
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase transfer of 1- and 2-dimensional Cd-based nanocrystals†
Torben
Kodanek
,
Hadeel M.
Banbela
,
Suraj
Naskar
,
Patrick
Adel
,
Nadja C.
Bigall
and
Dirk
Dorfs
*
Institute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstr. 3A, 30167 Hannover, Germany. E-mail: dirk.dorfs@pci.uni-hannover.de
First published on 28th October 2015
Abstract
In this work, luminescent CdSe@CdS dot-in-rod nanocrystals, CdSe@CdS/ZnS nanorods as well as CdSe–CdS core–crown nanoplatelets were transferred into aqueous phase via ligand exchange reactions. For this purpose, bifunctional thiol-based ligands were employed, namely mercaptoacetic acid (MAA), 3-mercaptopropionic acid (MPA), 11-mercaptoundecanoic acid (MUA) as well as 2-(dimethylamino)ethanthiol (DMAET). Systematic investigations by means of photoluminescence quantum yield measurements as well as photoluminescence decay measurements have shown that the luminescence properties of the transferred nanostructures are affected by hole traps (induced by the thiol ligands themselves) as well as by spatial insulation and passivation against the environment. The influence of the tips of the nanorods on the luminescence is, however, insignificant. Accordingly, different ligands yield optimum results for different nanoparticle samples, mainly depending on the inorganic passivation of the respective samples. In case of CdSe@CdS nanorods, the highest emission intensities have been obtained by using short-chain ligands for the transfer preserving more than 50% of the pristine quantum yield of the hydrophobic nanorods. As opposed to this, the best possible quantum efficiency for the CdSe@CdS/ZnS nanorods has been achieved via MUA. The gained knowledge could be applied to transfer for the first time 2-dimensional CdSe–CdS core–crown nanoplatelets into water while preserving significant photoluminescence (up to 12% quantum efficiency).
Introduction
In comparison to organic fluorophores, luminescent semiconductor nanoparticles exhibit many benefits, e.g. narrow emission lines, size-tunable optical properties and enhanced resistance against photobleaching.1,2 Due to these unique properties the materials are promising candidates for a variety of applications covering sensors, biomedical labels, and lasers.1–5 An established method to produce nanocrystals of high quality is the so-called “hot injection” approach.6,7 This technique provides access to a great number of highly luminescent semiconductor materials, which can be precisely controlled in shape and size. In this regard, CdSe@CdS dot-in-rod nanocrystals represent one of the most widely used nanostructures due to their simple and reproducible production. Till now, several works have dealt with this material, whereby their properties have been clarified to the greatest possible extent.8–12 Thus, CdSe@CdS dot-in-rod nanocrystals are an attractive alternative to conventional quantum dots. However, the drawback of the resulting nanoparticles is their insolubility in aqueous media due to the hydrophobic ligand shell present on their surface. Unfortunately, this fact restricts the utilization for prospective applications, since water-soluble nanomaterials are required in many fields e.g. labeling applications in biological systems. Hence, a major challenge is the transfer of hydrophobic luminescent nanoparticles into aqueous media without changing their optical and structural properties. Up to now, different techniques have been developed to obtain water-soluble nanocrystals including ligand exchange reactions13–17 as well as encapsulation into hydrophilic (e.g. silica18–20) or amphiphilic21–23 materials. In particular, the replacement of the pristine ligands with thiol-based molecules (e.g. 3-mercaptopropionic acid) describes an effective, cheap and uncomplicated phase transfer approach.13,24–26 Indeed, present reports have primarily investigated the transfer of 0-dimensional structures such as spherical CdSe@ZnS core–shell quantum dots.13,27,28 In contrast, investigations concerning higher-dimensional or in other words anisotropic nanocrystals such as nanorods and nanoplatelets are still lacking. In addition, systematic investigations of the influence of different thiol-based ligands are of great importance to give an insight into the mechanisms of the luminescence properties, which is not done so far for anisotropic nanocrystals.In comparison to luminescent quantum dots, anisotropic nanocrystals exhibit additional advantages e.g. larger absorption cross section,29 shorter radiative lifetime30 or in case of the nanoplatelets stronger quantum confinement.31 Thus, these materials are potentially more suitable for several applications than the spherical ones. Consequently, it is favorable to expand the repertoire of water-soluble nanoparticles by anisotropic nanostructures. Therefore, in this work, hydrophobic CdSe@CdS dot-in-rod nanocrystals, CdSe@CdS/ZnS nanorods (with the tips consisting of ZnS instead of CdS) as well as CdSe–CdS core–crown nanoplatelets as an example for a 2-dimensional nanostructure were transferred into aqueous media by ligand exchange reactions. In case of the CdSe@CdS dot-in-rod nanocrystals, it can be distinguished between the tips and the lateral surface. Due to the unlike facets as well as curvatures, these domains behave chemically differently. This raises the question, if the tips are crucial for the phase transfer in terms of the luminescence. To investigate their role, segmented CdSe@CdS/ZnS nanorods were additionally employed. Here, the nanorods are terminated by ZnS, which is known as electronic insulator. To expand this work by a 2-dimensional nanostructure, CdSe–CdS core–crown nanoplatelets were chosen. In those structures, a strong quantum confinement, depending only on the thickness of the nanoplatelets, exists. So far, there are no works reporting a successful phase transfer of these nanoplatelets, in which the luminescence is preserved. In this work, both, ligands and nanostructures, were varied systematically (see Scheme 1). Different hydrophilic thiol-based ligands were employed, namely mercaptoacetic acid (MAA), 3-mercaptopropionic acid (MPA), 11-mercaptoundecanoic acid (MUA) and 2-(dimethylamino)ethanethiol (DMAET). The influence of the ligands on the luminescence properties was investigated.
