
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface bioengineering of diatomite based nanovectors for efficient intracellular uptake and drug delivery†
Monica
Terracciano
abc,
Mohammad-Ali
Shahbazi
c,
Alexandra
Correia
c,
Ilaria
Rea
a,
Annalisa
Lamberti
d,
Luca
De Stefano
*a and
Hélder A.
Santos
*c
aInstitute for Microelectronics and Microsystems, National Research Council, Naples, 80131, Italy. E-mail: luca.destefano@na.imm.cnr.it; Tel: +390816132375
bFaculty of Pharmacy, University of Naples Federico II, Naples, 80131, Italy
cDivision of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: helder.santos@helsinki.fi; Tel: +358294159661
dDepartment of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, 80131, Italy
First published on 2nd November 2015
Abstract
Diatomite is a natural porous silica material of sedimentary origin. Due to its peculiar properties, it can be considered as a valid surrogate of synthetic porous silica for nano-based drug delivery. In this work, we exploit the potential of diatomite nanoparticles (DNPs) for drug delivery with the aim of developing a successful dual-biofunctionalization method by polyethylene glycol (PEG) coverage and cell-penetrating peptide (CPP) bioconjugation, to improve the physicochemical and biological properties of the particles, to enhance the intracellular uptake in cancer cells, and to increase the biocompatibility of 3-aminopropyltriethoxysilane (APT) modified-DNPs. DNPs-APT-PEG-CPP showed hemocompatibility for up to 200 μg mL−1 after 48 h of incubation with erythrocytes, with a hemolysis value of only 1.3%. The cytotoxicity of the modified-DNPs with a concentration up to 200 μg mL−1 and incubation with MCF-7 and MDA-MB-231 breast cancer cells for 24 h, demonstrated that PEGylation and CPP-bioconjugation can strongly reduce the cytotoxicity of DNPs-APT. The cellular uptake of the modified-DNPs was also evaluated using the above mentioned cancer cell lines, showing that the CPP-bioconjugation can considerably increase the DNP cellular uptake. Moreover, the dual surface modification of DNPs improved both the loading of a poorly water-soluble anticancer drug, sorafenib, with a loading degree up to 22 wt%, and also enhanced the drug release profiles in aqueous solutions. Overall, this work demonstrates that the biofunctionalization of DNPs is a promising platform for drug delivery applications in cancer therapy as a result of its enhanced stability, biocompatibility, cellular uptake, and drug release profiles.
Introduction
Nanomedicine is an innovative research field combining nanotechnology and medicine, radically changing the healthcare drug delivery landscape, in particular in cancer treatment.1 The application of nanotechnology in cancer therapy is expected to result from more patient compliance, making the therapy more efficient and painless, avoiding problems associated with conventional drug formulations. Thus, the aim of nanomedicine in cancer therapy is the production of nanoscale particles to enhance the bioavailability of drug molecules, improving the tumor-targeting ability and reducing the systemic drug toxicity.Over the last few decades, great efforts have been made in the development of innovative drug delivery systems currently used in clinical and preclinical studies.2–4 Nanoparticle (NP)-based drug delivery systems have provided many advantages over conventional drug formulations, including enhanced solubility of poorly-water soluble drugs, improved pharmacokinetic profiles of drugs, controlled drug release, and simultaneous delivery of drugs for combination therapy to reduce the drug resistance.5–7 Several types of organic, inorganic and hybrid NPs, including dendrimers, liposomes, polymer micelles, nanogels, carbon nanotubes, porous silicon (PSi)/silica NPs, gold NPs, and magnetic NPs have been exploited for drug delivery applications.8–13 Among them, PSi-based NPs are the most used inorganic NPs in biomedical applications due to their unique features such as controllable pore size, high surface area, thermal stability, chemical inertness, biocompatibility, high loading capability, excellent biodegradability, adaptable dissolution kinetics, and controllable drug release profiles.1,14,15
In recent years, emerging natural porous materials for biomedical applications have also been suggested to overcome the shortcomings of the synthetic porous materials, finding in diatomite a viable surrogate.16–19 Diatomite is a cheap fossil compound formed by fragments of diatom siliceous skeletons, with similar physicochemical properties as those of man-made fabricated PSi.20,21 Due to its peculiar properties, such as ordered pore structures, amorphous silica, high surface area, tailorable surface chemistry, high permeability, biocompatibility, non-toxicity, low cost, optical and photonic properties, diatomite has been used in different applications, including optics,22,23 photonics,23 filtration,24 sensing and biosensing,25 and protein separation.26 The main constituent of diatomite is amorphous silica, approved by the Food and Drug Administration (FDA) as Generally Recognized as Safe (GRAS, 21 CFR Section 573.340) for food and pharmaceutical production, and classified in the 3rd group of “Not classifiable as to its carcinogenicity to humans” by the International Agency for Research on Cancer (IARC).27 Surprisingly, its use in nanomedicine is still undervalued, and only recently diatomites have been explored as microcapsules for oral drug delivery, resulting in a non-cytotoxic biomaterial with high potential to improve the bioavailability of loaded oral drugs by sustaining the drug release and enhancing the drug permeability.19 Furthermore, diatom frustules reduced to NPs were explored as potential nanocarriers for biomedical applications.28,29 However, diatomite powder, due to its sedimentary origin, can contain some traces of impurities such as organic components and metallic oxides (MgO, Al2O3, Fe2O3) coming from the environment. As demonstrated in our previous work,28 a multistep procedure based on mechanical and chemical purification treatments was able to remove impurities from frustules, making diatomite NPs safer and more biocompatible vehicles for medical applications. In addition, the biocompatibility of the DNPs and their capability of transport through cellular membranes and their use as non-toxic carriers of siRNA inside cancer cells, has also been demonstrated.29
In the present work, we have investigated the potential of DNPs as a drug delivery system, improving their stability and biocompatibility by PEGylation, and cellular internalization by cell-penetrating peptide (CPP) bioconjugation. Several reports in the literature have demonstrated the enormous advantages arising from the use of polymers in the design of drug nanocarriers, such as the reduction of non-specific aggregation in aqueous medium and the increase of NPs’ stability, biocompatibility, drug loading, and cellular internalization.30–32 The enhancement of the NPs’ cellular uptake is one of the key issues in drug delivery; however, the cell membranes prevent drug carriers from entering the cells, unless an active transport mechanism is involved.33 An efficient approach to deliver NPs or molecules within the cells is to bind them to peptides that can cross the cellular membranes, enhancing their translocation inside the cells. CPP bioconjugation has been proved as a valid strategy to improve the intracellular drug delivery of conventional small drug molecules, NPs or oligonucleotides, and peptide-based therapeutics, increasing their systemic diffusion due to the CPP's property to overcome the lipophilic barrier of the cellular membranes and deliver these therapeutics inside the cells.34,35 Herein, for the first time, a valid biofunctionalization was able to improve the aqueous stability of DNPs, enhancing their hemocompatibility, minimizing their cytotoxicity, and increasing the solubility of a poorly water-soluble anticancer model drug, sorafenib.
Experimental
Production of diatomite NPs
Diatomite NPs (DNPs) were obtained by mechanical crushing, sonication, filtering and purification of natural diatomite (DEREF Spa, Italy), according to the method described in detail in the ESI and Fig. S1-S3.†DNP aminosilane functionalization
DNPs were amino-modified by 5% (v/v) 3-aminopropyltriethoxysilane (APT, from Sigma-Aldrich, USA) solution in absolute ethanol (EtOH).36,37 The silanization process was carried out for 1 h at room temperature (RT) with stirring. The NP dispersion was centrifuged for 30 min at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm and the supernatant was removed. The silanized NPs (DNPs-APT) were then washed twice with EtOH and re-suspended in phosphate buffer saline (PBS) (pH 7.4).
200 rpm and the supernatant was removed. The silanized NPs (DNPs-APT) were then washed twice with EtOH and re-suspended in phosphate buffer saline (PBS) (pH 7.4).
DNP PEGylation and peptide bioconjugation
The amino groups of DNPs-APT were covalently conjugated to the carboxyl groups of HOOC–poly(ethyleneglycol)–NH2 (HOOC–PEG–NH2, average MW ∼ 5000, Jenkem Technology, USA) by 1-ethyl-3-[3-dimethylaminopropyl]carbodiimidehydrochloride/N-hydroxysuccinimide (EDC/NHS, Sigma-Aldrich, USA), and vigorously stirred (800 rpm) overnight (ON) at RT (Scheme 1, I). The PEGylation of DNPs-APT was carried out by dispersing DNPs-APT in PEG solution with a ratio of 1:2 and adding EDC (20 mM)/NHS (12 mM) solution to promote the reaction (ON at RT, 800 rpm). In order to remove the excess of unconjugated polymer, the PEGylated-NPs (DNPs-APT-PEG) were extensively rinsed with EtOH and MilliQ-water, and re-suspended in 2-(N-morpholino)ethanesulfonic acid (MES) saline buffer. Finally, DNPs-APT-PEG were covalently conjugated to the carboxyl groups of a cell penetrating peptide (CPP, (aminooxy)acetyl-Lys-(Arg)9–COOH, GenicBio, China) in MES (NPs:CPP 40:1) by EDC/NHS, stirring ON at RT (Scheme 1, II). After the bioconjugation with the CPP-peptide, the NPs (DNPs-APT-PEG-CPP) were washed twice with MilliQ-water and re-suspended in Hank's balanced salt solution (HBSS)–(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer (pH 7.4).
