
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing local pH-based precipitation processes in self-assembled silica-carbonate hybrid materials†
Julian
Opel
a,
Mandy
Hecht
b,
Knut
Rurack
b,
Josef
Eiblmeier
c,
Werner
Kunz
c,
Helmut
Cölfen
*a and
Matthias
Kellermeier
*d
aPhysical Chemistry, University of Konstanz, Universitätsstrasse 10, D-78464 Konstanz, Germany. E-mail: helmut.coelfen@uni-konstanz.de; Fax: +49 7531 88 3139; Tel: +49 7531 88 4063
bDiv. 1.9 Chemical and Optical Sensing, BAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Strasse 11, D-12489 Berlin, Germany
cInstitute of Physical and Theoretical Chemistry, University of Regensburg, Universitätsstrasse 31, D-93053 Regensburg, Germany
dMaterial Physics, BASF SE, GMC/O – B007, Carl-Bosch-Strasse 38, D-67056 Ludwigshafen, Germany. E-mail: matthias.kellermeier@basf.com; Fax: +49 621 66 43388; Tel: +49 621 60 43388
First published on 28th September 2015
Abstract
Crystallisation of barium carbonate in the presence of silica can lead to the spontaneous assembly of highly complex superstructures, consisting of uniform and largely co-oriented BaCO3 nanocrystals that are interspersed by a matrix of amorphous silica. The formation of these biomimetic architectures (so-called silica biomorphs) is thought to be driven by a dynamic interplay between the components, in which subtle changes of conditions trigger ordered mineralisation at the nanoscale. In particular, it has been proposed that local pH gradients at growing fronts play a crucial role in the process of morphogenesis. In the present work, we have used a special pH-sensitive fluorescent dye to directly trace these presumed local fluctuations by means of confocal laser scanning microscopy. Our data demonstrate the existence of an active region near the growth front, where the pH is locally decreased with respect to the alkaline bulk solution on a length scale of few microns. This observation provides fundamental and, for the first time, direct experimental support for the current picture of the mechanism underlying the formation of these peculiar materials. On the other hand, the absence of any temporal oscillations in the local pH – another key feature of the envisaged mechanism – challenges the notion of autocatalytic phenomena in such systems and raises new questions about the actual role of silica as an additive in the crystallisation process.
Introduction
Silica biomorphs are a unique class of inorganic–inorganic hybrid materials, which form via bottom-up self-assembly of simple molecular species but nonetheless display remarkably complex shapes and structures,1–6 comparable to solid composite frameworks generated by organisms during biomineralisation,7 as well as artificial counterparts produced in vitro through state-of-the-art biomimetic crystal engineering and morphosynthesis.8 Just by precipitating alkaline-earth carbonates in the presence of dissolved silica (usually by slow diffusion of carbon dioxide into silica-containing solutions of corresponding metal salts),9–12 a broad variety of smoothly curved, non-crystallographic architectures can be obtained (Fig. 1). Characteristic morphologies range from flat sheets (Fig. 1a and b) over worm-like braids (Fig. 1c) to, most prominently, regular helicoids (Fig. 1d and e), but also include more elaborate 3D entities reminiscent of corals or flowers.1,5,13–15 The formation of these unusual structures essentially occurs in a spontaneous manner as long as certain boundary conditions are met, in first place that the pH of the growth medium is sufficiently, but not too alkaline (typically between 9 and 11).16–18 Under these circumstances, the components self-assemble into intricate superstructures that can otherwise only be achieved with the aid of relatively complex organic (macro)molecules or matrices, thus rendering silica biomorphs easy-to-handle model systems for investigating fundamental principles of self-organisation, in addition to their relevance in the context of primitive life detection.9,19 | ||
| Fig. 1 Typical morphologies of silica biomorphs: (a, e) polarised optical and (b–d, f) SEM micrographs of (a, b) even sheets, (c) worm-like aggregates, (d, e) twisted filaments, and (f) globular precursor particles. | ||
At the micrometre level, growth of BaCO3 biomorphs was reported to proceed along a sequence of phenomenological stages.1,3,4,12,18,20 First, an initially nucleated carbonate seed crystal experiences fractal branching21 induced by the poisoning influence of oligomeric silicate species, and thus evolves from a pseudohexagonal rod into increasingly bifurcated forms that eventually show raspberry- or cauliflower-like morphologies (Fig. 1f). With time, these globular clusters develop laminar excrescences, which usually grow flat and in contact with the walls of the vessel or the solution–air interface in the following. At some point, the sheets may curl and adopt scrolled margins, thus giving rise to the more complex curved ultrastructures (i.e. helicoids and worms),3,4 likely governed by the role of extrinsic and intrinsic surfaces in the process of morphogenesis.20
