
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intracellular bottom-up generation of targeted nanosensors for single-molecule imaging†
Yanyan
Hou
a,
Satoshi
Arai
a,
Tetsuya
Kitaguchi
ab and
Madoka
Suzuki
*ab
aWASEDA Bioscience Research Institute in Singapore (WABIOS), 11 Biopolis Way, #05-02 Helios, Singapore 138667, Singapore. E-mail: suzu_mado@aoni.waseda.jp; Fax: +65-6478-9416; Tel: +65-6478-9720
bOrganization for University Research Initiatives, Waseda University, Tokyo 162-0041, Japan
First published on 22nd December 2015
Abstract
Organic dyes are useful tools for sensing cellular activities but unfavorable in single-molecule imaging, whereas quantum dots (QDs) are widely applied in single-molecule imaging but with few sensing applications. Here, to visualize cellular activities by monitoring the response of a single probe in living cells, we propose a bottom-up approach to generate nanoprobes where four organic dyes are conjugated to tetravalent single-chain avidin (scAVD) proteins via an intracellular click reaction. We demonstrate that the nanoprobes, exhibiting increased brightness and enhanced photostability, were detectable as single dots in living cells. The ease of intracellular targeting allowed the tracking of endoplasmic reticulum (ER) remodeling with nanometer spatial resolution. Conjugating thermosensitive dyes generated temperature-sensitive nanoprobes on ER membranes that successfully monitored local temperature changes in response to external heat pulses. Our approach is potentially a suitable tool for visualizing localized cellular activities with single probe sensitivity in living cells.
Fluorescence imaging at the single-molecular level is indispensable for observing the basic cellular activities occurring in cells. Signal to noise (S/N) ratio is a critical parameter for single-molecule imaging, especially when it comes to living cells where background noise originating from biological structures such as mitochondria is high.1 One way to achieve a sufficient S/N ratio is to reduce the noise level by employing an optical setup that only images a thin volume2,3 or by limiting the number of unbound fluorophores.4 Another way is to use bright probes such as quantum dots (QDs). QDs have exhibited various applications in single-molecule imaging.5–7 However, these applications only utilize QDs as position markers that are able to emit fluorescence to reveal the position of biomolecules they are attached to. Except in a few cases, the applications of QDs as biosensors in living systems are rarely reported.8–10 In addition, QDs face challenges in intracellular delivery and targeting due to their large size.11
In contrast to QDs, organic dyes show advantages as biosensors.12–15 Many approaches of targeting organic dyes to proteins have been reported over the last two decades.16–18 When coupled to specific proteins, organic dyes act as reporters of protein functions, localizations and dynamics. However, the insufficient S/N ratio generated by a single dye molecule renders organic dyes less favorable than QDs in single-molecule imaging. To achieve a sufficient S/N ratio, studies either to improve the brightness of organic dyes19 or to develop new probes with multiple dyes conjugated to a molecular scaffold20 were reported in living cells and in in vitro, respectively.
In this study, to combine the generation of bright probes, as well as specific targeting and sensing in living cells, we present a convenient approach utilizing genetic engineering and subsequent chemical reaction to realize single-molecule imaging. The genetic engineering aspect involves overexpression of specifically-targeted single-chain avidin (scAVD) in cells. As a single polypeptide chain protein topologically modified from wild-type avidin,21 scAVD can be utilized as a fusion tag in recombinant protein technology. Similar to wild-type avidin, scAVD is functional with four biotin-binding sites with high affinity.21 This high affinity ensures efficient attachment of dibenzocyclooctyne (DBCO)–biotin to scAVD, creating a DBCO-functionalized scAVD conjugate. We subsequently incubate azide-functionalized organic dyes with DBCO-functionalized scAVD, generating a fluorescent nanoprobe containing four organic dyes by bioorthogonal reaction between DBCO and azide,22 known as the copper-free click reaction (Fig. 1). All the steps take place within living cells, thus there is no need to deliver nanocomposites through the plasma membrane.
