
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient siRNA delivery from core–shell mesoporous silica nanoparticles with multifunctional polymer caps†
Karin
Möller
a,
Katharina
Müller
b,
Hanna
Engelke
a,
Christoph
Bräuchle
a,
Ernst
Wagner
*b and
Thomas
Bein
*a
aDepartment of Chemistry and Center for NanoScience, University of Munich (LMU), Butenandtstrasse 5-13, 81377 Munich, Germany. E-mail: bein@lmu.de; Fax: (+49) 89-2180-77622
bPharmaceutical Biotechnology and Center for NanoScience, University of Munich (LMU), Butenandtstrasse 5-13, 81377 Munich, Germany. E-mail: ernst.wagner@cup.uni-muenchen.de
First published on 30th December 2015
Abstract
A new general route for siRNA delivery is presented combining porous core–shell silica nanocarriers with a modularly designed multifunctional block copolymer. Specifically, the internal storage and release of siRNA from mesoporous silica nanoparticles (MSN) with orthogonal core–shell surface chemistry was investigated as a function of pore-size, pore morphology, surface properties and pH. Very high siRNA loading capacities of up to 380 μg per mg MSN were obtained with charge-matched amino-functionalized mesoporous cores, and release profiles show up to 80% siRNA elution after 24 h. We demonstrate that adsorption and desorption of siRNA is mainly driven by electrostatic interactions, which allow for high loading capacities even in medium-sized mesopores with pore diameters down to 4 nm in a stellate pore morphology. The negatively charged MSN shell enabled the association with a block copolymer containing positively charged artificial amino acids and oleic acid blocks, which acts simultaneously as capping and endosomal release agent. The potential of this multifunctional delivery platform is demonstrated by highly effective cell transfection and siRNA delivery into KB-cells. A luciferase reporter gene knock-down of up to 80–90% was possible using extremely low cell exposures with only 2.5 μg MSN containing 0.5 μg siRNA per 100 μL well.
Introduction
In the past decade mesoporous silica nanoparticles (MSN) have been recognized as a powerful general tool for packaging fragile or toxic pharmaceuticals and ensuring their safe passage through cell membranes, to finally unload their cargo at the point of destination. Attractive features of these nanoparticles include extremely large surface areas associated with large pore volumes, adjustable pore sizes between 2 and about 20 nm, and great flexibility regarding the introduction of molecular functionalities in the pores and on the external particle surface. Particle sizes from about 100 nm to 300 nm are deemed optimal for cellular uptake by endocytosis. Additionally, the internal particle surface can be adapted by implementing basic, acidic or hydrophobic residues that are compatible with the cargo of choice. Surface functionalization can also be used to implement docking points for targeting ligands or dye labels enabling theranostic applications.While biocompatibility, cell uptake and effective delivery of therapeutic model cargos such as dye molecules were initially in the focus,1 in recent years MSNs have been investigated for different specific medical applications,2–4 including cancer therapy with large-pore mesoporous silica.5 In this context, the delivery of short interfering RNA (siRNA) raises hopes for achieving highly specific anti-cancer treatments with low side-effects. The siRNA molecules are double stranded short chain oligonucleotides with 20–23 base pairs that post-transcriptionally regulate protein synthesis by sequence-specific matching with mRNA molecules and thus triggering their degradation. The discovery of RNA interference in 1998 by Fire and Mellow, awarded with the Nobel Prize in 2006, has set off a surge in research activities trying to exploit synthetic siRNA as a therapeutic tool with first clinical trials underway.6,7 However, due to factors including their small size (leading to effective clearing from the body), their negative charge that is incompatible with the cell membrane, and their limited chemical stability, siRNA molecules have to be packaged or stabilized in order to successfully enter target cells via endocytosis. Several different packaging schemes have been investigated including the use of polymers, liposomes, dendrimers, hydrogels or inorganic host systems.6,8
Mesoporous silica nanoparticles are viewed to be promising candidates for stabilizing and packaging siRNA because they offer a chemically robust matrix and a pore system that can, in principle, accommodate even small oligonucleotides. However, due to the large molecular size of siRNA of approximately 2 × 8 nm and a hydrodynamic diameter of about 4.2 nm, sufficiently large pore entrances are necessary for an efficient uptake of siRNA molecules in the MSN pore system. Typical MSNs feature pore sizes at around 3 nm, and as a result such MSNs have been used to accommodate siRNA on their external surface, usually aided by large cationic polymers such as polyethyleneimine (PEI)9–11 or poly(2-dimethylaminoethyl methacrylate) (PDMAEMA).12 These polymers are known as endosomal escape agents, attributed to a proton-sponge effect, however, they are also known for their toxicity and are therefore sometimes used in combination with polyethylene glycol (PEG) to avoid these side effects.13,14
In order to offer improved protection as well as a higher loading capacity for siRNA, an uptake into the internal pore system of silica nanoparticles is highly desirable. Very few examples have been published where silica nanoparticles with pores larger than 10 nm were used as carrier system for siRNA. In an early study, Gao et al. showed that a new class of large-pore MSNs (LP-MSN) with 20 nm pores was able to absorb plasmid DNA and protect it against enzymatic degradation.15 Ashley et al. used microemulsion-derived LP-MSNs with 13–30 nm multimodal pores in combination with a supported lipid bilayer to host a 10–100-fold amount of siRNA as compared to liposomal carriers and showed their good efficacy against translation of different cyclin proteins.16,17 Na et al. synthesized sponge-like structured silica particles with about 23 nm pores that were subsequentially aminated and decorated with PEG. These particles, loaded with siRNA, efficiently reduced the green fluorescent protein (GFP) and the vascular endothelial growth factor (VEGF) in Hela cell xenografts upon direct injection.18 Other examples that used large-pore carriers for siRNA delivery needed the assistance of PEI to show good efficacy in gene knockdown.19,20 Lin et al. used 10 nm LP-MSN and cross-linked PDMAEMA via disulfide bonds to the host in order to achieve intracellular cleavage of the polymer and triggered siRNA delivery. They achieved silencing of luciferase and of the endogenous protein Lamin A/C of about 60% with low siRNA loadings of about 3 wt%.21
The group of Gu demonstrated that it is not a prerequisite to use extra-large pores in order to adsorb siRNA into the internal pores of silica nanoparticles. They used chaotropic salt solutions to shield the repulsive negative charges between silica surfaces and nucleic acids and were able to adsorb up to 27 μg per mg siRNA into the 3.7 nm pores of unfunctionalized MSN.22 These particles showed very good knockdown efficiency against GFP and VEGF when PEI coupled to the fusogenic peptide KALA was attached on the outside of their particles.23,24
Here we describe the development of a novel platform of medium-pore core–shell porous silica nanoparticles aimed at a high siRNA loading and release capacity as well as high siRNA knockdown efficiency. Internal siRNA uptake is achieved by using core–shell silica particles exhibiting a positively charged interior due to amino groups and a negatively charged exterior based on sulfhydryl groups to minimize external adsorption. In previous studies we could show that core–shell functionalized small-pore MSNs were efficient carriers for drug delivery of small molecules.25 Co-condensing functional groups in the particle core and chemically orthogonal groups on the particle surface allows for an efficient implementation of various functionalities such as particle labeling, attachment of pore-closing molecules and specific targeting agents in a spatially controlled manner. This strategy was successfully used for the selective delivery of cisplatin to cancerous cells in lung cancer tissue while sparing healthy cells.26
In this work we show that co-condensation derived core–shell nanoparticles with medium-sized pores (MP-MSN) of 4 nm can be charged with an extremely high load of up to 380 μg per mg siRNA/MSN even without using highly concentrated salt solutions. The subsequent siRNA desorption was quantitatively evaluated as a function of structural and process parameters in order to optimize the elution efficiency of siRNA for later applications in cell experiments. To enhance the potential for siRNA delivery, we developed a novel strategy for cell transfection by combining MSN particles with modularly designed block copolymers. The polymer presented here and more complex analogs of these modular polymers have been used successfully by some of us for siRNA delivery through polyplex formation.27,28 siRNA polyplexes often suffer from very variable, heterogeneous nanoparticle sizes. In the present work, polymers are attached to the exterior of our approximately 150 nm homogeneous MSN particles to inhibit premature release and to allow for endosomal escape after transfection into KB cells. These polymers contain segments with multiple succinoyl tetraethylene pentamine (stp) that can interact with the negatively charged silica surface. Lysine is used as a branching point for attachment of oleic acid units to establish hydrophobicity and membrane permeability.29 This combination of multifunctional MSNs and modularly designed block copolymers proved to be highly efficient in the down-regulation of a GFP-luciferase fusion reporter protein.