 | ||
| Scheme 1 Overview over the employed nanocrystals (top) and the bifunctional thiol-based ligands (bottom) for the phase transfer experiments. The shortcuts stand for mercaptoacetic acid (MAA), 3-mercaptopropionic acid (MPA), 11-mercaptoundecanoic acid (MUA) as well as 2-(dimethylamino)ethanethiol (DMAET). | ||
Experimental section
Materials
Cadmium oxide (CdO, 99.98%), selenium (Se, 99.999%, 200 mesh) and methanol (99.9%, anhydrous) were purchased from Alfa Aesar. Sulfur (S, 99.98%), tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(CH3CN)4]PF6, 97%), toluene (99.8%, anhydrous), zinc chloride (ZnCl2, 99.999%), cadmium acetate dihydrate (Cd(OAc)2·2H2O, 98%), mercaptoacetic acid (MAA, 99%), 3-mercaptopropionic acid (MPA, 99%), 11-mercaptoundecanoic acid (MUA, 95%), methanol (99.8%), n-hexane (99%), potassium hydroxide (KOH, 85%), chloroform (99.8%), hydrochloric acid (HCl, 37%), oleic acid (90%), cadmium nitrate tetrahydrate (99%), sodium myristate (99%) and toluene (99.7%) were purchased from Sigma-Aldrich. Tri-n-octylphosphine (TOP, 97%) and tri-n-octylphosphine oxide (TOPO, 99%) were purchased from ABCR. Octadecylphosphonic acid (ODPA, >99%) and hexylphosphonic acid (HPA, >99%) were purchased from PCI Synthesis. 1-Octadecene (ODE, 90%), oleylamine (OLAM, 80–90%) and 2-(dimethylamino)ethanethiol hydrochloride (DMAET·HCl, 95%) were purchased from Acros Organics. Ethanol (99.8%) was purchased from Roth. Rhodamine 6G was purchased from Lambda Physik.Synthesis of CdSe@CdS dot-in-rod nanocrystals
CdSe@CdS dot-in-rod nanocrystals were synthesized as described by Carbone et al.9 Briefly, CdSe seeds were prepared by mixing CdO (0.060 g), TOPO (3.0 g), and ODPA (0.280 g). After degassing at 150 °C under vacuum, the mixture was heated to 300 °C under nitrogen atmosphere, followed by the injection of 1.8 mL TOP. Subsequently, the mixture was heated to 380 °C. Once the mixture became clear, Se (0.058 g) dissolved in 1.8 mL TOP was injected quickly. The reaction was quenched rapidly after 45 s by the addition of 5 mL TOP. After a sufficient temperature decrease, 10 mL toluene was added. The obtained quantum dots were precipitated by adding an excess of methanol and separated by centrifugation at 3843g. The quantum dots were redispersed in toluene.To synthesize CdSe@CdS dot-in-rod nanocrystals, CdO (0.120 g), TOPO (6.0 g), ODPA (0.580 g), and HPA (0.160 g) were degassed under vacuum at 150 °C. Then, the mixture was heated to 300 °C under nitrogen atmosphere, followed by the addition of 3.6 mL TOP. Subsequently, the temperature was increased to 350 °C. After CdO has dissolved, CdSe (16 μmol) dispersed in a 2 M TOP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S solution (3.6 mL TOP:0.240 g S) was injected. The reaction was stopped after 8 min. After a sufficient temperature decrease, 10 mL toluene was added to the solution. The acquired CdSe@CdS nanorods were precipitated by adding an excess of methanol and centrifuged at 3843g. The nanoparticles were redispersed in 4 mL toluene under nitrogen for further steps.
:S solution (3.6 mL TOP:0.240 g S) was injected. The reaction was stopped after 8 min. After a sufficient temperature decrease, 10 mL toluene was added to the solution. The acquired CdSe@CdS nanorods were precipitated by adding an excess of methanol and centrifuged at 3843g. The nanoparticles were redispersed in 4 mL toluene under nitrogen for further steps.
Synthesis of CdSe@CdS/ZnS nanorods
CdSe@CdS/ZnS nanorods with a Zn2+ mole fraction of 0.25 were synthesized using the procedure from Adel et al.8 Initially, the prepared CdSe@CdS dot-in-rod nanocrystals had to be converted into CdSe@CdS/Cu2−xS. For this purpose, 1 ml of the CdSe@CdS dot-in-rod nanocrystals was diluted with anhydrous toluene to 20 mL. A solution of [Cu(CH3CN)4]PF6 in anhydrous methanol was prepared under inert conditions with exactly the same concentration as the Cd2+ concentration of the diluted CdSe@CdS nanorod solution (Cd2+ concentration was determined by atomic absorption spectroscopy). 10 mL of this [Cu(CH3CN)4]PF6 solution were added to the diluted CdSe@CdS nanorod solution under stirring. This way, the Cu+:Cd2+ ratio was 0.5 corresponding to 25% exchange of the original Cd2+ ions. After 5 min, the mixture was centrifuged at 3461g. The supernatant was discarded and the precipitate consisting of CdSe@CdS/Cu2−xS nanorods were redispersed in 0.5 mL anhydrous toluene and 7.2 mL TOP.
In order to obtain CdSe@CdS/ZnS nanorods, ZnCl2 (Cu:Zn ratio >1:20) together with 24 ml ODE and 16 mL OLAM were degassed under vacuum at 100 °C and then heated to 250 °C under nitrogen atmosphere. At this temperature, previously prepared CdSe@CdS/Cu2−xS nanorod solution was injected into the mixture and the reaction was carried out for 3 min. Then, the mixture was cooled down rapidly using first a water bath and then by adding 50 mL toluene at 150 °C. The nanorods were precipitated by adding 50 mL methanol and subsequently centrifuged at 3843g. Afterwards the CdSe@CdS/ZnS nanorods with a Zn2+ mole fraction of 0.25 were redispersed in toluene.