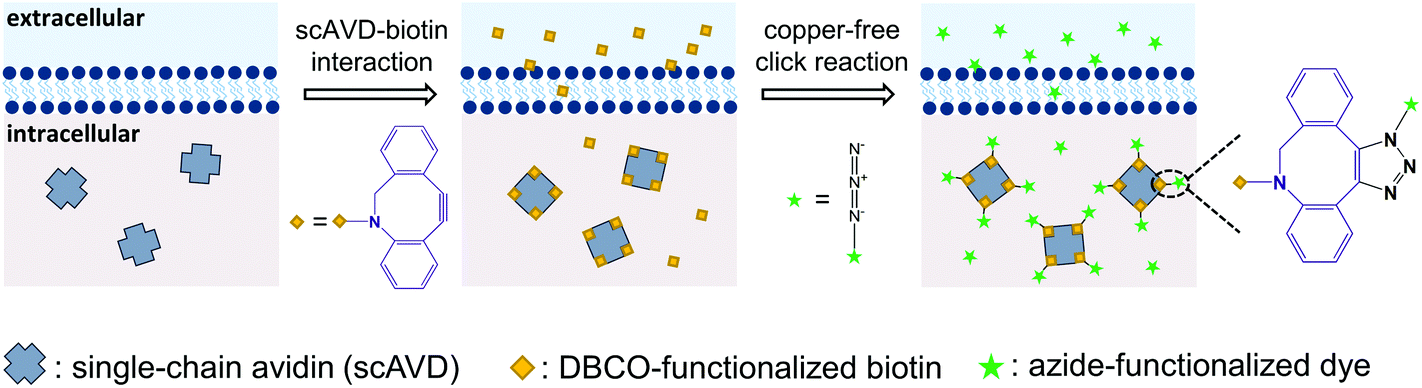 | ||
| Fig. 1 Bottom-up generation of a four-fluorophore nanoprobe in living cells in three steps. Step 1: overexpression of organelle-targeting scAVD in cells. Step 2: attachment of DBCO-functionalized biotin to scAVD via the scAVD–biotin interaction. Step 3: conjugation of azide-functionalized dye to the product in the second step via the copper-free click reaction. | ||
To evaluate the concept in vitro, mCherry-tagged scAVD (Fig. 2a) was purified from E. coli and reacted with DBCO–biotin and Cy5-azide sequentially. Bright Cy5 dots were detected in the mCherry–scAVD sample (Fig. 2b). Similar bright dots were also detected in a streptavidin sample – a commercial biotin-binding protein derived from bacteria – but were not found in the mCherry sample (Fig. 2b). Most of the dots in mCherry–scAVD and streptavidin samples were brighter than a single Cy5 dye under the same imaging conditions (Fig. 2b), suggesting multiple Cy5 dyes bound to each mCherry–scAVD/streptavidin molecule. The number of Cy5 dyes bound to a single dot is considered equivalent to the photobleaching steps. Some dots in the mCherry–scAVD sample showed a four-step fluorescence intensity decrease during photobleaching (Fig. 2c and Movie S1†), indicating scAVD is functional with four biotin-binding sites. Dots bound with four Cy5 dyes also showed improved localization precision (Fig. 2d), calculated as the standard deviation of position displacement of the same dot over a series of images.23,24 The localization precision for a Cy5 dot with four fluorophores in Fig. 2c was 6.6 nm for the x direction and 13.7 nm for the y direction over 5 s. As the fluorophore photobleached, the precision worsened (i.e. the standard deviation increased) (Fig. 2d and S1†). Quantification of the photobleaching steps showed that majority of the dots in the mCherry–scAVD sample bound to two to four Cy5-azide dyes (≈72%), which is similar to streptavidin (≈87%) (Fig. 2e). By contrast, only 2.5% dots in the free Cy5-azide sample showed multiple fluorophores.
 | ||
| Fig. 2 In vitro evaluation of four-fluorophore nanoprobes. (a) Schematic of mCherry and mCherry–scAVD constructs. Amino- and carboxyl-terminus are denoted by N and C, respectively. (b) Fluorescence images of nanoprobes. Scale bar, 1 μm. (c) Time courses of fluorescence intensity changes of the dot shown in ESI Movie S1† during photobleaching. Orange arrows indicate the photobleaching steps. (d) Position displacement of the dot shown in (c) before it was totally photobleached (time points that cannot be tracked by software were omitted). Dashed lines separate different photobleaching stages. Values indicate the localization precision at each stage. (e) A histogram showing the distribution of dots with different numbers of Cy5-azide fluorophores attached. The number of dots analyzed for mCherry–scAVD, streptavidin, mCherry and single Cy5-azide dye were 104, 106, 0 and 81, respectively. | ||
If nanoprobes are generated in living cells, they would be able to track or sense intracellular activities. We therefore next tested the binding ability of Cy5-azide to cytosol-localized scAVD in living HeLa cells. Transfected cells were incubated with Cy5-azide with or without DBCO–biotin pre-incubation. Incubation with DBCO-functionalized biotin and azide-functionalized dye at 0.1–100 μM did not compromise cell viability (Fig. S2†). Cy5 signals were observed in scAVD-overexpressing cells with DBCO–biotin pre-incubation as scattered dots (Fig. 3a), spreading across the whole cytoplasmic region inside cells (Fig. S3a†). These results suggested Cy5-azide is able to pass through the plasma membrane and the successful binding of Cy5-azide requires both the scAVD–biotin interaction and the click reaction. When incubated at room temperature (22 °C), only ∼5% of Cy5 dots colocalized with the endosome tracker pHrodo dextran (Fig. S3b†), suggesting Cy5-azide passes through the plasma membrane mainly by diffusion but not by endocytosis. Some dots showed discrete fluorescence decrease and a four-step photobleaching curve (Fig. 3b and Movie S2†), indicating scAVD is functional with four biotin-binding sites when overexpressed in cells. Some other dots with higher fluorescence intensity displayed gradual fluorescence decrease and indistinguishable photobleaching steps (Fig. 3c and Movie S3†). These dots probably consist of multiple nanoprobes due to the abundant overexpression of scAVD in cells where Cy5-azide dyes bind to closely-localized scAVD molecules that cannot be resolved by using a microscope. Single Cy5-azide dyes on a glass surface outside the cells showed weaker fluorescence and single-step photobleaching (Fig. 3d and Movie S4†). Quantification showed that Cy5 dots inside cells were brighter than single Cy5-azide dyes (Fig. 3e) and the duration of emission of Cy5 dots before being photobleached was also enhanced compared with single Cy5-azide dyes (Fig. 3f).