At the nanoscale, silica biomorphs were found to consist of a multitude of fairly uniform rod-like carbonate crystallites, typically about 200–300 nm and ∼50 nm wide,1,9,12 each carrying a greater or lesser amount of associated silica12,14 and being mutually arranged to establish some specific long-range orientational order.1,13 The driving force underlying the formation and continuous supply of these nanosized building units was suggested to rely on a dynamic interplay of carbonate and silicate species at molecular scales, based on the inverse pH-dependent solubilities of the components.1,3,4,17,18,22 The key point in the proposed model is that during crystallisation of any carbonate material in moderately alkaline solutions, the pH will be decreased at the surface of growing particles relative to the bulk due to dissociation of nearby bicarbonate ions,‡e.g. according to Ba2+ + HCO3− → BaCO3 + H+. This is thought to impact the speciation of silica by protonating charged silicate oligomers (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O− + H+ → Si–OH), hence promoting condensation reactions (Si–OH + HO–Si → Si–O–Si + H2O) and ultimately leading to precipitation of silica around the carbonate particles. Continued silica polycondensation is in turn expected to re-increase the local pH (owing to the consumption of acidic Si–OH groups) and, with it, raise the supersaturation of carbonate, eventually triggering a novel event of nucleation and thus maintaining an autocatalytic cycle of alternating co-mineralisation.23
Si–O− + H+ → Si–OH), hence promoting condensation reactions (Si–OH + HO–Si → Si–O–Si + H2O) and ultimately leading to precipitation of silica around the carbonate particles. Continued silica polycondensation is in turn expected to re-increase the local pH (owing to the consumption of acidic Si–OH groups) and, with it, raise the supersaturation of carbonate, eventually triggering a novel event of nucleation and thus maintaining an autocatalytic cycle of alternating co-mineralisation.23
While the described mechanism is able to explain many experimental observations1 and may be used to tailor or even program the morphological evolution in these systems,5 it has remained an essentially theoretical framework to date. Indeed, the notion that growth processes occur only at local scales in the vicinity of active fronts has been confirmed indirectly by microscopy analyses of the growth behaviour in stirred solutions,23 but still, concrete evidence supporting the proposed scenario is missing. Here we report, for the first time, experimental data that prove the envisaged local decrease in pH during the formation of silica biomorphs. For this purpose, we have used a newly developed pH-sensitive fluorescent dye with a suitable pH working range, to directly visualise subtle changes of alkaline pH in a spatially confined environment.
Analytical approach
In order to study local pH gradients during the formation of biomorphs, it is necessary to collect signals selectively from the growth front of single evolving crystal aggregates. Confocal laser scanning microscopy (CLSM) is a very promising technique in this context, due to its ability to deliver information from well-defined and highly limited depths of focus in the sample, thus avoiding superimposition of signal across the entire penetrated volume as in conventional optical microscopy. Sheets (Fig. 1a and b) are obviously the most suitable morphology for such analyses, because they grow in a quasi-two dimensional fashion along interfaces and hence, focusing on the bottom of the measurement cell should allow for facile monitoring of their development in situ. If, furthermore, a fluorescent dye is added to the mother liquor that responds to protons released (or consumed) during mineralisation by distinct changes in fluorescence intensity, such local variations of pH at the growth front should become directly visible. Fig. 2 illustrates the basic idea of the experiment. | ||
| Fig. 2 Sketch of the experiment: a growing biomorph sheet (top panel) decreases the pH in the vicinity of its active front (corresponding gradient indicated in green), as bicarbonate ions will dissociate to restore the local HCO3−/CO32− equilibrium during BaCO3 precipitation (middle panel). A suitable pH-sensitive fluorescent dye (green circles) will respond to the locally increased proton concentrations by changing its emission behaviour, ideally switching on its fluorescence at a given excitation wavelength in regions where the pH falls below a certain threshold (bottom panel). | ||