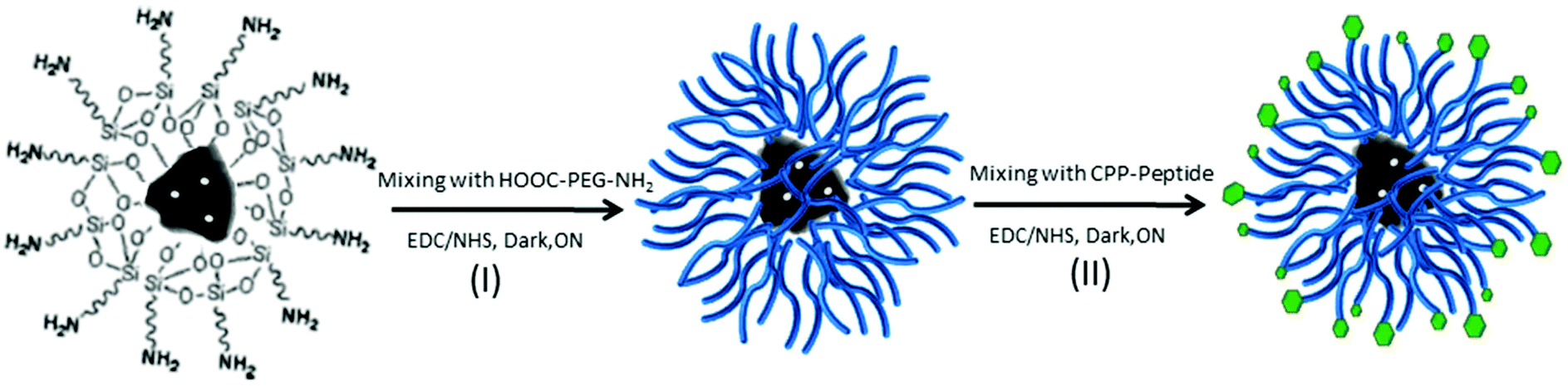 | ||
| Scheme 1 Schematic representation of the DNPs functionalization. Reaction I, the PEGylation of DNPs-APT (I) via EDC/NHS, under stirring ON at RT. Reaction II, CPP-peptide bioconjugation of DNPs-APT-PEG via EDC/NHS, under stirring ON at RT. The dual biofunctionalization is based on a covalent binding between the NPs’ surface and the biomolecules promoted by EDC/NHS chemistry. | ||
Characterization of the DNPs
The hydrodynamic diameter (Z-average), polydispersity index (PDI) and surface zeta (ζ)-potential of the modified-DNPs were measured using a Zetasizer Nano-ZS instrument (Malvern Instruments Ltd, UK). Modified-DNPs were centrifuged and re-dispersed in MilliQ-water before each measurement with a final concentration of 40 μg mL−1.The surface chemical composition of DNPs before and after modification was investigated by attenuated total reflectance Fourier transform infrared spectroscopy (ATR–FTIR). The ATR–FTIR spectra of all samples were obtained using a Bruker VERTEX 70 series FTIR spectrometer (Bruker Optics, Germany) with a horizontal ATR sampling accessory (MIRacle, Pike Technology, Inc.). The ATR–FTIR spectra were recorded in the wavenumber region of 4000–650 cm−1 with a resolution of 4 cm−1 at RT using OPUS 5.5 software. The measurements were carried out on dried DNP samples, left to dry prior to the measurements at RT for 48 h.
The morphology of the bare DNPs was studied using a transmission electron microscope (TEM, Jeol JEM-1400, Jeol Ltd, Japan). Samples were prepared in water and dropped on a carbon coated copper TEM grid before air-drying ON at RT.
DNP fluorescent labelling
Alexa-conjugated DNPs were obtained by using Alexa Fluor® 488 dye (100 μg mL−1 in MES) in a 70:1 ratio (DNPs:Alexa) by EDC/NHS chemistry, stirring for 2 h at RT in the dark. The labelled DNPs were washed twice with MilliQ-water and stored in HBSS–HEPES (pH 7.4). The NPs were labelled for confocal fluorescence microscopy as described below.
Cell culture
MCF-7 and MDA-MB-231 human breast cancer cells were used as cell models for the in vitro studies. The cell lines were grown in Dulbecco's modified Eagle's and Roswell Park Memorial Institute 1640 media, respectively, as described in detail in the ESI and Fig. S4.†Hemotoxicity and red blood cell (RBC) morphological studies
The studies of the hemolytic activity of modified-DNPs and the RBC morphological changes were carried out according to the method described in detail elsewhere.38 Heparin-stabilized fresh human blood was obtained from anonymous donors from the Finnish Red Cross Blood Service and used within 2 h. The hemotoxicity of modified-DNPs was estimated at final concentrations of 25, 50, 100, and 200 μg mL−1 (200 mL RBCs + DNPs) and studied at incubation times of 1, 4, 24, 48 h at RT.38 The morphological changes and DNP–cell interactions were evaluated by scanning electron microscope (SEM) imaging. 5% of RBC suspension was incubated with modified-DNPs (100 μg mL−1) at RT for 4 h. The samples were then fixed with 2.5% glutaraldehyde at RT for 1 h and treated with 0.1% osmium tetroxide in PBS for 1.5 h. The cells were then dehydrated in increasing concentrations of 50, 70, 96 and 100% of EtOH for 5, 10, 20, 15 min, respectively. The cell suspensions were then dropped onto coverslips, dried and sputter coated with platinum before SEM characterization (Zeiss DSM 962, Germany).Cell viability assay
The biocompatibility and toxicity of the DNPs were assessed by measuring the adenosine triphosphate (ATP) activity of the MCF-7 and MDA-MB-231 cells exposed to the bare and modified-DNPs.39 The ATP concentration was estimated quantitatively using the CellTiter-Glo® luminescent cell viability assay (Promega, USA). 100 μL of the cell suspensions with a concentration of 2 × 105 cells per mL in the cell media was seeded in 96-well plates and allowed to attach ON. Subsequently, the cell media were removed and replaced with 100 μL of the DNPs at concentrations of 25, 50, 100, 200 μg mL−1. After 6 and 24 h of incubation at 37 °C, 50 μL of the reagent assay and 50 μL of HBSS–HEPES (pH 7.4) were added to each well. The luminescence of the wells was measured using a Varioskan Flash (Thermo Fisher Scientific Inc., USA). Negative (HBSS–HEPES, pH 7.4) and positive (1% of Triton X-100) control wells were also used and treated similarly as described above. The viability of the negative control was taken as 100%. The results are shown as the average of three independent measurements.Confocal microscopy imaging
Inverted confocal fluorescence microscopy (Leica SP5 II HCS A, Germany) was used to evaluate the cellular uptake of the DNPs into two different breast cancer cell lines, MCF-7 and MDA-MB-231. In this experiment, each cell line (5 × 104) was seeded into Lab-Tek™ 8-Chamber Slides (Thermo Fisher Scientific, USA) in the cell culture media, and allowed to attach ON at 37 °C. After removing the media, 200 μL of Alexa conjugate DNP suspensions (50 μg mL−1) were added to each well before incubation at 37 °C for 6 (only MCF-7) and 12 h. The particle suspension was then carefully removed and the walls were washed twice with HBSS–HEPES buffer (pH 7.4). After that, the cell membrane was stained by adding 200 μL of CellMask™ Deep Red plasma membrane stain (3 μg mL−1; Invitrogen, USA) for 3 min at 37 °C and washed twice with HBSS–HEPES buffer (pH 7.4). Finally, the cells were fixed with 2.5% glutaraldehyde in 0.1 M PBS solution (pH 7.4) for 1 h at RT, washed twice with HBSS–HEPES buffer and analysed by confocal microscopy.Intracellular distribution by TEM imaging
TEM imaging was used to evaluate the cellular uptake of modified-DNPs after 12 h of incubation with MCF-7 and MDA-MB-231 breast cancer cells. For this experiment, 13 mm round shaped cover slips were placed at the bottom of 24-well plates (Corning Inc. Life Sciences, USA), and the cells were seeded in their media and allowed to attach ON. After that, the cells were washed twice with HBSS–HEPES (pH 7.4) and incubated for 12 h with modified-DNPs (50 μg mL−1) at 37 °C. The particle suspension was then removed and the cells were washed twice with HBSS–HEPES. The cells were fixed with 2.5% glutaraldehyde in 0.1 M PBS solution (pH 7.4) for 1 h at RT. After the fixation, the cells were washed with HBSS–HEPES (pH 7.4) and sodium cacodylate buffer (NaCac) for 3 min prior to post-fixation with 1% osmium tetroxide in 0.1 M NaCac buffer (pH 7.4). The cells were then dehydrated with 30–100% EtOH for 10 min each and embedded in epoxy resin. Ultrathin sections (60 nm) were cut parallel to the coverslip, post-stained with uranyl acetate and lead citrate, and then observed by TEM.Drug loading and release
The poorly water-soluble anticancer drug, sorafenib (MW 464.82; LC Laboratories, Boston, USA), was used as a model drug for the loading degree and release studies, before and after biofunctionalization of DNPs. The modified-DNPs were immersed in 10 mg mL−1 of SFN dissolved in acetone solution with a ratio of 1 mg of particles to 1 mL of drug solution, and stirred for 2 h at RT. After the drug-loading, the particles were washed twice with Milli-Q water and the loading degree was determined by immersing 200 μg of drug-loaded modified-DNPs in 1 mL of methanol, under vigorous stirring for 1 h. The amount of drug released was determined by high-performance liquid chromatography (HPLC, Agilent 1260, Agilent Technologies, USA) using a column Phenomenex Gemini (Nx, 3 μm, C18 110 Å). The mobile phase was composed of 0.2% trifluoroacetic acid (pH 2, 42%) and acetonitrile (58%) with a flow rate of 1.0 mL min−1 and UV-detection set at a wavelength of 254 nm (25 °C). The in vitro dissolution evaluation was performed dispersing 200 μg of SFN-loaded DNPs in 50 mL of HBSS–HEPES + 10% fetal bovine serum (FBS) at pH 7.4 and 5.5 and 37 °C, at a stirring speed of 100 rpm. 200 μL of samples was withdrawn from each dissolution test at different time points. The aliquots were then centrifuged for 3 min at 13500 rpm and the supernatant was analysed by HPLC as described above. All measurements were repeated at least three times.
