
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Size and surface chemistry of nanoparticles lead to a variant behavior in the unfolding dynamics of human carbonic anhydrase†
Irem
Nasir
*,
Martin
Lundqvist
 and
Celia
Cabaleiro-Lago
*
and
Celia
Cabaleiro-Lago
*
Department of Biochemistry and Structural Biology, Lund University, PO Box 124, SE 221 00, Lund, Sweden. E-mail: Celia.Cabaleiro-Lago@biochemistry.lu.se; Irem.Nasir@biochemistry.lu.se
First published on 30th September 2015
Abstract
The adsorption induced conformational changes of human carbonic anhydrase I (HCAi) and pseudo wild type human carbonic anhydrase II truncated at the 17th residue at the N-terminus (trHCAii) were studied in presence of nanoparticles of different sizes and polarities. Isothermal titration calorimetry (ITC) studies showed that the binding to apolar surfaces is affected by the nanoparticle size in combination with the inherent protein stability. 8-Anilino-1-naphthalenesulfonic acid (ANS) fluorescence revealed that HCAs adsorb to both hydrophilic and hydrophobic surfaces, however the dynamics of the unfolding at the nanoparticle surfaces drastically vary with the polarity. The size of the nanoparticles has opposite effects depending on the polarity of the nanoparticle surface. The apolar nanoparticles induce seconds timescale structural rearrangements whereas polar nanoparticles induce hours timescale structural rearrangements on the same charged HCA variant. Here, a simple model is proposed where the difference in the timescales of adsorption is correlated with the energy barriers for initial docking and structural rearrangements which are firmly regulated by the surface polarity. Near-UV circular dichorism (CD) further supports that both protein variants undergo structural rearrangements at the nanoparticle surfaces regardless of being “hard” or “soft”. However, the conformational changes induced by the apolar surfaces differ for each HCA isoform and diverge from the previously reported effect of silica nanoparticles.
1. Introduction
The development of nanomaterials for applications in medicine, industry and biotechnology has rapidly been accelerated in the past decades.1–4 Accordingly, casual exposure and the active use of such materials has escalated. Protein adsorption to surfaces of nanomaterials is a widely known phenomenon and can occasionally affect the stability of a protein.5 Introduction of nanoparticles into biological fluids like blood, often causes immediate protein adsorption.6,7 The composition of the protein layer on the nanoparticle changes with time, depending on different affinities and the exchange rates of the proteins present in solution.8,9 Accompanying adsorption of proteins onto nanoparticles can be beneficial or harmful depending on whether the proteins stay intact or not. Typically, a surface can influence a protein in three different ways and either of those can be desirable depending on the application of the bionanomaterial.10 First, a protein can stay intact in solution.11 Second, proteins may adsorb on the particle with no profound structural changes12 or third, adsorption causes structural rearrangement in the protein's native structure.13–16 Therefore, developing bionanomaterials requires a thorough investigation on the particular system in terms of protein adsorption and structural changes associated to it.The extent and type of interactions between a protein and a particle is dictated by their respective properties. Relevant nanoparticle properties include the surface chemistry, morphology and relative size to the protein, whereas the structural stability and the surface composition are crucial factors for a protein.17,18 Hence, the interactions between a protein and a nanoparticle are a blend of electrostatic forces, van der Waals attraction, hydrogen bonding and hydrophobic effects. Currently, predicting whether a certain type of nanoparticle destabilizes a particular protein is not achievable. For example, changing the size, thus the curvature of silica particles has opposite effects on different proteins.19 There are various studies in the literature investigating nanoparticle–protein interactions in terms of adsorption and conformational changes.20–27
Changes of structure and stability of proteins on nanoparticle surfaces are predominantly investigated using secondary structure determining methods. Circular dichorism spectroscopy (CD) is a widely used technique for this regard.13,28,29 However, subtle changes in the secondary structure is often compromised by fitting procedures that requires deconvolution of the spectra.30,31 Moreover, light scattering from nanoparticles is a common obstacle in far and near-UV CD measurements.18,32 Other methods include, Fourier transform infrared spectroscopy,33 surface-enhanced Raman spectroscopy34 and nuclear magnetic resonance (NMR).23,24,35,36 However, the aforementioned methods are not able to report on the kinetics of the structural changes on the nanoparticle surface in a rapid manner.
To date there is no detailed investigation of the effect of nanoparticle properties on the unfolding dynamics of proteins. Overall, unfolding dynamics are scarcely covered in the literature. Few examples include stopped-flow or continuous-flow rapid mixing techniques with optical detection.37,38 Recently, we have developed a high-throughput screening method based on fluorescence which allows to follow the adsorption induced unfolding events of a protein on a nanoparticle surface that happen as short as miliseconds timescale.39
In this paper, we report a detailed study concerning the effect of nanoparticle size on protein adsorption and unfolding dynamics. As a model system we used two human carbonic anhydrase (HCA) isoforms mixed with hydrophobic (polystyrene) and hydrophilic (silica) nanoparticles. HCA is a well-characterized enzyme with a metal cofactor that aids in the conversion of carbon dioxide to bicarbonate.40 Structural changes of the adsorbed proteins and the dynamics of these processes were determined by ANS fluorescence spectroscopy and cross-validated by near-UV CD spectroscopy. Our findings point out diverse effects of two different particle surface chemistries on HCA; hydrophilic surfaces lead to hour-timescale protein unfolding, whereas hydrophobic particles induce a milisecond-timescale structural change. Additionally, isothermal titration calorimetry (ITC) was used to calculate the stoichiometry of the protein–nanoparticle complex and revealed the influence of the nanoparticle size on the adsorption process.
2. Materials and methods
2.1. Materials
Polystyrene nanoparticles with carboxyl group surface modifications, henceforth referred as PS-COOH, (26, 49 and 94 nm) were purchased from Bangs Laboratories (Fishers, IN). Particles were dialyzed against water for a week to remove the stabilizers. The colloidal silica particles, henceforth referred as Si, (23, 34 and 90 nm) were kind gifts from Akzo Nobel. Particles were dialyzed against 10 mM Tris, pH 8.4 buffer (working buffer for all experiments unless stated otherwise) before use. 8-Anilino-1-naphthalenesulfonic acid ammonium salt (ANS) was purchased from Sigma Aldrich and used without further purification. ANS was dissolved in filtered water to reach a stock concentration of 1.3 mg ml−1. All other chemicals are of highest purity available.2.2. Human carbonic anhydrases
Wild type human carbonic anhydrase I (HCAi) and the plasmid of human carbonic anydrase II pseudo wild type truncated at position 17 at the N-terminal (trHCAii) were kind gifts from Prof. Bengt-Harald Jonsson. The protein was expressed and purified as described elsewhere.18 The protein powder was dissolved in working buffer, centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the pellet was discarded. Concentration determination was done spectroscopically by measuring A280 with extinction coefficients 46800 M−1 cm−1 and 44100 M−1 cm−1, for HCAi and trHCAii, respectively (Agilent 8453 spectrophotometer).
000 rpm and the pellet was discarded. Concentration determination was done spectroscopically by measuring A280 with extinction coefficients 46800 M−1 cm−1 and 44100 M−1 cm−1, for HCAi and trHCAii, respectively (Agilent 8453 spectrophotometer).