The dye chosen in this work is a boron–dipyrromethene (BODIPY) derivative bearing an acidic phenol group (1, cf. inset in Fig. 3, R = F). With increasing pH, this dye shows a decrease in fluorescence intensity over a working range of ca. 9.0–11.5 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol/water mixtures, with an emission maximum at 532 nm and a pKa of 9.98.24 This process is reversible and was ascribed to fluorescence quenching via efficient electron transfer induced by the electron-rich phenolate moiety, which results in a complete switching off of the fluorescence at sufficiently high pH values. Since growth of biomorphs does hardly tolerate the presence of any organic (co-)solvent,11 the first step in our study was to investigate the pH-dependence of the fluorescence of 1 in pure water, or rather in aqueous solutions containing barium ions and silicate oligomers at concentrations typical for biomorph syntheses (to check for potential interferences of these ions). A corresponding plot of the fluorescent intensity as a function of pH is given in Fig. 3 (black squares). It is evident that the dye still responds to changes in pH under these conditions, exhibiting a continuous decrease in relative intensity between pH 9.0 and 11.5. However, as can be expected for a change from H2O/EtOH to a neat aqueous medium, a slightly higher pKa of 10.34 is found for 1 in the biomorph growth solution. Nonetheless, the working range of the dye is in principle still suitable for probing the formation process of biomorphs, which usually occurs at bulk pH values of 10–11.18
:1 ethanol/water mixtures, with an emission maximum at 532 nm and a pKa of 9.98.24 This process is reversible and was ascribed to fluorescence quenching via efficient electron transfer induced by the electron-rich phenolate moiety, which results in a complete switching off of the fluorescence at sufficiently high pH values. Since growth of biomorphs does hardly tolerate the presence of any organic (co-)solvent,11 the first step in our study was to investigate the pH-dependence of the fluorescence of 1 in pure water, or rather in aqueous solutions containing barium ions and silicate oligomers at concentrations typical for biomorph syntheses (to check for potential interferences of these ions). A corresponding plot of the fluorescent intensity as a function of pH is given in Fig. 3 (black squares). It is evident that the dye still responds to changes in pH under these conditions, exhibiting a continuous decrease in relative intensity between pH 9.0 and 11.5. However, as can be expected for a change from H2O/EtOH to a neat aqueous medium, a slightly higher pKa of 10.34 is found for 1 in the biomorph growth solution. Nonetheless, the working range of the dye is in principle still suitable for probing the formation process of biomorphs, which usually occurs at bulk pH values of 10–11.18
 | ||
| Fig. 3 Normalised intensity of fluorescence displayed by the dyes 1 (black) and 2 (red) at different pH levels. Values were derived from the maximum of the emission peak at 532 nm (see Fig. S1 in the ESI† for corresponding spectra). Data were recorded at a constant dye concentration of 1 μM. Inset: molecular structure of the BODIPY dyes, where the acidic and thus pH-sensitive phenol–OH group is highlighted in green. | ||
Although it was possible to follow pH-dependent changes in the fluorescence of 1 in solution, we know from our earlier studies24 that this dye is prone to aggregation in water (as is evident from the broadened absorption spectrum shown in Fig. S1 of the ESI†). Since this behaviour is not only concentration- but also environment-dependent (ionic strength, nature of electrolyte, etc.) and can potentially lead to adsorption/deposition of the dye on solid surfaces (like vessel walls or biomorphs), 1 is practically not suitable for performing a precise analysis of the growth process of biomorphs. Therefore, the molecular structure of the dye was modified, replacing the two fluorine substituents by tetramers of ethylene glycol (EG4, see Section S2 in the ESI† for details on the synthesis), in order to obtain a water-soluble analogue without changing the optical properties (giving dye 2, with a structure as shown in the inset of Fig. 3 with R = EG4). Pegylation significantly reduced dye agglomeration (cf. Fig. S1†), again entailing straightforward spectroscopic BODIPY features while shifting the pKa towards a lower value of 9.29. From the pH-dependent fluorescence intensities shown in Fig. 3 (red circles), it can be deduced that 2 will essentially be non-fluorescent in the bulk growth solution (pH > 10, deprotonation of the dye), while its fluorescence should be switched on in local regions where the pH decreases below 10 due to proton release according to the proposed mechanism.
Thus, 2 is a promising candidate that meets the dye requirements (such as high brightness and switching ratio, suitable pH working range, and good water solubility) for an experiment as depicted in Fig. 2. Therefore, it was used for all analyses described in the following.
Before turning to the results of these investigations, it is worth noting that previous efforts with conventional optical pH indicators failed, primarily because dyes covering the relevant pH regime normally contain anionic groups (like carboxylates or sulfonates)25 that can strongly interact with Ba2+ ions in solution and form precipitates when added at concentrations typically required to give significant colouration and measurable effects (mM range). Here, the main advantage of 2 is its quasi-neutral character (the next-neighbour, formally zwitterionic group is not relevant in this sense) and high brightness, enabling its use at concentrations that are orders of magnitude lower (μM range).
Results and discussion
Fig. 4 shows laser scanning microscopy images representing the early stages of biomorph formation in the presence of the fluorescent dye 2. Immediately before the appearance of a distinct structure, a faint turbidity is discernible in the transmission image (Fig. 4a), indicating the onset of mineralisation, while the fluorescence channel (Fig. 4c) does not display any noticeable features (i.e.2 is in its OFF state and the pH is high). After some delay (20–30 s in this case), a raspberry-like particle is observed (Fig. 4b), which results from fractal branching of BaCO3 crystals in silica-containing solutions (cf. Fig. 1f) and is known to be a typical precursor of the more complex biomorphic morphologies.1,3,12,18 Interestingly, at this stage fluorescence can only be detected locally in the vicinity of the carbonate aggregate, as evidenced by Fig. 4d. These findings can be quantified by plotting the intensities of transmitted light and fluorescence as a function of time (Fig. 4e; the red squares in Fig. 4b and d mark the area from which signal was collected for the plot in Fig. 4e).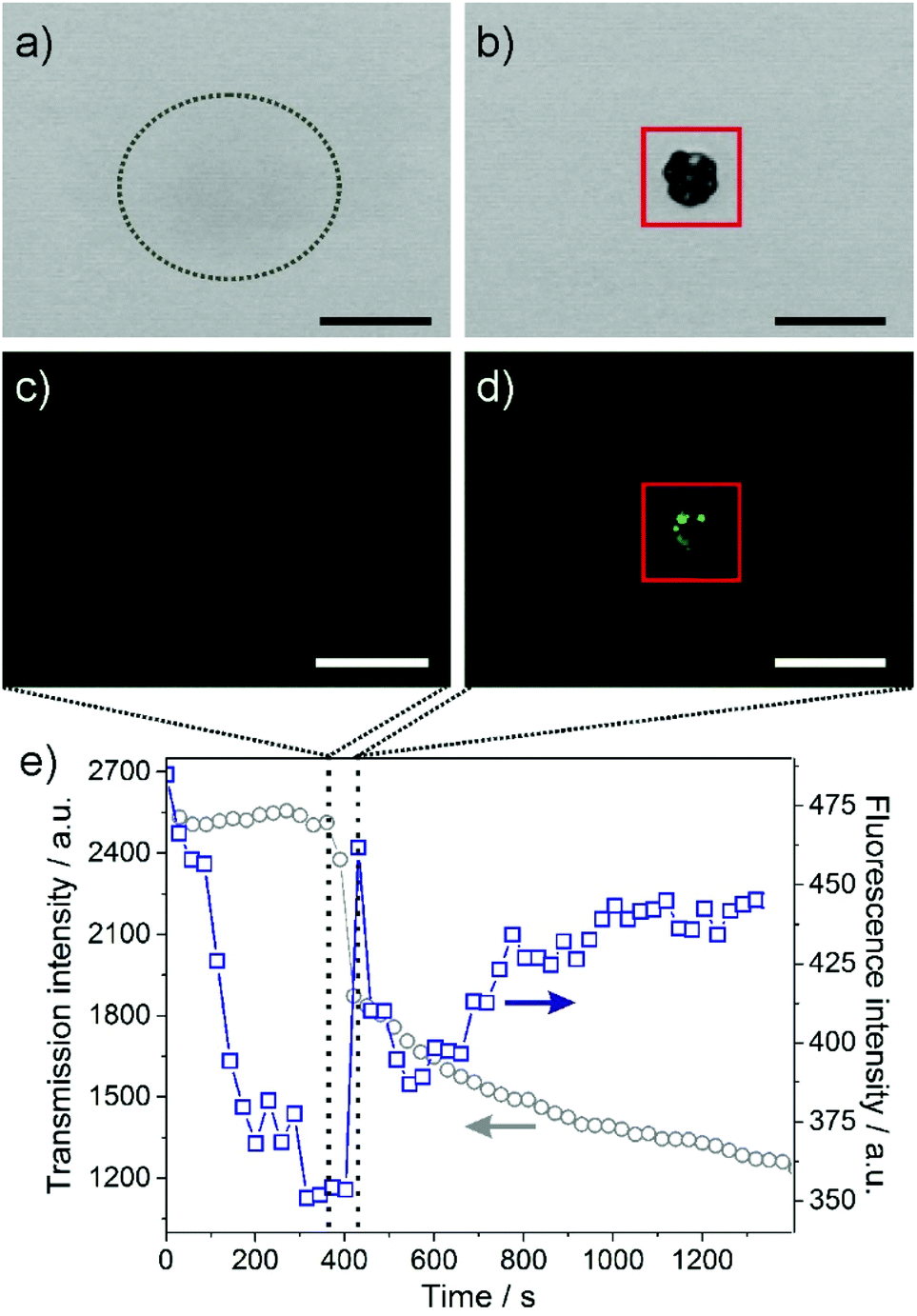 | ||
| Fig. 4 Growth of a fractal barium carbonate aggregate in the presence of silica, as viewed by CLSM in (a–b) transmission and (c–d) fluorescence mode. The dashed ellipsoid in (a) highlights a slightly blurred region, in which the structure appears about 30 s later (b). (e) Plot of the transmission (grey circles) and fluorescence (blue squares) intensities measured in the area delimited by the red boxes shown in (b) and (d) over time before, during and after the formation of the carbonate structure. The vertical dashed lines mark the times corresponding to the images above. Scale bars in (a–d) are 20 μm. | ||
As expected, the transmission signal is initially high and then decreases as the fractal structure nucleates and grows (which essentially occurs within a period of about 30 s). The fluorescence intensity, on the other hand, is generally rather low and decreases during the first 180 s prior to crystallisation. The reason for this behaviour is that the dye was added shortly after the experiment was started and deprotonation of 2 in the bulk alkaline growth medium occurs rather slowly. After this initial period of equilibration, there is a distinct peak in fluorescence that nearly coincides with the response in transmission and is well above background fluctuations (as is also clearly evident in the corresponding image, Fig. 4d). According to the pH-dependent fluorescence behaviour of 2 depicted in Fig. 3, this suggests that the pH around the globular particle is actually lower than in the surrounding bulk solution for a certain time, i.e. during the formation of the aggregate. These results indicate that precipitation of barium carbonate in alkaline media indeed leads to a local pH decrease at active surfaces, as envisaged in the current model for the formation mechanism of such materials.1,3,4,17,18,22,23
In order to confirm these observations and in particular to also show that this effect is a local phenomenon,23 further CLSM analyses were performed at higher magnification, mainly following the growth of flat sheets (for reasons outlined above). Fig. 5 shows one such structure at two distinct stages of evolution in the presence of 2.
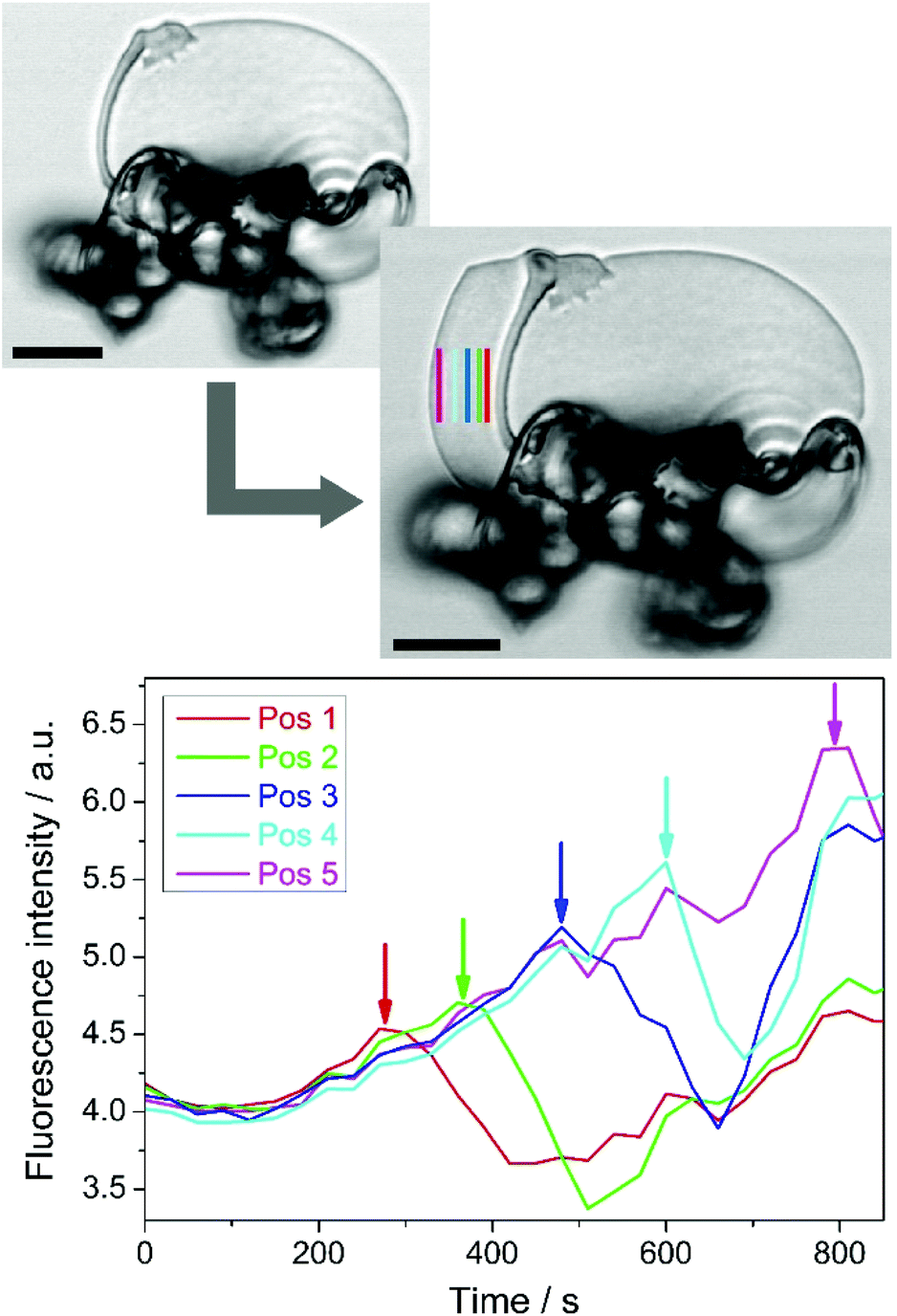 | ||
| Fig. 5 In situ monitoring of pH changes during the formation of silica biomorphs in solution. Top: confocal micrographs of a flat sheet at the beginning and at the end of the observed period (time lapse: 15 min). Bottom: Plots of the fluorescence intensity as a function of time, measured at different positions relative to the edge of the sheet in the first image (indicated by vertical lines with the same colour code). Scale bars are 10 μm. | ||
The coloured vertical lines mark different positions relative to the growth front (that in this example moves from right to left), at which the fluorescence intensity was continuously monitored. Time-dependent traces resulting from these measurements (graph in Fig. 5) again display a maximum in fluorescence; however, now the signal does not come from the entire structure, but only from a confined area (here about 3 μm wide) at the respective indicated spot. With growing distance from the initial position of the sheet edge, the maximum in fluorescence intensity continuously shifts to later times. Obviously, the fluorescence starts to increase when the growth front approaches the monitored position (at around 150–200 s in Fig. 5) and reaches the highest recorded value just before the sheet edge passes. Subsequently, a relatively sharp decrease in fluorescence intensity is observed. This suggests strongly that there is a zone close to the growth front in which the pH is locally reduced and gradually increases from the rim of the evolving aggregate towards the more alkaline bulk solution – well in line with the situation drawn schematically in Fig. 2. We note that there is a continuous drift of the fluorescence towards higher values after (and partly also before) the maxima in Fig. 5 (as well as in Fig. 4e). This can most likely be ascribed to the gradual lowering of the bulk pH upon continued uptake of atmospheric CO2 into the growth solution, as described in detail elsewhere.12 Another factor that might play a role here is dye deposition and/or agglomeration on the forming biomorph structure, although we expect this effect to be fairly limited due to the enhanced solubility of 2 compared to 1. In any case, the occurrence of a maximum in fluorescence intensity well above the aforementioned drift is clearly resolved by the data in Fig. 5.
Fig. 6 shows another example of a sheet growing in alkaline silica solution containing dye 2. Here, the local pH decrease can already be discerned in the fluorescence image (Fig. 6a) as a layer of slightly higher brightness in front of the rim of the aggregate (highlighted by the red arrows). To confirm this observation, we have performed fluorescence line scans along the normal to the surface from the solution towards and over the edge of the sheet (as shown by the yellow arrow in Fig. 6a). Integration over different azimuthal angles, as illustrated by the dashed blue arrow in Fig. 6a, led to the intensity–distance profile displayed in Fig. 6b. This data unambiguously demonstrates the existence of a pH gradient at the active front of growing biomorphs, most likely originating from the dissociation of bicarbonate ions during the formation of BaCO3 (as described above). From the results shown in Fig. 6, the thickness of this region can be estimated to ca. 2 μm (at the given level of sensitivity), which is in good agreement with indirect predictions made on the basis of simple fluid dynamics considerations in experiments at different stirring rates.23
 | ||
| Fig. 6 Local pH gradients in silica biomorphs. (a) Fluorescence image of the edge of a sheet growing from right to left. The blue and yellow arrows visualise how line scans of fluorescence intensity were performed along the normal to the surface at different azimuthal angles, while the red arrows point to a zone of slightly higher brightness near the rim of the structure. Note that the image has been converted to greyscale in order to better illustrate the higher brightness at the rim of the structure. (b) Resulting intensity–distance profile, clearly evidencing the local decrease in pH near the growing front. | ||
It is worth noting that we could not detect any hint for a cycling of the local pH in our assays, which one would expect if carbonate and silica were mineralised in an alternating fashion according to the idea of coupled co-precipitation.1,3,23 Although absolute pH measurements are not possible in a CLSM experiment with an ON/OFF dye such as 2, the intensity profiles strongly suggest that the pH at the front is continually lower than in the bulk. Indeed, the lack of any oscillating behaviour may be due to limited temporal resolution in the current setup. Nevertheless, our data indicates that carbonate crystallisation dominates the local pH and that there is little influence of silica in this context (i.e. co-condensation of silica does not lead to a measurable temporary increase of pH at the front) – at least under the conditions investigated in the present work. This conclusion is supported by the fact that the amount of silica co-precipitated with the carbonate phase during the growth process is relatively low, with Si/Ba atomic ratios in the core of biomorphs typically ranging from 0.05 to 0.10.12
Constant reduction of the pH at the growth front would imply that the supersaturation of silica is locally enhanced at all times (thus facilitating its incorporation into the forming aggregate), whereas the driving force for carbonate precipitation would in this case always be somewhat suppressed at the active surface as compared to the bulk. In our opinion, this is a plausible scenario as it was shown that the bulk mother solution of biomorphs is sufficiently supersaturated throughout the entire growth process18 and hence BaCO3 crystallisation does actually not need a local increase in supersaturation, while this is not necessarily true for silica. On the other hand, the high nucleation density observed in these materials (as manifested in the myriads of nanocrystals constituting the structures) could then no longer be explained on the basis of a locally increased driving force for precipitation.1,23 Most probably, this behaviour is caused by the presence of silica, which may act as an efficient inhibitor for carbonate growth (thus restricting the size of the crystallites to the nanoscale)26 and/or accelerate nucleation by lowering interfacial energies through epitaxial matching.27
Conclusions and outlook
In this work, we have developed a water-soluble fluorescent dye with pH sensitivity in the alkaline regime, in order to probe local changes in conditions during the formation of biomimetic silica-carbonate hybrid structures. Our results reveal the existence of a spatially confined region close to the front of growing aggregates, in which the pH is significantly reduced as compared to the bulk medium, thus lending direct experimental support to the postulated formation mechanism. This local pH gradient was found to extend over lengths of few microns into the solution and persisted over time periods of several minutes, while no oscillative behaviour could be detected. This finding challenges the notion of autocatalytic processes in these systems and stresses the role of silica as a growth-inhibiting additive. Although the present approach does not allow the quantification of local pH at every point in time, it nevertheless highlights the potential of fluorescent dyes to directly trace local precipitation phenomena in self-assembled silica-carbonate materials. Future work will be focused on the design of yet more suitable probes possessing two distinctly fluorescent species in their protonated and deprotonated form, the study of different experimental conditions in terms of pH and species concentrations, as well as on translating measured fluorescence intensities into actual local pH values.Acknowledgements
The authors thank Teresa Höβ (University of Regensburg) for performing parts of the experimental work, and the Bioimaging Center of the University of Konstanz for access to their instruments and advice concerning the analyses.Notes and references
- M. Kellermeier, H. Cölfen and J. M. Garcia-Ruiz, Eur. J. Inorg. Chem., 2012, 32, 5123 CrossRef PubMed.
- J. M. Garcia-Ruiz and J. L. Amoros, J. Cryst. Growth, 1981, 55, 379 CrossRef CAS; J. M. Garcia-Ruiz, J. Cryst. Growth, 1985, 73, 251 CrossRef.
- J. M. Garcia-Ruiz, E. Melero-Garcia and S. T. Hyde, Science, 2009, 323, 362 CrossRef CAS PubMed.
- W. Kunz and M. Kellermeier, Science, 2009, 323, 344 CrossRef CAS PubMed.
- W. L. Noorduin, A. Grinthal, L. Mahadevan and J. Aizenberg, Science, 2013, 340, 832 CrossRef CAS PubMed.
- M. Kellermeier, F. Glaab, E. Melero-Garcia and J. M. Garcia-Ruiz, Experimental Techniques for the Growth and Characterization of Silica Biomorphs and Silica Gardens, in Research Methods in Biomineralization Science, Academic Press, Burlington, 2013, vol. 532, pp. 225–256 Search PubMed.
- S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, Oxford, 2001 CrossRef CAS; P. Behrens and E. Bäuerlein, Handbook of Biomineralization, Wiley-VCH, Weinheim, 2009 CrossRef CAS; S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 41, 21 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332 CrossRef CAS PubMed; H. Cölfen and M. Antonietti, Mesocrystals and Non-Classical Crystallization, Wiley, Chichester, 2008 CrossRef PubMed; F. Nudelman and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582 CrossRef PubMed.
- J. M. Garcia-Ruiz, S. T. Hyde, A. M. Carnerup, A. G. Christy, M. J. Van Kranendonk and N. J. Welham, Science, 2003, 302, 1194 CrossRef CAS PubMed.
- E. Bittarello and D. Aquilano, Eur. J. Mineral., 2007, 19, 345 CrossRef CAS PubMed.
- A. E. Voinescu, M. Kellermeier, A. M. Carnerup, A. K. Larsson, D. Touraud, S. T. Hyde and W. Kunz, J. Cryst. Growth, 2007, 306, 152 CrossRef CAS PubMed.
- M. Kellermeier, E. Melero-Garcia, F. Glaab, J. Eiblmeier, L. Kienle, R. Rachel, W. Kunz and J. M. Garcia-Ruiz, Chem. – Eur. J., 2012, 18, 2272 CrossRef CAS PubMed.
- S. T. Hyde, A. M. Carnerup, A. K. Larsson, A. G. Christy and J. M. Garcia-Ruiz, Phys. A, 2004, 339, 24 CrossRef CAS PubMed.
- H. Imai, T. Terada, T. Miura and S. Yamabi, J. Cryst. Growth, 2002, 244, 200 CrossRef CAS; A. E. Voinescu, M. Kellermeier, B. Bartel, A. M. Carnerup, A. K. Larsson, D. Touraud, W. Kunz, L. Kienle, A. Pfitzner and S. T. Hyde, Cryst. Growth Des., 2008, 8, 1515 Search PubMed.
- T. Terada, S. Yamabi and H. Imai, J. Cryst. Growth, 2003, 253, 435 CrossRef CAS; M. Kellermeier, F. Glaab, A. M. Carnerup, M. Drechsler, B. Gossler, S. T. Hyde and W. Kunz, J. Cryst. Growth, 2009, 311, 2530 CrossRef PubMed.
- J. M. Garcia-Ruiz, Geology, 1998, 26, 843 CrossRef CAS.
- E. Melero-Garcia, R. Santisteban-Bailon and J. M. Garcia-Ruiz, Cryst. Growth Des., 2009, 9, 4730 CAS.
- J. Eiblmeier, M. Kellermeier, D. Rengstl, J. M. Garcia-Ruiz and W. Kunz, CrystEngComm, 2013, 15, 43 RSC.
- J. M. Garcia-Ruiz, A. M. Carnerup, A. G. Christy, N. J. Welham and S. T. Hyde, Astrobiology, 2002, 2, 353 CrossRef PubMed.
- M. Kellermeier, J. Eiblmeier, E. Melero-Garcia, M. Pretzl, A. Fery and W. Kunz, Cryst. Growth Des., 2012, 12, 3647 CAS.
- H. D. Keith and F. Padden, J. Appl. Phys., 1963, 34, 2409 CrossRef CAS PubMed.
- M. Kellermeier, E. Melero-Garcia, F. Glaab, R. Klein, M. Drechsler, R. Rachel, J. M. Garcia-Ruiz and W. Kunz, J. Am. Chem. Soc., 2010, 132, 17859 CrossRef CAS PubMed; J. Eiblmeier, M. Kellermeier, M. Deng, L. Kienle, J. M. Garcia-Ruiz and W. Kunz, Chem. Mater., 2013, 25, 1842 CrossRef.
- M. Kellermeier, E. Melero-Garcia, W. Kunz and J. M. Garcia-Ruiz, J. Colloid Interface Sci., 2012, 380, 1 CrossRef CAS PubMed.
- M. Hecht, W. Kraus and K. Rurack, Analyst, 2013, 138, 325 RSC.
- J. Han and K. Burgess, Chem. Rev., 2009, 110, 2709 CrossRef PubMed.
- J. Eiblmeier, U. Schürmann, L. Kienle, D. Gebauer, W. Kunz and M. Kellermeier, Nanoscale, 2014, 6, 14939 RSC.
- E. Bittarello, F. R. Massaro and D. Aquilano, J. Cryst. Growth, 2010, 312, 402 CrossRef CAS PubMed; E. Bittarello, F. R. Massaro, M. Rubbo, E. Costa and D. Aquilano, Cryst. Growth Des., 2009, 9, 971 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details (Section S1) and pH-dependent emission spectra of the used dyes (Fig. S1). See DOI: 10.1039/c5nr05399d |
| ‡ Note that the fraction of HCO3− ions existing in equilibrium with CO32− is significant at conditions typical for the formation of biomorphs, ranging from ca. 20% at pH 11 to >90% at pH 9.22 |
| This journal is © The Royal Society of Chemistry 2015 |