
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective fluorescence functionalization of dye-doped polymerized structures fabricated by direct laser writing (DLW) lithography†
Gustavo
de Miguel
*ab,
Giuseppe
Vicidomini
a,
Martí
Duocastella
a and
Alberto
Diaspro
ac
aNanoscopy, Department of Nanophysics, Istituto Italiano di Tecnologia, Via Morego 30, Genoa, 16163, Italy. E-mail: gustavo.demiguel@iit.it; gmiguel@uco.es
bInstitute of Fine Chemistry and Nanochemistry, Department of Physical Chemistry and Applied Thermodynamics, University of Cordoba, Campus Universitario de Rabanales, Edificio Marie Curie, Cordoba, 14014, Spain
cNIC@IIT, Istituto Italiano di Tecnologia, Via Morego 30, Genoa, 16163, Italy
First published on 4th November 2015
Abstract
The continuous development of the vast arsenal of fabrication techniques is a pivotal factor in the breakthrough of nanotechnology. Although the broad interest is generally focused on the reduction of the dimensions of the fabricated structures, localized functionalization of the nanomaterials emerges as a key factor closely linked to their potential applications. In particular, fabrication of spatially selective fluorescence nanostructures is highly demanded in nanophotonics, as for example in three-dimensional (3D) optical data storage (ODS), where massive storage capacity and fast writing–reading processes are promised. We have developed an innovative method to control the location and intensity of the fluorescence signal in dye-doped photopolymerized structures fabricated with Direct Laser Writing (DLW) lithography. Well-defined fluorescent pixels (area = 0.24 μm2) were written inside a polymer matrix with the help of a femtosecond pulsed laser (multiphoton absorption) via a thermally-induced di-aggregation of a fluorescent dye. Moreover, we have accomplished a fine control of the fluorescence intensity which can increase the storage capacity of ODS systems fabricated with this approach.
1. Introduction
Lithography is a widespread fabrication technique for micro- or nano-patterning of structuring materials, e.g. organic resins or inorganic semiconductors, which has been extensively employed in the microelectronics industry.1 Among the myriad of lithography approaches, multiphoton Direct Laser Writing (DLW) lithography has attracted a lot of attention in the last few years since the seminal paper of Kawata in 1997.2 DLW uses a tightly focused femtosecond (fs) pulsed laser to fabricate structures in a single processing step without any topological constraint, which renders it a unique fabrication tool among existing lithographic techniques.3,4 Because of the high concentration of photons needed for the multiphoton absorption process, the polymerization reaction is highly confined to the focal volume of the writing beam and hence, fabrication of three-dimensional (3D) structures is feasible. Moreover, sub-diffraction resolution can also be achieved due to the existence of a chemical threshold controlling the extension of the cross-linking in the polymer, which finely depends on the intensity of the writing beam.The unique features of DLW lithography renders this fabrication technique specially suitable for applications in many nanotechnological fields, including medicine, electronics and photonics.5,6 However, to take advantage of the 3D nanometer resolution, functionalization of the resin is required to provide the fabricated structure with the physical or chemical properties, e.g. conductivity, photoluminescence or ability to bind biomolecules, needed for each specific application. In the last few years, there have been many approaches to accomplish such resin functionalization in DLW. The traditional strategy involves the preliminary dispersion of the active material into the liquid monomer (doped-resin) and afterwards proceeds with the regular photopolymerization step.7 For example, organic dyes were incorporated into the polymer matrix to produce fluorescent structures.8,9 A second functionalization approach is to carry out an in situ synthesis of the active material during the photopolymerization process or in a post-treatment of resins containing precursors of the active materials. Fluorescence CdS-nanoparticles and gold metallic structures were successfully embedded into polymer nanocomposites with this method.10,11 Finally, a slightly different strategy consists of functionalizing the surface of the polymerized structure by covering it with a layer of the active material. An electroless plating metallization technique was employed after the nanofabrication with DLW to produce metal-coated woodpile photonic crystals at the near IR and optical regions.12
In all of these methods, a complete functionalization of the entire photopolymerized structure is accomplished since the active material is arbitrarily incorporated into the resin structure. Thus, these methods lack the ability to selectively incorporate the active material into areas of interest within the structure. Such control in the functionalization is a necessary step for the further development of DLW lithography, which can open the door to the fabrication of nanostructures with 3D spatially varying properties. An early example of this strategy is the gas-phase direct doping of the As2S3 photoresist with erbium, which can be spatially confined to layers as thin as 139 nm in a tri-layer photoresist stack.13 The 3D nanostructures prepared with this method are foreseen to be valuable in the fabrication of nanophotonic components. However, it is in the field of optical data storage (ODS) where the spatial control of the luminescence has a straightforward application.14 Data writing is normally accomplished with a laser beam and the mechanisms proposed to produce the optical changes in the ODS devices include many photochemical reactions such as photoisomerization, photodecomposition, and photopolymerization.15–17
Herein, we report a simple method to generate a spatially localized and intensity controlled enhancement of the fluorescence signal in a photopolymerized structure fabricated with DLW lithography, which has been previously doped with the fluorescent dye ATTO 565. Our approach is based on the selective control of the aggregation of the fluorescent dye upon multiple irradiation of the polymerized structure with a high peak-power femtosecond pulsed laser. This irradiation generates a local increase of the temperature in the resin, which affects the photophysics of the dye increasing the fluorescence signal. We have fabricated fluorescent dots or lines within a photopolymerized square with a minimum feature size of around 550 nm and a varying intensity signal, which can be exploited in optical data storage or photonic crystals.
2. Experimental
2.1 Materials
Pentaerythritol triacrylate (PETA, Sigma Aldrich) was utilized as the negative resin, employing isopropyl thioxanthone (ITX, Sigma Aldrich) as the photoinitiator. The ATTO 565 carboxy-derivative was purchased from Sigma Aldrich. All the chemicals were used without further purification. The resist employed for polymerization was prepared by dissolving ITX (0.16 wt%) and ATTO 565 (0.025 wt%) in PETA (2 g). In a typical lithography writing experiment, a glass cover slip was placed on an oil-immersion objective lens and a drop of the resin was deposited on top. After irradiation with the laser, the cover slip was immersed in methanol for 5 minutes and then rinsed with isopropanol to wash away the liquid monomer (development process).2.2 DLW lithography setup (writing setup)
The optical setup is based on a custom-made optical microscope which has been previously described.18 The two-photon polymerization beam is obtained from a mode-locked Ti:Sapphire laser (Chameleon Vision II, Coherent), which delivers pulses of ∼150 fs, with a wavelength of 760 nm and at a repetition rate of 80 MHz. The beam travels through an appropriate combination of dichroic mirrors (AHF Analysentechnik) and is then deflected by two fast galvanometric scanning mirrors (6215HM40B, Cambridge Technologies, USA), towards the objective lens (PL APO 100×/0.7–1.4, oil immersion, Leica Microsystems). The power of the beam is controlled with an acoustic optic modulator (AOM, AA Optoelectronics). The software package Imspector (Max-Planck Innovations) ensures the full control of the optical setup.2.3 Data read-out
The readout of the written information was carried out in a confocal fluorescence microscope incorporated into the lithography optical setup. The excitation of the ATTO 565 molecule is performed using a continuous-wave (CW) laser operating at 532 nm, and the fluorescence was detected in the spectral range of 540–640 nm.2.4 Fluorescence lifetime imaging setup
The experiments were performed on a home-built confocal fluorescence microscope.19 The excitation beam (488 nm) was provided by a supercontinuum source which is generated by pumping a photon-crystal-fibre (femtoWHITE-800, NKT Photonics) with a femtosecond mode-locked Ti:Sapphire laser of 150 fs pulse width and 80 MHz repetition rate (Chameleon, Vision II, Coherent). The excitation beam was deflected by two galvanometric scanning mirrors (6215HM40B, CTI-Cambridge) and directed towards the objective lens (HCX PL APO 100×/0.7–1.4, oil immersion, Leica Microsystems). The fluorescence light was collected by the same objective lens, descanned and passed through the dichroic mirrors as well as through a fluorescence band pass filter (ET Bandpass 540–640 nm, AHF analysentechnik) before being focused (focal length 60 mm, AC254-060-A-ML, Thorlabs) into a fibre pigtailed single photon avalanche diode (PDF Series, Micro Photon Devices). Photon-arrival times were detected at each pixel by a time-correlated-single-photon-counting-card (SPC-830, Becker & Hickl). Synchronization was obtained from the reference signal provided by the Ti:Sapphire laser. The acquisition time of the fluorescence signal for each pixel was maintained constant to fairly compare the intensity of the decays from the different pixels (100 μs). All imaging operations were automated and managed by the software Imspector (Max Planck Innovation).2.5 Steady-state spectra
The excitation and emission spectra were collected on an inverted Leica TCS SP5 AOBS confocal laser scanning microscope (Leica Microsystems CMS, Mannheim) equipped with a white light laser, 470–670 nm (SuperK EXTREME, NKT Photonics). The excitation beam was focused into the sample by using a PL APO 63×/1.4, oil immersion objective (Leica Microsystems CMS, Mannheim). The fluorescence signal was collected with high-sensitive hybrid detectors. The excitation spectra were recorded by tuning sequentially the excitation wavelength from 470 nm to 600 nm and the fluorescence was registered in the 610–700 nm spectral window. The emission spectra were recorded by exciting the sample at 488 nm (to be consistent with the time-resolved measurement) and the fluorescence was registered in 10 nm-width window from 520 to 750 nm.3. Results and discussion
The fluorescent dye selected to achieve the functionalization of the polymerized structure is a rhodamine derivative, the so-called ATTO 565. The steady-state absorption spectrum of ATTO 565 dissolved in the liquid monomer, pentaerythritol triacrylate (PETA), shows a peak centered at 550 nm and a weak shoulder at higher energy, ∼520 nm (Fig. S1†). The emission spectrum is a mirror image of the absorption one, with a maximum at 573 nm (Fig. S1†). The similar shape of absorption and emission spectra together with the small Stokes shift (725 cm−1) indicates a comparable configuration of the dye in the ground and excited state. ATTO 565 possesses a high molar extinction coefficient (120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and a fluorescence quantum yield of around 90%, which renders it a good candidate for fluorescence functionalization of polymers. Importantly, the fluorescent dye is mixed with the liquid monomer resin in a very high concentration, 0.2 g l−1, to assure the aggregation of the dye. DLW lithography is performed as usual: a droplet of the prepared dye-doped resin is placed on a cover slip and then it is irradiated with an ∼150 femtosecond pulsed laser at a repetition rate of 80 MHz (λexc = 760 nm) focusing the beam through a 100× oil immersion objective (NA = 1.4).
000 M−1 cm−1) and a fluorescence quantum yield of around 90%, which renders it a good candidate for fluorescence functionalization of polymers. Importantly, the fluorescent dye is mixed with the liquid monomer resin in a very high concentration, 0.2 g l−1, to assure the aggregation of the dye. DLW lithography is performed as usual: a droplet of the prepared dye-doped resin is placed on a cover slip and then it is irradiated with an ∼150 femtosecond pulsed laser at a repetition rate of 80 MHz (λexc = 760 nm) focusing the beam through a 100× oil immersion objective (NA = 1.4).
First, to control the possible influence of the fluorescent dye on the polymerization process, the same structures (20 μm length lines) were fabricated by using resins with or without the addition of ATTO 565 to the system. Neither changes in the needed power of the laser beam – no variation of the polymerization threshold – nor in the quality and size of the fabricated structures were observed. Therefore, the addition of ATTO 565 to the resin does not affect the chemical reactions occurring in the polymerization process. Then, before the development process, the lines fabricated with the dye-doped resin were imaged with a confocal fluorescence microscope upon irradiation at 532 nm and collecting the signal in the 540–640 nm range (Fig. S2A† is taken after polymerization). The photopolymerized lines are clearly defined with a good contrast with respect to the background, which indicates that the molecules of ATTO 565 remain mainly inside the solidified resin after the irradiation process. The measured thickness of the photopolymerized lines from the confocal image (∼500 nm) matches well with that obtained from the Scanning Electron Microscopy (SEM) image (∼550 nm), which confirms ATTO 565 is homogeneously distributed inside the lines.
Next, a 15 × 15 μm photopolymerized square was fabricated with the dye-doped resin and the fluorescence signal was measured before the development process, i.e. with the liquid monomer resin covering the structure. The square is clearly visible, but interestingly, its fluorescence intensity is lower compared to that from the surrounding liquid monomer (Fig. 1A). This behavior can be explained by three possible phenomena: a decrease in the fluorescence quantum yield (ϕfl) of ATTO 565 when the molecule is embedded within the polymer network; a reduction of the ATTO 565 concentration upon irradiation with the high power fs beam that might occur in parallel to the polymerization of the monomer; or a combination of both. To investigate in depth this behavior, the nanosecond time-resolved fluorescence decays were measured at the area corresponding to the liquid monomer resin as well as that of the photopolymerized square (Fig. S3†). Notably, the initial fluorescence signal at zero time is considerably lower at the square, which, assuming the same absorption capacity in both areas, indicates a lower concentration of ATTO 565 at the photopolymerized square. This can be explained due to photobleaching of ATTO 565 by the excitation beam. Thus, despite ATTO 565 exhibiting relatively high thermal- and photo-stability under mild conditions, photobleaching is expected to occur at the high powers used during the photopolymerization, causing a decrease in dye concentration. Moreover, the two fluorescence decays obey different decay kinetics, which surely implies a distinct ϕfl (Fig. S3†). In particular, the fluorescence signal from the polymerized square decays faster than that from the monomer resin, which can also explain the lower fluorescence intensity from the square (vide infra). To summarize, the origin of the “dark” photopolymerized square compared to the “bright” monomer resin can be ascribed to the combined effect of both parameters: lower ϕfl and the reduced concentration of the dye. Noticeably, after development of the system with methanol and ethanol, the monomer resin is washed out and the fluorescence signal from the square is more intense than that remaining from the cover slip (Fig. 1B).
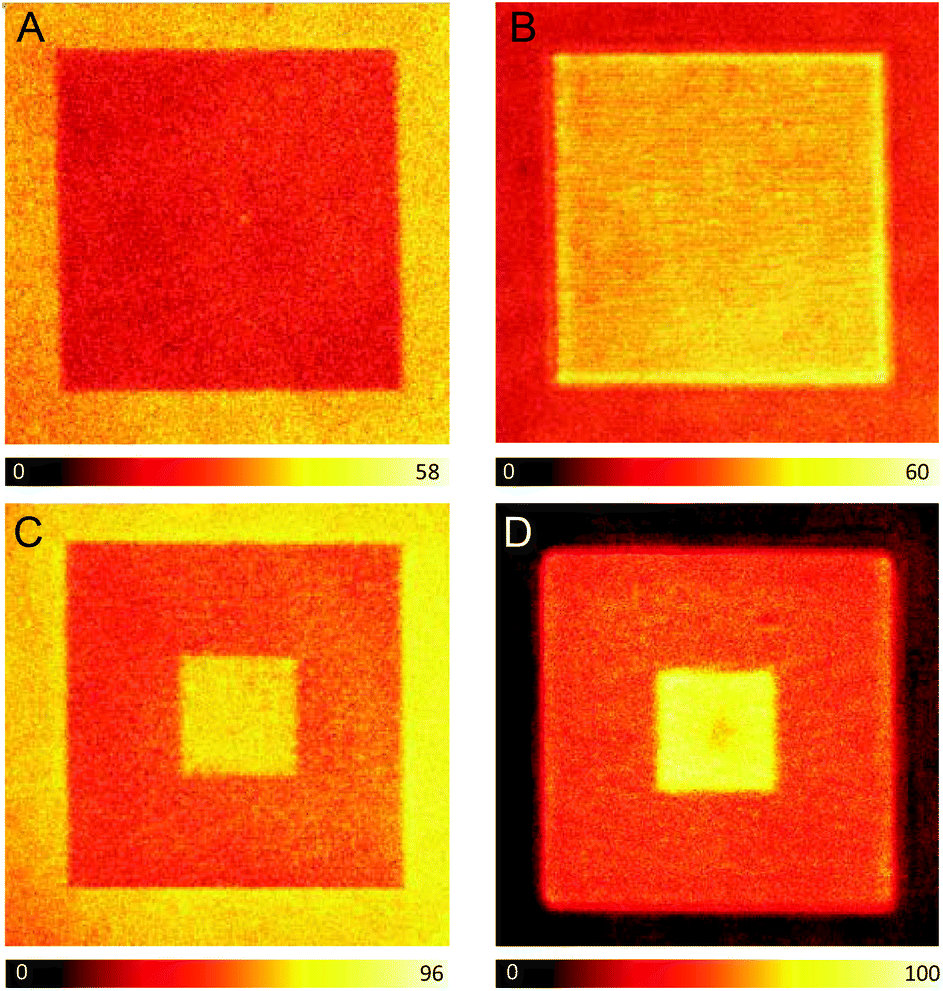 | ||
| Fig. 1 (A–D) Confocal fluorescence images of a photopolymerized square (15 × 15 μm) containing ATTO 565. (A and B) No further irradiation after polymerization. (C and D) The 5 × 5 μm central area is irradiated once with a 36 mW fs pulsed laser after polymerization. (A and C) Images were recorded before the development process. (B and D) Images were obtained after removing the remaining liquid monomer resin during the development step. λexc = 488 nm. The image size is 20 × 20 μm. | ||
At this point, to accomplish the spatially-selective enhancement of the fluorescence signal in the photopolymerized square, the 5 × 5 μm central area is irradiated a second time. This second irradiation step is performed before the development process, with the same fs pulsed beam used for the photopolymerization but at a much lower power (36 mW instead of 100 mW) – below the polymerization threshold. It has been previously reported that the second irradiation of a previously polymerized resin provokes the controlled axial expansion of the photopolymerized structures, which was attributed to a local increase of the temperature (heat accumulation) on the irradiated areas resulting in the vaporization of the cross-linked resin.18 The axial extent of this expansion can be controlled through the careful selection of the irradiation power and the number of exposures, with an expansion range of 40–250 nm.18 In this article, the conditions of the second irradiation were selected to produce a minimum expansion of the resin (∼40 nm). Fig. 1C shows the confocal fluorescence image of the 15 × 15 μm photopolymerized square whose central area (5 × 5 μm) is subsequently irradiated with a 36 mW fs pulsed beam. The image is measured before the development of the system. Interestingly, there is a 110–120% increase in the intensity of the fluorescence signal at the central 5 × 5 μm square, which provides a good contrast with respect to the rest of the photopolymerized resin. We have also studied the influence of the scanning velocity on the enhancement of the fluorescence signal. Thus, when decreasing the laser scanning velocity (longer irradiation of the polymerized area but preventing potential photodamage), an extra rise of the fluorescence signal is observed. Moreover, the intensity of the background, corresponding to the monomer resin, is comparable to that from the central 5 × 5 μm square. After washing out the monomer resin in the development step, the background fluorescence signal is negligible, however, the signal from the central square is still around 120% higher than that from the rest of the photopolymerized square (Fig. 1D).
To investigate the origin of the enhanced fluorescence signal after the second irradiation step, nanosecond time-resolved fluorescence decays and steady-state emission and excitation spectra were selectively recorded at the center of the photopolymerized square (double irradiation area), as well as the external part of the pattern (single irradiation area). Notably, the initial fluorescence signal (intensity at t = 0 s) is nearly the same for pixels at the central or external areas (Fig. 2B). This result is strong evidence of a similar concentration of ATTO 565 in both areas, since this initial value only depends on the concentration of the fluorescent excited state and not on the deactivation pathways.
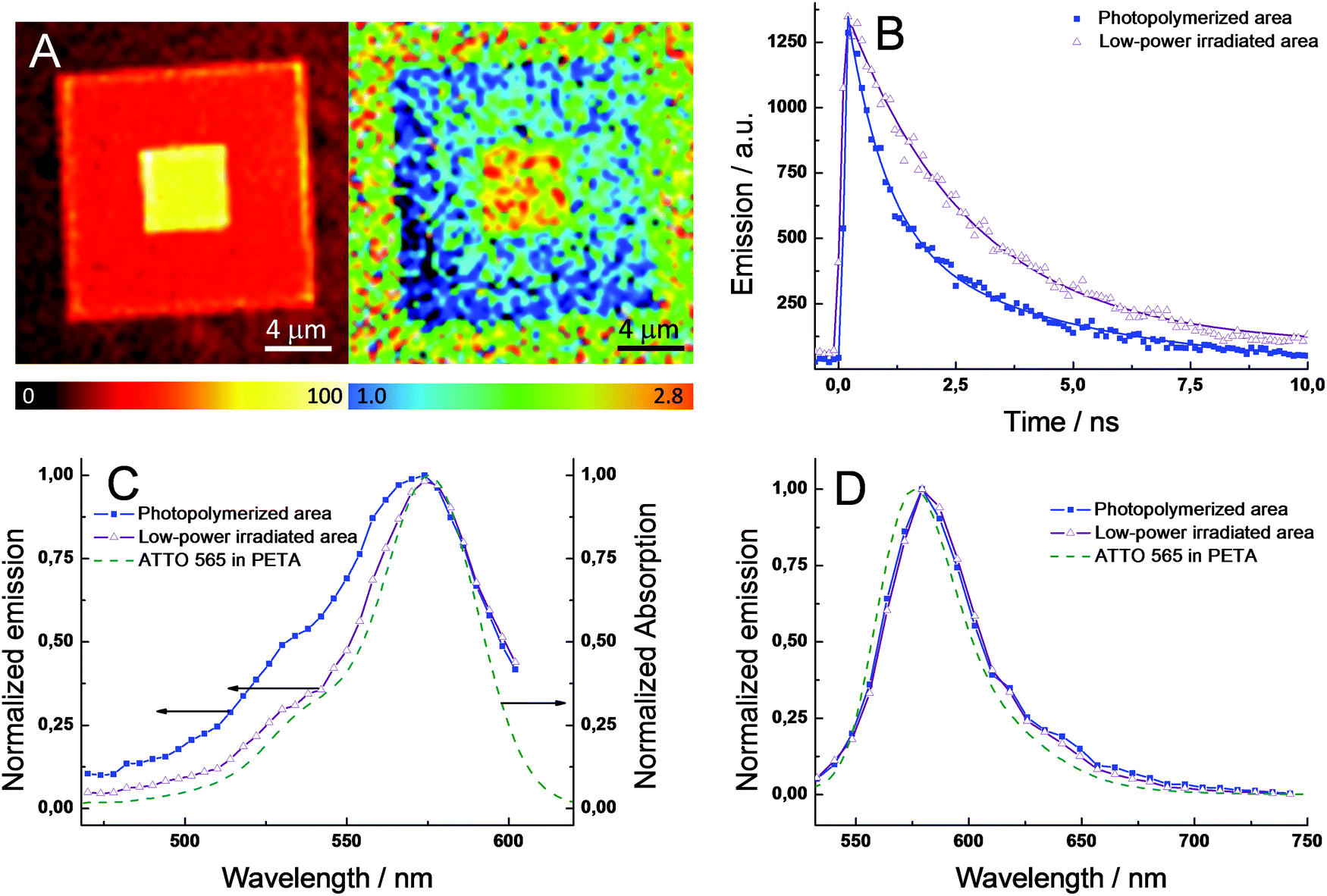 | ||
| Fig. 2 (A) Confocal image (left) and fluorescence lifetime mapping (right) of the same photopolymerized square (15 × 15 μm) containing ATTO 565 with the 5 × 5 μm central area irradiated once with a 36 mW fs pulsed laser. (B) Time profiles of the fluorescence signal (540–640 nm) at different positions in (A): external photopolymerized area (blue-filled squares) and the low-power irradiated central area (purple-emptied triangles). (C) Excitation and (D) emission spectra at the two areas as in (A). The solid lines in (A) correspond to the best fits of the experimental data with a mono- or bi-exponential function. λexc = 488 nm. The steady-state absorption and emission spectra for ATTO 565 dissolved in PETA are shown in (C) and (D) for comparison (green dashed line). | ||
Hence, a change in the ATTO 565 concentration upon the second irradiation step can be ruled out. Unlike the behavior shown in the photopolymerization of the monomer resin (vide supra), the low-power beam used during the second irradiation and the higher stability of this kind of dye when embedded in a solid matrix, photobleaching is not a major issue in this process. Fig. 2A exhibits the fluorescence lifetime mapping of a photopolymerized structure similar to the one shown in Fig. 1D. The lifetimes are calculated by fitting the experimental data from a group of pixels with a monoexponential function. Clearly, the central area presents a longer lifetime compared to the external area of the square, 2.7 vs. 1.1 ns. Interestingly, the lifetime at the central area is approximately 140% higher, which matches well with the increase of the fluorescence intensity in the confocal image (∼120%). The fluorescence quantum yield (ϕfl) can be expressed through the following equation, ϕfl = τS1kfl, where τS1 is the lifetime of the fluorescent excited state (S1) and kfl is the rate constant for the radiative deactivation (see ESI†). Thus, assuming kfl is constant for ATTO 565 in both areas – different chemical environments do not modify the radiative rate constant, the 140% difference in excited state lifetime (τS1) will produce the same increase in ϕfl in between both areas. In conclusion, the enhanced fluorescence signal upon the second irradiation cannot be ascribed to a higher concentration of ATTO 565 at the central area, but to an increase of ϕfl.
To continue with the photophysical investigations, Fig. 2B shows the fluorescence time profiles for large areas at the central and external areas. Both time profiles were fitted by using exponential functions. However, to obtain an accurate fit of the experimental data, mono- and bi-exponential functions were utilized to fit the central and external area, respectively. Table 1 collects the lifetimes (τS1) and relative fractional amplitudes (ai) for both fits: τ1 = 2.66 ns for the central area and τ1 = 2.55 ns and τ2 = 0.45 ns for the external area. The single lifetime found in the central area points out that only one species is responsible for the fluorescence signal. Interestingly, the value of 2.66 ns is close to the lifetime of the dye monomer in water (τS1 = 3.4 ns in water) – small differences are expected due to the distinct molecular environment of ATTO 565 when embedded in PETA or water – and hence it is possible to infer that the one species of the central area corresponds to the dye monomer. At the external area, the bi-exponential fit of the experimental data suggests a more complicated photophysical scheme, likely involving the presence of more than one fluorescent species. In particular, τ1 is very similar to that obtained for the central area, which might indicate the presence of the ATTO 565 monomer. The second lifetime (τ2) is very short indicating a fast deactivation of the emissive state compared to that in the monomer. An alternative hypothesis to explain the bi-exponential behavior is the existence of photophysical reactions between excited states (e.g. singlet–singlet annihilation), which might occur due to the high concentration of ATTO 565 inside the resin and the high peak-power used lasers.20 Indeed, to achieve the interaction of two molecules in their excited states (e.g. S1 state) it is necessary to maintain close contact between them, otherwise they decay through monomolecular reactions. Thereby, a high concentration of molecules in the excited state promotes the bimolecular reaction, which generates deactivation kinetics following the non-mono-exponential decay of the fluorescence signal. Thus, to assess the existence of excited state reactions, fluorescence time profiles at different intensities of the excitation laser were studied. However, no changes in the fluorescence decay kinetics were observed, which rejects the occurrence of excited state reactions. Next, the nature of the emissive species was investigated in detail. Fig. 2C and 2D exhibit the excitation and emission spectra for the two regions in the photopolymerized square, respectively. The spectra for ATTO 565 dissolved in PETA resin are also shown for comparison. No significant changes are observed in the shape of the emission spectra at the two areas, indicating a similar emissive species (Fig. 2D). Moreover, there is a good match with the emission spectrum of ATTO 565 in PETA. Assuming ATTO 565 appears as a monomer when dissolved in PETA, this proves the presence of a monomer in both areas of the polymerized structure. However, the excitation spectra are clearly different at the two areas (Fig. 2C). At the low-power irradiated central area, the excitation spectrum resembles the absorption spectrum of ATTO 565 in PETA, which supports the presence of merely a monomer in this area with a single fluorescence lifetime. In the photopolymerized external area, the excitation spectrum is broader with a notable shoulder centered around 530 nm (Fig. 2C). The development of new bands together with the broadening of the existing ones in the absorption and excitation spectra of dyes is typically ascribed to the formation of aggregates. In particular, several articles have demonstrated the appearance of shoulders at the high-energy region of the spectrum in different rhodamines due to the aggregation of the dye inside solid host materials, e.g. LAPONITE® clay.21 In our system, ATTO 565 molecules dissolved in PETA are present as monomers despite the high concentration added. However, after the polymerization process the solid resin tightly constrains the free movement of the ATTO 565 molecules inside the host material. As a consequence of this phenomenon and due to the close proximity of the molecules given the high dye loading, ATTO 565 molecules interact with each other inside the resin forming different types of aggregates. In particular, the high-energy aggregates (sandwich H-type dimers) of rhodamines operate as very efficient fluorescence quenchers due to the opening of non-radiative deactivation pathways, e.g. energy-transfer.21 Therefore, the increase of ϕfl in the central areas can be explained by the di-aggregation of the ATTO 565 during the second irradiation step. In fact, the partial evaporation of the resin matrix due to heat accumulation produces an expansion of the free volume available for the dye. Consequently, an increase in the mean distance between the dyes is expected preventing the interaction between molecules. Moreover, the energy available in the system due to the temperature increase can be used to directly break the aggregates into its respective monomers, producing a further di-aggregation of the dye. To further validate the hypothesis that attributes the enhancement in fluorescence signal to di-aggregation, we accomplished the fabrication of structures by varying the relative ratio of the H-aggregate/monomer. Thus, a decrease of the H-aggregate percentage is achieved by increasing the power of the fabricating beam. Fig. S4A† shows the excitation spectra for each of those structures showing a gradual growth of the shoulder at the high-energy region (530 nm), which is related to a higher percentage of H-aggregates in the sample. To correlate the relative ratio of H-aggregates with the photophysics of the ATTO 565, the fluorescence time profiles for each sample were studied. Fig. S4B† clearly exhibits a faster deactivation of the emissive state (S1) upon increasing the relative ratio of H-aggregates in the sample. Summarizing, the photopolymerized areas irradiated with the low power fs pulsed beam (large ϕfl) present a slower deactivation of the fluorescence signal (longer S1 lifetime), which is consistent with the presence of monomers of the dye. In fact, the excitation spectra of the non-irradiated areas feature a high-energy absorption associated with the formation of H-aggregates, which involve the appearance of non-radiative deactivation pathways for the excited state (shorter τS1/smaller ϕfl). Thus, the enhanced fluorescence signal upon irradiation with the fs beam is ascribed to a change in the aggregation of the ATTO 565, which resulted in an increase of the monomer percentage.
| τ 1/ns | a 1 | τ 2/ns | a 2 | |
|---|---|---|---|---|
| Center | 2.66 | |||
| Exterior | 2.55 | 0.52 | 0.46 | 0.48 |
To demonstrate the ability of our method to control the intensity and nanometer localization of the signal, three different fluorescent patterns were fabricated within a polymerized matrix. This selective enhancement of the fluorescence signal in the polymer is susceptible for employment in fabricating ODS devices due the ease in preparation, the good stability of the system, and the good resolution achieved with this method. The readout of the written information is carried out with a confocal fluorescence microscope using a continuous-wave (CW) laser operating at 532 nm to excite the ATTO 565 molecules and the fluorescence was detected in the spectral range of 540–640 nm. Fig. 3A shows 20 fluorescent dots created by irradiating eight times (dwell time = 300 μs) with a 20 mW fs pulsed beam on the previously photopolymerized square. It is clear that there is good reproducibility in the size and in the intensity of the fluorescent dots. Fig. 3B exhibits the fluorescence profiles for five of these dots, the maximum signal being more than twice that of the polymer matrix. The average radius of the fluorescent dots is around 270 nm, which matches well with the diffraction-limited spot size of the fs beam (λ = 760 nm, NA = 1.4). This is a particularly small value for fluorescence pixels that exhibit good contrast when compared to other ODS devices fabricated with a short-pulsed laser beam.13,16
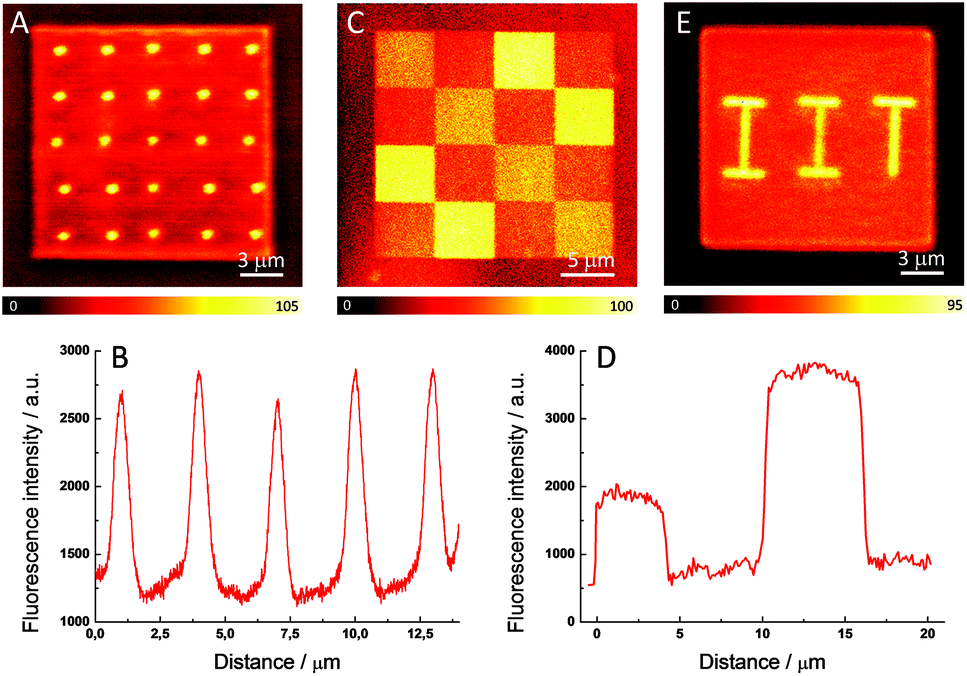 | ||
| Fig. 3 Confocal fluorescence images of a photopolymerized square fabricated from a ATTO 565-doped resin and subsequently irradiated with a 20 mW fs pulsed laser to produce (A) a set of 20 fluorescent dots, (C) chessboard-like fluorescent pattern and (E) the acronym of the Italian Institute of Technology. (B) and (D) Fluorescence profiles of five of the fluorescent dots (B) and the upper row in the chessboard-like fluorescent pattern (D). | ||
Thus, the enhancement of the fluorescence is clearly restricted to the irradiated areas. In addition to this, the extension of the fluorescence enhancement can also be controlled by varying the power and number of exposures of the laser beam. Thus, unlike other mechanisms of fluorescence enhancement, di-aggregation allows a precise adjustment of the percentage of aggregates with a linear control of the fluorescence intensity. Fig. 3C presents a confocal fluorescence image of a 20 × 20 μm photopolymerized square which is spatially-selectively irradiated with a 20 mW pulsed beam. The four 5 × 5 μm brighter areas are irradiated eight times and the other four 5 × 5 μm medium intensity areas located in the diagonal are exposed four times. There are another eight 5 × 5 μm dark areas which correspond to the fluorescence signal from the non-irradiated polymerized dye-doped matrix. Fig. 3D displays the fluorescence profiles with the three well-defined steps which demonstrate the achievement of a tunable modification of the intensity of the fluorescence signal. To the best of our knowledge, no additional parameters out of the three spatial coordinates has been demonstrated before to increase the amount of data that can be stored. This result clearly differentiates our device from other ODS systems since a non-binary code can be employed. Finally, as an illustrative example of the ability of this method to fabricate arbitrary fluorescent structures, Fig. 3E shows the acronym of the Italian Institute of Technology perfectly defined on a photopolymerized square.
4. Conclusions
In summary, we have developed a simple procedure to achieve the selective fluorescence functionalization of dye-doped photopolymerized structures fabricated with DLW lithography. This method relies on the thermally-induced di-aggregation of a fluorescent dye, ATTO 565, which generates an enhancement of the fluorescence signal in the polymer matrix. The di-aggregation is promoted via the spatially-selective irradiation of the polymer with the same fs pulsed beam employed for the polymerization, which involves an easy, fast and inexpensive functionalization of the polymer with this method. Well-defined fluorescence pixels (area = 0.24 μm2) were written inside a polymer matrix and the intensity of the signal can also be spatially-controlled by varying the power of the fs pulsed beam. The size of the pixel is comparable to the focal volume of the irradiating beam, suggesting that the use of sub-diffraction writing beams (superresolution approach) might decrease the pixel dimensions even more. The spatially- and intensity-selective fluorescence devices fabricated with this method can have a wide range of applications in the field of nanophotonics. In particular, the straightforward application is the utilization of these systems as ODS devices with a high storage capacity, due to the small dimensions of the fabricated pixels and the option of storing information in a non-binary code (intensity-controlled fluorescence).Acknowledgements
G. M. is grateful to the European Commission for a Marie Curie CIG grant (no. 631316).References
- G. M. Wallraff and W. D. Hinsberg, Chem. Rev., 1999, 99, 1801 CrossRef CAS PubMed.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697 CrossRef CAS PubMed.
- C. N. LaFratta, J. T. Fourkas, T. Baldacchini and R. A. Farrer, Angew. Chem., Int. Ed., 2007, 46, 6238 CrossRef CAS PubMed.
- M. Farsari, M. Vamvakaki and B. N. Chichkov, J. Opt., 2010, 12, 124001 CrossRef.
- Y.-L. Zhang, Q.-D. Chen, H. Xia and H.-B. Sun, Nano Today, 2010, 5, 435 CrossRef CAS.
- M. Malinauskas, M. Farsari, A. Piskarskas and S. Juodkazis, Phys. Rep., 2013, 553, 1 CrossRef.
- H.-B. Sun, T. Tanaka, K. Takada and S. Kawata, Appl. Phys. Lett., 2001, 79, 1411 CrossRef CAS.
- T. Oishi, M. Goto, A. Kasahara and M. Tosa, Appl. Phys. A, 2004, 79, 1733 CrossRef CAS.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I.-Y. Sandy Lee, D. McCord-Maughon, J. Qin, H. Roeckel, M. Rumi, X.-L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS.
- Z.-B. Sun, X.-Z. Dong, W.-Q. Chen, S. Nakanishi, X.-M. Duan and S. Kawata, Adv. Mater., 2008, 20, 914 CrossRef CAS.
- S. Shukla, X. Vidal, E. P. Furlani, M. T. Swihart, K.- T. Kim, Y.- K. Yoon, A. Urbas and P. N. Prasad, ACS Nano, 2011, 5, 1947 CrossRef CAS PubMed.
- Y. S. Chen, A. Tal, D. B. Torrance and S. M. Kuebler, Adv. Funct. Mater., 2006, 16, 1739 CrossRef CAS.
- S. Wong, O. Kiowski, M. Kappes, J. K. N. Lindner, N. Mandal, F. C. Peiris, G. A. Ozin, M. Thiel, M. Braun, M. Wegener and G. Von Freymann, Adv. Mater., 2008, 20, 4097 CrossRef CAS.
- A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, J. Phys. Chem. A, 2009, 113, 13633 CrossRef CAS PubMed.
- O. C. Yanez, C. D. Andrade, S. Yao, G. Luchita, M. V. Bondar and K. D. Belfield, ACS Appl. Mater. Interfaces, 2009, 10, 2219 Search PubMed.
- J. Lott, C. Ryan, B. Valle, J. R. Johnson III, D. A. Schiraldi, J. Shan, K. D. Singer and C. Weder, Adv. Mater., 2011, 23, 2425 CrossRef CAS PubMed.
- F. Gallego-Gomez, F. del Monte and K. Meerholz, Nat. Mater., 2008, 7, 490 CrossRef CAS PubMed.
- G. de Miguel, M. Duocastella, G. Vicidomini and A. Diaspro, Opt. Express, 2015, 23, 24850 CrossRef PubMed.
- G. Vicidomini, I. Coto Hernández, M. d'Amora, F. Cella Zanacchi, P. Bianchini and A. Diaspro, Methods, 2014, 66, 124 CrossRef CAS PubMed.
- G. de Miguel, M. Ziolek, M. Zitnan, J. A. Organero, S. S. Pandey, S. Hayase and A. Douhal, J. Phys. Chem. C, 2012, 116, 9379 CAS.
- V. Martínez Martínez, F. López Arbeloa, J. Bañuelos Prieto and I. López Arbeloa, J. Phys. Chem. B, 2005, 109, 7443 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr06071k |
| This journal is © The Royal Society of Chemistry 2015 |