
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical reduction of carbon dioxide to multicarbon (C2+) products: challenges and perspectives
Bin
Chang
 ab,
Hong
Pang
c,
Fazal
Raziq
ab,
Sibo
Wang
d,
Kuo-Wei
Huang
ab,
Jinhua
Ye
*c and
Huabin
Zhang
*ab
ab,
Hong
Pang
c,
Fazal
Raziq
ab,
Sibo
Wang
d,
Kuo-Wei
Huang
ab,
Jinhua
Ye
*c and
Huabin
Zhang
*ab
aChemistry Program, Physical Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: huabin.zhang@kaust.edu.sa
bKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal, Kingdom of Saudi Arabia
cInternational Center for Materials Nanoarchitectonics (WPI-MANA) National Institute for Materials Science (NIMS)1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: jinhua.ye@nims.go.jp
dState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350002, P. R. China
First published on 23rd June 2023
Abstract
Electrocatalytic CO2 reduction has been developed as a promising and attractive strategy to achieve carbon neutrality for sustainable chemical production. Among various reduction products, multi-carbon (C2+) compounds with higher energy density are desirable value-added products. Herein, we review and discuss the recent progress and challenges in preparing C2+ products. We start with the elaboration of the most recent advancement of carbon–carbon coupling results and the newly proposed mechanisms, which are much more complicated than that of single-carbon products. The complex scenarios involved in the initial CO2 activation process, the catalyst micro/nanostructure design, and mass transfer conditions optimization have been thoroughly discussed. In addition, we also propose the synergistic realization of high C2+ product selectivity through the rational design of the catalyst and elaborate on the influence of electrolytes (anion/cation/pH/ionic liquid) using theoretical calculation analysis and machine learning prediction. Several in situ/operando techniques have been elaborated for tracking the structural evolution and recording the reaction intermediates during electrocatalysis. Additional insights into the triphasic interfacial reaction systems with improved C2+ selectivity are also provided. By presenting these advances and future challenges with potential solutions related to the integral development of electrochemical reduction of carbon dioxide to C2+ products, we hope to shed some light on the forthcoming research on electrochemical carbon dioxide recycling.
 Bin Chang | Bin Chang received his PhD degree from Shandong University in 2020. Then, he worked as a postdoc under the supervision of Prof. Weijia Zhou at the University of Jinan (UJN) and Prof. Shuhui Sun at the Institut National de la Recherche Scientifique (INRS). He is currently a Postdoctoral Fellow in Huabin Zhang's group at KAUST Catalysis Center, King Abdullah University of Science and Technology (KAUST). His research interests focus on advanced catalysts for electrochemical energy conversion. |
 Jinhua Ye | Jinhua Ye received her PhD from the University of Tokyo in 1990. She is presently a Principal Investigator at the National Institute of Materials Science (NIMS) and a Professor of the Joint Doctoral Program at Hokkaido University, Japan. Her research interests focus on the research and development of photofunctional materials and their applications in the fields of environmental remediation and new energy production. She has published more than 600 high impact research papers with over 60 |
 Huabin Zhang | Huabin Zhang received his PhD degree in chemistry from Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (FJIRSM-CAS). After finishing his postdoc research in Japan (Supervisor: Jinhua Ye) at the National Institute of Materials Science (NIMS) and Singapore (supervisor: Xiong Wen Lou) at Nanyang Technological University, Singapore, he joined KAUST serving as an Assistant Professor in January 2021. His research interests focus on advanced catalysis for sustainable energy. |
Broader contextThe electrochemical CO2 reduction reaction (CO2RR) holds promise to revolutionize the chemical industry by producing value-added chemicals and fuels from CO2 and water while storing renewable energy and reducing anthropogenic CO2 emissions. However, electrocatalytic C–C coupling in aqueous electrolytes is still challenging due to low selectivity, activity, and stability. The optimization of catalysts, the reaction system including the electrolyte and reactors holds the key to addressing these challenges. We summarize the recent progress in achieving efficient C–C coupling for C2+ products, with emphasis on design strategies in electrocatalysts and electrocatalytic reactors, the influence of electrolytes and the theoretical investigations of corresponding mechanisms integrating with in situ/operando techniques. Moreover, the current challenges and future opportunities for C2+ product synthesis are discussed. We aim to provide a detailed review of the novel C–C coupling strategies for further development and inspiration in both fundamental understanding and technological applications of electrochemical carbon dioxide recycling. |
1. Introduction
Over the last century, energy has mainly been obtained through the combustion of fossil fuels, in the form of coal, oil, and gas, and the associated CO2 emissions have significantly increased the atmospheric CO2 level, resulting in drastic climate change, sea level rise, ocean acidification, and other problems. The concerns over the increase in CO2 emission and the demand for carbon-containing raw materials have accelerated the development of a variety of technologies for CO2 reduction conversion.1–5 The conversion of CO2 to chemicals and liquid fuels with low-carbon energy sources, particularly multicarbon (C2+) hydrocarbons and oxygenates, has emerged as a fundamental approach to alleviate extreme climate change and the increased energy demand.6–9 However, CO2 activation under mild conditions and the precise regulation of C–C coupling are the grand challenges in artificial carbon fixation and C2+ product synthesis. Electrocatalytic CO2 reduction reaction (eCO2RR) is considered a promising strategy from the perspectives of technical difficulty, maturity, and economy, and thus has attracted extensive research attention from both academia and industry.10–12The electrochemical reduction of carbon dioxide to highly selective C2+ products is the holy grail of electrochemical synthesis. Compared to C1 products (e.g., carbon monoxide, methane, formic acid, and methanol), C2+ products (e.g., ethylene, ethanol, acetic acid, and n-propanol) possess higher energy densities and economic value and can be further utilized as feedstocks for the synthesis of long-chain hydrocarbon fuels.13–17 Currently, *CO dimerization and *CO hydrogenation are believed to be the main C–C coupling pathways to realize the evolution of C2+ products, while the rate-determining steps of C2+ product synthesis can be attributed to the initial activation of CO2 molecules.18 Optimizing the *CO binding strength and the subsequent proton transfer-based formation of hydrogenated groups (e.g., *CHO, *COH, *OCCO, and *OCCOH) are pretty sensitive to the material structure and electrolyte composition. Focusing on the synthesis of the C2+ products, this review elaborates on the recent progress in eCO2RR and discusses the present challenges in promoting selectivity and efficiency (Fig. 1). Notably, we have summarized the fundamental principles for novel catalyst discovery and provided a comprehensive overview of the catalytic mechanisms, encompassing nearly all aspects for designing catalysts for C2+ synthesis. Various theoretical approaches and models for simulating the complicated C2+ synthesis process are systematically summarized. Moreover, we provide several strategies to construct more realistic models for theoretical simulation by considering the electrode–electrolyte interface, charge transfer, solvent effect, and kinetic factors. In addition, in situ/operando characterization-based research has been emphasized to clarify the structural evolution and the mutual interactions within the reaction interface. Meanwhile, we also summarize the current representative optimization strategies of electrolytes and electrolytic cells for the C2+ product system. Based on the triphasic interfacial reaction model, the corresponding design strategy has been put forward to overcome the diffusion and mass transfer limitations of traditional two-phase systems, thus improving eCO2RR efficiency at industrial current densities. It is highly expected that this review will deliver some new insights toward the understanding and engineering of eCO2RR and further accelerate the development of this important emerging research field.
 | ||
| Fig. 1 Recent development of electrochemical CO2 reduction. | ||
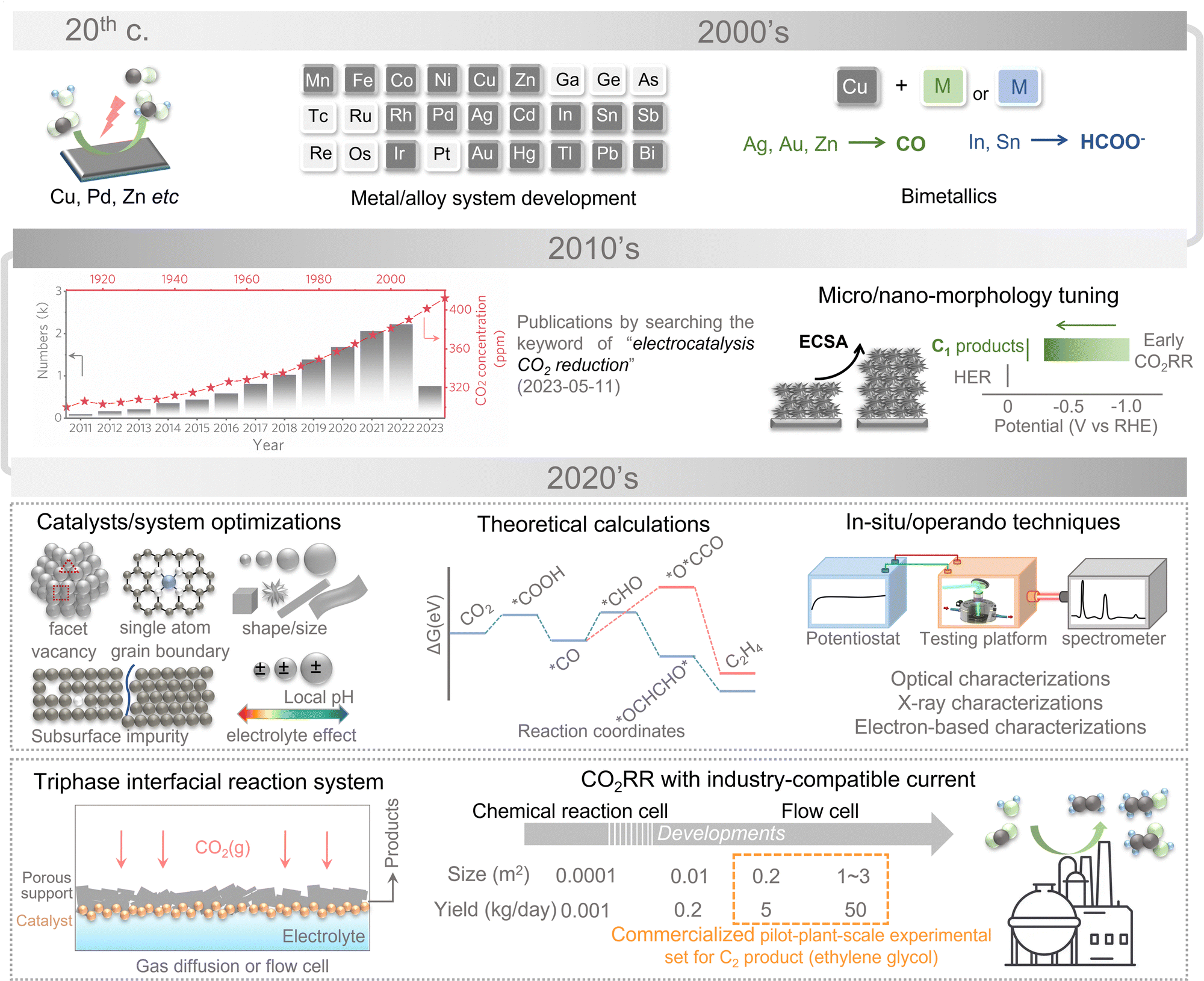
1.1. History and reality of electrochemical CO2 reduction
The first examples of electrochemical reduction of carbon dioxide are from the 19th century when carbon dioxide was reduced to carbon monoxide using a zinc cathode. Research in this field has been intensified in the 1980s following the oil embargoes of the 1970s, in which the electrocatalytic conversion of CO2 has been realized over copper, zinc, and lead electrodes with the main product being formate. Methanol has been further synthesized by resorting to ruthenium- and molybdenum-based catalysts and certain semiconductors, such as p-GaP and n-/p-GaAs.19–22 After entering the first decade of the 21st century, various metal/alloy systems have been developed to be capable of electrochemically reducing CO2 into hydrocarbons with decent efficiencies stimulating the explosive growth of subsequent studies on metallic electrodes with tuned micro/nano-morphology for eCO2RR. During this period, most of the research works focused on improving the product selectivity and efficiency of C1 products. Based on past experimental and theoretical studies, C2+ synthesis by CO2 electrolysis has become the focus of future research in recent years (the 2020s). In the first two years of the 2020s, the C2+ product conversion efficiency and selectivity have been greatly enhanced. The mechanisms are gradually revealed by optimized electrocatalysts, sophisticated theoretical considerations, novel reaction devices, and advanced in situ/operando characterization techniques.23–29eCO2RR exhibits very slow thermodynamic kinetics owing to the net zero dipole moment and the highly chemically inert linear CO2 molecule with short polar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (0.116 nm). The first step of eCO2RR is always believed to be CO2 activation, corresponding to the reduction of chemical bonds.30,31 The C–C coupling can be realized over the varied pathways, resulting in the emergence of the competing reaction and relatively low selectivity for particular products. Of course, the hydrogen evolution reaction (HER) may be accomplished and even be evolved into the primary reaction simultaneously, especially at high negative potentials, which bring additional barriers to promoting its selectivity for C2+ products. In such a scenario, realizing the efficient electrochemical reduction of CO2 into specific C2+ products with high selectivity is challenging.
O bonds (0.116 nm). The first step of eCO2RR is always believed to be CO2 activation, corresponding to the reduction of chemical bonds.30,31 The C–C coupling can be realized over the varied pathways, resulting in the emergence of the competing reaction and relatively low selectivity for particular products. Of course, the hydrogen evolution reaction (HER) may be accomplished and even be evolved into the primary reaction simultaneously, especially at high negative potentials, which bring additional barriers to promoting its selectivity for C2+ products. In such a scenario, realizing the efficient electrochemical reduction of CO2 into specific C2+ products with high selectivity is challenging.
It should also be noted that C2+ selectivity and activity are highly sensitive to multiple electron transfers during C2+ product formation. Unlike C1 synthesis, the *CO intermediate is considered the starting point for further C–C coupling. The subsequent complex reaction pathway and HER competition directly lead to the low Faraday efficiency (FE) of C2+ product formation.32,33 Presently, the FE of C2+ product formation can exceed 60%.34–36 However, the poor selectivity limits the synthesis and subsequent applications of C2+ products. Zheng and colleagues have designed defect-site-rich nanocatalysts with peak FE values of ethanol and n-propanol at 53% and 18%, respectively.37 The control sample with flat surfaces formed without *CO adsorbates produced C2H4 with a maximum FE of 60% at −1.23 V (vs. RHE), whereas the FEs of other C1 and C2+ products range from 5% to 15%. A similar phenomenon occurs in the gas-diffusion-electrode-based flow-cell system. The overall current densities of defect-site-rich nanocatalysts reached ∼200 mA cm−2 with the highest FE values of 52% and 15% for ethanol and n-propanol at −0.95 V (vs. RHE). Herein, the design of nanocatalyst structures and reaction systems is subject to stringent requirements, and C–C bond formation is still a fundamental chemical challenge for further C2+ synthesis.
1.2. Mechanism of C2+ product formation
| ΔfG° = ΔfH° − T°ΔfS° |
where i is the stoichiometric coefficient for species in the formation reactions, and the theoretical reactions form the compound from the constituent elements in the standard states. Liquid CO2 reduction products are also considered. The most reliable approach for obtaining the free energy of formation for aqueous products is tuning the solvation-free energy of the gas products (Δg→aqG°), which is related to Henry's law constant (KH) by
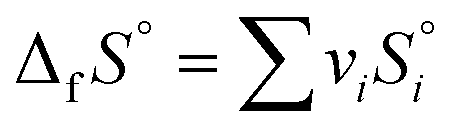
Δg→aqG° = RT°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(KH) ln(KH) |
| Reaction | C2+ products | E° (V vs. RHE) | ΔfG° (kJ mol−1) | ΔfH° (kJ mol−1) | ΔfS° (kJ mol−1) | K H (bar M−1) | |
|---|---|---|---|---|---|---|---|
| a Calculated data from Chem. Rev., 2019, 119, 7610. b John A. Dean, Langes Handbook of Chemistry, 15th edn, McGraw-Hill Inc, 1999. c NIST Chemistry Webbook, https://webbook.nist.gov/chemistry/. | |||||||
| 2CO2 + 2H+ + 2e− → (COOH)2 (s) | Oxalic acid | s | −0.47 | −698.9a | −829c | 116c | — |
| 2CO2 + 8H+ + 8e− → CH3COOH (aq) + 2H2O | Acetic acid | g | 0.11 | −374.9a | −433c | 282.8c | — |
| aq | −396.3a | — | — | 1.8210–4c | |||
| 2CO2 + 10H+ + 10e− → CH3CHO (aq) + 3H2O | Acetaldehyde | g | 0.06 | −133.0b | −166.1b | 263.8b | — |
| aq | −139.7a | — | — | 6.6710–2c | |||
| 2CO2 + 12H+ + 12e− → C2H5OH (aq) + 3H2O | Ethanol | g | 0.09 | −167.9b | −234.8b | 281.6b | — |
| aq | −181.3a | — | — | 4.5510–3c | |||
| 2CO2 + 12H+ + 12e− → C2H4 (g) + 4H2O | Ethylene | g | 0.08 | 68.3a | 52.4c | 219.3c | — |
| 3CO2 + 16H+ + 16e− → C2H5CHO (aq) + 5H2O | Propionaldehyde | g | 0.09 | −127.0a | −188.7c | −304.4c | — |
| aq | −133.3a | — | — | 7.6910–2c | |||
| 3CO2 + 18H+ + 18e− → C3H7CHO (aq) + 5H2O | 1-Propanol | g | 0.10 | −160.7a | −256c | 322.5c | |
| aq | −173.0 | — | — | 7.1410–3c | |||
Note that Henry's law constant in this equation must be dimensionless. The normal state of gas and solvated compounds is 1 bar and 1 M, respectively.
In general, the electrochemical reduction of CO2 is described by the equation
| xCO2 + nH+ + ne− → products + yH2O |
The free energy formation of a proton–electron pair is
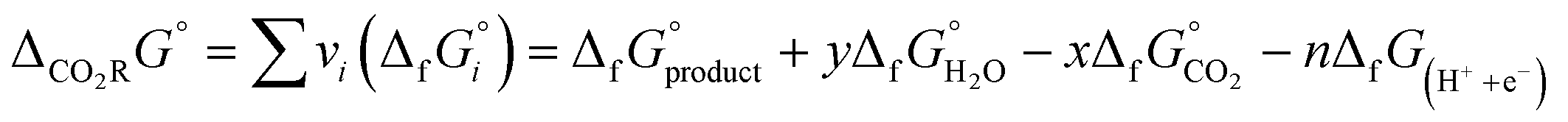
| ΔfG(H++e−) = −FURHE |
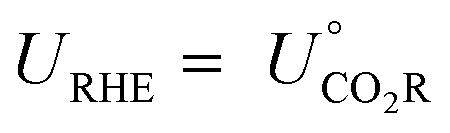 , and the free energy (ΔCO2RG°) is zero. Herein, UCO2R can be solved by
, and the free energy (ΔCO2RG°) is zero. Herein, UCO2R can be solved by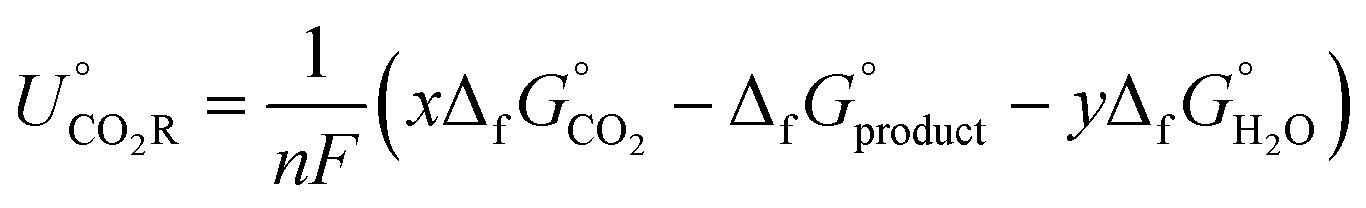
All standard equilibrium potentials are calculated from the free energy formation of the reactants and products. As detailed in Table 1, except the equilibrium potentials of solid oxalic acid which are negative (−0.47 V vs. RHE), all other equilibrium potentials of gas or liquid products range from 0.06 V to 0.11 V vs. RHE. Regardless of the type of C2+ products formed at the cathode, the electron and proton stoichiometry coefficients of the eCO2RR and the oxygen evolution reaction (OER) must be equal in a continuous eCO2RR system. Theoretically, the difference between the equilibrium potentials of eCO2RR and OER, which is the minimum potential for driving the entire reaction, is above 1 V.39 The thermodynamics of eCO2RR pathways is affected by the different chemical potentials of electrons. The surface structure of electrocatalysts influences the reaction kinetics at low current densities or overpotentials. Meanwhile, mass transfer plays a significant role at high overpotentials or current densities.40 Beyond the above limit, further increased overpotential enhances HER side reactions and decreases the selectivity of C2+ synthesis. Therefore, eCO2RR currently requires a relatively large overpotential exceeding the thermodynamic potential. It is also urgent to achieve higher current densities and overpotentials for eCO2RR.
Recently, the *CO dimerization mechanism has been well accepted by the catalytic community, in which the production of OC*–*CO is followed by subsequent hydrogenation reaction (Fig. 2, purple line highlighted pathway).11,15,16,42,43 Theoretical simulations of *CO dimerization on several copper facets have confirmed the lowest energy barrier on the (100) facet.44 *CO dimerization is exothermic on the Cu(100) facet but endothermic on the Cu(111) facet. The energy barrier of subsequent hydrogenation on the Cu(100) facet is 0.3–0.4 eV lower than that on the control Cu(111) facet. In addition, *OC–CO is adsorbed on the quadruple site of the Cu(100) facet via two carbon ends and is more strongly bound than in the case of the Cu(111) facet, where the carbon atoms are confined to a triple site. In situ spectroscopy has detected the characteristic absorption bands of *OC–CO species.45,46 The possibility of subsequent hydrogen assisted C–C coupling increases at a more negative potential.47 This mechanism involves the formation of *CHO via hydrogenation and its subsequent reaction with *CO to form *COCHO. Bell and colleagues have confirmed the presence of *CHO species on the fluoro-modified catalyst by in situ spectroscopy.48 The fluorine in the modified catalyst enhanced the hydrogenation of *CO to CHO intermediates, which are efficiently coupled to obtain *COCHO and thus improve the selectivity of C2–4 products. *HOCHCH2 derived from the hydrogenation of *COCHO is also a critical intermediate, which can directly desorb from the reactive centers to generate acetaldehyde and further converted to ethanol under hydrogenation. Qiao and colleagues have reported a novel silver-modified copper oxide catalyst with a significant FE of 40.8% for ethanol production.49 Both top and bridge configurations of *CO adsorption on the catalyst surface trigger asymmetric C–C coupling to ethanol intermediates *HOCHCH2. In addition to the above hydrogenation path, *HOCHCH2 dehydration is another kinetically more favorable path to forming acetaldehyde under neutral conditions. Sun and colleagues have developed ferromagnetic hexagonal-close-packed (hcp) Co nanosheets for selective CO2RR to acetaldehyde with an FE of 60% in 0.5 M KHCO3 solution.50 On the hcp Co surface, the C2 pathway toward acetaldehyde shows a lower overall energy barrier than other competitive pathways to form C2H4, CH3OH, and CH3CH2OH. The above reactions follow the path of *CO–CO coupling before hydrogenation/dehydration. Another C–C coupling path is based on the coupling of hydrocarbon groups. Dismukes and colleagues have introduced iron phosphide for the C2 product (ethylene glycol) synthesis by (H2CO)* coupling.51 In this reaction system, formate is more likely to undergo protonation to form the (O–CHO)* intermediate rather than desorb to produce C1 formic acid. Moreover, hydride transfer to the carbon of (O–CHO)* and the resulting *H2CO–OH2 release a water molecule to form (H2CO)*, which easily undergoes carbon–carbon coupling to form C2+ products.
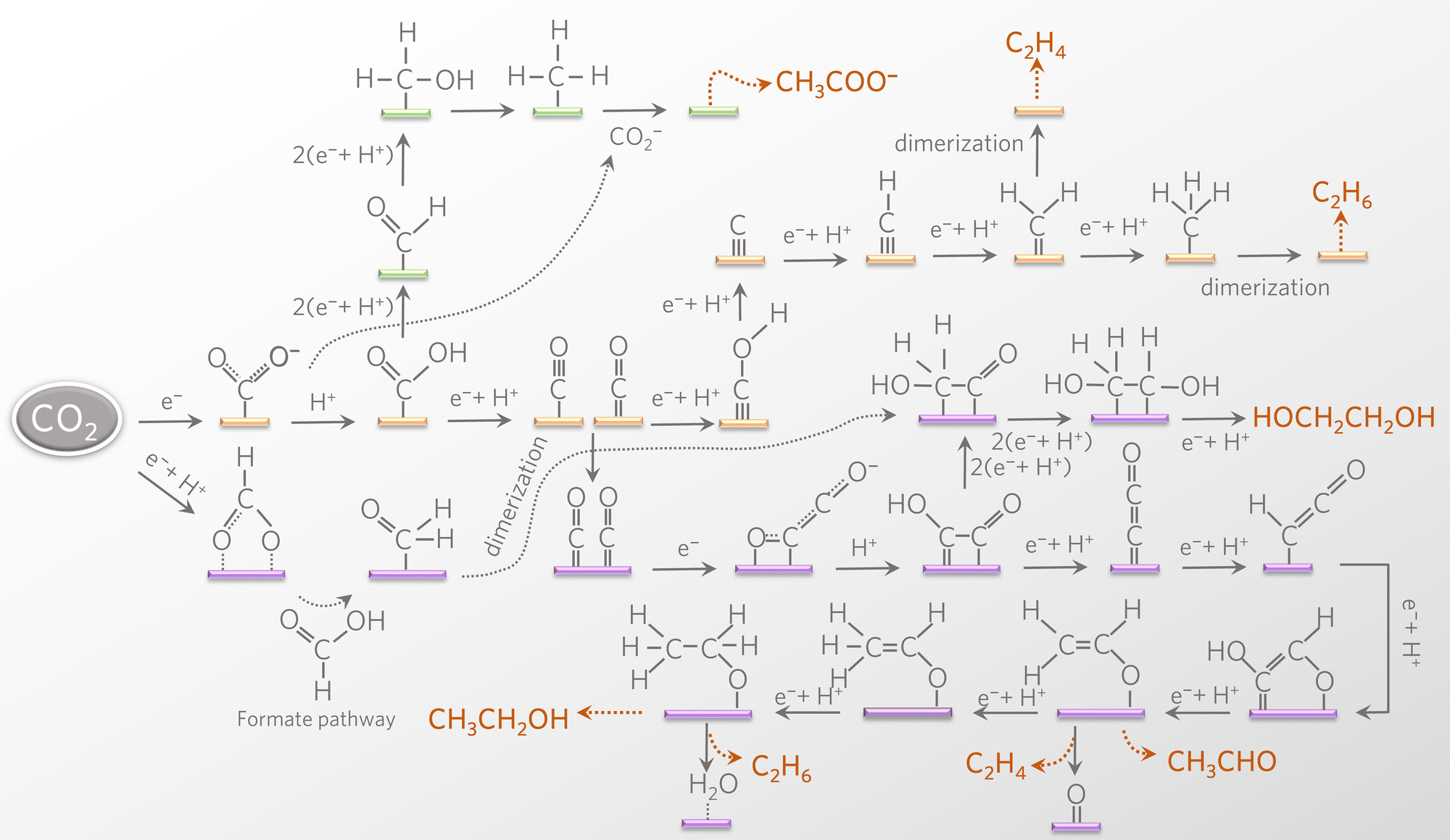 | ||
| Fig. 2 Overview of reaction pathways for eCO2RR towards different C2+ products. | ||
Carbene (CH2) dimerization (yellow line highlighted pathway) and *CO dimerization under hydrogenation (purple line highlighted pathway) are the two main mechanisms for ethylene synthesis (Fig. 2).5,6,52 The adsorbed *CO is hydrogenated to form *COH intermediates, which further produce *C and *CH2 carbene intermediates.53 Ethylene (or ethane) is generated from *CH2 (or *CH3) dimerization under acidic conditions.54,55 Buonsanti and colleagues have confirmed the carbene coupling mechanism of ethylene synthesis via the tunable tandem catalysts comprising iron porphyrin and Cu nanocubes.56 Furthermore, another procedure for ethylene synthesis is *CO/CO dimerization and hydrogenation. Jaramillo and colleagues have directly converted vapor-fed CO2 to ethylene on a tandem electrocatalyst.57 The enhancements of ethylene yield are attributed to the increased *CO/CO concentration near the copper surface via effective CO2 to CO conversion on neighboring nickel-coordinated nitrogen-doped carbon, in which the as-produced *CO–CO intermediate is further hydrogenated to ethylene.