Synthesis of CdSe–CdS core–crown nanoplatelets
5 monolayers thick CdSe–CdS core–crown nanoplatelets were synthesized according to the procedure, described by Tessier et al. with slight modifications.31 Initially, CdSe nanoplatelets with 5 monolayers, acting as seeds for the following growth step, were prepared. Briefly, cadmium myristate (0.170 g) in 14 mL ODE were degassed under vacuum at 100 °C. Afterwards, the temperature was increased to 250 °C under argon atmosphere. Subsequently, Se (0.012 g) dispersed in 1 mL ODE was quickly injected into the reaction mixture. After 1 minute of Se injection, Cd(OAc)2·2H2O (0.120 g) was furthermore added. The reaction was allowed to continue at 250 °C for 10 min, followed by the addition of 0.5 mL oleic acid. After cooling down to room temperature, the obtained particles were precipitated by the addition of 25 mL of a hexane–ethanol-mixture (3:1) and centrifuged for 15 min at 3800g. The supernatant containing all the byproducts was discarded and the dark red precipitate of the CdSe nanoplatelets was dispersed in 2 mL hexane.
To grow a crown around the CdSe nanoplatelets, a growth solution had to be prepared. For this purpose, Cd(OAc)2·2H2O (0.480 g), 340 μL oleic acid and 2 mL ODE were heated to 150 °C under aerobic condition and kept at this temperature for 15 min resulting in a grey gel. Subsequently, 3 mL of S dissolved in ODE (0.1 M) was inserted into the flask. The whole mixture was then filled inside a syringe.
In order to obtain CdSe–CdS core–crown nanoplatelets, 2 mL of previously synthesized CdSe nanoplatelets (in hexane) together with 10 mL ODE were degassed at 100 °C for 30 min under vacuum. The temperature was raised to 240 °C under argon atmosphere and the growth solution was then continuously injected at a rate of 8 mL h−1 for 7.5 min. After cooling down to room temperature, the CdSe–CdS core–crown nanoplatelets were precipitated by adding 15 mL ethanol followed by centrifugation at 3800g for 15 min. The supernatant was discarded and the precipitate containing the nanoplatelets was dispersed in 4 mL hexane.
Phase transfer of CdSe@CdS and CdSe@CdS/ZnS nanorods
Phase transfer of CdSe–CdS core–crown nanoplatelets
For the phase transfer, MUA (0.030 g) was dissolved in 4 mL of methanol containing KOH (0.05 g) in a glass vial. The CdSe–CdS core–crown nanoplatelets, dispersed in 4 mL hexane, were added in the same vial. Afterwards, the mixture was shaken overnight. The colorless hexane phase was separated from methanol containing the nanoplatelets. After centrifugation of the methanol phase, the precipitate was dispersed in 3 mL of aqueous KOH (0.1 M).Elemental analysis
Quantitative cadmium elemental analysis was performed by atomic absorption spectroscopy (AAS) using a Varian AA 140 spectrometer. For the measurements, the samples were dissolved in aqua regia and then diluted with deionized water. The measurements were carried out at 228.8 nm using an acetylene/air flame.Transmission electron microscope (TEM) analysis
TEM analyses were performed using a FEI Tecnai G2 F20 microscope equipped with a field emission gun operated at 200 kV. Generally, the samples were prepared by dropping a diluted sample solution on a carbon coated copper grid (QUANTIFOIL, 300 mesh). In case of the hydrophilic nanoparticles, the samples were washed several times with water to get rid of KOH.Optical spectroscopy
UV/vis extinction spectra were recorded on a Cary 5000 spectrophotometer from Agilent. For this purpose, a quartz glass cuvette with a path length of 10 mm was employed. The sample stabilized with mercaptocarboxylic acids were diluted with aqueous KOH (0.1 M), while the DMAET stabilized samples were measured in deionized water. The hydrophobic samples were diluted with the same solvent, in which they were dispersed.Emission spectra of the same samples were recorded using a Horiba Fluoromax-4 spectrophotometer. To calculate the quantum yield, the reference standard rhodamine 6G dissolved in ethanol was used. The quantum yield of the nanoplatelets was determined in absolute mode using a Dual-FL spectrophotometer equipped with a Quanta-ϕ integrating sphere from Horiba.
Emission lifetime measurements were also carried out with the Fluoromax-4 spectrophotometer equipped with a time correlated single photon counting (TCSPC) accessory and an ns-pulsed LED as excitation source (full width half maximum of the pulse is about 1.2 ns, used pulse wavelength: 368 nm for nanorods and 454 nm for nanoplatelets).
Zeta potential and dynamic light scattering (DLS) measurements
Zeta-potential and DLS measurements were recorded with a Zetasizer Nano ZSP instrument from Malvern equipped with a HeNe laser (Wavelength: 632.8 nm).Results and discussion
The synthesis of hydrophobic CdSe@CdS dot-in-rod nanocrystals as well as CdSe–CdS core–crown nanoplatelets was carried out according to a seeded growth approach at high temperature reported by Carbone et al.9 and Tessier et al.31 respectively. The CdSe@CdS dot-in-rod nanocrystals were further used to produce CdSe@CdS/ZnS nanorods with a Zn2+ mole fraction of 0.25 adapted from a sequential two step cation exchange described by Adel et al.8 In this paper, they have shown that the cation exchange occurs regio-selectively at the tips of the rods resulting in a segment like structure. This means that a distinct interface exists between the CdS and the ZnS phase instead of alloy formation.A detailed discussion about the optical and structural properties of the hydrophobic CdSe@CdS dot-in-rod nanocrystals as well as CdSe@CdS/ZnS nanorods employed in this work can be found in the ESI.† One important fact not reported by Adel et al.8 which was confirmed by transmission electron microscope (TEM) analysis and extinction measurements, concerns the shrinking of the nanorods diameter from 5.3 ± 0.5 nm to 4.6 ± 0.4 nm after the cation exchange. This finding plays a crucial role for the luminescence properties with regard to the phase transfer, as discussed later.