Statistical analysis
Results of the assays are expressed as mean ± standard deviation (s.d.) of three independent experiments. Results were evaluated by means of one-way analysis of variance (ANOVA) with the level of significance set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001 using Origin 8.6 (Origin Lab. Corp., USA).Results and discussion
Preparation and characterization of diatomite NPs
The main peculiarities of NPs as drug delivery systems are their low toxicity, high stability, biocompatibility, and suitability for cellular uptake. In this work, the surface biofunctionalization of DNPs, in order to improve their physicochemical and biological properties, such biocompatibility and cellular internalization, was investigated. Purified DNPs were characterized before biofunctionalization by TEM imaging (Fig. 1), showing a nanometric size and an irregular shape. However, it was previously demonstrated that the irregular shape of DNPs does not affect the cell proliferation and morphology.29 As shown in Fig. 1c and d, the porous structure of the NPs is preserved even after the breaking of the diatomite powder in nanoparticles, where it is clearly visible in the hierarchical pore organization on the surface of the diatomite NPs and the mesopores (10 nm < pores diameter < 50 nm) inside macropores (pores diameter > 50 nm). Due to their peculiar porous nature, diatomite NPs can be very promising for the loading40,41 of a wide size range of molecules from small molecules to peptides, oligonucleotides, proteins, and antibodies for the preparation of targeted NPs for drug delivery applications.41 See the ESI† for more detailed information on the diatomite NP preparation procedure and characterization. | ||
| Fig. 1 TEM images of bare DNPs (a–c) and increasing zoom of the pores of the NPs (d). | ||
Polymers are very versatile materials widely used in drug delivery studies in order to improve the nanocarriers’ properties such as stability and biocompatibility.42 In order to obtain an effective DNP PEGylation, the bare NPs were hydroxylated by Piranha solution, thus increasing the reactivity of their silica surface, by the introduction of –OH groups. The covalent bond between PEG and DNPs requires DNP salinization using an APT solution,28,29 which introduces the highly reactive amino groups (–NH2) onto the NP surface that can be covalently conjugated with the carboxyl groups of PEG molecules using EDC/NHS chemistry (Scheme 1, I). Without this double-step chemical surface treatment, it would not be possible to obtain stable covalently-bonded PEG- or CPP-DNP complexes. Due to the polymer solubility in water and in a wide variety of organic solvents used during the NP modification, it is crucial that the polymer is covalently bound onto the NP surface in order to avoid its untimely detachment.43,44 To improve the DNPs’ cellular uptake, the free amino groups of DNPs-APT-PEG were further chemically conjugated with the carboxyl groups of CPP-peptide, known to highly facilitate the delivery of cargos (e.g., peptides, proteins, genes, and even nanoparticles) across the cell membrane,45 by EDC/NHS chemistry (Scheme 1, II).
The DNPs were characterized before and after the surface modification by DLS, analyzing the hydrodynamic diameter, the PDI, and the surface charge ζ-potential of the particles. An increase of the particles’ size from 317 ± 8 nm to 364 ± 3 nm (DNPs-APT) after the APT solution treatment was observed, confirming the success of the silanization process. A progressive decrease of the nano-aggregates’ size was observed after PEGylation and CPP bioconjugation. The NPs’ size decreased from 364 ± 3 nm (DNPs-APT) to 346 ± 4 nm after PEGylation (DNPs-APT-PEG), and to 340 ± 8 nm after CPP-conjugation (DNPs-APT-PEG-CPP). This result is due to an increase of the DNPs’ surface repulsion forces of the modified surface (DNPs-bare, −19.2 ± 2.0 mV; DNPs-APT, +19.8 ± 3.0 mV; DNPs-APT-PEG, +35.6 ± 1.5 mV; DNPs-APT-PEG-CPP, +40 ± 2 mV), which can be attributed to the positive charge of PEG and CPP-peptide onto the NPs’ surface. As a result, the DNP stability was improved in aqueous solution and a decrease in the PDI of the NPs was also observed (Fig. 2).
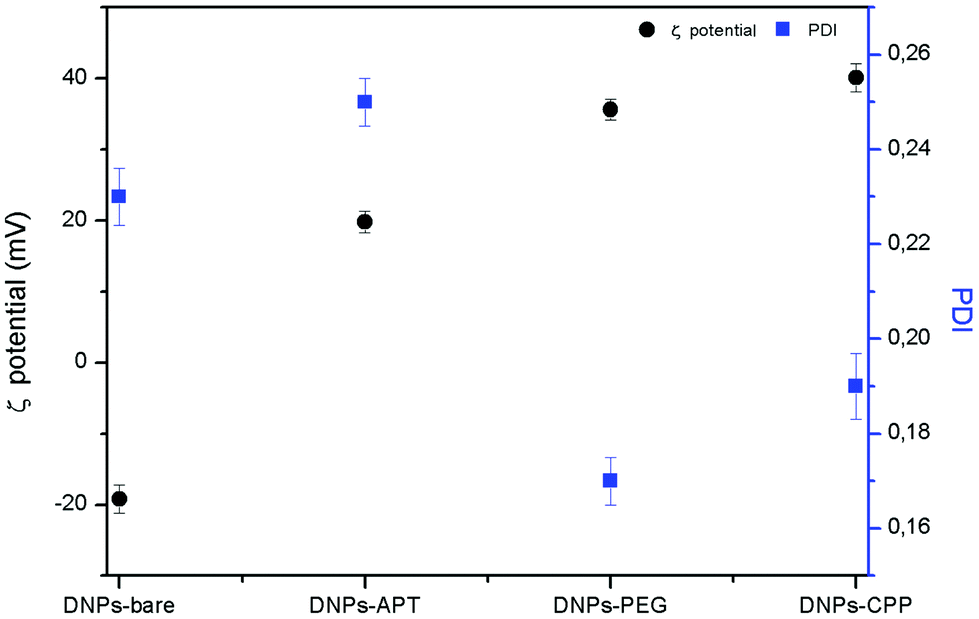 | ||
| Fig. 2 DNPs ζ-potential and PDI before and after each step of functionalization determined by DLS at RT. | ||
The chemical silanization, PEGylation and CPP-conjugation of DNPs were further analysed by ATR–FTIR spectroscopy. Fig. 3 shows the progressive change of DNP FTIR spectra after each modification step. All spectra of DNPs showed an intense band at 1100 cm−1 that corresponds to Si–O–Si bonds, because silica is the main constituent of the diatomite frustules (Fig. S5, ESI†). After the silanization process, the DNPs-APT displayed typical bands of APT corresponding to the CHx stretching at 2941–2570 cm−1, the bending mode of the free NH2 at 1630–1470 cm−1, and the C–N stretching at 1385 cm−1.46,47 After the PEGylation, the DNPs-APT-PEG showed the stretching bands of CHx at 2960–2849 cm−1, the C–H bending vibrations at 2160–1722 cm−1, the amide I band at 1640 cm−1 associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, the amide II resulting from the N–H bending vibration, and the C–N stretching vibration at 1580 and 1360 cm−1, respectively, thus confirming the covalent binding of the PEG molecules onto the NPs’ surface.42,47 After incubation with the CPP-peptide, the DNPs-APT-PEG-CPP displayed a band at 2984–2881 cm−1 corresponding to the CHx stretching, and at 1930 cm−1 assigned to the C–N stretching of amide II, confirming the successful CPP-peptide bioconjugation to the surface of the NPs.47,48