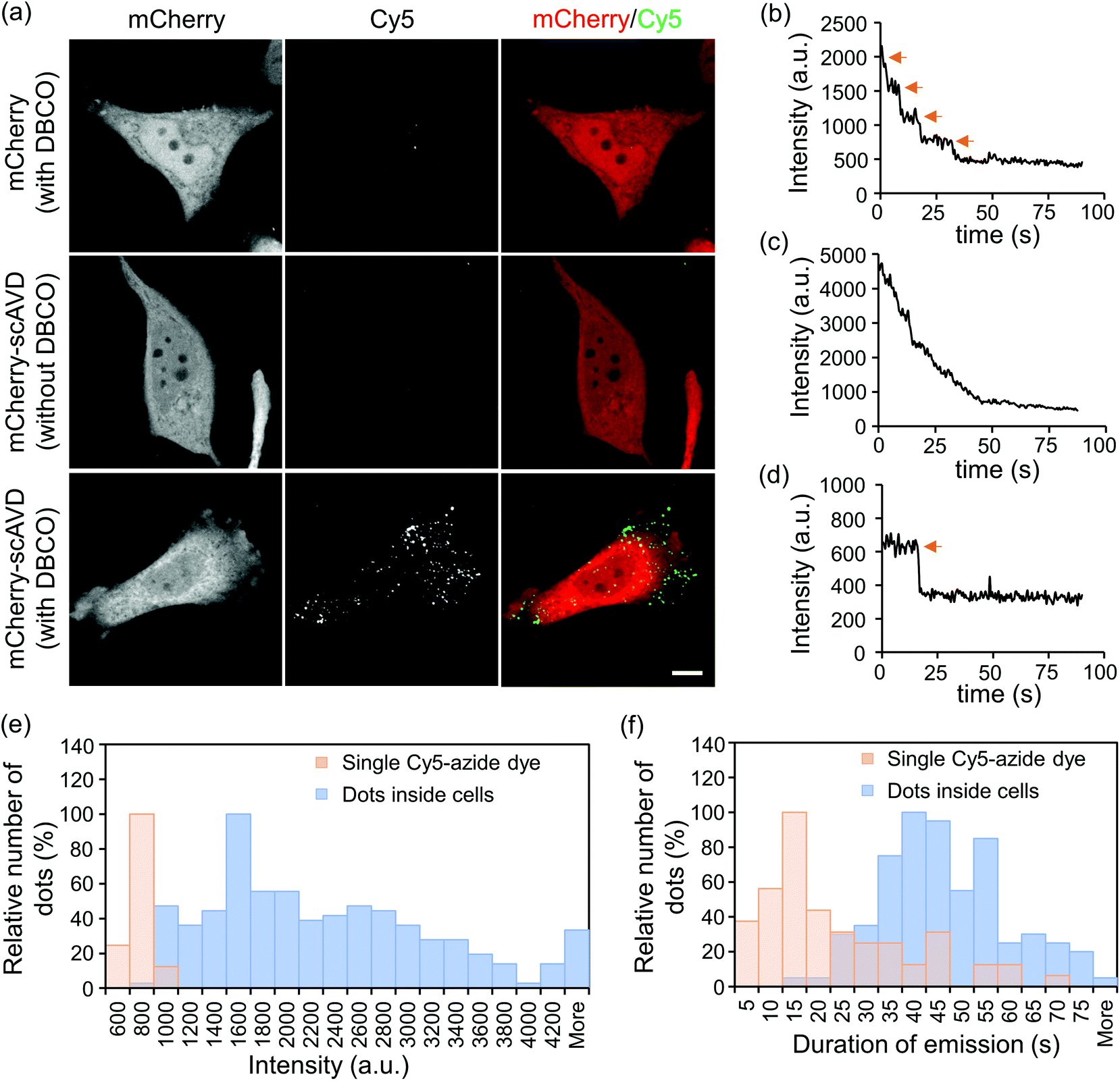 | ||
| Fig. 3 Fluorescent nanoprobes showed increased brightness and enhanced photostability. (a) Confocal microscopy showed Cy5 dots formed in scAVD-overexpressing cells. Scale bar, 10 μm. (b–d) Time courses of fluorescence intensity changes of dots inside cells (b and c) and a single Cy5-azide dye on a glass bottom dish located outside cells (d) during photobleaching. Orange arrows indicate the photobleaching steps. (e) Histogram showing the distribution of fluorescence intensity of Cy5 dots (n = 248) and single Cy5-azide dye (n = 156). (f) Histogram showing the distribution of the duration of emission of Cy5 dots (n = 118) and single Cy5-azide dye (n = 63) before they were photobleached. | ||
Our approach can achieve targeting specificity by tagging scAVD to a targeting signal. To target scAVD onto the endoplasmic reticulum (ER) membrane at the cytosolic side (Fig. 4a), scAVD was fused to the C-terminus of truncated mouse STIM1 (mSTIM1Δ2), a single-pass ER transmembrane protein with an ER-targeting signal at the luminal N-terminus.25 Cy5 dots formed on ER in scAVD-overexpressing cells (mSTIM1Δ2–mCherry–scAVD) but not in mCherry cells (mSTIM1Δ2–mCherry) (Fig. 4a). The Cy5 signals observed in mCherry cells were probably from endocytosed and/or unwashed dyes (cf. Fig. S3b†). ER remodeling is a dynamic and complex process involving tubule branching, ring formation/closure and junction sliding.26 The study of ER remodeling was traditionally limited to ER tracker dyes.27 Considering the pleomorphic nature of ER remodeling, Cy5 dots that can indicate specific tubules/junctions rather than a whole network would provide more detailed information of the remodeling process. Indeed, Cy5 dots exhibited different travel paths (Fig. 4b–d), instant velocities and distances (Fig. 4e) during remodeling. Some dots showed extremely slow movement with velocities of 0.2–29.7 nm s−1 (average 7.2 ± 4.6 nm s−1, n = 30, Fig. S4a†) during the observation period (Fig. 4b), indicating that there are parts of ER structures remaining relatively static during the remodeling process within certain periods. ER tubule branching involves forces generated by microtubules.26,27 However, the forces mediating ER ring formation/closure or junction sliding are not well established. Cy5 dots attached to moving ER rings/junctions (Fig. 4c and d) showed velocities of 0.2–226.0 nm s−1 (average 37.1 ± 35.0 nm s−1, n = 49, Fig. S4b†), which is slower than motor protein-mediated microtubule movement26,28 but within the range of microtubule polymerization-driven ER remodeling,27 indicating the involvement of microtubule in mediating ER polygon remodeling. It is noteworthy that the velocities of dots rapidly changed (Fig. 4e). The velocities of different dots varied, suggesting local environment such as cytoplasm viscoelasticity may affect ER remodeling.29 The detailed dynamics obtained with Cy5 dots would potentially facilitate the understanding of mechanisms underlying the intricate and complex ER remodeling process.