In order to transfer the TOPO/TOP capped nanoparticles into the aqueous phase, two kinds of bifunctional hydrophilic ligands were employed. On the one hand, mercaptocarboxylic acids (MAA, MPA and MUA) differing in their alkyl chain length (2, 3 and 11 carbon atoms, respectively) were used. The other type was an aminothiol (DMAET), which features a ternary amino function instead of a carboxyl group. Generally, the principle of the phase transfer bases on the replacement of the nonpolar ligands by an excess of hydrophilic molecules. Thereby, the thiol function of the hydrophilic ligand binds to the nanoparticle surface. The second group ensures colloidal stability in water through the formation of surface charges caused by deprotonation or protonation processes. Since the stabilization depends on the pH value, the transferred samples are better soluble in either alkaline (MAA, MPA, MUA) or slightly acidic (DMAET) solution. Of course, this fact is of great importance for all possible applications requiring colloidal stability. For instance, pH values play a decisive role for biomedical examinations. Hence, the right choice of the ligand is essential to ensure the solubility of nanoparticles in different aqueous media.
The extinction spectra of the phase transferred CdSe@CdS and CdSe@CdS/ZnS nanorods are illustrated in Fig. 1A and B. To allow a better comparison, the extinction curves of the hydrophobic species are included in the same images (see Fig. 1A and B, black curves). Here, it should be noted, that the spectra are not normalized. Instead, the solutions were adjusted to the same concentration so that possible effects of the dilution on the photoluminescence quantum yield and/or photoluminescence decay behavior are of no concern. In addition, the insets of both figures show a magnification of the spectral part attributed to the CdSe core for all samples. As can be seen, the nanorods dispersed in water exhibit exactly the same characteristics at similar wavelengths in comparison to their hydrophobic counterparts. Hence, it is assumed that the phase transfer procedure with the different hydrophilic ligands does not provoke any drastic changes concerning the size, structure or composition of the initial nanocrystals. The absence of scattering background in the spectra clearly indicates the colloidal stability in aqueous media. As mentioned above, the hydrophilic CdSe@CdS and CdSe@CdS/ZnS remain dispersed owing to the electrostatic repulsion between equal charges. For further validation, zeta potential measurements were performed. As expected, the MAA, MPA and MUA stabilized nano-heterostructures (dispersed in 0.1 M aqueous KOH) possess zeta potentials between −31 mV and −55 mV due to the deprotonated carboxyl groups, whereas the DMAET ligand (dispersed in deionized water) effects a value of around +31 mV resulting from the protonated ternary amino functions. According to the extinction spectra, the values of these measurements imply stable colloidal solutions.
 | ||
| Fig. 1 Extinction spectra of CdSe@CdS (A) and CdSe@CdS/ZnS (B) before and after the phase transfer (spectra are not normalized). For the phase transfer MAA (red), MPA (blue), MUA (purple) and DMAET (green) were employed. The insets show a magnification of the particular extinction referred to the CdSe core (the spectra have an offset for clarity). The lower part shows TEM images of CdSe@CdS (C) as well as CdSe@CdS/ZnS (D) before (left) and after (right) the water transfer with MAA. | ||
In accordance with the afore-mentioned results, TEM analysis (see Fig. 1C and D as well as Fig. S2 and S3 in ESI†) reveals no apparent changes after the water transfer concerning the morphology and size. For instance, MAA covered CdSe@CdS dot-in-rod nanocrystals exhibit mean dimensions of 24.5 ± 1.7 nm times 5.5 ± 0.5 nm, which do not differ from the hydrophobic starting material. Similar findings are observed in case of the transferred CdSe@CdS/ZnS samples compared to their hydrophobic counterpart.
The photoluminescence (PL) spectra of the hydrophilic as well as hydrophobic CdSe@CdS dot-in-rod nanocrystals are shown in Fig. 2A. As can be seen, the emission maxima of the transferred samples exhibit a negligible shift of around 3 nm to shorter wavelengths (see Table 1). To calculate the quantum yield (QY), the reference standard rhodamine 6G dissolved in ethanol was used (QY = 95%). Generally, it can be ascertained that the QY reduces in all cases after the phase transfer (see Table 1). The QYs of the CdSe@CdS nanorods drop from initially 40% (hydrophobic sample) to 23% after exchanging the ligand shell to MAA or MPA. Here, similar PL intensities are expected due to the small chemical differences between MAA and MPA. The strongest emission quenching is observed for the MUA stabilized nanorods (with QY of 14%), whereas the DMAET sample exhibits the highest QY of 34%.
 | ||
| Fig. 2 Panel A shows the PL spectra of the hydrophilic as well as hydrophobic CdSe@CdS dot-in-rod nanocrystals of the same solution, which were used for the extinction measurements. The emission was recorded at an excitation wavelength of 466 nm. The corresponding PL lifetime measurements are illustrated in panel B. The emission decay was recorded at 628 nm in all cases. | ||
| Sample | Ligand | λ em/nm | QY | τ/ns |
|---|---|---|---|---|
| CdSe@CdS | TOPO/TOP | 628 | 40% | 19.4 |
| MAA | 626 | 23% | 15.9 | |
| MPA | 625 | 23% | 16.4 | |
| MUA | 626 | 14% | 12.3 | |
| DMAET | 625 | 34% | 22.2 |
To achieve a deeper understanding, PL lifetime measurements were performed as well (see Fig. 2B). The determination of the lifetime τ was approximated by means of a mono-exponential fit. In accordance with the QYs, the lifetimes shorten with a similar trend after the phase transfer (see Table 1). For example, the MUA capped CdSe@CdS nanorods exhibit not only the lowest QY, but also the shortest lifetime of 12.3 ns. The phase transfer via MAA induces, in contrast, a higher QY in combination with a longer lifetime (15.9 ns). This correlation can be interpreted in a way that the emission properties of the present system are specified by a homogeneous part. In this context, homogeneous means, that all particles behave the same. Thereby, the shortened lifetimes as well as the lower QY after the phase transfer are caused by an increased nonradiative decay rate in all particles of the sample. The observed tendency seems to result from a uniform impact of the respective hydrophilic ligands on the nonradiative decay channels. On closer examination, it is apparent that the lifetime and the QY do not decrease to the same degree, which is referred to an additional inhomogeneous influence. For example, the QY drops by a factor of 1.7 after exchanging the hydrophobic ligand to MAA, while the corresponding lifetime shortens only by a factor of 1.2. This effect appears even more drastically for the DMAET sample. The reason for the inhomogeneity can be explained by different degrees of emission quenching of individual nanorods (up to complete quenching of some particles). At this point, it is important to know that minor discrepancies regarding e.g. defects at the nanoparticle surface are already able to affect the PL properties of this individual particle drastically. As a result, such differences lead to an inhomogeneous component as observed in this work (e.g. some particles are completely quenched while others stay unaffected).