O stretching vibration, the amide II resulting from the N–H bending vibration, and the C–N stretching vibration at 1580 and 1360 cm−1, respectively, thus confirming the covalent binding of the PEG molecules onto the NPs’ surface.42,47 After incubation with the CPP-peptide, the DNPs-APT-PEG-CPP displayed a band at 2984–2881 cm−1 corresponding to the CHx stretching, and at 1930 cm−1 assigned to the C–N stretching of amide II, confirming the successful CPP-peptide bioconjugation to the surface of the NPs.47,48
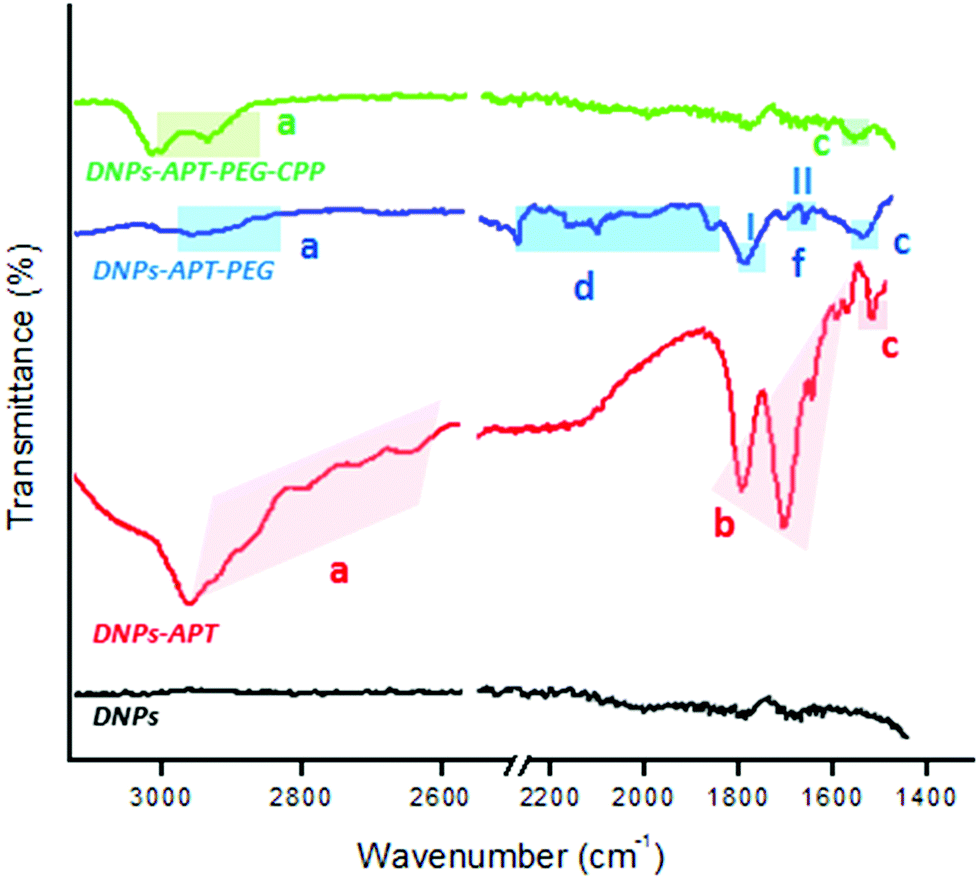 | ||
| Fig. 3 ATR–FTIR spectra of the modified-DNPs. DNPs were characterized before the biofunctionalization (black line), after the silanization process (red line), after the PEGylation (blue line), and also after the CPP-peptide bioconjugation (green line). (a) indicates CHx stretching vibration, (b) the bending mode of the free NH2, (c) the C–N stretching, (d) the C–H bending vibrations, and (f-I and f-II) N–H bending vibration and the C–N stretching vibration, respectively. | ||
Modified-DNP hemotoxicity
The hemotoxicity study, based on the %-lysed RBCs, is an important preclinical study to evaluate the level of the NPs’ hemocompatibility, in order to avoid serious risks to human health (e.g., after intravenous injection of NPs), considering that the erythrocytes constitute a large volume of the blood. Moreover, if NPs induce hemolysis, there is a higher risk that blood constituents can react immunologically to inactivate the NPs, and thus, affecting their function and increasing their elimination by macrophages.49,50In order to evaluate the impact of the modified-DNPs on RBCs, the hemocompatibility and the morphological studies of RBCs were determined after exposure to the modified-DNPs at increasing incubation times (1, 4, 24, and 48 h) and different NP concentrations (25, 50, 100, and 200 μg mL−1).38 The NP hemotoxicity was qualitatively determined by naked-eye colour evaluation of the RBCs’ supernatant incubated with modified-DNPs. The hemotoxicity degree of the DNPs-APT was higher than that observed for PEG and CPP modified-DNPs, since the red colour intensity of the DNPs-APT-RBC supernatant was closer to that of the positive control one (water), as shown in Fig. 4.
 | ||
| Fig. 4 Representative pictures of the RBCs after interaction with the modified-DNPs. The DNPs were incubated with the cells for 48 h and at different concentrations (25, 50, 100, and 200 μg mL−1). | ||
The %-hemolysis determined by spectrophotometric analysis of the supernatants after 48 h of incubation at the maximum concentration of modified-DNPs (200 μg mL−1) was 34% for DNP-APT, 7% for DNP-PEG, and 1.3% for CPP-DNP. This result clearly demonstrated that the biofunctionalization of the NPs improved the DNP hemocompatibility (Fig. 5).
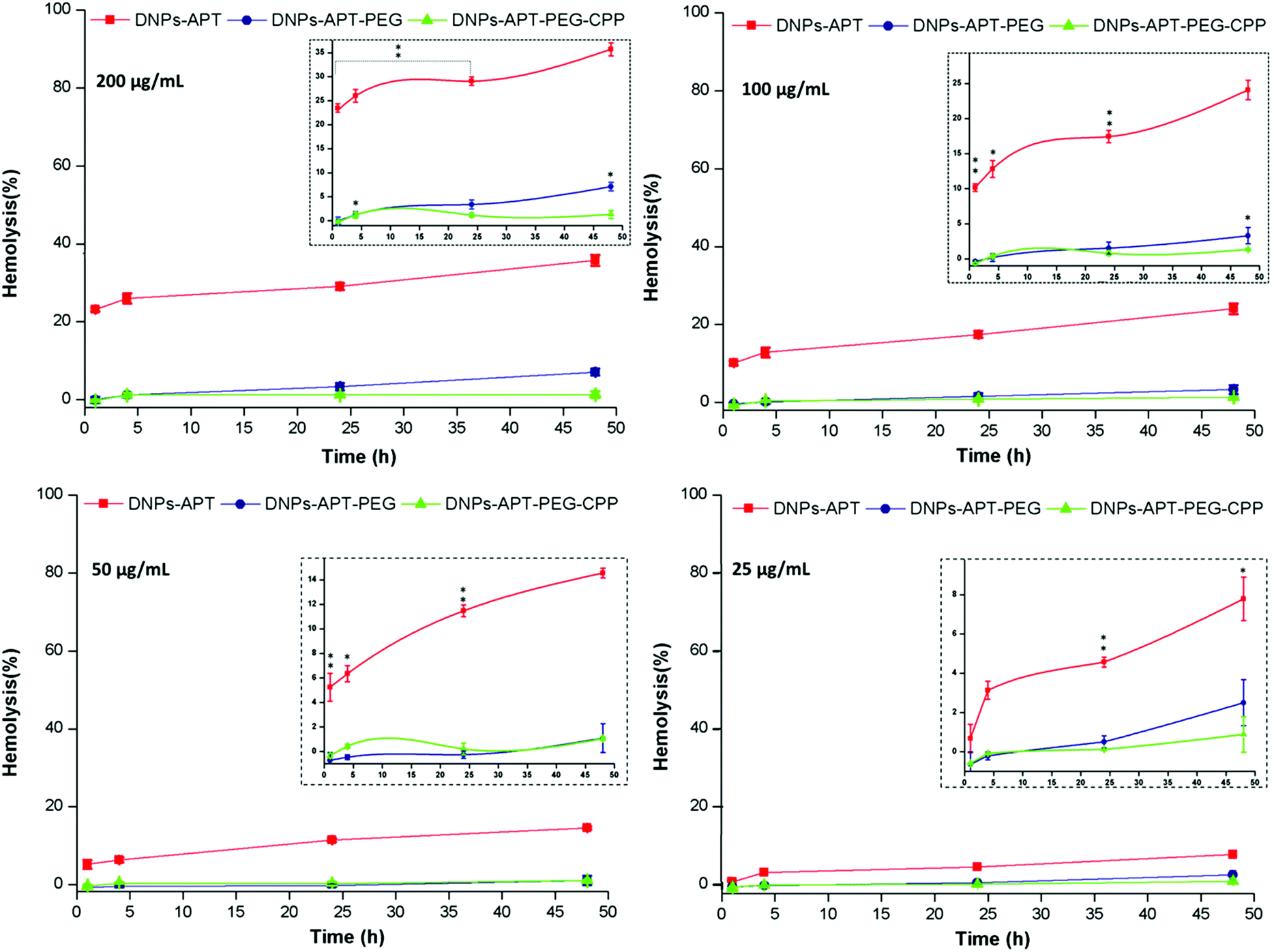 | ||
| Fig. 5 Hemocompatibility of the modified-DNPs. Hemotoxicity of APT-, PEG- and CPP- modified DNPs incubated for 48 h at different concentrations (25, 50, 100, and 200 μg mL−1) with RBCs, estimated by spectrophotometric methods (577 nm) to analyse the amount of lysed-hemoglobin in the supernatants. The insert figures show the magnifications of the graphs. Statistical analysis was performed by ANOVA. The level of significance from the negative control was set as probabilities of *p < 0.05, **p < 0.01 and ***p < 0.001. Error bars represent s.d. (n = 3). | ||
The morphological changes of the RBCs after exposure to the modified-DNPs (200 μg mL−1 for 4 h, at RT) were examined by SEM characterization. The RBCs, in the presence of DNP-APT, completely altered their morphology, changing from the biconcave-like disks to a shrunken shape, with consequent hemolysis (Fig. 6). The DNPs-APT hemotoxicity is attributed to the free positive amine groups existing on the surface of the NPs, which strongly interact with the negative charge surface of the RBCs, resulting in hemolysis and a change of the cell's shape.38 In the case of PEGylated particles, no severe change was observed in the RBC morphology, but the cell membrane was locally wrapped around with the appearance of small holes without significant hemolysis. The relevant decrease of the DNPs-APT hemotoxicity after PEGylation is due to the improved biocompatibility of the NPs as a result of the presence of PEG on their surface, which is known to be relatively non-cytotoxic, non-immunogenic, non-antigenic, and to decrease the protein interaction.51,52 In the case of DNPs-APT-PEG-CPP, there was no relevant change observed in the RBCs’ morphology due to the low cytotoxicity of the CPP-peptide,52 which improved the DNPs’ biocompatibility. Therefore, both the amino function and also the charge density (the number and spatial arrangements of the cationic residues) are important factors for toxicity.53 A three-point attachment is necessary for eliciting a biological response on cell membranes and the activity of a molecule decreases when the space between reactive amino groups increases in the primary structure.54 The arrangement of cationic charges on the molecular structure determines the accessibility of the charges to the cell surface.53 Our study confirms these observations, where APT showed more toxicity than PEG and CPP, due to the simple molecular structure formed by the short amino-alkyl chains of APT, and thus, more accessibility of amino groups to interact with the cells. As shown in Fig. 6, very small amounts of APT- and PEG-modified DNPs (indicated by white arrows) were adsorbed onto the RBCs’ surface, while a considerable amount was observed for CPP-DNPs, thus this biofunctionalization significantly improved the DNPs–cell membrane interactions.
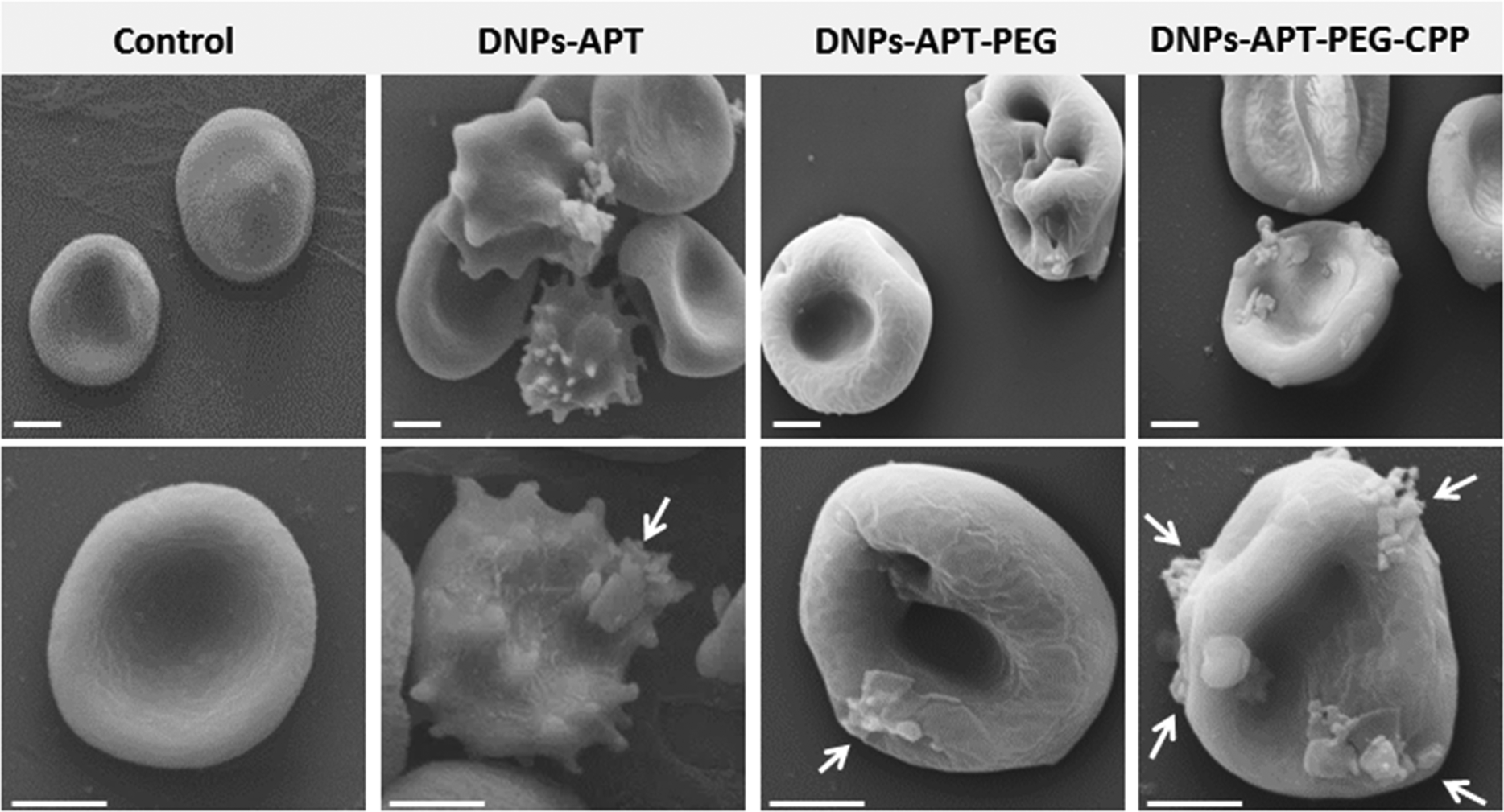 | ||
| Fig. 6 SEM pictures of the RBCs’ morphological modification after the exposure to the modified-DNPs. The modified DNPs (100 μg mL−1) were incubated with RBCs for 4 h at RT. The DNPs-APT showed the higher toxicity than the PEG and CPP-modified DNPs, resulting in severe morphological changes in the cell. The CPP-bioconjugation improved significantly the DNP–cell membrane interactions, as indicated by white arrows. Scale bars are 3 μm. | ||
In vitro cell viability assay
The ATP-content was used for the evaluation of the bare and modified-DNP cytotoxicity on MCF-7 and MDA-MB-231 breast cancer cells, after 6 and 24 h of incubation time at different DNP concentrations (25, 50, 100, and 200 μg mL−1). The exposure of MCF-7 and MDA-MB 231 cells to increasing concentrations of bare diatomite NPs within 24 h of incubation time induced very low toxicity, demonstrating their potential applicability as nanovectors in nanomedicine (Fig. S4, ESI†). The modified-NPs showed the same degree of cytotoxicity on both cell lines, as shown in Fig. 7. The DNPs-APT showed significant cytotoxic activity after 6 h of incubation and at lower concentrations (25 μg mL−1). Increasing the incubation time and the NP concentration resulted in an increased cytotoxicity of the NPs. After PEGylation and CPP-bioconjugation to the DNPs-APT, the cytotoxicity was significantly decreased after 24 h of incubation compared to the negative control (HBSS–HEPES, pH 7.4). In addition, no significant dependency on the exposure time and NP concentration was observed for the cytotoxicity of the PEGylated and CPP-biofunctionalized NPs. The toxicity of DNPs-APT can be attributed to the positive charge of free amino groups of APT, as discussed previously. These results are in agreement with the hemotoxicity data (Fig. 4), confirming an increase in the DNPs’ biocompatibility after the functionalization with PEG and biofunctionalization with the CPP-peptide. These results further suggest that DNPs-APT-PEG-CPP could be used as nanocarriers for long incubation times and high concentrations.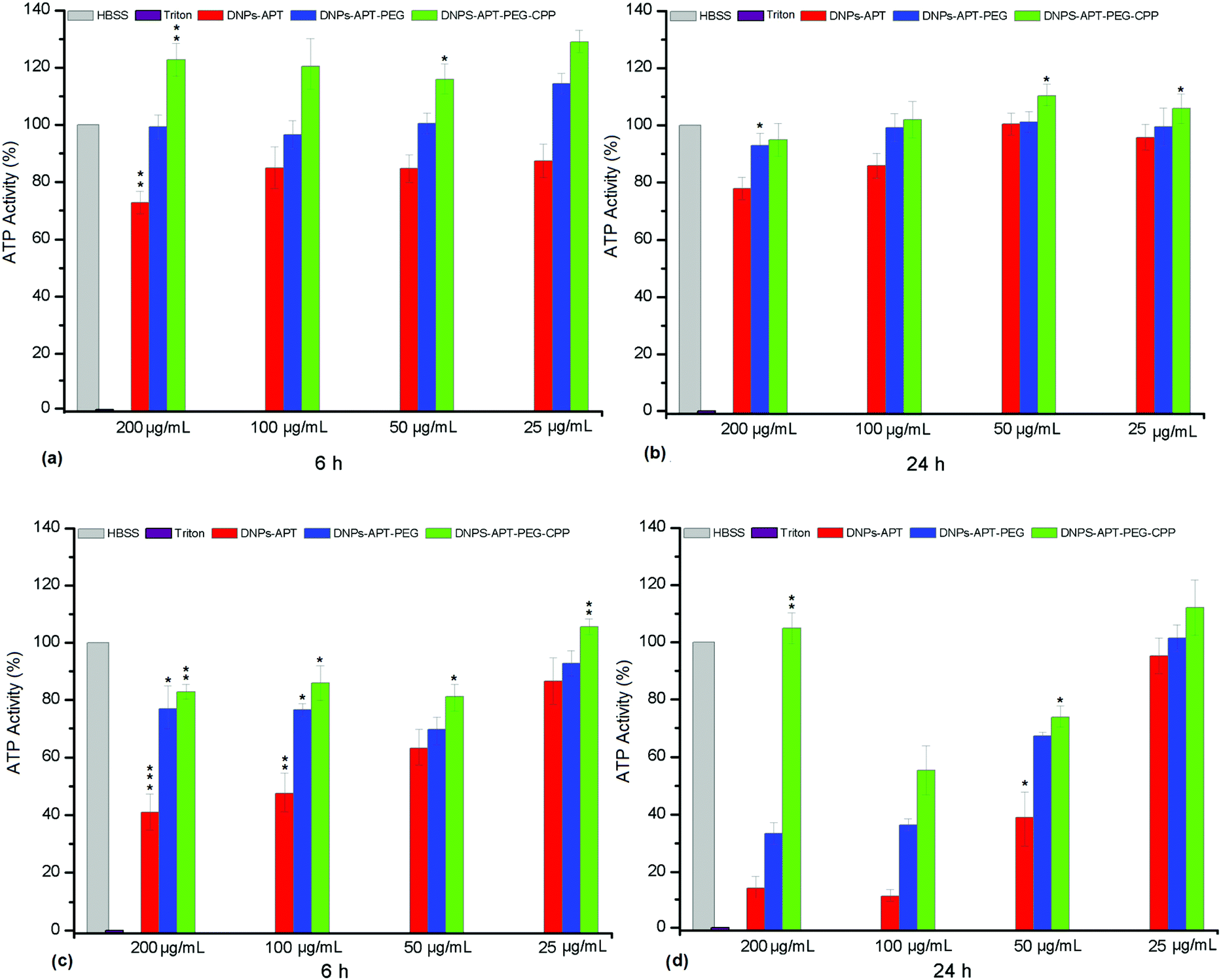 | ||
| Fig. 7 Cell viability of MCF-7 (a and b) and MDA-MB-231 (c and d) cells after exposure to the modified-DNPs at different concentrations. Statistical analysis was made by ANOVA comparing all data sets to the negative control HBSS (pH 7.4). The HBSS–HEPES (pH 7.4) and Triton X-100 were used as negative and positive controls, respectively. The level of significance was set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001. Error bars represent s.d. (n = 3). | ||
Cellular uptake of the modified-DNPs
Another key requirement of the nanocarriers for drug delivery is the cellular internalization into cancer cells without damaging the cellular integrity of healthy cells; therefore, the surface chemistry of the NPs has a significant impact on the NP performance in biomedical applications. Several peptides, capable of translocation in cellular membranes, have been used to improve the NPs’ cellular uptake.55,56 The CPP-peptide used in the present study consists of a short basic amino acid sequence with a net positive charge sequence and has the ability to facilitate the delivery into the cells of various molecular cargos, such as oligonucleotides, small molecules, siRNA, NPs, peptides and proteins.57The cellular uptake was evaluated by confocal fluorescence microscopy after DNP and cellular membrane labelling with Alexa Fluor-488® and CellMask™ Deep Red, respectively. Initially, the cellular internalization of modified-DNPs was evaluated on MCF-7 at two different incubation times (6 and 12 h) in order to determine the time required for a satisfactory uptake. The best incubation time, regardless of the modified-DNP type was 12 h, while the NPs were localized prevalently in the cellular membranes after 6 h. In both the cell lines, an increase in the NPs’ cellular uptake after CPP-bioconjugation was observed, as shown in Fig. 8. The merge images of the CPP modified-DNPs showed a yellow colour, resulting from the co-localization of the green labelled-DNPs and the red colour of cell membranes, which indicates the presence of the NPs inside the cells. In the case of the APT-modified NPs, more cellular uptake was observed compared to the NPs functionalized with PEG, due to the presence of the free amino groups of APT.
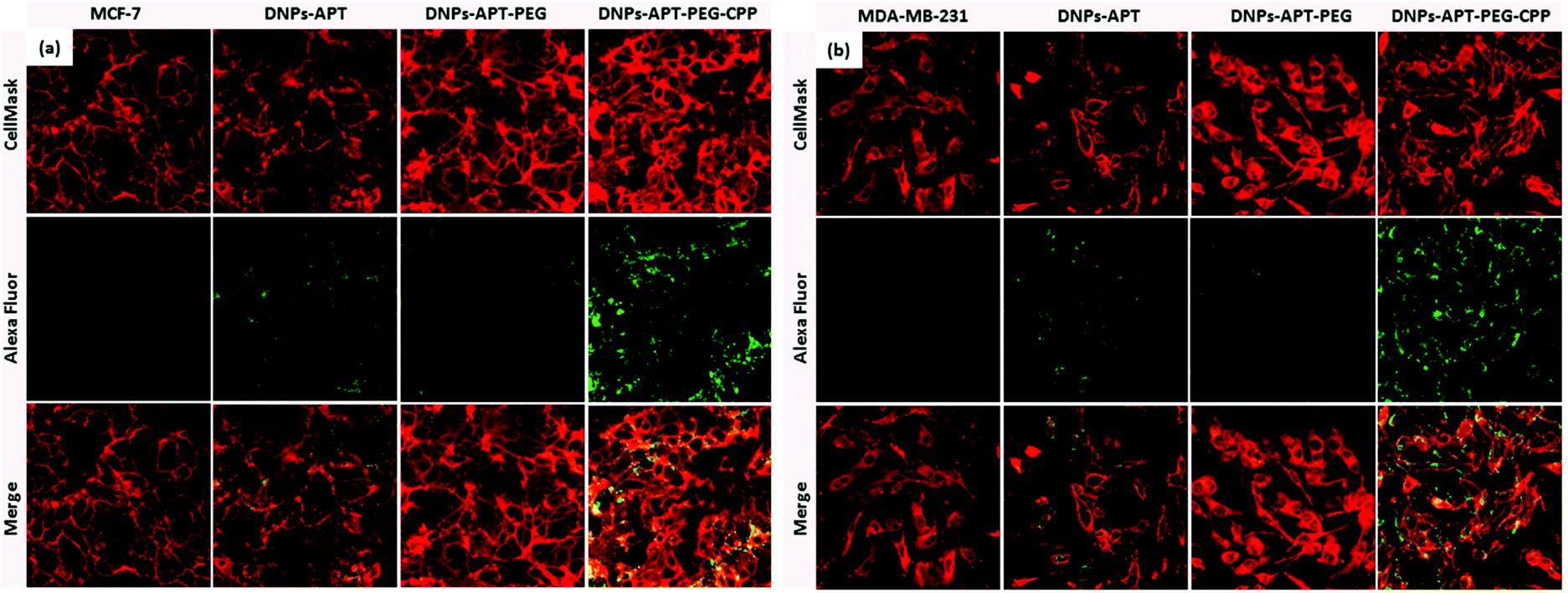 | ||
| Fig. 8 Confocal fluorescence microscopy of MCF-7 (a) and MDA-MB-231 (b) cells treated with APT, APT-PEG, APT-PEG-CPP modified-DNPs for 12 h at 37 °C. CellMask® (red) and Alexa Fluor-488® (green) were used to label the cell membrane and the DNPs, respectively. The merge figures are obtained by overlapping the DNP and the cell membrane images, allowing the determination of whether the NPs are located outside (green colour) or inside (yellow colour) the cells. | ||
The cellular uptake was also evaluated by TEM imaging of both cancer cells after 12 h of incubation with the modified-DNPs (50 μg mL−1), confirming the results obtained for the confocal fluorescence microscopy studies. In Fig. 9, the APT-modified DNPs were mainly localized in the proximity of the cell membrane, while in the case of the DNPs-APT-PEG no significant cellular uptake was observed. For the CPP-modified NPs, a considerable amount of DNPs was internalized into the cells with a homogeneous distribution in the cytoplasm and very close to the nucleus. All these results confirmed that CPP bioconjugation is a valid functionalization strategy to increase the cell penetration of diatomite NPs. The benefit of CPP on the surface of the NPs is the ability to translocate into the intracellular compartment without causing any cell membrane damage, resulting in low cytotoxicity and high uptake efficiency.58–60
 | ||
| Fig. 9 TEM images of MCF-7 (a) and MDA-MB-231 (b) cells treated with 50 μg mL−1 of DNPs-APT, DNPs-APT-PEG, and DNPs-APT-PEG-CPP for 12 h at 37 °C. A very small amount of APT- and PEG-modified DNPs was found inside the MCF-7 and MDA-MB-231 cells. In the case of DNPs-APT-PEG-CPP, a considerable amount of the NPs was observed inside the cells. Scale bars are 10 μm. | ||
Drug loading and release studies
The ultimate goal of targeted drug delivery is to deliver the administered drug to the target, while eliminating or minimizing the accumulation of the drug at any non-target site with minimal side effects.61 In this context, the poorly water-soluble anticancer drug, sorafenib (SFN), was used as a model drug to investigate the loading efficacy in the modified-DNPs and the release profiles in aqueous buffer solutions at pH 7.4 and 5.5, mimicking the physiological and intratumoral pH, respectively. The multikinase inhibitor, SFN, is considered a promising targeted protein kinase agent for the treatment of a broad range of cancers due to its significant in vitro and in vivo antitumor activity, resulting in the tumor growth inhibition and disruption of tumor microvasculature through antiproliferative, antiangiogenic, and/or proapoptotic effects.62,63 Firstly, the loading degree of SFN in APT-, PEG-, CPP-modified DNPs was evaluated in order to determine the influence of the surface biofunctionalization on the drug loading. The loading degree of SFN was 10.4 ± 1.1% in the DNPs-APT, 22 ± 2% in the DNPs-APT-PEG, and 17 ± 2% in the DNPs-APT-PEG-CPP. The surface biofunctionalization improved the drug loading, entrapping higher amounts of the drug into the modified DNPs than in APT alone. In the case of PEG-modified DNPs, an higher amount of the drug was loaded than in DNPs-APT, probably due to weak interactions (e.g., van der Waals forces) between PEG and the SFN.64 The CPP-conjugation step, even if it requires ON bioconjugation time and several washes after the reaction, causes only a very small decrease in the drug loading, possibly due to some removal of the adsorbed PEG molecules that are not covalently bound to the NP surface. The release profiles of SFN were assessed in HBSS–HEPES + 10% FBS solution at pH 7.4 and 5.5 (Fig. 10). The dissolution of pure SFN within 24 h (data not shown) in aqueous solution (pH 7.4 and 5.5) was negligible. In the case of SFN-loaded in APT- and CPP-modified DNPs, the drug release in both buffer solutions (pH 7.4 and 5.5) was gradual and constant within 4 h. SFN is characterized by its very poor water solubility, and the results show that the surface modification of DNPs improved the solubility of the drug in aqueous solution, but the functionalization of DNPs with CPP did not significantly influence the release profile of the loaded drug. However, in view of DNPs’ use for in vivo drug release and anticancer studies, it should be recommended that CPP-modified DNPs be used for the remarkable results of stability, biocompatibility and cellular uptake obtained here.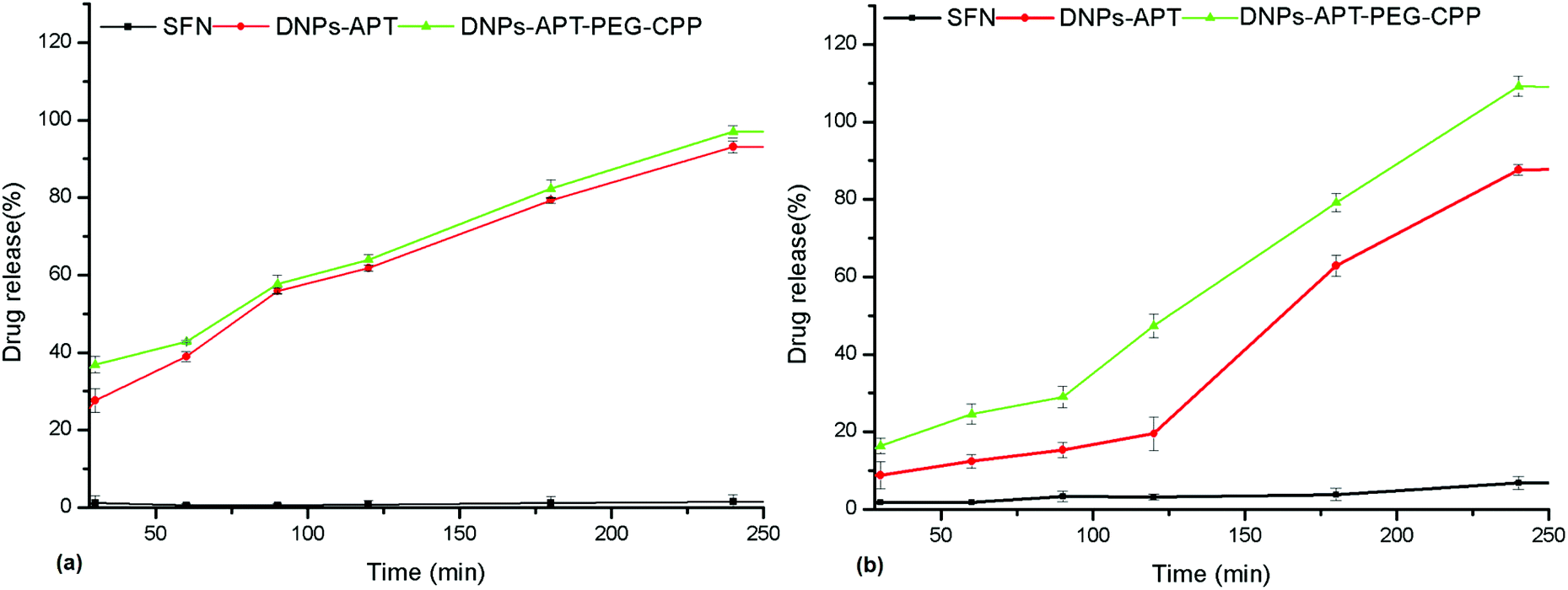 | ||
| Fig. 10 Drug release profiles of the SFN-loaded DNPs in HBSS–HEPES + 10% FBS at pH 7.4 (a) and pH 5.5 (b) at 37 °C. Values represent the mean ± s.d. (n = 3). | ||
Conclusions
In this work, we exploited the great potential of a natural porous silica NP derived from diatomite as a drug delivery system for cancer therapy applications. The preparation of NPs with suitable biocompatibility, stability in aqueous solution and cellular uptake by covalent conjugation of PEG and the CPP-peptide onto the NPs’ surface, was reported. Two step surface modifications of DNPs-APT by covalent attachment of PEG and bioconjugation with the CPP peptide allowed not only the reduction in the PDI and the enhancement of the NPs’ stability in aqueous solution, but also the improvement of the NPs’ cellular uptake and their enhanced biocompatibility, resulting in a decreased hemotoxicity and cytotoxicity of RBCs and breast cancer cells (MCF-7 and MDA-MB-231), respectively. The drug loading and drug release studies indicated that the NPs’ surface functionalization improved the loading capacity of DNPs (up to 22%) and provided the sustained release of the poorly water-soluble anticancer drug SFN. Overall, we demonstrated that the low toxicity, the optimal cellular uptake, and the drug loading and release properties make DNPs-APT-PEG-CPP a promising nanovector for cancer therapy. In conclusion, herein we demonstrated the potential of cheap, natural and biocompatible DNPs as a valid alternative to synthetic NPs and effective biofunctionalized nanovectors for superior intracellular localization and drug delivery in cancer cells, with future perspectives to develop bioengineered DNPs for localized drug delivery applications.Acknowledgements
The authors thank DEREF S.p.A. for kindly providing the diatomite earth sample and the Italian National Operative Program PON01_02782 which partially supported this work. Dr H. A. Santos acknowledges financial support from the Academy of Finland (grant no. 252215 and 281300), the University of Helsinki Research Funds, the Biocentrum Helsinki, and the European Research Council (FP/2007–2013, Grant No. 310892).Notes and references
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS PubMed.
- R. van der Meel, L. J. Vehmeijer, R. J. Kok, G. Storm and E. V. van Gaal, Adv. Drug Delivery Rev., 2013, 65, 1284–1298 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2015, 19, 311–330 CrossRef PubMed.
- M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu and Q. Gu, ACS Nano, 2013, 7, 1918–1928 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- H. A. Santos, Porous silicon for biomedical applications, Elsevier, 2014 Search PubMed.
- M. J. Sailor and J. H. Park, Adv. Mater., 2012, 24, 3779–3802 CrossRef CAS PubMed.
- B. Herranz-Blanco, D. Liu, E. Mäkilä, M. A. Shahbazi, E. Ginestar, H. Zhang, V. Aseyev, V. Balasubramanian, J. Salonen, J. Hirvonen and H. A. Santos, Adv. Funct. Mater., 2015, 25, 1488–1497 CrossRef CAS.
- L. M. Bimbo, L. Peltonen, J. Hirvonen and H. A. Santos, Curr. Drug Metab., 2012, 13, 1068–1086 CrossRef CAS PubMed.
- L. Russo, F. Colangelo, R. Cioffi, I. Rea and L. De Stefano, Materials, 2011, 4, 1023–1033 CrossRef CAS.
- F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo and G. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS PubMed.
