
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving DNA double-strand repair inhibitor KU55933 therapeutic index in cancer radiotherapy using nanoparticle drug delivery
Xi
Tian†
a,
Haydee
Lara†
a,
Kyle T.
Wagner
a,
Srinivas
Saripalli
a,
Syed Nabeel
Hyder
a,
Michael
Foote
a,
Manish
Sethi
a,
Edina
Wang
a,
Joseph M.
Caster
a,
Longzhen
Zhang
b and
Andrew Z.
Wang
*ab
aLaboratory of Nano- and Translational Medicine, Department of Radiation Oncology, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: zawang@med.unc.edu; Fax: +1 919-966-7681; Tel: +1 919-966-7700
bDepartment of Radiation Oncology, Affiliated Hospital of Xuzhou Medical College, Cancer Institute of Xuzhou Medical College, Jiangsu, China
First published on 10th November 2015
Abstract
Radiotherapy is a key component of cancer treatment. Because of its importance, there has been high interest in developing agents and strategies to further improve the therapeutic index of radiotherapy. DNA double-strand repair inhibitors (DSBRIs) are among the most promising agents to improve radiotherapy. However, their clinical translation has been limited by their potential toxicity to normal tissue. Recent advances in nanomedicine offer an opportunity to overcome this limitation. In this study, we aim to demonstrate the proof of principle by developing and evaluating nanoparticle (NP) formulations of KU55933, a DSBRI. We engineered a NP formulation of KU55933 using nanoprecipitation method with different lipid polymer nanoparticle formulation. NP KU55933 using PLGA formulation has the best loading efficacy as well as prolonged drug release profile. We demonstrated that NP KU55933 is a potent radiosensitizer in vitro using clonogenic assay and is more effective as a radiosensitizer than free KU55933 in vivo using mouse xenograft models of non-small cell lung cancer (NSCLC). Western blots and immunofluorescence showed NP KU55933 exhibited more prolonged inhibition of DNA repair pathway. In addition, NP KU55933 leads to lower skin toxicity than KU55933. Our study supports further investigations using NP to deliver DSBRIs to improve cancer radiotherapy treatment.
Introduction
Radiotherapy is a key component of cancer treatment, with nearly two-thirds of all cancer patients receiving radiotherapy sometime during their illness.1 Because of its importance, there has been long standing interest in the development of novel approaches to improve the therapeutic index of radiotherapy. One of the main strategies has been to administer radiosensitizers, agents that can sensitize tumor cells to radiotherapy, in conjunction with radiotherapy. Today, the most commonly utilized radiosensitizers are chemotherapeutics.2 The addition of these radiosensitizers to radiotherapy has resulted in improved local tumor control, survival, and the rates of cancer cure for many cancers.2,3 Despite the success with chemotherapeutics as radiosensitizers, they are not without limitations. The combined treatment cannot always eradicate the primary tumor, especially in diseases such as pancreatic cancer. Adding chemotherapeutics to radiotherapy have also significantly increased overall treatment toxicity.4,5 Therefore, there is a strong need for the development of novel radiosensitizers that can further improve radiotherapy treatment.Among the many classes of agents that can improve radiotherapy, DNA double-strand repair inhibitors (DSBRIs) is the most promising. DSBRIs, as their name suggest, inhibit the repair of DNA double-strand breaks (DSB) in cells. DSBs can result spontaneously or from exposure to ionizing radiation and/or certain chemotherapeutic agents such as topoisomerase inhibitors. If DSBs are not repaired, the result is severe genomic instability that generally leads to cell death. Since the induction of DSBs is the principal mechanism of action of radiotherapy, DSBRIs hold high potential in improving chemoradiotherapy.6 There are three key proteins involved in the detection and repair of DNA DSBs: ataxia-telangiectasia mutated (ATM), ATM and Rad3 related (ATR), and DNA-dependent protein kinase (DNA-PK).7 All three belong to a family of proteins called phosphatidylinositol 3-kinase related kinases (PIKKs). ATM and ATR signal to the cell cycle and apoptotic pathways that DNA DSBs have occurred.8 In response to DSB, ATM phosphorylates histone H2AX which can be visualized as foci and considered as a marker of induction of DSB. DNA-PK repairs DSBs through a process called non-homologous end-joining.9 Over the past five decades, several DSBRIs have been developed for clinical application.10–12 None of them, however, have been translated clinically due to their potential toxicity to normal tissues and poor solubility. These inhibitors are so effective at inhibiting DNA DSBs that if systemically administered, in addition to sensitizing tumor cells, they would greatly sensitize normal cells to the effects of radiotherapy. Hence, the key challenge in utilizing DSBRIs is identifying methods to deliver them selectively to tumors while minimizing drug concentrations in normal tissues. While such differential drug delivery is generally not possible with traditional drug delivery techniques, the development of nanoparticle (NP) drug delivery carriers offers an unprecedented opportunity. NPs preferential accumulation in tumors and their low distribution in normal tissue are characteristics that are well suited for delivering DSBRIs.13,14 Compared to small molecules, the differential drug concentration between tumor and its surrounding tissue of NPs should lead to higher therapeutic efficacy and lower toxicity when combined with radiotherapy (Fig. 1A and B). We hypothesized that NP delivery of DSBRIs can reduce the toxicity of DSBRIs and enable their clinical translation as radiosensitizers. To demonstrate the proof-of-principle of this approach, we engineered NP formulation of an ATM inhibitor, KU55933, and evaluated its efficacy and toxicity as a radiosensitizer using a mouse xenograft model of non-small cell lung cancer.