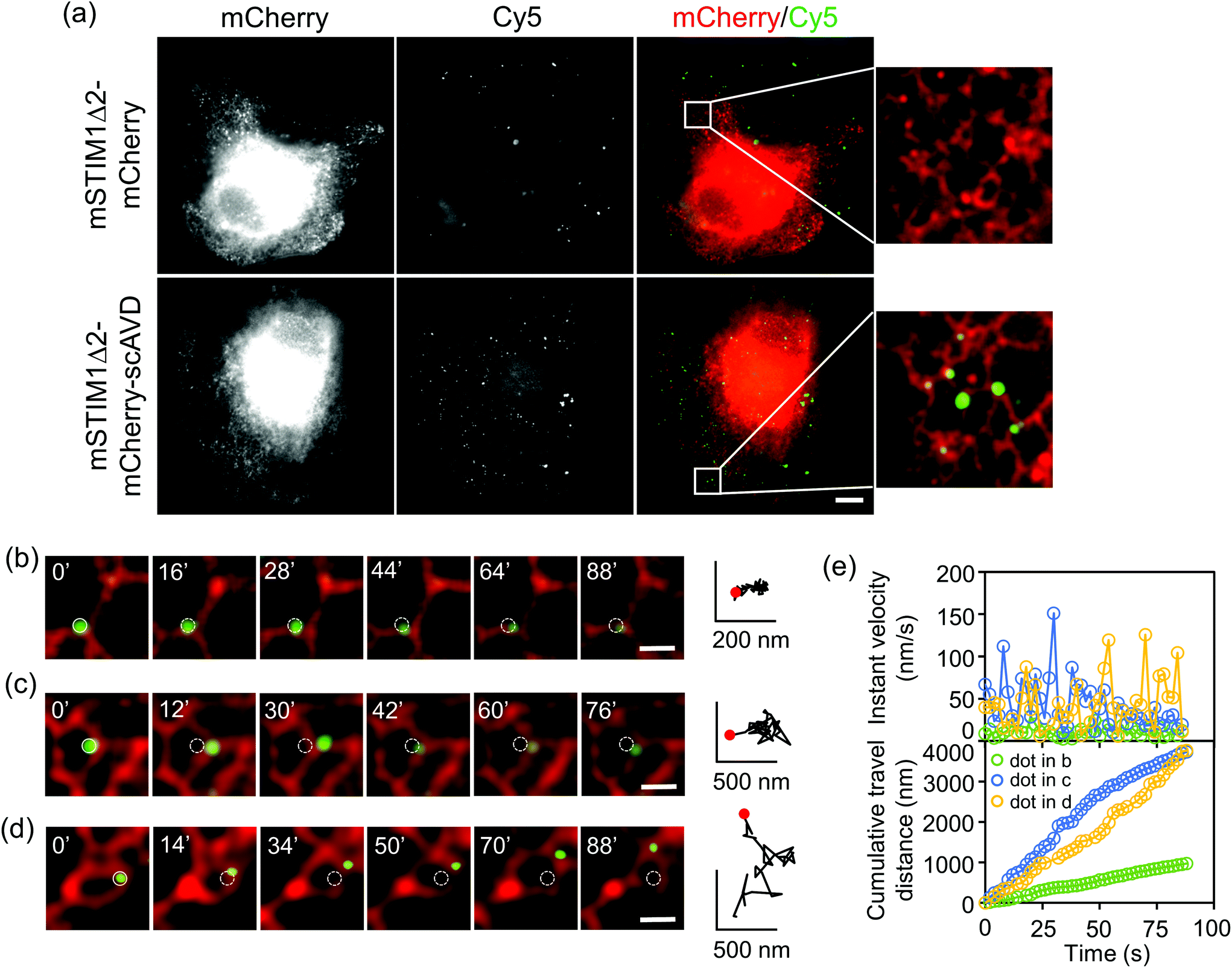 | ||
| Fig. 4 Fluorescent nanoprobes targeting ER track ER dynamic remodeling with nanometer spatial resolution. (a) Epifluorescence microscopy showed Cy5 dots assembled on ER in scAVD-overexpressing cells. Scale bar, 10 μm. (b–d) Fluorescent dots with different velocities colocalized with ER following ER remodeling. Both solid and dashed circles indicate original positions of dots. Scale bar, 1 μm. Red dots indicate the starting points. (e) Instant velocity and cumulative travel distance of dots in (b–d) were analyzed. | ||
Our approach can also conjugate other organic dyes aside from Cy5-azide with biosensor functions. Thermosensitive TAMRA-azide (azide-PEG3-5(6)-carboxytetramethylrhodamine), a cell permeable dye with supreme cytosolic diffusivity30 was tested next. Although TAMRA-azide localized to mitochondria in HeLa cells, the amount was trivial compared with the substantial presence in cytosol (Fig. S5†). To induce ER localization, scAVD was tagged to mSTIM1Δ3,31 another truncated form of mSTIM1. To shorten plasmid size and increase protein expression, we deleted the fluorescent protein and fused scAVD directly to the C-terminus of STIM1Δ3 (mSTIM1Δ3–scAVD). TAMRA-azide efficiently passed through the plasma membrane and distributed in whole cells without accumulation on ER in vector cells (mSTIM1Δ3–EGFP) before being gradually released out of cells, but localized to ER in scAVD-overexpressing cells, shown by colocalization with an ER tracker (Fig. 5a).