- Y. Zhao, B. G. Trewyn, I. Slowing and V. S. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS PubMed.
- D. Losic, J. G. Mitchell and N. H. Voelcker, Adv. Mater., 2009, 21, 2947–2958 CrossRef CAS.
- M. S. Aw, S. Simovic, Y. Yu, J. Addai-Mensah and D. Losic, Powder Technol., 2012, 223, 52–58 CrossRef CAS.
- M. Sumper and E. Brunner, Adv. Funct. Mater., 2006, 16, 17–26 CrossRef CAS.
- H. Zhang, M. A. Shahbazi, E. Mäkilä, T. H. da Silva, R. L. Reis, J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 9210–9219 CrossRef CAS PubMed.
- M. A. Ferrara, P. Dardano, L. De Stefano, I. Rea, G. Coppola, I. Rendina, R. Congestri, A. Antonucci, M. De Stefano and E. De Tommasi, PLoS One, 2014, 9, 103750 Search PubMed.
- L. De Stefano, I. Rendina, M. De Stefano, A. Bismuto and P. Maddalena, Appl. Phys. Lett., 2005, 87, 233902 CrossRef.
- J. Parkinson and R. Gordon, Trends Biotechnol., 1999, 17, 230–232 CrossRef.
- T. Fuhrmann, S. Landwehr, M. El Rharbi-Kucki and M. Sumper, Appl. Phys. B, 2004, 78, 257–260 CrossRef CAS.
- P. J. Lopez, J. Descles and A. E. Allen, Curr. Opin. Biotechnol., 2006, 16, 180–186 CrossRef PubMed.
- S. Lettieri, A. Setaro, L. De Stefano, M. De Stefano and P. Maddalena, Adv. Funct. Mater., 2008, 18, 1257–1264 CrossRef CAS.
- F. Xu, Y. Wang, X. Wang, Y. Zhang, Y. Tang and P. Yang, Adv. Mater., 2003, 15, 1751–1753 CrossRef CAS.
- IARC Monographs, No. 68, 1997.
- I. Ruggiero, M. Terracciano, N. M. Martucci, L. De Stefano, N. Migliaccio, R. Taté, I. Rendina, P. Arcari, A. Lamberti and I. Rea, Nanoscale Res. Lett., 2014, 9, 1–7 CrossRef CAS PubMed.
- I. Rea, N. M. Martucci, L. De Stefano, I. Ruggiero, M. Terracciano, P. Dardano, N. Migliaccio, P. Arcari, R. Taté, I. Rendina and A. Lamberti, Biochim. Biophys. Acta, 2014, 1840, 3393–3403 CrossRef CAS PubMed.
- M. A. Shahbazi, P. V. Almeida, E. Mäkilä, M. Kaasalainen, J. Salonen, J. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 7488–7500 CrossRef CAS PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 246–255 CrossRef.
- Q. Gao, Y. Xu, D. Wu, Y. Sun and X. Li, J. Phys. Chem. C, 2009, 113, 12753–12758 CAS.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H. S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2013, 12, 588–595 CrossRef.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- M. C. Shin, J. Zhang, K. A. Min, K. Lee, Y. Byun, A. E. David, H. He and V. C. Yang, J. Biomed. Mater. Res., Part A, 2014, 102, 575–587 CrossRef PubMed.
- M. Terracciano, I. Rea, J. Politi and L. De Stefano, J. Eur. Opt. Soc.: RP, 2013, 8, 1375–1376 Search PubMed.
- L. De Stefano, O. Oliviero, J. Amato, N. Borbone, G. Piccialli, L. Mayol, I. Rendina, M. Terracciano and I. Rea, J. R. Soc., Interface, 2013, 10, 20130160 CrossRef PubMed.
- M. A. Shahbazi, M. Hamidi, E. Mäkilä, H. Zhang, P. V. Almeida, M. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 7776–7789 CrossRef CAS PubMed.
- H. A. Santos, J. Riikonen, J. Salonen, E. Mäkilä, T. Heikkilä, T. Laaksonen, L. Peltonen, V. P. Lehto and J. Hirvonen, Acta Biomater., 2010, 6, 2721–2727 CrossRef CAS PubMed.
- L. De Stefano, P. Maddalena, L. Moretti, I. Rea, I. Rendina, E. De Tommasi, V. Mocella and M. De Stefano, Superlattices Microstruct., 2009, 46, 84–89 CrossRef CAS.
- J. O. Martinez, B. S. Brown, N. Quattrocchi, M. Evangelopoulos, M. Ferrari and E. Tasciotti, Chin. Sci. Bull., 2012, 57, 3961–3971 CrossRef CAS.
- M. A. Shahbazi, P. V. Almeida, E. Mäkilä, A. Correia, M. Ferreira, M. Kaasalainen, J. Salonen, J. Hirvonen and H. A. Santos, Macromol. Rapid Commun., 2014, 35, 624–629 CrossRef CAS PubMed.
- J. Milton Harris, Poly(ethylene glycol) chemistry: biotechnical and biomedical applications, Springer, US, 1992 Search PubMed.
- N. A. Alcantar, E. S. Aydil and J. N. Israelachvili, J. Biomed. Mater. Res., 2000, 51, 343–351 CrossRef CAS PubMed.
- Y. Z. Huang, Y. F. Jiang, J. X. Wang, M. C. Shin, Y. Byun, H. He, Y. Liang and V. C. Yang, Adv. Drug Delivery Rev., 2013, 64, 1299–1315 CrossRef PubMed.
- E. T. Vandenberg, L. Bertilsson, B. Liedberg, K. Uvdal, R. Erlandsson, H. Elwing and I. Lundström, J. Colloid Interface Sci., 1991, 147, 103–118 CrossRef CAS.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, 2004 Search PubMed.
- D. Liu, H. Zhang, E. Mäkilä, J. Fan, B. Herranz-Blanco, C. F. Wang, R. Rosa, A. J. Ribeiro, J. Salonen, J. Hirvonen and H. A. Santos, Biomaterials, 2015, 39, 249–259 CrossRef CAS PubMed.
- D. Schaer, A. I. Alayash and P. W. Buehler, Antioxid. Redox Signalling, 2007, 9, 991–999 CrossRef CAS PubMed.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717–5728 CrossRef CAS PubMed.
- H. Wu, G. Liu, S. Zhang, J. Shi, L. Zhang, Y. Chen, F. Chena and H. Chen, J. Mater. Chem., 2011, 21, 3037–3045 RSC.
- Z. Zhang, M. Zhang, S. Chen, T. A. Horbett, B. D. Ratner and S. Jiang, Biomaterials, 2008, 29, 4285–4291 CrossRef CAS PubMed.
- D. Fischera, Y. Lib, B. Ahlemeyerc, J. Krieglsteinc and T. Kissela, Biomaterials, 2003, 24, 1121–1131 CrossRef.
- H. J. P. Ryser, Nature, 1967, 215, 934–936 CrossRef CAS PubMed.
- H. He, J. Ye, Y. Wang, Q. Liu, H. S. Chung, Y. M. Kwon, M. C. Shin, K. Lee and V. C. Yang, J. Controlled Release, 2014, 176, 123–132 CrossRef CAS PubMed.
- E. Jin, B. Zhang, X. Sun, Z. Zhou, X. Ma, Q. Sun, J. Tang, Y. Shen, E. Van Kirk, W. J. Murdoch and M. Radosz, J. Am. Chem. Soc., 2013, 135, 933–940 CrossRef CAS PubMed.
- J. Jo, S. Hong, W. Y. Choi and D. R. Lee, Sci. Rep., 2014, 4, 4378 Search PubMed.
- L. Vasconcelos, F. Madani, P. Arukuusk, L. Pärnaste, A. Gräslund and Ü. Langel, Biochim. Biophys. Acta, 2014, 1838, 3118–3129 CrossRef CAS PubMed.
- M. Zorko and Ü. Langel, Adv. Drug Delivery Rev., 2005, 57, 529–545 CrossRef CAS PubMed.
- S. W. Jones, R. Christison, K. Bundell, C. J. Voyce, S. M. V. Brockbank, P. Newham and M. A. Lindsay, Br. J. Pharmacol., 2005, 145, 1093–1102 CrossRef CAS PubMed.
- F. Madani, S. Lindberg, Ü. Langel, S. Futaki and A. Gräslund, Biophys. J., 2011, 2011, 1–10 CrossRef PubMed.
- Y. S. Kim, H. O. Jin, S. K. Seo, S. H. Woo, T. B. Choe, S. An, S. I. Hong, S. J. Lee, K. H. Lee and I. C. Park, Biochem. Pharmacol., 2011, 82, 216–226 CrossRef CAS PubMed.
- M. A. Shahbazi, N. Shrestha, E. Mäkilä, F. Araújo, A. Correia, T. Ramos, B. Sarmento, J. Salonen, J. Hirvonen and H. A. Santos, Nano Res., 2015, 8, 1505–1521 CrossRef CAS.
- Y. C. Li, S. Rissanen, M. Stepniewski, O. Cramariuc, T. Róg, S. Mirza and A. Bunker, J. Phys. Chem. B, 2012, 116, 7334–7341 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr05173h |
| This journal is © The Royal Society of Chemistry 2015 |