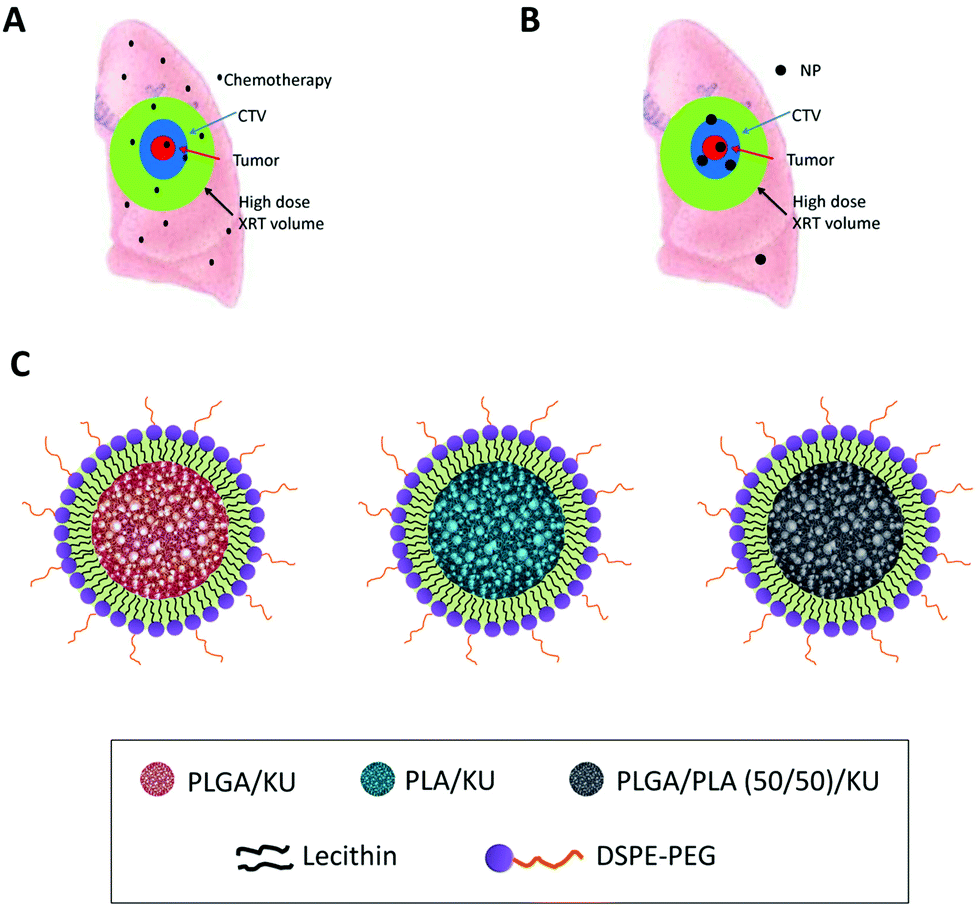 | ||
| Fig. 1 Diagram of chemoradiotherapy for lung cancer. The red area is the gross tumor. Blue area is CTV (clinical target volume), which is the area of lung that should receive high dose radiotherapy and radiosensitizer. Green area indicates the area of lung tissue that receives high dose radiotherapy (XRT) because of motion, entrance and exit radiotherapy dose. In conventional treatment (A) with small molecule chemotherapeutics such as KU55933, the green area receives both high dose radiotherapy and radiosensitizers. In contrast, NP therapeutics (B) concentrate in tumors and minimize the area of normal lung that receives both radiotherapy and radiosensitizer/chemotherapy. (C) Cartoon of NP KU55933 with different lipid polymers. | ||
Material and methods
Mice
Nu/Nu mice (male, 6–8 weeks old) were obtained from the animal colony at the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center (LCCC). NOD SCID mice (male, 16 week old) were purchased from Charles River Laboratories (Wilmington, MA). All animal experiments were approved by and in accordance with guidelines provided by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee.Formulation and characterization of NP KU55933
The lipid/polymeric KU55933 nanoparticle was synthesized using modified a nanoprecipitation technique that has been previously reported.15,16 Three lipid polymers have been used as nanoparticle carrier for drug delivery: PLGA (poly(D,L-lactide-co-glycolide), with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 monomer ratio, ester terminated, Mw 50000–75000), PLA (poly(D,L-lactide), ester terminated, Mw 18000–28000), PLGA/PLA (50/50) (Fig. 1C). The core consists of either PLGA, PLA or PLGA/PLA (50/50) and KU55933 (Selleck Chemicals, Houston, TX). KU55933 was dissolved into the polymer/acetonitrile solution (10 mg mL−1 of PLGA, PLA, or PLGA/PLA in acetonitrile) before the nanoprecipitation. The outer shell consists of lecithin containing 90–95% phosphatidylcholine (Alfa Aesar, Ward Hill, MA) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG2000) (Avanti Polar Lipids, Alabaster, AL). Nanoparticle size and surface charge were determined with a Malvern Zetasizer. TEM images were obtained at the Microscopy Services Laboratory Core Facility at the University of North Carolina School of Medicine.