Theoretically, it is also feasible to synthesize acetate with the coexistence of *CO intermediates and hydrocarbon intermediates. However, the acetate selectivity on traditional inorganic catalysts is generally limited by the competitive reaction of ethylene and ethanol. Schöfberger and colleagues have developed a molecular MnIII-corrole complex with an acetate selectivity of 63%.58 Such a high acetate selectivity originates from the Lewis acidity of the MnIII center, which tends to bind with the Lewis basic O-site of the carboxyl group, hence facilitating the C–C dimerization leading to an oxalate-type intermediate. To significantly increase the coverage of carboxyl intermediates, Liao and colleagues have developed a stable and conductive phthalocyanine-based covalent-organic framework (COF) for acetate synthesis with an FE of 90.3%.59 The isolated copper-phthalocyanine active sites with elevated electron density are conducive to the critical C–C coupling step of *CH3 with carboxyl intermediates to produce acetate (Fig. 2, green line highlighted pathway). Herein, the construction of molecular catalysts sheds light on the rational design of highly efficient electrocatalysts for highly valuable C2+ products.
2. Electrocatalyst design for C2+ product synthesis
The structure and composition of the catalyst play a decisive role in determining the substrate activation and reduction process and thus significantly influence the final product selectivity. The desired high-value C2+ products can be obtained by optimizing the surface electronic structure and the geometric environment by regulating the morphology, surface structure, supports, and active sites (Fig. 3).11,60,61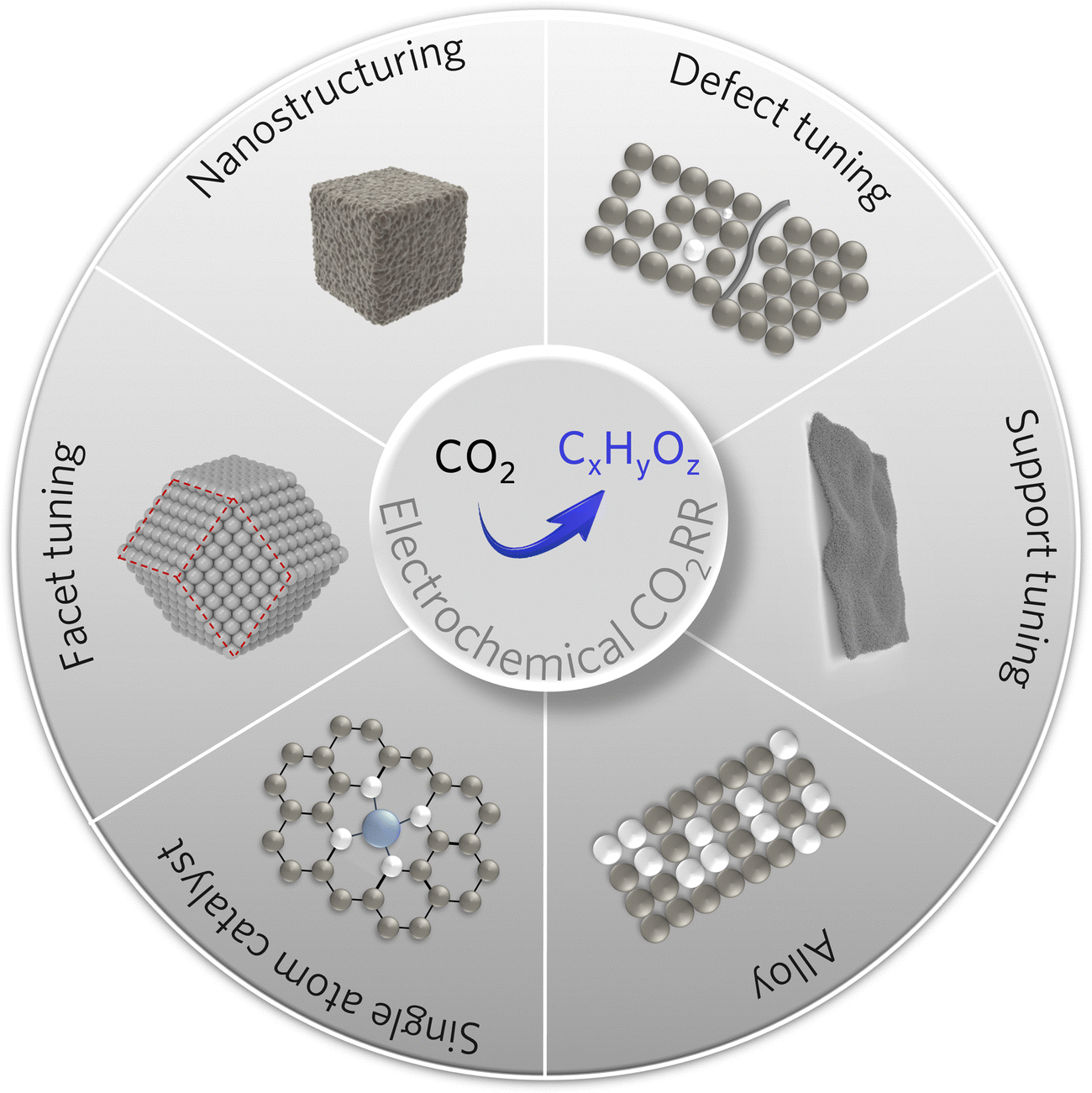 | ||
| Fig. 3 Scheme of various strategies for designing copper-based electrocatalysts for C2+ products. | ||
2.1. Single-atom catalyst design
Single-atom electrocatalysts are widely investigated owing to the merits of efficient atomic utilization and excellent activity.62–65 The type of central metal species, electron configuration, and the surrounding coordination environment affect their geometric and electronic structure for tuning the reaction pathways and product distribution of eCO2RR.66–68 Appropriate central metal atoms with low Gibbs free energy of eCO2RR are selected to enhance the electrocatalytic performance, which include Fe, Co, Ni, Cu, Zn, Sn, and Sb. Xu and colleagues have prepared a copper single-atom catalyst anchored on a carbon support by a copper-lithium hybrid method. The as-synthesized catalyst has achieved a single-product FE of 91% at −0.7V vs. RHE and an onset potential as low as −0.4V vs. RHE for electrocatalytic CO2-to-ethanol conversion.41 The FE of ethanol formation is highly sensitive to the dispersion of copper atoms. The dynamic reversible conversion between copper single atoms and nanoparticles/clusters has been observed using operando X-ray absorption spectroscopy (XAS), which promotes an understanding of real catalytically active sites. Similarly, single-atom electrocatalysts have attracted tremendous attention.69 Chen and colleagues have achieved CO2 reduction on single-atom copper with acetic acid, ethanol, and acetone production at low overpotential (Fig. 4A).70 Adjusting the cooperative structure of single copper atoms and exploring reversible reconstruction under negative reduction voltages is a promising research direction to achieve effective C2+ formation.
 | ||
| Fig. 4 (A) Morphological characterization of single copper atom electrocatalysts. Reproduced with permission from Chen et al.70 Copyright 2020 Springer Nature. (B) Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of CuO micropore nanowires. Reproduced with permission from Yu et al.85 Copyright 2020 American Chemical Society. (C) SEM images and (D) corresponding structure of OD-Cu derived from Cu2O crystals. Reproduced with permission from Gao et al.82 Copyright 2022 American Chemical Society. | ||
Currently, specific coordination configurations can be induced by representative strategies of single-atom alloys and tandem catalysts. The doped atoms in single-atom alloys are diluted to avoid bond formation between neighboring atoms, which appropriately tunes the surface structure to achieve moderate binding with the reaction intermediates. The introduction of the atom with low binding energy to CO may provide enough free CO molecules for further C2+ synthesis. Grätzel and colleagues have prepared CuZn single-atom alloys for eCO2RR with 41% FE for C2+ liquids.71 Based on the strong CO adsorption on Cu sites, the incorporated heteroatom Zn enhances the CO production and favors the release of free CO molecules to Cu sites. The as-formed *CH3 intermediates with the free CO further form C2H5OH with high selectivity. Furthermore, the tandem catalysts also significantly expand the applications of single-atom electrocatalysts in multi-electron eCO2RR.
Strasser and colleagues have investigated the mechanistic aspects of the eCO2RR reactivity of CO2/CO co-feeds on Cu/Ni single-atom N-doped carbon tandem catalysts.72 They have confirmed the *CO (from CO2)–*CO (from CO) cross-coupling pathway, implying the existence of separate CO2/CO adsorption sites. In this tandem catalyst, the Ni single-atom acts as an efficient CO producer, and CuOx nanoparticles work for further C–C coupling. The ethylene yield rate of this tandem catalyst is much higher than that of the sole component catalyst. The results verify the efficient tandem system for local CO co-feeding, which can enhance the ethylene yield and provide a basis for subsequent extended research in eCO2RR. Integration of copper and other adjacent active sites provides the opportunity for efficient C2+ synthesis, such as tuning the adjacent active site spacing, single atom loading density, coordination of the electronic environment around single atoms, etc. Despite the above research progress, there is still a big gap and challenge in developing single-atom CO2RR catalysts for further industrial applications.
2.2. Morphology regulation
The particular morphology of nanostructured catalysts and distinct electronic and chemical surface properties usually confer improved eCO2RR activity and selectivity compared to bulk materials.73,74 In this sense, the reactivity of nanocatalysts can be rationally tuned by modifying the fundamental parameters: size, shape, surface composition, or loading on the support.4,5,10,75As for the 0D nanoparticles, the direct influence of particle size on catalyst reactivity is mainly related to the significant increase in the ratio of the surface atoms to the volume atoms. With the decrease in the particle size, the surface curvature increases, and the corresponding average coordination number decreases.76 In addition, owing to the minimal size of nanoparticles, their surface atoms experience significant strain, which changes the d-band position, thus affecting the reactivity of intermediate groups. Copper nanoparticles exhibit a larger effective area for the further reduction of *CO and favor the formation of C2+ products. The increased CO coverage on cuboid copper nanoparticles provides more C–C coupling opportunities, enabling C2+ selectivity at low overpotential.77,78 Moreover, the *COOH and *OCHO intermediates also play a decisive role in determining the C2+ selectivity. Amal and colleagues have achieved the controllable generation of crystal defects, vacancies, and coordinatively unsaturated metal sites by regulating the voltage of the reduction–reoxidation–reduction pretreatment method. Thus, high formic acid selectivity has been achieved with *COOH as the intermediate on as-synthesized copper nanoparticles.79,80 The oxidation–reduction/amorphous–recrystallization process has been observed using operando XAS, which has revealed dynamic changes in the oxidation state and grain boundary formation during catalyst reconstruction.
Nanostructured materials with various morphologies also present unprecedented catalytic activity, namely, lower onset potentials and enhanced selectivity toward C2+ products.81 Such trends have been assigned to their rough morphology and exposed active facets, as well as to possible changes in the chemical state of the active species. Gao and colleagues have proved that cubic copper nanocrystals with major exposed (100) facets tend to reduce CO2 to ethylene, whereas octahedral copper nanocrystals with predominantly (111) facets convert CO2 into CO and CH4 (Fig. 4C and D).82–84 Hexagonal polyhedral copper enhances ethanol synthesis owing to the regulated binding energy of surface-adsorbed *O on numerous edge sites. Furthermore, the catalyst morphology also affects the surface strain, thus influencing the adsorption of key intermediates (Fig. 4B).85 This relationship has inspired the development of suitable 3D nanostructures to further improve the C2+ activity and selectivity by maximizing the number of active sites and controlling proton transfer/reaction pathways.
Hollow nanostructures play a critical role in the eCO2RR process, exhibiting the following advantages: affluent surface areas, an excellent interface, a strong synergistic effect between different components, and protection of metal active sites. Zhang and colleagues have prepared hollow Cu/CeO2 nanotubes with a high ethylene FE of 78.3% in the flow cell at a low applied potential of −0.7 V vs. RHE.86 The high reduction efficiency can be attributed to the synergistic effects of the inseparable interface structure between Cu and CeO2, which promote the effective adsorption of intermediates. Furthermore, Wang and colleagues have chosen hollow mesoporous carbon spheres for protecting copper clusters to achieve high C2+ selectivity.87 The projected active sites promoted eCO2RR performance with the enhanced formation of *CHO, thus facilitating the C–C bond coupling to form C2H4 and C2H5OH. We propose that the unique hollow structure plays a significant role in the future development of electrochemical C2+ synthesis.
2.3. Surface structure tuning
The engineering of surface defects or surface ligand structure has achieved electronic structure regulation and surface/interface optimization, which provide suitable adsorption sites for reaction intermediates and favorable conditions for further C–C coupling.88,89 Meanwhile, defect engineering achieves the optimization of catalytic surface activity by precisely tuning the defects, including type, concentration, and spatial distribution.90 In carbon-based metal-free materials, the defects are divided into intrinsic and externally induced defects. Intrinsic defects can be purposely introduced to enhance CO2 adsorption by regulating the edge sites, holes, or topological defects. Unlike the intrinsic defects, the defects induced by the covalent bonding of heteroatom doping can break the original sp2 carbon lattice. Defect configurations (such as pyridine nitrogen, pyrrole nitrogen, heteroatom coordination, etc.) can perturb the electron symmetry in the aromatic ring and change the local charge distributions, thus leading to acceleration of the CO2RR process and suppression of the HER activity.As a typical defect existing in metal-based catalysts, vacancies can effectively regulate the electronic structure and mass transfer performance of the active sites, thus optimizing the electrochemical reaction kinetics of C–C coupling (Fig. 5). Oxygen vacancies can be easily introduced and controlled in the form of anionic vacancies. Zheng and colleagues have reported an oxygen vacancy-containing copper-based electrocatalyst with high eCO2RR activity. The number of oxygen vacancies directly affected the selectivity to ethylene and eCO2RR current density.91 The CuOx catalyst with abundant oxygen vacancies exhibited a high ethylene yield at −1.4 V vs. RHE, while the corresponding FE has been confirmed as 63%. As demonstrated by the authors, the ethylene yield is consistent with the change in oxygen vacancies. The oxygen vacancy-rich CuOx surfaces provide strong binding affinity to the intermediates of *CO and *COH, but weak affinity to *CH2, thus leading to efficient formation of ethylene. Moreover, the positive effects of sulfur and selenium vacancies over metal chalcogenide electrocatalysts for eCO2RR have also been well investigated and explored.92–94 In another study by Zheng and colleagues, a copper-based electrocatalyst with disulfide vacancies has been designed and synthesized via lithium regulation. Both theoretical calculations and experiments have confirmed that these disulfide vacancies act as catalytically active sites for enhanced performance of n-propanol synthesis (Fig. 6A and B).92 Previous studies have successfully constructed disulfide vacancies. These defects exert synergistic effects via negative charge enrichment by the adsorption of three *CO intermediates, the provision of a closer Cu−Cu distance for *CO–*CO coupling, and the provision of suitable space for charge repulsion caused by OCCOCO* formation. The adsorption of *CO cannot be completed without vacancies. Although the dimerization of *CO can be achieved on single sulfur vacancies, further OCCO–CO coupling is not feasible. Lastly, CuSx containing double sulfur vacancies has demonstrated an FE of 15.4% for forming n-propanol. Although tremendous efforts have been made to design anionic vacancies, the C2+ selectivity is still unsatisfactory.95 The design of multi-vacancies or coupling of multiple type vacancies (anionic and cationic vacancies) to optimize the migration pathway of reaction intermediates (*COH, *CHO, *COOH) represents a promising research direction for enhanced performance toward C2+ synthesis.
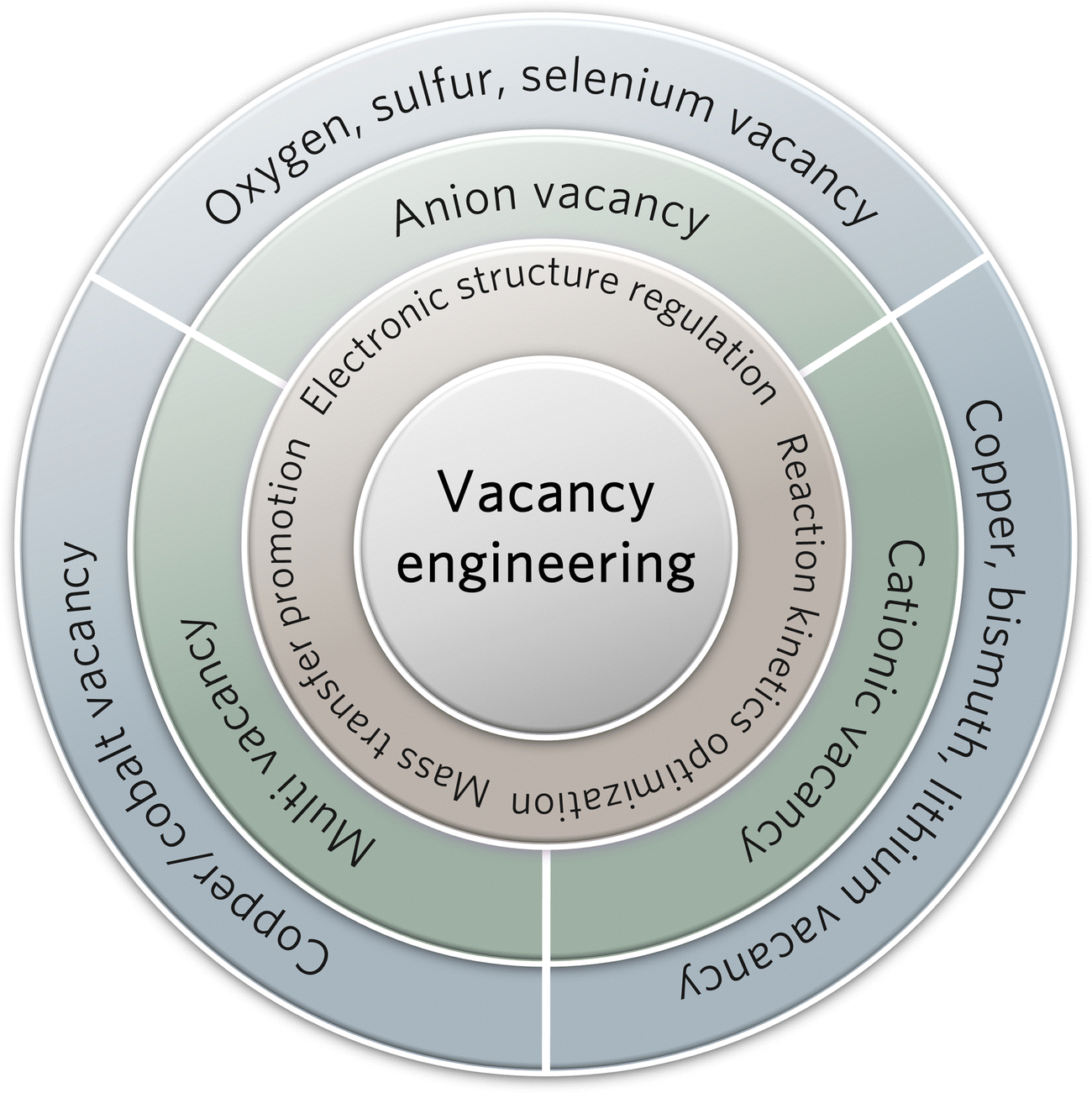 | ||
| Fig. 5 Scheme of vacancy engineering with properties and classifications. | ||
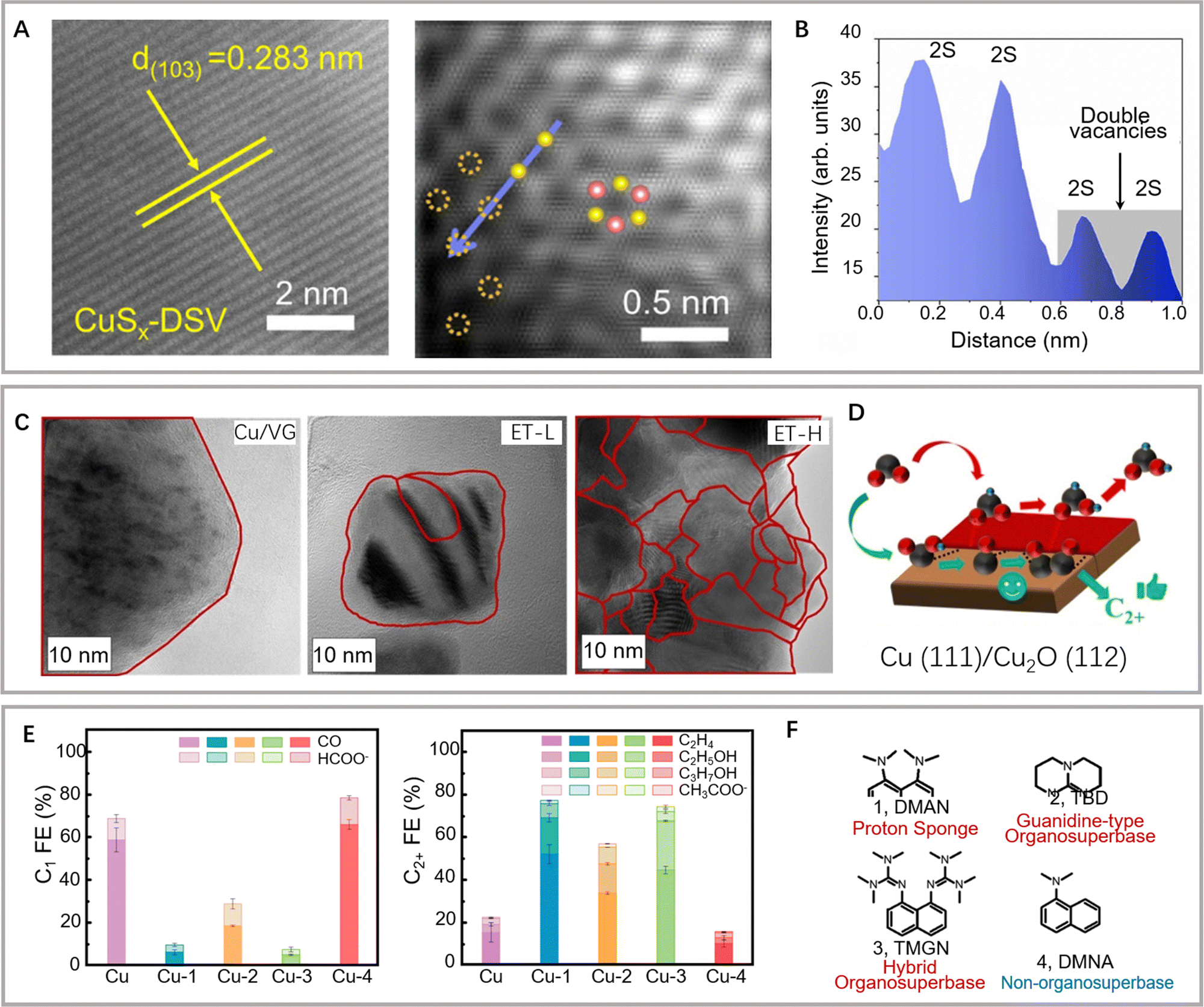 | ||
| Fig. 6 (A and B) Structural characterization of the double sulfur vacancy-rich CuSx catalyst. (C) Intensity profiles extracted from the blue line in (B). Reproduced with permission from Zheng et al.92 Copyright 2021 Springer Nature. (C) HR-TEM images of Cu/VG, ET-L, and ET-H (grain boundaries are highlighted in red spline). (D) Schematic of the preferred CO2RR pathway on the Cu(111)/Cu2O (112) interface. Reproduced with permission from Rose Amal et al.98 Copyright 2022 American Chemical Society. (E) eCO2RR performance of C1 products and C2+ products on different catalysts. (F) Different organic/non-organosuperbases. Reproduced with permission from Wang et al.18 Copyright 2022 Elsevier. | ||
Furthermore, as there is no preferred facet orientation on nanostructured materials, high-density defects, grain boundaries, and surface roughness are considered to determine the final catalytic behavior.93,96,97 As mentioned previously, the residence time of intermediates is critical for the selectivity of C2+ products. Although adsorbed *CO is the crucial intermediate for C2+ synthesis, the correlation between the grain boundaries and the low overpotential does not guarantee similar eCO2RR mechanisms. Rose and colleagues explored a reduction–oxidation–reduction (ROR) electrochemical treatment to advisedly reconstruct copper nanoparticles. Rich grain boundaries were observed, which were attributed to different phases of Cu or CuxO (Fig. 6C).98 In particular, rich grain boundaries formed on the ROR catalyst with Cu(0)/Cu(I) interfaces promoted *COOH/*OCCO adsorption, thus facilitating C–C coupling and leading to *COOH-derived products (Fig. 6D). Most copper-based catalysts inevitably undergo structural reconstruction processes at reaction potentials. The structure and chemical state of the original catalyst (pre-catalyst) may undergo significant changes under the action of the applied potential and reaction intermediates, such as fragmentation, aggregation, and morphology reshaping.99–101 On the one hand, structural reconstruction may lead to the deactivation of copper-based catalysts. On the other hand, it may also induce the formation of active sites that promote C–C coupling to produce C2+ products. The uncertainty brought by this structural reconstruction constrains the development and application of copper-based catalysts. Therefore, it is crucial to design and select pre-catalysts and study their structural evolution under electroreduction conditions. Superparticles have complex assembly structures and undergo unique structural reconstruction processes under electroreduction conditions, which may play a positive role in C2+ product synthesis. Xiong and colleagues have explored the assembly structure of Cu2O superparticles that underwent complex structural evolution during eCO2RR.102 The high C2+ selectivity has been confirmed by in situ spectroscopic tests and electron microscopy characterization. The internal building blocks of these superparticles have produced many grain boundaries, whereas the outer building blocks are separated to form nanostructures. The above structural evolution effectively limits OH− and induces a high local pH around the active sites. The synergy of these extraordinary structural characteristics and the reaction environment on the catalyst surface provides important favorable factors for promoting C–C coupling. Although lattice strain effects in electrocatalysis have been widely investigated,103 the exact relationship between grain boundary-related micro strains and the binding energy of reaction intermediates remains unclear.
