg, 65%) was produced as a yellow solid after chromatography purification (DCM–MeOH 0.75% MeOH), mp 203–204 °C (hexane–EtOAc).
:1 + 2% MeOH), mp 189–190 °C. (Found: C, 63.70; H, 5.26; N, 10.87; S, 4.95; C34H33N5O6S requires: C, 63.83; H, 5.20; N, 10.95; S, 5.01%); νmax (KBr)/cm−1 3166, 1657. δH (300 MHz, DMSO-d6): 1.43–1.58 (6H, m, 3CH2), 2.66–2.73 (1H, m, ½CH2Ph), 2.79–2.86 (1H, m, ½CH2Ph), 3.42–3.49 (1H, m, CH), 3.76–3.85 (1H, m, OCH2, isomer a), 3.93–3.98 (1H, m, OCH2, isomer b), 4.38 (0.5H, s, O–CH–O, isomer a), 4.57 (0.5H, s, O–CH–O, isomer b), 7.08–7.21 (10H, m, ArH), 7.41–7.46 (2H, m, ArH), 7.78–7.82 (2H, dd, J 8.6, 2,1, ArH), 7.89 (2H, d, J 8.6, ArH), 8.03 (2H, d, J 7.4, ArH), 8.5 (1H, bs, SO2NH), 9.39 (1H, s, triazole), 11.32 (1H, bs, CONH); δC (75.4 MHz, DMSO-d6) 18.2, 24.5, 27.6, 38.5, 55.2, 61.4, 101.1, 118.93, (118.98 b), 119.2, 119.7, 123.7, 125.1, 126.4, 127.2, 128.0, 128.15, 129.2 (129.3 b), 130.1, 136.5, 136.8, 138.7, 140.7, 147.1, 156.3, 156.9, 166.5 (166.7 b). MS (ESI+): m/z 662.26 [M + Na]+.
:1, 5 mL), (R)-6f (170 mg, 79%) was produced as a white solid after chromatography purification (DCM–MeOH 0.6% MeOH), mp 169–170 °C (hexane–EtOAc).
:1, 5 mL), (S)-6f (175 mg, 81%) was produced as a white solid after chromatography purification (DCM–MeOH 0.6% MeOH), mp 169–170 °C (hexane–EtOAc).
:1 + 2% MeOH), mp 189–190 °C. (Found: C, 63.72; H, 6.43; N, 11.32; S, 5.17; C33H39N5O5S requires: C, 64.16; H, 6.36; N, 11.34; S, 5.19; νmax (KBr)/cm−1 3291, 1690. δH (300 MHz, DMSO-d6) 0.87 (3H, t, J 6.8, CH3), 1.30–1.63 (12H, m, 6CH2), 2.62–2.73 (3H, t, J 7.6, CH2Ar + m, ½CH2Ph), 2.79–2.89 (1H, m, ½CH2Ph), 3.42–3.49 (1H, m, CH), 3.78–3.85 (1H, m, OCH2, isomer a), 3.92–3.99 (1H, m, OCH2, isomer b), 4.37 (0.5H, s, O–CH–O, isomer a), 4.57 (0.5H, s, O–CH–O, isomer b), 7.01–7.21 (5H, m, ArH), 7.34 (2H, d, J 8.1, ArH), 7.80 (2H, dd, J 8.7, 2.1, ArH), 7.87 (2H, d, J 8.0, ArH), 8.03 (2H, dd, J 8.6, 1.6, ArH), 8.52 (1H, bs, SO2NH), 9.38 (1H, s, triazole), 11.24 (0.5H, bs, CONH isomer a), 11.32 (0.5H, bs, CONH, isomer b); δC (75.4 MHz, DMSO-d6) 13.9, 18.2, 21.9, 24.5, 27.7, 30.5, 30.8, 34.8, 38.5, 55.3, 61.4, 101.0, 119.1 (119.7 b), 125.3, 126.4, 127.4, 128.04, 128.1, 128.9, 129.2 (129.3 b), 136.5, 136.8, 138.7, 140.79, 142.7, 147.7, 166.5 (166.7 b). MS (ESI+): m/z 640.33 [M + Na]+.
Cleavage of the tetrahydropyrane protecting group: general procedure 2
To a suspension of the corresponding tetrahydro-2H-pyranyl derivative in DCM (5 mL) were added 4 M HCl in dioxane (4 equivalents) and MeOH (0.1 mL). After stirring at room temperature for 1 h the reaction mixture was concentrated under vacuum and the obtained solid was washed with DCM–hexane 1:1.