2.3. Particle characterization
After each dialysis, the electrophoretic mobility of the particles was measured using Malvern Zetasizer Nano S operating with a 632.8 nm laser and at 173° scattering angle at 30° C. The ζ-potential of the particles were calculated using the Smoluchowski equation. Particles were dispersed either in water or in 10 mM Tris buffer pH 8.4. Three measurements of each nanoparticle dispersion were measured using a folded capillary cell (Malvern). The hydrodynamic diameters of nanoparticles was checked using a DLS plate reader (DynaPro Plate Reader II, Wyatt Technology, Santa Barbara, CA) operating with a 158° scattering angle at 30 °C using a 96-well black plate with clear bottom (Costar). 3–6 replicates of each sample were measured with 10 acquisitions per sample. Each acquisition was set to five seconds. A general purpose analysis model (cumulant fit for monomodal dispersions) was employed to determine the hydrodynamic diameter of the particles.2.4. Isothermal titration calorimetry (ITC)
ITC measurements were performed on a VP-ITC instrument from Microcal. The proteins were titrated into particle dispersions at various concentrations to achieve the best titration curve. The titration involved generally 30 injections of 10 μl of protein solution at sufficient long intervals to ensure the recovery of the baseline before the next injection (usually between 5 to 7 minutes). During the experiment, the cell was continuously stirred at 300 rpm. At least two experiments were performed for each protein–nanoparticle pair. Control experiments to determine the heats of dilution of particles and proteins into buffer were performed. The integrated heats for the titration of protein into particles were calculated and corrected with the corresponding heats of dilution in buffer. The molar concentration of the particles was calculated from the weight/volume concentration using the DLS hydrodynamic radius and the density of the particles assuming that the particles are spherical and homogeneous. The data was analyzed using the coupled Origin software (OriginLab Corporation, Northampton, MA) to obtain the binding stoichiometry of the complex formed. The single set of identical sites model or the two sets of identical sites model was used depending on the heat profile observed.2.5. ANS fluorescence experiments in plate reader
Fluorescence measurements were done in FLUOstar plate reader (BMG Labtech, Germany) at 30 °C using 96-well black plates with clear bottom (Costar). The final ANS concentration in each sample was 0.195 mg ml−1 unless it is stated otherwise. Excitation and emission filters were: λex: 320 nm, λem: 460 nm. For Si experiments, the protein concentration was 40 μM and total surface area of the particle was 6 × 10−5 m−2 ml−1 (total volume per well: 100 μl). For the automated injection experiments, wells were filled with 80 μl aliquots of PS-COOH nanoparticles of different size with ANS (equal total surface area for different sizes, 9 × 10−2 m−2 ml−1 in each well). 20 μl of a protein stock solution with ANS was injected to each well to reach a final concentration of 3.5 and 3.7 μM HCAi and trHCAii, respectively. Each well was read individually with an interval time of 0.2 seconds for a total of 40 seconds. Injections were made at 1.1 s (between two consecutive intervals) in order to avoid the alterations in the fluorescence signal from mixing. Nanoparticle controls were prepared in 100 μl in two different concentrations as the concentration prior and after the injection. Buffer control was subtracted from each sample and the nanoparticle controls were subtracted from the nanoparticle–protein sample for the sake comparison with the protein control.2.6. Near-UV circular dichorism (CD)
Near-UV CD spectra were collected using a JASCO-720 spectrometer equipped with a JASCO PTC-343 Peltier thermostat cell holder at 30 °C. The final volume was set to 600 μl in a 1 mm path-length quartz cuvette. The concentration of human carbonic anhydrases were 35 and 37 μM for HCAi and trHCAii, respectively. Two concentrations of 26 nm nominal size PS-COOH nanoparticles were used (0.08 and 0.1 mg ml−1). Spectra were recorded between 320 to 240 nm with a data pitch of 1 nm and a response time of 4 s. Bandwidth was set to be 1 nm at a scanning speed of 10 nm min−1. Three spectra per sample were accumulated. Buffer control was subtracted from each sample and nanoparticle control spectra were subtracted from protein–nanoparticle spectra.3. Results
3.1. Particle characterization
For determining the effect of nanoparticle size on protein adsorption, it is crucial to have particles with very narrow size distribution. Dynamic light scattering (DLS) measurements were done in working buffer to determine the sizes of particles together with the polydispersity index (PDI). Furthermore, ζ-potential of the particles were determined in order to make sure that the particles are sufficiently charged at the experimental conditions. Results indeed show that all particles have a narrow size distribution and possess large negative ζ-potentials that favor colloidal stability and do not aggregate in the chosen buffer as seen in Table S1.†Additionally, the nanoparticle sizes were measured after the addition of protein to ensure a stable colloidal mixture. In our experiments, none of the nanoparticle–protein pairs caused visible agglomeration. Fig. 1 shows that the nanoparticle size slightly increases upon protein adsorption for most of the nanoparticles. (For further details on the difference in sizes, see Fig. S1†). PDI is found to be constant within the nanoparticle control value.