A further observation becomes apparent in the trend that the QY decreases with increasing alkyl chain length of the hydrophilic ligand (see Table 1). To interpret this finding, two influencing factors should be considered: first, the charge carrier traps induced by the ligand and on the other hand the passivation against the environment.
Generally, charge carrier traps occur, if electronic states are introduced within the band gap of a semiconductor material. Those states can arise from e.g. ligands or surface defects. For thiols like MAA or MUA, it has been discovered that they are able to generate hole traps in terms of CdSe and CdS nanoparticles.32,33 After the formation of excitons, those states can trap holes which are not available anymore for the radiative recombination. Instead, they create new channels for nonradiative processes being in competition with the radiative recombination. Here, the intensity of the hole trapping depends on the alignment of the trap states generated by the ligands relative to the position of the top valence band of the semiconductor.32,33 In this context, Algar et al. have found out via voltammetry measurements that the hole affinity of mercaptocarboxylic acids raises with increasing alkyl chain length.13 In other words, the efficiency to trap holes is expected to increase from MAA to MUA. Consequently, the emission quenching of MUA capped emitters must be stronger compared to the MAA stabilized nanoparticles (if only hole trapping is considered as quenching source).
Besides hole trapping, passivation defects can be taken into account as well. Concerning this matter, a poor passivation against the environment through ligands has a huge impact on the emission quenching due to e.g. surface reactions, which was reported by Blum et al.27 In terms of the ligands employed, the MUA stabilizer is supposed to improve the separation between the CdSe@CdS nanorods and the outer media due to its long alkyl chain length (11 carbon atoms). Furthermore, it is assumed that the coverage at the nanoparticle surface is denser and comparable with a self-assembled monolayer owing to the strong van der Waals interaction between the carbon frameworks.27,34 In contrast, MAA or DMAET exhibit the shortest alkyl chain and thus the poorest shielding against the surrounding. Concerning the QY, this implies, in turn, that the emission intensity of MUA stabilized CdSe@CdS nanorods is expected to be more intense than in the other cases (if only passivation defects are considered as quenching source).
Considering the two possible behaviors as explained above and the obtained findings that the quantum yield decreases with increasing alkyl chain length, passivation defects seem to make little contribution to the emission quenching. Instead, the observed tendency can be well explained by the hole affinity of the individual ligands. Hence, the trap states are expected to play a major role for the emission quenching in case of the hydrophilic CdSe@CdS dot-in-rod nanocrystals.
In addition to the understanding of the quenching mechanism, the knowledge about the PL stability is of fundamental importance, particularly with regard to potential applications. Indeed, it is senseless to employ materials, which lose their emission before their application ends. For this purpose, the QYs as well as the PL lifetimes of the water-soluble CdSe@CdS dot-in-rod nanocrystals were monitored again after 8 days. Here, the samples stabilized by MAA, MPA or MUA exhibit no significant changes concerning their luminescence properties. As opposed to this, the QY of the DMAET covered nanorods reduces drastically from 34% to 16%. In connection with the emission quenching, a lifetime shortening can be ascertained as well. It is assumed that surface processes play a major role for this finding. One possible explanation would be the decomposition or at least the desorption of a few ligand molecules over time leading to the formation of dangling bonds at the nanoparticle surface. In turn, these dangling bonds can act as charge carrier traps, which induce a decrease of the quantum efficiency. However, the exact reason behind this drastic change is not completely understood at that point. With these findings in mind, the mercaptocarboxylic acids enclosed CdSe@CdS nanorods seem to be suitable for such applications, which require a constant emission behavior over a long period of time. However, if the stability plays a minor part, then the DMAET stabilized nanorods are supposed to be more favorable due to the higher QY right after the phase transfer.
As many potential applications of nanoparticles are in aqueous media, the aim was to functionalize the CdS surface of the nanorods with hydrophilic groups. Thereby, it has to be considered, that the rods exhibit a lateral surface area and two tips with different reactivities. The reason for the different behavior lies in the unlike crystallographic planes as well as in the curvature. Thus, it is possible, that in the case that the PL quenching occurs, it depends on the tips due to the different amounts of ligands. To detect this change, CdSe@CdS/ZnS nanorods are employed. In these nanorods the tips are exchanged by ZnS, which acts as electronic insulator and thus passivates the tips. That way, it is possible to analyze the influence of the functionalized tips on the quenching process.
The PL spectra as well as the corresponding lifetime measurements of the hydrophilic CdSe@CdS/ZnS nanorods are illustrated together with their hydrophobic counterpart in Fig. 3A and B. As before, the emission maxima remain basically unchanged after the phase transfer (see Fig. 3A and Table 2). However, large differences can be observed regarding the quantum yield. In this context, the short-chain ligands MAA, MPA as well as DMAET cause the lowest emission intensities, which stands in contrast to the hydrophilic CdSe@CdS nanorods. In other words, the development of the emission intensities behaves reversed compared to the CdSe@CdS nanorods. In connection with the QY, a lifetime shortening was also ascertained with decreasing alkyl chain length of the ligand (see Table 2).