Results and discussion
Particle synthesis and analysis
Mesoporous silica nanoparticles have been proposed as host materials for truly internal adsorption of nucleic acids since 2006, when Solberg et al. used micron-sized mesoporous silica to adsorb linear DNA with 760 to 2000 bp.30 To facilitate the adsorption of negatively charged, large plasmid DNA (pDNA) molecules of over 5000 base pairs, Gao et al.15 adapted a new synthesis route that resulted in exceptionally large pore diameters between 12 and 20 nm while maintaining particle sizes between 70–300 nm by using a fluorinated polymer FC-4 as particle growth inhibitor. The silica surface was subsequently grafted with aminoproylsilane (APTES) in order to create a cationic layer that could interact with the phosphate moieties of the pDNA. A good protection against endonucleases was demonstrated through this encapsulation. Porous hosts that are able to internally adsorb nucleic acid molecules offer both protection from external attack and a large pore volume that promises high loading capacities. This is expected to be advantageous compared to electrostatic attachment on the outside of inorganic particles, as often performed for RNA delivery with small-pore MSNs.Here we investigated functionalized mesoporous silica hosts with large pore sizes of about 10 nm (LP-MSN), in addition to nanoparticles with medium-sized pores between 4 and 5 nm (MP-MSN). The LP-MSNs were made according to a previous report15 by a multistep acid-catalyzed synthesis using F123 as a template accompanied by trimethylbenzene as swelling agent and FC-4 as particle growth retardant. These purely siliceous particles were subsequently grafted with APTES or a mixture of APTES and phenyltriethoxysilane (PhTES), resulting in a homogenous distribution of amino groups throughout the particle body. Mesoporous particles sized between 100 and 200 nm are formed as shown in the transmission electron micrograph (TEM) in Fig. 1a. TEM and nitrogen sorption data in combination indicate that the pores have a bottleneck morphology. When the pore-size distribution is analyzed by using the adsorption branch we observe a size-distribution around 11 nm. However, when the NLDFT-equilibrium model is used we observe a pore-size distribution around 7.8 nm reflecting the presence of smaller pore windows (see Fig. 1b).
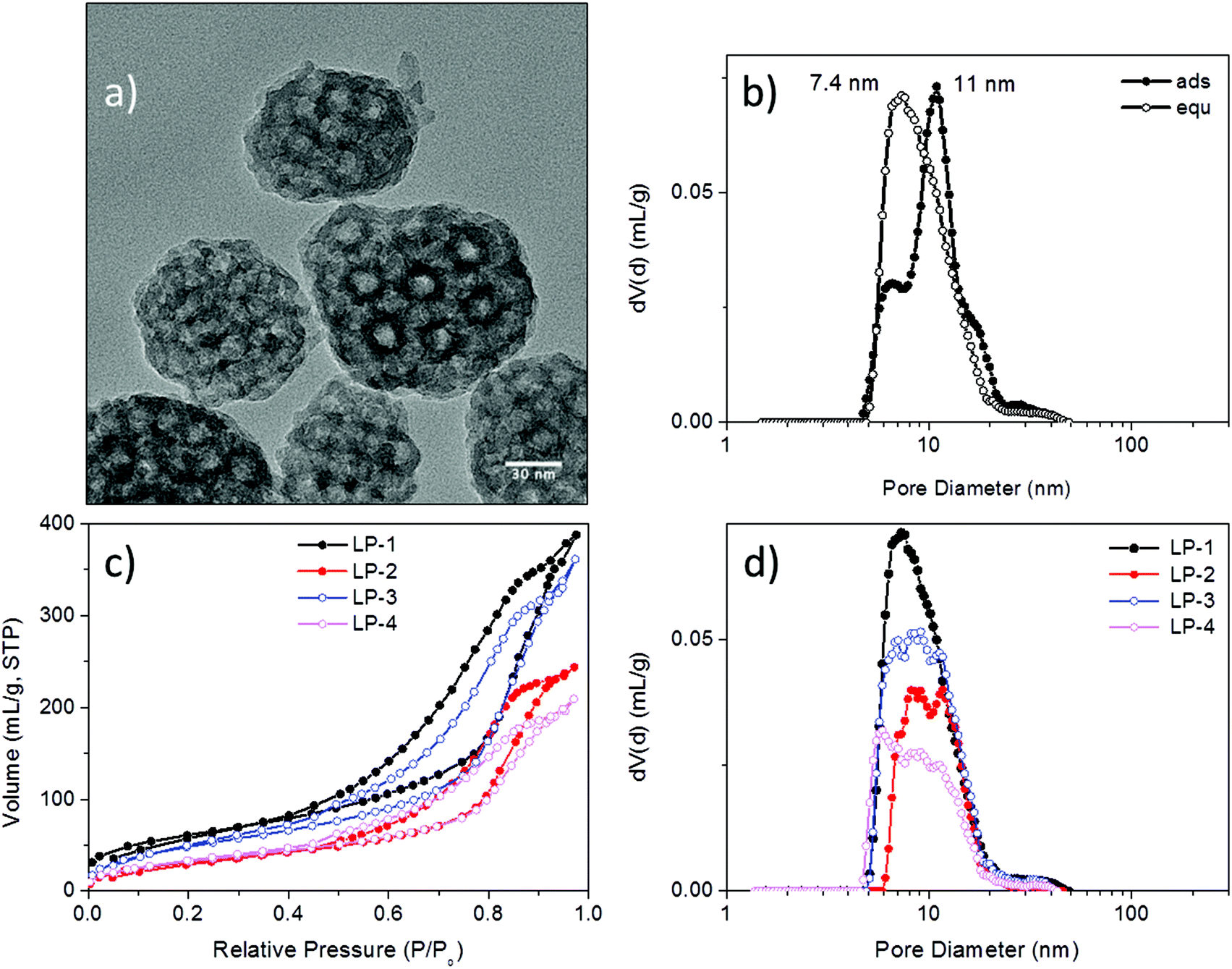 | ||
| Fig. 1 Characterization of large-pore silica nanoparticles (LP-MSN). (a) Transmission electron micrograph, (b) pore-size distribution of sample LP-1 derived from the NLDFT adsorption branch model and NLDFT equilibrium model of the respective nitrogen sorption isotherm, (c) nitrogen sorption isotherms derived from parent siliceous sample LP-1 and samples LP-2 to LP-4 after grafting of different amounts of PhTES and APTES (see Table 1), and (d) corresponding pore-size distributions derived from the NLDFT equilibrium kernel for cylindrical pores. | ||
Due to their large pore size and corresponding wall thickness, the parent LP-MSNs show a relatively small surface area of 220 m2 g−1, which is further reduced to 120 m2 g−1 after grafting with increasing amounts of phenyl and amino residues. The pore volume is concomitantly reduced as shown in the associated pore-size distributions (Fig. 1d).
In addition to LP-MSN, we created MP-MSN nanoparticles with a pore size sufficient for small oligonucleotide adsorption and with orthogonal surface functionalities that drive the siRNA into their inner void volume. For this purpose, a core–shell co-condensation reaction was performed that results in a positively charged interior lining as well as a negatively charged outer shell. Medium-sized pores were obtained by using a modification of our previously established synthesis route.31–33 In brief, a preheated solution containing cetyltrimethylammonium chloride (CTAC) as template was enriched by the pore-expanding agent triisopropylbenzene (TiPB) and mixed with a preheated solution containing tetraethoxysilane (TEOS) and APTES at elevated temperature for 20 minutes, followed by addition of the ingredients for the second particle layer (containing TEOS and mercaptopropylsilane (MPTES)). The amounts of the silane coupling agents were varied to achieve different relative surface concentrations of amino- and mercapto-groups (for further details see Table 1 and ESI†).
| Sample | Compositiona (mol%) | Surface area (m2 g−1) | Pore size (cavity/pore) (nm) | Pore volume at 0.8 p/p0 (mL g−1) | Particle size (nm) (TEM) | IEP (pH) | NH2 (mmol g−1) | NH2 (μmol m−2) |
|---|---|---|---|---|---|---|---|---|
| a Determined from elemental analysis. | ||||||||
| LP-1 | Pure silica | 220 | 11/7.4 | 0.60 | 70–170 | 4.0 | ||
| LP-2 | 10.7% NH2 | 120 | 8–12/9.9 | 0.38 | 10.1 | 1.84 | 15 | |
| LP-3 | 3.5% Ph | 186 | 7–12/9.8 | 0.56 | 9.9 | 1.39 | 7.5 | |
| 8% NH2 | ||||||||
| LP-4 | 6.6% Ph | 123 | 6–13/9.6 | 0.32 | 8.6 | 0.67 | 5.5 | |
| 3.9% NH2 | ||||||||
| LP-5 | 1.1% Ph | 294 | 7–13/10 | 0.34 | 60–120 | 7.7 | 0.23 | 0.8 |
| 1.4% NH2 | ||||||||
| LP-6 | 4.6% Ph | 437 | 10/8 | 0.60 | 90–200 | 6.4 | 0.09 | 0.2 |
| 0.5% NH2 | ||||||||
| MP-1 | 8.5% NH2 | 670 | 4.7 | 0.75 | 150 | 5.9 | 1.44 | 2.2 |
| 1.3% SH | ||||||||
| MP-2 | 5.7% NH2 | 694 | 3.9 | 0.66 | 150 | 6.3 | 0.94 | 1.45 |
| 1.2% SH | ||||||||
| MP-3 | 3.3% NH2 | 937 | 4.0 | 0.85 | 150 | 4.8 | 0.52 | 0.6 |
| 3.7% SH | ||||||||
Nanosized MP-MSN particles of around 150 nm were obtained as shown in the transmission electron micrographs in Fig. 2 for the different particle compositions. These range from around 9 to 3 mol% amino groups (of the combined silica precursors) in the core and from around 1 to 4 mol% mercapto groups in the shell (see also Table 1). Surface areas varying between about 900 and 700 m2 g−1 reflect the different concentrations of functional groups present, with the smaller amount of aminopropyl groups in sample MP-3 resulting in a higher surface area as well as a larger pore volume. A comparable pore size of 4 nm was obtained in sample MP-3 and MP-2 despite changes in composition, while a pore-widening to 4.7 nm was achieved in sample MP-1 by changes in sample treatment before template extraction (see Experimental part). The pore morphology can be described as an ordered stellate arrangement with conically widening mesopores, with pore-size distributions derived from the adsorption and desorption branches being nearly identical.