:50 monomer ratio, ester terminated, Mw 50000–75000), PLA (poly(D,L-lactide), ester terminated, Mw 18000–28000), PLGA/PLA (50/50) (Fig. 1C). The core consists of either PLGA, PLA or PLGA/PLA (50/50) and KU55933 (Selleck Chemicals, Houston, TX). KU55933 was dissolved into the polymer/acetonitrile solution (10 mg mL−1 of PLGA, PLA, or PLGA/PLA in acetonitrile) before the nanoprecipitation. The outer shell consists of lecithin containing 90–95% phosphatidylcholine (Alfa Aesar, Ward Hill, MA) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG2000) (Avanti Polar Lipids, Alabaster, AL). Nanoparticle size and surface charge were determined with a Malvern Zetasizer. TEM images were obtained at the Microscopy Services Laboratory Core Facility at the University of North Carolina School of Medicine.
Drug release
NP KU55933 solutions of 1 mg mL−1 were added into Slide-A-Lyzer Mini Dialysis Device with a molecular weight cutoff of 2 k MWCO (ThermoScientific, Rockford, IL) and subjected to dialysis against 4 L of PBS 1× with a gentle stirring at 37 °C. Since KU55933 has extremely low solubility in water, this dialysis method using large quantity of PBS (pH7.4) to ensure that KU55933 inside the nanoparticles is less than 10% of its solubility limit in PBS. The PBS was changed daily during the dialysis process to avoid reaching the equilibrium of the drug in the buffer. At the indicated times, 50 μL of solution from the microtubes was removed and mixed with 50 μL of acetonitrile. To determine KU55933 content, the samples were subjected to quantitative analysis in a Shimadzu UFLC System with a Chromolith Fast Gradient column (Merck KGaA, Germany) and read at 254 nm. KU55933 retention time was 9.2 minutes in a gradient elution of 11.0 minutes with water and acetonitrile at 0.25 mL min−1.Cell culture
The lung cancer cell lines H460, A549 and H23 were obtained from the Tissue Culture Facility at the University of North Carolina. Unless otherwise stated, all the mediums and supplements were obtained from Gibco (Carlsbad, CA). H460 was cultured in RPMI medium supplemented with 2 mM glutamine, 4.5 g L−1 glucose (Sigma, St Louis, MO), 1.5 g L−1 sodium bicarbonate, 10 mM HEPES, 1 mM sodium pyruvate, 10% FBS and 1× Penicillin/Streptomycin. A549 was cultured in DMEM high glucose supplemented with 10% FBS and 1× Penicillin/Streptomycin. H23 was cultured in RPMI supplemented with 10% FBS and 1× Penicillin/Streptomycin. The cells were incubated at 37 °C in a humidified 5% CO2 atmosphere.X-Ray irradiation
Cells and mice were irradiated using a Precision X-RAD 320 (Precision X-Ray, Inc) machine operating at 320 kVp and 12.5 mA. During irradiation, a 5 mm lead shield protected the mice's vital organs while the left flank remained exposed. For hair loss toxicity, a 0.5 cm thick bolus covered the left dorsal region of the mice during irradiation to enhance the skin dose.Clonogenic survival assay
Plating efficiency (PE) of each cell line was determined first for each cell line. Tumor cells were plated in 60 mm plates at a density of 3 × 105 cells per plate the day before the KU55933 treatment. The cells were treated with 5 to 10 μM of KU55933 as a free drug formulation (KU) or nanoparticles (NP KU). The free drug formulation consists of the drug dissolved 10 mg mL−1 in a 1:1 solution of Tween80 (Sigma, St Louis, MO) and ethanol. Six hours after the drug treatment, the cells were treated with ionizing radiation (0, 1, 2, 4, 6, and 8 Gy). The dose rate at a source-subject distance of 50 cm was ∼2.5 Gy min−1. Eighteen hours after irradiation (and a total of twenty-four hours of KU55933 treatment), the cells were trypsinized and plated at various densities to determine the surviving fraction via the clonogenic assay. The cells were incubated for fourteen days after plated and then fixed with a 1:1 methanol/acetone solution and stained with trypan blue (Sigma, St Louis, MO). All colonies with over 50 cells were counted. Surviving fraction (SF) was calculated using the formula # of colonies/(# of plated cells)(PE). SF were then plotted against radiation dose on a log scale.17
Western blot
H460 cells (2.5 × 106 cells in 2 mL RPMI 1640) were treated with 10 μM KU55933, NP KU55933, or NP for 3 hours, washed with PBS, and incubated with fresh media before 1 Gy irradiation. Cells were lysed in RIPA lysis and extraction buffer (25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitor cocktail (Thermo Scientific) at the indicated times post irradiation. Protein concentration was determined using bicinchoninic acid protein assay (Pierce). Protein (15 μg) from each sample was mixed with 2× Laemmli sample buffer (Bio-Rad), boiled at 100 °C for 5 min, loaded onto SDS-PAGE gel and transferred to PVDF membrane. The membrane was incubated at 4 °C overnight with primary antibodies against p-AKT (1:1000, cell signaling #4060), p-ATM (1:1000, cell signaling #4526), and β-actin (1:1000, cell signaling #4970). The secondary antibodies used were anti-rabbit or anti-mouse IgG HRP-linked antibody (1:5000, cell signaling). The intensity of protein bands on the western blot image was quantified by Image J software. Ratios of p-ATM and p-AKT protein levels after β-actin normalization were calculated from three independent experiments.