Generally, surface ligands are considered harmful to catalysis during surface structure optimization as they occupy other active surface sites.104,105 However, several studies have revealed that surface ligands can help improve the catalytic environment and performance through various mechanisms.104,105 Ligands can perform functions similar to those of protein structures surrounding the active site of enzymes (e.g., by acting as selective permeation membranes, regulating interface solvation structures, regulating the microenvironment on the electrode surface, and participating in chemical activation and the selection of template active sites).106,107 Agapie and colleagues have optimized the surface of nanostructured copper to significantly promote C–C coupling and attenuate H2 and CH4 generation based on the combination of an organic halide additive and a polycrystalline copper structure.108 Mechanistic studies have revealed several effects of organic additives, including the formation of a specific cubic nanostructure by copper surface corrosion, the stabilization of nanostructures via the formation of protective organic layers, and the promotion of C2+ formation. Wang and colleagues have regulated the interfacial microenvironment by modifying the surface of the copper catalyst using a water-insoluble organic super naphthene proton sponge (Fig. 6E and F).18 The adsorbed *CO intermediate has been stabilized by the locally enhanced electrostatic field and the protonated organic superbase. However, the chemical activation mechanism of the surface ligands is yet to be unified. Further experimental/theoretical works should investigate the interaction between surface functionalized ligands and the key intermediates. Moreover, the dynamic structural evolution analysis over surface strain, surface defects, and surface functional groups provides a new opportunity to realize the precise structure regulation for achieving higher eCO2RR activity and selectivity.
2.4. Support modification
Typically, supports effectively inhibit the agglomeration of nanomaterials, increase the surface area, and optimize the electrochemical properties. The introduction of appropriate supports can not only be conducive to achieving a high nanocatalyst dispersion but can also change the electronic structure.109–111 The rational utilization of suitable supports also promotes chemical coupling with the supported catalysts, changes the electronic interface structure, and improves C2+ selectivity.Carbon-based metal-free materials have been widely utilized as carriers for eCO2RR catalysts due to their high conductivity and stability. Carbon supports with a high specific surface area and porosity can be realized through morphology regulation, providing more catalyst anchor sites and accelerating mass transfer.112–115 Nanostructured carbon supports with various morphology are commonly used to support active substances and prevent the aggregation of nanostructures during reactions.116,117 Moreover, the intrinsic physicochemical properties of carbon supports can be regulated using doping and surface functionalization, thus improving the total charge and increasing CO2 concentration on the electrode surface.118
Zhou and colleagues have theoretically investigated the activity and selectivity of metal trimer clusters anchored on N-doped carbon supports to form C2–C3 hydrocarbons and alcohols (Fig. 7A).119 The space-constrained triatomic metal centers have been observed to synchronously immobilize multiple CO2, thus providing electrons and reaction channels for promoting C–C coupling (Fig. 7B). In addition, the mediated cluster–substrate interactions are known to regulate C2+ selectivity. Xu and colleagues have prepared a carbon-supported copper catalyst using the copper–lithium mixed synthesis method and achieved the exclusive formation of ethanol with an FE of 91%.90
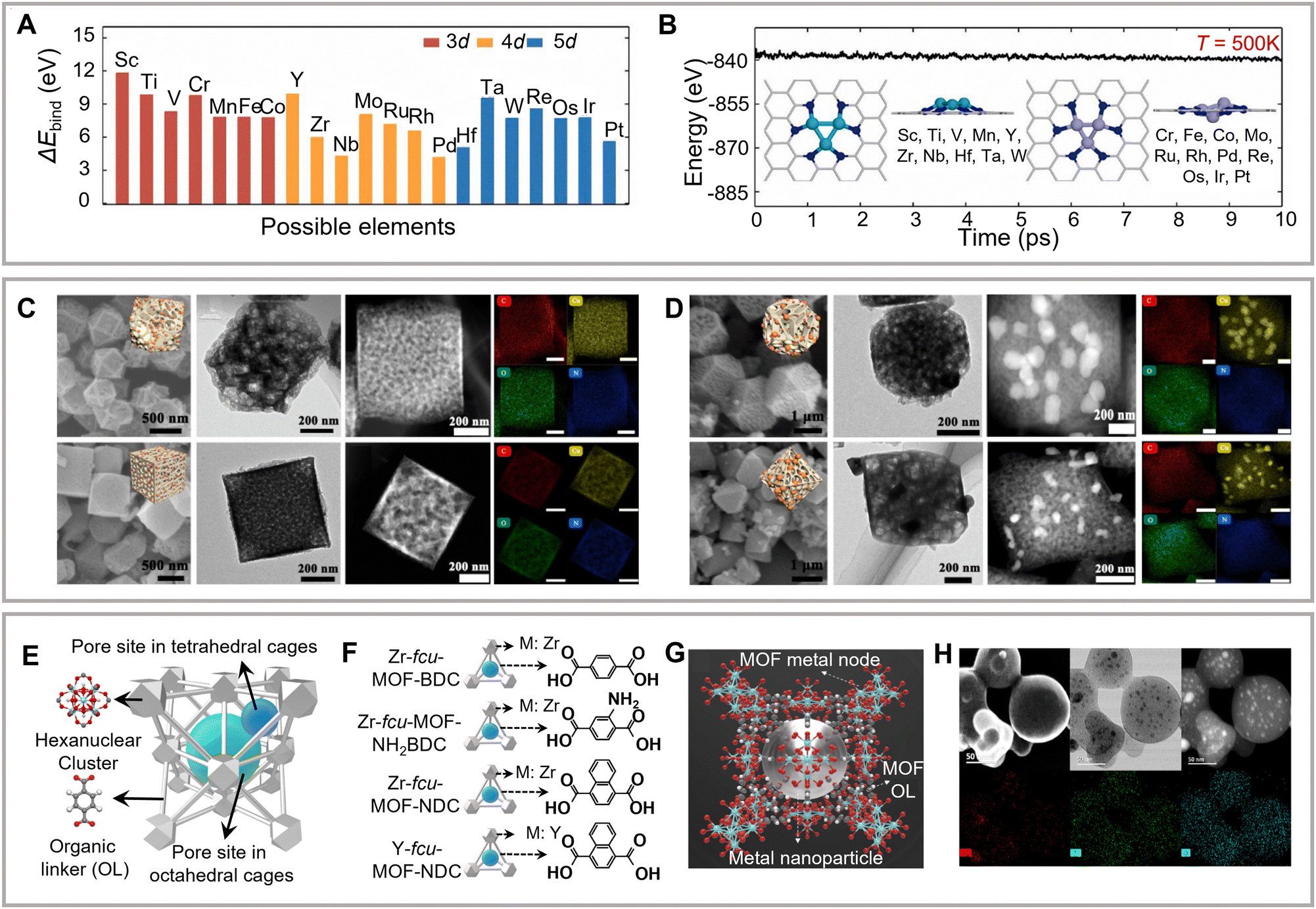 | ||
| Fig. 7 (A) Binding energies between the metal trimer and carbon supports. (B) The corresponding ab initio molecular dynamics (AIMD) simulations and the equilibrium structures. Reproduced with permission from Dou et al.119 Copyright 2020 Elsevier. (C–D) Morphological characterization of shape-tuned MOF support copper catalysts. Reproduced with permission from Cao et al.127 Copyright 2022 Elsevier. (E–G) Schematic of the metal nanoparticle incorporated MOF structure. (H) Morphological characterization of the above MOF structure. Reproduced with permission from Sargent et al.128 Copyright 2020 American Chemical Society. | ||
In summary, the beneficial effect of carbon supports on eCO2RR activity and selectivity is attributed to the following aspects. Firstly, surface functional groups can affect the surrounding electron density. Secondly, optimizing interactions between catalysts and carbon supports is beneficial for forming various products at different overpotentials. Thirdly, the micropores of carbon supports affect the proton transport and CO2 concentration on the catalyst surface, thus affecting the eCO2RR activity. Porous carbon supports optimized by doping, functionalization, or defects have been demonstrated to be ideal substrates for improving the C2+ selectivity.
Due to the electrocatalytic stability and conductivity, metal-based inorganic materials have collectively been widely explored with carbon-based materials as supports for eCO2RR in the past two decades.120 Compared with carbon, metal-based inorganic materials and metal active sites exert a more significant synergistic effect during eCO2RR. For example, theoretical studies have confirmed that the active centers of metal oxides adsorb and activate CO2 at the interface between the metal oxides and metal active centers. Owing to the high affinity of transition metals to reaction intermediates, intermediates can be optimized via oxygen binding. Goddard and colleagues have constructed a copper-embedded oxide matrix model.121 The thermodynamics and kinetics of CO2 activation and *CO dimerization have been significantly improved by the synergistic interaction between surface Cu+ and surface Cu0, thus increasing the selectivity and efficiency of C2+ product formation. Moreover, they have identified the crystallinity of metal oxides and the interaction between active substances and supports as crucial factors for improving activity and selectivity. Suominen and colleagues have successfully achieved efficient CO2 electroreduction to value-added products on Cu/metal oxide heterostructure.122,123 Modified metal oxides can stabilize the key intermediates and decrease the Gibbs free energy of the C–C coupling step. To confirm the surface adsorbed intermediates, Wu and colleagues have conducted in situ characterization based on Cu/ZrO2 electrodes with a high FE of 85% for C2+ products. Experimental results have confirmed the enhanced adsorption of CO* on the Cu/ZrO2 electrode, while theoretical calculations reveal the decreased energy barriers of the C–C coupling process at the Cu/ZrO2 interface. The C–C coupling process is kinetically favored over the Cu/ZrO2 interfacial boundaries, while the competing C1 pathway reactions are significantly suppressed. Current studies tend to utilize MXene materials as carriers for tuning surface functional groups or loading active components in electrocatalysis.124–126 Theoretical calculations have proved that appropriate removal of hydroxyl groups (OH) from MXene-Tx (T = OH, O) can promote surface adsorption capacity for CO2, making activation of CO2 on its interfacial sites easier. The stable existence of CO2δ−, the reduction of the free energy of *CO, *CHO or *COH intermediate formation and the improvement of adsorption stability are more conducive to promoting the C–C coupling process on the above MXene-Tx. Such simple functional group modifications on MXene systems cannot achieve satisfactory C2+ product selectivity in practical experiments. Herein, loading CO2RR active groups (heterojunction, functional group, cluster, single atom, etc.) to prepare MXene-based composite catalysts is an effective way to reduce the reaction barrier and promote the C–C coupling reaction in the future.
Metal–organic frameworks (MOFs) and COFs are commonly used as porous supports in heterogeneous molecular complexes. Compared to traditional carbon-based materials, these species provide highly ordered porous networks, which may enhance electrolyte permeability. Meanwhile, organic frameworks with redox-active components as linkages provide a high specific surface area and tunable porosity (Fig. 7C and D).127,128 MOFs and COFs with optimized structures have been reported to facilitate charge transfer. Chang and colleagues have achieved a breakthrough in the modularized optimization of COF construction using a cobalt porphyrin catalyst organically supported and connected by imine bonds as the construction unit.129 The eCO2RR activity of the synthesized catalyst was 26-fold higher than that of the corresponding control compounds. In addition, XAS data further revealed surface environments’ influence on the electronic structure of the metal center in the COF catalysts. Subsequently, several studies have confirmed the feasibility of using the metal–organic framework structure as a fixed active center and possible support for efficient eCO2RR in aqueous solutions.82,130–132 However, the proposed systems mainly afford the formation of C1 products. To promote the C2+ selectivity, both inorganic substrate (carbon or metal oxide/carbide) and organometallic materials (MOF or COF) should provide well-defined active sites with precise tunability of steric and electronic properties and prevent the active sites from aggregation and demetallation. Moreover, changing the electronic environment of the support around active sites is also effective in promoting CO2RR selectivity.
2.5. Multi-metal cooperation
The formation of bimetallic alloys is commonly applied to alter eCO2RR activity and selectivity. The catalytic performance of bimetallic catalysts varies due to their surface composition and structure.133–136 It is generally assumed that the distinct activity and selectivity of bimetallic alloy catalysts might be attributed to changes in their electronic and geometric structure. The incorporation of a different atom in the lattice modifies the interatomic distance of surface atoms and the lattice strain. The synergy between the two metals can be exploited to regulate the binding (adsorption) strength and configuration of specific intermediates on the catalyst surface, thus promoting C–C coupling and C2+ product formation. Theoretical calculations have revealed that the Pd atom promotes *CO desorption by optimizing the electronic structure of the neighboring copper sites. Sargent and colleagues have introduced palladium into copper catalysts to enhance local *CO coverage and C–C coupling.137 The high affinity of the *CO intermediate enables its competition with H* on active sites. The Pd–Cu bimetallic catalysts achieved high selectivity for C2+ products (FE = 89 ± 4%) by maximizing *CO and CO2 adsorption to weaken H* binding and inhibit HER.
The atomic order of metal components in bimetallic catalysts is also an essential factor in determining their selectivity. Kenis and colleagues have prepared Cu–Pd bimetallic nanocatalysts with equal atomic ratios but different atomic arrangements and examined their electrocatalytic eCO2RR performances.138 Compared to ordered and disordered Cu–Pd particles, the FE for ethylene and ethanol formation is significantly higher, whereas the FE for methane formation is significantly lower. In phase-separated Cu–Pd nanoparticles, adjacent copper atoms promoted *CO dimerization. In the case of alloy nanoparticles, *CO adsorbed on copper atoms may combine with oxygen atoms adsorbed on adjacent palladium atoms to form *CHO, which is further hydrogenated and reduced to methane. The utilization of co-catalysts is another approach for promoting pathways involving *CO by contributing or optimizing *CO formation sites, thus enhancing the production of C2+ products. Accordingly, Qiao and colleagues have demonstrated that the redispersion of silver in copper significantly optimizes the coordination environment and oxidation state of copper.139 The *CO binding strength is changed to form a hybrid adsorption structure, which induces asymmetric C–C coupling and stabilizes the ethanol intermediate to increase ethanol yield. The reactivity and selectivity of bimetallic eCO2RR catalysts are easily affected by various factors. Herein, the modification of the intra-particle atomic arrangement and spillover effects must be considered in the rational design of advanced catalysts to achieve the effective synthesis of specific C2+ products. The multi-metal catalysts should be designed with a donor of the spillover species and a corresponding acceptor of reactive adsorbates (CO2). The spillover effects promote the *CO intermediate coverage and diffusion at the interface of C2+ favored catalysts (copper) and CO-favored catalysts (Ag, Au). This conclusion has been further confirmed by Jaramillo and colleagues, who decorated gold nanoparticles onto polycrystalline copper foil for efficient alcohol synthesis under alkaline conditions.140 CO2 reduction on gold generates a high *CO concentration on nearby copper, where *CO is further coupled to alcohols. The bimetallic electrocatalyst exhibits synergistic activity and selectivity and opens new possibilities for developing CO2RR electrodes exploiting tandem catalysis mechanisms.
Cu-based catalysts with moderate bonding strength for *CO and *H currently only exhibit satisfactory FE and current density in preparing C2 products such as ethylene and ethanol. The development of catalysts for long-chain products is still limited, and nickel has shown specific potential among the materials that have already demonstrated certain effects. For example, nickel phosphide exhibits CO2RR selectivity for C3 and C4 hydroxyalkanes (methylglyoxal and 2,3-furandiol) under an extremely low overpotential and current density.141 However, its stability and selectivity are not satisfactory, which is attributed to nickel susceptibility to CO poisoning and promotion of HER side reactions.142,143 Yeo and colleagues have reported that catalysts derived from inorganic nickel oxide exhibit unexpected activity and stability for the preparation of multi-carbon products, with a total FE of approximately 30% for carbonaceous products.144 The FE of C3 to C6 hydrocarbon products reached 6.5%, with a partial current density of 0.91 mA cm−2. This excellent activity and stability are attributed to the presence of Niδ+ with stable Ni–O bonds during the CO2RR reaction. Unlike Ni0, Niδ+ sites have moderate binding energy with CO, which prevents CO poisoning of the catalyst. Meanwhile, the CH/CH2 insertion mechanism facilitated by Niδ+ leads to the generation of long-chain hydrocarbons. This mechanism of long-chain hydrocarbon synthesis is different from copper-based catalytic systems, providing more options and possibilities for C2+ synthesis.
The strategy for designing highly active catalysts for C2+ products should focus on enhancing the activation of CO2 and stabilizing some key intermediates, in which surface structure engineering plays a crucial role. In this regard, organic molecular functionalization-based surface modification has been achieved by different additives, such as N-containing compounds (amino acids, phenanthroline and the corresponding derivatives), S-containing compounds (alkanethiols) or P-containing compounds.145–147 All the above-mentioned organic molecules can dramatically change the electronic structure of active metal sites, resulting in the optimization of *CO/*H coverage, hydrogen bond interactions and hydrophobic effects. Another effective approach is heteroatom doping for tailoring the spin state and electronic structure of active sites. For metal-based catalysts, the introduction of electron-withdrawing doping sites enables effective bonding with CO2 and regulation of surface metal valence states, thereby improving the kinetics process of CO2 activation and *CO dimerization. For carbon-based metal-free catalysts, the polarization of Lewis basic doping sites can optimize the electronic distribution of adjacent carbon active centers and the bonding ability with reaction intermediates.
Electrochemical CO2RR activity and selectivity are significantly dependent on the size, exposed active facets and morphology of electrocatalysts. The size of nanoparticles determines the density of low coordination atoms on the surface, which directly affects the binding strength of different reaction intermediates. Reducing the size of nanoparticles is conducive to increasing the number of surface-active sites. However, when the nanoparticle size is reduced to below 15 nm, the binding strength between the catalyst surface and H* gradually becomes stronger, which directly induces a weaker binding strength of reaction intermediates (e.g., *CO, *CHO, *COH).141 Therefore, the size of nanoparticles and C2+ synthesis activity follows a volcano-like relationship, suggesting a recommended nanoparticle size of approximately 25 nm. This can provide enough active sites and atoms with different coordination numbers and chemical interaction energy on the corners, edges, and crystal facets. On this basis, the surface binding energy of adsorbed intermediates can be regulated, thereby optimizing the reaction pathways towards different products and improving the selective synthesis of C2+ products. Moreover, considering the different Lewis acidity and polarization abilities of different active facets, optimizing the active facets with the lowest activation barrier and high adsorption energy is beneficial for enhancing CO2 adsorption and activation. The relationship between the facet structure and reaction pathways can be achieved by regulating the single active facet. Accordingly, control over selectivity can be achieved by maximizing the exposure of potential active facets, such as Cu(100) and high-index facets of other transition metals. To maximize the C2+ activity and selectivity on an active crystal facet, ultrathin 2D nanosheets can be prepared as a new form of the CO2RR catalyst. The surface structure sensitivity of CO2RR implies the benefits of utilizing nanoparticles with controlled morphologies to investigate the facet effects and fabricate customized materials with optimized key parameters for improving C2+ selectivity, such as high-density defects (strain, step edge, and grain boundary) and surface roughness.
3. Computational simulation of the formation of C2+ products during eCO2RR
The development of ab initio methods for simulating electrochemical reactions has enabled the investigation of theoretical mechanisms and guided the design of catalysts. Generally, a computational hydrogen electrode model has been used to probe the thermodynamics of electrochemical CO2 reactions.148,149 When a proton/electron is transferred to the *CO intermediate to construct the *CHO intermediate, the free energy change is calculated as| ΔG = μ(CHO*) − μ(CO*) − [1/2μ(H2) − eU] + ΔGsolv + ΔGfield |
3.1. Mechanism prediction
Among all metal cathode catalysts, copper exhibits high selectivity for hydrocarbons. The theoretical mechanism for forming C2+ products on copper is one of the most thoroughly investigated topics in the eCO2RR field. Recently developed theoretical research models for eCO2RR mainly include the implicit solvent model, explicit solvent model, and H-shuttling model.152 These physical models enable the calculation of the pathways of surface eCO2RR. The obtained surface physicochemical properties (such as the electrode–electrolyte interface structure, adsorption energy of active groups, the position of the band edge, heterogeneous reduction potential, and acidity/hydrogenation of chemical substances in solution or adsorbed on the surface) lay the foundation for the further understanding of reaction mechanisms and reaction rate estimation (Fig. 8A and B).153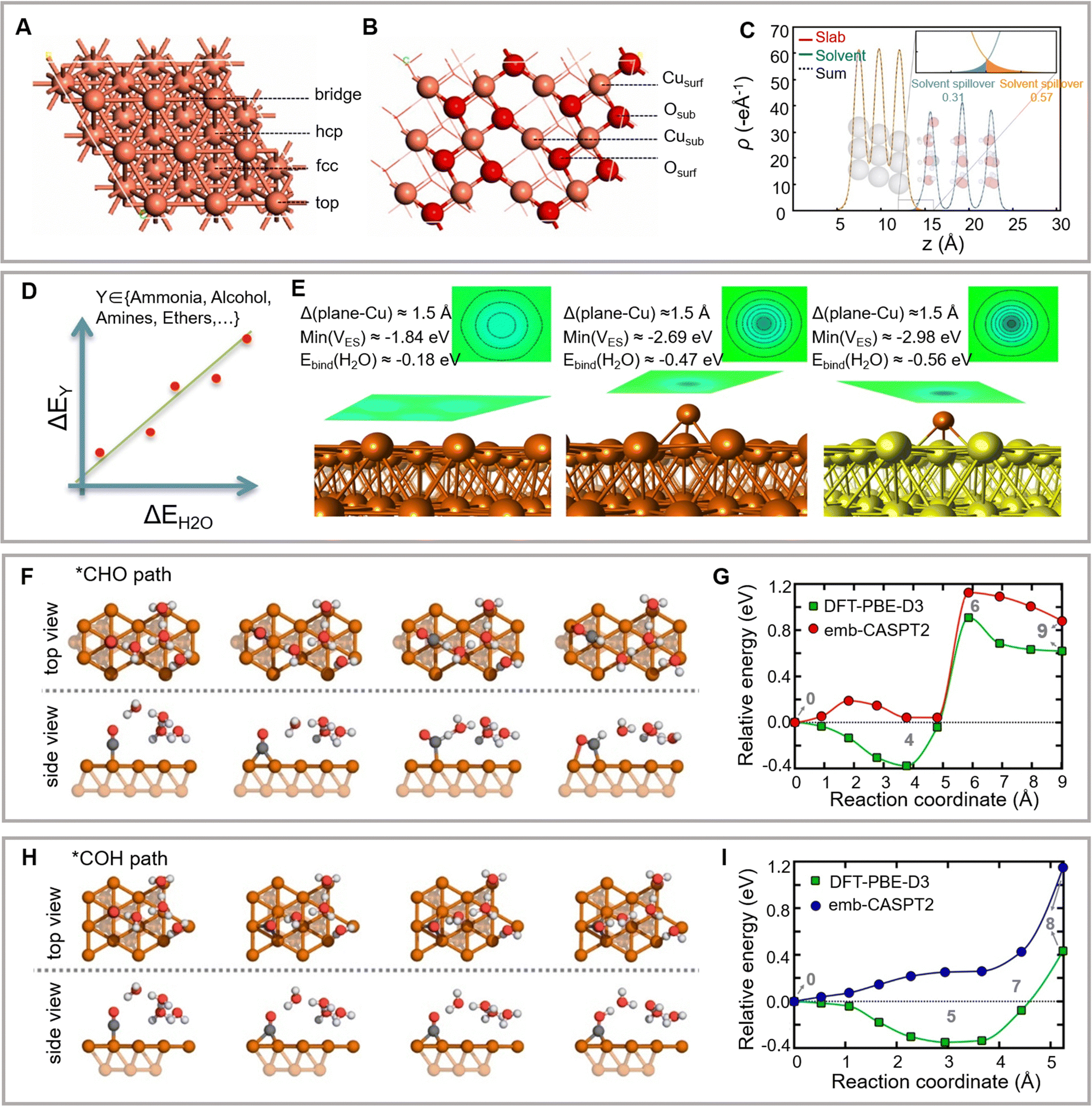 | ||
| Fig. 8 (A and B) Surface structures of Cu(111) and CuO (111) facets with labeled adsorption sites. Reproduced with permission from Liu et al.153 Copyright 2021 Elsevier. (C) Effective fractional charge of a proton in the outer Helmholtz plane. Reproduced with permission from Nørskov et al.160 Copyright 2018 Spring Nature. (D–E) Electron density redistribution on copper surfaces. Reproduced with permission from Nørskov et al.167 Copyright 2018 American Chemical Society. Critical structures optimized in vacuum from the *CHO path (F) and *COH path (H) on the Cu(111) facet. Energy curves for forming *CHO (G) and *COH (I). Reproduced with permission from Carter et al.168 Copyright 2021 American Chemical Society. | ||
Compared to the implicit model, the explicit solvent model is used to investigate electrolyte effects at the atomic level. As ions are explicitly modeled in a finite unit cell, the potentials are not variable. In addition, the simulated proton–electron transfer process induces significant changes in the pathway potential. The generalized gradient approximation level functionals predict the poor band alignment of the solvent's highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) and the metal's Fermi energy levels, which induces artificial charge transfer across the interface and the corresponding effects on the reaction energetics.158 Thus, the artificial interface charge transfer further impacts the reaction energy.159 On the Helmholtz plane, the partial charge generated by the overlap of the solvent and metal charges is independent of the band arrangement (Fig. 8C).160 The nudged elastic band, meta-dynamics, and average force potential determine the transition state.161–164
In the H-shuttling model, H2O and adsorbed *H molecules are shuttled from the surface to the adsorbent. The proton–electron transfer during the shuttle process provides a potential barrier to the hydrogenation of eCO2RR intermediate groups. Meanwhile, compared to the explicit solvent model, potential transfer resulting from charge transfer along the pathway is not taken into consideration. Although the simplicity of the H-shuttling model is convincing, more detailed models are urgently required to evaluate the feasibility and accuracy of these assumptions. The explicit solvent model combines the central molecule and its surrounding solvent molecules as a system for processing. Similar to general cluster models, accurate theoretical calculation results can be obtained by using more solvent molecules. Due to a large number of solvent molecules, it is often necessary to first construct a cluster structure and perform molecular dynamics simulations to obtain a satisfactory initial structure before carrying out quantum chemical calculations. Relying entirely on the introduction of explicit solvents to reflect solvent effects will inevitably result in an extensive calculation system. Moreover, the conformation of explicit solvents is extremely complicated, which cannot achieve correct handling by separately studying the static potential energy surface.