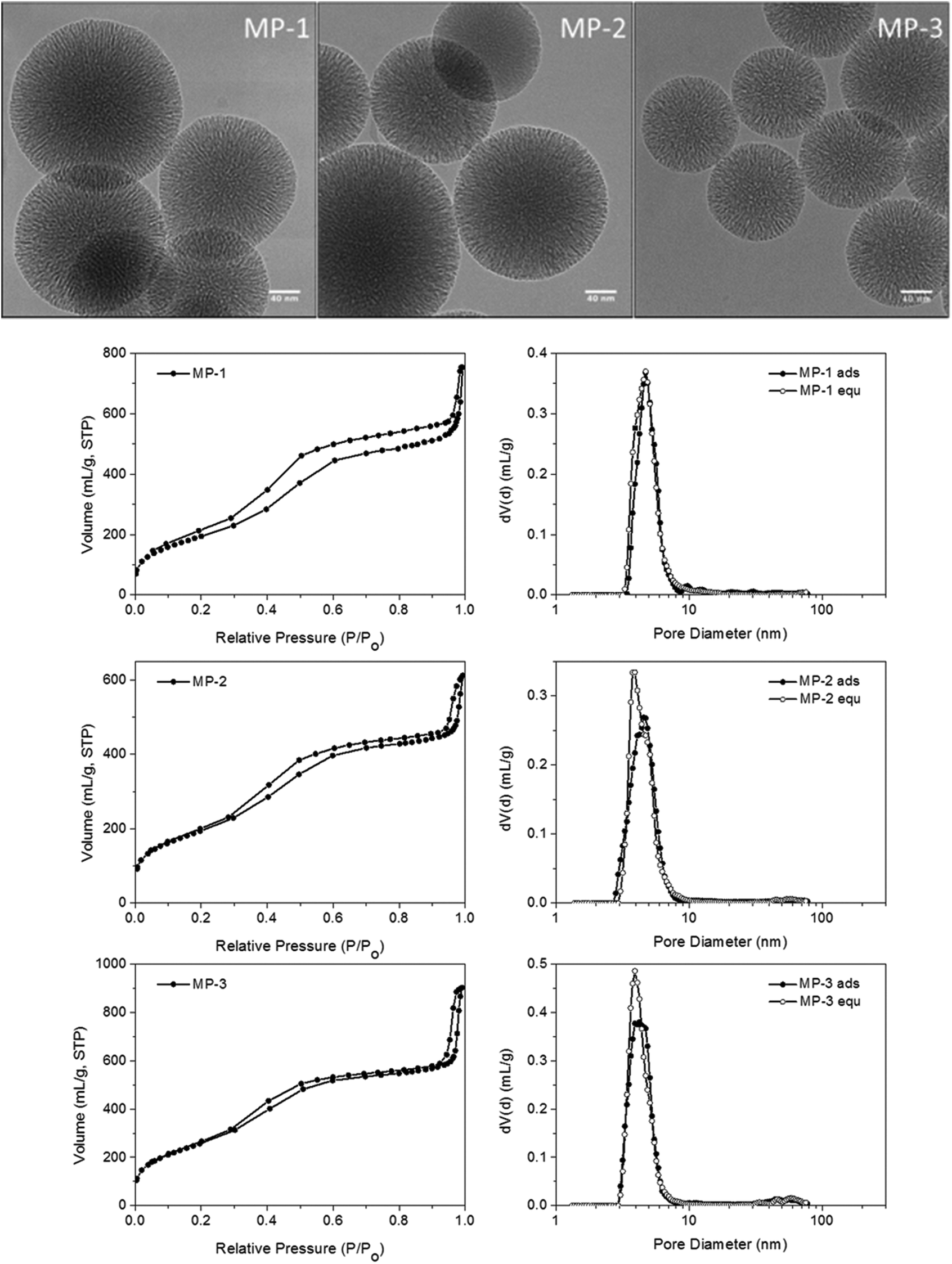 | ||
| Fig. 2 Transmission electron micrographs of core–shell samples MP-1 to MP-3 and the corresponding nitrogen sorption isotherms with pore-size distribution curves derived from the NLDFT equilibrium kernel for cylindrical pores (the NLDFT adsorption branch model and NLDFT equilibrium model superimposed). | ||
siRNA adsorption and release studies with LP-MSN
A prerequisite for any efficient siRNA delivery system is a sufficient loading capacity and high desorption yield, as well as an adequate rate of release of the cargo under conditions that resemble cellular environments. To investigate the behavior of our different LP-MSN and MP-MSN carrier systems, we undertook siRNA adsorption and desorption studies under varying conditions.It has been assumed that the relatively large siRNA oligonucleotides require a minimum pore size of 5 nm or more for unhindered diffusion into the particle interior.15 Furthermore, it is recognized that the negative charge of siRNA prohibits its inclusion into siliceous materials that are negatively charged at neutral pH, and that a surface decoration with positively charged groups is necessary. In order to introduce surface amino groups into LP-MSN we performed a post-synthetic grafting with APTES since a direct cocondensation of more than 1 mol% APTES disrupts the silica network under the acidic reaction conditions. We used APTES in combination with PhTES as additional shielding against silanol surface groups. This way we were able to change the surface properties of LP-MSN systematically, as reflected in the zeta potential curves in Fig. 3a. The isoelectric point (IEP) shifts over the whole range between pH 4 to pH 10. We included our purely siliceous LP-MSN parent particles for comparison (LP-1). These siliceous mesoporous particles have an isoelectric point around 4, thus at neutral pH they feature a negative charge of −10 to −20 mV that prevented any significant adsorption of siRNA.
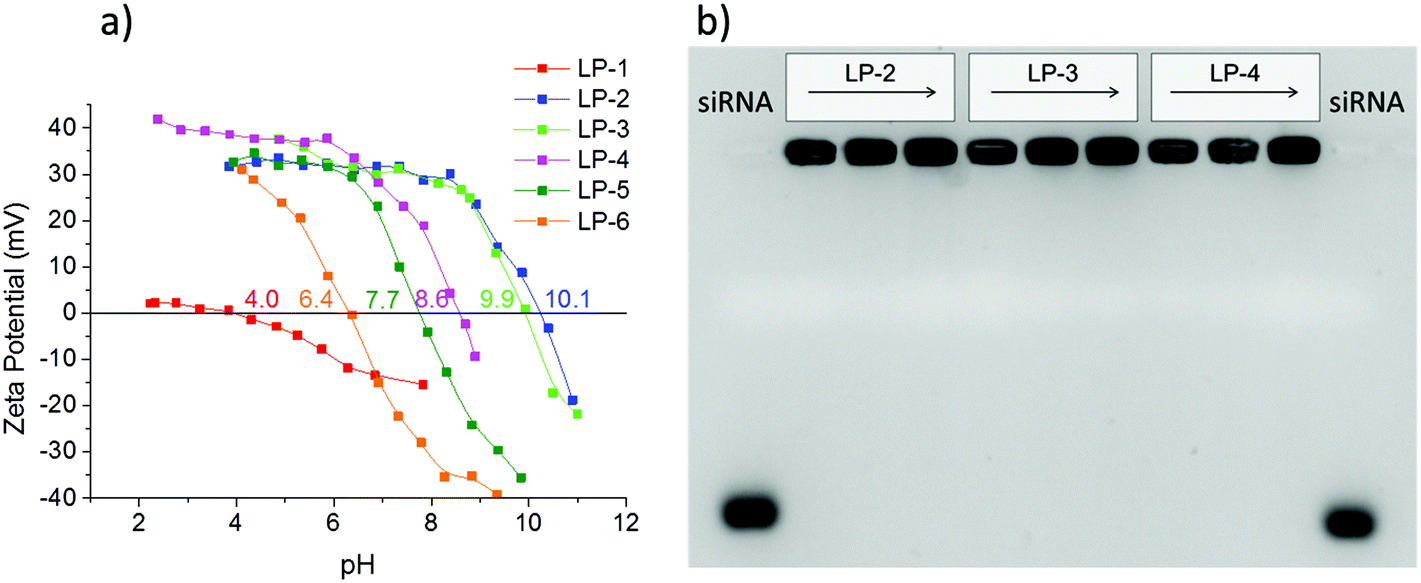 | ||
| Fig. 3 (a) Zeta potential measurements of LP-MSN with varying surface functionalization, (b) gel electrophoresis measurements with samples LP-2, LP-3 and LP-4 loaded with either 2.5, 6.6, or 12 μg siRNA per mg MSN (increasing concentration from left to right). | ||
When 0.5 mol% amino groups were included into the internal particle surface in addition to 2.5 mol% phenyl groups, the IEP was shifted from 4.0 to 6.4 (LP-6). However, siRNA adsorption was only minimal. For any significant amount of adsorption, a 1/1 mol% amino/phenyl content was necessary, having an IEP of 7.7 (LP-5). Higher siRNA loadings were achieved with amino-group contents exceeding 1 mol% throughout the particle body. Increasing the APTES content from about 4 to 11 mol% in LP-4 to LP-2 we obtained particles with very high IEP's and thus high positive zeta potentials at pH = 7 between 30 and 40 mV. These surface charges were sufficient for an uptake of siRNA between 32 to 40 μg siRNA per mg MSN (see ESI Fig. S1†). Surprisingly, when the desorption process was studied by exchanging the solution for a fresh aqueous solution, nearly no elution was observed even after several hours under stirring. However, offering a cytosol-simulating PBS buffer solution at pH 7.4, the release was slightly improved to about 5% RNA after 1 h, or 23% after 1 day (fraction of loaded RNA released; see ESI Fig. S1†). The tight binding of siRNA in these samples is reflected in gel shift results (Fig. 3b). Here, samples LP-2, 3 and 4 were each loaded with 2.5, 6.6 or 12 μg siRNA per mg MSN. The siRNA was retained tightly in all samples even without the presence of any pore-capping systems on the particles. No siRNA elution is visible when 100 V was applied for 1.5 h to the gel.