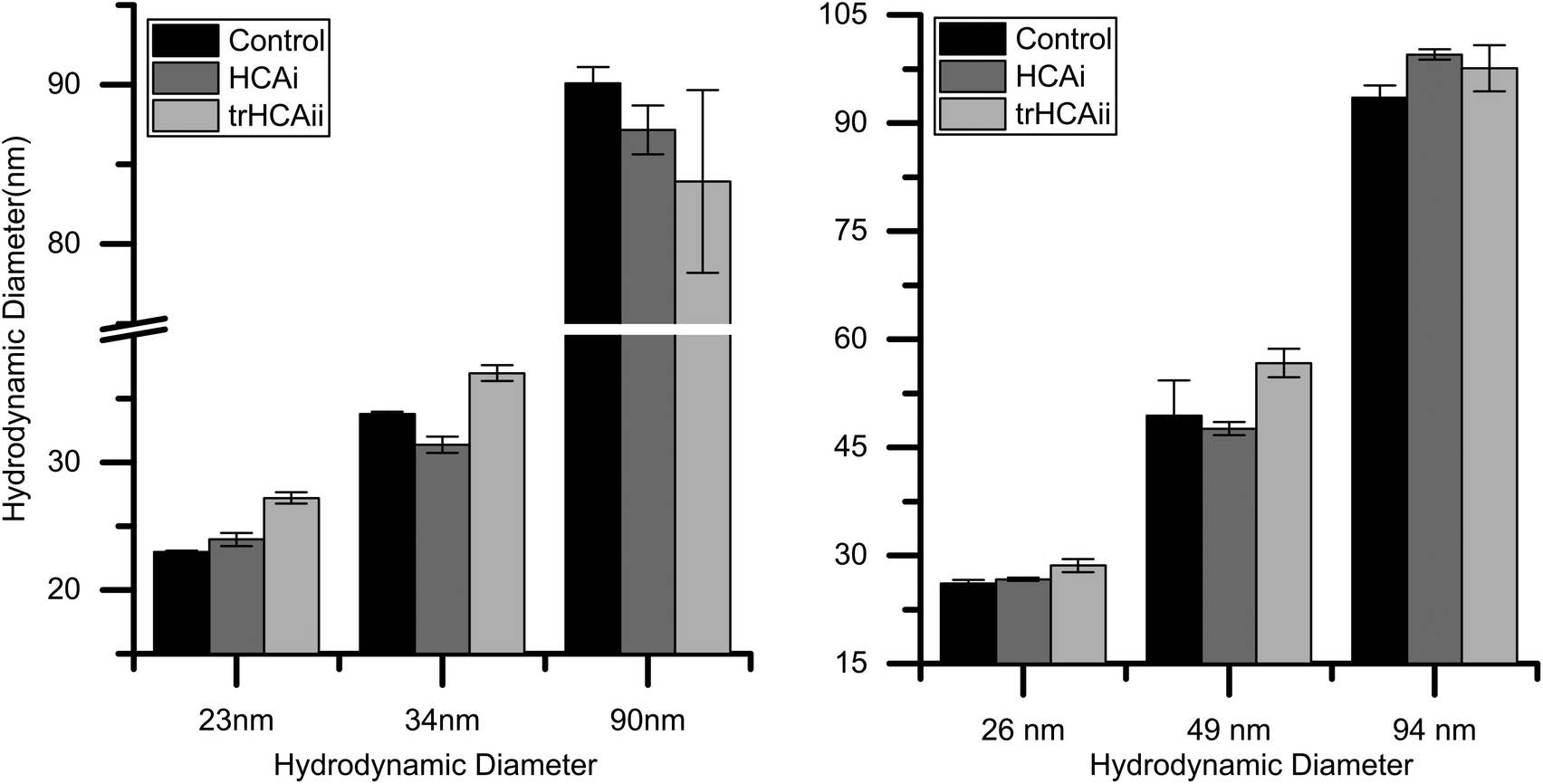 | ||
| Fig. 1 Hydrodynamic diameter of nanoparticles before and after HCA addition. (Left) Silica, (Right) PS-COOH. | ||
3.2. Stoichiometry of the nanoparticle protein complex is determined by ITC
The stoichiometry of the nanoparticle–protein complex is an important factor to be considered when analyzing the extent of unfolding of HCAs on nanoparticles. ITC is an effective technique to study the adsorption of proteins on the nanoparticle surfaces.41 To address whether the nanoparticle size influences the density of the adsorbed HCA layer, ITC experiments using three different PS-COOH nanoparticles (26, 49 and 94 nm diameter) and the two HCA isoforms were performed.The integrated heats of the titration of HCA isoforms to the different PS-COOH nanoparticles are shown in Fig. 2. For the sake of comparison, the integrated heats are plotted against the number of proteins per nanoparticle surface area. Heat flows and integrated heats versus the protein molar ratio can be found in Fig. S2a and b.†
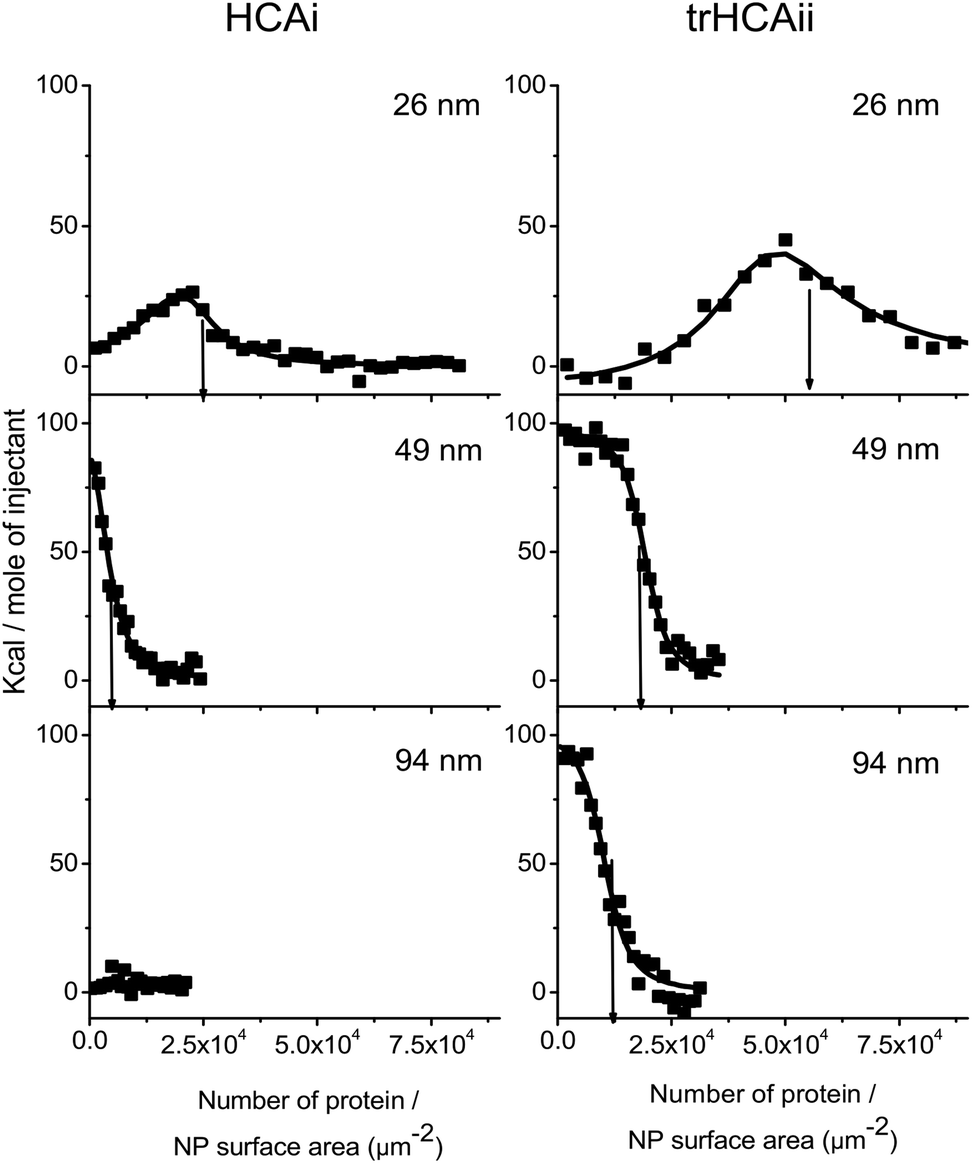 | ||
| Fig. 2 Integrated heat values after the correction of dilution heats against the number of proteins per nanoparticle surface area of PS-COOH nanoparticles of different size. PS-COOH is titrated with (Left) HCAi and (Right) trHCAii at various concentrations (for details see Fig. S2 a and b†). The line shows the best fit of the one set of identical sites or two sets of identical sites model. The arrows indicate the amount of adsorbed proteins per nanoparticle surface area (Nsurf) derived from the stoichiometry value (n) obtained from the fitting procedure. | ||
Endothermic peaks are observed in all nanoparticle–protein pairs except of HCAi titration into 94 nm PS-COOH, which indicates binding for all nanoparticle–protein pairs except of HCAi and 94 nm PS-COOH couple. The heat profiles for the 26 nm particles are different from those observed for the bigger particles (49 and 94 nm). The thermograms obtained for the latter ones could successfully be fitted to a single site binding model. However, in case of the 26 nm particles, a two-site binding model must be used.
The stoichiometry of binding of the protein–nanoparticle complex (n) can be obtained from the fitted model, and the number of proteins, normalized by the nanoparticle surface area (Nsurf) was calculated. As indicated by arrows in Fig. 2, the number of protein adsorbed per nanoparticle surface area decreases as the particle size increases even though the particles have identical material properties. In all cases, Nsurf is higher for trHCAii than HCAi as seen in Table 1, indicating an overall higher affinity to the nanoparticles for trHCAii. Interestingly, no heat signal was measured upon titration of HCAi into 94 nm particles. Consequently, for this mixture the stoichiometry cannot be calculated.
| HCAi | trHCAii | |||
|---|---|---|---|---|
| n | N surf/(10−3 μm) | n | N surf/(10−3 μm) | |
| 26 nm | 56 ± 6 | 24 ± 3 | 133 ± 29 | 58 ± 13 |
| 49 nm | 48 ± 5 | 4.1 ± 0.4 | 218 ± 19 | 19 ± 1 |
| 94 nm | — | — | 332 ± 20 | 10 ± 1 |
The stability of the colloidal dispersions was checked after each ITC experiment to rule out undesirable heat changes originating from the nanoparticle agglomeration. After the titrations, all particle–protein mixture showed an increase in size between 5 and 10%, as measured by DLS. Thus, the integrated heats observed here are purely the result of the association process between nanoparticles and proteins.
3.3. Dynamics of the conformational changes of HCAs on Si and PS-COOH particles
To analyze on what time scale protein unfolding on nanoparticle surfaces occurs, time-resolved fluorescence spectroscopy using the solvatochromic dye ANS was used. ANS fluorescence and the peak maximum changes upon non-covalent binding to hydrophobic patches on the protein.42 Increased fluorescence intensity indicates the increase in the exposure of the hydrophobic patches of the HCAs upon unfolding which can be either local or global.Differently sized particles were used to elucidate the influence of the size on the dynamics of the adsorption. The Si nanoparticles, 23 and 34 nm, have no effect on the integrity of the HCAi, as the ANS intensity does not change over the time (Fig. 3a). For the 90 nm particles a higher ANS intensity is observed. Moreover, the signal progressively increases for 90 nm Si particle, indicating that HCAi is most affected by the presence of the biggest particle. trHCAii on the other hand, is impaired by all Si particles drastically as seen in Fig. 3b. The ANS intensity is equal to the protein control initially and increases continuously for 40 hours. As expected, particles with a similar size, 23 and 34 nm, contribute to the unfolding almost identically, whereas 90 nm has a profound effect compared to aforementioned sizes.
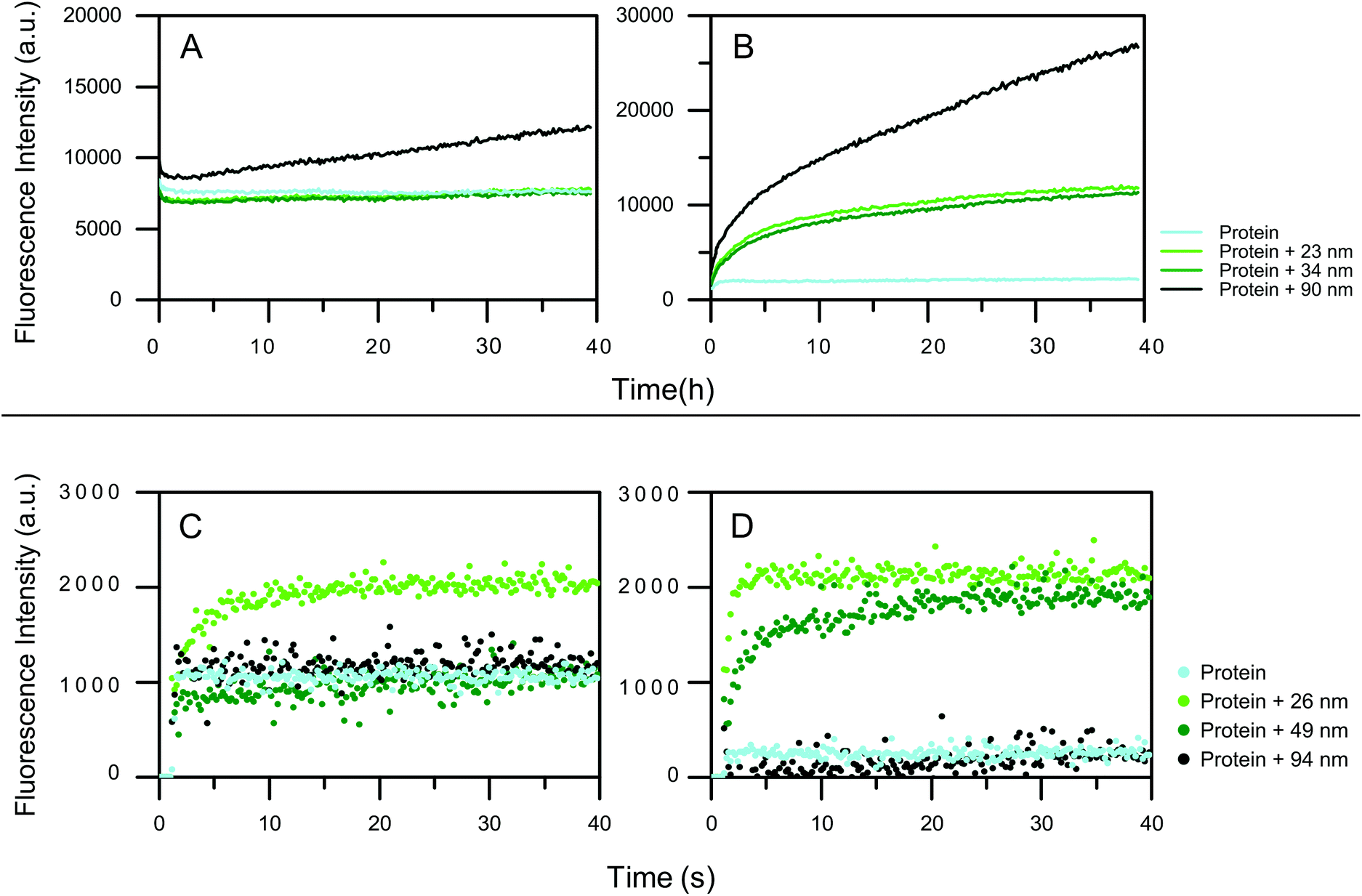 | ||
| Fig. 3 ANS intensity change over the time for proteins in presence of Top) silica particles of different sizes. The particle surface is 6 × 10−5 m2 ml−1 for each size and the protein concentration is 40 μM of (a) HCAi (b) trHCAii (Bottom) PS-COOH particles of different sizes. The total particle surface area is 9 × 10−2 m2 ml−1 for all sizes and protein concentration is (c) 3.5 μM HCAi (d) 3.7 μM trHCAii. The protein is injected at 1.1 s after the measurement has started. | ||
The effect of hydrophobic PS-COOH size on HCA variants is also monitored using ANS fluorescence. Fig. 3 shows the ANS fluorescence measurements (for a detailed plot with marked injections, see Fig. S3†). Given the rapid conformational change that happens immediately at the mixing of the components,39 automated injection mode of the plate reader was used. For HCAi, an immediate fluorescence intensity response is observed when injected into 26 nm nanoparticles which is an indication of the unfolding of the protein whereas 49 and 94 nm nanoparticles cause no ANS signal change compared to the protein control as seen in Fig. 3c. Equivalently, the quick conformational changes also take place for the trHCAii when injected into the 26 and 49 nm PS-COOH nanoparticles as presented in Fig. 3d. Finally, trHCAii stays almost intact in presence of 94 nm PS-COOH nanoparticles as in the case of HCAi.
3.4. Tertiary structure determination using near-UV CD
Near-UV CD experiments were performed with the two HCA variants and two different concentrations of 26 nm PS-COOH particles, in order to get a deeper insight on the local unfolding and explore the systematic effects imposed by the nanoparticles. Changes in the near-UV spectra are mainly attributed to changes in the environment around Trp residues therefore the tertiary structure of the protein. Thus, it has been used in the past for decoding the interactions between particles and proteins.43–45There is a clear systematic effect of the nanoparticle concentration on the CD spectra of both isozymes, as shown in Fig. 4. The CD spectra for HCAi in presence of PS-COOH is not affected in the wavelength region ≈285–300 nm. The signal is decreased compared to the native spectrum in between wavelengths ≈260–285 nm. Finally an increase in the CD signal in the wavelength region ≈240–260 nm is observed. In contrast, trHCAii spectrum is altered in a different way than HCAi; the signal in the whole spectrum decreases as shown by the arrows in Fig. 4. Moreover, the characteristic 296 nm minima is blue-shifted for trHCAii on nanoparticles together with a significant loss of the 287 nm minima. Unfortunately, the larger PS-COOH particles induce heavy scattering at the lower wavelengths of the near-UV range therefore monitoring the tertiary structure differences by CD is greatly hindered.
 | ||
| Fig. 4 Near-UV CD spectra of HCAi (35 μM) and trHCAii (37 μM) with three different PS-COOH (26 nm) concentrations. Continuous lines are for native protein (black), 0.1 mg ml−1 PS-COOH (red) and 0.08 mg ml−1 PS-COOH (blue). The arrows indicate the direction of the protein signal change in presence of nanoparticles. | ||
4. Discussion
Here, we have studied the effects of Si and PS-COOH nanoparticles of different sizes, on the unfolding dynamics of wild type HCAi and pseudo wild type trHCAii. Silica and polystyrene nanoparticles have been widely used in the past as model systems for studying bionano interactions.18,22,46–49 Both types of particles bear net negative charge at the experimental conditions and have similar ζ-potential values (Table S1†). However the hydrophobicity of the surfaces differs. This difference allows us to study the influence of the surface polarity on the adsorption process. HCA is a well characterized enzyme and the mentioned two isoforms differ 40% in sequence whereas the overall structure overlaps in a larger extent. Despite the sequence and structure similarity and the conservation of the enzymatic function, the two isoforms are marginally different in terms of inherent stability. HCAi is 6–7 kcal mol−1 more stable than trHCAii.50,51 Accordingly, HCAi and trHCAii were shown to have different tendencies to unfold at the surfaces in accordance with the definition of “hard” and “soft” proteins respectively.23,52 In general, carbonic anhydrases were reported to have a molten-globule state, which may be the reason that these proteins are prone to have induced conformational changes.53,54 Nanoparticle adsorption as an external factor, may lower the energy barrier between the native and molten-globule state even further, making the molten-globule state more accessible.Exploring the destabilizing effect of nanoparticles on proteins requires stable particles with defined physicochemical properties. Here, the dialyzed particles were observed to be stable for weeks according to visual examination and DLS measurements. Moreover, the nanoparticle stability in presence of HCA is also an essential issue to be considered when analyzing any type of experimental data; agglomeration of nanoparticles can make the data interpretation complicated. Agglomeration of the nanoparticles may happen due to the screening of the surface charges of the nanoparticle by the adsorbed protein which may trigger the process, acting as a glue to stick nanoparticles together. We found that the addition of the proteins to the nanoparticle solutions provokes an increase of the mean hydrodynamic diameter of the particle without significant broadening of the size distribution in most of the cases (Fig. 1 and S1†). The size increase is less than 4 nm, and in general bigger for trHCAii than HCAi for both types of nanoparticles. This may imply that trHCAii forms a more extended layer (into the bulk solution) than HCAi. The increase in the hydrodynamic diameter is compatible with the formation of a protein monolayer around the nanoparticles in which the proteins may partially have lost their native structure. Monolayer formation is in agreement with previous reports for different nanoparticle–protein systems.55,56
Protein adsorption to surfaces and interfaces is a widely explored phenomenon that can be explained in four steps: (1) bulk diffusion of the protein, (2) docking to the surface/interface, (3) conformational changes of the protein dictated by the mutual characteristics of the system, (4) lateral diffusion and interactions of the proteins on the surface/interface.57 Hence, in order to understand the overall adsorption effect of the nanoparticle size and the two distinct surface chemistries on the dynamics of protein unfolding upon adsorption, the process needs to be broken into its steps and must be analyzed independently.
4.1. Adsorption to the nanoparticle surface
To determine whether the equilibrium number of protein per particle surface area changes with respect to particle size, ITC was used. The calorimetry data shows that the binding capacity per surface changes with PS-COOH nanoparticles′ size (Table 1). The 26 nm particles harbor the most trHCAii per surface area whereas the number drastically falls for 49 nm particles. The least Nsurf value is found for the 94 nm particles for trHCAii. A similar trend is observed for HCAi although Nsurf values are less than half the trHCAii values, indicating a lower stoichiometry for the nanoparticle-HCAi system. Data analysis for the n values for 94 nm PS-COOH and HCAi system could not be performed due to the lack of heat signals. However, this fact does not exclude the existence of interaction for this nanoparticle–protein pair. For example, if the nanoparticle–protein association process was driven purely by entropy, no enthalpic heat profile would be observed. From taking into account the trend observed for trHCAii, it can be concluded that HCAi most likely does not bind to the 94 nm particles.The density of the protein layer at the PS-COOH nanoparticle surface increases as the size of the nanoparticle decreases. Opposite trends have often been observed for other systems. Lindman et al. showed that the Nsurf reaches the saturation level for hydrophobic polymeric nanoparticles over 100 nm but smaller nanoparticles (70 nm) has lower degree of saturation.41 The studies on the interaction of albumin on gold nanoparticles report the increase of protein adsorbed per surface area as the nanoparticle size increases, accompanying the formation of multilayers on nanoparticle surfaces that are larger than 10 nm.34,58 Similarly, the binding of IgG and protein A to gold nanoparticles shows the same trend regarding the size of gold nanoparticles.59
Given that we observe an opposite effect of size on the number of protein adsorbed per surface area for the hydrophobic PS-COOH nanoparticles compared to the reports in the literature, the difference must arise from very fundamentals of the process; the driving forces that act on the adsorption of the protein on the nanoparticle. Adsorption can be simplified as a two step process; first the protein approaches to the surface and binds loosely in the native state at random positions on the surface. After the initial docking, the protein can undertake conformational changes and form non-covalent protein–nanoparticle bonds that help to stabilize the protein on the surface therefore leading to a stronger adsorption.60 Yet, the initial binding and following reorganization can be affected by lateral interactions between the adsorbed proteins. The equilibrium coverage on the surface is therefore determined by the balance of the stabilization of the protein at the surface and the intermolecular protein interactions on the surface.
A denser HCA layer was observed for both variants on hydrophobic 26 nm PS-COOH nanoparticles in our experiments. Regardless of the polarity of the surface, a denser protein layer on the nanoparticle surface suggests that the lateral interactions are minimized as smaller the particle gets. Taking into account that the main driving force for the adsorption of proteins on hydrophobic surfaces is the hydrophobic effect, formation of polar contacts (hence extensive conformational changes) is not essential for the adsorption. Given the limited conformational changes of both proteins on 26 nm hydrophobic PS-COOH surface (discussed in the next section), the charge distribution of the protein may presumably not be altered. Therefore repulsive protein–protein interactions can play an important role on the formation of the adsorbed protein layer and may lead to a scarce layer flatter the particle gets. On the other hand, protein–protein repulsions are geometrically minimized due to the increased curvature in the 26 nm particles, allowing a denser protein layer. A general scheme of adsorption to hydrophilic and hydrophobic surfaces can be seen in Fig. 5. This hypothesis is supported by the thermodynamics of the adsorption process as observed in the ITC experiments (Fig. S2a and b†). For each HCAs, the association on 26 nm nanoparticles at low protein coverage exhibits a favorable exothermic process that competes with an endothermic process. Initially dominating exothermic process can be the formation of non-covalent bonds between the COO− groups of the nanoparticles and available residues in the protein. However the contribution of this process is compensated at high protein coverage probably by the endothermic contribution of the intermolecular lateral repulsions. Moreover, the initial exothermic process is only seen at very low coverage for PS-COOH 49 nm indicating that for bigger nanoparticles, contribution of the lateral repulsions is considerably larger resulting in low surface density. Long range lateral repulsions at low protein coverage have also been reported previously by Duinhoven and coworkers.61
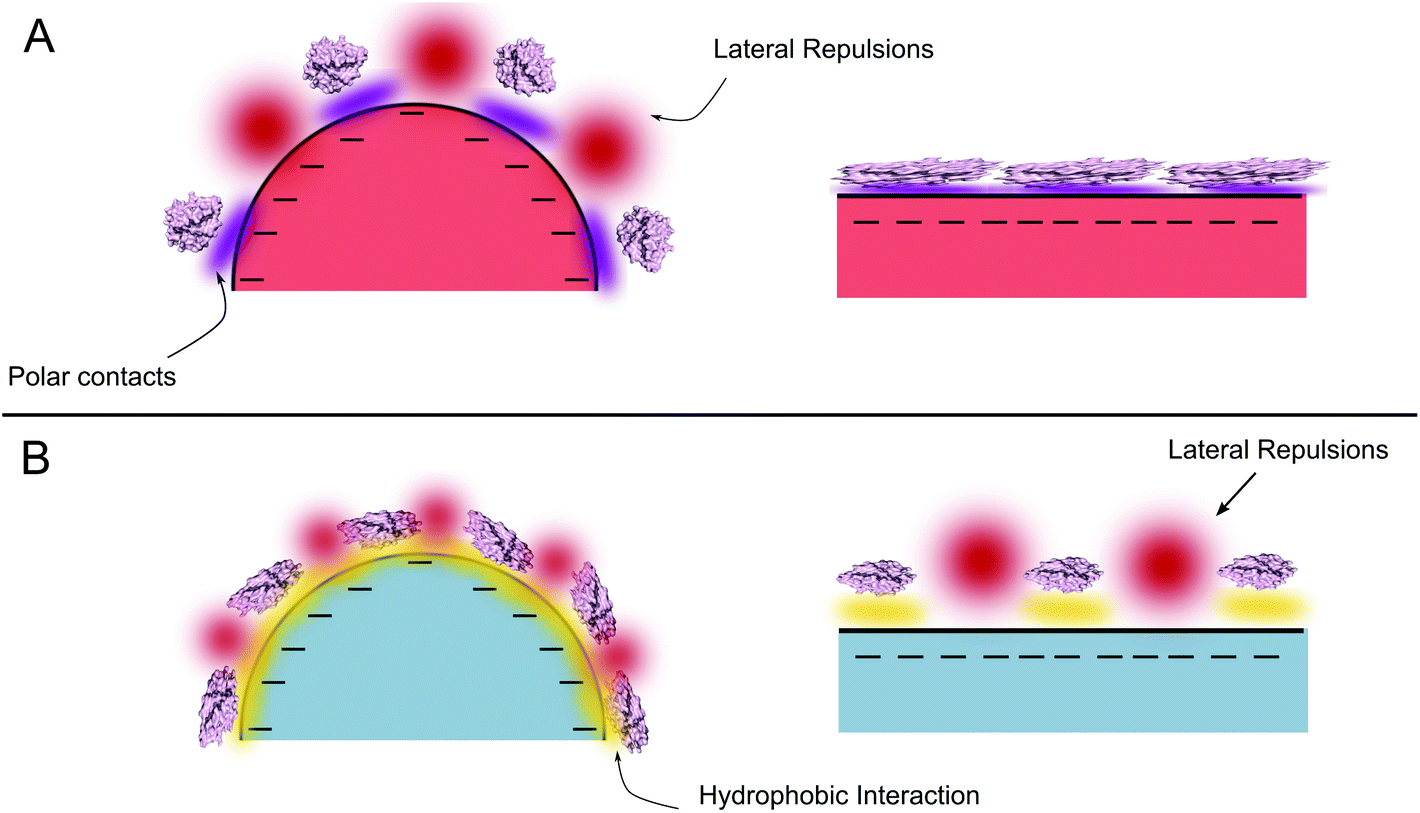 | ||
| Fig. 5 Scheme of the interactions that determine the conformational change and the adsorption for curved and flat surfaces. (a) Polar (b) apolar surfaces. | ||
Protein adsorption to the polar surfaces is postulated to happen via replacing the intramolecular polar contacts (that stabilize the native fold of the protein) with the formation of new polar contacts with the surface.7 Accordingly, a flatter surface with respect to the protein provides the largest contact area for interactions, therefore presumably facilitating a stronger protein adsorption and coverage on the nanoparticle surface, as demonstrated in Fig. 5. The significant difference between the adsorption of trHCAii and HCAi to the PS-COOH nanoparticles mimics the behavior observed previously for trHCAii and HCAi adsorption on Si nanoparticles and presumably the determining factor is the difference in the stabilities of the proteins. Therefore the poor adsorption of HCAi onto negatively charged particles is probably due to the smaller gain in the conformational entropy at the surface (since HCAi is a “hard” protein) that cannot compensate the electrostatic penalty which originates from the same sign net charges of the HCAi and PS-COOH nanoparticles. trHCAii on the other hand, classified as a “soft” protein, can partially unfold at the nanoparticle surface with a subsequent gain in the conformational entropy that favors the adsorption process.5,6
4.2. Conformational changes are dependent on protein and the particle nature
Conformational changes or rearrangements of a protein on the nanoparticle surface often occur and contribute to the protein adsorption process. ANS fluorescence is an ensemble method that reports the sum of the unfolding of the protein on the particle surface together with the unbound and intact protein. Therefore, to break the cumulative signal into individual contributions solely result from conformational changes, the amount of bound protein to each nanoparticle size at the equilibrium must be known which is determined by the association constant in a reversible adsorption process. Consequently, the extent of protein structural changes for each particle size can be correctly estimated. It is important to note that a quantitative measure of the unfolding extent cannot be achieved by using ANS as a reporter. This is because of the amount of hydrophobic patches exposed is not linearly correlated to the extent of unfolding. Especially at the extreme cases of unfolding, where protein exists as a linear polypeptide chain, the hydrophobic patches are nonexistent.The conformational changes of HCA variants on Si nanoparticles have been widely investigated by using a variety of methods. An earlier study by Lundqvist and coworkers showed that the HCAi and trHCAii are destabilized by Si nanoparticles of 6, 9 and 15 nm and the effect gets pronounced with decreased curvature of nanoparticle.23 This is in line with our results even though the particle sizes are significantly bigger. The ANS signals for trHCAii on the Si surfaces indicate that the protein undergoes conformational changes after the initial docking event regardless of the size of the nanoparticle. On the other hand, HCAi is more resistant to surface-induced unfolding and only the biggest Si nanoparticle causes a significant conformational change as shown by the ANS fluorescence intensity over time. The lack of the difference between the protein–nanoparticle and protein in bulk signal for the smallest nanoparticles in the case of HCAi could be due to the low binding of this variant to the nanoparticle surface.
Further studies on the Si particle curvature effect on the conformational changes of human serum albumin,19 ribonuclease A62 and cytochrome c63 share the same observations as our findings, that the curvature decrease leads to a greater unfolding of the protein. Norde has proposed a model for the adsorption mechanism where the unfolding of a protein is beneficial in terms of the ability of forming polar contacts with the surface. Eventually, new hydrogen bonds are formed between amino acid residues that were previously involved in forming a secondary structure, and the hydrophilic surface stabilizing the protein in the new conformation.7 This process is more favorable the flatter the surface gets compared to the protein size, as mentioned in the adsorption section because of the larger contact area. We believe that the driving forces of conformational changes seen here are the result of a combination of the model proposed by Norde and a slight difference of ζ-potential of Si particles as seen in Table S1.†
HCAi and trHCAii adsorbs on the PS-COOH nanoparticles with a clear dependence on the particle size. The conformational changes detected by ANS fluorescence also show the very same correlation between the nanoparticle size and unfolding. At the early stages of the experiment, trHCAii on 26 nm PS-COOH particle surfaces are disrupted the most, followed by 49 nm and 94 nm. The initial difference between trHCAii-26 nm and trHCAii-49 nm system is compensated by time and gradually converge to the same ANS intensity. If we take into account that the Nsurf for 26 nm particles is more than double the value of the Nsurf for 49 nm particles, reaching the same ANS intensity plateau could be an indication of a different extent of conformational changes; such that trHCAii eventually relaxes to a higher extent on the surface of the 49 nm particles than in the 26 nm particles. Another evidence for the difference in the unfolding behavior of the trHCAii on 26 and 49 nm is the time that is required to reach equilibrium in ANS intensity; in trHCAii-26 nm case, equilibrium is reached rapidly at a single step whereas in the case of trHCAii-49 nm system, a fast process is accompanied by a slow process that eventually reaches the plateau much later. The mentioned difference in dynamics of unfolding and the additional slow process could be a requisite to further unfold and create new contacts with the surface which in turn compensate the repulsive lateral interactions that have a bigger role for 49 nm nanoparticle as discussed in the previous section. The low signal evolution for 94 nm particles compared to 49 nm particles may be mainly due to the low stoichiometry of the trHCAii-PSCOOH 94 nm complex (Nsurf = 10 ± 1) combined with the detection limits of the assay. Likewise, HCAi is also affected the most by 26 nm particles. However, no detectable changes were observed for HCAi and 49 nm PS-COOH nanoparticle system. This may again be associated to the detection limit of the ANS fluorescence method because the Nsurf value is smaller than 10. As discussed in the adsorption section, HCAi presumably does not adsorb to the 94 nm nanoparticles hence it is not surprising that the ANS intensity is equal to the bulk protein control intensity over the time.
In order to validate the structural changes in the protein on the nanoparticle surfaces, CD experiments were done. There is a vast majority of the studies in the literature that employ far-UV CD for this purpose. However, near-UV CD is advantageous over far-UV CD because slight relaxation of the tertiary structure can be pointed out which may not necessarily be obvious by the secondary structure determining methods.31,64 Moreover, artifacts to the signal originated from the light scattering of the colloidal dispersions are much less severe in the near-UV range. The spectra of trHCAii in the presence of PS-COOH particles in Fig. 4 show uniform alterations in the whole wavelength region, 240–300 nm. The effect on the whole wavelength region resembles previously observed CD spectra of the interaction between trHCAii and Si particles, note that the referred spectra included contributions only from 100% bound protein.44 CD is an ensemble method, in which the resulting signal comprises the signal from bound proteins on nanoparticles and free proteins in bulk. This fact should be taken into consideration when interpreting the data, as experimental conditions can have a direct impact on the signal output. Taking into account the experimental conditions in our CD experiments, only less than 10% of the total protein is expected on the nanoparticle surface. Therefore the difference in the signal of the native and the nanoparticle bound protein is significant (Fig. 4), considering that 90% of the protein is in native state in bulk.
A closer inspection of the trHCAii CD spectra in Fig. 4 reveals that the 296 nm minima is blue-shifted and a significant loss of the 287 nm minima is observed. These observations can provide information about the structural effects of trHCAii caused by PS-COOH. Freskgard et al. characterized individual Trp contributions to the native HCAii near and far-UV CD spectrum.65 The authors found that all seven Trp residues contribute to the entire near-UV spectrum, to different extents. The major contributions to the native HCAii spectrum were found to be the product of two individual residues, Trp97 and Trp245, which give rise to the two distinct negative minima around 287 and 296 nm. In our study, the variant that is studied (trHCAii) has Trp5 and Trp16 removed but in the light of what is reported in the literature, those Trp residues are among the intermediate contributors to the fingerprint near-UV CD spectrum of HCAii. Observed blue-shift at 296 nm and signal loss at 287 nm indicates that the environment has changed around Trp245 and Trp97. Trp245 is located at the surface of the protein (Fig. S4†) hence a blue-shift indicates a greater solvent accessibility of this residue when adsorbed on PS-COOH nanoparticles. Trp97, however is inside the hydrophobic core of the protein (Fig. S4†), thus the disappearance of the 287 nm minima indicates that Trp97 is more mobile when adsorbed on PS-COOH nanoparticles. Our observations differ from previously published structural changes for HCAii in presence of Si nanoparticles followed over time by Lundqvist and coworkers66 and may indicate different conformational changes induced by PS-COOH compare to Si nanoparticles.
The CD spectra for HCAi in the presence of PS-COOH particles do not show the same spectral alteration as trHCAii. Furthermore, it differs from the spectral alterations caused by Si particles on HCAi, as reported in a previous study.23 The CD spectra for HCAi in presence of PS-COOH are not affected in wavelength region ≈285–300 nm and CD signal decreases (i.e. loss of native structure) in wavelength region ≈260–285 nm. Moreover, CD signal increases in wavelength region ≈240–260 nm, meaning that the structure around one or several Trp residues change in a way that the side chain gets more restricted. HCAi's individual Trp contributions to the CD signal has not been investigated in the same way as HCAii. However, two native near-UV spectra may be speculated to resemble each other as the positions of the individual Trp residues are highly conserved as seen in Fig. S4† except of the additional Trp245 in HCAii. Therefore assuming the contributions of the different Trp residues in the two isoforms are similar, Trp 98, 124, 193 and 210 of HCAi may be speculated to significantly contribute to the signal at the wavelength region 240–260 nm whereas Trp 6 and 17 have a much lower contribution, due to their higher rotational freedom in the native structure (see Fig. S4†). Thus, an increase of signal in this region may indicate a restriction in the mobility of Trp 6 and 17, suggesting a preferential orientation for the adsorption of HCAi on PS-COOH.
In general, the near-UV CD spectra further supports the findings of ANS fluorescence for HCA and 26 nm PS-COOH system. Furthermore, the CD data also support that the proteins interacts differently with Silica and PS-COOH.
4.3. Time scales
Despite the net negative charge of both Si and PS-COOH nanoparticles and HCA variants at the experimental conditions, the proteins adsorb and unfold on the nanoparticle surface (except of HCAi 49 and 94 nm pairs). However, the difference between the dynamics of unfolding of HCAs upon adsorption to Si and PS-COOH are striking. PS-COOH induces a milisecond timescale conformational change on the HCAs whereas Si induces hour timescale conformational changes.The driving forces of the protein adsorption to surfaces and interfaces have been discussed broadly as a result of many pioneering work in the field. However, the driving forces towards the hydrophobic surfaces are scarcely covered compared to hydrophilic surfaces. In many cases, the proteins are found to be readily adsorbed on the hydrophobic nanoparticles rather than hydrophilic particles.5,67 Proteins are amphiphilic molecules therefore not only polar but apolar patches exist on the surface. This facilitates the adsorption of a protein to a hydrophobic surface, which is mainly driven by the hydrophobic effect and possibly enhanced by other favorable non-covalent interactions. Even though the protein stays attached to the surface, higher degrees of freedom upon unfolding and the contribution of solvation of water and counter-ions from the surface makes it entropically favorable.17
On the other hand, the adsorption of proteins on hydrophilic surfaces is greatly influenced by electrostatic interactions. The adsorption of a protein on a same charge sign hydrophilic nanoparticles requires extensive conformational changes to overcome the unfavorable surface-protein repulsions. Moreover the energy barrier that is needed to be overcome for the protein to dock on the surface is exceptionally higher compared to the hydrophobic surfaces therefore the final equilibrium is reached much slower (Fig. 6). The energy barrier for a protein to dock on a hydrophobic surface (despite of the same charges) is much lower due to the favorable contribution of the hydrophobic effect. Furthermore, subtle conformational changes on the hydrophobic surfaces may lead to a faster overall adsorption process. Therefore, we hypothesize that the difference in the time-scales of unfolding for both HCAs on different types of surfaces arise from the simple fact that the time that is needed for a protein to form favorable contacts and minimize its energy differs depending on the polarity of the surface.
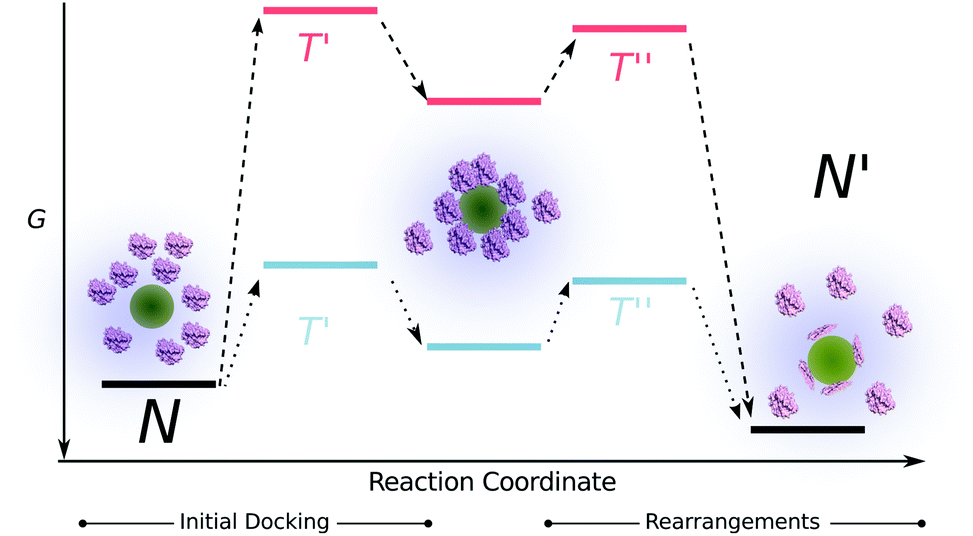 | ||
| Fig. 6 Scheme of the energy profile with respect to the reaction coordinate for the adsorption process of a “soft” protein on a surface. Pink and blue lines represent the energy levels for the adsorption on the polar and apolar surfaces, respectively. | ||
The variation of the unfolding times between the two HCA variants on the same surface is a result of the inherent protein stability, as discussed earlier. HCAi has a stronger resistance to adapt to a surface therefore the energy barrier corresponding to the protein rearrangements at the surface that is depicted in Fig. 6, is higher for HCAi than trHCAii, eventually leading to slower dynamics. The difference between the unfolding dynamics of HCAi and HCAii has been observed earlier by Lundqvist et al. with NMR measurements.68 The authors followed the loss of NMR signals from the two isozymes on 6 and 9 nm Si nanoparticles over a time period and found that the HCAii immediately loses its native structure whereas much prolonged incubation of HCAi and Si nanoparticles lead to the loss of native NMR signals. The authors report a fast on-off exchange of HCAi on Si particles as well, pointing to a low affinity between HCAi and the nanoparticle surface. The combination of low affinity to surface and the resistance to conformational changes results in slow dynamics for the “hard” HCAi compared to “soft” trHCAii.
5. Conclusions
Here, we have performed a detailed study on the effect of different sizes of the negatively charged silica (hydrophilic) and polystyrene with carboxyl functional groups (hydrophobic) nanoparticles on the unfolding behavior of two isoforms of human carbonic anhydrase enzyme. We show that the dynamics of the binding-induced structural rearrangements of HCAs on hydrophobic nanoparticles take place in seconds whereas hydrophilic particles impose an hour-scale process. Here, we propose a simple model for the adsorption process in order to provide a combined explanation to the effects of size and polarity of nanoparticles together with inherent protein stability. The discrepancy between the timescales for different surfaces are dependent on the relative weight of two main processes involved in the adsorption, namely the initial docking and structural rearrangements. The timescale of the initial docking is determined by the energy barrier of the process, which is tightly regulated by the surface properties of the nanoparticles. Furthermore, alongside the effect of the surface properties, the inherent stability of the protein contributes to the dynamics of the structural rearrangements. Higher tendency to unfold at a surface renders faster adsorption dynamics. We show that the adsorbed protein per nanoparticle surface area inversely correlates with the size of PS-COOH nanoparticles and the unfolding extent at the particle surface varies. Considering a reversible process and the homogeneity of the particle surface, this finding indicates a higher coverage of the surface due to the reduction of unfavorable interprotein interactions arising from the conformational rearrangements. Finally, the size of Si nanoparticles (polar) is found to be directly proportional to the extent of adsorption induced structural rearrangements. This finding further supports the model, in which a higher contact area for bigger sized nanoparticles leading to extensive conformational changes. Mentioned changes in turn may decrease the interprotein interactions.Acknowledgements
This work was supported by Swedish Research Council (VR) and the Crafoord Foundation.References
- D. F. Emerich, Expert Opin. Biol. Ther., 2005, 5, 1–5 CrossRef CAS PubMed.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef CAS PubMed.
- C. Buzea, I. Pacheco and K. Robbie, Biointerphases, 2007, 2, MR17–MR71 CrossRef.
- G. Y. Tonga, K. Saha and V. M. Rotello, Adv. Mater., 2014, 26, 359–370 CrossRef CAS PubMed.
- W. Norde, Adv. Colloid Interface Sci., 1986, 25, 267–340 CrossRef CAS.
- C. A. Haynes and W. Norde, J. Colloid Interface Sci., 1995, 169, 313–328 CrossRef CAS.
- W. Norde, Colloids Surf., B, 2008, 61, 1–9 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggård, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- A. A. Shemetov, I. Nabiev and A. Sukhanova, ACS Nano, 2012, 6, 4585–4602 CrossRef CAS PubMed.
- A. Reisch, J. Hemmerle, J.-C. Voegel, E. Gonthier, G. Decher, N. Benkirane-Jessel, A. Chassepot, D. Mertz, P. Lavalle, P. Mesini and P. Schaaf, J. Mater. Chem., 2008, 18, 4242–4245 RSC.
- J. S. Bee, D. Chiu, S. Sawicki, J. L. Stevenson, K. Chatterjee, E. Freund, J. F. Carpenter and T. W. Randolph, J. Pharm. Sci., 2009, 98, 3218–3238 CrossRef CAS PubMed.
- R. Cukalevski, M. Lundqvist, C. Oslakovic, B. Dahlback, S. Linse and T. Cedervall, Langmuir, 2011, 27, 14360–14369 CrossRef CAS PubMed.
- I. Lynch, K. A. Dawson and S. Linse, Sci. STKE, 2006, 2006, pe14 Search PubMed.
- G. Anand, S. Sharma, A. K. Dutta, S. K. Kumar and G. Belfort, Langmuir, 2010, 26, 10803–10811 CrossRef CAS PubMed.
- S. Linse, C. Cabaleiro-Lago, W.-F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- M. Malmsten, J. Colloid Interface Sci., 1998, 207, 186–199 CrossRef CAS PubMed.
- A. Assarsson, I. Pastoriza-Santos and C. Cabaleiro-Lago, Langmuir, 2014, 30, 9448–9456 CrossRef CAS PubMed.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 128, 3939–3945 CrossRef CAS PubMed.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244–6256 CrossRef CAS PubMed.
- L. B. Koh, I. Rodriguez and S. S. Venkatraman, Phys. Chem. Chem. Phys., 2010, 12, 10301–10308 RSC.
- J. Wang, X. Y. Chen, M. L. Clarke and Z. Chen, J. Phys. Chem. B, 2006, 110, 5017–5024 CrossRef CAS PubMed.
- M. Lundqvist, I. Sethson and B. H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- M. Lundqvist, I. Sethson and B. H. Jonsson, Biochemistry, 2005, 44, 10093–10099 CrossRef CAS PubMed.
- D.-H. Tsai, F. W. DelRio, A. M. Keene, K. M. Tyner, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2011, 27, 2464–2477 CrossRef CAS PubMed.
- X.-C. Shen, X.-Y. Liou, L.-P. Ye, H. Liang and Z.-Y. Wang, J. Colloid Interface Sci., 2007, 311, 400–406 CrossRef CAS PubMed.
- P. Sabatino, L. Casella, A. Granata, M. Iafisco, I. G. Lesci, E. Monzani and N. Roveri, J. Colloid Interface Sci., 2007, 314, 389–397 CrossRef CAS PubMed.
- A. Kondo, F. Murakami and K. Higashitani, Biotechnol. Bioeng., 1992, 40, 889–894 CrossRef CAS PubMed.
- P. Billsten, U. Carlsson, B. H. Jonsson, G. Olofsson, F. Hook and H. Elwing, Langmuir, 1999, 15, 6395–6399 CrossRef CAS.
- S. Khrapunov, Anal. Biochem., 2009, 389, 174–176 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- S. Koutsopoulos, K. Patzsch, W. T. E. Bosker and W. Norde, Langmuir, 2007, 23, 2000–2006 CrossRef CAS PubMed.
- A. M. Baty, P. A. Suci, B. J. Tyler and G. G. Geesey, J. Colloid Interface Sci., 1996, 177, 307–315 CrossRef CAS.
- S. Goy-Lopez, J. Juarez, M. Alatorre-Meda, E. Casals, V. F. Puntes, P. Taboada and V. Mosquera, Langmuir, 2012, 28, 9113–9126 CrossRef CAS PubMed.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
- M. F. M. Engel, A. J. W. G. Visser and C. P. M. van Mierlo, Langmuir, 2004, 20, 5530–5538 CrossRef CAS.
- M. F. M. Engel, C. P. M. van Mierlo and A. Visser, J. Biol. Chem., 2002, 277, 10922–10930 CrossRef CAS PubMed.
- H. Pan, M. Qin, W. Meng, Y. Cao and W. Wang, Langmuir, 2012, 28, 12779–12787 CrossRef CAS PubMed.
- I. Nasir, W. Fatih, A. Svensson, D. Radu, S. Linse, C. Cabaleiro Lago and M. Lundqvist, PLoS One, 2015, 10, e0136687 Search PubMed.
- T. H. Maren, Physiol. Rev., 1967, 47, 595–781 CAS.
- S. Lindman, I. Lynch, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Nano Lett., 2007, 7, 914–920 CrossRef CAS PubMed.
- L. Stryer, J. Mol. Biol., 1965, 13, 482–495 CrossRef CAS.
- J. Wang, U. B. Jensen, G. V. Jensen, S. Shipovskov, V. S. Balakrishnan, D. Otzen, J. S. Pedersen, F. Besenbacher and D. S. Sutherland, Nano Lett., 2011, 11, 4985–4991 CrossRef CAS PubMed.
- P. Billsten, P. O. Freskgard, U. Carlsson, B. H. Jonsson and H. Elwing, FEBS Lett., 1997, 402, 67–72 CrossRef CAS.
- M. Karlsson, L. G. Martensson, B. H. Jonsson and U. Carlsson, Langmuir, 2000, 16, 8470–8479 CrossRef CAS.
- M. Malmsten, Colloids Surf., B, 1995, 3, 297–308 CrossRef CAS.
- B. Bharti, J. Meissner and G. H. Findenegg, Langmuir, 2011, 27, 9823–9833 CrossRef CAS PubMed.
- P. Billsten, M. Wahlgren, T. Arnebrant, J. McGuire and H. Elwing, J. Colloid Interface Sci., 1995, 175, 77–82 CrossRef CAS.
- E. Sanfins, C. Augustsson, B. Dahlback, S. Linse and T. Cedervall, Nano Lett., 2014, 14, 4736–4744 CrossRef CAS PubMed.
- G. Aronsson, L. G. Martensson, U. Carlsson and B. H. Jonsson, Biochemistry, 1995, 34, 2153–2162 CrossRef CAS.
- P. O. Freskgard, U. Carlsson, L. G. Mårtensson and B. H. Jonsson, FEBS Lett., 1991, 289, 117–122 CrossRef CAS.
- T. Arai and W. Norde, Colloids Surf., 1990, 51, 1–15 CrossRef CAS.
- D. A. Dolgikh, A. P. Kolomiets, I. A. Bolotina and O. B. Ptitsyn, FEBS Lett., 1984, 165, 88–92 CrossRef CAS.
- M. Svensson, P. Jonasson, P. O. Freskgård, B. H. Jonsson, M. Lindgren, L. G. Mårtensson, M. Gentile, K. Borén and U. Carlsson, Biochemistry, 1995, 34, 8606–8620 CrossRef CAS.
- C. Röcker, M. Pötzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580 CrossRef PubMed.
- X. Jiang, S. Weise, M. Hafner, C. Röcker, F. Zhang, W. J. Parak and G. U. Nienhaus, J. R. Soc., Interface, 2010, 7(Suppl 1), S5–S13 CrossRef CAS PubMed.
- B. C. Tripp, J. J. Magda and J. D. Andrade, J. Colloid Interface Sci., 1995, 173, 16–27 CrossRef CAS.
- J. H. Teichroeb, J. A. Forrest and L. W. Jones, Eur. Phys. J. E, 2008, 26, 411–415 CrossRef CAS PubMed.
- K. Kaur and J. A. Forrest, Langmuir, 2012, 28, 2736–2744 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- S. Duinhoven, R. Poort, G. Van der Voet, W. Agterof, W. Norde and J. Lyklema, J. Colloid Interface Sci., 1995, 170, 340–350 CrossRef CAS.
- W. Shang, J. H. Nuffer, J. S. Dordick and R. W. Siegel, Nano Lett., 2007, 7, 1991–1995 CrossRef CAS PubMed.
- W. Shang, J. H. Nuffer, V. A. Muniz-Papandrea, W. Colon, R. W. Siegel and J. S. Dordick, Small, 2009, 5, 470–476 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- P. O. Freskgard, L. G. Martensson, P. Jonasson, B. H. Jonsson and U. Carlsson, Biochemistry, 1994, 33, 14281–14288 CrossRef CAS.
- M. Lundqvist, C. Andresen, S. Christensson, S. Johansson, M. Karlsson, K. Broo and B. H. Jonsson, Langmuir, 2005, 21, 11903–11906 CrossRef CAS PubMed.
- C. A. Haynes, E. Sliwinsky and W. Norde, J. Colloid Interface Sci., 1994, 164, 394–409 CrossRef CAS.
- M. Lundqvist, I. Sethson and B. H. Jonsson, Langmuir, 2005, 21, 5974–5979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Particle characterization, Fig. S1: Hydrodynamic diameter difference between protein:nanoparticle complex and nanoparticle control, Fig. S2: ITC raw data, Fig. S3: ANS fluorescence in detail with injection, Fig. S4: Structural representations of proteins. See DOI: 10.1039/C5NR05360A |
| This journal is © The Royal Society of Chemistry 2015 |