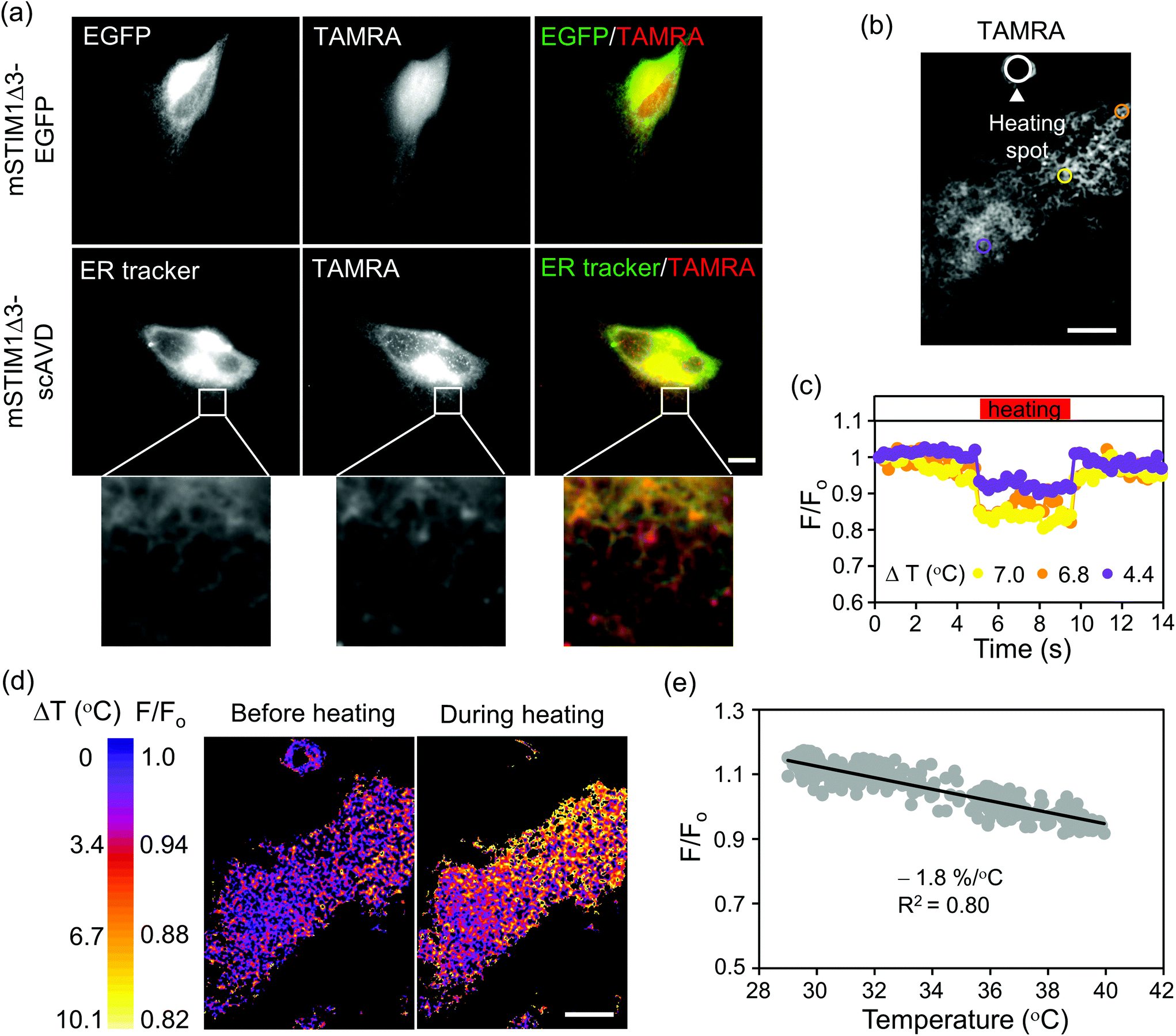 | ||
| Fig. 5 Generation of nanothermometer on ER. (a) Epifluorescence microscopy showed colocalization of TAMRA-azide with ER in scAVD-overexpressing cells. Scale bar, 10 μm. (b) A HeLa cell with TAMRA-azide on ER. Regions (colored circles) with different distances from the heating spot (white circle) were chosen and analyzed in (c). Scale bar, 10 μm. (c) ‘Square wave’ of TAMRA-azide generated in response to laser heating and re-cooling. (d) Temperature gradient (ΔT) mapped based on fluorescence intensity change of TAMRA-azide. Scale bar, 10 μm. (e) Temperature sensitivity of TAMRA-azide on ER, −1.8% per °C, was determined from the measurements as in (d) (24 cells were analyzed). The medium temperature in the cell culture dish (T0) was maintained at 28.5 °C or 34.5 °C. The fluorescence intensity was normalized to 37 °C and plotted versus temperature (T0 + ΔT). | ||
TAMRA-azide has a temperature sensitivity of −1.9% per °C in cuvette (relative to 36 °C, Fig. S6†). We therefore examined the potential thermosensor functions of TAMRA-azide in cells. The temperature sensitivity was −2.1% per °C in fixed HeLa cells overexpressing cytosol-localized scAVD (relative to 37 °C, Fig. S7†), similar to the result obtained in cuvette. We next tested the temperature sensitivity of TAMRA-azide on ER in living cells. A confocal fluorescence microscopy system equipped with a near infrared laser (1064 nm) was employed to generate a localized heat pulse.28,32,33 The microheater was placed in close vicinity of a scAVD-overexpressing cell with TAMRA-azide binding (Fig. 5b). When the microheater was turned on, the fluorescence intensity of TAMRA-azide decreased and responded to heating and re-cooling with a ‘square wave’ (Fig. 5c). The change of fluorescence intensity in a specific region depends on its distance from the heating spot. The microheater generated a local temperature gradient across the cell (Fig. 5d). However, the gradient was not immediately visible in ER tracker green (Fig. S8c†). The plot of normalized fluorescence intensity against temperature showed that the temperature sensitivity of ER-targeted TAMRA-azide was −1.8% per °C (Fig. 5e), which is very close to the sensitivity tested in cuvette and in the cytosol of fixed cells. With a much lower temperature sensitivity of −0.46% per °C, ER tracker green is considered insensitive to temperature changes (Fig. S8†). These results demonstrate the potential ability of TAMRA-conjugated probes for monitoring cellular temperature changes at specific organelles in living cells.
Conclusions
In summary, we reported a three-step method to generate bright and photostable fluorescent nanoprobes that can target the arbitrary object in living cells for sensing and single-molecule imaging by means of genetic engineering and chemical reactions. The nanoprobe exhibited increased brightness compared with the single organic dye both in vitro and in living cells, which contributed to an increased S/N ratio. The nanoprobe can be observed at the single-molecular level in living cells with a conventional fluorescence microscope. This improvement together with the enhanced photostability, as well as improved localization precision will make single-molecule imaging which looks into biological processes at the nanometer scale more achievable. Specific targeting of the probe circumvents the limitation of traditional organic dyes that usually lack site-specificity or localize to the lumen of organelles. At the same time, the probe can utilize the advantage of organic dyes as trackers or biosensors to monitor biological processes. Synthesizing azide-functionalized organic dyes with the ability of sensing cellular parameters would facilitate the observation of different biological processes at localized regions of organelles in future.Methods
Cell culture
HeLa cells were cultured in Dulbecco's modified Eagle medium (DMEM), containing 10% fetal bovine serum (FBS), 100 units per ml penicillin and 100 μg per ml streptomycin at 37 °C in the presence of 5% CO2. All the components were purchased from Invitrogen (CA, USA). For microscopy experiments, the cells were cultured in 3.5 cm glass-based dishes (IWAKI, Tokyo, Japan) at 37 °C with 5% CO2.Plasmids
pmCherry-C3 vector was generated by replacing EGFP in pEGFP-C3 (BD Biosciences Clontech, SD, USA) with mCherry. Synthesized scAVD DNA21 (Integrated DNA Technologies, IA, USA) was cloned into pmCherry-C3 or pEGFP-C3 vector, and named mCherry–scAVD and EGFP–scAVD, respectively. To create ER-targeting scAVD, DNA fragments of mSTIM1Δ2 (1–343 a.a.) and mSTIM1Δ3 (1–425 a.a.) amplified by PCR from mouse brain cDNA were inserted into pmCherry–scAVD-C3 and pEGFP–scAVD-C3 vectors, respectively, and named mSTIM1Δ2–mCherry–scAVD and mSTIM1Δ3–EGFP–scAVD. To increase the expression of scAVD, the EGFP fragment was removed from mSTIM1Δ3–EGFP–scAVD, and named mSTIM1Δ3–scAVD.Materials
DBCO-PEG4-Biotin, Cy5-azide and TAMRA-azide were purchased from Click Chemistry Tools (AZ, USA). ER tracker green was purchased from Invitrogen. Catalase and glucose oxidase were purchased from Sigma-Aldrich (St. Louis, MO, USA), glucose and DTT were purchased from Wako Pure Chemical Industries (Osaka, Japan).Protein production in E. coli and purification
mCherry and mCherry–scAVD were subcloned into pRSETA vector. The production and purification of protein were performed as described previously.34 Briefly, the plasmid was introduced into E. coli JM109 (DE3) and cultured at 20 °C for 4 days. Bacteria were harvested by centrifugation and lysed by three freeze–thaw cycles and sonication. Bacteria lysates were mixed with Ni-NTA agarose (Qiagen, Hilden, Germany) and incubated at 4 °C for 1 hour with rotation. Ni-NTA agarose was then collected by centrifugation and applied to a column for elution. The eluent was further purified by a PD-10 gel-filtration column (GE Healthcare, Buckinghamshire, UK). Purified protein was kept in Hepes buffer (150 mM KCl and 50 mM Hepes–KOH, pH 7.4). The purified protein concentration was determined by Bradford protein assay.In vitro reaction
Purified mCherry, mCherry–scAVD and a bacterially-derived commercial product, streptavidin (Invitrogen) were used for in vitro reactions. Protein was mixed with DBCO-PEG4-Biotin with a mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and incubated at room temperature for 1 hour. Cy5-azide was then added to the mixture with a mole ratio of 2:1 to DBCO-PEG4-Biotin. The mixture was further incubated at room temperature for 1 hour. Protein concentration was 10−5 M in 50 μl reaction mixture. The reaction product was applied to a PD-10 column for purification and eluted in Hepes buffer.
:10 and incubated at room temperature for 1 hour. Cy5-azide was then added to the mixture with a mole ratio of 2:1 to DBCO-PEG4-Biotin. The mixture was further incubated at room temperature for 1 hour. Protein concentration was 10−5 M in 50 μl reaction mixture. The reaction product was applied to a PD-10 column for purification and eluted in Hepes buffer.
Epifluorescence microscopy
For in vitro observation, glass slides and coverslips were sonicated in 0.1 M KOH for 30 min followed by sonication in 100% ethanol for another 30 min to remove dust from the surfaces. The cleaned coverslips were rinsed thoroughly with dH2O and air-dried. The purified reaction product was diluted in an oxygen scavenging system (catalase 50 U ml−1, glucose 4.5 mg ml−1, glucose-oxidase 50 U ml−1, DTT 10 mM in PBS) to improve dye stability. Then, 1 μl diluted reaction product was applied on top of a glass coverslip and covered by another glass coverslip.Cells were incubated with 1 μM DBCO-PEG4-Biotin at 37 °C for 1 hour, followed by incubation with 1 μM Cy5-azide at room temperature for 1 hour or 1 μM TAMRA-azide at room temperature for 30 min. Incubations were performed in serum-free DMEM. Cells were washed with PBS three times before observation. For observing ER in cells overexpressing mSTIM1Δ3–scAVD, the cells were incubated with 125 nM ER tracker green for 30 min at room temperature after incubation with TAMRA-azide. Images were taken with an Olympus IX83 inverted microscope equipped with an electron multiplying charge-coupled device (EMCCD) camera (iXon3, 1024 × 1024 pixels, Andor Technology, Belfast, Antrim, UK). An UPlanSAPO 100× NA 1.40 objective lens or a PlanApo 60× NA 1.40 objective lens (both from Olympus, Tokyo, Japan) was used. A Xenon lamp (U-LH75XEAPO, Olympus) was used as a light source. Cy5-azide images were acquired with a FF02-632/22 excitation filter, a FF660-Di02 dichroic mirror and a FF01-692/40 emission filter (all from Semrock, Rochester, NY, USA). mCherry and TAMRA-azide images were acquired with a FF01-549/15 excitation filter, a FF585-Di01 dichroic mirror and a F01-607/36 emission filter (all from Semrock). EGFP and ER tracker green images were acquired with a BP470-495 excitation filter, a DM505 dichroic mirror and a BP510-550 emission filter (all from Olympus). MetaMorph NX software (Molecular Devices, CA, USA) was used to acquire images. Images were captured at 1.12 s per frame with 1 s exposure time for in vitro observations, 750 ms per frame with 500 ms exposure time for Cy5 dots in cytosol and 2 s per frame with 500 ms exposure time for Cy5 dots on ER.
Confocal microscopy
Images were acquired using an Olympus confocal microscope (FluoView FV1000). A PlanApo N 60× NA 1.42 (Olympus) objective was used. Microscopic images of Cy5-azide and mCherry were obtained with Cy5 (633 nm laser) and Texas red (543 nm laser) filter sets, respectively.Microscopic laser heating system on a spinning disk confocal microscope
Evaluation of the temperature sensitivity of TAMRA-azide on ER in living HeLa cells was performed on a spinning disc confocal microscope equipped with a microscopic heat generating system based on an inverted microscope (IX81, Olympus) and a spinning disc confocal unit (CSU-10, Yokogawa), as previously reported.32,33 Briefly, a near infrared laser beam (1064 nm, KPS-KILAS-COOL-1064-02-P, Keopsys) was focused by using an objective (APON 60XOTIRF, Olympus) on an aggregate of aluminum particles fixed at the tip of a glass micro needle. This microheater was positioned close to cells overexpressing mSTIM1Δ3–scAVD with TAMRA-azide binding. ER tracker green was excited by using a 488 nm laser (CUBE 488, Coherent) and the emission was collected through a band pass filter FF01-520/35 (Semrock), whereas TAMRA-azide was imaged using a 561 nm laser (Sapphire 561, Coherent) and a filter FF01-607/36 (Semrock). Images were captured at 229 ms per frame with 221 ms exposure time. Cell medium was maintained at 28.5 °C or 34.5 °C by a KRi stage top incubator (Tokai Hit, Shizuoka, Japan).Data analysis
All fluorescence images obtained were analyzed with ImageJ software (National Institute of Health, MD, USA). In analyzing the localization precision of Cy5 dots on glass slides and tracking the movement of Cy5 dots attached to ER, the Particle Tracker plug-in of ImageJ was used.35 In determining the relationship between temperature and fluorescence ratio of TAMRA-azide in living cells overexpressing ER-targeting scAVD, the fluorescence intensity of a small region encompassing ER structures (Fig. 5b) was obtained. Regions with different distances from the heating spot were chosen. The temperature at each region was calculated from tetramethylrhodamine-conjugated dextran.32 Briefly, tetramethylrhodamine-conjugated dextran was added to a culture dish that was maintained at 28.5 °C or 34.5 °C (T0) and the same laser heating setup for TAMRA-azide was applied. With known temperature sensitivity of −1.2% per °C and measured fluorescence change of tetramethylrhodamine-conjugated dextran, the temperature change (ΔT) at each region was then calculated. The actual temperature (T) at each region T = T0 + ΔT. Movies were created using the Walking Average plug-in of ImageJ with 4 frames averaged. In photobleaching graphs, the actual fluorescence intensity of each frame was shown.Acknowledgements
This research was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 26107717 (to M. S.).Notes and references
- H. Andersson, T. Baechi, M. Hoechl and C. Richter, J. Microsc., 1998, 191, 1–7 CrossRef CAS PubMed.
- M. Tokunaga, N. Imamoto and K. Sakata-Sogawa, Nat. Methods, 2008, 5, 159–161 CrossRef CAS PubMed.
- H. Tadakuma, Y. Ishihama, T. Shibuya, T. Tani and T. Funatsu, Biochem. Biophys. Res. Commun., 2006, 344, 772–779 CrossRef CAS PubMed.
- N. Watanabe and T. J. Mitchison, Science, 2002, 295, 1083–1086 CrossRef CAS PubMed.
- T. Serizawa, T. Terui, T. Kagemoto, A. Mizuno, T. Shimozawa, F. Kobirumaki, S. Ishiwata, S. Kurihara and N. Fukuda, Am. J. Physiol. Cell Physiol., 2011, 301, C1116–C1127 CrossRef CAS PubMed.
- H. Tada, H. Higuchi, T. M. Wanatabe and N. Ohuchi, Cancer Res., 2007, 67, 1138–1144 CrossRef CAS PubMed.
- T. M. Watanabe and H. Higuchi, Biophys. J., 2007, 92, 4109–4120 CrossRef CAS PubMed.
- Y. S. Liu, Y. Sun, P. T. Vernier, C. H. Liang, S. Y. Chong and M. A. Gundersen, J. Phys. Chem. C, 2007, 111, 2872–2878 CAS.
- J. M. Yang, H. Yang and L. Lin, ACS Nano, 2011, 5, 5067–5071 CrossRef CAS PubMed.
- I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- H. Benink, M. McDougall, D. Klaubert and G. Los, BioTechniques, 2009, 47, 769–774 CrossRef CAS PubMed.
- M. Kamiya and K. Johnsson, Anal. Chem., 2010, 82, 6472–6479 CrossRef CAS PubMed.
- Y. Kurishita, T. Kohira, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 18779–18789 CrossRef CAS PubMed.
- E. Tomat, E. M. Nolan, J. Jaworski and S. J. Lippard, J. Am. Chem. Soc., 2008, 130, 15776–15777 CrossRef CAS PubMed.
- M. J. Hinner and K. Johnsson, Curr. Opin. Biotechnol., 2010, 21, 766–776 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- Y. Kim, S. H. Kim, M. Tanyeri, J. A. Katzenellenbogen and C. M. Schroeder, Biophys. J., 2013, 104, 1566–1575 CrossRef CAS PubMed.
- H. R. Nordlund, V. P. Hytonen, J. Horha, J. A. Maatta, D. J. White, K. Halling, E. J. Porkka, J. P. Slotte, O. H. Laitinen and M. S. Kulomaa, Biochem. J., 2005, 392, 485–491 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- T. Nishizaka, R. Seo, H. Tadakuma, K. Kinosita Jr. and S. Ishiwata, Biophys. J., 2000, 79, 962–974 CrossRef CAS PubMed.
- M. Suzuki, H. Fujita and S. Ishiwata, Biophys. J., 2005, 89, 321–328 CrossRef CAS PubMed.
- P. B. Stathopulos, L. Zheng, G. Y. Li, M. J. Plevin and M. Ikura, Cell, 2008, 135, 110–122 CrossRef CAS PubMed.
- C. Lee and L. B. Chen, Cell, 1988, 54, 37–46 CrossRef CAS PubMed.
- C. M. Waterman-Storer and E. D. Salmon, Curr. Biol., 1998, 8, 798–806 CrossRef CAS PubMed.
- K. Oyama, M. Takabayashi, Y. Takei, S. Arai, S. Takeoka, S. Ishiwata and M. Suzuki, Lab Chip, 2012, 12, 1591–1593 RSC.
- C. Lin, Y. Zhang, I. Sparkes and P. Ashwin, Biophys. J., 2014, 107, 763–772 CrossRef CAS PubMed.
- C. W. Cunningham, A. Mukhopadhyay, G. H. Lushington, B. S. Blagg, T. E. Prisinzano and J. P. Krise, Mol. Pharm., 2010, 7, 1301–1310 CrossRef CAS PubMed.
- Z. Li, J. Lu, P. Xu, X. Xie, L. Chen and T. Xu, J. Biol. Chem., 2007, 282, 29448–29456 CrossRef CAS PubMed.
- S. Arai, S. C. Lee, D. Zhai, M. Suzuki and Y. T. Chang, Sci. Rep., 2014, 4, 6701 CrossRef CAS PubMed.
- V. Zeeb, M. Suzuki and S. Ishiwata, J. Neurosci. Methods, 2004, 139, 69–77 CrossRef PubMed.
- T. Kitaguchi, M. Oya, Y. Wada, T. Tsuboi and A. Miyawaki, Biochem. J., 2013, 450, 365–373 CrossRef CAS PubMed.
- I. F. Sbalzarini and P. Koumoutsakos, J. Struct. Biol., 2005, 151, 182–195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr08012f |
| This journal is © The Royal Society of Chemistry 2016 |