The implicit solvation model allows for the consideration of solvation effects without directly studying a large number of solvent molecules, but instead focusing on the target molecules. The current popular implicit solvent models based on the polarizable-continuum model ignore the structure and distribution of solvent molecules. The solvent is abstracted as an infinitely extended continuum dielectric, surrounding the hole occupied by the solute molecule. By treating solvation as a polar interaction between the surface of the solute molecule and the solvent background, the solvation effect can be approximately described by the dielectric constant. However, the disadvantage of implicit solvent models is that they cannot handle systems in which the solvent participates in the reaction or systems with strong interactions between the solute and solvent (such as hydrogen bonds). The influence of implicit solvent models on the geometric structure and vibration frequency is minimal for neutral molecules or local non-ionic conditions. For cases with significant local charges, the optimized structure without considering the solvent model may differ significantly from the actual structure in the solvent environment. Herein, it is recommended to add implicit solvent models in the CO2RR system to avoid the risk of obtaining qualitatively incorrect results for specific systems. Considering the possible differences in the potential energy surface of the reaction path under the solvent and gas phase at the three-phase interface of the CO2RR reaction, all calculations should add an implicit solvent model, and explicit solvent models should be considered when necessary.
The linear correlation between the adsorption energies of similar intermediates enables the reduction of the complexity of theoretical models. Moreover, Sabatier-type activity diagrams are utilized for simultaneously analysis of multiple materials. According to the linear relationship of the binding energies between eCO2RR intermediate groups and surface atoms, the UL (ΔG) of proton–electron transfer is a linear function of the single intermediate binding energy. The breakage or circumvention of the linear relationship between eCO2RR and the intermediates of electrocatalytic oxygen evolution and reduction reactions has recently attracted significant attention. For example, Nørskov and colleagues have demonstrated that the electrostatic well on a metal electrode surface may polarize the overhanging solitary pair in the case of a lone pair surface bonding to induce strong electrostatically driven bonding (Fig. 8D).167 These strong electrostatic interactions can break the linear proportionality between the binding energies of unsaturated intermediates and molecular species. The robust polymerization of the CO dimer enables the formation of *C2O2, which further explains the preference of the Cu(100) facet for the synthesis of ethylene and ethanol over methane (Fig. 8E). This study opens a novel avenue for designing a new approach for breaking the linear relationships of the adsorbed intermediates on the catalyst surface.
Compared to the methods mentioned above, the water-solvation H-shuttling model considers the proton-coupled electron transfer step and the kinetic barrier of explicit water molecules. This model has been used to simulate eCO2RR on the Cu(111) facet (Fig. 8F–I).78,168,169 Owing to the low barrier, the H2O-assisted *CO → *COH H-shuttling favors *CO reduction over *CHO formation compared to the computational hydrogen electrode model. However, this difference may also be attributed to the reactivity of the simulated copper facets in the above two studies. The computational hydrogen electrode model uses the Cu(211) facet, whereas the water-solvation H-shuttling model uses the Cu(111) facet.
3.2. Kinetics and pathways of C2 product formation
The related theoretical research has been conducted based on different models and approaches due to the complex pathway. This increased complexity can be attributed to the cation-induced field at the interface significantly affecting the C2+ intermediates and the corresponding additional degrees of freedom. *OCCO possesses an insurmountable dimerization barrier of >1 eV on different active facets in a vacuum.171,172 However, electrolyte effects render *OCCO stable concerning 2*CO, thus facilitating dimerization. The Cu(211) facet features exposed under-coordinated surface copper atoms and is, therefore, less stable than Cu(100) and (111).173,174 Under standard electrochemical conditions, the corresponding stability order is Cu(100) facet > Cu(111) facet > Cu(211) facet. The eCO2RR activity order is Cu(211) facet > Cu(100) facet > Cu(111) facet, indicating that the Cu(211) facet exhibits lower barriers for the formation of C2+ products.175 The charge and field distributions of the implicit solvent model will not be localized. However, localization is still necessary owing to the explicit cations. Gordon and colleagues have reported that the dimerization barrier at 0 V on the Cu(100) facet is only 0.6 eV in the case of continuous charge distribution in the solvent.154 Generally, the sensitivity of water solvation and the cation-induced field may induce significant changes in the barrier of CO dimerization on different copper facets, with values of 0.53–1.3 eV obtained for the Cu(100) facet and values of 0.89–1.7 eV obtained for the Cu(111) facet.
The first factor is the local pH surrounding the active site, which directly affects the eCO2RR pathways and product distributions in different theoretical calculation systems.176–178 Under neutral conditions, *CO is coupled with *COH to form a new C–C bond, whereas *CO dimerization is dominant under alkaline conditions. It is worth noting that other factors, such as the ion buffer effect and mass transfer, also influence the local pH value. The buffer solutions (CO2/H2CO3/HCO3−/CO32− equilibria or KHPO4) are sensitive to pH, electrolyte components, and buffer capacity, which cast a vital impact on the generation of different concentrations of carbonaceous intermediate species.179–182 Dunwell and colleagues have explored the primary carbon source using in situ Fourier transform infrared experiments with isotope labeling, which excludes the controversy about the natural source of CO2RR products.183 The buffer anions enhance CO2RR performance by increasing the effective reducible CO2 concentration in solution through rapid equilibrium exchange between CO2 and bicarbonate. Meanwhile, the mass transfer of reactants is of great significance for improving C2+ synthesis.184 Nanostructure optimization and active site engineering still cannot effectively improve mass transfer. It is crucial to supply CO2 diffusion to the catalyst surface by gas-diffusion electrodes based on CO2 electrolyzers.
The second factor is the overpotential, which determines the activation energy barrier and selectivity of the C1 and C2+ pathways. Theoretical studies suggest that C2H4 is formed by *CO dimerization at low overpotential, whereas *CHO/*CO coupling reactions dominate at high overpotential.185 Presently, the recognized mechanism involves the formation of C–C bonds via *CO coupling on the catalyst surface or CO molecules in solution with *CHO adsorbed on the electrode surface.154 Although the form of CO is still controversial, the applied potential certainly affects C2+ product selectivity.
The third factor is the oxidation state of surface metal atoms. Numerous experiments have proven that the oxidation state of surface Cu atoms plays an important role in eCO2RR.102,186,187 For example, Goddard and colleagues have theoretically predicted that the coupling of Cu+ and Cu0 sites promotes CO2 activation and *CO dimerization in oxide-matrix-embedded metals.161 In addition, *CO2 at Cu0 sites can be stabilized by *H2O at adjacent Cu+ sites further enhancing the activation of CO2. Meanwhile, *CO species adsorbed at Cu+ and Cu0 sites possess opposite charges (positive in the former case and negative in the latter case), making them electrostatically attracted to each other.
The fourth factor is the exposed active facet. The energy distributions depend on the atomic structure of the catalyst surface.188,189 Moreover, it is essential to consider surface reconstruction to achieve C–C coupling during theoretical analysis. The last factor is dissolved cations and anions. Section 3.3 focuses on the analysis of electrolyte-ion accelerators and buffering effects.
Electrochemical kinetics characterizes the rate, selectivity, and intermediate coverage, which are all critical functions of reaction conditions.192,193 However, owing to the dependence of the rate index on energy, the prediction of the rate-determining steps is subject to significant uncertainty. Therefore, general trends are considered more reliable than absolute ratios. The C2 dynamic model tends to the initial C–C coupling step. The coupling barrier for *CO dimerization increases with the increasing cathode potential, whereas the coupling barrier of *CO–*CHO dimerization decreases with increasing overpotential.154 Kinetic analysis has revealed that an appropriate overpotential can optimize the C2+ product yield, consistent with the experimental results obtained for the Cu(100) facet. Owing to the pH modification of the models, all hydrogen transfers have been observed to be less favorable than C–C coupling under alkaline conditions, which may explain the higher C2 selectivity at high pH.
Meanwhile, rate-limiting electron transfer in the eCO2RR process has been a recurring and controversial topic in various theoretical studies.154,194 Experiments have revealed that C2+ products with increased pH exhibit a lower initial potential; thus, *CO dimerization was assumed to exhibit rate-limited electron–proton transfer decoupling. Theoretical simulation has revealed that the limited electron transfer uncoupled from electron transfer exhibits absolute potential dependence, and the reaction process is independent of the pH.154 The dependence on the absolute potential (vs. SHE) may result from (i) proton–electron transfer from the proton source (H2O) during the increase in pH or (ii) field stabilization through chemical dipole steps. Particularly, the initial potential decreases with an increase in the pH towards alkalinity. The current density of HER on the metal surface depends only on the potential.176
The effect of pH on C2+ selectivity has been investigated using kinetic models.136 C2+ activity increases at a high pH value and depends on the absolute potential.195 Jaramillo and colleagues have explored the complete dependence of C2+ product activity, which is ascribed to the earlier determination of the proton–electron transfer rate. Generally, the main qualitative characteristics of the experiment are consistent with those obtained theoretically. At high overpotentials, the decrease in C2+ product yield is due to the decrease in CO coverage at the beginning of the formation of C1 products. As C2+ productivity is synchronized with CO coverage, the effect of the reduced coverage on C2+ production activity is more significant than that on C1 production activity.
3.3. Electrolyte effects
The influence of electrolytes cannot be neglected in theoretical simulations as the electrolyte participates in the reaction by interacting with reactants, catalyst surfaces, intermediates, and products.A consistent theoretical explanation of the effects of cation size requires the exclusion of mass transport constraints (Fig. 9A). For example, Bell and colleagues have explored the effects of cation size on catalyst activity and selectivity at low overpotentials (Fig. 9B and C).199 In addition, another study has evaluated the effects of cation size on the energetics of eCO2RR intermediates using the Bayesian error estimation functional (vdW).200 They have further found that the cation-induced electric fields on the outer Helmholtz surface significantly stabilized *CO2 intermediates. Moreover, an increase in the size of the cations in the outer Helmholtz plane is the crucial factor for enhancing the C2+ product, whereas larger cations prefer to be located on the catalyst surface (Fig. 9D).201 Another study has revealed that the mechanism of a similar cationic accelerator effect changes with the variation of overpotential and active facets.202 At low overpotential, the cation size strongly affects the onset potential of C2H4 synthesis. CH4 production is favored at higher potential with a cation size-independent onset potential. Moreover, DFT Perdew–Burke–Ernzerhof calculations have revealed that cations firmly stabilize OCCO* and OCCOH* adsorbents, thus promoting the preparation of C2+ products.199
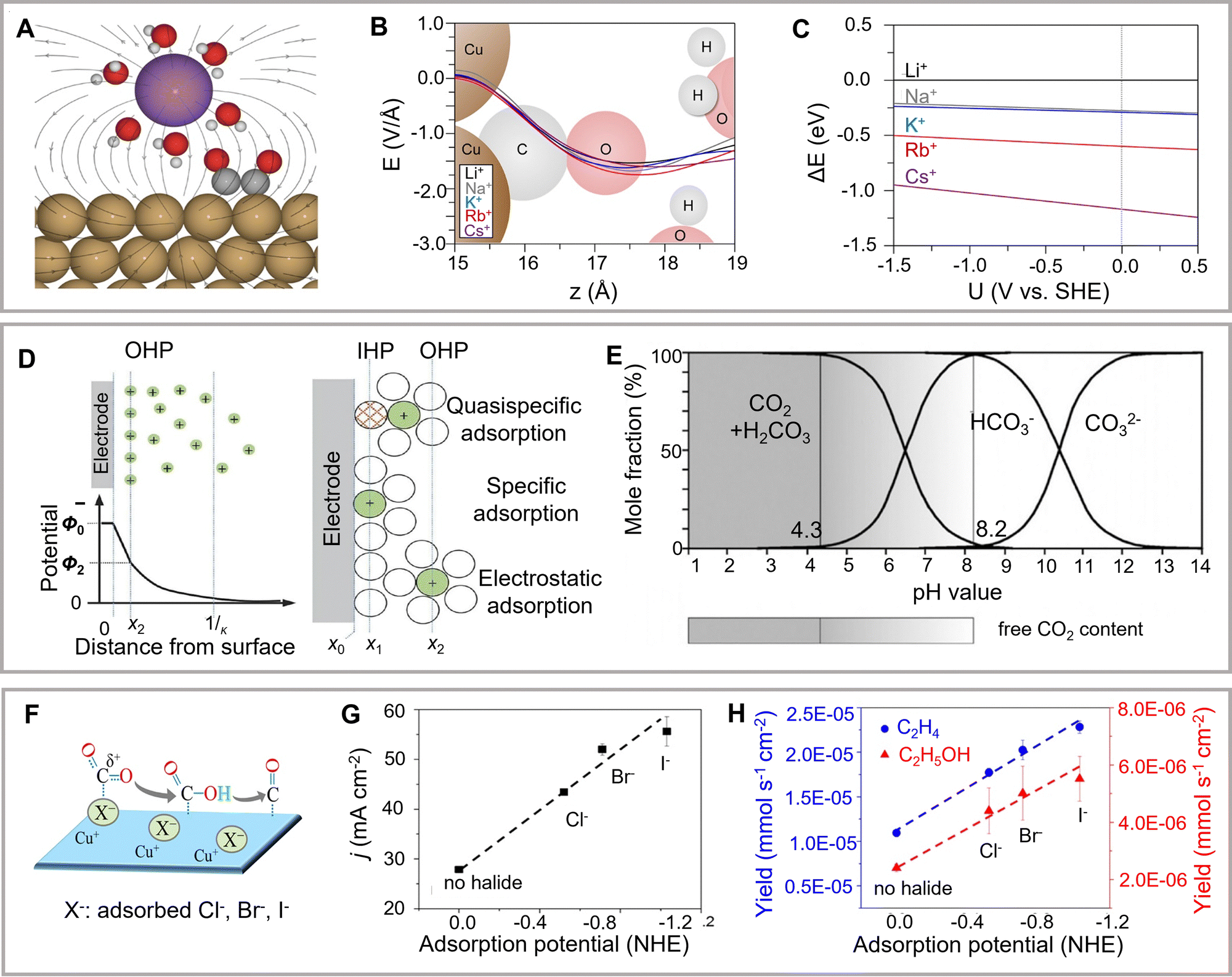 | ||
| Fig. 9 (A) Scheme of cation effects. (B) Electric field distributions near the center. (C) The energy change for bringing solvated cations at the Cu(111) facet. Reproduced with permission from Bell et al.199 Copyright 2017 American Chemical Society. (D) Model showing accumulated cations and depleted anions in the vicinity of the electrode. (E) The modified model involves specific adsorption at the inner Helmholtz plane. Reproduced with permission from Dong et al.201 Copyright 2021 American Chemical Society. (F) Scheme showing the effect of anions. (G) Current density and (H) production rates of ethylene and ethanol in different electrolytes. Reproduced with permission from Cuenya et al.207 Copyright 2017 American Chemical Society. | ||
Electrolyte concentration is the primary consideration. To further evaluate the impact of different electrolytes on CO2RR, it is necessary to deeply understand the interaction among the electrolyte, matrix, and intermediate species. Single properties of the relative dielectric constant or pKa are insufficient to describe the effect of the solvent on CO2RR activity and selectivity. The potential, solvent, and ion models should be clarified to investigate the electrochemical activation energy, which may influence the reaction kinetics and selectivity. Numerous corresponding theoretical methods have been developed.185 However, the same processes measured using various methods have comparatively different amounts of energy. CO2RR energetics have been theoretically investigated by implicit solvent models, which include explicit solvent models with atomized water and ions and H-shuttling models with protons shuttling back and forth from the surface. Goddard and colleagues have applied quantum mechanics methods to develop a mechanistic understanding of the processes of copper.176 The energetics of reaction pathways on Cu(111) with variable pH have been further investigated using a hybrid of H-shuttling and implicit solvent models. C2+ production is kinetically suppressed under acidic conditions (pH = 1). The selectivity for C2+ products arises by kinetically blocking C1 pathways under alkaline conditions (pH = 12). However, the theoretical model shows higher C2+ selectivity under alkaline conditions, which is similar to the experimental results. The application of solvent models needs to be further optimized and explored in the future theoretical simulations.
Traditional simulations often employ the NEB method to describe reaction processes at a static level, which cannot accurately capture systems with dynamic solvent behavior. To solve the above issues, full dynamic simulations can be applied to sample free energy surfaces. However, if the intermediate state is separated by a high kinetic energy barrier, standard unbiased ab initio molecular dynamics (AIMD) simulations will be impractical. Various algorithms (including constrained molecular dynamics, umbrella sampling, and metadynamics) have been introduced to promote sampling potential energy surfaces during CO2RR processes.217–219 Explicit solvent models incorporating the above algorithms can provide a relatively straightforward solution to evaluate ionic effects. Liu and colleagues have investigated the influence of different alkali metal cations on C2+ product synthesis on the Cu(100) facet using a combination of explicit solvent models, AIMD simulations, and free energy sampling techniques.220 As the radius of alkali metal cations increases, the reaction free energy and kinetic barriers of key steps involved in the production of C2+ species (such as CO dimerization to OC–CO) gradually decrease.
Another challenge in current kinetic simulations is the limitation of the electron reservoir in the simulations. Most simulations are conducted under constant charge conditions, resulting in the lack of potential variation throughout the reaction process. Nørskov and colleagues have proposed a straightforward method based on the ideal capacitor model, which only needs to calculate the barriers and the corresponding surface charges at the initial, transition, and final states under constant charge simulations.221 The energy at different potentials can be corrected using the following equation:
| ΔE = 1/2(Q2 − Q1)(Φ2 − Φ1) |
3.4. High-throughput calculations
Artificial intelligence and big data analysis play an increasingly important role in modern technology, presenting new development opportunities in several fields due to the rapid development of computer hardware and software.222,223 Recently, machine learning models have been applied to atomic simulations and electronic property predictions.224,225 Machine learning enables the solving of the traditional Schrödinger equation to obtain the formation enthalpy, density function, basis group effect, recombination energy, chemical reactivity, atomization energy, dynamic density function, electronic ground state properties, transition state partition surface, polymer properties, electron transfer coefficient, crystal properties, atomic charge, dipole moment, electronic excitation energy, and electronic density function.226 Developing density functional techniques has led to an unprecedented atomic-level understanding of catalyst surface reactions to develop suitable catalysts. A large number of prospective materials and a system composed of dozens of atoms may require several hours of calculations. It is impossible to achieve high-throughput catalyst screening through quantitative calculations. The rapid development of machine learning enables the performance of such screening using reasonable chemical models of specific reactions.227,228 The atomic-level quantum chemical mechanisms enable the search for a suitable single or small descriptor to evaluate catalyst reactivity.Zhou and colleagues have predicted the eCO2RR performance of single-atom catalysts using a machine-learning model.229 Based on two simulated materials (M-C2S2 and M-CNO2), ΔGCO heat maps have been predicted using machine learning, considering various bonding environments between metal and non-metal atoms. Taking the ΔGCO of materials experimentally confirmed to be good electrocatalysts as a reference, the authors have preliminarily screened the most promising doping atoms and the most suitable non-metallic coordination environments for CO2 reduction. Furthermore, considering the HER competition, catalytic systems with improved eCO2RR selectivity (such as Sc-CN3, Ti-C2S2, V-NP3, Fe-C2S2, Co-CS3, Ni-C2NP, and Zr-CN2S) have been screened by overlapping ΔGCO and ΔGH heat maps. This machine-learning research provides an effective method for optimizing the composition of single-atom catalysts and guiding the experimental exploration of eCO2RR electrocatalysts.
4. In situ/operando spectroscopic investigation of the eCO2RR
Reaction rates and selectivity determined by computational studies are well correlated with the corresponding experimental data. However, theoretical models focus on well-ordered electrocatalytic systems, whereas actual reaction systems are complex. Therefore, there is an urgent need to apply advanced in situ/operando techniques to explore eCO2RR mechanisms under various operating conditions. Many vital issues of eCO2RR (such as catalyst stability, critical intermediate identification and configuration, determination of catalytically active sites, and the influence of the reaction environment) can be clarified using the in situ/operando approach.230–233 These techniques can capture the subtle physicochemical details of materials. The real challenge is in coupling eCO2RR occurrence and meeting the requirements of operating conditions.4.1. Optical characterization techniques
Optical spectroscopy employs non-ionizing radiations ranging from far infrared to deep ultraviolet. In situ catalytic processes, such as electron transfer and bonding, have been explored with excellent sensitivity and rates using inelastic light scattering. In the eCO2RR process, the structural evolution of catalysts and the vibration frequency of the adsorbed intermediates can be monitored by various characterization methods. For example, optical Raman and infrared spectroscopy are fingerprint techniques used to identify the vibration patterns of key intermediates.234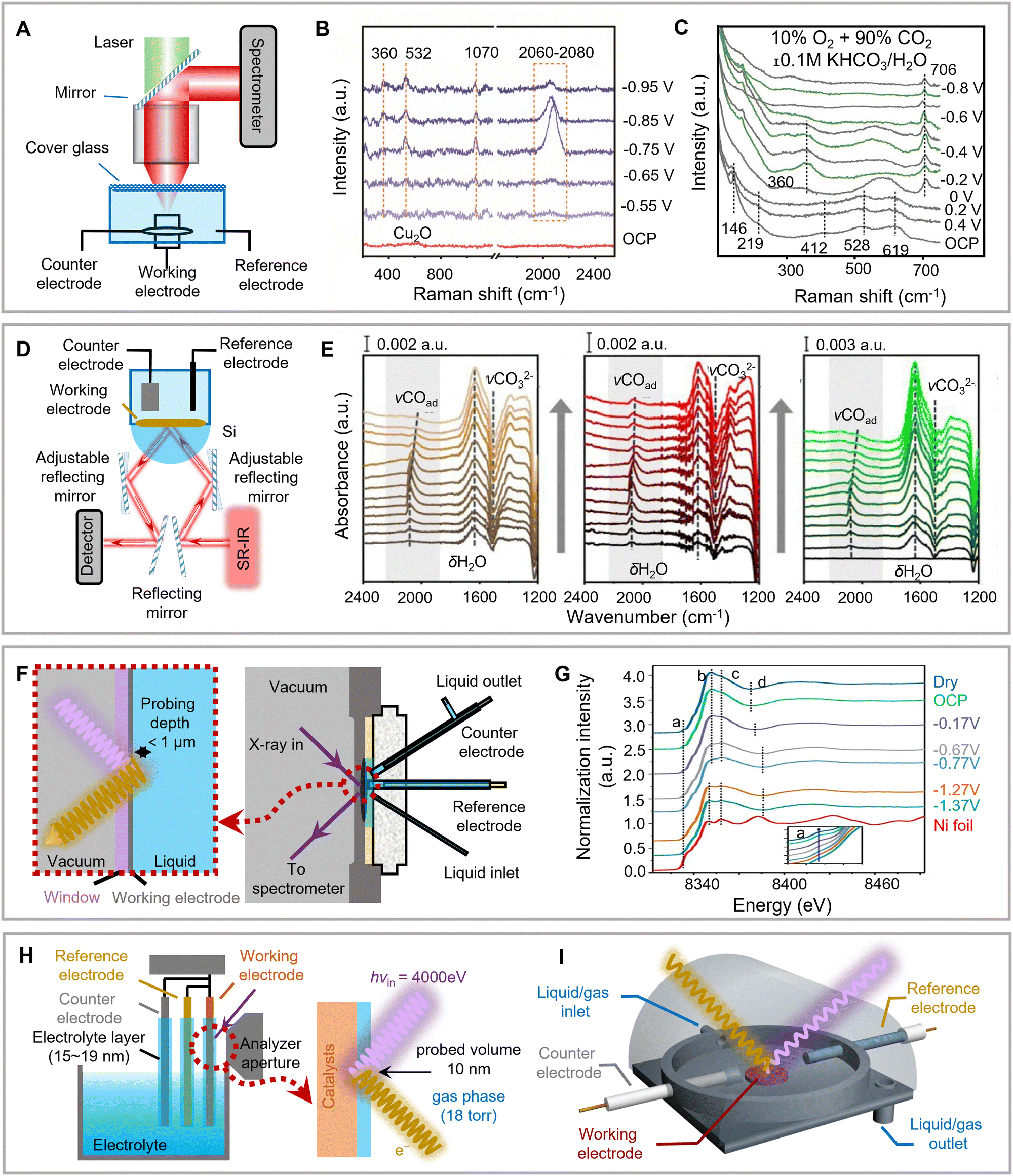 | ||
| Fig. 10 (A) Schematic illustration of the operando Raman spectroscopy setup. (B) In situ Raman spectra of Cu2O superparticle-CP3 during eCO2RR. Reproduced with permission from Xiong et al.102 Copyright 2022 WileyVCH Verlag GmbH & Co. KGaA, Weinheim. (C) In situ surface-enhanced Raman spectroscopy of the copper catalyst in 10% O2/90% CO2 feed solvent. Reproduced with permission from Lu et al.239 Copyright 2020 Spring Nature. (D) Schematic illustration of the operando infrared spectroscopy setup. (E) In situ ATR-SEIRAS spectra on different copper oxides as a function of the applied potential. Reproduced with permission from Gao et al.82 Copyright 2022 American Chemical Society. (F) Schematic illustration of the operando XAS setup. (G) XANES spectra of metal–N–C at various applied potentials in a CO2-saturated solvent. Reproduced with permission from Jiang et al.251 Copyright 2022 American Chemical Society. Schematic illustrations of the operando XPS (H) and XRD (I) setup. | ||
In situ Raman spectroscopy has been utilized to explore the surface structure catalysts. Xiong and colleagues have resorted to in situ Raman for proving a large number of grain boundaries on Cu2O superparticles.102 The peaks of in situ Raman spectra observed at 2060–2080 cm−1 are assigned to the C–O stretching mode of the *CO intermediate (Fig. 10B). The *CO peak is enhanced with decreased potential, and the intensity of this peak becomes lost with a further decrease in the overpotential (−0.95 V vs. RHE). This behavior has indicated the conversion of *CO into C2H4 and other products. The *CO peak of the control Cu2O cube is weak, which is consistent with the eCO2RR results. HCOOH/H2 has been observed as the main product, and the *CO intermediate cannot be formed at low overpotentials. Considering the direct impact of local pH changes near the electrode surface on electrochemical CO2RR, Francisco and colleagues have integrated in situ Raman technology with a continuous flow cell to achieve real-time detection of local pH near the GDE under reaction conditions.235 The CO2–OH− neutralization formed CO32− and HCO3− have been selected as the pH probes for in situ Raman characterization. The above pH probes have distinguishable Raman features and can be independently quantified. Meanwhile, the acid–base equilibrium between CO32− and HCO3− can be utilized to derive the pH. As the in situ Raman tested, the HCO3− distribution extends 120 μm into the electrolyte, and the local pH on the cathode surface is 7.2. This demonstrates that the nominal overpotential reduction originates from the Nernst potential energy of the pH gradient layer at the cathode/electrolyte interface. Therefore, in situ microarea Raman spectroscopy has great potential for investigation of the local pH value near the GDE under working conditions. Although the spatial resolution and Raman sensitivity of in situ microarea region Raman are still limited, the above issues can be overcome by techniques such as surface-enhanced Raman.