:1) to give 9 (0.69 g, 74%) as a white solid, mp. 118–119 °C (DCM–hexane). (C, 60.74; H, 5.96; N, 3.50; S, 7.66; C21H25NO4SSi requires: C, 60.69; H, 6.06; N, 3.37; S, 7.72%). ]+. νmax (KBr)/cm-1 3239, 2154, 1745, 1587. δH (300 MHz, CDCl3) 0.27 (9H, s, (CH3)3Si), 2.97–3.09 (2H, m, CH2Ar), 3.52 (3H, s, OCH3), 4.17–4.24 (1H, m, CH), 5.29 (1H, d, J 9.2, SO2NH), 7.05–7.23 (2H, m, ArH), 7.24–7.26 (3H, m, ArH), 7.49 (2H, d, J 8.3 Hz, ArH), 7.66 (2H, d, J 8.3, ArH); δC (75.4 MHz, CDCl3) −0.3, 39.1, 52.4, 56.6, 98.4, 102.9, 126.8, 127.2, 127.7, 129.3, 132.2, 134.7, 138.9, 171.0. MS (ESI+) m/z 438.10 [M + Na]+.
:8, 1:9) to give 10 as a white solid (0.165 g, 83%), mp 136–137 °C. νmax (KBr)/cm−1 3335, 3261, 2108, 1705, 1590. δH (300 MHz, CDCl3) 2.98 (1H, dd, J 13.9, 7.3, ½CH2Ar), 3.15 (1H, dd, J 13.9, 4.8, ½CH2Ar), 3.26 (1H, s, ArCCH), 4.19–4.27 (1H, m, CH), 5.28 (1H, d, J 8.3, SO2NH), 7.08–7.09 (2H, m, ArH), 7.24–7.27 (3H, m, ArH), 7.48 (2H, d, J 8.3, ArH), 7.67 (2H, d, J 8.3, ArH); δC (75.4 MHz, CDCl3) 38.7, 56.5, 80.8, 81.9, 126.8, 126.9, 127.4, 128.7, 129.3, 132.5, 134.5, 139.5, 176.0. MS (ESI+) m/z 352.04 [M + Na]+.
:3) to afford 11 (0.135 g, 69%) as a white solid. This compound is present as a diastereoisomeric mixture (55:45), mp 148–149 °C. (C, 61.86; H, 5.69; N, 6.61; S, 7.34; C22H24N2O5S requires: C, 61.67; H, 5.65; N, 6.54; S, 7.48%). νmax (KBr)/cm−1 3276, 3202, 2110, 1660. δH (300 MHz, CDCl3) 1.57–1.77 (6H, m, 3 × CH2), 2.88–3.07 (2H, m, CH2Ar), 3.27 (1H, s, ArCCH), 3.55–3.59 (1H, m, CH), 3.80–3.89 (2H, m, CH2O), 4.7 (0.55H, s, O–CH–O, isomer a), 4.79 (0.45H, s, O–CH–O, isomer b), 5.43 (1H, bs, SO2NH), 6.99 (2H, d, J 7.32, ArH), 7.18–7.22 (3H, m, ArH), 7.48 (2H dd, J 8.3, 2.8, ArH) 7.58 (2H dd, J 8.3, 2.8, ArH), 8.79 (0.55H, s, CONH, isomer a), 9.09 (0.45H, s, CONH, isomer b); δC (75.4 MHz, CDCl3) 18.4, 24.8, 27.8, 38.9, 56.4, 62.4, 80.9, 82.2, 102.5, 126.8, 126.9, 127.3, 128.8, 128.9, 129.1, 132.7, 134.9, 162.7. MS (ESI+) m/z = 451.14 [M + Na]+.
:1) to give 15 (4.05 g, 86%) as a yellowish solid, mp 75.5–77.4 °C. νmax (KBr)/cm−1 3271, 2135, 2106, 1730, 1589. δH (400 MHz, DMSO-d6) 1.15 (9H, s, (CH3)3), 2.91 (1H, dd, J 6.4, 1.3, CH2Ar), 3.94–4.04 (1H, m, CH), 5.43 (1H, d, J 9.3 NH), 6.93 (2H, d, J 8.7 ArH), 7.01–7.08 (2H, m, ArH), 7.09–7.20 (3H, m, ArH), 7.64 (2H, d, J 8.7, ArH); δC (100.6 MHz, DMSO-d6) 27.7, 39.5, 57.1, 82.7, 119.3, 127.1, 128.4, 129.2, 129.6, 135.4, 136.2, 144.6, 170.0. MS (ESI+): m/z = 401.2 [M − H]+.