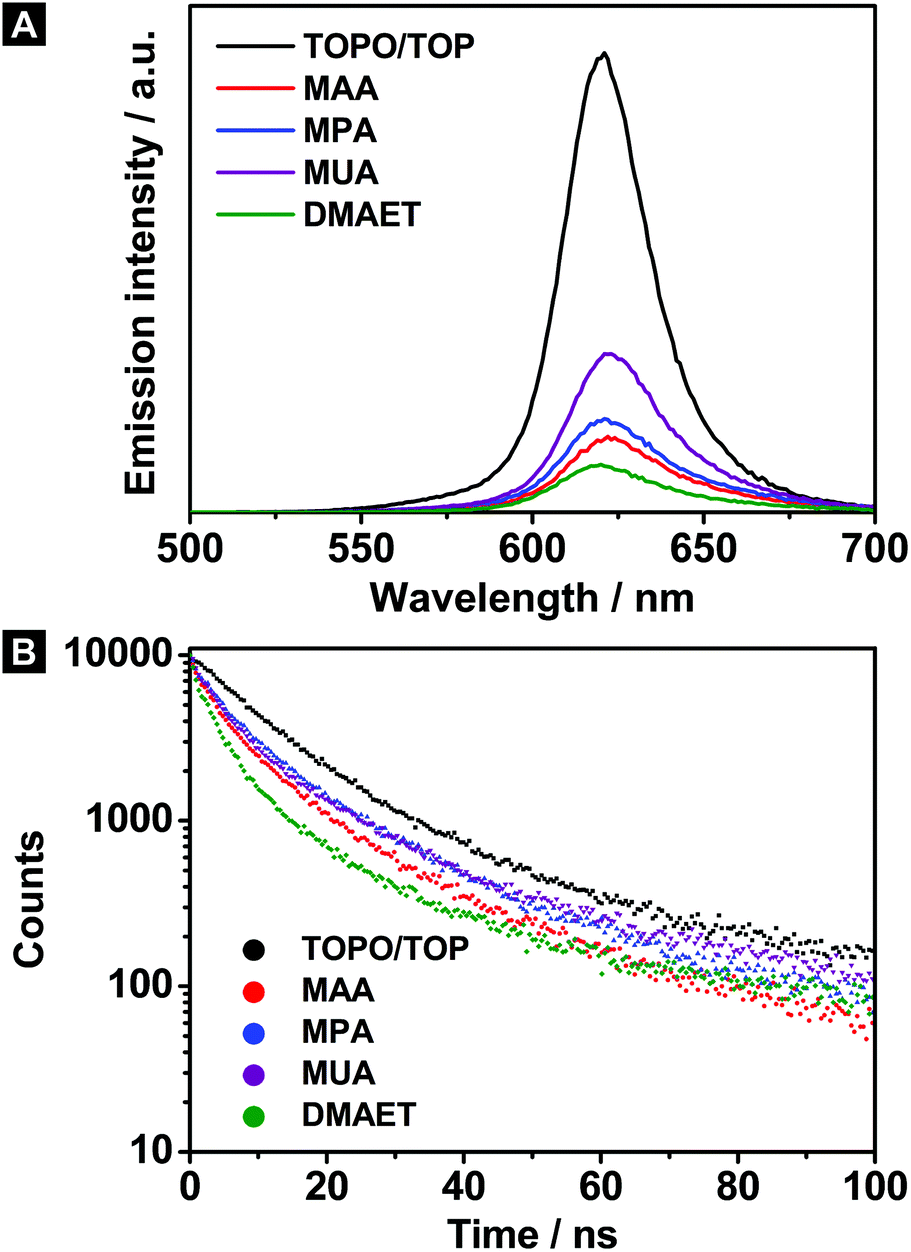 | ||
| Fig. 3 Panel A shows the PL emission spectra of the hydrophilic and hydrophobic CdSe@CdS/ZnS nanorods of the same solution, which were used for the extinction measurements. The emission was measured at an excitation wavelength of 466 nm. Panel B shows the corresponding PL lifetime measurements of the same samples. The emission decay was recorded at 621 nm in all cases. | ||
| Sample | Ligand | λ em/nm | QY | τ/ns |
|---|---|---|---|---|
| CdSe@CdS/ZnS | TOPO/TOP | 621 | 29% | 13.1 |
| MAA | 623 | 6% | 7.0 | |
| MPA | 622 | 8% | 8.8 | |
| MUA | 623 | 12% | 8.1 | |
| DMAET | 621 | 4% | 4.7 |
However, if hole traps are considered to be the only quenching source for the ion exchanged samples, the same QY behavior would be expected as in the case of the hydrophilic CdSe@CdS nanorods. On that account, an explanatory approach, which only considers the hole affinity of the individual ligands, is insufficient. Thus, the chemical shielding against the environment has to be taken into account as well. As already shown, the cation exchange procedure leads to a shrinking of the nanorod diameter. This finding can be ascribed to etching processes occurring at the CdS body. This fact supports the assumption that the CdSe core is less protected against the environment than before. Accordingly, the radiative recombination within the core reacts more sensitive to surface processes. The influence of the ligands is supposed to be reflected in the extent of the protection against the outer media. Regarding the alkyl chain length, MUA is expected to form a more effective protective cover than the short-chain ligands, since the separation between the nanorod surface and the aqueous media is maximized. Furthermore, the MUA can pack densely around the nanorod like a self-assembled monolayer resulting from van der Waals interaction between the carbon frameworks.27,34 Such an arrangement hinders diffusion processes through the ligand shell. Instead, the protection given by short-chain ligands is insufficient due to the poor separation between nanoparticle and surrounding. Therefore, surface processes exert a raised influence on the core, resulting in an increased emission quenching for the CdSe@CdS/ZnS nanorods stabilized by MAA, MPA or DMAET.
In general, it turned out, that the water-soluble segmented CdSe@CdS/ZnS nanorods behave inferior to the hydrophilic CdSe@CdS dot-in-rod nanocrystals regarding their luminescence properties. Consequently, the tips of the nanorods play only a negligible role for the phase transfer. Instead, the passivation of the mantle facets either by an inorganic shell or by organic ligands is of far greater importance for the emission behavior. Accordingly, a poor protection against the environment leads to a strong emission quenching. Hence, the insufficient passivation must be compensated by long-chain ligands like MUA.
In the last part of this work, the investigation is further expanded to 2-dimensional structures using CdSe–CdS core–crown nanoplatelets. The lateral dimensions of the employed nanoplatelets are 38.2 ± 4.4 nm in length and 13.7 ± 1.5 nm in width (see Fig. 4D left side). The thickness is a much more important parameter, since this dimension determines the quantum confinement of these nanocrystals. In this case, the thickness should be 1.5 nm, corresponding to 5 monolayers. Here, a monolayer consists of one sulfur/selenium as well as one cadmium sublayer. At this point, it is noteworthy, that only the MUA ligand is able to ensure a certain preservation of emission. In contrast, the transfer via short-chain ligands like MPA results in a complete quenching of the emission. The corresponding extinction spectra of the hydrophobic as well as MUA stabilized CdSe–CdS core–crown sheets are illustrated in Fig. 4B (solid lines). As expected, the hydrophobic sample exhibits typical characteristics of core–crown nanoplatelets with a thickness of five monolayers (black solid line), which was already described by Tessier et al.31 The CdSe core provokes the local maximum at 554 nm, while the shoulder at around 430 nm is associated with the band gap absorption of the CdS crown. Similar features are also observed for the MUA stabilized nanoplatelets. However, the extinction in this case is overall shifted about 18 nm to longer wavelengths (red solid line). Two reasons could be taken into account: the formation of superlattices and the epitaxial growth of sulfur layers induced by the ligand. To give an accurate explanation, dynamic light scattering (DLS) measurements were additionally performed (see Fig. S5 in ESI†). It turns out, that the hydrodynamic sizes of the hydrophobic and the MUA stabilized sample do not differ significantly from each other (aside of a few agglomerates). This finding allows the assumption that the observed shift to longer wavelengths in the extinction spectrum originates from the thiolate linkage of the MUA to the nanoparticle surface. The thickness of the core–crown nanoplatelets is enlarged by two additional sulfur layers, which reduces the exciton confinement. To understand this fact, it is important to know, that the facets of the nanoplatelets are terminated with cadmium.4,35 These findings agree very well with the results reported by Mahler et al.36 Within the scope of their investigations in hydrophobic environment, ligand exchange experiments have been carried out with dodecanethiol instead of MUA causing a similar shift to longer wavelengths in organic media.
 | ||
| Fig. 4 Panel A shows a scheme of the CdSe–CdS core–crown nanoplatelets with a thickness of 5 monolayers. The corresponding extinction (solid line) and emission (dashed line) spectra of the hydrophobic as well as MUA stabilized nanocrystals are illustrated in Panel B. Panel C shows the PL lifetime curves of the same samples. Panel D illustrates TEM images of the CdSe–CdS core–crown nanoplatelets before (left) and after (right) the phase transfer with MUA. | ||
In line with the previous outcome, the emission spectra (see Fig. 4B, dashed lines) likewise reveal a spectral shift of the emission wavelength from 556 nm (hydrophobic sample) to 580 nm (MUA stabilized sample). Besides that, the quantum yield decreases by a factor of 3 after the phase transfer procedure from 36% to 12%. In contrast, the luminescence has completely disappeared using the short-chain ligands MPA, MAA as well as DMAET for the transfer of the nanoplatelets into aqueous media. To give an explanation for this observation, additional TEM images of the hydrophilic samples have been recorded (see Fig. 4D right side and Fig. S4 in ESI†). As expected, the MUA stabilized nanoplatelets do not exhibit any drastic change in their morphology compared to the hydrophobic sample (see Fig. 4D). Instead, the transfer by means of MPA, MAA as well as DMAET results in a nearly complete decomposition of the core–crown structure (see Fig. S4†). Especially in case of the MPA as well as DMAET stabilized sample, it is clearly visible that a great number of the CdSe cores are partially or fully dissolved. Since the CdSe core acts as the recombination side for electron and hole, it is reasonable that the luminescence in all cases has vanished completely. Tentatively, we assume that in case of the samples which were transferred via short-chained ligands, the transferred particles are much more affected by oxidative processes (which naturally occur at the easier oxidizable CdSe core of the particles) caused by e.g. ambient oxygen.
A comparison between the PL decay curves shows that the lifetime increases from 4.8 ns to 13.8 ns after the phase transfer with MUA (see Fig. 4C). Furthermore, both samples exhibit similar long-time components. In contrast to the nanorods, the increase of the PL lifetime connected with the reduced quantum yield cannot be simply explained by a variation of the nonradiative decay rates. Here, it is more reasonable to assume that the radiative lifetime of the emitting species is increased by the MUA attachment. This finding can be expected due to the additional sulfur layers produced by the thiol groups of the ligand. In other words, this means that the direct binding of MUA on the CdSe surface reduces both the exciton confinement and the kinetics of the radiative recombination within the core.
Conclusion
In this work, 1-dimensional CdSe@CdS dot-in-rod nanocrystals, CdSe@CdS/ZnS nanorods as well as 2-dimensional CdSe–CdS core–crown nanoplatelets were transferred into aqueous media. One main finding is that for the quantum rod samples, the tips of the structures play an inferior role for fluorescence quenching during phase transfer. We found that for samples with a better inorganic passivation, short-chained thiol ligands are the best choice to obtain high quantum yields due to reduced hole trapping. Instead, for samples with a poorer inorganic passivation, long chained organic ligands appear as the best choice for high quantum yields, since in this case spatial passivation against the environment becomes a more important factor than hole trapping. As a consequence, for the 2-dimensional CdSe–CdS core–crown particles which are the inorganically worst passivated samples investigated and which were never reported to be luminescent in aqueous media to date, luminescence after phase transfer (up to 12% quantum efficiency) could be achieved using a long-chained ligand for phase transfer.Acknowledgements
D.D. wants to thank the German research foundation (DFG, research grants DO1580/2-1 and DO1580/3-1) and the Volkswagen foundation (lower Saxony/Israel cooperation, Grant ZN2916) for funding. N. B. and S. N. are thankful for the financial support to the German Ministry of Education and research (BMBF, NanoMatFutur, support code 03X5525). Moreover, T. K. is grateful to the Hannover School for Nanotechnology (hsn) for financial support. We are also thankful to Laboratory of Nano and Quantum Engineering of the Leibniz Universität Hannover for its support. Finally, the authors wish to thank L. Diestel and J. Poppe for proofreading.References
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- A. Fu, W. Gu, B. Boussert, K. Koski, D. Gerion, L. Manna, M. Le Gros, C. A. Larabell and A. P. Alivisatos, Nano Lett., 2007, 7, 179–182 CrossRef CAS PubMed.
- C. She, I. Fedin, D. S. Dolzhnikov, A. Demortière, R. D. Schaller, M. Pelton and D. V. Talapin, Nano Lett., 2014, 14, 2772–2777 CrossRef CAS PubMed.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- R. K. Čapek, K. Lambert, D. Dorfs, P. F. Smet, D. Poelman, A. Eychmüller and Z. Hens, Chem. Mater., 2009, 21, 1743–1749 CrossRef.
- P. Adel, A. Wolf, T. Kodanek and D. Dorfs, Chem. Mater., 2014, 26, 3121–3127 CrossRef CAS.
- L. Carbone, C. Nobile, M. de Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- H. Li, R. Brescia, R. Krahne, G. Bertoni, M. J. P. Alcocer, C. D'Andrea, F. Scotognella, F. Tassone, M. Zanella, M. de Giorgi and L. Manna, ACS Nano, 2012, 6, 1637–1647 CrossRef CAS PubMed.
- M. G. Lupo, F. Della Sala, L. Carbone, M. Zavelani-Rossi, A. Fiore, L. Lüer, D. Polli, R. Cingolani, L. Manna and G. Lanzani, Nano Lett., 2008, 8, 4582–4587 CrossRef CAS PubMed.
- S. Sánchez-Paradinas, D. Dorfs, S. Friebe, A. Freytag, A. Wolf and N. C. Bigall, Adv. Mater., 2015, 27, 6152–6156 CrossRef PubMed.
- W. R. Algar and U. J. Krull, ChemPhysChem, 2007, 8, 561–568 CrossRef CAS PubMed.
- F. Dubois, B. Mahler, B. Dubertret, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2007, 129, 482–483 CrossRef CAS PubMed.
- L. Li, T. J. Daou, I. Texier, T. T. Kim Chi, N. Q. Liem and P. Reiss, Chem. Mater., 2009, 21, 2422–2429 CrossRef CAS.
- S. Tamang, G. Beaune, I. Texier and P. Reiss, ACS Nano, 2011, 5, 9392–9402 CrossRef CAS PubMed.
- J. Zylstra, J. Amey, N. J. Miska, L. Pang, C. R. Hine, J. Langer, R. P. Doyle and M. M. Maye, Langmuir, 2011, 27, 4371–4379 CrossRef CAS PubMed.
- N. R. Jana, C. Earhart and J. Y. Ying, Chem. Mater., 2007, 19, 5074–5082 CrossRef CAS.
- E. M. Hutter, F. Pietra, R. J. A. van Dijk-Moes, D. Mitoraj, J. D. Meeldijk, C. de Mello Donegá and D. Vanmaekelbergh, Chem. Mater., 2014, 26, 1905–1911 CrossRef CAS.
- M. Darbandi, R. Thomann and T. Nann, Chem. Mater., 2005, 17, 5720–5725 CrossRef CAS.
- M. Booth, R. Peel, R. Partanen, N. Hondow, V. Vasilca, L. J. C. Jeuken and K. Critchley, RSC Adv., 2013, 3, 20559 RSC.
- T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, A. L. Rogach, S. Keller, J. Rädler, G. Natile and W. J. Parak, Nano Lett., 2004, 4, 703–707 CrossRef CAS.
- F. Zhang, E. Lees, F. Amin, P. RiveraGil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- S. F. Wuister, I. Swart, F. van Driel, S. G. Hickey and C. de Mello Donegá, Nano Lett., 2003, 3, 503–507 CrossRef CAS.
- V. V. Breus, C. D. Heyes and G. U. Nienhaus, J. Phys. Chem. C, 2007, 111, 18589–18594 CAS.
- H. G. Bagaria, E. T. Ada, M. Shamsuzzoha, D. E. Nikles and D. T. Johnson, Langmuir, 2006, 22, 7732–7737 CrossRef CAS PubMed.
- A. S. Blum, M. H. Moore and B. R. Ratna, Langmuir, 2008, 24, 9194–9197 CrossRef PubMed.
- M. J. Ruedas-Rama, A. Orte, E. A. H. Hall, J. M. Alvarez-Pez and E. M. Talavera, ChemPhysChem, 2011, 12, 919–929 CrossRef CAS PubMed.
- H. Htoon, J. A. Hollingworth, A. V. Malko, R. Dickerson and V. I. Klimov, Appl. Phys. Lett., 2003, 82, 4776 CrossRef CAS.
- A. Shabaev and A. L. Efros, Nano Lett., 2004, 4, 1821–1825 CrossRef CAS.
- M. D. Tessier, P. Spinicelli, D. Dupont, G. Patriarche, S. Ithurria and B. Dubertret, Nano Lett., 2014, 14, 207–213 CrossRef CAS PubMed.
- S. F. Wuister, C. de Mello Donegá and A. Meijerink, J. Phys. Chem. B, 2004, 108, 17393–17397 CrossRef CAS.
- A. Rogach, T. Franzl, T. Klar, J. Feldmann, N. Gaponik, V. Lesnyak, A. Shavel, A. Eychmuller, Y. Rakovich and J. Donegan, J. Phys. Chem. C, 2007, 111, 14628–14637 CAS.
- U. Dürig, O. Züger, B. Michel, L. Häussling and H. Ringsdorf, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 1711–1717 CrossRef.
- S. J. Lim, W. Kim and S. K. Shin, J. Am. Chem. Soc., 2012, 134, 7576–7579 CrossRef CAS PubMed.
- B. Mahler, B. Nadal, C. Bouet, G. Patriarche and B. Dubertret, J. Am. Chem. Soc., 2012, 134, 18591–18598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further TEM images, further extinction spectra, particle size distribution and discussion about optical properties of the hydrophobic nanostructures. See DOI: 10.1039/c5nr06221g |
| This journal is © The Royal Society of Chemistry 2015 |