Meanwhile, the extensive application of Raman spectroscopy begins with observing the surface-enhanced Raman scattering (SERS) effects.236–238 To date, operando SERS has been used to monitor the metastable status of the catalyst and identify intermediates during the eCO2RR process. In situ SERS has proved that surface hydroxyl species can significantly improve the eCO2RR activity of Cu microparticle systems during co-electrolysis at a low O2 concentration (Fig. 10C).239 Cu microparticle catalysts can easily exhibit a surface-enhanced Raman signal, reducing the introduction of SERS-induced particles. Multiple peaks attributable to surface Cu2O (Cu2Osurf) have been observed at the open circuit potential. An additional peak appeared at 360 cm−1 after Cu2Osurf has been removed at 0 V vs. RHE in the CO2 atmosphere and disappeared at −0.4 V vs. RHE. Therefore, the intermediate species are unlikely to affect eCO2RR significantly. In the O2 atmosphere, a prominent surface hydroxyl peak appeared below 0 V vs. RHE. The SER spectra have demonstrated the characteristics of O2 and CO2 atmospheres. The critical difference in the SER spectra of CO2 and O2 + CO2 atmospheres has been identified as the presence of surface hydroxyl species, which are responsible for the different eCO2RR pathways observed with or without O2 in the reaction atmosphere. Furthermore, Weckhuysen and colleagues have integrated operando SERS with sub-second time resolution and atomic force microscopy (AFM) to successfully monitor the dynamics of CO2RR intermediates and Cu surfaces.240 AFM results have demonstrated the SERS-active nanoparticles formed on the Cu surface after anodic treatment. Besides, a characteristic vibration band below 2060 cm−1 in operando SERS spectra has been observed, which should be ascribed to the dynamic *CO intermediate related to C–C coupling and ethylene production. Further investigations have concluded that anodic treatment and subsequent surface oxide reduction induced greatly enhanced roughness of the Cu electrode surface, resulting in fourfold improved CO2RR efficiency toward ethylene. The detailed examples above demonstrate that in situ Raman/SERS can easily provide the microstructure information of the molecules on the electrode surface (interface). This technology has incomparable advantages in tracking phase structure transition and reaction intermediate transformation, which contribute to exploring the eCO2RR reaction mechanisms.
Furthermore, ATR-SEIRAS has been employed to investigate the eCO2RR mechanism of oxide-derived Cu nanocrystals.69,170,245 Three notable characteristic peaks have been observed in the open circuit voltage range of −1.2 V (vs. RHE). Characteristic peaks observed at −1520 and −1620 cm−1 are assigned to the desorption of HCO3− and the bending vibration of water molecules, respectively. The peak at 2040–2049 cm−1 corresponds to the stretching vibration of *CO linearly adsorbed on the interface (Fig. 10E).82 It is worth noting that this characteristic peak experiences a slight red shift at negative potentials, which is caused by the Stark tuning effect under the action of a more negative electric field. Electron transfer from the catalyst to the 2π* orbital of *CO exhibits a negative correlation with the wavenumber of the ATR-SEIRAS peak. Further, the stretching band frequency of *CO adsorbed on OD-Cu-III has always been lower than those of OD-Cu-I and OD-Cu-V at different potentials. The interaction between the adsorbed *CO species and the OD-Cu-III catalyst is concluded to be stronger, promoting *CO dimerization. The study of eCO2RR kinetics requires in situ/operando experimental investigations of the quantitative correlation between the surface-mediated electrochemical reaction rate and interfacial intermediate concentration. Electrochemical in situ FTIR technology monitors the adsorption and desorption behavior of the intermediates, the electrode structure evolution, and the micro-environment of the electrode surface at the molecular level. This is of great significance to the rational design of catalyst structure and the exploration of new reaction mechanisms. Although in situ FTIR techniques have numerous advantages in studying the catalytic reaction at the electrode surface/interface, a series of problems will occur in the actual test process, such as the absence of any intermediate absorption peak, the strong infrared absorption peak of H2O, and the red or blue shift of the absorption peak position. Therefore, it is necessary to improve specific test methods according to the existing system to obtain objective and accurate in situ FTIR results.
4.2. X-Ray characterization techniques
X-Ray characterization techniques complement optical characterization and help to overcome the limitations of catalyst surface analysis. Operando X-ray techniques have demonstrated outstanding potential for exploring the active sites and mechanisms of catalysts.246,247 However, no direct applications of these techniques to eCO2RR systems have been reported owing to installation complexity or limited central facilities.Jiang and colleagues have integrated operando XAS analysis and used an atom-dispersed nickel catalyst as a model, in which the isolated Ni sites are stabilized by pyrrole nitrogen in the form of Ni–N4. A complete view of potential-induced structural changes at the atomic level during eCO2RR has been achieved by the in situ investigation (Fig. 10G).251 The XAS test during the CO2RR electrolysis shows no obvious changes in Ni XANES under different potentials applied, suggesting the high stability of those isolated Ni active sites over graphene and thus ensuring a practical use in long-term electrolysis. Furthermore, Sargent and colleagues have systematically altered the organic linkers and metal nodes of a face-centered cubic MOF to regulate its CO2 adsorption ability, porosity, and Lewis acidity.127Operando XAS has revealed the stability of MOFs under in situ operating conditions. With an increase in the CO2 concentration, the above regulation plays an essential role in optimizing the binding mode of the *CO intermediate on the surface. Hwang and colleagues have conducted the XANES analyses of Cu-based nanoparticles during the eCO2RR process.252Operando XANES spectra measured at −1.1 V vs. RHE have clearly shown that the oxidation state of the catalyst (Cu2O) reduces toward metallic Cu. This observation is consistent with the other previously reported structural evolutions of oxide-derived Cu catalysts.253 The reduced metallic Cu0 has been fragmented, while producing the highest FE (73%) for C2 + C3 chemicals. The authors have also concluded that the fragmented Cu-based nanoparticles for CO2RR deriving from the initially generated metallic Cu cast significant influence on the final catalytic performance. To sum up, operando XAS is able to capture and analyze the coordination environment of atoms and valence configuration of the catalysts during the eCO2RR. The change of interface electrons, atomic structures, and oxidation state of active species can be tracked. However, operando XAS is a bulk detection technique by measuring the ensemble catalyst film and hence cannot precisely reflect the structural information of the catalytic reaction occurring at the solid–liquid interface. The new XAS technique, combined with other in situ/operando approaches and theoretical calculations, opens a novel approach to clarify the mechanism of eCO2RR.
Fundamental analysis of the chemical structures can be achieved by transferring materials from the electrocatalytic reaction reactor to the XPS chamber under vacuum. Such ex situ experiments have been widely performed for oxide-derived eCO2RR catalysts. However, during sample transfer under environmental conditions devoid of electrochemistry, the rapid interaction between the reactants and solvent may affect the outcome of measurements. These ex situ XPS tests are conducted during the inevitable exposure of the sample to air, which induces changes in the oxidation state of the catalyst surface. Therefore, the natural catalyst surface structure cannot be captured.
A feasible strategy to solve this problem is constructing a reactor with an optoelectronic transparent graphene film as a pressure barrier. Using a technique similar to operando photoemission electron microscopy, a membrane-sealed flow cell electrochemical reactor can be employed for operando XPS characterization in an ultrahigh vacuum.255,256 Another strategy is to separate the sample chamber from the electronic analyzer using a differential pumping and electrostatic lens system. This method enables the placement of an unsealed reactor in a sample chamber at near-atmospheric pressure (Fig. 10H). Ambient-pressure X-ray photoelectron spectroscopy (APXPS) recommends the application of synchrotron radiation sources owing to their balance of incident photon energy, photoelectron IMFP, and photoionization cross-sections.112 However, the electrolyte layer must be sufficiently thin (<20 nm) for adequate characterization under these conditions. Hence, future works must focus on the effective combination of vacuum XPS and in situ eCO2RR tests or the development of in situ/operando APXPS. Further development of X-ray based in situ XRD characterization tests is required to detect the reaction stability of metal materials (Fig. 10I).257,258
4.3. Electron microscopy characterization
Electron microscopy characterization, including transmission/scanning electron microscopy (TEM/SEM), has provided atomic-level information on the morphological evolution and compositional changes of catalysts during a reaction. An in-depth study of the target reaction mechanism can be achieved by tracking subtle changes in the catalysts and electron transfer processes on the surface.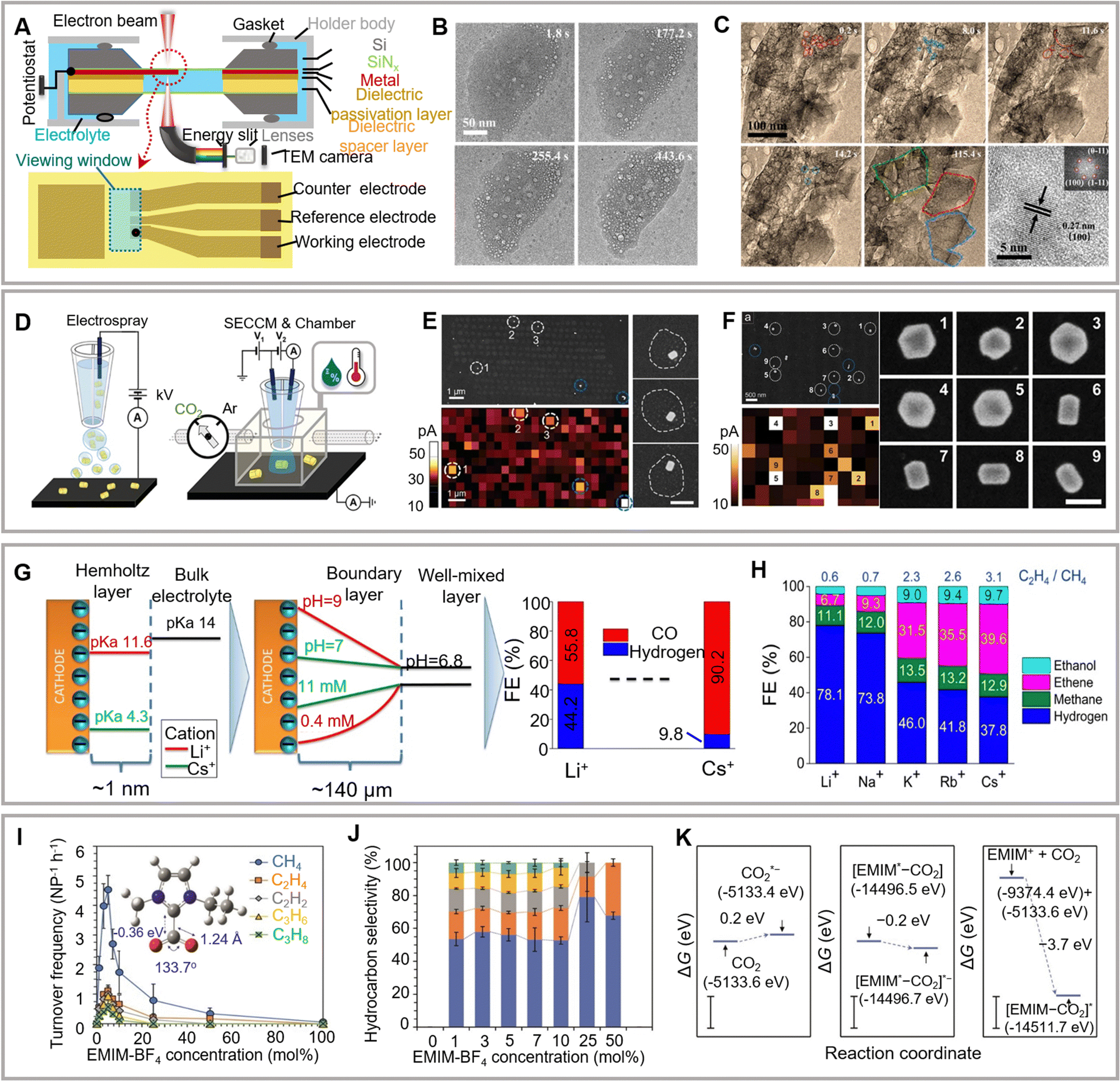 | ||
| Fig. 11 (A) Schematic illustration of the operando TEM setup. Sequential TEM images tracking the transformation process of materials during the catalyst synthesis (B) and eCO2RR (C). Reproduced with permission from Liao et al.262 Copyright 2022 Elsevier. (D) Schematic illustration of the operando scanning electrochemical microscopy (SECCM) setup. SEM image and the corresponding SECCM map of metal nanoparticles under an Ar atmosphere (E) and CO2 atmosphere (F). Reproduced with permission from Ye et al.214 Copyright 2022 American Chemical Society. (G) pKa of hydrolysis of hydrated Li+ and Cs+ inside the Helmholtz layer and in the bulk electrolyte and distribution of pH and CO2 concentration in the boundary layer. (H) FE of eCO2RR products with different electrolytes. Reproduced with permission from Bell et al.197 Copyright 2016 American Chemical Society. (I) Turnover frequencies and (J) selectivity of hydrocarbon products formed plotted as a function of the EMIM-BF4 concentration. (K) DFT-computed free energy cost, ΔG, of formation of different intermediates. Reproduced with permission from Jain et al.282 Copyright 2019 Spring Nature. | ||
Morphological changes under high-resolution operating conditions can be instantly recorded by in situ LPTEM, thus providing valuable information on the surface transformation mechanisms during the catalytic eCO2RR. Liao and colleagues have observed the etching of MOFs during eCO2RR using in situ LPTEM and identified the mechanism of the stability-controlled transformations from ZIF-67 to transition metal cobalt hydroxide (Fig. 11B and C).262 Under slow diffusion conditions, the nanobubbles in the materials gradually move out. Transition metal cobalt hydroxide clusters are formed on the interface, which favors the formation of the porous structures (Fig. 11C). Furthermore, diffusion results in the rapid formation, aggregation, and remodeling of nanobubbles, thus inducing the formation of layered structures. Although LPTEM is not currently available for fully operando tests, this technique holds great promise for further applications. The demand of the ultra-high vacuum condition, the difficulty in the design of in situ TEM cells, and the possible damage from the electron beam to the catalysts are the existing challenges for developing operando TEM techniques.
5. Optimization of the triphasic interfacial reaction system
The efficiency of eCO2RR typically depends on the multistep proton–electron transfer process and the interfacial diffusion mass transfer kinetics. The former process is mainly affected by the crystal structure and active sites of the electrocatalyst, whereas the latter is affected by the wettability and physicochemical properties of the interface structure. The promising achievements in regulating the composition and structure of electrocatalysts are summarized in the previous sections. In traditional solid–liquid two-phase reaction systems, high selectivity for CO2 reduction products can be achieved at a low current density. However, the low solubility of CO2 in common electrolytes and the low liquid diffusion coefficient of the gas therein limit further improvements in the current density. In recent years, gas–solid–liquid triphasic interface models have been increasingly applied to study eCO2RR.266–269 The design and regulation of the micro-/nanostructure and interface properties of catalysts enable the adequate enrichment of the catalyst surface with CO2 molecules in the form of adsorption bubbles or a continuous flowing gas phase. The optimization of the above reaction system enables overcoming the diffusion and mass transfer limitations of traditional two-phase systems, thus improving eCO2RR efficiency at high current densities.5.1. Optimization of electrolytes
Electrolytes participate in eCO2RR by interacting with catalysts, reactants, intermediates, and products. Common aqueous electrolytes mainly contain inorganic salts, alkali metal cations, carbonate/bicarbonate ions, and halogen ions. A deep mechanistic understanding of the electrolyte effect helps to improve eCO2RR activity and selectivity through efficient electrolyte design. The theoretical calculations summarized in previous sections indicate that different anions and cations exhibit different eCO2RR activities.270 Therefore, the influence of the electrolyte on the catalyst structure and the catalytic reaction should be further explored to improve eCO2RR activity and C2+ selectivity through practical electrolyte design.The interpretation of cations effects has been further investigated. Bell and colleagues have proposed a theory for this buffering effect.273 The pKa of the cations decreases with increasing ion size. Suppose the pKa is lower than the pH of the adjacent solution, the solvated alkali cations release H+ as a buffer to adjust the pH on the surface, reduce the CO2 conversion into CO32− and HCO3−, and improve the CO2 solubility at the interface. Meanwhile, Xu and colleagues have explored the effect of cation size on interfacial CO2 concentration using in situ transmission infrared spectroscopy.183 The interfacial CO2 concentration decreases and depends on the rate of OH− generation by electrolysis rather than on the buffering effect of alkali metal cations.
It is difficult to determine the degree of interaction between cations and electrode surfaces from an experimental perspective. Only some macroscopic physical parameters, such as electrode surface pH and interfacial CO2 concentration, can be experimentally determined. The experimental determination of mechanisms is extraordinarily challenging and prone to conflicting theories. This pending problem requires applying in situ characterization techniques and computational simulation methods to gain a deeper understanding.
In addition to being a pH buffer and a proton source, bicarbonate can function as a CO2 source. Dunwell and colleagues investigated the eCO2RR mechanism in a bicarbonate electrolyte on a gold electrode using isotope labeling.183 Owing to the dynamic chemical equilibrium between CO2 and HCO3−, bicarbonate acts as a carbon source for converting CO2 to *CO. OH− is another commonly found anion in electrolytes, with alkaline environments favoring the formation of C2+ products. Both theoretical calculations and experimental explorations by Sargent and colleagues have revealed that OH− not only reduces the binding energy of *CO dimerization for the formation of *OCCO but also promotes the charge imbalance between carbon atoms in *OCCO. The stronger dipole attraction in this intermediate and the reduction of the activation energy of *CO dimerization are finally realized.111,272
In recent years, the excellent performance of halogen ions in eCO2RR has been demonstrated.276,277 During the deposition process, these ions change the surface structure and morphology, generate unique crystal facets, and improve the roughness of the electrode material. The doping of halogen ions enables the activation of H2O via the regulation of the electronic structure, which promotes the formation of critical active intermediates. Halogen ions can also be used as electrolyte additives to affect the adsorption of intermediates and active species. After adsorption on the inner Helmholtz layer, halogen ions engage in strong van der Waals interactions with active intermediates. Anions mainly affect the morphology of electrode materials during deposition, regulating the transfer of electrons from the electrode to CO2. Zhai and colleagues optimized the different steps of C2+ product synthesis by leaching out halogen ions on the AgI–CuO tandem catalyst under CO2RR conditions.278 The leaching of iodine ions inhibits the reduction of CuO nanosheets to obtain stable active Cu0/Cu+ species, promoting *CO overflow. After the in situ leaching of iodine ions, the I-modified Ag structure tandem catalyst promotes the production of CO, and the Cu–Cu2O heterojunction structure facilitates the formation of the key intermediate *OCCO for C–C coupling. Presently, progress has been made to elucidate the interaction mechanisms of both cations and anions. However, deeper microscopic interaction mechanisms should be further investigated with the development of in situ characterization technologies and computational simulation methods.
A comparison of the structural characteristics of CO2 activated by water and 1-ethyl-3-methylimidazolium tetrafluoroborate has revealed that the interaction energy between carbon atoms in the imidazole ring and carbon atoms in CO2 was −0.36 eV. On the other hand, the interaction energy between water molecules and CO2 was only −0.11 eV (Fig. 11I).282 CO2 molecules undergo significant bending under the action of ionic liquids, and the resulting elongation of C–O bonds favors CO2 activation (Fig. 11J). The influence of ionic liquids on the stability of intermediates and reaction pathways has also been explored (Fig. 11K). Sha and colleagues have found that the FE of C2H4 formation significantly increased with the modification of the copper electrode surface by using 1-ethyl-3-methylimidazolium nitrate.283 For the decisive step of C–C coupling to produce C2H4, the presence of an ionic liquid reduces the energy barrier of *CO dimerization into *OCCO by 0.35 eV, thus favoring the formation of C2H4.
The interface between a metal electrode and an ionic liquid is very different from that between a metal electrode and an aqueous solution. At the applied potential, the dynamic transformation of the ionic liquid structure at the interface is slow. The reconstruction of the multilayer structure enables the formation of a significant double-layer structure. Characterization by vibrational sum-frequency generation spectroscopy has revealed that their structural transformation controls the reduction of CO2 in ionic liquids.284 However, it is difficult to directly detect the interfacial reaction mechanism in the structure of interfacial double electric layers with thicknesses of only several nanometers. At this stage, the formation and action mechanism of the interface structure have been discussed extensively using simulation calculations. In conclusion, although introducing ionic liquids into eCO2RR systems may be a promising approach to explore, the insufficient stability of ionic liquids during eCO2RR may complicate product detection and limit the practicality of large-scale reuse.
5.2. Optimization of the triphasic interface
The formation rate of eCO2RR products is mainly limited by the following two factors. (a) Generally, a large overpotential is required to provide a high current density for eCO2RR. However, HER dominates the reaction with increasing current density. (b) The solubility of CO2 in water is relatively low. Beyond a certain threshold, the current density is no longer controlled by the chemical reaction kinetics but is determined by the proton diffusion process of CO2. Therefore, the mass transfer of CO2 molecules at the interface of catalytic materials greatly influences the reaction efficiency. Reduction in traditional solid–liquid two-phase systems is limited by slow diffusion and mass transfer, which results in low catalytic activity and product selectivity. These problems cannot be effectively solved by optimally regulating catalysts alone. Therefore, triphasic systems are introduced to solve the problem of the speed limit in the mass transfer process and ensure the effective progress of highly selective eCO2RR at high current densities.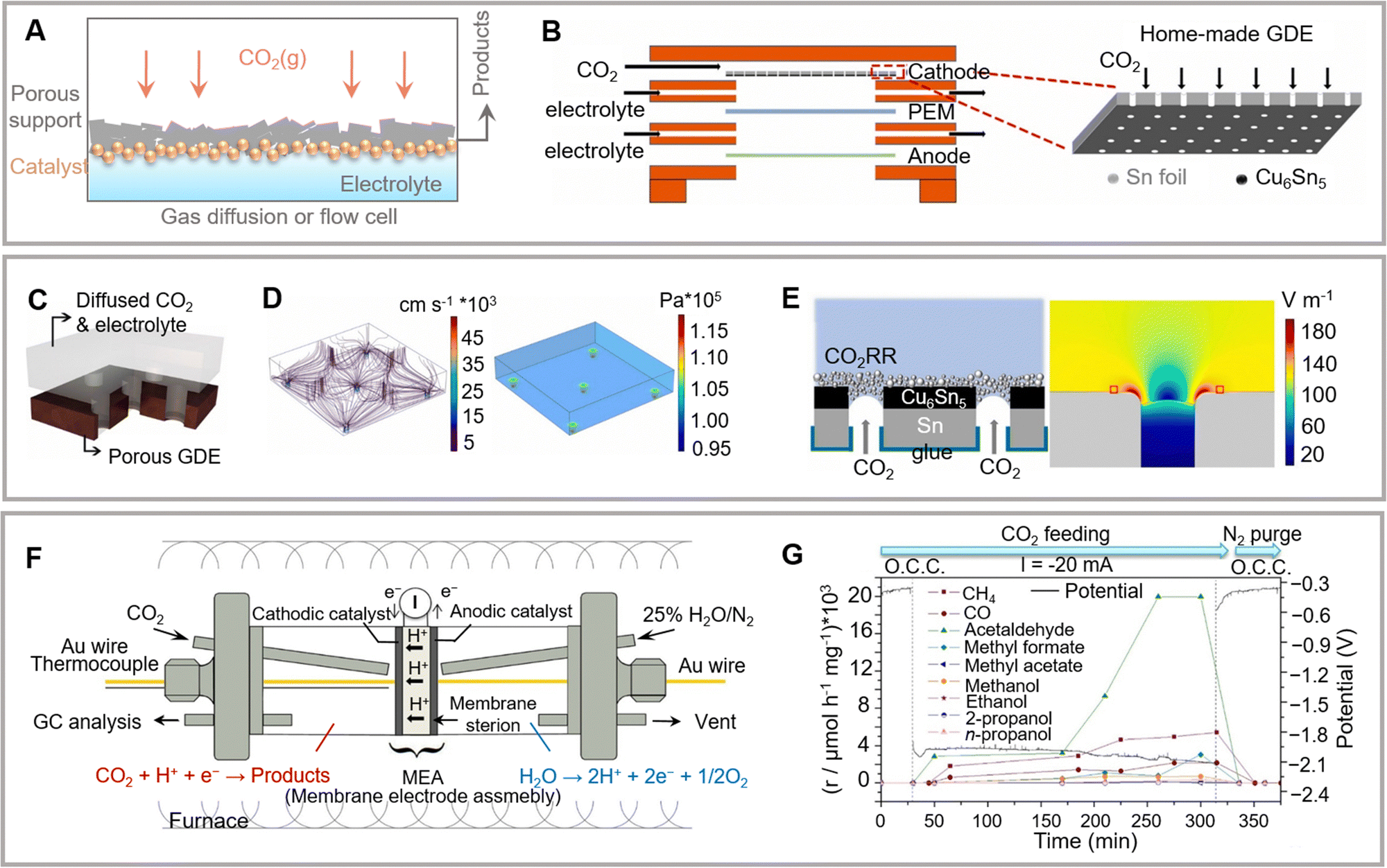 | ||
| Fig. 12 (A) Schematic illustration of the triphasic electrocatalysis system. (B) Schematic diagram of the flow cell and gas diffusion layer-based electrolyzer (GDE). (C) 3D schematic of porous GDE diffused CO2 and the electrolyte. (D) Simulation results of the velocity and pressure field of diffused CO2 in the 3D model. (E) 2D schematic of the liquid–gas interface and the corresponding electric field distributions. Reproduced with permission from Zhou et al.310 Copyright 2022 Elsevier. (F) Schematic illustration of the eCO2RR reactor. (G) Time-on-stream variation of different products and applied potential. Reproduced with permission from Lucas-Consuegra et al.312 Copyright 2016 Elsevier. | ||
GDL-based cells have achieved higher current density than traditional H-type cells. Sargent and colleagues have reported a copper-based electrocatalyst at the mutagenesis interface with a high FE (70%) of ethylene and a maximum current density of ∼750 mA cm−2.111 Catalysts deposited on the GDL significantly increase the local concentration of CO2 in the electrolytic cell. Hydroxyl ions on the copper surface reduce the energy barriers for CO2 activation and *CO–CO coupling. In this study, a polymer-based GDL has been introduced to enhance operational stability. PTFE and carbon nanoparticles are divided into two layers, and the copper catalyst layer is sandwiched between them to form a graphite/carbon nanoparticle/copper/PTFE electrode. This adjustment effectively prevents liquid leakage from the GDL. Moreover, the as-designed GDL is sandwiched at the interface between separate hydrophobic and conductive carriers. This optimized structure can ensure constant and efficient ethylene selectivity during long-term electrolysis.
Based on this study, Sargent and colleagues have further optimized a novel ionomer heterojunction structure.288 The decoupling of gas, ion, and electron transmission enables the effective electrolysis of CO2 in the gas phase to produce C2+ products at a high current density (>1 A cm−2). The ionomer heterojunction comprises a copper nanoparticle catalyst layer and a perfluorosulfonic acid ionomer layer. A 3D morphology has been formed with metal and ionomer permeation pathways. This structure is both hydrophilic and hydrophobic. The separate transmission of gas, ions, and electrons increased gas diffusion and transmission paths. Herein, the elevated current density and significantly improved reaction efficiency enable a maximum ethylene yield of 65–75%. Although the catalyst is the core of electrocatalysis, a GDL-based catalytic reaction system featuring a gas–solid–liquid triphasic interface can also play an influential role by improving the GDL and increasing the concentration of reactants around the catalyst.32,289–291
Various liquid electrolytes (such as salt solutions, ionic liquids, etc.) have been utilized in different CO2RR electrolyzers for C2+ synthesis. The drawback of these catalytic systems is that the resulting liquid products are mixed with the electrolyte liquid phase, leading to significant energy consumption during the subsequent separation process. The introduction of solid polymer electrolytes or porous solid ion conductors can theoretically solve the above issues. The water gas hydrophobic layer formed at the gas–solid interface can not only kinetically limit the dissociation of H2O to suppress HER but also stabilize the formation of CO2 reaction intermediates to facilitate the formation of C2+ products through a relay catalysis model. The successful construction of solid-state electrolytes has the following advantages: optimization of the three-phase interface reaction environment, reduction of the ohmic loss of the entire device, avoidance of corrosion and electrolyte consumption issues, and solution of the product separation problems.