Immunofluorescence
Cells were fixed in 4% formaldehyde for 15 min, rinsed with PBS, and incubated in goat blocking solution (2% goat serum, 1% BSA, 0.1% Triton X-100, and 0.05% Tween 20) for 1 hour. After blocking, cells were immunostained with Anti-phospho-Histone H2AX (Ser139) antibody (Millipore, Cat #05-636) for 1 hour at room temperature. Cells were then washed with PBS and further incubated with secondary antibody Alexa Fluor 594 Donkey Anti-Mouse IgG Antibody (Molecular Probes #A21203) for 30 min. After washing, cells were mounted and coverslipped using ProLong Gold Antifade Mountant with DAPI (Molecular Probes #P36935). Images were acquired using a Zeiss LSM 700 laser scanning confocal microscope with z-stack sectioning (63× oil immersion objective lens) and stacked by maximum intensity projection. The number of γH2AX foci per cell was counted using Image J software.Tumor growth experiment
Six to eight week old Nu/Nu mice were subcutaneously inoculated in the left flank with 1 × 106 H460 or A549 cells resuspended in 200 μL of a 1:1 solution of matrigel (BD Biosciences, San Jose, CA) and RPMI 1640 medium (Gibco, Carlsbad, CA). Eleven days after inoculation and when the tumors reached about 100 mm3, the animals received a 500 μg kg−1 dose of KU55933 as a free drug formulation or NP through the tail vein. Six hours after the injection, the animals received the first dose of irradiation. The mice will receive a total of 15 Gy in three daily fractions. The dose rate at a source-subject distance of 70 cm was ∼0.5 Gy min−1. The mice were lead shielded to protect most of the organs and to allow radiation of the tumor site. The tumor growth was assessed starting on the day of the last irradiation treatment (Day 1) and every two days by measuring two perpendicular diameters with a caliper. The volume was calculated with the formula V = 0.5 × a × b2 where a is the larger and b is the smaller diameter.
Hair toxicity
Approximately sixteen week old NOD SCID white mice were injected with a 150 μg kg−1 dose of KU55933 as a free drug formulation or NP through the tail vein. Three hours after the injection, the animals received 14 or 16 Gy of irradiation from an X-RAD 320 machine operating at 320 kV and 12.5 mA. The dose rate at a source-subject distance of 70 cm was ∼0.5 Gy min−1. The mice were shielded with lead (5 mm) to protect most of the body and to allow radiation in the left leg. The hair loss and skin damage was assessed in a scale previously reported18 and recorded by images.Statistical analysis
γH2AX foci number, and clonogenic results were analyzed by either one-way or two-way ANOVA followed by Tukey's honestly significant difference post hoc test if a positive F test was detected. For tumor measurements, a method known as ‘response features analysis’ was used to analyze serial measures of tumor growth.19 This method allows for the comparison of individual growth delay profiles by using ‘summary measures’. The particular summary measure we used was the AUC (Area Under the Curve), which was particularly appropriate for the growth delay profiles we encountered. The Wilcoxon two-group method (using Van der Waerden normal scores) was used to test whether there was a significant difference in AUCs. Statistical analyses were performed using R software. P-Values less than 0.05 were considered significant.Results
Characterization of NP KU55933
NP KU55933 was formulated using a lipid-polymer NP platform.15,16 We formulated NPs using different lipid: polymer ratios and with different levels of drug loading. We found that a lipid/polymer weight ratio of 0.26 and 10% (wt/wt) drug loading was optimal for KU55933 encapsulation. Higher drug loading had lower encapsulation efficiency and did not lead to significantly higher KU55933 within the NPs. Compared with other lipid polymer formulations using PLA and PLGA/PLA, the lipid – coated PLGA gives the best loading efficacy and slowest release profile (Fig. 2). As shown in Fig. 2A, the encapsulation efficiency of KU55933 using PLGA was approximately 8.4% as compared to 6.8% with PLA and 7.2% with PLGA/PLA. Thus, we chose to use PEG-PLGA NP platform to encapsulate KU55933. Dynamic light scattering analysis of the NP KU55933 revealed NP sizes of 86.73 ± 0.57 nm and a polydispersity index of 0.1 ± 0.004. Transmission electron microscopy (TEM) of the NPs (Fig. 2B) showed well defined spherical NPs and confirmed the NP sizes. ζ-Potential analysis demonstrated that the NPs possess a negatively charged surface −36.1 ± 0.9 mV. In vitro drug release of KU55933 from NP is slow and in first-order release kinetic, with a half-life of 18.4 hours (Fig. 2).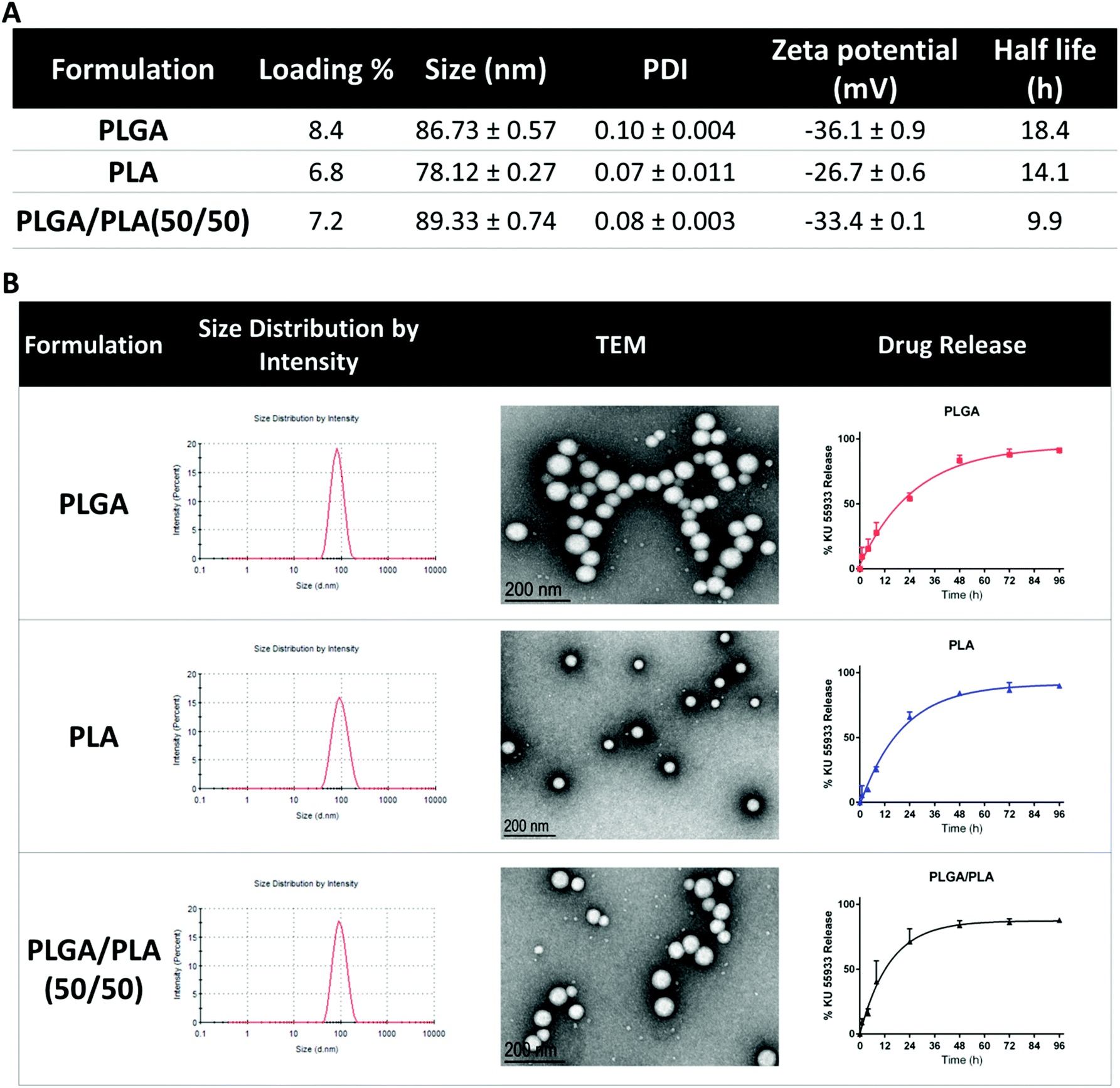 | ||
| Fig. 2 Characterization of NP KU55933 with different lipid polymers. (A) Comparison of different lipid polymer formulations of NP KU55933 in drug loading efficacy, size, zeta potential and half-life. (B) Nanoparticle size distribution, TEM image and release profile of NP KU55933. Values are represented as mean ± standard error (n = 3). | ||
NP KU55933 is an excellent radiosensitizer in vitro
NP KU55933 was evaluated as a radiosensitizer in vitro using three non-small cell lung cancer (NSCLC) cell lines (H460, A549 and H23). Radiation survival curves were then generated using surviving fractions at different radiation doses. NP KU55933 was found to be a potent radiosensitizer in all three cell lines, indicated by the downward shift of survival curves when NP KU55933 is given with radiotherapy (Fig. 3). NP KU55933 is not significantly better than free KU55933 in these in vitro studies, as expected and consistent with our previous studies.16,20 | ||
| Fig. 3 Clonogenic survival curves of cancer cell lines cultured with NP KU55933 or KU55933 for 6 hours, treated with various dose of radiation (XRT). H460 and A549 were treated with 10 μM and H23 with 5 μM of the drug. | ||
NP KU55933 leads to longer inhibition of DNA repair signaling pathways than that of KU55933
KU55933 is known to inhibit ATM and also prevent its downstream substrate AKT phosphorylation at Ser473.21 To compare the difference of NP KU55933 and KU55933 in inhibiting the DNA damage response pathways, we examined the effect on irradiation-induced phosphorylation of ATM and its target AKT in H460 cells. To avoid extensive cell death, we irradiated the cells with 1 Gy. As shown in Fig. 4A, untreated H460 cells had increased levels of phosphorylation of ATM, whereas NP KU55933 and free KU55933 suppressed p-ATM (S1981) expression at 10 μM until 24 hours after irradiation, suggesting NP KU55933 has similar target effects with free KU55933 against p-ATM. Since ATM is a major upstream activator of AKT through serine 473 phosphorylation in response to gamma-radiation,22 we further tested if NP KU55933 affects this response after radiation. Immediately followed by radiotherapy (1 Gy), both NP KU55933 and free KU55933 at 10 μM completely inhibited p-AKT (Ser473) (Fig. 4A). In contrast, AKT phosphorylation at Ser473 was restored in free KU55933-treated cells 3 hours after irradiation, while cells treated with NP KU55933 still showed a lower level of p-AKT (Ser473) (Fig. 4A). | ||
| Fig. 4 Effect of KU55933 formulations and radiation on phosphorylation of ATM, AKT and H2AX. H460 cells were pretreated with 10 μM KU or NP KU (containing 10 μM KU) for 3 h and subsequently irradiated (1 Gy). (A) Cell lysates were collected at indicated time points for western blot. The western blot band intensity was quantified after normalized to β-actin based on three independent experiments. (B) Cells were fixed at indicated time points for immunofluorescence of γH2AX. (C) Average numbers of γH2AX foci per cell were presented. | ||
One downstream mediator of ATM is H2AX. Upon DSBs, phosphorylation of H2AX at Ser 139 (γH2AX) is quickly recruited to damage sites and thus γH2AX foci form. When DNA is repaired, γH2AX can be dephosphorylated and foci disappear. Consistent with western blot results, confocal microscopy showed that cells treated with NP KU55933 had significantly higher level of γH2AX foci (p < 0.01) as compared to cells treated with free KU55933 after 1 and 24 h after radiation (Fig. 2B and C), suggesting NP KU55933 leads to longer inhibition of DNA repair signaling pathways than that of free KU55933.
NP KU55933 is more effective than KU55933 as a radiosensitizer in vivo
To validate our in vitro results and to evaluate the efficacy of NP KU55933 as a radiosensitizer in vivo, mice bearing either H460 or A549 flank xenograft tumors were treated with NP KU55933, free KU55933, or PBS (control) followed by radiotherapy. Radiotherapy was given in a fractionate regimen (5 Gy × 3). Tumor growth delay curves were generated and utilized to compare the therapeutic efficacy of NP KU55933 and free KU55933. We found that NP KU55933 lead to significantly longer tumor growth delay when compared to free KU55933 or control groups (H460: p = 0.004; A549: p = 0.03) (Fig. 5). Our in vivo data confirmed that NP KU55933 is a potent radiosensitizer with potential for clinical translation. | ||
| Fig. 5 In vivo efficacy of NP KU55933 as a radiosensitizer for non-small cell lung cancer. Mice bearing flank tumor xenografts (A) H460 and (B) A549 were intravenous injected with NP KU55933, KU55933 and saline followed by radiation therapy (XRT). Tumors were irradiated in 3 daily fractions of 5 Gy followed by chemotherapy injection. | ||
NP KU55933 has less hair toxicity than KU55933 when given concurrently with radiotherapy
To compare the potential toxicity of NP KU55933 to that of KU55933 with radiotherapy, we utilized the hair toxicity assay. Mice were given either NP KU55933 or KU55933 followed by a single dose of 16 Gy, a typical dose for hair toxicity studies. Hair loss was determined and scored based on an established method approximately four weeks after treatment. We found that NP KU55933 had significantly less skin toxicity when combined with radiotherapy than that of KU55933 (Fig. 6). | ||
| Fig. 6 In vivo toxicity of KU55933. (A) Hair loss in mice treated with radiation (XRT) and free drug (KU) or the nanoparticle formulation of KU55933 (NP KU). The animals were injected with a single dose of 150 μg kg−1 of KU and treated with 14 and 16 Gy on the day of the injection. The representative pictures were taken on day 21. (B) Skin reaction score on the mice treated with radiation (14 Gy) and KU. The animals were treated as indicated in (A) and a score was given based on the work published by Douglas and Fouler (1976). | ||
Discussion
The aim of our study is to demonstrate that NP delivery of DSBRIs can improve their therapeutic efficacy and, more importantly, lower their toxicity. We chose to use KU55933 as the model drug. KU55933 was chosen as it is a potent and selective inhibitor of ATM with IC50 of 13 nmol L−1 and a Ki of 2.2 nmol L−1.10 Since ATM is the key protein in sensing DSBs and initiating DSB repairs, we theorized that NP KU55933 will have high potential in improving radiotherapy.KU55933 is a hydrophobic molecule and is insoluble in water. Therefore, we utilized a polymeric micelle NP platform for its delivery. The hydrophobic polymeric core is used to encapsulate KU55933 and enable its controlled release in aqueous environments. Since polymer composition determines the hydrophilicity and rate of degradation which further affect the nanoparticle size, loading efficacy as well as drug release rate, we compared several different lipid polymer nanoparticle formulations of KU55933. NP KU55933 using PLGA formulation has best loading efficiency as well as prolonged drug release profile. The NP KU55933 size of 86.73 nm is excellent for tumor targeting.23 Since KU55933 is a very potent inhibitor of ATM, the 8.4% loading of KU55933 in NPs is more than adequate for ATM inhibition in tumors. Because the hydrophobicity of KU55933, it is released very slowly from NP and more prolonged than other hydrophobic molecules such as docetaxel.20 We do want to acknowledge that our method of conducting drug release (using PBS) likely under-estimate the true drug release kinetic of these NPs. Past literature have shown that in vivo release tend to be more rapid due to the interactions between NPs and serum proteins.24,25
To evaluate NP KU55933's therapeutic efficacy and toxicity, we chose to use NSCLC as a model disease. Chemoradiotherapy is an important treatment paradigm in the management of NSCLC.26 It has been shown to improve survival when compared to sequential treatment or single modality treatments.27 Despite its success, the overall overcome for NSCLC patients after radiotherapy remains poor.28 Furthermore, chemoradiotherapy treatment is very toxic with treatment mortality of 5–10%.28 Therefore, one of the key research objectives in lung cancer research has been to develop agents that can improve the therapeutic index of radiotherapy.
Our in vitro evaluations of KU55933 showed that NP KU55933 is as potent as KU55933. In general, NP therapeutics exhibits lower efficacy than their small molecule counterparts in in vitro studies due to pegylation of the NPs, which leads to lower intracellular uptake by tumor cells.20,29 The unique and favorable properties of nanotherapeutics are also not manifest under in vitro conditions. For these therapeutics, the higher therapeutic efficacy are seen in vivo despite the lower efficacy data in vitro. It is also important to note that the survival curves with KU55933 as radiosensitizer are linear in shape, consistent with KU55933's mechanism of action as a DNA repair inhibitor.
The potential mechanism of NP KU55933's excellent in vitro data is that NP KU55933 can provide more prolonged inhibition of DNA repair pathways, which can lead to higher therapeutic efficacy. Indeed, our results on AKT inhibition and ATM inhibition support this hypothesis. Notably, γH2AX foci persist 24 hours after radiation suggesting NP KU55933 leads to longer defects in DNA repair. The prolonged the suppression of DNA repair pathways can cause more tumor cells undergo mitotic death, the principle mechanism of death from cancer cells after radiotherapy.
Normal tissue toxicity from radiotherapy is difficult to study. One of the most well-established assays to assess radiation toxicity is hair toxicity in mice. It is a good surrogate for skin and mucosal toxicity, which is a one of the major toxicities of clinical radiotherapy. As expected, we found NP KU55933 led to lower hair toxicity than that of small molecule KU55933. This is consistent with our hypothesis that NP KU55933 has much less normal tissue distribution. Our data also suggest that the overall toxicity profile of NP KU55933 with radiotherapy is less than that of KU55933 with radiotherapy.
Conclusion
In summary, we have formulated a NP formulation of KU55933, a DSBRI, and evaluated its therapeutic efficacy and toxicity as a radiosensitizer. We demonstrated that NP formulation of KU55933 improved its therapeutic efficacy as well as lowered its toxicity. Our work demonstrates the proof-of-principle of using NPs to deliver DSBRI, a potent class of anti-cancer therapeutic, to renew their clinical translation potential.Future perspective
One of the key applications of nanomedicine will be in radiation oncology.30 Preclinical data have consistently shown that nanotherapeutics are more effective than their small molecule counterparts when utilized in concurrent chemoradiation. One nanotherapeutic, CRLX101, is already under investigation in chemoradiation in rectal cancer (NCT02010567). Future research will aim to translate novel radiosensitizers, such as DSBRI, to further improve radiotherapy treatment for cancer.Executive summary
• DNA double strand break repair inhibitors (DSBRIs) hold high potential in improving radiotherapy treatment for cancer• Clinical translation of DSBRIs has been limited by potential toxicity and drug delivery challenges
• Polymeric NP delivery of KU55933, a DSBRI, improves its efficacy in vitro and in vivo with radiotherapy in non-small cell lung cancer model
• NP KU55933 has less skin toxicity than that of small molecule KU55933
• NP delivery of DSBRI is a potential clinical translation strategy for this class of agents
Disclosure
The author reports no conflicts of interest in this work.Acknowledgements
This work was supported by the University Cancer Research Fund from the University of North Carolina and R01CA178748-01 from the National Institutes of Health/National Cancer Institute. AZW was also supported by Career Development Award 5-K12-CA120780-01-05 and National Institutes of Health Center for Nanotechnology Excellence Grant 1-U54-CA151652-01.References
- A. S. f. R. Oncology, Fast Facts About Radiation Therapy, Accessed 1/14/2014, 2014.
- T. Y. Seiwert, J. K. Salama and E. E. Vokes, Nat. Clin. Pract., 2007, 4, 86–100 CrossRef CAS PubMed
.
- P. J. Eifel, Semin. Radiat. Oncol., 2006, 16, 177–185 CrossRef PubMed
- J. S. Cooper, T. F. Pajak, A. A. Forastiere, J. Jacobs, B. H. Campbell, S. B. Saxman, J. A. Kish, H. E. Kim, A. J. Cmelak, M. Rotman, M. Machtay, J. F. Ensley, K. S. Chao, C. J. Schultz, N. Lee and K. K. Fu, N. Engl. J. Med., 2004, 350, 1937–1944 CrossRef PubMed
- A. A. Forastiere, H. Goepfert, M. Maor, T. F. Pajak, R. Weber, W. Morrison, B. Glisson, A. Trotti, J. A. Ridge, C. Chao, G. Peters, D. J. Lee, A. Leaf, J. Ensley and J. Cooper, N. Engl. J. Med., 2003, 349, 2091–2098 CrossRef CAS PubMed
-
A. G. Eric and J. Hall, Radiobiology for the Radiologist, Lippincott Williams & Wilkins, Philadelphia, PA, 2011 Search PubMed
- J. H. Hoeijmakers, Nature, 2001, 411, 366–374 CrossRef CAS PubMed
- R. T. Abraham, Genes Dev., 2001, 15, 2177–2196 CrossRef CAS PubMed
- B. L. Mahaney, K. Meek and S. P. Lees-Miller, Biochem. J., 2009, 417, 639–650 CrossRef CAS PubMed
- I. Hickson, Y. Zhao, C. J. Richardson, S. J. Green, N. M. Martin, A. I. Orr, P. M. Reaper, S. P. Jackson, N. J. Curtin and G. C. Smith, Cancer Res., 2004, 64, 9152–9159 CrossRef CAS PubMed
- Y. Zhao, H. D. Thomas, M. A. Batey, I. G. Cowell, C. J. Richardson, R. J. Griffin, A. H. Calvert, D. R. Newell, G. C. Smith and N. J. Curtin, Cancer Res., 2006, 66, 5354–5362 CrossRef CAS PubMed
- D. Chirnomas, T. Taniguchi, M. de la Vega, A. P. Vaidya, M. Vasserman, A. R. Hartman, R. Kennedy, R. Foster, J. Mahoney, M. V. Seiden and A. D. D'Andrea, Mol. Cancer Ther., 2006, 5, 952–961 CrossRef CAS PubMed
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith, V. P. Torchilin and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4607–4612 CrossRef CAS
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed
- L. Zhang, J. M. Chan, F. X. Gu, J. W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed
- S. Karve, M. E. Werner, R. Sukumar, N. D. Cummings, J. A. Copp, E. C. Wang, C. Li, M. Sethi, R. C. Chen, M. E. Pacold and A. Z. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8230–8235 CrossRef CAS PubMed
- A. Munshi, M. Hobbs and R. E. Meyn, Methods Mol. Med., 2005, 110, 21–28 Search PubMed
- V. Holler, V. Buard, M. H. Gaugler, O. Guipaud, C. Baudelin, A. Sache, R. Perez Mdel, C. Squiban, R. Tamarat, F. Milliat and M. Benderitter, J. Invest. Dermatol., 2009, 129, 1280–1291 CrossRef CAS PubMed
- J. N. Matthews, D. G. Altman, M. J. Campbell and P. Royston, BMJ, 1990, 300, 230–235 CrossRef CAS PubMed
- M. E. Werner, J. A. Copp, S. Karve, N. D. Cummings, R. Sukumar, C. Li, M. E. Napier, R. C. Chen, A. D. Cox and A. Z. Wang, ACS Nano, 2011, 5, 8990–8998 CrossRef CAS PubMed
- Y. Li and D. Q. Yang, Mol. Cancer Ther., 2010, 9, 113–125 CrossRef CAS PubMed
- J. G. Viniegra, N. Martinez, P. Modirassari, J. Hernandez Losa, C. Parada Cobo, V. J. Sanchez-Arevalo Lobo, C. I. Aceves Luquero, L. Alvarez-Vallina, S. Ramon y Cajal, J. M. Rojas and R. Sanchez-Prieto, J. Biol. Chem., 2005, 280, 4029–4036 CrossRef CAS
- E. A. Sykes, J. Chen, G. Zheng and W. C. Chan, ACS Nano, 2014, 5696–5706 CrossRef CAS PubMed
- L. Feng, H. Wu, P. Ma, R. J. Mumper and S. R. Benhabbour, Int. J. Nanomed., 2011, 6, 2545–2556 CAS
- S. Modi and B. D. Anderson, Mol. Pharm., 2013, 10, 3076–3089 CrossRef CAS PubMed
- W. Eberhardt, C. Pottgen and M. Stuschke, Nat. Clin. Pract. Oncol., 2006, 3, 188–199 CrossRef CAS PubMed
- R. O. Dillman, J. Herndon, S. L. Seagren, W. L. Eaton Jr. and M. R. Green, J. Natl. Cancer Inst., 1996, 88, 1210–1215 CrossRef CAS
- P. Fournel, G. Robinet, P. Thomas, P. J. Souquet, H. Lena, A. Vergnenegre, J. Y. Delhoume, J. Le Treut, J. A. Silvani, E. Dansin, M. C. Bozonnat, J. P. Daures, F. Mornex and M. Perol, J. Clin. Oncol., 2005, 23, 5910–5917 CrossRef CAS PubMed
- M. E. Werner, N. D. Cummings, M. Sethi, E. C. Wang, R. Sukumar, D. T. Moore and A. Z. Wang, Int. J. Radiat. Oncol., Biol., Phys., 2013, 86, 463–468 CrossRef CAS PubMed
- A. Z. Wang and J. E. Tepper, J. Clin. Oncol., 2014, 32, 2879–2885 CrossRef CAS
Footnote |
| † These authors contributed equally to the work of this manuscript. |
| This journal is © The Royal Society of Chemistry 2015 |