Similar difficulties regarding the release of pDNA from comparable particles were encountered already by Gao et al.15 who used a 2 M NaCl solution at 50 °C to free the DNA, conditions that are clearly not compatible with cell experiments. Only one other research group used these bottleneck LP-MSN and modified them with either APTES or by covalently attaching polylysine to the surface. This way, Hartono et al. increased the loading capacities for RNA-mimicking oligo-DNA to 57 μg mg−1. Nevertheless, when applied in cell experiments with functional siRNA, only low knockdown efficacies of 15 or 30% were reached when using very high siRNA concentrations of 100 nM.34 In follow-up studies they adsorbed the endosomolytic agent chloroquine into the pores while siRNA was attached via poly(2-dimethylaminoethyl methacrylate) (PDMAEMA) on the outside of the silica particles.19 Good efficacies were still only reached when the LP-MSNs were equipped with covalently bound PEI instead of PDMAEMA.35 In light of these collective findings we suggest that not only the pore size and surface charge are important in determining the loading capacity, but that the pore morphology might play a pivotal role in efficient siRNA release. Nitrogen sorption data of LP-MSN show a bottleneck-type pore morphology that features smaller openings than pore diameters, which might compromise an efficient diffusion of the highly charged molecules. These examples also show the importance of studying the siRNA elution process as a basis for choosing the appropriate hosts for siRNA.
siRNA adsorption and release studies with MP-MSN
For reasons discussed in the previous section, we developed and evaluated stellate-morphology medium-pore (MP-MSN) samples for loading and release of siRNA. We offered siRNA at different concentrations to aliquots of MP-MSN samples and followed the uptake after 15 minutes and 1 h by measuring the remaining siRNA concentrations in the supernatants. Similarly, we eluted the adsorbed RNA by removing the supernatant and exchanging the solution by equal amounts of PBS buffer solution at pH = 7.4. Fig. 4 compares results obtained with samples MP-1 and MP-2 having slightly different pore sizes of 4.7 and 3.9 nm, respectively.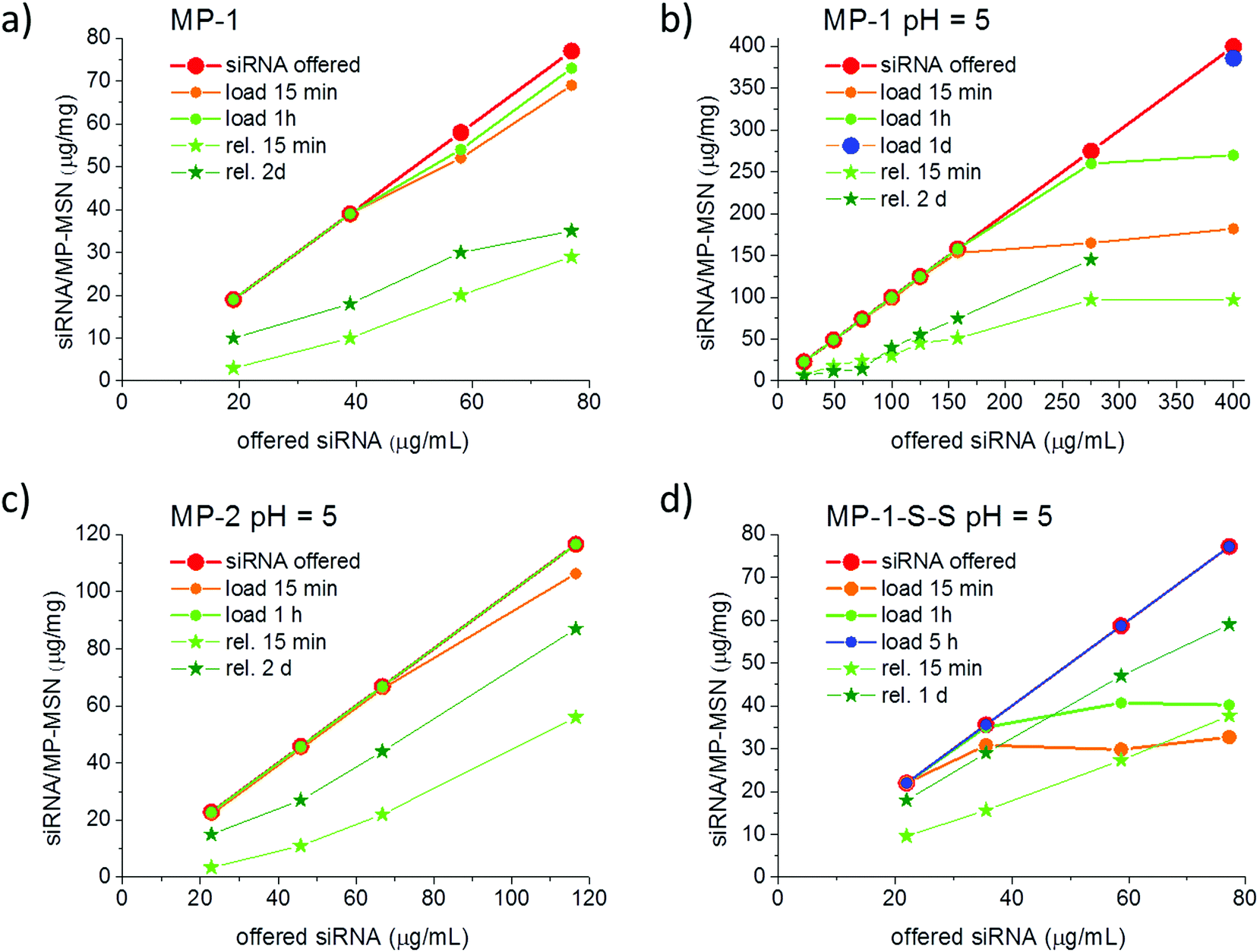 | ||
| Fig. 4 Kinetic development of siRNA uptake and release with stellate MP-MSN. Aliquots of 100 μg of MSN were exposed to 100 μL of siRNA solutions with increasing concentrations. (a) Uptake in MP-1 from water, and (b–d) uptake from MES buffer at pH = 5 into samples (b) MP-1, (c) MP-2, and (d) MP-1-S-S. Release experiments were performed in 100 μL PBS buffer at pH = 7.4 in all samples. The red curve indicates the offered siRNA amount, and orange and green dots indicate the adsorbed siRNA after 15 minutes and 1 h, respectively. Light green and dark green stars are the cumulatively released amounts of RNA after 15 min and 2 days, respectively. | ||
The siRNA amount offered is indicated by the red graph in Fig. 4, and the actual siRNA uptake after 15 minutes and after 1 h was color-coded with orange and green dots, respectively. The eluted siRNA amounts are depicted in green colored stars after the indicated time. It is evident from the data in Fig. 4a that in sample MP-1 with pores of only 4.7 nm diameter a rapid uptake of siRNA within 15 min takes place up to an siRNA loading concentration of 40 μg siRNA per mg when the siRNA is offered in an aqueous solution. Even larger amounts to at least 78 μg mg−1 are rapidly adsorbed after 1 hour of exposure. When siRNA is offered in an MES-buffer solution at pH = 5 (Fig. 4b), the uptake after 15 min can be increased to 150 μg RNA per mg MSN. At very high siRNA concentrations, the adsorption slows down. However, even when 275 or 400 μg per mL RNA is offered as loading solution, a nearly complete uptake occurs after 1 h exposure in the former case, while a very high loading of 386 μg mg−1 is reached when measured after 1 day. To our knowledge, these are the highest amounts of siRNA that have been included in such narrow-pore nanoparticles. A rapid and efficient adsorption takes also place in the sample MP-2 with narrower pores of only 4 nm, which when exposed up to 120 μg per mL siRNA solution showed a complete adsorption after 1 hour (comparative adsorption isotherms are shown in the ESI, Fig. S5†).
Since an equally important feature for later cell experiments is the efficient release of siRNA, we studied these samples after stirring in a PBS buffer solution at pH = 7.4 and followed the elution in time. As shown with the green-starred graphs in Fig. 4, the sample MP-1 eluted between 16 and 40% of the adsorbed siRNA within 15 min and up to 55% after 2 days. In contrast, the narrow-pore sample MP-2 eluted a remarkably higher content of siRNA after 2 days amounting to between 66 and 75%, depending on the siRNA loading.
To also achieve a higher release of siRNA in sample MP-1, we coupled amino groups in MP-1 via a redox-reactive disulfide bridge to the particle body. Such a procedure was proposed by Zhang et al., who argued that a stimuli-responsive coverage of the inner and outer surface could result in a triggered and thus more efficient release of small oligonucleotides.36 They transformed the amino-covered surfaces of MP-MSN into disulfide-coupled amino groups that are then cleavable under the reductive action of glutathione (GSH), which is present in concentrations of up to 10 mM in the cytosol. They observed the highest release of oligo DNA under the action of GSH with particles having 4.5 nm pores. We adapted their procedure in order to compare the release behavior and finally the transfection efficiency between our samples with and without disulfide-coupled amino groups. Thus, in a first step surface amino groups were converted via succinic anhydride coupling into carboxy groups. These were subsequently reacted with cystamine catalyzed by the EDC/NHS-sulfo reaction to result in amino-terminated cleavable residues (sample MP-1-S-S). The progress of this reaction was confirmed by FTIR spectroscopy and zeta potential measurements, as shown in the ESI (see ESI, Fig. S2†).
siRNA adsorption into MP-1-S-S turned out to be markedly slower than in the purely co-condensed sample MP-1, most likely due to the pore size reduction caused by the long residual linker. In this case, instead of 1 hour it took 5 hours for a complete uptake of the administered siRNA when measured with concentrations of up to 80 μg mL−1 (see Fig. 4d). On the other hand, the desorption was more efficient, showing a release of 43–49% already after 15 minutes and between 76 and 81% after 1 day (depending on siRNA loading). Notably, no reducing agent was used here for the release.