First, the crossover of products and reactants is a prominent issue. However, AEMs can alleviate cathode flooding and improve CO2RR performance.297 Negatively charged CO2RR products are easily transported through the positively charged AEM, while neutral products (such as ethanol) can crossover the membrane. Another operational issue is the mechanical and chemical stability of commercial AEMs. The discovery of novel AEMs has been realized with the recent developments of polymers. Polystyrene tetramethyl imidazolium chloride (PSTMIM), commercialized as Sustainion, is an AEM designed for gas-fed CO2RR electrolyzers in neutral solutions. This thin hydrophilic membrane features elevated OH− conductivity and ion exchange capacity. Such outstanding properties of Sustainion ensure 93% faradaic efficiency of the CO2RR electrolyzer at the practical current of 350 mA cm−2 for 40 h.298 However, Sustainion is prone to CO2RR products’ crossover, particularly at the high current densities relevant to commercial operation. Furthermore, quaternary ammonium poly(N-methyl-piperidine-co-pterphenyl) (QAPPT) has been introduced as another candidate material for AEM construction.299–301 QAPPT exhibits better chemical stability than Sustainion in high pH solutions or at elevated temperatures (>80 °C). The high conductivity of QAPPT eliminates the need to humidify the CO2 feed or use ionic anode electrolytes. With an operating temperature of 60 °C, the CO2RR electrolyzer involving QAPPT demonstrated a high current density with an FE of 90% and a cell voltage of 3 V. These results motivate further research on incorporating Sustainion, QAPPT or other novel AEMs for utilization in C2+ producing systems.
The chemical stability, ionic conductivity, size and mechanical stability of the AEM directly determine the performance and lifetime of CO2RR electrolyzers. The main issues with the AEM are chemical stability and low ion conductivity. For example, in an anion exchange membrane, the ion conductivity of OH− in water is much lower than that of H+. Some strategies have been developed accordingly to boost the chemical stability and ion conductivity. Polystyrene and its perfluorinated polymer, as well as polybenzimidazole polymers with excellent mechanical properties, can be introduced to construct semi-interpenetrating network membranes to improve chemical stability. Meanwhile, the membrane with one-chain multiple functional groups can be designed to promote the conductivity of the membrane with C–H-based main chain of poly(styrene-ethylene-butene). Another issue that should not be ignored in the membrane structure optimization process is the thickness of the AEM, which should be specially adjusted to achieve synchronous enhancement of stability and conductivity.
The mass transfer of *CO is still crucial for tuning eCO2RR activity and selectivity. The operating current density and target product selectivity are highly dependent on the CO2 flow rate, CO2 partial pressure, or even the thickness of the catalyst layer. When using the GDL, the thickness of the mass transfer boundary layer decreases from 60–160 μm in the H-type cell to 0.01–10 μm in the GDE, and the current density is significantly improved.303,304 The local pH value is enhanced at high current densities to inhibit methane formation and promote C2+ product selectivity. Furthermore, high CO2 conversion rates can be realized using low CO2 flow rates. The optimal flow rate should exceed ten sccm for optimal C2+ product formation at current densities of 100–150 mA cm−2.305 The partial pressure of CO2 favors the selective formation of C2H4 rather than CO. Moreover, the 250 nm-thick layer is more selective than the 50 nm-thick layer, suggesting that the optimized thickness enhances C–C coupling.306 Owing to the increase of OH− concentration, the dipole interaction becomes stronger, and the binding of *CO has been improved. In addition, the increased *CO mass transfer and *CO dimer stability promote the C2+ selectivity.307,308
Sargent and colleagues have calculated the GDL thickness using parameters such as bubble separation diameter, pressure, fluid velocity, and current density. The bubble separation diameter depends on the catalyst surface morphology and wettability.309 By comparing the influence of nanowires, nanorods, and nanoparticle catalysts on the bubble separation diameter, the authors have found that tiny bubbles generated by nanowires produce a smaller diffusion layer thickness and promote the interfacial mass transfer of CO2. Thus, the electrode morphology is concluded to influence the long-distance transportation of CO2 profoundly. Zhou and colleagues employed a laser to prepare channels on a Cu6Sn5 alloy electrode as a GDE and applied it in a flow cell to obtain a high eCO2RR FE and catalytic stability (Fig. 12B).310 The distributions of CO2 concentration and an electric field near the electrode are simulated using COMSOL Multiphysics. A high CO2 concentration and a strong electric field around the electrode surface favor the occurrence of eCO2RR (Fig. 12C and D). In addition, Kelvin probe force microscopy has been utilized to measure the actual electric field distribution around the channel, and the results are consistent with simulation data (Fig. 12E). Zhang and colleagues have explored the conditions and influencing factors of CO2 mass transfer by changing the wetting characteristics of a typical GDE surface.311 Interfacial structure plays a crucial role in stabilizing CO2 concentration during eCO2RR. The Cassie–Wenzel coexistence state is an ideal triphasic structure with continuous CO2 supply on active sites at a high current density. This study provides the mechanism of the triphasic electrocatalytic reactions of other gaseous substances. However, more understanding of the actual mass transfer at the micro-interface is required. Therefore, more in-depth mechanistic research and accurate and intuitive in situ/operando characterization methods are essential to overcome the limitations of CO2 interfacial mass transfer.
Kenis and colleagues have successfully designed a microfluidic CO2 cell system using a GDE with a thin (1 mm) channel. Compared to MEA systems, the supplied CO2 diffused through the porous GDL to naturally form an electrolyte–catalyst–gas triphasic interface even in the absence of water vapor (Fig. 12F).312 The eCO2RR can occur at the boundary between the catalyst and the cathode liquid. The reactor is not sensitive to the ion transfer rate and can achieve a high current density (Fig. 12G).306 It is essential to introduce a membrane between the electrolyte channels to achieve the separation and anti-reoxidation of liquid C2+ products on the anode side. Over the past few years, various electrolytic cells have been developed using shared high-performance catalysts.313 Presently, the volatilization of gas products and the shuttling of liquid products across multiple flow cells remain challenging.314 In addition, research on catalyst–electrolyte interface tuning (e.g., the development of different ion-exchange membranes, optimization of electrode surface/interface structures, and innovation in electrolytic cell design) requires further exploration.
In the traditional flow cell, the gas–liquid cathode chambers of the hydrophobic GDE are relatively well separated.286,316,317 The cathode is exposed to the feed gas to realize the coexistence of the liquid and gas phases in the catalyst layer, while CO2RR occurs at this triphasic boundary of the GDE (Fig. 13A). This structure can promote mass transfer and significantly improve the stability of CO2RR in alkaline electrolytes at industrial-grade current density. For the catalyst layer, the optimized catalyst material powder is generally loaded onto the GDE by brush coating, air spraying, or ion sputtering technology.318,319 The composition and proportion of solvent, the selection of binders, and the coating method of the catalyst may all affect the performance of electrolyzers. For the microporous layer, the appropriate microporous layer can achieve gas–liquid two-phase separation (especially in the microfluidic device) and prevent electrode flooding.286,320 The smooth and tight microporous layer decreases the contact resistance between the catalyst layer and the carbon substrate and prevents the catalyst from entering the microporous or substrate layer. The PTFE in the microporous layer affects the porosity, conductivity, and water distribution of the electrode.321 Kim and colleagues have introduced a thin liquid pH buffer layer between the cathode GDE and the ion exchange membrane to further improve the CO2RR selectivity.322 The microfluidic flow cell with a PTFE content of 20% achieves low charge transfer resistance and enhanced performance (Fig. 13B). Moreover, the hydrophobicity of the carbon substrate also influences the performance of the electrolyzer. Park and colleagues have constructed a microfluidic flow cell with different hydrophobic carbon substrates. The current and selectivity are finally optimized by tuning the hydrophobicity.323 This microfluidic structure is shown in Fig. 12C. By coordinating gas and liquid flow rates to prevent gas diffusion or electrode flooding, the pressure balance can be well tuned. The gas at the anode side can be directly diffused into the air without sealing treatment. The electrolyte solution flows through an extremely tiny channel. The narrow electrode spacing reduces the ohmic polarization loss of the microfluidic cell. Based on the above examples, suitable GDE structures can promote CO2 electroreduction for their fast mass transfer or high stability. Despite the tremendous progress made with GDEs and liquid flow-cell electrolyzers, numerous problems still need to be well addressed for scalable applications, such as gas diffusion electrode flooding, salt precipitation, reduction product purification, and single-path conversion of CO2.
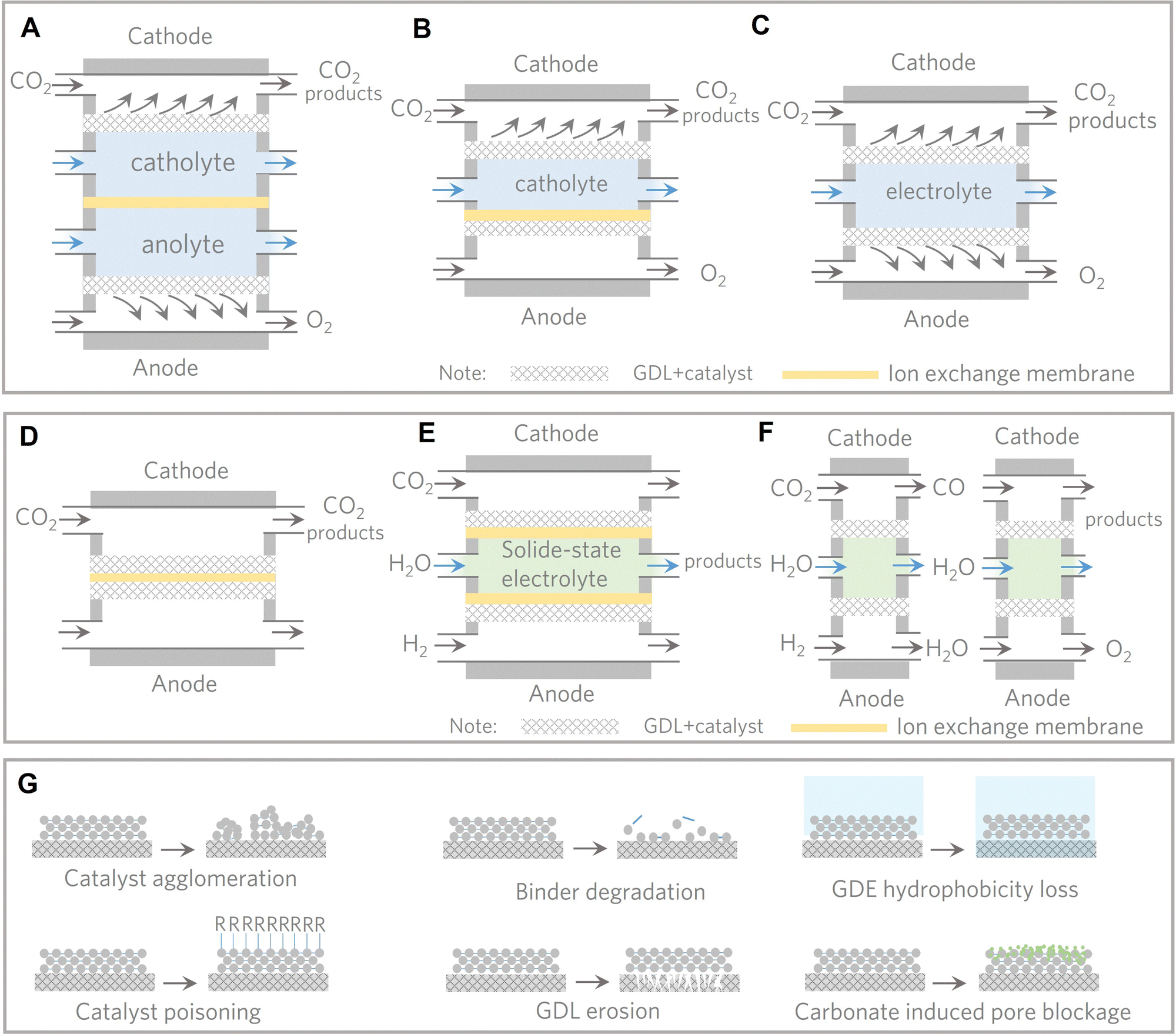 | ||
| Fig. 13 Schematic of the typical electrochemical flow cells for CO2 reduction. (A) Traditional flow cell; (B) flow cell with a thin liquid layer; (C) microfluidic cell; (D) gas phase membrane electrode assembly cell; (E) all-solid-state cell; (F) two-step tandem cell. (G) Schematic of degradation mechanisms. | ||
Compared with liquid flow-cell electrolyzers, the gas phase MEA electrolyzer laminates the gas diffusion layer, ion exchange membrane, and catalysts into one unit (Fig. 12D). In a continuous flow MEA cell, the anodic catalyst and the cathodic GDE are directly assembled on both sides of the ion-exchange membrane with lower ohmic resistance, further leading to excellent CO2RR efficiency.111,306,324 The direct contact between the catalyst layer and the ion exchange membrane can significantly reduce the resistance and improve the stability of the catalytic system. During the operation, there is no flowing electrolyte in the gas phase MEA, and the CO2 sampling methods are as follows: humidified carbon dioxide with anode electrolyte, humidified anode gas or anode open without any gas or electrolyte; dry carbon dioxide with humidified anode gas or pure water.325–327 This cathode electrolyte-free property efficiently alleviates the problem of carbonate deposition.328,329 Meanwhile, accurate flow cell design with the circular or serpentine channel has proven significant in developing MEA cells with efficient reactant delivery. Berlinguette and colleagues have assembled a commercial molecular CoPc electrocatalyst in MEA with the serpentine channel, where the cathode side is fed with wet CO2 gas flow while the anode is immersed in the KOH solution.330 A current density of 150 mA cm−2 can be well maintained during a long-term operation time of more than 100 h, which adequately illustrates the advantages of the MEA cell in terms of high current density and stability.
The all-solid-state electrolyzer has been developed to collect pure liquid C2+ products.331,332 The solid electrolyte cell employs a central solid electrolyte between anion-exchange and cation-exchange membranes (Fig. 13E). Membrane optimization is one of the crucial ways to improve mass transfer in the solid-state electrolyzer. Asadi and colleagues have employed a Dioxide Materials Sustainion™ anion exchange membrane in a solid electrolyte cell.333 The CO2RR performance is improved for more than 700 h at a current of 420 mA cm−2. Furthermore, the porous solid electrolyte of solid-state electrolyzers has also been investigated. Unlike conventional liquid electrolytes, the porous solid electrolyte layer facilitates ion conduction without introducing impurity ions. Wang and colleagues have designed a novel CO2 reduction cell with a solid electrolyte, which is capable of collecting pure C2+ liquid solution.334 Different forms of solid electrolytes, such as ceramics, polymer/ceramic hybrids, or solidified gel, can also be employed for C2+ synthesis in the future.
The single-pass conversion of CO2 to carbon-based products is a crucial indicator for practical applications. This index remains low (∼50%) for an alkaline electrolyte environment, where locally generated hydroxide anions accelerate carbonate formation at the electrode–electrolyte interface. Tandem processes (including tandem electrocatalysts and electrolyzers) are potential options for efficient CO2 utilization, which will significantly increase the yield and FE of C2+ products. The current works mainly focus on the cathode for different tandem electrode and electrolyzer designs.335,336 Tandem and resistive sandwich structures have been designed for the electrodes. Several attractive tandem devices have been developed for electrolyzers to increase CO2 solubility and achieve high CO2/CO conversion for further C2+ product synthesis (Fig. 13F). Despite the exciting progress of tandem electrolyzers, it is vital to note that studies on the anodic reactions still need to be completed, which are necessary for a complete system.
For flow cells, CO2 crossover has gradually become a significant challenge to improving CO2RR performance. In neutral or alkaline solutions, the carbonate crossover and precipitation induce low single-pass conversion efficiency and limited device lifetimes. Recent research has shifted towards acidic electrolytes to avoid carbonate, although circumventing the HER becomes a severe challenge. Sargent and colleagues have reported one representative example of interfacial engineering in proton exchange membrane MEA. The concentrated potassium ions at the catalyst–membrane interface primarily improve CO2RR performance and mitigate the CO2 crossover.211 Furthermore, Hu and colleagues have achieved efficient CO2RR in an acidic medium by suppressing the predominant HER using alkali cations and uncovered the essential role of the balance between carbonate formation rates and H+ diffusion.337,338 Hydrated alkali cations physiosorbed on the cathode modify the electric field distribution in the double layer. It impedes HER by suppressing hydronium ion migration, thus promoting CO2RR by stabilization of critical intermediates. Considering the restriction of CO2 crossover to the anode side, Kim and colleagues have reported a porous solid electrolyte reactor to recover the carbon losses efficiently. A permeable and ion-conducting sulfonated polymer electrolyte has been constructed as a buffer layer between the cathode and the anode. The crossed over carbonate can combine with protons to reform CO2 gas for reuse. Future studies can focus on optimizing the porous solid electrolyte reactor for practical CO2 recovery, including optimizing the thickness of the solid electrolyte layer and improving ion conduction by designing different solid ion conductors.
Furthermore, the investigation of different degradation mechanisms is vital to improving the stability of flow cells. A continuous supply of electrolytes and CO2 is required to ensure the cell operation in the kinetically limited state rather than the mass-transfer-limited state.339,340 The most common GDE degradation mechanism is shown in Fig. 13G, mainly including electrode degradation and electrolyte-related degradation. Physical changes in the catalyst structure, such as agglomeration or pulverization of catalyst particles, induce the coverage or loss of available active sites in the catalyst layer. This physical change is irreversible. However, chemical changes are reversible through specific mitigation strategies. The catalytic active site on the surface is covered by adsorbed metal impurities, thus inducing catalyst poisoning. Purifying electrolytes can eliminate the above problems.341 Einaga and colleagues have applied a positive potential to re-oxidize the material and desorb the adsorbed species.342,343 Meanwhile, the binders holding the catalyst layers may also suffer various forms of chemical degradation during prolonged exposure to electrolytes and CO2. The following are the main degradation mechanisms of GDL components: compression-force effects, dissolution, gas flow erosion, and carbon corrosion.344 The GDE flooding may be caused by macroscopic pressure imbalances in microfluidic devices, electrode hydrophobicity disruption, carbonate deposition, etc. After the GDE is flooded, the HER side reaction is more likely to occur with the decrease of the FE of C2+ product synthesis. Furthermore, some components of the electrolyzer may also be oxidized. The carbon material of the anode flow channel may be oxidized and decomposed. It is worth noting that no evident corrosion phenomenon is observed using titanium as the flow channel.345 To sum up, the design and optimization of electrolytic cell components to suppress the above degradation is an essential prerequisite for improving stability for industrial applications in the future.
Currently the most commonly used microflow cell is a three-chamber configuration, which can effectively prevent cathodic liquid products from being oxidized in the anode. Since the liquid electrolyte layer allows the use of a reference electrode, the microflow electrolytic cell can accurately control the cathode potential. However, the main factor limiting the performance of this configuration is the stability of the gas–liquid–solid three-phase interface on the GDE surface. Once the balanced three-phase interface is disrupted, excessive electrolytes penetrate the GDE channel and water flooding occurs. Mass transfer will be severely inhibited, and the reaction rate will significantly decrease. MEA cells, especially for the anion exchange membrane-based MEA cells, have been developed in which the direct contact between the GDE and the ion exchange membrane is realized, which may largely resolve the above-mentioned issues. However, carboxylic acid anionic products can also migrate to the anode through the anion exchange membrane, causing product loss and separation difficulties. In addition, the poor stability of the anion exchange membrane leads to a shorter operating life of this configuration, which limits its further application. The bipolar membrane-based MEA cells can significantly improve the conversion rate of CO2 and suppress the carbonate concentration on the electrode surface. The challenge lies in the fact that high concentrations of hydrogen ions on the electrode surface may promote the occurrence of the side HER reaction. Moreover, bipolar membrane configurations often require higher voltage and lower energy efficiency compared to other configurations. Research can be conducted in the following areas based on the advantages, disadvantages, and challenges of the above-mentioned electrolytic cells. The selection and amount of binder and embedded ionomers directly affect the stability of the catalyst layer and the operational life of the reactor. The most direct approach for GDE structural optimization is tuning feeding parameters, including the selection of the feeding method, gas humidification, degree of humidification, and flow rate optimization. In addition, the preparation of the carbon fiber substrate (hydrophobicity, thickness, etc.), adjusting the PTFE loading or adding a porous membrane layer can improve the hydrophobicity of the diffusion electrode. For practical application, exploring the failure and repair mechanism of electrolytic cells is conducive to realizing long-term stability tests under high currents, scaling up CO2 flow cells (including individual electrolyzers and stacks of multiple electrolyzer units). A reasonable flow channel design of the electrode plate should ensure uniform fluid distribution and maintain a low voltage drop.
Several studies have revealed that alkaline conditions are conducive to the formation of C2+ products. CO2 tends to form carbonate under strongly alkaline conditions. Sargent and colleagues have achieved the electrocatalytic production of ethylene from CO2via a composite electrode under 10 M KOH (strong base conditions), with a stable FE of 70% (at 750 mA cm−2).111 First a 25 nm-thick copper nanocatalyst has been sputtered on a porous polytetrafluoroethylene film with an aperture of approximately 220 nm. Subsequently, a carbon nanoparticle layer has been sprayed on the catalyst layer as a conductive layer. Lastly, a graphite nanoparticle layer has been added as the base of the collector and the entire structure. This electrode structure exhibits the following advantages: (1) the PTFE porous diffusion protective layer reduces the diffusion rate of CO2 to ensure the reduction of CO2 before the side reaction with the strong alkaline electrolyte occurs. In alkaline electrolytes, the competitive HER reaction rate also decreases, which further improves the selectivity of the electrocatalytic reduction to olefin. (2) As a large amount of CO2 has been reduced before contacting OH−, the surface of the copper catalyst can absorb a large amount of OH−, which reduces the activation energy barrier of CO–CO coupling and further enhances ethylene selectivity. (3) The PTFE porous diffusion protective layer significantly improves the stability of copper nanocatalysts and can operate stably for 150 h under test conditions.
Subsequently, Sargent and colleagues have also proposed a method for designing a hybrid catalyst. By decoupling the gas, ion, and electron transmission, CO2 can be effectively electrolyzed to generate C2 products in the gas phase electrolysis at current densities of >1 A cm−2.288 An ionomer layer with hydrophobic and hydrophilic functions facilitates the transport of gases and ions across metal surfaces. Thus, the reaction interface of these three components (gaseous reactants, ions, and electrons) has been located at the catalytic active site, thus increasing in length from the submicron range to several microns. Perfluorinated sulfonic acid (PFSA) ions with hydrophobic and hydrophilic functions have attracted significant attention. PFSA ions exhibit a high dependency on their structure/function. Based on the excellent advantages of the triphasic reaction, a novel catalyst has been designed to utilize gas–electrolyte separation beyond that of 2D catalysts. Typically, this catalyst with a maximized triphasic interface enables the system to operate efficiently at a higher current. The 3D catalyst has been prepared on a PTFE/Cu/ionomer (CIPH) gas diffusion layer support. With an increase in the loading and the corresponding thickness, the total eCO2RR current increases monotonously, exceeding 1 A cm−2 at a low load of 3.33 mg cm−2. When the load is higher, saturation is reached, and the current density significantly increases, after which the energy efficiency decreases. Under the top current operating condition, the maximum ethylene yield of the optimized catalyst is 65–75%, and the peak off-current density reaches up to 1.34 A cm−2 with a high cathode energy efficiency (46.3%). At 1.1 A cm−2 without iR compensation, the energy efficiency of C2+ products in the full electrolytic cell has been estimated to be 20%. However, current research still needs to be improved, such as the separation of C2+ products, the economic feasibility of the process, stability, and selectivity under industry-compatible current.
To summarize, the prominent challenges facing the commercialization of electrochemical CO2RR technology are energy efficiency, selectivity, low current density, and stability. For industrial applications, the current density should be higher than 300 mA cm−2 with FE above 80%, cell voltage below 1.8 V and stability over 80000 hours (Fig. 14). The biggest challenge currently faced is low energy efficiency and CO2 utilization efficiency. As a heterogeneous catalysis electrochemical process, it is anticipated that increasing the total catalyst surface area in the reactor or increasing the intrinsic reaction rate should be conducive to boosting the chemical productivity, which is undoubtedly necessary for scaling up and long-term reactor design. To break through the above challenges, the FE of the C2+ product synthesis system should be further enhanced, while the overvoltage of the electrolytic cell should be kept at a low level. Meanwhile, another influencing factor for CO2RR commercialization is the duration of stable operation. The minimum stable operation time required for profit scale expansion should exceed 10000 hours at least. The lifetime of cells mainly depends on the key components of the electrodes (or MEAs), including the form of the electrocatalyst, the regulation of functional groups in the polymer membrane, the selection of polymer binders, optimization of GDLs, and configuration type (tandem catalytic system or multilayer system).
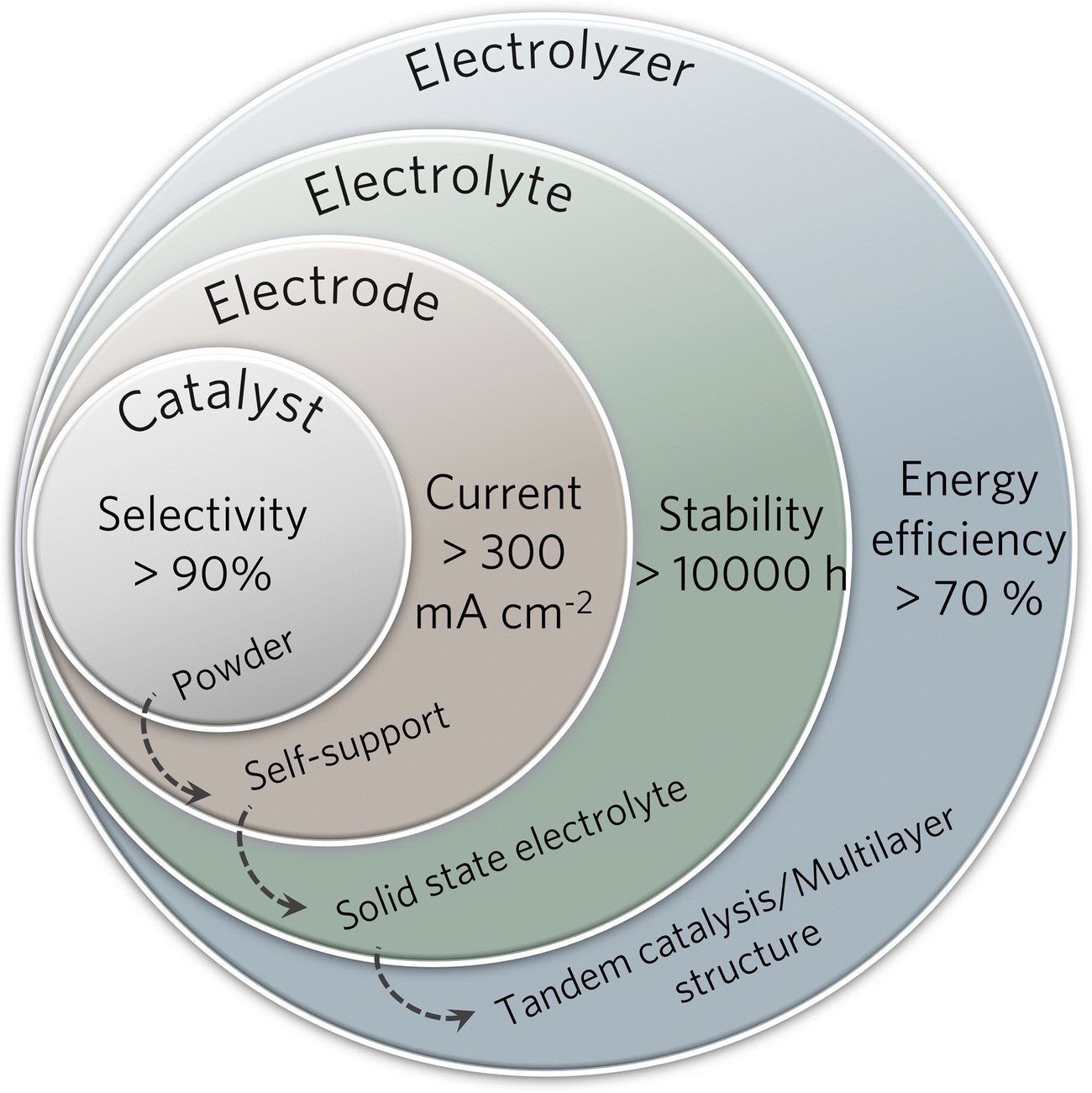 | ||
| Fig. 14 Scheme of the challenges involved in a CO2RR system toward high-performance commercialization. | ||
6. Summary and outlook
Significant progress has been made during the past few decades regarding the design of highly active or selective electrocatalysts and their eCO2RR mechanism. In this review, we have summarized the fundamental principles for catalyst design and provided a comprehensive overview of the catalytic mechanism, which covers almost all aspects of this emerging field with a perspective on C2+ synthesis. There is no doubt that eCO2RR is a promising approach for realizing the carbon cycle balance and addressing the current clean energy crisis simultaneously soon. On the other hand, we should be aware that there are still many challenges in the exploration and practical application of the electrochemical conversion of CO2 into C2+ products. We summarize the following perspectives and possible resolutions to address the current challenges in the electrochemical CO2 conversion into C2+ products.6.1. Novel catalyst discovery
Designing and regulating the intrinsic catalytic activity of advanced nanocatalysts are essential. The electronic structure of the catalyst surface can be adjusted by controlling the crystal facets, defects, and surface stress. Thus, the adsorption energy of *CO, *CHO, and other primary intermediates can be changed to break the scaling relationship and achieve high C2+ product selectivity. Meanwhile, the C2+ product synthesis is closely related to the coverage and concentration of *CO intermediates. Several catalysts with high CO selectivity have been reported (such as Au, Ag, and Zn). Therefore, CO2 → CO → C2+ tandem catalysis via multi-metal cooperation is another practical approach. In this approach, the CO2 feed gas is first reduced to CO on the surface of the second component metal, after which it overflows to the adjacent metal interface for C–C coupling and further reduction. In this strategy, the high CO coverage on the catalyst surface is conducive to inhibiting side reactions and promoting C2+ product preparation.Single-atom catalysis is a hot topic in the field of energy conversion. However, the large distance between the active centers of single atoms complicates the effective promotion of C–C coupling. C2+ product formation may be enhanced by increasing the surface density of homogeneously dispersed active sites. Moreover, catalysts for C–C coupling are not limited to metal-based catalysts. Several metal-free carbon-based materials have also been demonstrated to exhibit potential eCO2RR activity for C2+ products (such as graphene quantum dots and metal-doped nanodiamonds).
Furthermore, new opportunities have emerged in traditional molecular catalysts in recent years. Heterogeneous immobilization is an effective solution for addressing the problems of low solubility, low utilization, and difficult recovery of molecular catalysts. In this approach, the catalyst molecules are fixed to specific carriers as electrode materials through specific interactions (such as covalent bonds, electrostatic forces, and π–π interactions).
6.2. Profound computational approaches exploration
The complex C2+ reaction pathway in theoretical research illustrates the inherent complexity of pathways, the sensitivity of intermediates, and the controversy related to the pH effect. Presently, experimental evidence cannot obtain reaction information from catalyst–intermediate interactions at the atomic or even electronic level. Theoretical calculations can effectively remedy the above deficiencies. The theoretical calculations can be utilized to investigate the transformation of critical intermediates, the structural evolution of reaction centers, and the ion and electrolyte effects. However, at present the application of theoretical simulations in exploring C2+ synthesis mechanisms still faces several challenges. The electrolyte–electrode interface treatment is the critical issue. It is time-consuming to construct the explicit solvent model with numerous water molecules, whereas the implicit model may lose hydrogen bonds or other interactions of intermediates. Moreover, the constant charge model with a changeable Fermi level needs to be improved with the charge transfer between electrode and catalysts. It is necessary to take the solvent effect into consideration to better simulate the electrostatic interactions of the electric double layer in the working environment.Furthermore, based on theoretical calculations, the combination of machine learning can accelerate the prediction and screening of better eCO2RR electrocatalysts. By optimizing a machine learning model, a high-throughput calculation can be performed on the critical data of the electronic structure of catalytic materials. Future research on machine learning may focus on the following aspects to address some of the most important constraints of eCO2RR. (a) Current machine learning methods are rather limited in mechanism research and the prediction of C2+ products. The key characteristics should be investigated, and appropriate models for exploring the optimal reaction pathway for C2+ products should be explored. (b) The combination of machine learning calculations and advanced in situ/operando characterization exhibits significant potential for designing novel catalysts and elucidating the eCO2RR mechanism. (c) Combined with the solvent effect mechanism, screening suitable solvents through machine learning has broad prospects for developing high-performance eCO2RR systems.
6.3. Original in situ/operando technique development
eCO2RR possesses abundant products and complex reaction pathways, and the catalyst structure may change during the reaction process. The application of in situ/operando characterization techniques in eCO2RR is highly desirable to establish a more precise “structure–performance” relationship by exploring the interactions between substrate molecules, intermediates, and catalytic active sites. Despite impressive progress in in situ/operando characterization, there are still many problems to be solved in this field. Given the limited catalytic information provided by an individual in situ/operando technique, different in situ/operando tests need to be performed on the same in situ/operando reactor to simultaneously detect the catalyst structure change and the evolution of intermediates. In addition, the signal intensities of several in situ/operando techniques still require improvement owing to the low coverage of the key reaction intermediates. Such experimental results obtained at the same time and space scales by tightly coupling multiple in situ/operando representations are more convincing for exploring eCO2RR mechanisms. Another major challenge of in situ/operando experiments is the optimal design of in situ/operando electrolytic cells. To conform to the harsh conditions of in situ/operando testing, the electrolytic cell undergoes targeted structural changes, which results in a mismatch between the in situ/operando testing conditions and the actual working conditions. Therefore, it is urgent to develop optimized electrolytic cells to truly bridge the gap between in situ/operando testing and actual working conditions and to obtain effective information that fully reflects the real catalytic eCO2RR. Moreover, the close integration of machine learning, in situ/operando experiments, and theoretical calculations is a practical approach to exploring reaction thermodynamics/kinetics and pathways.6.4. Electrolyte and electrode optimization
Although materials are the key to eCO2RR, the catalytic performance is also affected by other factors, such as the chemical composition of electrolytes and structural design of the electrodes. The optimization of selectivity and corresponding mechanism analysis can be achieved by tuning the cation size and concentration. Meanwhile, ionic liquids have also been proven to be suitable media for CO2 dissolution, activation, and stabilization of free radicals and electrochemically active ionic substances from aqueous solutions. Unlike conventional electrolytes, most anisotropic ionic liquids display properties of an extended cooperative network of supramolecular species. The future challenges mainly include (1) the influence mechanism of cations/anions on surface groups and material stability; (2) the relationship between the structure and organization of ionic liquids and the eCO2RR.Focusing on the electrolyte system and addressing the above challenges, the following research will be focused on in the future. (1) The influence mechanism of the ionic liquid electrolyte structure on eCO2RR needs to be clarified. The reduction law between the ionic liquid structure and eCO2RR activity can be thoroughly understood by the cooperation of theoretical simulations and in situ/operando characterization techniques. Furthermore, it is necessary to clarify the synergistic mechanism between the proton/electron transfer process in an ionic liquid system and the interface structure of catalytic materials. (2) An ionic liquid electrolyte system for synthesizing C2+ products needs to be developed to activate CO2 molecules and facilitate the functionalization of C–C coupling in the intermediate states of the reaction. (3) It is urgent to study the flow mass transfer and stability of CO2 reduction in ionic liquid systems. The internal structure, electrode structure, and electrolyte flow type of eCO2RR devices with different structures must be optimized to promote the C2+ activity and selectivity. (4) The CO2 crossover issue should be resolved. Future studies can focus on designing porous solid electrolyte reactors for practical CO2 recovery, including optimizing the thickness of the solid electrolyte layer for minimized ohmic drop and improving ion conduction between the cathode and the anode by designing different solid ion conductors.
As mentioned above, the selectivity and the yield of C2+ products can be further improved by the rational design of catalytic electrodes and devices. The utilization of the GDE has helped to bridge the gap between laboratory experimental findings and industrial needs. However, achieving long-term stability with such high selectivity and activity remains a significant challenge. At the macro level, eCO2RR activity can be enhanced by simple process-strengthening techniques, such as increasing the partial pressure of CO2 and forced electrolyte flow. At the micro level, it is urgent to develop the technology to realize process enhancement with the help of the GDE microporous structure and hydrophilic and hydrophobic adjustment. The main research direction of the cathode microstructure is to expand and form a stable three-phase interface. Furthermore, the selectivity for specific C2+ products should also be optimized to reduce the cost of product separation and purification. Thus, further research is needed to investigate and explore effective methods for improving reactant concentrations around active sites and the turnover and selectivity of adsorbed intermediates at high current densities. It is urgent to design high-throughput reactors with economic reactant capture technologies to increase industrial production potential.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the King Abdullah University of Science and Technology (KAUST).References
- Z. H. Xue, D. Y. Luan, H. B. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- X. Wu, H. B. Zhang, J. Zhang and X. W. Lou, Adv. Mater., 2021, 33, 2008376 CrossRef CAS PubMed.
- C. Feng, Z. P. Wu, K. W. Huang, J. H. Ye and H. B. Zhang, Adv. Mater., 2022, 34, 2200180 CrossRef CAS PubMed.
- M. G. Lee, X.-Y. Li, A. Ozden, J. Wicks, P. Ou, Y. Li, R. Dorakhan, J. Lee, H. K. Park, J. W. Yang, B. Chen, J. Abed, R. dos Reis, G. Lee, J. E. Huang, T. Peng, Y.-H. Chin, D. Sinton and E. H. Sargent, Natl. Catal., 2023, 6, 310–318 CrossRef CAS.
- W. H. Huang, C. Y. Su, C. Zhu, T. T. Bo, S. W. Zuo, W. Zhou, Y. F. Ren, Y. N. Zhang, J. Zhang, M. Rueping and H. B. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202304634 CrossRef.
- J. Du, B. Cheng, H. Yuan, Y. Tao, Y. Chen, M. Ming, Z. Han and R. Eisenberg, Angew. Chem., Int. Ed., 2023, 62, e202211804 CrossRef CAS PubMed.
- W. Ahmad, P. Koley, S. Dwivedi, R. Lakshman, Y. K. Shin, A. C. T. van Duin, A. Shrotri and A. Tanksale, Nat. Commun., 2023, 14, 2821 CrossRef CAS PubMed.
- H. W. Lin, S. Q. Luo, H. B. Zhang and J. H. Ye, Joule, 2022, 6, 294–314 CrossRef CAS.
- H. B. Zhang, W. R. Cheng, D. Y. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 13177–13196 CrossRef CAS PubMed.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Natl. Catal., 2022, 5, 564–570 CrossRef CAS.
- S. D. Rihm, M. K. Kovalev, A. A. Lapkin, J. W. Ager and M. Kraft, Energy Environ. Sci., 2023, 16, 1697–1710 RSC.
- X. F. Zhang, W. H. Huang, L. Yu, M. García-Melchor, D. S. Wang, L. J. Zhi and H. B. Zhang, Carbon Energy, 2023, e362 CrossRef.
- Y. J. Shi, Y. J. Wang, J. Y. Yu, Y. K. Chen, C. Q. Fang, D. Jiang, Q. H. Zhang, L. Gu, X. W. Yu, X. Li, H. Liu and W. J. Zhou, Adv. Energy Mater., 2023, 13, 2203506 CrossRef CAS.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- Y. Zhu, P. Li, X. Yang, M. Wang, Y. Zhang, P. Gao, Q. Huang, Y. Wei, X. Yang, D. Wang, Y. Shen and M. Wang, Adv. Energy Mater., 2023, 13, 2204243 Search PubMed.
- J. Li, K. Xu, F. Liu, Y. Li, Y. Hu, X. Chen, H. Wang, W. Xu, Y. Ni, G. Ding, T. Zhao, M. Yu, W. Xie and F. Cheng, Adv. Mater., 2023, e2301127 CrossRef PubMed.
- J. Qin, T. Wang, M. Zhai, C. Wu, Y. A. Liu, B. Yang, H. Yang, K. Wen and W. Hu, Adv. Funct. Mater., 2023, 2300697 CrossRef.
- L. Fan, C.-Y. Liu, P. Zhu, C. Xia, X. Zhang, Z.-Y. Wu, Y. Lu, T. P. Senftle and H. Wang, Joule, 2022, 6, 205–220 CrossRef CAS.
- E. Fujita, D. C. Grills, G. F. Manbeck and D. E. Polyansky, Acc. Chem. Res., 2022, 55, 616–628 CrossRef CAS PubMed.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CAS.
- Z. Li, Y. Gao, X. Meng, B. Sun, K. Song, Z. Wang, Y. Liu, Z. Zheng, P. Wang, Y. Dai, H. Cheng and B. Huang, Cell Rep. Phys. Sci., 2022, 3, 100972 CrossRef CAS.
- M. Esmaeilirad, A. Kondori, N. Shan, M. T. Saray, S. Sarkar, A. M. Harzandi, C. M. Megaridis, R. Shahbazian-Yassar, L. A. Curtiss, C. U. Segre and M. Asadi, Appl. Catal., B, 2022, 317, 121681 CrossRef CAS.
- F. Dattila, R. R. Seemakurthi, Y. Zhou and N. Lopez, Chem. Rev., 2022, 122, 11085–11130 CrossRef CAS PubMed.
- C. Han, V. Kundi, Z. Ma, C. Y. Toe, P. Kumar, C. Tsounis, J. Jiang, S. Xi, Z. Han, X. Lu, R. Amal and J. Pan, Adv. Funct. Mater., 2023, 33, 2210938 CrossRef CAS.
- S. H. Li, S. Hu, H. Liu, J. Liu, X. Kang, S. Ge, Z. Zhang, Q. Yu and B. Liu, ACS Nano, 2023, 17, 9338–9346 CrossRef CAS PubMed.
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y. H. Chou, X. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. Han and R. Amal, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- D. Giusi, M. Miceli, C. Genovese, G. Centi, S. Perathoner and C. Ampelli, Appl. Catal., B, 2022, 318, 121845 CrossRef CAS.
- Y. Zou and S. Wang, Adv. Sci., 2021, 8, 2003579 CrossRef CAS PubMed.
- Q. S. Wang, Y. C. Yuan, C. F. Li, Z. R. Zhang, C. Xia, W. G. Pan and R. T. Guo, Small, 2023, e2301892 CrossRef PubMed.
- K. Xiang, F. Shen, Y. Fu, L. Wu, Z. Wang, H. Yi, X. Liu, P. Wang, M. Liu, Z. Lin and H. Liu, Environ. Sci.: Nano, 2022, 9, 911–953 RSC.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- W. X. Nie, G. P. Heim, N. B. Watkins, T. Agapie and J. C. Peters, Angew. Chem., Int. Ed., 2023, 135, e202216102 CrossRef.
- Z. Lin, Z. Jiang, Y. Yuan, H. Li, H. Wang, Y. Tang, C. Liu and Y. Liang, Chin. J. Catal., 2022, 43, 104–109 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu, D. D. Zhou and J. P. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng, L. Yin, Z. Weng and Q. H. Yang, Nat. Commun., 2022, 13, 3158 CrossRef CAS PubMed.
- X. Lv, Q. Liu, J. Wang, X. Wu, X. Li, Y. Yang, J. Yan, A. Wu and H. B. Wu, Appl. Catal., B, 2023, 324, 122272 CrossRef CAS.
- Z. Wei, J. Ding, X. Duan, G.-L. Chen, F.-Y. Wu, L. Zhang, X. Yang, Q. Zhang, Q. He, Z. Chen, J. Huang, S.-F. Hung, X. Yang and Y. Zhai, ACS Catal., 2023, 13, 4711–4718 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- Y. Liang, J. Zhao, Y. Yang, S. F. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang, D. Appadoo, L. Zhang, Z. Geng, F. Li and J. Zeng, Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS PubMed.
- T. Zhang, B. Yuan, W. Wang, J. He and X. Xiang, Angew. Chem., Int. Ed., 2023, 135, e202302096 CrossRef.
- C. Kim, L.-C. Weng and A. T. Bell, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- J. Yin, Z. Yin, J. Jin, M. Sun, B. Huang, H. Lin, Z. Ma, M. Muzzio, M. Shen, C. Yu, H. Zhang, Y. Peng, P. Xi, C. H. Yan and S. Sun, J. Am. Chem. Soc., 2021, 143, 15335–15343 CrossRef CAS PubMed.
- K. U. D. Calvinho, A. W. Alherz, K. M. K. Yap, A. B. Laursen, S. Hwang, Z. J. L. Bare, Z. Clifford, C. B. Musgrave and G. C. Dismukes, J. Am. Chem. Soc., 2021, 143, 21275–21285 CrossRef CAS PubMed.
- P. Chen, Y. Wu, T. E. Rufford, L. Wang, G. Wang and Z. Wang, Mater. Today Chem., 2023, 27, 101328 CrossRef CAS.
- C. Li, Y. Ji, Y. Wang, C. Liu, Z. Chen, J. Tang, Y. Hong, X. Li, T. Zheng, Q. Jiang and C. Xia, Nano-Micro Lett., 2023, 15, 113 CrossRef CAS PubMed.
- P. Wei, D. Gao, T. Liu, H. Li, J. Sang, C. Wang, R. Cai, G. Wang and X. Bao, Nat. Nanotechnol., 2023, 18, 299–306 CrossRef CAS PubMed.
- F. Chang, Y. Liu, J. Wei, L. Yang and Z. Bai, Inorg. Chem. Front., 2023, 10, 240–249 RSC.
- M. Wang, V. Nikolaou, A. Loiudice, I. D. Sharp, A. Llobet and R. Buonsanti, Chem. Sci., 2022, 13, 12673–12680 RSC.
- Y. R. Lin, D. U. Lee, S. Tan, D. M. Koshy, T. Y. Lin, L. Wang, D. Corral, J. E. Avilés Acosta, J. A. Zamora Zeledon, V. A. Beck, S. E. Baker, E. B. Duoss, C. Hahn and T. F. Jaramillo, Adv. Funct. Mater., 2022, 32, 2113252 CrossRef CAS.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U. P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schofberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- X. F. Qiu, J. R. Huang, C. Yu, Z. H. Zhao, H. L. Zhu, Z. Ke, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CAS.
- J. B. Pang, B. Chang, H. Liu and W. J. Zhou, ACS Energy Lett., 2022, 7, 78–96 CrossRef CAS.
- C. E. Creissen and M. Fontecave, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, J. Zhu, C. Ye, Y. Mao, B. Wang, G. Zhou, L. Mai, Z. Wang, X. Liu and D. Wang, Nano Lett., 2023, 23, 2312–2320 CrossRef CAS PubMed.
- K. Rossi and R. Buonsanti, Acc. Chem. Res., 2022, 55, 629–637 CrossRef CAS PubMed.
- H. Zhang, W. Zhou, X. F. Lu, T. Chen and X. W. Lou, Adv. Energy Mater., 2020, 10, 2000882 CrossRef CAS.
- X. Wu, H. Zhang, S. Zuo, J. Dong, Y. Li, J. Zhang and Y. Han, Nano-Micro Lett., 2021, 13, 136 CrossRef CAS PubMed.
- K. Lakshmanan, W. H. Huang, S. A. Chala, B. W. Taklu, E. A. Moges, J. F. Lee, P. Y. Huang, Y. C. Lee, M. C. Tsai, W. N. Su and B. J. Hwang, Adv. Funct. Mater., 2022, 32, 2109310 CrossRef CAS.
- X. Li, J. Wang, X. Lv, Y. Yang, Y. Xu, Q. Liu and H. B. Wu, Nano-Micro Lett., 2022, 14, 134 CrossRef CAS PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Gratzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS PubMed.
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- X. Wu, H. Zhang, J. Zhang and X. W. D. Lou, Adv. Mater., 2021, 33, e2008376 CrossRef PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- S. Kim, D. Shin, J. Park, J. Y. Jung and H. Song, Adv. Sci., 2023, 10, e2207187 CrossRef PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Natl. Catal., 2020, 3, 478–487 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 125, 2519–2522 CrossRef.
- Y. T. Guntern, V. Okatenko, J. Pankhurst, S. B. Varandili, P. Iyengar, C. Koolen, D. Stoian, J. Vavra and R. Buonsanti, ACS Catal., 2021, 11, 1248–1295 CrossRef CAS.
- H. Lin, K. Wei, Z. Yin and S. Sun, iScience, 2021, 24, 102172 CrossRef CAS PubMed.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 134, e202207252 CrossRef.
- Z. Z. Wu, X. L. Zhang, Z. Z. Niu, F. Y. Gao, P. P. Yang, L. P. Chi, L. Shi, W. S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M. R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed.
- C. Obasanjo, A. Shayesteh Zeraati, H. S. Shiran, T. N. Nguyen, M. G. Kibria, S. M. Sadaf and C. T. Dinh, J. Mater. Chem. A, 2022, 10, 20059–20070 RSC.
- G. O. Larrazabal, V. Okatenko, I. Chorkendorff, R. Buonsanti and B. Seger, ACS Appl. Mater. Interfaces, 2022, 14, 7779–7787 CrossRef CAS PubMed.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- D. Tan, B. Wulan, X. Cao and J. Zhang, Nano Energy, 2021, 89, 106460 CrossRef CAS.
- Y. Pan, H. Li, J. Xiong, Y. Yu, H. Du, S. Li, Z. Wu, S. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 306, 121111 CrossRef CAS.
- C. E. Creissen and M. Fontecave, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei, Z. Jiang, J. Zhong, L. Li and G. Zheng, Small Methods, 2019, 3, 1800449 CrossRef.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- Q. Li, Y.-C. Wang, J. Zeng, X. Zhao, C. Chen, Q.-M. Wu, L.-M. Chen, Z.-Y. Chen and Y.-P. Lei, Rare Met., 2021, 40, 3442–3453 CrossRef CAS.
- F.-Y. Gao, Z.-Z. Wu and M.-R. Gao, Energy Fuels, 2021, 35, 12869–12883 CrossRef CAS.
- Y. Sun, G. Li, W. Sun and X. Zhou, J. CO2 Util., 2023, 67, 102344 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- J. Wang, C. Cheng, B. Huang, J. Cao, L. Li, Q. Shao, L. Zhang and X. Huang, Nano Lett., 2021, 21, 980–987 CrossRef CAS PubMed.
- Z. Ma, C. Tsounis, C. Y. Toe, P. V. Kumar, B. Subhash, S. Xi, H. Y. Yang, S. Zhou, Z. Lin, K.-H. Wu, R. J. Wong, L. Thomsen, N. M. Bedford, X. Lu, Y. H. Ng, Z. Han and R. Amal, ACS Catal., 2022, 12, 4792–4805 CrossRef CAS.
- D. Tan, B. Wulan, J. Ma, X. Cao and J. Zhang, Chem. Catal., 2023, 3, 100512 CrossRef CAS.
- H. Shen, Y. Zhao, L. Zhang, Y. He, S. Yang, T. Wang, Y. Cao, Y. Guo, Q. Zhang and H. Zhang, Adv. Energy Mater., 2022, 13, 2202818 CrossRef.
- Q. Ren, N. Zhang, Z. Dong, L. Zhang, X. Chen and L. Luo, Nano Energy, 2023, 106, 108080 CrossRef CAS.
- Y. Jiang, X. Wang, D. Duan, C. He, J. Ma, W. Zhang, H. Liu, R. Long, Z. Li, T. Kong, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Sci., 2022, 9, e2105292 CrossRef PubMed.
- Y. Du and W. An, J. Phys. Chem. C, 2021, 125, 9138–9149 CrossRef CAS.
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Angew. Chem., Int. Ed., 2021, 133, 17394–17407 CrossRef.
- D. Song, Y. B. Lian, M. Wang, Y. H. Su, F. L. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- S. Yu, D. Kim, Z. Qi, S. Louisia, Y. Li, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2021, 143, 19919–19927 CrossRef CAS PubMed.
- A. Thevenon, A. Rosas-Hernandez, J. C. Peters and T. Agapie, Angew. Chem., Int. Ed., 2019, 58, 16952–16958 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- J. Yuan, M.-P. Yang, W.-Y. Zhi, H. Wang, H. Wang and J.-X. Lu, J. CO2 Util., 2019, 33, 452–460 CrossRef CAS.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. Garcia de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- L. Li, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- H. Li, Y. Pan, Z. Wang, Y. Yu, J. Xiong, H. Du, J. Lai, L. Wang and S. Feng, Nano Res., 2021, 15, 3056–3064 CrossRef.
- H. Dong, M. Lu, Y. Wang, H.-L. Tang, D. Wu, X. Sun and F.-M. Zhang, Appl. Catal., B, 2022, 303, 120897 CrossRef CAS.
- K. A. Adegoke and N. W. Maxakato, Mater. Today Chem., 2022, 24, 100838 CrossRef CAS.
- Q. Lu, C. Chen, Q. Di, W. Liu, X. Sun, Y. Tuo, Y. Zhou, Y. Pan, X. Feng, L. Li, D. Chen and J. Zhang, ACS Catal., 2022, 12, 1364–1374 CrossRef CAS.
- J. Zhou, B. An, Z. Zhu, L. Wang and J. Zhang, Inorg. Chem., 2022, 61, 6073–6082 CrossRef CAS PubMed.
- W. Pei, S. Zhou, J. Zhao, X. Xu, Y. Du and S. X. Dou, Nano Energy, 2020, 76, 105049 CrossRef CAS.
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Adv. Energy Mater., 2021, 12, 2003990 CrossRef.
- H. Xiao, W. A. Goddard, 3rd, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- M. Suominen and T. Kallio, ChemElectroChem, 2021, 8, 2397–2406 CrossRef CAS.