:2) to give 16 (872 mg, 85%) as a yellow oil. νmax (KBr)/cm−1 2971, 2853, 2806, 2129, 2106, 2106, 1730, 1589. δH (400 MHz, DMSO-d6) 1.25 (9H, s), 2.38–2.56 (5H, m), 2.57–2.65 (1H, m), 2.97 (1H, dd, J = 14.0, 7.2), 2.97 (1H, dd, J = 14.0, 8.2), 3.34–3.53 (2H, m), 3.68 (4H, t, J 4.6), 4.67 (1H, t, J 7.7), 7.04 (2H, d, J 8.7 ArH), 7.18–7.31 (5H, m, ArH), 7.73 (2H, d, J 8.7, ArH); δC (100.6 MHz, DMSO-d6) 27.7, 37.3, 42.7, 53.9, 58.8, 61.7, 66.9, 82.2, 119.2, 126.9, 128.5, 129.2, 129.4, 136.3, 136.6, 144.6, 169.4. MS (ESI+): m/z = 516.1 [M + H]+.
:1) to afford 17 (580 mg, 80%) as a white foam, mp 160.2 °C (dec.). νmax (KBr)/cm−1 3430, 2926, 2131, 2101, 1617, 1591. δH (400 MHz, DMSO-d6) 2.70–3.05 (7H, m), 3.45–3.60 (3H, m), 3.60–3.80 (4H, m), 4.79 (1H, dd, J = 9.7, 3.9), 6.94 (2H, d, J 8.4 ArH), 7.12–7.23 (5H, m, ArH), 7.60 (2H, d, J 8.4, ArH); δC (100.6 MHz, DMSO-d6) 36.9, 41.0, 52.4, 56.9, 62.9, 64.5, 119.1, 126.6, 128.5, 128.9, 129.3, 135.6, 137.8, 144.5, 174.7. MS (ESI+): m/z = 460.1 [M + H]+.
:3) to afford 18 (600 mg, 91%). This compound is present as a diastereoisomeric a:b mixture (60:40), mp 59.8–61.5 °C. νmax (KBr)/cm−1 3206, 2126, 2101, 1693, 1592; δH (400 MHz, CDCl3) 1.50–1.65 (3H, m), 1.68–1.85 (3H, m), 2.35–2.47 (2H, m), 2.50–2.65 (4H, m), 2.65–2.85 (1H, m), 3.25–3.52 (2H, m), 3.54–3.82 (6H, m), 3.94–4.02 (0.6H, m, isomer a), 4.05–4.13 (0.4H, m, isomer b), 4.42–4.49 (0.4H, m, isomer b), 4.50–4.55 (0.6H, m, isomer a), 4.84 (0.6H, d, J 2.6, isomer a), 4.97 (0.4H, s, isomer b), 6.90–7.03 (4H, m, ArH), 7.08–7.18 (3H, m, ArH), 7.58 (1.2H, d, J 8.6, ArH, isomer a), 7.64 (0.8H, d, J 8.5, ArH, isomer b), 11.01 (0.4H, bs, CONH, isomer b), 11.13 (0.6H, bs, CONH, isomer a); δC (100.6 MHz, CDCl3): 18.9 (isomer b), 19.5 (isomer a), 25.0, 28.1 (isomer b), 28.3 (isomer a), 34.4 (isomer a), 34.7 (isomer b), 41.6 (isomer a), 42.0 (isomer b), 53.5, 57.0 (isomer b), 57.2 (isomer a), 59.9 (isomer a), 60.0 (isomer b), 62.9 (isomer b), 63.4 (isomer a), 66.4 (isomer a), 66.5 (isomer b), 102.5 (isomer b), 103.3 (isomer a), 119.4, 126.7, 128.6, 128.8, 128.9, 135.4 (isomer a), 135.6 (isomer b), 136.7 (isomer a), 137.0 (isomer b), 144.7, 167.9 (isomer a), 168.1 (isomer b). MS (ESI) m/z 559.1 [M + H]+.
:b mixture (60:40), mp 196.1 °C (dec.). νmax (KBr)/cm−1 3374, 2951, 2854, 2814, 1704, 1597; δH (400 MHz, CDCl3) 1.50–1.65 (3H, m), 1.70–1.88 (3H, m), 2.40–2.54 (2H, m), 2.56–2.70 (4H, m), 2.75–2.85 (1H, m), 3.35–3.85 (8H, m), 3.94–4.02 (0.6H, m, isomer a), 4.05–4.13 (0.4H, m, isomer b), 4.50–4.59 (1H, m), 4.89 (0.6H, d, J 2.7, isomer a), 5.01 (0.4H, s, isomer b), 6.92–7.00 (2H, m, ArH), 7.04–7.08 (3H, m, ArH), 7.36 (1H, t, J 7.3, ArH), 7.45 (2H, t, J 7.5, ArH), 7.63 (2H, d, J 7.5, ArH), 7.62–7.78 (6H, m, ArH), 7.99 (2H, d, J 8.2, ArH), 8.34 (1H, s, triazole), 11.12 (0.4H, bs, CONH, isomer b), 11.24 (0.6H, bs, CONH, isomer a); δC (100.6 MHz, CDCl3): 19.0 (isomer b), 19.6 (isomer a), 25.0, 28.2 (isomer b), 28.4 (isomer a), 34.4, 41.8 (isomer a), 42.2 (isomer b), 53.4, 57.0 (isomer b), 57.2 (isomer a), 60.4 (isomer a), 60.5 (isomer b), 63.0 (isomer b), 63.6 (isomer a), 66.5 (isomer a), 66.6 (isomer b), 102.8 (isomer b), 103.5 (isomer a), 114.1, 117.3 (isomer a), 117.4 (isomer b), 120.3, 123.6, 126.4, 126.9, 127.0, 127.7, 128.6, 128.7, 128.8, 128.9, 128.9, 129.0, 136.6 (isomer a), 136.9 (isomer b), 139.2, 139.3, 139.3, 139.7, 140.3, 140.5, 141.5, 148.7, 168.0 (isomer a), 168.2 (isomer b). MS (ESI) m/z 737.1 [M + H]+.
:b mixture (60:40), mp 81.5–82.9 °C. νmax (KBr)/cm−1 2956, 1693, 1597; δH (400 MHz, CDCl3) 1.50–1.65 (3H, m), 1.70–1.85 (3H, m), 2.44–2.52 (2H, m), 2.56–2.72 (4H, m), 2.75–2.86 (1H, m), 3.38–3.85 (8H, m), 3.97–4.03 (0.6H, m, isomer a), 4.10–4.15 (0.4H, m, isomer b), 4.47–4.59 (1H, m), 4.89 (0.6H, d, J 2.7, isomer a), 5.01 (0.4H, s, isomer b), 6.94–7.00 (2H, m, ArH), 7.04–7.11 (7H, m, ArH), 7.15 (1H, t, J 7.4, ArH), 7.33–7.40 (2H, m, ArH), 7.71–7.80 (4H, m, ArH), 7.89 (2H, d, J 8.5, ArH), 8.27 (1H, s, triazole), 11.12 (0.4H, bs, CONH, isomer b), 11.25 (0.6H, bs, CONH, isomer a); δC (100.6 MHz, CDCl3): 19.0 (isomer b), 19.6 (isomer a), 25.0, 28.2 (isomer b), 28.4 (isomer a), 34.4 (isomer a), 34.6 (isomer b), 41.7 (isomer a), 42.2 (isomer b), 53.4, 57.0 (isomer b), 57.1 (isomer a), 60.4 (isomer a), 60.5 (isomer b), 63.1 (isomer b), 63.6 (isomer a), 66.5 (isomer a), 66.6 (isomer b), 102.8 (isomer b), 103.5 (isomer a), 114.1, 116.8 (isomer a), 116.9 (isomer b), 119.0, 119.3, 120.3, 123.8, 124.6, 126.9, 127.5, 128.7, 128.8, 128.8, 128.9, 128.9, 129.0, 136.5 (isomer a), 136.8 (isomer b), 139.1, 139.3, 139.7, 148.6, 156.6, 158.0 168.0 (isomer a), 168.2 (isomer b). MS (ESI) m/z 753.1 [M + H]+.
Conclusions
We have used a click chemistry approach to synthesize a series of phenylalanine derived hydroxamates. The substitution pattern in the resulting triazole seems to be crucial to obtain a high degree of selectivity between both gelatinases. The best compound 1b displayed subnanomolar IC50 against MMP-2 and was 95-fold less potent against MMP-9, while the closely structurally related compound 7 was devoid of selectivity. We have used docking and molecular dynamics tools to shed light on the reason for this difference in selectivity between 1b and 7. The difference in the energetic profile of the dihedral angle that describes the torsion around Csp2–Csp2 and Csp2–Nsp2 induces a change in the dynamic behaviour of the P1′-segment that allows 7 to establish good interactions with both MMP-2 and MMP-9.Notes and references
- M. Yamamoto, S. Ikeda, H. Kondo and S. Inoue, Bioorg. Med. Chem. Lett., 2002, 12, 375–378 CrossRef CAS.
- S. Rivera, M. Khrestchatisky, L. Kaczmarek, G. A. Rosenberg and D. M. Jaworski, J. Neurosci., 2010, 30, 15337–15357 CrossRef CAS PubMed.
- J. Huxley-Jones, T.-K. Clarke, C. Beck, G. Toubaris, D. L. Robertson and R. P. Boot-Handford, BMC Evol. Biol., 2007, 7 CrossRef PubMed , 63.
- W. Bode, F. X. Gomisruth and W. Stockler, FEBS Lett., 1993, 331, 134–140 CrossRef CAS.
- R. Roy, J. Yang and M. A. Moses, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS PubMed.
- B. Fingleton, Curr. Pharm. Des., 2007, 13, 333–346 CrossRef CAS.
- T. Klein and R. Bischoff, Amino Acids, 2011, 41, 271–290 CrossRef CAS PubMed.
- T. P. Theruvath, J. A. Jones and J. S. Ikonomidis, J. Card. Surg., 2012, 27, 81–90 CrossRef PubMed.
- M. L. Lindsey and R. Zamilpa, Cardiovasc. Ther., 2012, 30, 31–41 CrossRef CAS PubMed.
- G. Siasos, D. Tousoulis, S. Kioufis, E. Oikonomou, Z. Siasou, M. Limperi, A. G. Papavassiliou and C. Stefanadis, Curr. Top. Med. Chem., 2012, 12, 1132–1148 CrossRef CAS.
- P. S. Burrage, K. S. Mix and C. E. Brinckerhoff, Front. Biosci., 2006, 11, 529–543 CrossRef CAS PubMed.
- A. Davey, D. F. McAuley and C. M. O'Kane, Eur. Respir. J., 2011, 38, 959–970 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- F. Vasaturo, F. Solai, C. Malacrino, T. Nardo, B. Vincenzi, M. Modesti and S. Scarpa, Oncol. Lett., 2013, 5, 316–320 Search PubMed.
- D. Stellas and E. Patsavoudi, Anti-Cancer Agents Med. Chem., 2012, 12, 707–717 CrossRef CAS.
- B. Bauvois, Biochim. Biophys. Acta, Rev. Cancer, 2012, 1825, 29–36 CrossRef CAS PubMed.
- T. Itoh, M. Tanioka, H. Yoshida, T. Yoshioka, H. Nishimoto and S. Itohara, Cancer Res., 1998, 58, 1048–1051 CAS.
- C. M. Overall and O. Kleifeld, Nat. Rev. Cancer, 2006, 6, 227–239 CrossRef CAS PubMed.
- P. Serra, M. Bruczko, J. M. Zapico, A. Puckowska, M. A. Garcia, S. Martin-Santamaria, A. Ramos and B. de Pascual-Teresa, Curr. Med. Chem., 2012, 19, 1036–1064 CrossRef CAS.
- M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735–2776 CrossRef CAS PubMed.
- R. K. Yadav, S. P. Gupta, P. K. Sharma and V. M. Patil, Curr. Med. Chem., 2011, 18, 1704–1722 CrossRef CAS.
- R. P. Verma and C. Hansch, Bioorg. Med. Chem., 2007, 15, 2223–2268 CrossRef CAS PubMed.
- J. M. Zapico, P. Serra, J. Garcia-Sanmartin, K. Filipiak, R. J. Carbajo, A. K. Schott, A. Pineda-Lucena, A. Martinez, S. Martin-Santamaria, B. de Pascual-Teresa and A. Ramos, Org. Biomol. Chem., 2011, 9, 4587–4599 CAS.
- B. Fabre, K. Filipiak, J. M. Zapico, N. Diaz, R. J. Carbajo, A. K. Schott, M. P. Martinez-Alcazar, D. Suarez, A. Pineda-Lucena, A. Ramos and B. de Pascual-Teresa, Org. Biomol. Chem., 2013, 11, 6623–6641 CAS.
- B. Fabre, K. Filipiak, N. Diaz, J. M. Zapico, D. Suarez, A. Pineda-Lucena, A. Ramos and B. de Pascual-Teresa, ChemBioChem, 2014, 15, 399–412 CrossRef CAS PubMed.
- Y. Tamura, F. Watanabe, T. Nakatani, K. Yasui, M. Fuji, T. Komurasaki, H. Tsuzuki, R. Maekawa, T. Yoshioka, K. Kawada, K. Sugita and M. Ohtani, J. Med. Chem., 1998, 41, 640–649 CrossRef CAS PubMed.
- M. L. Di Gioia, A. Leggio and A. Liguori, J. Org. Chem., 2005, 70, 3892–3897 CrossRef CAS PubMed.
- F. R. Bou-Hamdan, F. Levesque, A. G. O'Brien and P. H. Seeberger, Beilstein J. Org. Chem., 2011, 7, 1124–1129 CrossRef CAS PubMed.
- Y. Q. Feng, J. J. Likos, L. M. Zhu, H. Woodward, G. Munie, J. J. McDonald, A. M. Stevens, C. P. Howard, G. A. De Crescenzo, D. Welsch, H. S. Shieh and W. C. Stallings, Biochim. Biophys. Acta, Proteins Proteomics, 2002, 1598, 10–23 CrossRef CAS.
- L. J. MacPherson, E. K. Bayburt, M. P. Capparelli, B. J. Carroll, R. Goldstein, M. R. Justice, L. Zhu, S. Hu, R. A. Melton, L. Fryer, R. L. Goldberg, J. R. Doughty, S. Spirito, V. Blancuzzi, D. Wilson, E. M. O'Byrne, V. Ganu and D. T. Parker, J. Med. Chem., 1997, 40, 2525–2532 CrossRef CAS PubMed.
- D. P. Becker, T. E. Barta, L. J. Bedell, T. L. Boehm, B. R. Bond, J. Carroll, C. P. Carron, G. A. DeCrescenzo, A. M. Easton and J. N. Freskos, J. Med. Chem., 2010, 53, 6653–6680 CrossRef CAS PubMed.
- T. Tuccinardi, A. Martinelli, E. Nuti, P. Carelli, F. Balzano, G. Uccello-Barretta, G. Murphy and A. Rossello, Bioorg. Med. Chem., 2006, 14, 4260–4276 CrossRef CAS PubMed.
- M. D. Martin and L. M. Matrisian, Cancer Metastasis Rev., 2007, 26, 717–724 CrossRef CAS PubMed.
- S. A. Kolodziej, S. L. Hockerman, T. L. Boehm, J. N. Carroll, G. A. DeCrescenzo, J. J. McDonald, D. A. Mischke, G. E. Munie, T. R. Fletcher, J. G. Rico, N. W. Stehle, C. Swearingen and D. P. Becker, Bioorg. Med. Chem. Lett., 2010, 20, 3557–3560 CrossRef CAS PubMed.
- B. Fabre, K. Filipiak, C. Coderch, J. M. Zapico, R. J. Carbajo, A. K. Schott, A. Pineda-Lucena, B. de Pascual-Teresa and A. Ramos, RSC Adv., 2014, 4, 17726–17735 RSC.
- S. H. Hilal, S. W. Karickhoff and L. A. Carreira, Quant. Struct-Act. Relat., 1995, 14, 348–355 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- J. Aaqvist, J. Phys. Chem., 1990, 94, 8021–8024 CrossRef CAS.
- C. Hansch, J. Comput. Aided Mol. Des., 2011, 25, 495–507 CrossRef CAS PubMed.
- M. A. Lill, Biochemistry, 2011, 50, 6157–6169 CrossRef CAS PubMed.
- J. Klett, A. Nunez-Salgado, H. G. Dos Santos, A. Cortes-Cabrera, A. Perona, R. Gil-Redondo, D. Abia, F. Gago and A. Morreale, J. Chem. Theory Comput., 2012, 8, 3395–3408 CrossRef CAS.
- A. Morreale, R. Gil-Redondo and A. R. Ortiz, Proteins, 2007, 67, 606–616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Torsional energies of the Csp2–Csp2 and Csp2–Nsp2 scan; and per-residue energy decomposition of the four systems. 1H NMR and 13C NMR spectra of compounds 1a–g, 7, 13 and 14. See DOI: 10.1039/c4ob01516a |
| This journal is © The Royal Society of Chemistry 2015 |