We believe that the observed differences in diffusivity are controlled to a large part by electrostatic interactions between the charged pore walls and the siRNA. The higher the concentration of protonated amino groups on the internal surface the more efficient and faster is the adsorption process. Purely siliceous mesoporous silica is not able to adsorb siRNA, and neither did we observe any significant uptake when the surface in LP-MSNs was covered with cyano, glycidoxy or with mercapto groups. More than 1 mol% amino groups (of the silica precursor) are necessary for any significant uptake of siRNA. Additionally, pore size and pore morphology influence the distribution of short oligonucleotides with respect to the pore walls. In LP-MSN a side-by side adsorption of RNA molecules may be feasible by the large size of the entrance pores. In contrast, the pore-size in MP-MSNs is close to the hydrodynamic radius of siRNA and a single-file diffusion is more likely to occur. Reducing the pore-size in these samples brings the siRNA molecules into a closer contact with the pore surface and its positive charge so that the progressing adsorption is slowed down. This might explain the slower diffusion in sample MP-2, which has a pore-size reduced by 0.8 nm compared to MP-1, the latter showing a complete uptake already after 15 min even at a high RNA concentration of 150 μg mL−1. However, when the pore size in this sample is reduced by extending the size of the functional groups (sample MP-1-S-S), the uptake of siRNA is again slowed down.
Considering the desorption process, a wide pore size allowing for facile accommodation of the large guest molecules does not guarantee efficient release kinetics. The pore morphology of the large-pore samples required a high amino-group density in order to achieve substantial siRNA adsorption. This in turn favors a tight binding of the molecules, inhibiting an efficient release in these samples. In contrast, a tighter fit of the surrounding pore surface with the highly negatively charged phosphate groups of the double stranded siRNA and a lower amino-group-density in the straight channels of MP-MSN samples is apparently advantageous. Desorption is favored when the internal surface charge of the MSN is reduced, such as in sample MP-3. In this case, with only 4 mol% amino groups in the sample core we observe fast and efficient desorption of 60 to 70% of the adsorbed siRNA already after 15 minutes (see ESI, Fig. S3a†).
Cell transfection and GFP-luciferase knockdown
Since a very high siRNA uptake and efficient release behavior was established in this work, we chose samples MP-1-S-S and MP-1 as candidates for comparative siRNA delivery studies with KB-cells. To reduce untimely elution of siRNA and simultaneously add endosomal escape functionality to our system, we combined the siRNA-loaded MP-MSN particles with a modularly designed block copolymer. This polymer 454 has been used previously for siRNA polyplex formation.28 It is composed of multiple succinoyl tetraethylene pentamine (stp) units that carry three basic amino groups, while the terminal amino and carboxy groups are used for coupling to additional lysine (K) or tyrosine (Y) units. The latter are further terminated with cysteine modules (C) that potentially can undergo disulfide bridging through their mercapto groups or could be used for adding other functionalities such as targeting modules. Lysine groups are used as branching points for the attachment of oleic acid segments. The latter are included for establishing pH-dependent endosomolytic properties that are most pronounced at decreasing pH as has been shown before37 (see Scheme 1). This feature is especially important since most of the delivering vectors are taken up via endocytosis. However, if a timely escape of the RNA cargo from the endosome cannot be established degradation sets in and the delivery will fail.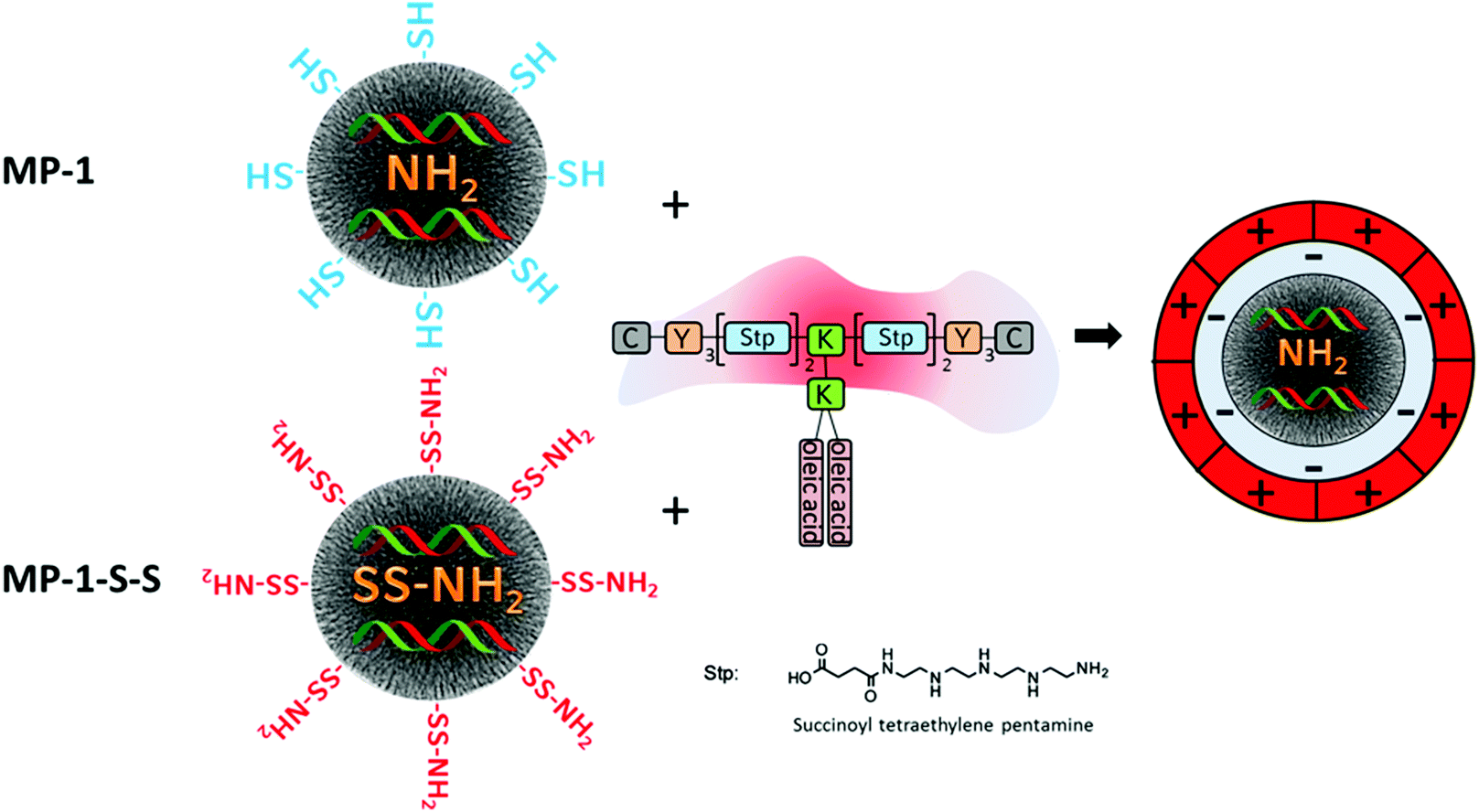 | ||
| Scheme 1 Construction of an siRNA delivery platform by combining multifunctional core–shell nanoparticles with modularly designed block copolymer 454. We show medium-pore MSN particles MP-1 lined internally with amino groups and externally with mercapto groups, and sample MP-1-S-S where all functional groups were transformed into disulfide-bridged amino groups. Particles are loaded with siRNA and capped with the block-copolymer that covers the final particles with a positively charged layer (chemical moieties depicted in this scheme are abbreviated for clarity; C: cysteine, Y: tyrosine, K: lysine). | ||
Attachment of this cationic polymer to the siRNA-loaded MSN particles occurs likely through electrostatic interactions with sample MP-1 as well as in MP-1-S-S, even though the surface layer is different in both cases. In MP-1-S-S, the originally mercapto-terminated outer surface is transformed into amino groups through reaction with the cystamine molecules. This was established by Raman spectroscopy and zeta potential measurements and is detailed in the ESI in Fig. S4.† Thus, in this sample some of the adsorbed siRNA is likely bound to the external surface, rendering it negatively charged again. Coulomb interactions with the cationic polymer are thus possible in both samples, and binding of the polymer is confirmed by zeta potential measurements (see ESI; Fig. S6†). Initially, the IEP in sample MP-1 was relatively high (5.9) since the high loading with amino groups was not completely screened by the 1 mol% terminating mercapto groups in the particle shell. This effect was partially neutralized after siRNA adsorption and the IEP shifted to a lower value of 5.1. However, upon exposure to the cationic polymer the surface charge increased again and the IEP reached a value of 6.4. This is still much lower than the IEP of 7.6 of the pure polymer, confirming its binding to the particle surface (zeta potential of MP-1 at pH = 7 is −29 mV, −24 mV after siRNA sorption and −11 mV after polymer capping, respectively).
Cell transfection with these siRNA delivery systems was next verified with confocal microscopy, shown in Fig. 5. Wild-type KB cells were first incubated for 45 minutes with sample MP-1 and unbound particles were removed by a medium exchange. Microscopy was then performed after 24 h and 48 h. To visualize the internalization of siRNA, we labeled 20% of the siRNA with the dye CY5 before adsorbing it into the carrier MP-MSN. A significant uptake of siRNA is observed in the green stained KB cells at both times (see top row in Fig. 5). To track the location of the siRNA within the MSN particles, we labeled the MP-MSN carrier to some degree with NHS-ATTO-488 before adsorbing the partially labeled siRNA. A Z-stack maximum projection is shown in the middle row in Fig. 5 illustrating the large uptake of MSN particles in the cells. The green cell membrane staining was omitted here to optimize the visibility of the green MSN particles and the red siRNA. Significant co-localization between MSN carrier and siRNA is visible both after 24 h and 48 h. We note that the released fraction of siRNA is not observed due to dilution effects. In the bottom row of Fig. 5 we show micrographs in which the particles were labeled green again with NHS-ATTO-488, but now part of the polymer was labeled with Mal-ATTO-633 to confirm the sustained attachment of the polymer to the particle surface. This is confirmed through the yellow (additive) coloring.
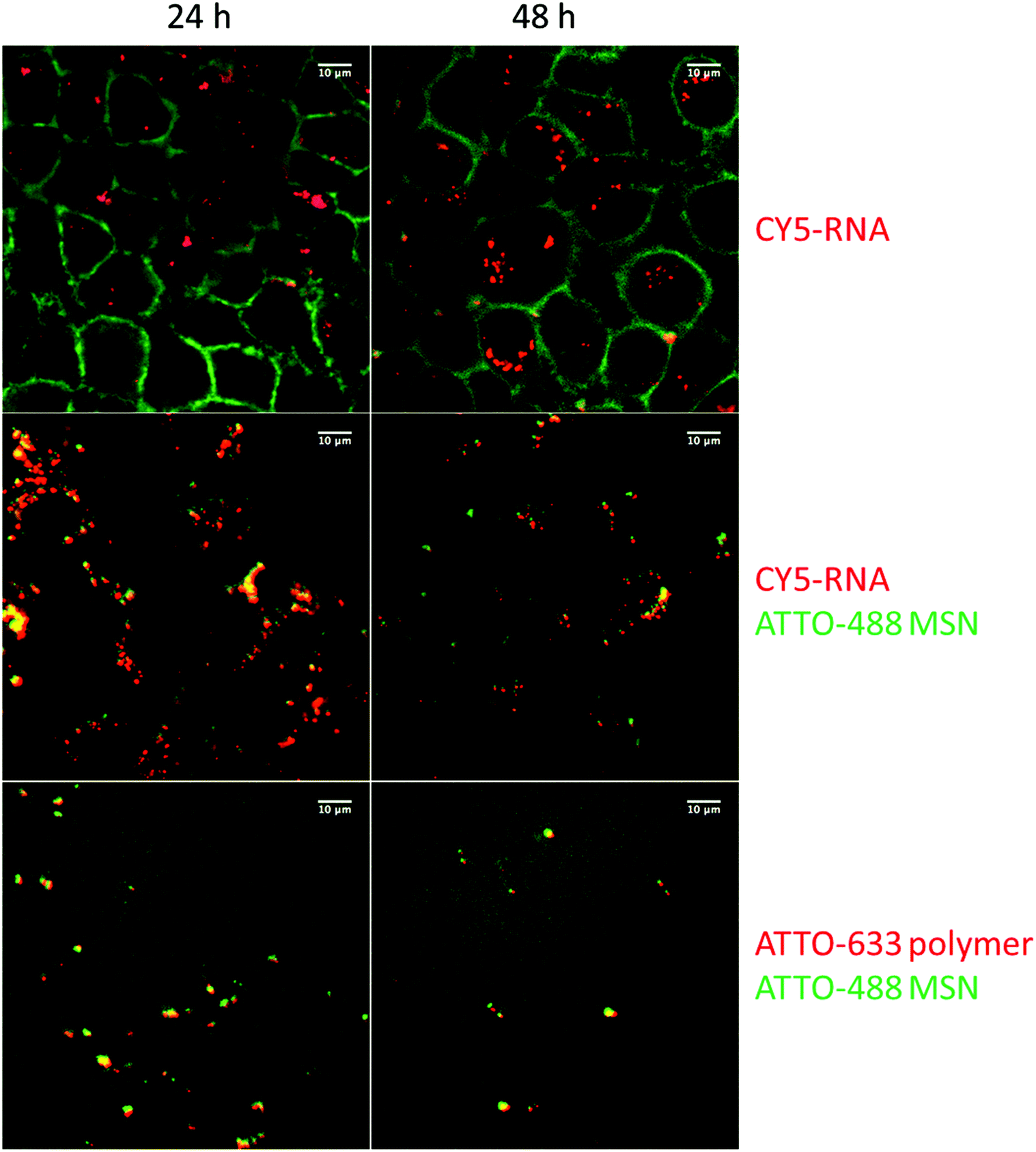 | ||
| Fig. 5 Confocal microscopy of the transfection of sample MP-1 in KB cells. Left column: after 24 h incubation, right column: after 48 h incubation. Top row: particles with partially CY5-labeled siRNA and green stained KB cells. Middle row: Z-stack maximum projection; NHS-ATTO-488 stained MP-MSN (green) loaded with partially CY5-labeled siRNA (red). Bottom: NHS-ATTO-488 stained MP-MSN (green) loaded with unlabeled siRNA and partially Mal-ATTO-633 labeled 454 polymer (red). | ||
Further cell experiments were undertaken to study the biological activity of the MP-MSN carrier system. If not stated differently, we used the same siRNA loading of 5 wt% siRNA for this purpose as for the cell microscopy studies. First, the complete uptake of siRNA into the MP-MSN host was confirmed by measuring the supernatant solution, then samples were redispersed and the block copolymer was added at increasing amounts to evaluate the best transfection conditions. After incubating the polymer-containing samples for one hour at 37 °C under shaking, samples were centrifuged in order to replace the supernatant with a PBS buffer solution and to remove any surplus block copolymer or free siRNA. Cells were usually exposed to 10 μg MSN per well; each well had a volume of 100 μL. Each well thus contained 0.5 μg siRNA. This siRNA concentration was kept constant in all experiments. Gel electrophoresis was performed with the same samples that were used for cell transfection.
In Fig. 6 we show the results for sample MP-1-S-S, which was prepared using increasing concentrations of polymer/MP-MSN samples with a w/w ratio ranging from 1/100 to 1.5/1.
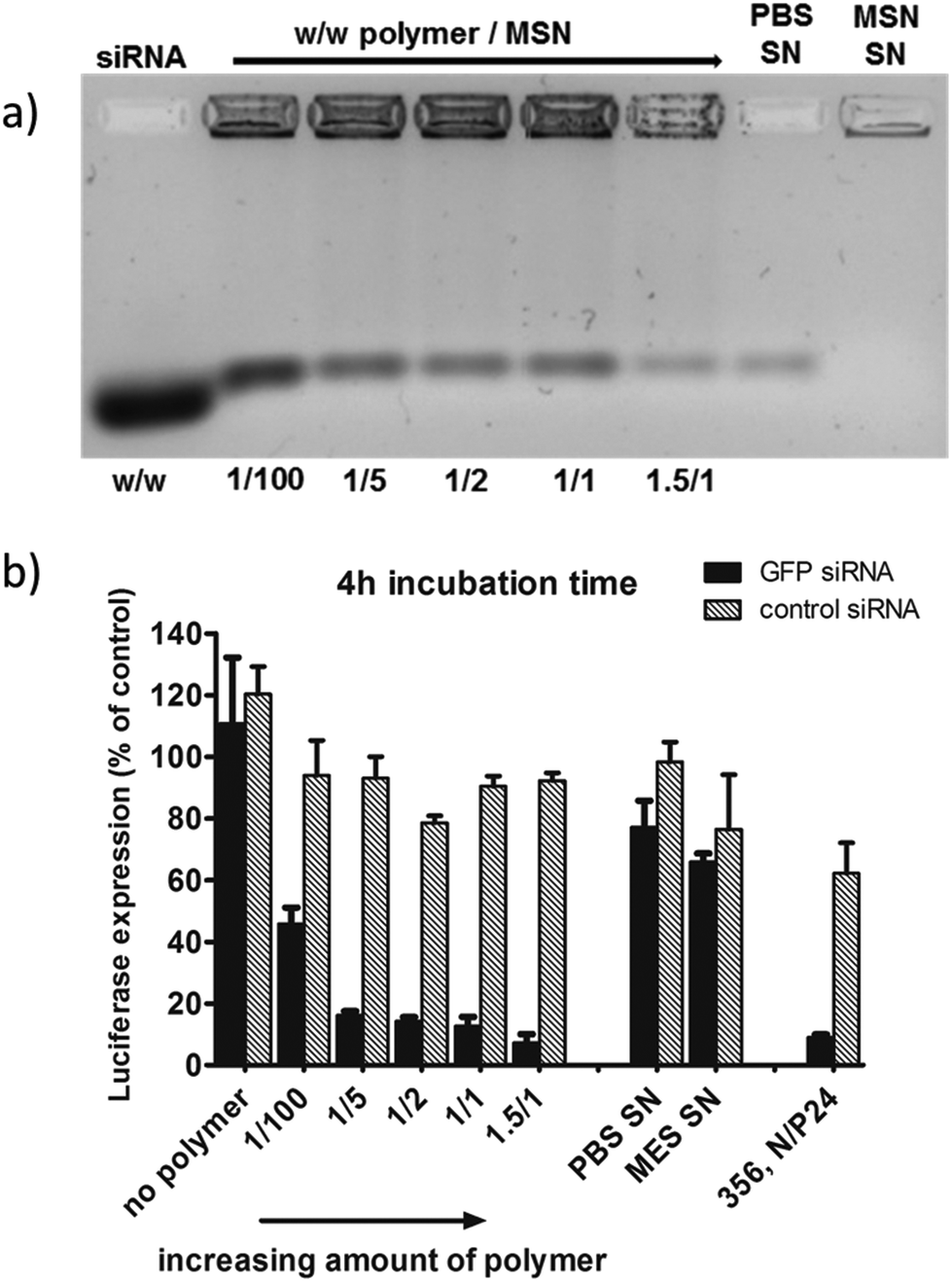 | ||
Fig. 6 (a) Gel electrophoresis of sample MP-1-S-S (5 wt% siRNA loading in the MSN) with an increasing amount of block copolymer ranging from 1/100 up to 1.5/1 w/w polymer to particle. The first and the second supernatant solutions (SN) after sample preparation are included. (b) The corresponding KB/eGFPLuc cell transfections after a particle incubation time of 4 h on cells, luciferase read out after 48 h. A sample without polymer capping and a positive control consisting of a block copolymer 356![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 siRNA polyplex formulation are also included for comparison. 27 siRNA polyplex formulation are also included for comparison. | ||
In the gel-electrophoresis experiments (Fig. 6a) we compared the release of siRNA from the MP-MSN particles as a function of polymer concentration. It can be seen that all samples do retain large quantities of siRNA, but that some minor elution of siRNA is observed, decreasing with increasing polymer content. Electrophoresis showed that the supernatants of the MES loading solution (MSN-SN) and the first wash in PBS buffer (PBS-SN) contain only negligible amounts of siRNA.
The above samples were used in experiments with KB/eGFPLuc cells which stably express a GFP-luciferase fusion reporter gene. Cells were incubated for 4 hours with samples containing either GFP siRNA (black bars, for GFP-luciferase silencing) or irrelevant control siRNA (hatched bars, no gene silencing) before they were replaced with fresh medium. The luciferase expression was measured after an additional 48 h and its activity is depicted in Fig. 6b. A striking concentration-dependent down-regulation can be observed, starting with an efficacy of almost 60% with the smallest polymer addition, up to over 90% knock-down with the highest polymer addition. Importantly, we note that without the polymer coating, no significant knockdown activity was observed. Between a polymer/MSN weight ratio of 1/5 and 1.5/1 the differences are only small, and for further experiments we used a 1/2 w/w ratio. We note that the supernatant solutions were also analyzed (with the nanodrop protein 280 routine), where significant quantities of the polymer were found that did not bind to the MSN particles. We included the supernatant solution in the cell experiments and observed that only a minor knockdown of 35% occurs (MES SN), and even less with the second PBS washing solution (PBS SN) containing small amounts of free siRNA as seen in the gel electrophoresis. In our experiments we also included a positive control. It is a folate-containing polyplex formulation consisting of the block copolymer 356 and either siGFP-RNA modified with the lytic peptide Inf7 or its control sequence Inf7-siCtrl. This 356 siRNA polyplex was previously shown to be highly efficient in siRNA delivery27 (356; structural composition see ESI, Scheme 1†). These results clearly show that the high knockdown efficacy is brought about by a concerted action of the endocytosed MP-MSN particles releasing their siRNA cargo and the endosomolytic activity of the attached block copolymer.
These results were obtained with an siRNA-loading of only 5 wt%, and the question arises if we can take advantage of the high loading capacity of our MSNs. In Fig. 7a we show cell transfection experiments where we used sample MP-1 and increased the loading concentration of RNA from 5 wt% up to 20 wt%. This way we could decrease the amount of MSN particles administered to the cells while keeping the total amount of siRNA at 0.5 μg per well. Here we used a short particle incubation time on cells of only 45 minutes. Luciferase activity was measured after 48 h as before. It can be seen that in all cases we still observe a very high efficiency that allows us to reduce the particle concentration from 100 μg mL−1 down to only 25 μg mL−1, which translates to 2.5 μg MSN per well. To our knowledge, these efficiencies are the highest that have been achieved with mesoporous silica carriers, since usually greater particle concentrations are needed to show a substantial knockdown.18,20,23 In order to investigate the limiting concentration of siRNA for good efficiency we further decreased the total amount of RNA per well (see Fig. 7b). Starting from 0.5 μg RNA per well applied with the highest loaded sample (20 wt%), we reduced the particle/RNA amount stepwise down to 0.063 μg per well. It is evident that 0.5 μg siRNA is the optimal concentration needed for a 80–90% knock-down efficiency.
 | ||
| Fig. 7 (a) KB/eGFPLuc cell transfections with sample MP-1 with increasing particle loadings of 5 wt%, 10 wt%, 13 wt% and 20 wt% siRNA, while keeping the total amount of siRNA at 0.5 μg per well. (b) Cell transfections with sample MP-1 with decreasing siRNA concentrations/well. Cell transfections after a short particle incubation time of only 45 min on cells, read out after 48 h. | ||
How versatile and robust is the above design concept? In the following we briefly discuss several examples where the general construction principle was varied. This relates (i) to the siRNA interactions with the interior of the MSNs, (ii) to the MSN pore size, and (iii) to the nature of the polymer coat on the MSN.
Firstly, considering siRNA–MSN interactions, we ask about the importance of internal MSN charge density. Here, sample MP-3-S-S having a lower concentration of amino groups (4 mol%) was used in combination with the block copolymer. Despite the high siRNA loading capacity (see ESI, S3a†), the weaker electrostatic interactions with the host lead to a more facile extraction of the siRNA from the pore system as soon as the polymer is added. Hence, these samples cannot be effective transfection agents (see ESI, S3b†). How important is the disulfide-bridged attachment of the amino groups for the siRNA functionality? In Fig. 7 we show the luciferase knockdown when siRNA is administered via sample MP-1, which does not contain disulfide coupled amino groups. When the particles are covered with the polymer, they again induce a successful knockdown of over 80%, showing that with the efficient MP-MSN hosts the additional reductive cleavage in the cytosol is not required for effective knockdown.
Secondly, we examined the impact of MSN pore size. Even with the smaller 3.9 nm pore size in sample MP-2, the same efficient luciferase knockdown was observed as with the larger pore system MP-1 having 4.7 nm pore size (see ESI, S7†). However, when we investigated the large-pore sample LP-2 we could only reach an efficiency of maximal 60% under comparable conditions (see ESI, Fig. S8†). We believe that this is related to the difference in pore and particle morphology. LP samples possess much smaller surface areas and thus high areal amino-group densities (see Table 1), which in turn reduce the driving force for RNA desorption. On the other hand, a high functionalization with amino groups is necessary to achieve reasonable RNA loadings. A maximum capacity of only 88 μg RNA per mg was found for LP-2 as compared to 380 μg RNA per mg MP-1. This renders the medium-pore samples even more preferable.
Third, we ask about the importance of the polymer capping system. We also examined the siRNA-delivery system with a standard cationic lipid by using DOTAP instead of the block copolymer. DOTAP adds a similar endosomolytic effect to our MP-MSN carrier system and shows again very high efficacies of over 80% (see Fig. 8 and ESI, Fig. S9†).
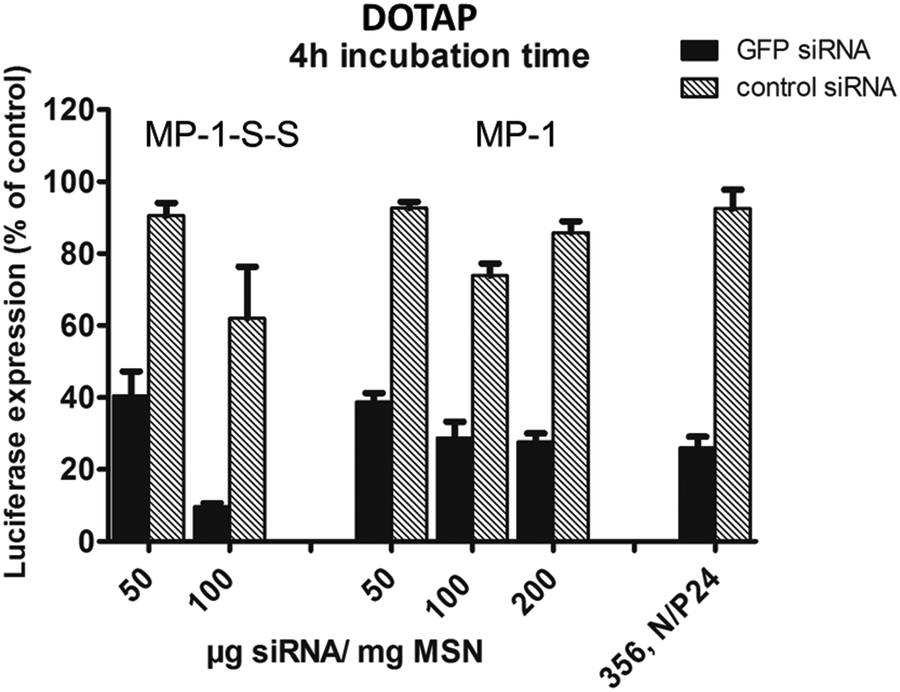 | ||
| Fig. 8 KB/eGFPLuc cell transfections performed with DOTAP-capped MSN samples. Comparison of MP-1-S-S and MP-1 with increasing particle loadings of 5 wt%, 10 wt% and 20 wt% siRNA, while keeping the total amount of siRNA at 0.5 μg per well. Cell transfections are shown after 4 h particle incubation time, read out after 48 h. | ||
While similar high efficacies could be achieved with this cationic lipid especially with sample MP-1-S-S, it does not offer the chemical versatility and robustness of our novel block copolymer capping agent. Through its functional groups, the latter offers numerous opportunities for future covalent modification with complementary functionalities aimed at effective drug delivery.
Forth, we evaluated the transfection efficiency of our MSN particles with the MCF-7 cell line, capped with DOTAP as well as with our novel modular polymer (see ESI, Fig. S10†). While the DOTAP experiment showed only minor silencing effects, we could achieve a knock-down of up to about 60% with our novel, highly loaded polymer capped MSN system.
Conclusion
We have introduced a new carrier system for siRNA by capping medium-size pore MSN particles with a modularly designed cationic block copolymer, resulting in excellent gene knockdown. For this purpose novel stellate core–shell mesoporous silica nanoparticles were introduced, which prove to be ideal hosts for siRNA due to their versatile surface properties. The possibility to selectively modify their internal and external surface chemistry allowed us to optimize the conditions for siRNA delivery. By tuning the internal surface charge as well as the pore size and morphology in the MSNs we demonstrate that the adsorption and desorption of siRNA is driven by electrostatic interactions and is optimized with medium-pore silica nanoparticles featuring a stellate pore morphology. A high internal surface concentration of amino groups proved to be critical for achieving extremely large siRNA loading capacities of up to 380 μg mg−1. A complementary external coverage with mercapto groups both prevents external siRNA adsorption and allows for favorable interactions with cationic polymers. In several examples we show that siRNA knockdown efficacies better than 80% can be achieved with a very low exposure of the cells to mesoporous silica (2.5 μg MSN per 100 μL well for only 45 minutes), however, only when the MP-MSN carriers are combined with cationic polymers. In summary, we have established a new, highly efficient, non-toxic (see ESI, Fig. S11†) platform for siRNA delivery.We assume that DOTAP as well as the block copolymer via its oleic acid units assist in transfection by destabilizing the endosomal/lysosomal membrane, thus allowing for the release of RNA into the cytosol. Here, the novel modularly designed block copolymers are preferable since they can serve as multifunctional agents. They are robust in handling, function as biocompatible capping agents and ensure endosomal escape, respectively. Additionally, they are adaptable to specific cell environments and targeting requirements through covalent modifications. Previously we could show that the 454 polymer28 as well as related MSN particles38 were well tolerated in vivo. Hence it is anticipated that MSN particles featuring optimized morphology and internal charge density in combination with such modular block copolymer capping agents offer numerous opportunities for future in vivo biomedical applications of RNA-based oligonucleotides.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for financial support (SFB 1032). Additional support is gratefully acknowledged from the Excellence Cluster Nanosystem Initiative Munich (NIM) and from the Center for NanoScience Munich (CeNS). We owe our thanks to Philipp Klein for supplying us with the block copolymers 454 and 356.References
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- V. Mamaeva, C. Sahlgren and M. Linden, Adv. Drug Delivery Rev., 2013, 65, 689–702 CrossRef CAS PubMed.
- N. Z. Knezevic and J.-O. Durand, Nanoscale, 2015, 7, 2199–2209 RSC.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. S. Au, AAPS J., 2010, 12, 492–503 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- M. S. Draz, B. A. Fang, P. Zhang, Z. Hu, S. Gu, K. C. Weng, J. W. Gray and F. F. Chen, Theranostics, 2014, 4, 872–892 CrossRef CAS PubMed.
- J. Shen, H.-C. Kim, D. Kirui, J. Mai, C. Mu, H. Su, L.-N. Ji, Z.-W. Mao, F. Wang, J. Wolfram and H. Shen, Theranostics, 2014, 4, 487–497 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS PubMed.
- M. Wang, X. Li, Y. Ma and H. Gu, Int. J. Pharm., 2013, 448, 51–57 CrossRef CAS PubMed.
- S. R. Bhattarai, E. Muthuswamy, A. Wani, M. Brichacek, A. L. Castaneda, S. L. Brock and D. Oupicky, Pharm. Res., 2010, 27, 2556–2568 CrossRef CAS PubMed.
- W. Ngamcherdtrakul, J. Morry, S. Gu, D. J. Castro, S. M. Goodyear, T. Sangvanich, M. M. Reda, R. Lee, S. A. Mihelic, B. L. Beckman, Z. Hu, J. W. Gray and W. Yantasee, Adv. Funct. Mater., 2015, 25, 2646–2659 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- F. Gao, P. Botella, A. Corma, J. Blesa and L. Dong, J. Phys. Chem. B, 2009, 113, 1796–1804 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, K. E. Epler, D. P. Padilla, G. K. Phillips, R. E. Castillo, D. C. Wilkinson, B. S. Wilkinson, C. A. Burgard, R. M. Kalinich, J. L. Townson, B. Chackerian, C. L. Willman, D. S. Peabody, W. Wharton and C. J. Brinker, ACS Nano, 2012, 6, 2174–2188 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- H.-K. Na, M.-H. Kim, K. Park, S.-R. Ryoo, K. E. Lee, H. Jeon, R. Ryoo, C. Hyeon and D.-H. Min, Small, 2012, 8, 1752–1761 CrossRef CAS PubMed.
- S. B. Hartono, N. T. Phuoc, M. Yu, Z. Jia, M. J. Monteiro, S. Qiao and C. Yu, J. Mater. Chem. B, 2014, 2, 718–726 RSC.
- X. Du, L. Xiong, S. Dai, F. Kleitz and S. Z. Qiao, Adv. Funct. Mater., 2014, 24, 7627–7637 CrossRef CAS.
- D. Lin, Q. Cheng, Q. Jiang, Y. Huang, Z. Yang, S. Han, Y. Zhao, S. Guo, Z. Liang and A. Dong, Nanoscale, 2013, 5, 4291–4301 RSC.
- X. Li, Q. R. Xie, J. Zhang, W. Xia and H. Gu, Biomaterials, 2011, 32, 9546–9556 CrossRef CAS PubMed.
- X. Li, Y. Chen, M. Wang, Y. Ma, W. Xia and H. Gu, Biomaterials, 2013, 34, 1391–1401 CrossRef CAS PubMed.
- Y. Chen, D. S.-Z. Zhang, H. Gu, X. Wang, T. Liu, Y. Wang and W. Di, Int. J. Nanomedicine, 2015, 10, 2579–2594 CAS.
- S. A. Mackowiak, A. Schmidt, V. Weiss, C. Argyo, C. von Schirnding, T. Bein and C. Bräuchle, Nano Lett., 2013, 13, 2576–2583 CrossRef CAS PubMed.
- S. H. van Rijt, D. A. Bölükbas, C. Argyo, S. Datz, M. Lindner, O. Eickelberg, M. Königshoff, T. Bein and S. Meiners, ACS Nano, 2015, 9, 2377–2389 CrossRef CAS PubMed.
- C. Dohmen, D. Edinger, T. Fröhlich, L. Schreiner, U. Lächelt, C. Troiber, J. Rädler, P. Hadwiger, H.-P. Vornlocher and E. Wagner, ACS Nano, 2012, 6, 5198–5208 CrossRef CAS PubMed.
- C. Troiber, D. Edinger, P. Kos, L. Schreiner, R. Kläger, A. Herrmann and E. Wagner, Biomaterials, 2013, 34, 1624–1633 CrossRef CAS PubMed.
- T. Fröhlich, D. Edinger, R. Klager, C. Troiber, E. Salcher, N. Badgujar, I. Martin, D. Schaffert, A. Cengizeroglu, P. Hadwiger, H. P. Vornlocher and E. Wagner, J. Controlled Release, 2012, 160, 532–541 CrossRef PubMed.
- S. M. Solberg and C. C. Landry, J. Phys. Chem. B, 2006, 110, 15261–15268 CrossRef CAS PubMed.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef.
- J. Kecht, A. Schlossbauer and T. Bein, Chem. Mater., 2008, 20, 7207–7214 CrossRef CAS.
- J. Kobler, K. Möller and T. Bein, ACS Nano, 2008, 2, 791–799 CrossRef CAS PubMed.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- S. B. Hartono, M. Yu, W. Gu, J. Yang, E. Strounina, X. Wang, S. Qiao and C. Yu, Nanotechnology, 2014, 25, 055701 CrossRef PubMed.
- J. Zhang, M. Niemelae, J. Westermarck and J. M. Rosenholm, Dalton Trans., 2014, 43, 4115–4126 RSC.
- D. Schaffert, C. Troiber, E. E. Salcher, T. Fröhlich, I. Martin, N. Badgujar, C. Dohmen, D. Edinger, R. Kläger, G. Maiwald, K. Farkasova, S. Seeber, K. Jahn-Hofmann, P. Hadwiger and E. Wagner, Angew. Chem., Int. Ed., 2011, 50, 8986–8989 CrossRef CAS PubMed.
- S. Niedermayer, V. Weiss, A. Herrmann, A. Schmidt, S. Datz, K. Müller, E. Wagner, T. Bein and C. Bräuchle, Nanoscale, 2015, 7, 7953–7964 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: MSN synthesis and analysis, sample preparation for cell transfections as well as additional studies including experiments with a second cell line and a toxicity assay. See DOI: 10.1039/c5nr06246b |
| This journal is © The Royal Society of Chemistry 2016 |