- X. Li, Q. Liu, J. Wang, D. Meng, Y. Shu, X. Lv, B. Zhao, H. Yang, T. Cheng, Q. Gao, L. Li and H. B. Wu, Chem, 2022, 8, 2148–2162 CAS.
- F. Yu, Z. Zhou, Y. You, J. Zhan, T. Yao and L. H. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 24346–24353 CrossRef CAS PubMed.
- N. Li, X. Chen, W. J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- M. Abdinejad, S. Subramanian, M. K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, D. Ripepi, D. Pinto, M. Li, K. Tang, J. Middelkoop, A. Urakawa, O. Voznyy, H. B. Kraatz and T. Burdyny, Adv. Energy Mater., 2023, 13, 2300402 CrossRef CAS.
- W. Xiong, D. Si, J. Yi, Y. Huang, H. Li and R. Cao, Appl. Catal., B, 2022, 314, 121498 CrossRef CAS.
- D. H. Nam, O. Shekhah, G. Lee, A. Mallick, H. Jiang, F. Li, B. Chen, J. Wicks, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 21513–21521 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Zhang, X. Sun, Q. Zhu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202202607 CAS.
- X. Xie, X. Zhang, M. Xie, L. Xiong, H. Sun, Y. Lu, Q. Mu, M. H. Rummeli, J. Xu, S. Li, J. Zhong, Z. Deng, B. Ma, T. Cheng, W. A. Goddard, 3rd and Y. Peng, Nat. Commun., 2022, 13, 63 CrossRef CAS PubMed.
- C. F. Wen, M. Zhou, P. F. Liu, Y. Liu, X. Wu, F. Mao, S. Dai, B. Xu, X. L. Wang, Z. Jiang, P. Hu, S. Yang, H. F. Wang and H. G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202111700 CAS.
- L. Cheng, P. Zhang, Q. Wen, J. Fan and Q. Xiang, Chin. J. Catal., 2022, 43, 451–460 CrossRef CAS.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Mater., 2022, 1, 100012 CrossRef.
- L. Wan, X. Zhang, J. Cheng, R. Chen, L. Wu, J. Shi and J. Luo, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS.
- W. Lai, Z. Ma, J. Zhang, Y. Yuan, Y. Qiao and H. Huang, Adv. Funct. Mater., 2022, 32, 2111193 CrossRef CAS.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Natl. Catal., 2022, 5, 564–570 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Natl. Catal., 2018, 1, 764–771 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- M. Qu, Z. Chen, Z. Sun, D. Zhou, W. Xu, H. Tang, H. Gu, T. Liang, P. Hu, G. Li, Y. Wang, Z. Chen, T. Wang and B. Jia, Nano Res., 2022, 16, 2170–2176 CrossRef.
- J. Meng, Z. Miao, J. Zhang, Z. Wang, R. Zhang, L. Xu, L. Diao, J. Zhou and S. Zhuo, J. Alloys Compd., 2023, 939, 168798 CrossRef CAS.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Natl. Catal., 2022, 5, 545–554 CrossRef CAS.
- J. Du, B. Cheng, L. Jiang and Z. Han, Chem. Commun., 2023, 59, 4778–4781 RSC.
- Y. Fang, X. Liu, Z. Liu, L. Han, J. Ai, G. Zhao, O. Terasaki, C. Cui, J. Yang, C. Liu, Z. Zhou, L. Chen and S. Che, Chem, 2023, 9, 460–471 CAS.
- N. C. Ramos, J. W. Medlin and A. Holewinski, ACS Appl. Mater. Interfaces, 2023, 15, 14470 CAS.
- F. Dattila, R. R. Seemakurthi, Y. Zhou and N. Lopez, Chem. Rev., 2022, 122, 11085–11130 CrossRef CAS PubMed.
- N. Karmodak, S. Vijay, G. Kastlunger and K. Chan, ACS Catal., 2022, 12, 4818–4824 CrossRef CAS PubMed.
- J. Santatiwongchai, K. Faungnawakij and P. Hirunsit, ACS Catal., 2021, 11, 9688–9701 CrossRef CAS.
- Y. A. Alsunni, A. W. Alherz and C. B. Musgrave, J. Phys. Chem. C, 2021, 125, 23773–23783 CrossRef CAS.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- Y. Guan, W. Suo, Z. Zhang, Y. Wang, S. Sun and G. Liu, Mol. Catal., 2021, 511, 111725 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- F. Grun, M. Jardat, P. Turq and C. Amatore, J. Chem. Phys., 2004, 120, 9648–9655 CrossRef CAS PubMed.
- M. F. Kling and M. J. Vrakking, Annu. Rev. Phys. Chem., 2008, 59, 463–492 CrossRef CAS PubMed.
- S. Ali, G. Yasin, R. Iqbal, X. Huang, J. Su, S. Ibraheem, Z. Zhang, X. Wu, F. Wahid, P. M. Ismail, L. Qiao and H. Xu, Mol. Catal., 2022, 524, 112285 CrossRef CAS.
- M. E. Björketun, Z. Zeng, R. Ahmed, V. Tripkovic, K. S. Thygesen and J. Rossmeisl, Chem. Phys. Lett., 2013, 555, 145–148 CrossRef.
- L. D. Chen, M. Bajdich, J. M. P. Martirez, C. M. Krauter, J. A. Gauthier, E. A. Carter, A. C. Luntz, K. Chan and J. K. Norskov, Nat. Commun., 2018, 9, 3202 CrossRef PubMed.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Norskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, 3rd, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- C. Shi, K. Chan, J. S. Yoo and J. K. Nørskov, Org. Process Res. Dev., 2016, 20, 1424–1430 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard, 3rd, J. Phys. Chem. Lett., 2015, 6, 4767–4773 CrossRef CAS PubMed.
- C.-C. Chang and M.-S. Ku, J. Phys. Chem. C, 2021, 125, 10919–10925 CrossRef CAS.
- Y. Feng, W. An, Z. Wang, Y. Wang, Y. Men and Y. Du, ACS Sustainable Chem. Eng., 2019, 8, 210–222 CrossRef.
- A. Kakekhani, L. T. Roling, A. Kulkarni, A. A. Latimer, H. Abroshan, J. Schumann, H. AlJama, S. Siahrostami, S. Ismail-Beigi, F. Abild-Pedersen and J. K. Norskov, Inorg. Chem., 2018, 57, 7222–7238 CrossRef CAS PubMed.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- Y. Tian, T. Zhao, C. Zhao and Y. Likai, Appl. Surf. Sci., 2022, 597, 153724 CrossRef CAS.
- M. Wan, Z. Gu and F. Che, ChemCatChem, 2021, 14, e202101224 Search PubMed.
- H. Dong, Y. Li and D.-E. Jiang, J. Phys. Chem. C, 2018, 122, 11392–11398 CrossRef CAS.
- C. Zhu, Z. Zhang, L. Zhong, C.-S. Hsu, X. Xu, Y. Li, S. Zhao, S. Chen, J. Yu, S. Chen, M. Wu, P. Gao, S. Li, H. M. Chen, K. Liu and L. Zhang, Chem, 2021, 7, 406–420 CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Natl. Catal., 2018, 1, 111–119 CrossRef CAS.
- H. Xiao, T. Cheng, W. A. Goddard, 3rd and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 125, 7423–7426 CrossRef.
- D. Raciti, M. Mao, J. H. Park and C. Wang, J. Electrochem. Soc., 2018, 165, 799–804 CrossRef.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 133, 20795–20816 CrossRef.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- H. Hashiba, L.-C. Weng, Y. Chen, H. K. Sato, S. Yotsuhashi, C. Xiang and A. Z. Weber, J. Phys. Chem. C, 2018, 122, 3719–3726 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- K. Li, W. Wang, H. Zheng, X. Wang, Z. Xie, L. Ding, S. Yu, Y. Yao and F. Y. Zhang, Mater. Today Phys., 2021, 19, 1000427 Search PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang and B. Han, Nat. Commun., 2022, 13, 1965 CrossRef CAS PubMed.
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y. M. Yan, Angew. Chem., Int. Ed., 2022, 134, e202205832 Search PubMed.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Norskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio, J. H. Montoya, D. B. Trimarco, C. Roy, S. Meier, J. Wendland, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2016, 128, 1472–1476 CrossRef.
- N. Abidi and S. N. Steinmann, Curr. Opin. Electrochem., 2022, 33, 100940 CrossRef CAS.
- X. Zhang and Z. Zhou, J. Phys. Chem. C, 2022, 126, 3820–3829 CrossRef CAS.
- C. Xu, X. Zhi, A. Vasileff, D. Wang, B. Jin, Y. Jiao, Y. Zheng and S.-Z. Qiao, Small Structures, 2020, 2, 2000058 CrossRef.
- W. Lai, Z. Ma, J. Zhang, Y. Yuan, Y. Qiao and H. Huang, Adv. Funct. Mater., 2022, 32, 2111193 CrossRef CAS.
- W. Deng, P. Zhang, B. Seger and J. Gong, Nat. Commun., 2022, 13, 803 CrossRef CAS PubMed.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager, 3rd and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- X. Zhou, H. Liu, B. Y. Xia, K. Ostrikov, Y. Zheng and S. Z. Qiao, Smart Mater., 2022, 3, 111–129 CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B, 2012, 85, 235149 CrossRef.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2021, 12, 331–362 CrossRef.
- E. Perez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef CAS PubMed.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- K. Ogura, J. CO2 Util., 2013, 1, 43–49 CrossRef CAS.
- S. K. Shaw, A. Berna, J. M. Feliu, R. J. Nichols, T. Jacob and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2011, 13, 5242–5251 RSC.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- D. Gao, F. Scholten and B. Roldan Cuenya, ACS Catal., 2017, 7, 5112–5120 CrossRef CAS.
- Y. Huang, C. W. Ong and B. S. Yeo, ChemSusChem, 2018, 11, 3299–3306 CrossRef CAS PubMed.
- M. K. Kim, H. Lee, J. H. Won, W. Sim, S. J. Kang, H. Choi, M. Sharma, H. S. Oh, S. Ringe, Y. Kwon and H. M. Jeong, Adv. Funct. Mater., 2021, 32, 2107349 CrossRef.
- S. Banerjee, C. S. Gerke and V. S. Thoi, Acc. Chem. Res., 2022, 55, 504–515 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. Garcia de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- M. Löffler, P. Khanipour, N. Kulyk, K. J. J. Mayrhofer and I. Katsounaros, ACS Catal., 2020, 10, 6735–6740 CrossRef.
- S. Jeong, M. H. Choi, G. S. Jagdale, Y. Zhong, N. P. Siepser, Y. Wang, X. Zhan, L. A. Baker and X. Ye, J. Am. Chem. Soc., 2022, 144, 12673–12680 CrossRef CAS PubMed.
- Y. Shi, Y. Wang, C. L. Dong, T. T. T. Nga, D. Wei, J. Wang, X. Zhao, M. Wang, K. Zhang, M. Li, F. Dong and S. Shen, Adv. Energy Mater., 2023, 13, 2203896 CrossRef CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- H. Cao, Z. Zhang, J.-W. Chen and Y.-G. Wang, ACS Catal., 2022, 12, 6606–6617 CrossRef CAS.
- X. Zhang and Z. Zhou, J. Phys. Chem. C, 2022, 126, 3820–3829 CrossRef CAS.
- X. Qin, T. Vegge and H. A. Hansen, J. Am. Chem. Soc., 2023, 145, 1897–1905 CrossRef CAS PubMed.
- H. Liu, J. Liu and B. Yang, ACS Catal., 2021, 11, 12336–12343 CrossRef CAS.
- K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS PubMed.
- A. Chen, X. Zhang and Z. Zhou, InfoMat, 2020, 2, 553–576 CrossRef CAS.
- J. L. Hitt, Y. C. Li, S. Tao, Z. Yan, Y. Gao, S. J. L. Billinge and T. E. Mallouk, Nat. Commun., 2021, 12, 1114 CrossRef CAS PubMed.
- Z. Yang, W. Gao and Q. Jiang, J. Mater. Chem. A, 2020, 8, 17507–17515 RSC.
- S. Gusarov, S. R. Stoyanov and S. Siahrostami, J. Phys. Chem. C, 2020, 124, 10079–10084 CrossRef CAS.
- R. Qi, B. Zhu, Z. Han and Y. Gao, ACS Catal., 2022, 12, 8269–8278 CrossRef CAS.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Norskov, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- R. Juneja and A. K. Singh, J. Mater. Chem. A, 2020, 8, 8716–8721 RSC.
- A. Chen, X. Zhang, L. Chen, S. Yao and Z. Zhou, J. Phys. Chem. C, 2020, 124, 22471–22478 CrossRef CAS.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS.
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC.
- L. Liu, W. Li, X. He, J. Yang and N. Liu, Small, 2022, 18, e2104205 CrossRef PubMed.
- S. Zuo, Z. P. Wu, H. Zhang and X. W. Lou, Adv. Energy Mater., 2022, 12, 2103383 CrossRef CAS.
- H. An, L. Wu, L. D. B. Mandemaker, S. Yang, J. de Ruiter, J. H. J. Wijten, J. C. L. Janssens, T. Hartman, W. van der Stam and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 16576–16584 CrossRef CAS PubMed.
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed.
- H. An, L. Wu, L. D. B. Mandemaker, S. Yang, J. de Ruiter, J. H. J. Wijten, J. C. L. Janssens, T. Hartman, W. van der Stam and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 16576–16584 CrossRef CAS PubMed.
- P. B. Joshi, N. Karki and A. J. Wilson, ACS Energy Lett., 2022, 7, 602–609 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kuhl, R. Garcia-Muelas, N. Lopez and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- M. He, C. Li, H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, 3rd, M. J. Cheng, B. Xu and Q. Lu, Nat. Commun., 2020, 11, 3844 CrossRef CAS PubMed.
- H. An, L. Wu, L. D. B. Mandemaker, S. Yang, J. de Ruiter, J. H. J. Wijten, J. C. L. Janssens, T. Hartman, W. van der Stam and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 16576–16584 CrossRef CAS PubMed.
- X. Cao, D. Tan, B. Wulan, K. S. Hui, K. N. Hui and J. Zhang, Small Methods, 2021, 5, e2100700 CrossRef PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 133, 15472–15475 CrossRef.
- H. Liu, Z. Qi and L. Song, J. Phys. Chem. C, 2021, 125, 24289–24300 CrossRef CAS.
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed.
- C. Liu, Y. Wu, J. Fang, K. Yu, H. Li, W. He, W.-C. Cheong, S. Liu, Z. Chen, J. Dong and C. Chen, Chin. J. Catal., 2022, 43, 1697–1702 CrossRef CAS.
- B. Wang, S. Chu, L. Zheng, X. Li, J. Zhang and F. Zhang, Small Sci., 2021, 1, 2100023 CrossRef CAS.
- Y. Yang, I. Roh, S. Louisia, C. Chen, J. Jin, S. Yu, M. B. Salmeron, C. Wang and P. Yang, J. Am. Chem. Soc., 2022, 144, 8927–8931 CrossRef CAS PubMed.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- S. C. Lin, C. C. Chang, S. Y. Chiu, H. T. Pai, T. Y. Liao, C. S. Hsu, W. H. Chiang, M. K. Tsai and H. M. Chen, Nat. Commun., 2020, 11, 3525 CrossRef CAS PubMed.
- N. Kornienko, Nanoscale, 2021, 13, 1507–1514 RSC.
- B. Mei, C. Liu, F. Sun, S. Lu, X. Du, X. Li, F. Song, W. Xu and Z. Jiang, ACS Catal., 2022, 12, 8676–8686 CrossRef CAS.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y. W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- H. Y. Wang, M. Soldemo, D. Degerman, P. Lomker, C. Schlueter, A. Nilsson and P. Amann, Angew. Chem., Int. Ed., 2022, 61, e202111021 CAS.
- C. Long, J. Han, J. Guo, C. Yang, S. Liu and Z. Tang, Chem. Catal., 2021, 1, 509–522 CrossRef CAS.
- R. Arrigo, R. Blume, A. I. Large, J. J. Velasco-Velez, M. Havecker, A. Knop-Gericke and G. Held, Faraday Discuss., 2022, 236, 126–140 RSC.
- X. Feng, H. Zou, R. Zheng, W. Wei, R. Wang, W. Zou, G. Lim, J. Hong, L. Duan and H. Chen, Nano Lett., 2022, 22, 1656–1664 CrossRef CAS PubMed.
- Y. Zhu, T.-R. Kuo, Y.-H. Li, M.-Y. Qi, G. Chen, J. Wang, Y.-J. Xu and H. M. Chen, Energy Environ. Sci., 2021, 14, 1928–1958 RSC.
- K. A. Fichthorn and T. Yan, J. Phys. Chem. C, 2021, 125, 3668–3679 CrossRef CAS.
- A. F. Beker, H. Sun, M. Lemang, J. T. van Omme, R. G. Spruit, M. Bremmer, S. Basak and H. H. Perez Garza, Nanoscale, 2020, 12, 22192–22201 RSC.
- C. Zhu, S. Liang, E. Song, Y. Zhou, W. Wang, F. Shan, Y. Shi, C. Hao, K. Yin, T. Zhang, J. Liu, H. Zheng and L. Sun, Nat. Commun., 2018, 9, 421 CrossRef PubMed.
- L. Xiao, G. Wang, X. Huang, S. Zhou, R. Zhou, Y. Jiang, S. Liu, G. Li, H. Zheng, S.-G. Sun and H.-G. Liao, Appl. Catal., B, 2022, 307, 121164 CrossRef CAS.
- M. C. O. Monteiro, A. Mirabal, L. Jacobse, K. Doblhoff-Dier, S. C. Barton and M. T. M. Koper, JACS Au, 2021, 1, 1915–1924 CrossRef CAS PubMed.
- A. Preet and T.-E. Lin, Catalysts, 2021, 11, 594 CrossRef CAS.
- C. H. Ryu, Y. Nam and H. S. Ahn, Chin. J. Catal., 2022, 43, 59–70 CrossRef CAS.
- Y. Wang, Y. Zou, L. Tao, Y. Wang, G. Huang, S. Du and S. Wang, Nano Res., 2019, 12, 2055–2066 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Natl. Catal., 2018, 1, 592–600 CrossRef CAS.
- Y. Jiang, X. Zhang, D. Xu, W. Li, M. Liu and X. Qiu, Chem. Commun., 2021, 57, 6011–6014 RSC.
- S. Li, W. Chen, X. Dong, C. Zhu, A. Chen, Y. Song, G. Li, W. Wei and Y. Sun, Nat. Commun., 2022, 13, 3080 CrossRef CAS PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- M. C. O. Monteiro, F. Dattila, N. Lopez and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. Garcia de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- J. Resasco, Y. Lum, E. Clark, J. Z. Zeledon and A. T. Bell, ChemElectroChem, 2018, 5, 1064–1072 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- X. Wang, K. Klingan, M. Klingenhof, T. Moller, J. Ferreira de Araujo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- R. Yang, J. Duan, P. Dong, Q. Wen, M. Wu, Y. Liu, Y. Liu, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2022, 61, e202116706 CAS.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Natl. Catal., 2021, 4, 654–662 CrossRef CAS.
- R. Yang, J. Duan, P. Dong, Q. Wen, M. Wu, Y. Liu, Y. Liu, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2022, 61, e202116706 CAS.
- A. Mota-Lima, M. L. Alcantara, F. J. Pérez-Sanz, R. C. Bazito, P. Vidinha, R. M. B. Alves and C. A. Oller Nascimento, J. Electrochem. Soc., 2021, 168, 086502 CrossRef CAS.
- G. R. Zhang, S. D. Straub, L. L. Shen, Y. Hermans, P. Schmatz, A. M. Reichert, J. P. Hofmann, I. Katsounaros and B. J. M. Etzold, Angew. Chem., Int. Ed., 2020, 59, 18095–18102 CrossRef CAS PubMed.
- W. Ren, X. Tan, X. Chen, G. Zhang, K. Zhao, W. Yang, C. Jia, Y. Zhao, S. C. Smith and C. Zhao, ACS Catal., 2020, 10, 13171–13178 CrossRef CAS.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 2022 CrossRef PubMed.
- Y. Sha, J. Zhang, X. Cheng, M. Xu, Z. Su, Y. Wang, J. Hu, B. Han and L. Zheng, Angew. Chem., Int. Ed., 2022, 61, e202200039 CAS.
- G. Neri, J. J. Walsh, G. Teobaldi, P. M. Donaldson and A. J. Cowan, Natl. Catal., 2018, 1, 952–959 CrossRef CAS.
- P. Kamat and P. Christopher, ACS Energy Lett., 2022, 7, 1469–1472 CrossRef CAS.
- T. Möller, T. Ngo Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC.
- T. Zhang, R. Shi and Y. Ma, Acta Chim. Sin., 2021, 79, 369 CrossRef.
- F. P. Garcia de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef CAS PubMed.
- D. Raciti, T. Braun, B. M. Tackett, H. Xu, M. Cruz, B. J. Wiley and T. P. Moffat, ACS Catal., 2021, 11, 11945–11959 CrossRef CAS.
- L. Xue, X. Wu, Y. Liu, B. Xu, X. Wang, S. Dai, P. Liu and H. Yang, Nano Res., 2021, 15, 1393–1398 CrossRef.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Natl. Catal., 2020, 4, 20–27 CrossRef.
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrődi, C. Janáky and D. Deng, Angew. Chem., Int. Ed., 2023, 135, e202302789 CrossRef.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- N. Sikdar, J. R. C. Junqueira, S. Dieckhofer, T. Quast, M. Braun, Y. Song, H. B. Aiyappa, S. Seisel, J. Weidner, D. Ohl, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 23427–23434 CrossRef CAS PubMed.
- Z. Qiu, Y. Yun, M. He and L. Wang, Chem. Eng. J., 2023, 456, 140942 CrossRef CAS.
- A. Prajapati, R. Sartape, M. T. Galante, J. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Rebouças, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- Z.-Y. Wu, P. Zhu, D. A. Cullen, Y. Hu, Q.-Q. Yan, S.-C. Shen, F.-Y. Chen, H. Yu, M. Shakouri, J. D. Arregui-Mena, A. Ziabari, A. R. Paterson, H.-W. Liang and H. Wang, Nat. Synth., 2022, 1, 658–667 CrossRef.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455–2462 RSC.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2021, 7, 55–64 CrossRef.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- L. C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. Garcia de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, e1804867 CrossRef PubMed.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Natl. Catal., 2018, 1, 103–110 CrossRef CAS.
- K. Jiang, P. Kharel, Y. Peng, M. K. Gangishetty, H.-Y. G. Lin, E. Stavitski, K. Attenkofer and H. Wang, ACS Sustainable Chem. Eng., 2017, 5, 8529–8534 CrossRef CAS.
- Y. Wang, Y. Chen, Y. Zhao, J. Yu, Z. Liu, Y. Shi, H. Liu, X. Li and W. Zhou, Appl. Catal., B, 2022, 307, 120991 CrossRef CAS.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Appl. Catal., B, 2016, 188, 272–282 CrossRef.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. Kenis, Phys. Chem., 2016, 18, 7075–7084 CAS.
- Z.-Z. Niu, L.-P. Chi, R. Liu, Z. Chen and M.-R. Gao, Energy Environ. Sci., 2021, 14, 4169–4176 RSC.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li, S. Smart, Z. Zhu, H. Wang and Z. Yuan, Appl. Catal., B, 2021, 298, 121362 CrossRef.
- S. Alinejad, J. Quinson, G. K. H. Wiberg, N. Schlegel, D. Zhang, Y. Li, S. Reichenberger, S. Barcikowski and M. Arenz, ChemElectroChem, 2022, 9, e202200341 CrossRef CAS.
- M. C. O. Monteiro, S. Dieckhofer, T. Bobrowski, T. Quast, D. Pavesi, M. T. M. Koper and W. Schuhmann, Chem. Sci., 2021, 12, 15682–15690 RSC.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, J. Catal., 2022, 408, 1–8 CrossRef CAS.
- J.-B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Engin. J., 2019, 364, 89–101 CrossRef CAS.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- G. Park, S. Hong, M. Choi, S. Lee and J. Lee, Catal. Today, 2020, 355, 340–346 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li, J. Wicks, A. Johnston, D. Sinton, M. Eddaoudi and E. H. Sargent, Adv. Mater., 2022, 34, e2207088 CrossRef PubMed.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- G. Wang, J. Pan, S. P. Jiang and H. Yang, J. CO2 Util., 2018, 23, 152–158 CrossRef CAS.
- B. Endrodi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Torok, A. Danyi and C. Janaky, ACS Energy Lett., 2019, 4, 1770–1777 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- S. Ren, D. Joulie, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- J. Zou, C.-Y. Lee and G. G. Wallace, ACS Sustainable Chem. Eng., 2021, 9, 16394–16402 CrossRef CAS.
- Y. Xu, R. K. Miao, J. P. Edwards, S. Liu, C. P. O’Brien, C. M. Gabardo, M. Fan, J. E. Huang, A. Robb, E. H. Sargent and D. Sinton, Joule, 2022, 6, 1333–1343 CrossRef CAS.
- M. Esmaeilirad, A. Baskin, A. Kondori, A. Sanz-Matias, J. Qian, B. Song, M. Tamadoni Saray, K. Kucuk, A. R. Belmonte, P. N. M. Delgado, J. Park, R. Azari, C. U. Segre, R. Shahbazian-Yassar, D. Prendergast and M. Asadi, Nat. Commun., 2021, 12, 5067 CrossRef CAS PubMed.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Natl. Catal., 2022, 5, 288–299 CrossRef CAS.
- A. N. Biswas, Z. Xie, R. Xia, S. Overa, F. Jiao and J. G. Chen, ACS Energy Lett., 2022, 7, 2904–2910 CrossRef CAS.
- U. Savino and A. Sacco, J. CO2 Util., 2021, 52, 101697 CrossRef CAS.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Natl. Catal., 2022, 5, 268–276 CrossRef CAS.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- C. Yang, Z. Gao, D. Wang, S. Li, J. Li, Y. Zhu, H. Wang, W. Yang, X. J. Gao, Z. Zhang and W. Hu, Sci. China Mater., 2021, 65, 155–162 CrossRef.
- X. Pang, S. Verma, C. Liu and D. V. Esposito, Joule, 2022, 6, 2745–2761 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- N. Ikemiya, K. Natsui, K. Nakata and Y. Einaga, ACS Sustainable Chem. Eng., 2018, 6, 8108–8112 CrossRef CAS.
- Irkham, S. Nagashima, M. Tomisaki and Y. Einaga, ACS Sustainable Chem. Eng., 2021, 9, 5298–5303 CrossRef CAS.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2017, 3, 193–198 CrossRef.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2017, 20, 208–217 CrossRef CAS.
- M. Zhang, W. Wei, S. Zhou, D.-D. Ma, A. Cao, X.-T. Wu and Q.-L. Zhu, Energy Environ. Sci., 2021, 14, 4998–5008 RSC.
- S. G. Han, M. Zhang, Z. H. Fu, L. Zheng, D. D. Ma, X. T. Wu and Q. L. Zhu, Adv. Mater., 2022, 33, e2202830 CrossRef PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS PubMed.
- W. Liu, S. Wei, P. Bai, C. Yang and L. Xu, Appl. Catal., B, 2021, 299, 120661 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |