
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extracting local surface charges and charge regulation behavior from atomic force microscopy measurements at heterogeneous solid-electrolyte interfaces
Cunlu
Zhao
,
Daniel
Ebeling
,
Igor
Siretanu
,
Dirk
van den Ende
and
Frieder
Mugele
*
Physics of Complex Fluids Group and MESA+ Institute, Faculty of Science and Technology, University of Twente, PO Box 217, 7500 AE Enschede, The Netherlands. E-mail: f.mugele@utwente.nl
First published on 10th September 2015
Abstract
We present a method to determine the local surface charge of solid–liquid interfaces from Atomic Force Microscopy (AFM) measurements that takes into account shifts of the adsorption/desorption equilibria of protons and ions as the cantilever tip approaches the sample. We recorded AFM force distance curves in dynamic mode with sharp tips on heterogeneous silica surfaces partially covered by gibbsite nano-particles immersed in an aqueous electrolyte with variable concentrations of dissolved NaCl and KCl at pH 5.8. Forces are analyzed in the framework of Derjaguin–Landau–Verwey–Overbeek (DLVO) theory in combination with a charge regulation boundary that describes adsorption and desorption reactions of protons and ions. A systematic method to extract the equilibrium constants of these reactions by simultaneous least-squared fitting to experimental data for various salt concentrations is developed and is shown to yield highly consistent results for silica-electrolyte interfaces. For gibbsite-electrolyte interfaces, the surface charge can be determined, yet, an unambiguous identification of the relevant surface speciation reactions is not possible, presumably due to a combination of intrinsic chemical complexity and heterogeneity of the nano-particle surfaces.
1. Introduction
In recent years, high resolution imaging and spectroscopy techniques in Atomic Force Microscopy (AFM) have generated unprecedented insights into structure and dissipation in liquids in the vicinity of solid surfaces. Certain organic liquids have attracted specific attention because of their model character and the simplicity of the dominant molecular interaction forces (e.g. van der Waals interactions), which gives rise – amongst others – to very pronounced and characteristic oscillatory solvation forces.1–5 Compared to these systems, water and aqueous electrolytes are much more complex for several reasons including the strongly dipolar character of water molecules, the role of hydrogen bonding, the hydration of surfaces, and the almost unavoidable presence of ions.6–12 In addition, solid surfaces, including AFM tips, typically acquire finite surface charges upon immersion into water. These surface charges give rise to rather long range electrostatic forces that decay exponentially with a decay length ranging from approximately 1 nm to 100 nm, depending on the salt concentration. Technically, long range electrostatic forces generate a background force that is superimposed onto the more short-ranged chemical forces such as surface and ion hydration forces that play a crucial role in atomic resolution imaging in aqueous environment. More importantly, long range electrostatic forces also provide the physical background field that controls the adsorption of ions, which has been found to have a strong effect not only on the average surface charge but also on the strength of oscillatory hydration forces in water.7,8 A decent understanding and quantitative characterization of electrostatic interactions is therefore crucial for the interpretation of high resolution AFM experiments in aqueous environment.In colloid science, the general principles controlling surface charge, ion adsorption, and electrostatic interaction forces are well established. Surface charge and ion adsorption are generally governed by an equilibrium between desorption and adsorption of protons and ions from and to specific sites on the surface.13,14 To first approximation, the binding energies involved in these processes are governed by short range molecular forces that can be described by the equilibrium constants K (or their counterpart pK = −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) of individual adsorption/desorption reactions, or so-called speciation reactions. In addition to the equilibrium constants, the actual fractional coverage of adsorbed/desorbed species for a given situation depends on the local concentration of the ions next to the surface. The latter is proportional to the bulk concentration but it is modified by the local electrostatic potential at the surface. Because the latter itself is generated by the surface charge, determining the equilibrium charge density of solid-electrolyte interfaces requires a self-consistent solution of both adsorption/desorption equilibria and the electrostatic potential distribution in the vicinity of the interface. In the classical mean field picture, the solution is obtained by coupling the Poisson–Boltzmann (PB) equation for the distribution of ions and electrostatic potential in the diffuse part of the electric double layer to surface speciation reactions of a variable degree of complexity15,16 for the adsorbed ions in the Stern part of the electric double layer. Colloidal and AFM force measurements necessarily involve the presence of two solid-electrolyte interfaces in close proximity. Electrostatic forces only arise once the diffuse parts of the double layers overlap. As a consequence, the electrostatic potential and the local concentration of ions that determine surface charge and fractional coverage of the surfaces change upon varying the distance between tip and sample. This phenomenon is known as charge regulation (CR) and was first described in detail by Ninham and Parsegian.17 Since then, numerous colloidal surface force measurements using the surface forces apparatus (SFA)18–21 as well as colloidal probe AFM force measurements22–27 have established that the surface charge typically changes upon approaching two solid surfaces in an ambient electrolyte because of the CR behavior. In particular, the group of Borkovec (see ref. 28 and refs. therein) invested substantial effort to implement charge regulation models in colloid probe AFM force microscopy and to quantify the degree of charge regulation for a wide variety of materials. Because their primary interest was to describe colloidal interaction forces and not the specific surface chemistry, they introduced a so-called constant regulation approach that allows for describing force distance curves and extracting the net surface charges without explicitly specifying the individual surface speciation reactions.
K) of individual adsorption/desorption reactions, or so-called speciation reactions. In addition to the equilibrium constants, the actual fractional coverage of adsorbed/desorbed species for a given situation depends on the local concentration of the ions next to the surface. The latter is proportional to the bulk concentration but it is modified by the local electrostatic potential at the surface. Because the latter itself is generated by the surface charge, determining the equilibrium charge density of solid-electrolyte interfaces requires a self-consistent solution of both adsorption/desorption equilibria and the electrostatic potential distribution in the vicinity of the interface. In the classical mean field picture, the solution is obtained by coupling the Poisson–Boltzmann (PB) equation for the distribution of ions and electrostatic potential in the diffuse part of the electric double layer to surface speciation reactions of a variable degree of complexity15,16 for the adsorbed ions in the Stern part of the electric double layer. Colloidal and AFM force measurements necessarily involve the presence of two solid-electrolyte interfaces in close proximity. Electrostatic forces only arise once the diffuse parts of the double layers overlap. As a consequence, the electrostatic potential and the local concentration of ions that determine surface charge and fractional coverage of the surfaces change upon varying the distance between tip and sample. This phenomenon is known as charge regulation (CR) and was first described in detail by Ninham and Parsegian.17 Since then, numerous colloidal surface force measurements using the surface forces apparatus (SFA)18–21 as well as colloidal probe AFM force measurements22–27 have established that the surface charge typically changes upon approaching two solid surfaces in an ambient electrolyte because of the CR behavior. In particular, the group of Borkovec (see ref. 28 and refs. therein) invested substantial effort to implement charge regulation models in colloid probe AFM force microscopy and to quantify the degree of charge regulation for a wide variety of materials. Because their primary interest was to describe colloidal interaction forces and not the specific surface chemistry, they introduced a so-called constant regulation approach that allows for describing force distance curves and extracting the net surface charges without explicitly specifying the individual surface speciation reactions.
While providing excellent average forces on a mesoscopic scale, colloidal probe AFM force measurements do not provide the lateral resolution that is required to characterize heterogeneous surfaces nor do they provide a sufficiently well-defined confinement geometry to help bridging the gap from the colloidal scale to atomic scale imaging of solid surfaces. To bridge this gap, we recently extended the principle of electrostatic surface characterization from colloidal science to dynamic AFM measurements with sharp cantilever tips and corresponding high lateral resolution of the order of the tip radius (several tens of nanometers).12,29 Experiments with solutions of chloride salts of Na, K, Ca, and Mg demonstrated that the surface charge of silica and gibbsite surfaces strongly depends on the concentration and valency of the cations. Local surface charge density was extracted based on the asymptotic forces measured at distances much larger than the Debye screening length using solutions of the Poisson–Boltzmann equation under the classical constant charge (CC) or constant potential (CP) boundary condition. In that range, however, the measured forces are inherently small, which limits the accuracy of the measured charge densities. At smaller tip-sample separations, measured forces were found to fall in between the classical CC and CP solutions of the PB equation, indicating the occurrence of charge regulation. In the present work, we implement a data analysis procedure that includes charge regulation to the force measurements presented in ref. 12. Fig. 1(a) presents a typical silica/gibbsite composite sample and the corresponding measured forces for 10 mM NaCl solution at pH 5.8. The novelty of our approach is thus twofold: (i) we demonstrate the applicability and significancy of the charge regulation concept for AFM measurements with sharp tips and correspondingly high lateral resolution. (ii) We go beyond the common constant regulation approach30,31 and extract directly equilibrium constants of surface speciation reactions using the full non-linear Poisson–Boltzmann equation and charge regulation boundary condition. We discuss the applicability and reliability of our approach for common silica-electrolyte and for less common gibbsite-electrolyte interfaces.
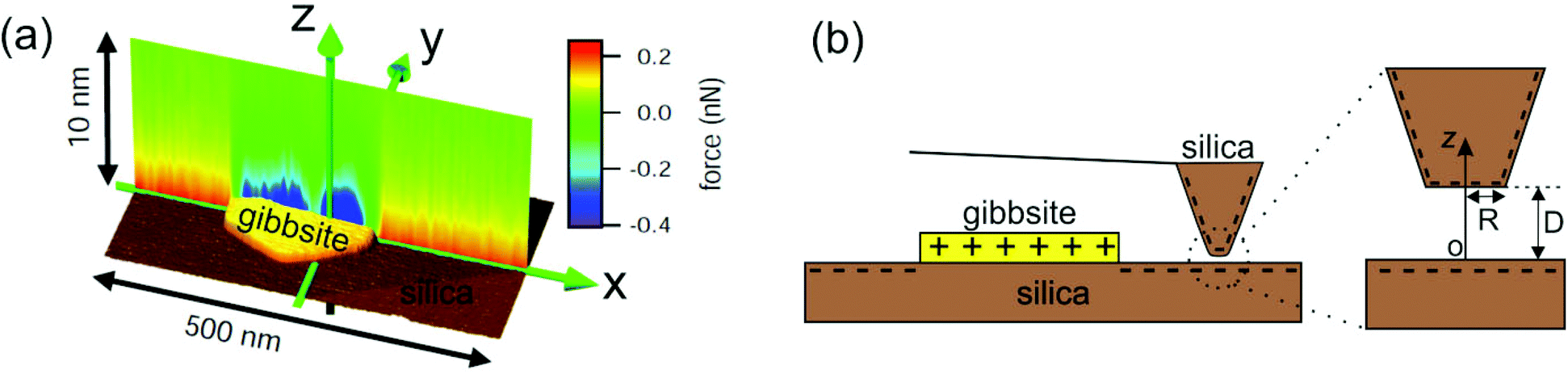 | ||
| Fig. 1 (a) 3D view of a gibbsite nanoparticle adsorbed onto a silica surface along with force-vs.-distance curves (color coded) along a line section through the particle. Repulsive forces (red) upon approaching the silica surface indicate negative surface charge and attractive forces (blue) on the gibbsite particle indicate positive surface charge. Data are acquired with a negatively charged oxidized silicon tip in 10 mM NaCl solution at pH ≈ 5.8. Data adapted from ref. 12. (b) Schematic of the sample system (dimensions not to scale) consisting of a gibbsite platelet immobilized on a silica substrate. The magnification of the tip apex in the right part of the figure gives a more detailed view of the used tip geometry which is modeled as a truncated cone with a flat end having radius of R ≈ 30 nm. | ||
The manuscript is organized as follows. In section 2, we recapitulate the general charge regulation scheme of coupling the PB description of the ion distribution in the diffuse layer to the surface complexation reactions and discuss specific aspects of its implementation for symmetric and asymmetric material combinations of tip and sample. In section 3, we describe the specific data analysis procedure, which involves the simultaneous optimization of parameters by a least-squared fitting of the charge regulation model to the experimental data obtained for variable fluid compositions. We identify well-defined equilibrium constants for deprotonation and cation adsorption reactions on silica. In section 4 we discuss the consequences of our results for AFM measurements in aqueous electrolytes in general. In particular, we extrapolate our results for silica to a wider range of fluid compositions and discuss possible limitations of the mean field approach inherent to our PB description of the electrolyte.
2 Theoretical framework
2.1 DLVO theory
Tip-sample interaction forces are analyzed in the framework of DLVO theory. In DLVO theory the disjoining pressure between two adjacent surfaces at distance D is decomposed into contributions from van der Waals interaction ΠvdW and electrostatic double layer forces Πel.| Π(D) = ΠvdW + Πel | (1) |
Additional contributions to the disjoining pressure due to short range interactions such as hydration forces only become important at tip-sample separations of ≲1–2 nm. In the present analysis, we disregard these contributions. This implies that our model will only be applicable for tip sample separations beyond 1–2 nm.
Once the disjoining pressure is known, the force on the tip is calculated by integrating Π over the tip surface. For spherical probes as in colloidal probe AFM this is typically done using the Derjaguin approximation (see e.g.ref. 32). In our experiments, the AFM tips are slightly flattened leading to a local parallel plate geometry with a rather small contribution from the adjacent cone,12,29 as sketched in Fig. 1(b). We therefore approximate the total force by
| F(D) = πR2Π(D) | (2) |
We estimate the absolute uncertainty of the procedure to be of order 10%.29 Relative trends and the dependence of the force on the fluid composition, however, are not affected by these geometric uncertainties.
 | (3) |
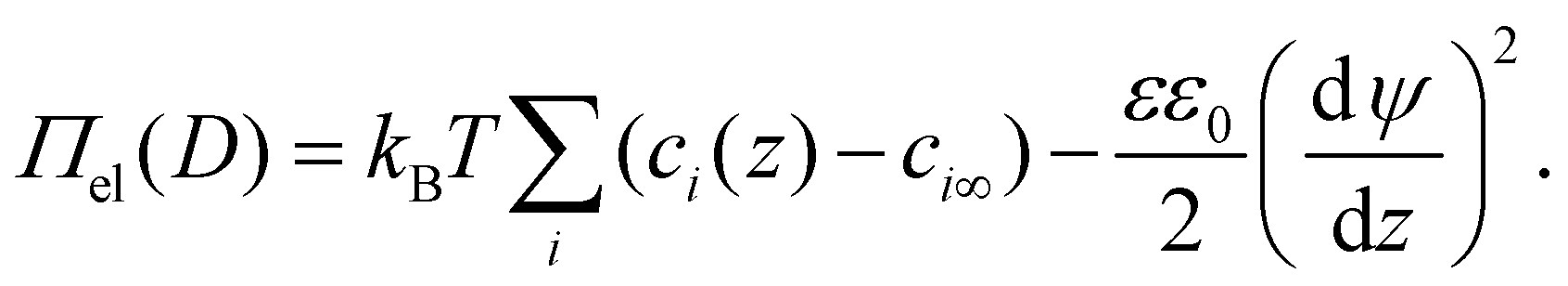 | (4) |
Π el consists of a first contribution due to osmotic repulsion caused by local variations of the ion concentration and a second one due to direct electrostatic attraction (Maxwell stress). Here kB is the Boltzmann constant, T is temperature, εε0 the dielectric permittivity of water. In the first term, the sum runs over all ionic species i in the system. ci∞ is the bulk number concentration of corresponding ions. The solution of eqn (4) depends on the unknown functions ci(z) and ψ(z), i.e., the concentration profiles of all ionic species and electrostatic potential in the electrolyte at an arbitrary position ds < z < D − ds between the two solid surfaces, where ds is the thickness of the Stern layer. Making use of the fact that the ions follow the Boltzmann distribution, i.e. ci(z) = ci∞ exp(−Zieψ(z)/kBT), we can calculate the potential distribution ψ(z) by numerically solving the Poisson–Boltzmann (PB) equation
 | (5) |
 | (6) |
However, ψ(ds) and ψ(D − ds) (or equivalently σI and σII) are not known a priori and need to be determined self-consistently as part of the solution procedure. For an electric double layer at a single solid electrolyte interface this equation reduces to Grahame's equation14 that relates the total diffuse layer charge density and the potential drop in a double layer.
2.2 Surface charge and charge regulation
As discussed above, the surface charge is controlled by adsorption and desorption equilibrium of protons and salt ions from the solution at the interface. We first recapitulate the problem for a single surface reaction (i.e. deprotonation) at the interface following the scheme originally described by Ninham and Parsegian17 to explain the coupling between surface chemistry and diffuse layer physics using the so-called Gouy–Chapman model of double layer. Subsequently, we generalize this scheme to the actual situation of our experiments that involves several surface reactions including the adsorption of salt ions and the formation of a Stern layer.| S− + H+ ⇌ SH | (7) |
The reaction is characterized by an equilibrium constant K1H with a corresponding pK value pK1H = −logK1H. For the silica surfaces to be described below, SH would be simply a silanol group SiOH. The location of the equilibrium of the chemical reaction eqn (7) follows the law of mass action
| {S−}[H+]0 = K1H{SH} | (8) |
| {S−} + {SH} = Γ | (9) |
Eqn (8) and (9) form a set of linear equations for the surface concentration {S−} and {SH} that we can formally rewrite as a matrix equation
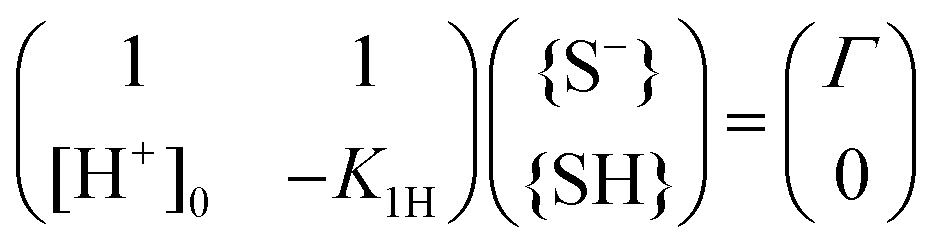 | (10) |
lnK1H/[H+]∞.33
The above procedure results in a surface charge density that is given by
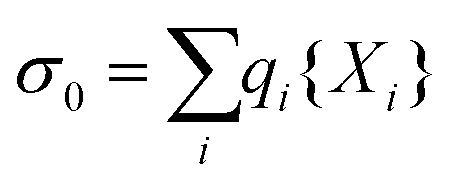 | (11) |
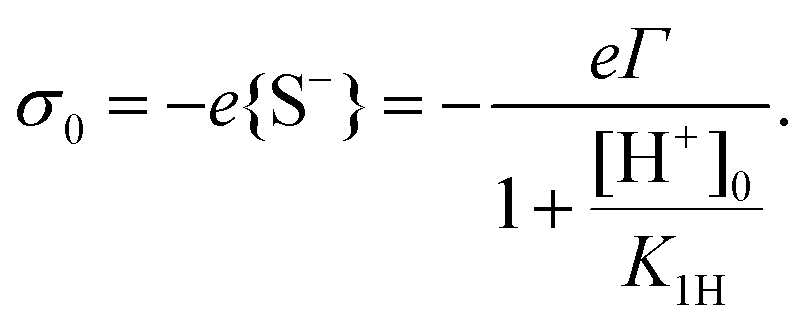 | (12) |
Eqn (12) provides the surface charge as a function of the local proton concentration [H+]0 at the surface. [H+]0 deviates from the bulk concentration [H+]∞ (which is fixed by the pH = −log[H+]∞ of the solution) because of the unknown electrostatic potential on the surface, ψ0. [H+]0 is assumed to follow a Boltzmann distribution
| [H+]0 = [H+]∞e−eψ0/kBT | (13) |
Together, eqn (12) and (13) lead to an expression σ0 = σ0(ψ0), i.e. an equation that connects the potential at the surface to the surface charge via the surface chemistry. This relation is the counterpart of eqn (6), which expresses the charge in the diffuse layer as a function of the potential at the surface. Together they assure charge neutrality, i.e. the charge on the surfaces of tip and sample have to be compensated by the charge in the diffuse layer.
| σI(ψ0I) + σII(ψ0II) + σd(ψ(ds),ψ(D − ds)) = 0 | (14) |
For the simple case of a symmetric system with tip and surface both made of the same material (e.g. silica in our experiments), we can write down the same potential-charge relations, eqn (12), for both surfaces. Using the simple Gouy–Chapman model of the electric double layer, we assume that the ionizable groups are located directly at the surface and that the Poisson–Boltzmann description of the diffuse layer extends all the way to surface. Hence, we identify ψ0I = ψ(0) and ψ0II = ψ(D). (In this case there is no Stern layer, so ds = 0.) Solving eqn (14) we thus obtain the self-consistent distribution of the electrostatic potential, the salt ions in the solution and the ad/desorption of protons on the surface. The results are inserted into eqn (4) and (2) to calculate the total force, which is then expressed as a function of the parameters that characterize the charging reaction of the surface, i.e. the site density Γ and the equilibrium constant K1H. Because Γ is usually fixed by the crystallography of the surface, K1H is the parameter of primary interest that is determined by fitting to the experimental data.
| S− + Czc+ ⇌ S−Czc+ | (15) |
 | (16) |
If lateral interactions between adsorbed species are ignored, as usual in first order approximations, the location of the chemical equilibria is determined for each ionic species separately by a law of mass action using the relevant local concentration of the respective ion in the electrolyte.
While ions are treated as point-like in the Poisson–Boltzmann description of the diffuse layer, their finite size is usually taken into account when considering the position of the adsorbed ions in the Stern layer. Fig. 2 illustrates the Stern models of electric double layer for both surfaces considered in current study. Sophisticated implementations of this idea involve several planes away from the actual surface. Here we consider two planes the “0” plane and the “s” plane, which divides the double layer into a Stern layer and the diffuse layer. The (de-) protonation reaction takes place at the “0” plane, while electrolyte ions are adsorbed at the “s” plane.
 | ||
| Fig. 2 Schematic of the basic Stern layer model of electric double layer on (a) silica and on (b) gibbsite. Protonated and deprotonated silanol and aluminol groups are located at the “0” plane. Adsorbed cations (Na+, K+) on silica and Cl− anions on gibbsite from the solution are located at the “s” plane to neutralize partially the deprotonated silanol and protonated doubly coordinated aluminol sites. The region between “0”and “s” planes is the so-called Stern layer, and potential drop inside it is linear because it does not contain any space charge. Beyond the “s” plane is the diffuse part of electric double layer, and potential drop is exponential because of non-zero space charge density in the diffuse layer. | ||
This approach results in a generalization of the matrix eqn (10)
 | (17) |
2.3 Specific implementation
| σ0 = −e(Γ − {SiOH}) = −e{SiO−} − e{SiO−C+} | (18) |
| σs = e{SiO−C+} | (19) |
Because the space charge density between the “0” plane and the “s” plane vanishes, the potential drop in the Stern layer is linear and can be expressed as
 | (20) |
Together, σ0 and σs define the net or ‘effective’ charge of each surface in the AFM experiments, i.e. we can write σI = σ0I + σsI for the tip and σII = σ0II + σsII, respectively. These two expressions are inserted in eqn (14) to obtain the solution. Because the charge in the diffuse layer compensates the net charges σI and σII and the Poisson–Boltzmann equation is only applied between the s planes of the two surfaces, we identify ψ(ds) = ψsI and ψ(D − ds) = ψsII in eqn (14) in the presence of a Stern layer with adsorbed ions. If tip and sample are of the same material (e.g., AFM silica tip over silica substrate), we use the additional simplification σI = σII and ψ(ds) = ψ(D − ds). The specific chemical reactions and parameter values required for the data analysis are summarized in Table 1.
| Surfaces | ||
|---|---|---|
| Parameters | Silica | Gibbsite basal plane |
| “0” plane reaction | SiOH ⇌ SiO− + H+ with pK1H | Al2OH2+ ⇌ Al2OH + H+ with pK2H |
| “s” plane reaction | SiO−Czc+ ⇌ SiO− + Czc+ with pKC | Al2OH2+Aza− ⇌ Al2OH2+ + Aza− with pKA |
| Stern layer capacitance Cs | 2.9 F m−2 (ref. 59) | 1.49 F m−2 (ref. 38) |
| Site density of surface group Γ | 8 nm−2 (ref. 59) | 13.8 nm−2 (ref. 38 and 59) |
| Hamaker constant A | 0.65 × 10−20 J (ref. 26 and 29) | 1.2 × 10−20 J (ref. 29 and 39) |
| Stern layer thickness ds | 0.4 nm (ref. 35 and 60) | |
| AFM Tip diameter 2R | 52 ± 5 nm (SEM measurement) | |
| pH of solution | 5.8 (measurement) | |
Al2OH2+ ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Al2OH + H+ with K2H Al2OH + H+ with K2H | (21) |
| Al2OH2+Cl− Al2OH2+ + Cl− with KA | (22) |
The charge densities at the “0” and “s” planes are then given by
| σ0 = e{Al2OH2+} + e{Al2OH2+Cl−} | (23) |
| σs = −e{Al2OH2+Cl−} | (24) |
The structure of the electric double layer near gibbsite is sketched in Fig. 2(b). Because the charge density between the “0” plane and the “s” plane vanishes, the electrostatic potential ψs at the “s” plane can be calculated again from the potential and charge density at “0” plane using eqn (20).
For the calculation of the charge distribution and the forces, we now use the σI = σ0I + σsI for the silica tip as obtained in the preceding section and combine it with σII = σ0II + σsII for the gibbsite surface. The characterization of the surface charge and surface chemistry of the AFM tip, first using a silica substrate, is thus a necessary prerequisite to characterize the substrate of interest.
3 Results and discussion
3.1 Least-squared fitting
The force vs. distance curves calculated using the theoretical model depend on a number of parameters, including the radius of the AFM tip R, the Hamaker constant A, the site density Γ, the capacitance of the Stern layer Cs, and the equilibrium constants Ki of the surface speciation reactions considered. The last are the primary parameters of interest here. Therefore, we use reasonable estimates for the former ones based on tip calibration measurements and literature values as described in Table 1. Only the equilibrium constants Ki are used as free parameters to optimize the agreement between experimental data and calculated model curves. We define a merit function | (25) |
where Ft and Fexp denote the theoretically calculated and the experimentally measured force value at the distance Dj. Best fit values for the fit parameters are calculated by maximizing Q within a reasonably chosen range of values for the Ki’s (or the corresponding pKi's) under consideration. Such ranges are chosen based on literature data and refined manually in the course of the fitting procedure. To increase the speed of the optimization procedure, we evaluate Ft(D) on a reduced number of points, N = 15, chosen equidistantly within the range 2 < D < 15 nm and determined Ft(Dj) by interpolating between the adjacent Ft(D) values. Empirical tests showed that the use of a larger number of evaluation points had no significant effect on the fit quality and the resulting optimum parameter values, while significantly reducing the speed of the fit process because the optimization involves the solution of the non-linear Poisson–Boltzmann equation at each distance. The minimum separation of 2 nm was chosen to minimize the influence of short range forces such as hydration forces that are not included in the physical model. The maximum value of 15 nm is based on the rapid decay and the dominance of noise beyond that distance for the conditions of the present set of experimental data.
3.2 Surface charge and optimization of pK values
Very good fits of individual force curves are easily obtained upon optimizing the merit function Q for each individual fluid composition both on silica and on gibbsite surfaces (Fig. 3). For data acquired on silica, the deviation between model curve and experimental data can be reduced below the symbol size in Fig. 3 for the entire parameter range of interest, i.e. for 2 < D < 15 nm. The optimized model curves that include the CR boundary condition (solid lines in Fig. 3) describe the experimental data of a significantly wider range than the approximate solutions for constant potential (CP) and constant charge (CC) solutions (dashed lines in Fig. 3). For gibbsite, the description of the data by the CR solution is also much better than for the CC and CP solutions. Yet, the quality is not quite as good as in the case of silica and deviations are seen already at tip sample separations of ≈2 nm. | ||
| Fig. 3 A comparison of experimental tip-sample force curves (taken from ref. 12) with theoretical force curves for the silica-silica (coded in red) and silica-gibbsite(coded in blue) interaction in aqueous solutions of two monovalent salts (NaCl: first row, KCl: second row) under three different concentrations (1 mM: left column, 10 mM: middle column and 100 mM: right column). Symbols: laterally averaged AFM forces; solid lines: force prediction using charge regulation (CR) with globally optimized pK values (see Fig. 5); dashed lines: constant charge (CC) and constant potential (CP) force predictions. | ||
The fit curves shown in Fig. 3 result from a simultaneous global optimization of the data for all salt concentrations. If optimized individually for each concentration, much better fits than shown in the figure can be obtained. Yet, such an approach would be inconsistent with our modeling that assumes concentration-independent equilibrium constants for each reaction. However, it turns out that good agreement between model curve and experimental data for any single salt concentration is not sufficient to determine a unique set of equilibrium constants. Rather, a whole range of combinations of fit parameters provides fits of similar quality. Fig. 4(a) illustrates this observation for a specific data set, where deprotonation of the silanol group and adsorption of Na+ ions were taken into account in the modeling. This representation of the merit function Q clearly demonstrates that the optimum values of pK1H and pKNa are highly correlated. Fits of equally excellent quality can be obtained for the wide range of parameters shown by the narrow ridge forming the maximum of Q(pK1H, pKNa). These results clearly show that a reliable measurement of pK values based on force curves for a single fluid composition is impossible when several surface reactions occur simultaneously.
 | ||
| Fig. 4 Correlation of fit parameters pK1H and pKC on silica surface for a single salt concentration (10 mM NaCl). (a) Contour plot of the fit quality merit function Q (inverse of squared error). (b) Contour plot of net surface charge, σ0 + σs = −σd, in the limit D → ∞, which illustrates that the quantity probed by the AFM experiment is the diffuse layer charge. Any pK pairs, (pK1H, pKC), falling on the cyan thick solid line produce a same diffuse layer charge density of −0.068e/nm2, and thus a same force. | ||
In fact, this result does not come as a surprise. As the theoretical analysis described in the preceding section showed, the electrostatic part of the disjoining pressure, eqn (4), experienced by the AFM tip depends on the surface chemistry only via the boundary conditions, ψ(ds) and ψ(D − ds) or via the corresponding charge density of the diffuse layer, i.e. via the global charge neutrality condition, eqn (14). Any combination of surface chemical reactions with adequate fractional adsorption that generates the same surface charge gives rise to the same force in the AFM measurement and can thus not be distinguished, as shown in Fig. 4(b). For the present situation, deprotonation and adsorption of monovalent Na+ (or K+) cations give rise to the same surface charge density and thus to the same force. The small difference arising from the different locations in the “0” plane and in the “s” plane is apparently insufficient to create a significant difference in the forces. Specifically, Fig. 4a shows that the experimental data can be explained by assuming exclusively (de-) protonation with a value of pK1H ≈ 7.05 with negligible Na+ adsorption with arbitrary pKNa < 0.5. If pKNa is assumed to be larger than 0.5, pK1H has to be decreased slightly to compensate for the weak adsorption of Na+.
Similar results are obtained for other concentrations and for KCl, both on silica and on gibbsite surfaces (data not shown). For each combination of materials, the correlation curves on silica have the same qualitative shape. A differently shaped family of correlation curves is obtained on gibbsite. Yet, the exact location of the curves in the parameter space depends on the salt concentration: for higher salt concentrations the contribution of adsorbing Na cations is more pronounced, for lower salt concentrations the effect of deprotonation is dominant. The fact that the exact location of the correlation curves depends on the salt concentration is a direct indication that the adsorption of Na+ ions does indeed contribute to the surface charge on silica. Because the surface chemistry should be same for all conditions, we can further constrain the equilibrium constants by requiring that one consistent set of pK values – pK1H and pKC for silica, and pK2H and pKA for gibbsite – should be obtained by simultaneous optimization of Q for all salt concentrations investigated. Fig. 5 shows the merit function Qall for both silica and gibbsite surfaces determined from all the concentrations in the range of 1 mM to 100 mM (i.e., 1, 3, 10, 30, 100 mM). Forces obtained at the lowest (0.5 mM) salt concentration were excluded from the analysis because the measured forces were overall too low for reliable fitting within the distance range of interest.
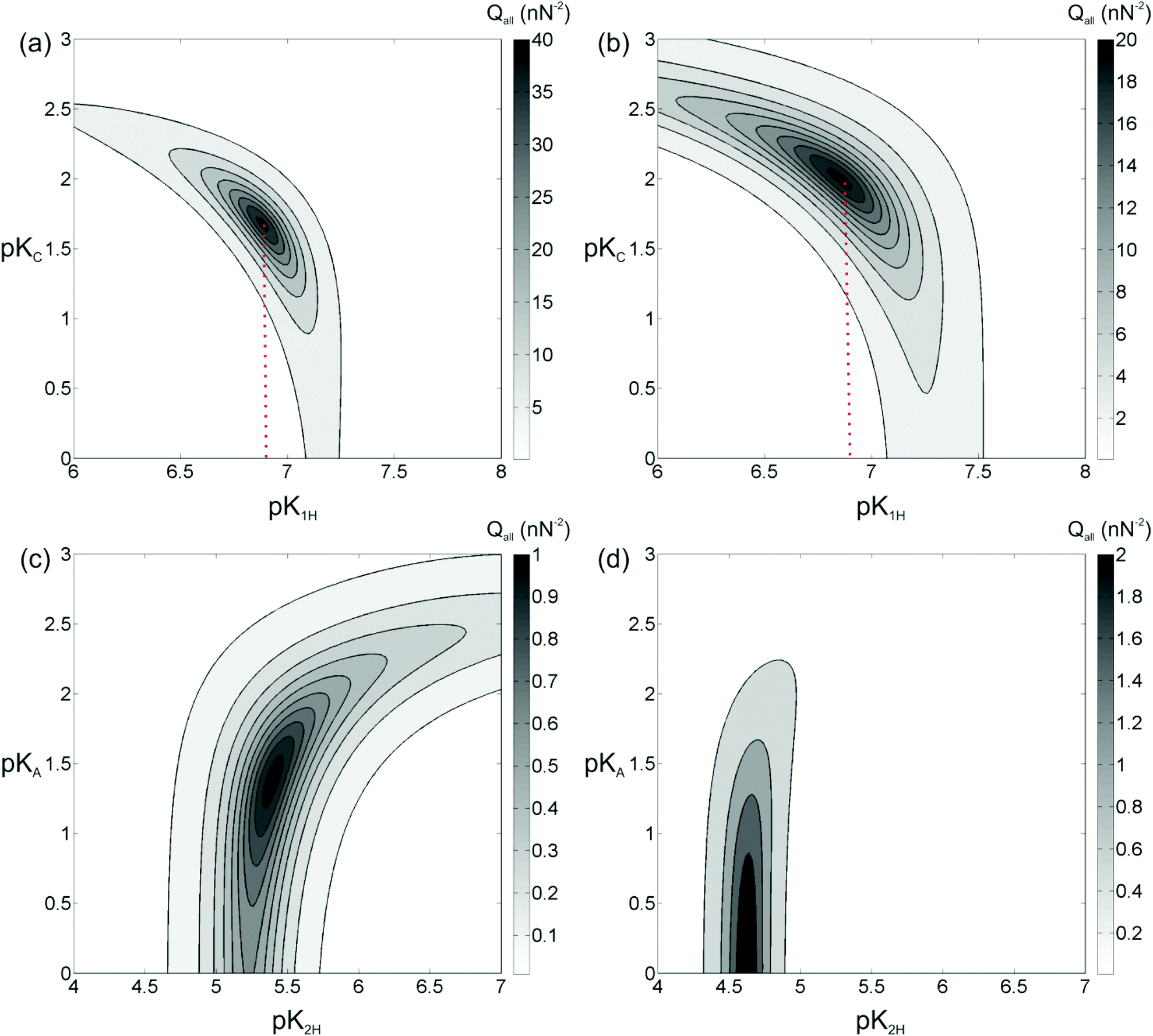 | ||
| Fig. 5 Contour of the inverse of squared error (Qall) in the parameter space defined by pK values of surface reactions. (a) Qall in parameter space (pK1H, pKC) for silica in NaCl solution. (b) Qall in parameter space (pK1H, pKC) for silica in KCl solution. (c) Qall in parameter space (pK2H, pKA) for gibbsite in NaCl solution. (d) Qall in parameter space (pK2H, pKA) for gibbsite in KCl solution. For silica surface, local maxima in (a) and (b) provide best estimates of pK1H = 6.9 ± 0.3, pKNa = 1.65 ± 0.1, pKK = 2.0 ± 0.2. For gibbsite surface, local maxima in (c) and (d) provide best estimates of pK2H = 5.4 ± 0.3/pKCl = 1.4 ± 0.6 for NaCl and pK2H = 4.6 ± 0.2/pKCl < 0.7 for KCl, respectively. | ||
Fig. 5a shows the merit function for a superposition of data acquired for the silica surface at different NaCl concentrations. Indeed, it yields a well-defined combination pK1H = 6.9 ± 0.3 and pKNa = 1.65 ± 0.1 for which the model curves simultaneously describe all experimental force curves. From the measurements with KCl solutions the corresponding optimum values are pK1H = 6.9 ± 0.3 and pKK = 2.0 ± 0.2, as shown in Fig. 5b. Note that the value for pK1H obtained for the two different salts coincides as one should expect. The deprotonation of silanol groups is one of the most widely studied surface reactions in the literature and the reported pK1H values (obtained with potentiometric titration) typically fall in the range 7 to 7.5.33,41–44 Our result includes the lower part of this range. The exact values are known to depend on the origin and the specific preparation conditions of the silica surfaces. For instance, the surface chemistry of our oxidized surfaces of bulk silicon both on the tip and on the sample surface is expected to differ from bulk amorphous silica such as the one precipitated from silanes in a Stöber reaction that is frequently used in colloidal studies of silica. In addition, we note that deviations of the order of 0.5 pK units may also result from uncertainties in other experimental parameters such as the exact geometry and radius of the AFM tip and physical assumptions related to the Poisson–Boltzmann mean field approach.
Comparison between NaCl and KCl solutions shows a somewhat stronger adsorption for K+ than for Na+ ions on silica. This difference, which was already visible in our approximate analysis of the data based on the constant charge model,12 is consistent with earlier reports in the literature based on SFA,21 colloidal probe AFM,26 and electrokinetic measurements.45 It is consistent with the idea that the slightly larger K+ ions are more polarizable than Na+ and hence experience a somewhat stronger dispersion attraction towards the surface.46 Simultaneously, the hydration shell of K+ is somewhat less strongly bound. This may lead to somewhat weaker hydration repulsion between the hydrated ion and the hydrated silica surface.
Similar results can be obtained for gibbsite surfaces. From the plots, we extract optimum values of pK2H = 5.4 ± 0.3 and pKCl = 1.4 ± 0.6 for the measurements with NaCl (Fig. 5c) and pK2H = 4.6 ± 0.2 and pKCl < 0.7 for the measurements with KCl (Fig. 5d). The quality of the analysis for gibbsite is less satisfying than in the case of silica discussed above. First, the maximum value of Qall for the optimum combination of pK values is more than an order of magnitude lower, showing that the quality of the fits is not as good as for silica. Second, if the surface speciation reactions, eqn (21) and (22), chosen to model the data are correct, the values of pK2H and pKCl should actually be the same for both NaCl and KCl solutions. The analysis shows that this is not quite the case. Given the fact that the experimental data are obtained in the course of the same measurement as the very satisfying results on silica, we conclude that the quality of the experimental data as such cannot be the cause of these deviations. This suggests that the specific surface speciation model chosen to describe the data is not appropriate. In fact, we tested a few other possibilities of surface complexation, such as the last two reactions given in eqn (16). None of them yielded more consistent results than the reactions chosen here. Possibly, the actual surface chemistry is in fact much more complex and requires, e.g. the involvement of hydration water. The latter was found to be necessary to understand the adsorption of divalent Mg2+ and Ca2+ cations onto the same surface, as atomically resolved images of these ions on gibbsite in combination with extensive density functional theory (DFT) calculations showed.12 For those ions, the DFT calculations suggested that six partially deprotonated molecules of hydration water play a crucial role for the observed effective surface charge. For monovalent cations of Na+ and K+, the same type of calculation did not reveal a comparable ordered arrangement of adsorbed ions. Yet, it is clear, also from molecular dynamics simulations,11,47 that rather complex configurations of cations and water molecules can appear at clay-electrolyte interfaces that may be too complex to be captured by a few simple surface complexation reactions as eqn (21) and (22). Moreover, the observed heterogeneity of the force curves on the gibbsite particles points to an intrinsic heterogeneity of the surface charge of the particles that might be caused by intrinsic structural and/or chemical defects on the surface. In this context it is worth noting that the absolute value of the surface charge densities in the present experiments is rather low in all cases, typically of the order of 0.1e/nm2 (see Fig. 6). For a typical tip-sample interaction area of the order of 500 nm2, this means that the tip typically probes no more than a few tens of charges on each surface. Surface defects carrying – say – 10 elementary charges therefore already produce substantial deviations that undermine the idealized approach of the surface speciation reactions discussed in section 2.3. These considerations also highlight the remarkable degree of homogeneity of the silica surfaces.
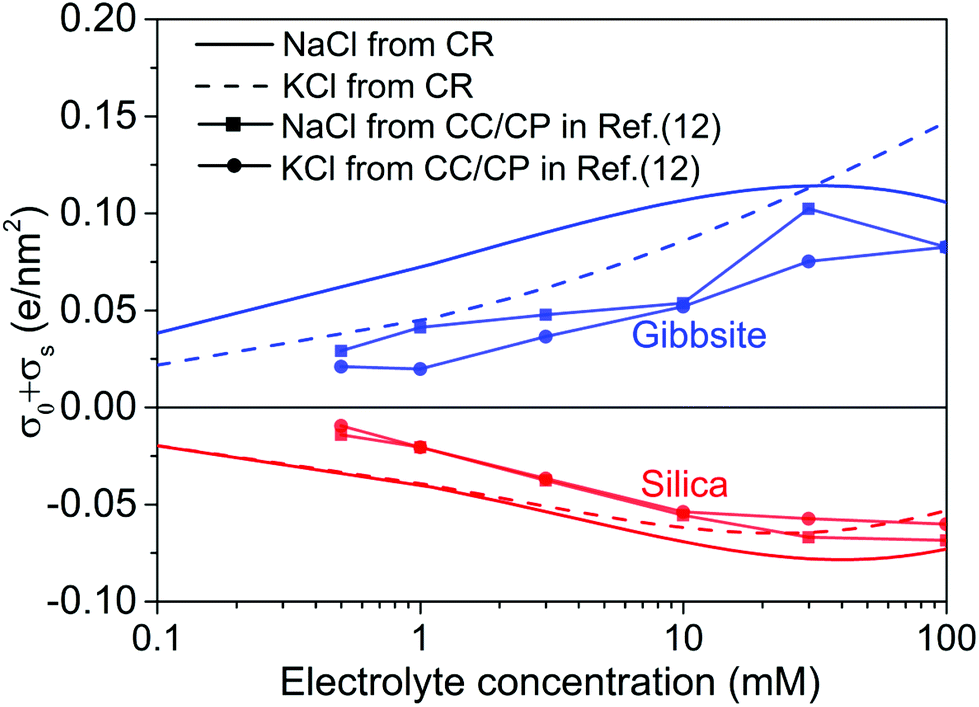 | ||
| Fig. 6 The unregulated (no interaction between two surface, i.e., D → ∞) diffuse layer charge density, σ0 + σs, as a function of electrolyte concentration for two monovalent electrolytes used in the current study. For comparison purposes, the results determined from CC/CP boundaries in ref. 12 is also included in the plot. | ||
4 Discussion
4.1 Surface charge density
Fig. 6 shows the effective surface charge density σ0 + σs of silica and gibbsite in both NaCl and KCl solutions as calculated based on the surface speciation reactions with optimized pK values for individual solid-electrolyte interfaces, i.e. extrapolated to infinite tip-sample separation. On both surfaces, the absolute value of the surface charge density is found to increase with increasing salt concentration. This is due to the fact that increased concentration improves the electrostatic screening and thereby reduces the cost in free energy involved in the creation of surface charge.Next to the solid and dashed lines representing the results with current charge regulation model, the graph also shows as symbols the results from approximate analysis of the tails of the force–distance curves with constant charge/constant potential model reported earlier in ref. 12. The earlier data display the same trends as the present more sophisticated analysis, yet, that analysis clearly underestimated the absolute values of the charge density. Considering the fact that the charge regulation model produces much better fits of the experimental force curves than the constant charge/constant potential model (see Fig. 3) does, it is thus reasonable to believe that the surface charge extracted from charge regulation model is indeed more reliable.
Note that the surface charge densities obtained for gibbsite should be trusted notwithstanding the uncertainties discussed in the preceding section. As our discussion of Fig. 4 showed, good fits of the force curves imply a correct measurements of the surface charge density. Yet, they do not guarantee the correct identification of the surface chemistry. For the rest of the discussion, we will focus on silica surfaces and explore the consequences of the specific surface speciation reactions that we can trust.
4.2 Charge regulation and local fluid composition
Using the optimized combination of pK values, we can analyze the composition of the fluid and the coverage of specific species on the silica surface as a function of the tip-sample separation. Fig. 7 summarizes some in general terms well-known (see e.g.ref. 14) basic results for the distribution of protons that arises from the self-consistently determined potential ψ(z) using the Boltzmann distribution. If the separation of tip and sample is large compared to the Debye length, the system displays a typical behavior of the so-called thin double layer limit. The surface charges are screened in a diffuse layer extending a distance of the order of the Debye screening length. In the middle of the gap, the electrolyte has its bulk composition. Under these reference conditions, charge regulation has no effect, as shown by the overlapping blue curves on the top of Fig. 7. For tip-sample gaps of the order of the Debye length or less, the effect of charge regulation becomes apparent. The local concentration of protons increases with decreasing tip-sample separation. Charge regulation then leads to a partial re-adsorption of protons to the solid surface, as a comparison between the dotted lines for the CC boundary and the solid lines for the CR boundary shows. A CP boundary condition (dashed lines) would lead to even more pronounced re-protonation of the surface. At the smallest separation (black curves), the proton concentration is almost constant all across the gap, as expected for the thick double layer limit. Note that the Na+ and the K+ ions follow exactly the same Boltzmann distribution, except for the generally different limiting concentration in the bulk. | ||
| Fig. 7 Effect of charge regulation on pH/proton concentration distribution between tip and silica sample within 10 mM NaCl solution for tip-sample separations of D = 50 nm (green), 5 nm (red) and 1 nm (black) corresponding to weak, intermediate and strong double layer overlap (Debye length: 1/κ = 3 nm). | ||
It is also interesting to consider explicitly the consequences for the composition of the surface. It turns out in the first place that the total degree of deprotonation, Γ − {SiOH}, is rather low as expected for the conditions of our experiments at pH ≈ 6, see Fig. 8(a). Given the typical site density of 8 sites per nm2, this low degree of deprotonation implies that the average separation between charged sites on the surface is of the order of a few nanometers, which is comparable to the diameter of supersharp AFM tips that are typically used for high resolution AFM imaging. From that perspective, it is not surprising that supersharp tips hardly feel the presence of such small degrees of surface charge. One may also question, whether the use of a continuous surface charge density is still appropriate under such conditions. The second notable feature in Fig. 8(a) is that most of the deprotonated silanol groups on the surface directly adsorb a cation from the solution. Addition of salt thus promotes the replacement of surface-bound protons by cations. This observation holds for all conditions shown in Fig. 3, and is slightly more pronounced for the slightly more strongly adsorbing K+ ions than for Na+. The majority of the surface charge is thus compensated directly in the Stern layer rather than in the diffuse part of the double layer. This conclusion is consistent with X-ray reflectivity studies,48,49 optical measurements,50,51 and recent molecular simulations.47,52 One consequence of this observation is that the surface charge as determined from an AFM (or SFA) force measurement, which is based on the ion distribution in the diffuse layer, is always lower than the charge density determined by a titration measurement that measures the total number of protons or ions adsorbing to or desorbing from a surface.53
 | ||
| Fig. 8 (a) Fraction of total deprotonated and ion-occupied surface sites in solutions of NaCl and KCl at 10 mM for silica/silica interaction. (b) Schematic illustration of proton and ion transfer during the course of charge regulation as the AFM tip move towards the silica sample. Upon decreasing the tip-sample distance both protons and cations migrate from the solution to the silica surfaces. | ||
As a final remark, Fig. 8(a) also shows that the coverage of the various species on the surface does not depend very strongly on the tip-sample separation. As expected, both protons and cations condense onto charged SiO− sites as tip and sample are brought closer together and the total residual surface charge decreases, as shown schematically in Fig. 8(b). Yet, the total variation between infinite separations and a minimum separation D ≈ 1 nm, down to which the model is reasonably applicable, is typically of the order of several percent. This implies that the chemical composition of the surface in this range is not dramatically altered by the presence of the AFM tip. This conclusion is crucial for the interpretation of AFM measurements in general.7,11,12,54–57 It illustrates that the structure of a solid–electrolyte interface as probed by AFM in high resolution spectroscopy or in high resolution imaging mode is hardly affected by the presence of the tip. While charge regulation has a strong effect on the net surface charge and hence the measured forces, as shown in Fig. 3, the fraction of adsorbed ions still provides a fairly good representation of a single interface in contact with a bulk electrolyte for all the conditions studied here.
Some caveats apply. Obviously, this conclusion holds within the limitations of the present mean field Poisson–Boltzmann model. Under conditions of atomic resolution imaging, where short range chemical and hydration forces play an important role, the picture may be altered. Moreover, the present considerations are limited to a symmetric system, in which tip and sample are made of the same material. If the material of tip and sample behave very differently, e.g. one acting as a proton donor and the other one as a proton acceptor, the effect of bringing of close proximity may be more pronounced. Yet, our results obtained so far all suggest that the tip can be reasonably well considered as a moderate perturbation of a state that is overall governed by the properties of the individual solid–electrolyte interface.
4.3 Extrapolation to variable pH and limitations of Poisson–Boltzmann approach
The good consistency of the analysis presented so far for silica surface, encourages us to extrapolate our data to a broader range of fluid compositions, including in particular conditions of variable pH. Substituting the globally optimized pK values of deprotonation and Na+ and K+ adsorption into our numerical scheme, we calculate the concentration of surface species in the limit D → ∞ for silica surface, and the results are shown in Fig. 9. The calculations display several expected qualitative trends. For both NaCl and KCl, the fraction of free deprotonated SiO− groups monotonically increases with increasing pH at all concentrations (Fig. 9a). In contrast, the behavior as a function of the salt concentration at fixed pH is non-monotonic because two opposing processes compete. On the one hand, the increasing salt concentration improves the screening of electric fields and thereby reduces the energetic cost for the system to increase the surface charge by deprotonation. This trend prevails for low salt concentrations. This process competes, however, with the formation of SiO−C+ complexes on the surface according to eqn (15). At higher salt concentrations, the latter process dominates and causes a decrease of the fractional coverage of SiO−, along with an increase of {SiO−C+}. As expected, {SiO−C+} increases monotonically both with increase pH and with increasing salt concentration, see Fig. 9b. | ||
Fig. 9 Calculated fraction of surface complexes for individual silica-electrolyte interfaces as a function of pH and salt concentration. (a) {SiO−}/Γ in NaCl (left) and KCl (right) solutions. (b) {SiO−Na+}/Γ complexes (left) and {SiO−K+}/Γ complexes (right) in the corresponding salt solutions. The pK values are those globally optimized in Fig. 5. Plot (b) also includes the contour lines of two dimensionless parameters,  (white dash lines) and (white dash lines) and 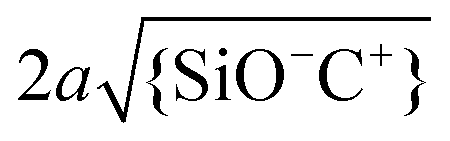 (red dash lines). (Note the difference in gray scale in a) and b).). (red dash lines). (Note the difference in gray scale in a) and b).). | ||
This extrapolation of the model predictions to a broader range of fluid compositions also allows for a systematic discussion of the limitations of the applicability of the Poisson–Boltzmann treatment applied in this study. Various deviations from the simple Poisson–Boltzmann picture have been discussed in the literature.58 The most important corrections include the breakdown of the mean field approach due to direct electrostatic correlations between adjacent ions and the neglect of the finite radius a of the ions. A criterion for the validity of the mean field approach can be determined by comparing the average separation between adjacent adsorbed cations C+ on the surface to the Debye screening length. The white solid lines in Fig. 9b show iso-lines of fixed values of the ratio  . If this ratio is not too large, many screening charges are found between adjacent surface charges and hence the mean field picture is expected to hold – and vice versa. Fig. 9b thus shows that the mean field approach primarily becomes questionable at high pH for low salt concentrations, i.e. for conditions of poor screening but nevertheless high degrees of deprotonation.
. If this ratio is not too large, many screening charges are found between adjacent surface charges and hence the mean field picture is expected to hold – and vice versa. Fig. 9b thus shows that the mean field approach primarily becomes questionable at high pH for low salt concentrations, i.e. for conditions of poor screening but nevertheless high degrees of deprotonation.
Similarly, we can consider the ratio between the diameter 2a of the (hydrated) ions and the average distance between adsorbed ions, i.e. . The corresponding red iso-lines in Fig. 9b show that neglecting the finite ion radius is acceptable provided that the pH and salt concentration are not simultaneously high. For the specific conditions of our experiments (pH ≈ 6 and c∞ = 5 × 10−4 to 0.1 M) the two criteria are indeed decently fulfilled, as Fig. 9b shows.
. The corresponding red iso-lines in Fig. 9b show that neglecting the finite ion radius is acceptable provided that the pH and salt concentration are not simultaneously high. For the specific conditions of our experiments (pH ≈ 6 and c∞ = 5 × 10−4 to 0.1 M) the two criteria are indeed decently fulfilled, as Fig. 9b shows.
5 Conclusions
We used AFM force measurements with sharp tips to probe the surface chemistries/charging behavior of two interacting solid surfaces in aqueous solutions of NaCl and KCl of variable concentration. In agreement with earlier studies using colloidal probe force microscopy, we find that the measured force–distance curves between tip and sample can be described quantitatively down to tip-sample separations of 1–2 nm by taking into account charge regulation (CR), i.e. adsorption/desorption of protons and salt ions as induced by the proximity of tip and sample. For silica surfaces, our systematic approach of simultaneously analyzing experimental data obtained for a variety of salt concentrations allows for quantifying the equilibrium constants for the deprotonation of silanol groups and for the adsorption of Na+ and K+ cations. Given the fact that the vast majority of AFM experiments are carried out with tips made of oxidized silicon, our results can be used to quantify the charge of most AFM tips in some of the most common aqueous electrolyte solutions. Our measurements on the gibbsite surface illustrate the usefulness of this knowledge. Although the identification of the correct surface speciation reactions fails on the more complex gibbsite surfaces, the ability to quantify the charge density on the AFM tip nevertheless enables the measurements of the net surface charge density and thus provides a useful characterization of the material. In contrast to earlier colloidal probe AFM measurements, this is now possible in AFM measurements with sharp tips and a lateral resolution of the order of the tip diameter (in nanometers).Acknowledgements
We thank David Andelman for interesting discussions. This work was financially supported by the Exploratory Research (ExploRe) program of BP plc.References
- A. Maali, T. Cohen-Bouhacina, G. Couturier and J.-P. Aimé, Phys. Rev. Lett., 2006, 96, 086105 CrossRef.
- G. B. Kaggwa, J. I. Kilpatrick, J. E. Sader and S. P. Jarvis, Appl. Phys. Lett., 2008, 93, 011909 CrossRef PubMed.
- S. H. Khan, G. Matei, S. Patil and P. M. Hoffmann, Phys. Rev. Lett., 2010, 105, 106101 CrossRef.
- S. de Beer, D. van den Ende and F. Mugele, Nanotechnology, 2010, 21, 325703 CrossRef PubMed.
- F. Liu, S. de Beer, D. van den Ende and F. Mugele, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2013, 87, 062406 CrossRef.
- J. J. Kuna, K. Voitchovsky, C. Singh, H. Jiang, S. Mwenifumbo, P. K. Ghorai, M. M. Stevens, S. C. Glotzer and F. Stellacci, Nat. Mater., 2009, 8, 837–842 CrossRef CAS PubMed.
- T. Fukuma, Sci. Technol. Adv. Mater., 2010, 11, 033003 CrossRef.
- J. I. Kilpatrick, S.-H. Loh and S. P. Jarvis, J. Am. Chem. Soc., 2013, 135, 2628–2634 CrossRef CAS PubMed.
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954–5960 CrossRef CAS PubMed.
- E. T. Herruzo, H. Asakawa, T. Fukuma and R. Garcia, Nanoscale, 2013, 5, 2678–2685 RSC.
- M. Ricci, P. Spijker and K. Voïtchovsky, Nat. Commun., 2014, 5, 4400 CAS.
- I. Siretanu, D. Ebeling, M. P. Andersson, S. L. S. Stipp, A. Philipse, M. C. Stuart, D. van den Ende and F. Mugele, Sci. Rep., 2014, 4, 4956 CAS.
- R. J. Hunter, Zeta Potential in Colloid Science: Principles and Applications, Academic Press, 1981 Search PubMed.
- H. J. Butt, K. Graf and M. Kappl, Physics and Chemistry of Interfaces, Wiley, 2006 Search PubMed.
- J. A. Davis, R. O. James and J. O. Leckie, J. Colloid Interface Sci., 1978, 63, 480–499 CrossRef CAS.
- J.-F. Boily, J. Lützenkirchen, O. Balmès, J. Beattie and S. Sjöberg, Colloids Surf., A, 2001, 179, 11–27 CrossRef CAS.
- B. W. Ninham and V. A. Parsegian, J. Theor. Biol., 1971, 31, 405–428 CrossRef CAS.
- J. N. Israelachvili and G. E. Adams, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 975–1001 RSC.
- R. M. Pashley and J. N. Israelachvili, J. Colloid Interface Sci., 1984, 97, 446–455 CrossRef CAS.
- V. E. Shubin and P. Kékicheff, J. Colloid Interface Sci., 1993, 155, 108–123 CrossRef CAS.
- J. P. Chapel, Langmuir, 1994, 10, 4237–4243 CrossRef CAS.
- H. J. Butt, Biophys. J., 1991, 60, 1438–1444 CrossRef CAS.
- I. Larson, C. J. Drummond, D. Y. C. Chan and F. Grieser, J. Am. Chem. Soc., 1993, 115, 11885–11890 CrossRef CAS.
- P. Kékicheff and O. Spalla, Phys. Rev. Lett., 1995, 75, 1851–1854 CrossRef.
- R. Pericet-Camara, G. Papastavrou and M. Borkovec, Macromolecules, 2009, 42, 1749–1758 CrossRef CAS.
- M. Dishon, O. Zohar and U. Sivan, Langmuir, 2009, 25, 2831–2836 CrossRef CAS PubMed.
- I. Popa, P. Sinha, M. Finessi, P. Maroni, G. Papastavrou and M. Borkovec, Phys. Rev. Lett., 2010, 104, 228301 CrossRef.
- F. J. Montes Ruiz-Cabello, G. Trefalt, P. Maroni and M. Borkovec, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2014, 90, 012301 CrossRef.
- D. Ebeling, D. van den Ende and F. Mugele, Nanotechnology, 2011, 22, 305706 CrossRef PubMed.
- S. L. Carnie and D. Y. C. Chan, J. Colloid Interface Sci., 1993, 161, 260–264 CrossRef CAS.
- R. Pericet-Camara, G. Papastavrou, S. H. Behrens and M. Borkovec, J. Phys. Chem. B, 2004, 108, 19467–19475 CrossRef CAS.
- H.-J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS PubMed.
- T. Hiemstra, J. C. M. De Wit and W. H. Van Riemsdijk, J. Colloid Interface Sci., 1989, 133, 105–117 CrossRef CAS.
- T. Hiemstra, P. Venema and W. H. V. Riemsdijk, J. Colloid Interface Sci., 1996, 184, 680–692 CrossRef CAS.
- T. Hiemstra, H. Yong and W. H. Van Riemsdijk, Langmuir, 1999, 15, 5942–5955 CrossRef CAS.
- B. R. Bickmore, C. J. Tadanier, K. M. Rosso, W. D. Monn and D. L. Eggett, Geochim. Cosmochim. Acta, 2004, 68, 2025–2042 CrossRef CAS PubMed.
- M.-C. Jodin, F. Gaboriaud and B. Humbert, J. Colloid Interface Sci., 2005, 287, 581–591 CrossRef CAS PubMed.
- J. Rosenqvist, P. Persson and S. Sjöberg, Langmuir, 2002, 18, 4598–4604 CrossRef CAS.
- Y. Gan and G. V. Franks, Langmuir, 2006, 22, 6087–6092 CrossRef CAS PubMed.
- G. V. Franks and Y. Gan, J. Am. Ceram. Soc., 2007, 90, 3373–3388 CrossRef CAS PubMed.
- M. L. Hair and W. Hertl, J. Phys. Chem., 1970, 74, 91–94 CrossRef CAS.
- K. R. Marshall, G. L. Rochester and C. H. Simpson, J. Chem. Ind., 1974, 19, 775–776 Search PubMed.
- N. Sahai and D. A. Sverjensky, Geochim. Cosmochim. Acta, 1997, 61, 2801–2826 CrossRef CAS.
- S. H. Behrens and D. G. Grier, J. Chem. Phys., 2001, 115, 6716–6721 CrossRef CAS PubMed.
- G. V. Franks, J. Colloid Interface Sci., 2002, 249, 44–51 CrossRef CAS PubMed.
- B. W. Ninham and V. Yaminsky, Langmuir, 1997, 13, 2097–2108 CrossRef CAS.
- I. C. Bourg and G. Sposito, J. Colloid Interface Sci., 2011, 360, 701–715 CrossRef CAS PubMed.
- Z. Zhang, P. Fenter, L. Cheng, N. C. Sturchio, M. J. Bedzyk, M. Předota, A. Bandura, J. D. Kubicki, S. N. Lvov, P. T. Cummings, A. A. Chialvo, M. K. Ridley, P. Bénézeth, L. Anovitz, D. A. Palmer, M. L. Machesky and D. J. Wesolowski, Langmuir, 2004, 20, 4954–4969 CrossRef CAS.
- C. Park, P. A. Fenter, K. L. Nagy and N. C. Sturchio, Phys. Rev. Lett., 2006, 97, 016101 CrossRef.
- M. Porus, C. Labbez, P. Maroni and M. Borkovec, J. Chem. Phys., 2011, 135, 064701 CrossRef PubMed.
- L. Wang, C. Zhao, M. H. G. Duits, F. Mugele and I. Siretanu, Sens. Actuators, B, 2015, 210, 649–655 CrossRef CAS PubMed.
- O. Kroutil, Z. Chval, A. A. Skelton and M. Předota, J. Phys. Chem. C, 2015, 119, 9274–9286 CAS.
- J. Lyklema, Fundamentals of Interface and Colloid Science: Liquid-Fluid Interfaces, Elsevier Science, 2000 Search PubMed.
- K.-i. Fukui, H. Onishi and Y. Iwasawa, Chem. Phys. Lett., 1997, 280, 296–301 CrossRef CAS.
- R. García and R. Pérez, Surf. Sci. Rep., 2002, 47, 197–301 CrossRef.
- S.-H. Loh and S. P. Jarvis, Langmuir, 2010, 26, 9176–9178 CrossRef CAS PubMed.
- K. Voïtchovsky, Nanotechnology, 2015, 26, 100501 CrossRef PubMed.
- D. Ben-Yaakov, D. Andelman, D. Harries and R. Podgornik, J. Phys.: Condens. Matter, 2009, 21, 424106 CrossRef PubMed.
- T. Hiemstra, W. H. Van Riemsdijk and G. H. Bolt, J. Colloid Interface Sci., 1989, 133, 91–104 CrossRef CAS.
- J. Newman and K. E. Thomas-Alyea, Electrochemical Systems, John Wiley & Sons, 2004 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |