
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic interpretation of selective catalytic hydrogenation and isomerization of alkenes and dienes by ligand deactivated Pd nanoparticles†
Jie S.
Zhu
and
Young-Seok
Shon
*
Department of Chemistry and Biochemistry, California State University, Long Beach, Long Beach, CA, 90840 USA. E-mail: ys.shon@csulb.edu
First published on 5th October 2015
Abstract
Unsupported thiolate-capped palladium nanoparticle catalysts are found to be highly substrate-selective for alkene hydrogenation and isomerization. Steric and poisoning effects from thiolate ligands on the nanoparticle surface control reactivity and selectivity by influencing alkene adsorption and directing either di-σ or mono-σ bond formation. The presence of overlapping p orbitals and α protons in alkenes greatly influences the catalytic properties of deactivated palladium nanoparticles leading to easily predictable hydrogenation or isomerization products.
Introduction
Alkene transformation reactions significantly impact industrial processes, including those utilized for pharmaceutical, polymer, and petrochemical synthesis.1,2 Diene transformations represent an especially interesting problem due to the similar reactivity of both alkenes.3,4 To overcome this problem, synthetic methods utilizing alkene masking and modification of the other alkene followed by demasking have been demonstrated.5–8 Despite the value of these methods, protection–deprotection is generally atom inefficient and lengthens synthesis by at least two steps. In terms of catalytic hydrogenation, metal complexes such as Wilkinson's9,10 or Crabtree's11,12 catalysts and various Pd complexes13–15 are very efficient for regioselective or chemoselective hydrogenation. However, their ligand dissociation mechanism and the risk of residue metal toxicity limit their modern synthetic utility,16,17 especially in pharmaceutical applications. Palladium based catalytic materials circumvent some of these challenges and have been adopted by many industries.1,2 One of the most challenging problems associated with palladium, however, has been the poisoning and deactivation of these materials.18–21Recently, the use of nanomaterials as catalysts gained significant attention owing to their enhanced catalytic activity and recyclability. Many groups have taken advantage of functionalized supports and ligands or designer surfactants to tune the properties of various nanomaterials.22,23 Our laboratory demonstrated that the catalytic activity and recyclability of palladium nanoparticles (PdNP) can be controlled with thiolate-capping agents by selectively converting allyl alcohol to propanal via isomerization instead of 1-propanol via hydrogenation.24–26 Our previous work also demonstrated that the isomerization mechanism occurs through π coordination followed by the formation of a mono-σ Pd-alkyl intermediate.27,28 With this type of mechanism in mind, the application of these PdNP catalysts for alkene transformation reactions is further exploited. Herein, this paper reports a new discovery showing the influence thiolate ligands on PdNP surfaces have on substrate adsorption and surface hydrogen reactivity, leading to selective hydrogenation or isomerization of dienes and monoenes.
Results and discussion
Supported bare PdNPs and various Pd complexes are known to hydrogenate pentene isomers (Table 1).29–33 It is well known that higher alkenes such as pentene are known to only undergo complete hydrogenation via the di-σ-bonded species on palladium surfaces.34,35 However, when 1-pentene (1) was subjected to unsupported, octanethiolate-capped PdNP (2.6 ± 1.1 nm, 0.52 surface ligands/Pd surface atoms) in CDCl3 under H2 conditions for 24 h, the isomerization product (2-pentene, 2) with only minimal amounts of pentane (3) was observed (entry 1). When H2 was replaced with N2 as a control experiment, the catalytic con version of 1-pentene did not occur (entry 2). SilPdCl2 is the only catalyst with somewhat similar catalytic activity to PdNP (entry 5), however it completely loses its catalytic ability upon expo sure to air.31 The lack of reaction in the absence of H2 shows that isomerization cannot be due to the π allyl mechanism because that reaction mechanism does not require H2 to proceed. Instead, isomerization of 1-pentene (1) on these PdNPs must occur through the mono-σ bonded Pd-alkyl intermediate followed by β-hydride elimination as we previously proposed.27,28 This mechanism is consistent with trans-2-pentene hydrogenation on Pd/Al2O3 under ultrahigh-vacuum conditions.34 Since 2 reluctantly reacted with thiolate-capped PdNP, it is likely that the thiolate ligands are preventing the di-substituted alkene from forming the prerequisite di-σ-bonded species for hydrogenation. On large supported bare PdNP catalysts, catalytic activity is heavily reduced over time because the alkene substrate forms too many strong di-σ-bonded species on the catalytic surface.18,19 The thiolate capping agents on our PdNP catalyst help reduce substrate oversaturation, thereby limiting the poisonous effects.27,28 The presence of partial PdSx layer on the surface of PdNP after thiolate monolayer formation, which has been observed by others, might also be the reason for deactivation of PdNP catalyst.36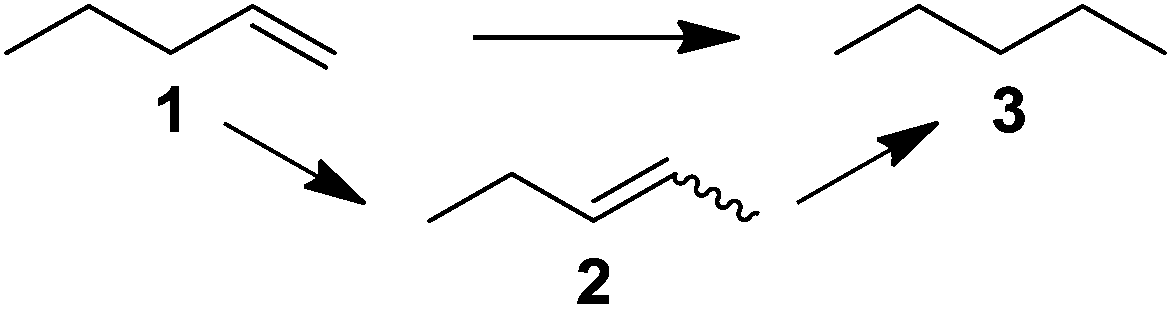
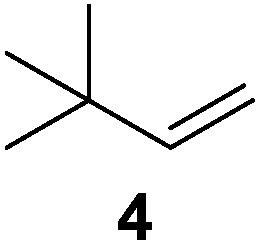
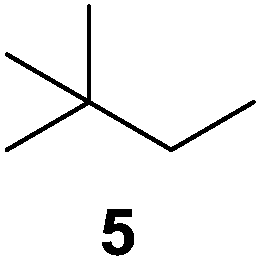
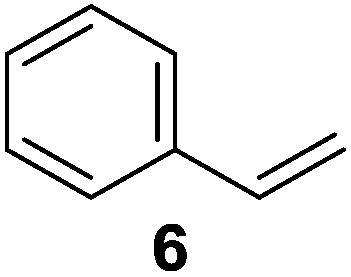
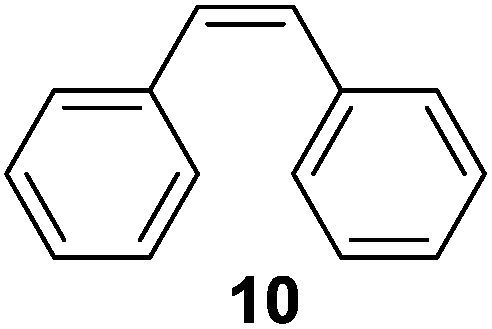
To understand whether hydrogenation could be forced to occur by blocking isomerization (Table 2), 3,3-dimethylbut-1-ene (4) and styrene (6) were subjected to PdNP under H2 conditions. No reaction between PdNP and 4 took place (entry 1) while 6 was completely converted to ethylbenzene (entry 2). Even though styrene's steric bulk is quite considerable, the p orbitals and planar geometry of the benzene ring must be aiding di-σ-bond formation, which allows hydrogenation to proceed. Comparatively, the t-butyl group in 4 does not contribute to, but hinders di-σ-bond formation, preventing hydrogenation to take place. The effect of substitution pattern around nonaromatic sp2 carbons was also studied using isomers of stilbene. Under the same reaction conditions, both trans-stilbene (8) and cis-stilbene (10) were hydrogenated to 1,2-diphenylethane (9) with 4% and 9% conversion, respectively. While no isomerization of 8 was observed (entry 3), partial isomerization (29%) of 10 to 8 (cis to trans) clearly took place (entry 4). Concerned that the isomerization of 10 was taking place due to ambient light,38–40 a control experiment without PdNP was performed and no isomerization resulted. By extending the reaction time to 48 h with additional H2 after 24 h, 10 was converted to a mixture of 54% isomerization product (8) and 21% hydrogenation product (9) (Fig. S3†). Because of stilbenes (8 and 10) being bulkier than styrene (6), the extended p orbital overlap from two benzene rings would not be enough to allow 8 and 10 to be hydrogenated with higher conversion. Furthermore, the preference of isomerization over hydrogenation for 10 is strongly indicative of the formation of mono-σ bonded Pd-alkyl species. As the steric bulk of these species increases, the addition of the second H atom must be slower than the isomerization of cis to trans on PdNP surface.





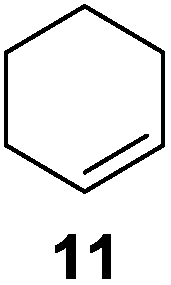
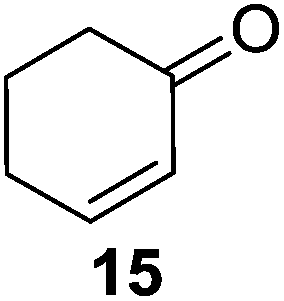
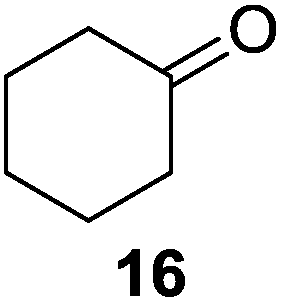
Further expansion of the substrate scope of the thiolate-capped PdNPs was attempted using cyclic alkenes (Table 3). First, the hydrogenation of cyclohexene (11) was examined and as expected, only a low 5% conversion to cyclohexane (12) was obtained (entry 1). In the case of 1,4-cyclohexadiene (13), 33% conversion to 11 resulted (entry 2). Its isomerization product, 1,3-cyclohexadiene, was not observed. This is likely due to the near planar geometry of 13 which allows all of its p-orbitals to contribute to di-σ bond formation. Instead of forming the mono-σ bonded species and undergoing isomerization, this geometry makes the direct addition of both H atoms possible. Conjugation with carbonyls was also considered. For 2-cyclohexenone (15), only the alkene was expected to be hydrogenated because thiolate-capped PdNPs are not active enough to reduce carbonyls.24–26 Indeed, 15 underwent 83% conversion to cyclohexanone (16) (entry 3). Similarly, maleic anhydride (17) was hydrogenated to succinic anhydride (18) with 64% conversion after 24 h. Extending the reaction time to 48 h, with hydrogen gas added after the first 24 h, increases the conversion to 87% (entry 4). Even though the conversions of these cyclic alkenes range from 33%–83% after 24 h, the selectivity remains excellent in all cases.




Next, the reactivity of isolated dienes with PdNP was investigated. In contrast to stilbene, a pentadiene system is not as sterically hindered while still having strong interaction with the PdNP surface. It was projected that symmetric 1,4-pentadiene (19) might first isomerize to conjugated 1,3-pentadiene (20), followed by hydrogenation of the terminal alkene, forming 2-pentene (2) (Scheme 1). Alternatively, it was also possible that 19 would be directly hydrogenated to 1-pentene without the intermediate 20, similar to 1,4-cyclohexadiene (13).
 | ||
| Scheme 1 Possible pathways to 2. | ||
The reaction progress was followed with 1H NMR spectroscopy (Fig. 1). The gradual decrease in the αCH2 signals at ∼2.8 ppm of 19 directly correlates to the consumption of 19. Since the characteristic vinyl proton signals at 6–7 ppm of 20 is not observed while the new characteristic αCH2 signals at ∼1.4 ppm of 1 is detected, the hydrogenation pathway must be the mechanism by which 2 forms. Therefore, we propose the mechanism (Fig. 2) of 1,4-pentadiene (19) isomerization and hydrogenation to 2-pentene (3) on thiolate-capped PdNP to be direct di-σ-bond formation after substrate adsorption via two Pd–π bond formation. This allows two hydrogen atom insertions to occur simultaneously, resulting in the formation of 1 from the di-σ bonded intermediate A. With only a single alkene moiety, 1 can either desorb or isomerize to 2via mono-σ bonded Pd-alkyl intermediate C. This is followed by β-hydride reductive elimination to form Pd–π bond intermediate D and 2 desorbs to create free space on the PdNP surface to restart the catalytic cycle.
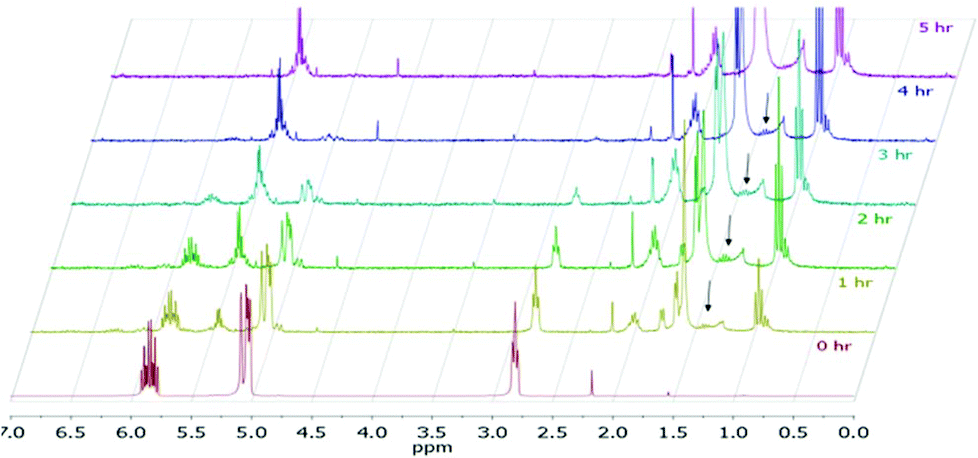 | ||
| Fig. 1 1H NMR of 19 conversion to 2 over time. | ||
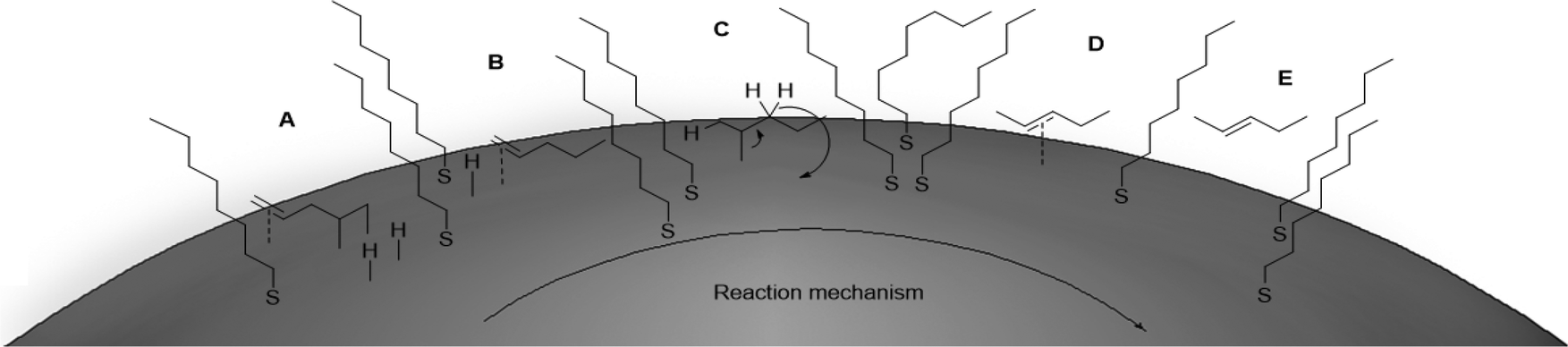 | ||
| Fig. 2 Proposed mechanism of 19 reacting with PdNP. | ||
Isolated dienes with αCH2 such as 1,4-cyclohexadiene (13) and 1,4-pentadiene (19) were directly hydrogenated without initial isomerization while larger activated alkenes such as stilbenes (8 and 10) were much less reactive, with 10 preferring cis-to-trans isomerization over hydrogenation. It would therefore be interesting to examine the reactivity of alkenes with both an activating phenyl ring and αCH2 (Table 4). trans-β-Methylstyrene (21) only underwent 10% conversion (entry 1) while allylbenzene (22) underwent 92% conversion (entry 2). This result confirms a low reactivity of di-substituted alkenes, especially trans-alkenes, for the catalytic reaction of deactivated PdNPs. The conversion of 1-phenyl-3-butene (23) was >99% with 96% selectivity for its isomerization products (entry 3), 1-phenyl-1-butene (25) and 1-phenyl-2-butene (26), with the selectivity of 13% and 87%, respectively.



The results showed that the proportion of isomers in the equilibrium mixture of alkenes depended largely on the structure of starting alkenes as indicated by the isomerization of 22 and 23 to the corresponding disubstituted alkenes. The ratio of trans/cis isomers for the reaction of 22 was 10.1, which was much greater than the trans/cis ratio for the reaction of 23 (the trans/cis ratio of 25 and 26 were 2.7 and 3.5, respectively). This indicated the location of phenyl group near β-H elimination site might decrease the activation energy for more thermodynamically stable trans products. The overall results prove that the deactivated PdNPs favor single isomerization of mono-substituted alkenes to trans-di-substituted alkenes. Despite the steric bulk of the benzene ring, the reactivity of these 1-phenyl, α-proton containing alkenes (22 and 23) on PdNP is comparable to the reactivity of terminal monoalkenes, such as 1-pentene (1) producing mostly isomerization products.
In general, acyclic alkenes hydrogenation or isomerization can be predicted (Scheme 2). Diene directly hydrogenates to monoene without isomerization; monoene's reactivity with PdNP depends on its structure. If it is mono-substituted with αCH2 (1, 22, 23) or cis di-substituted (10), isomerization is favored. If it is both unactivated and too bulky (4) or trans di-substituted (8, 21), low reactivity is predicted. If it is mono-substituted but activated and does not contain αCH2 (6), hydrogenation occurs.
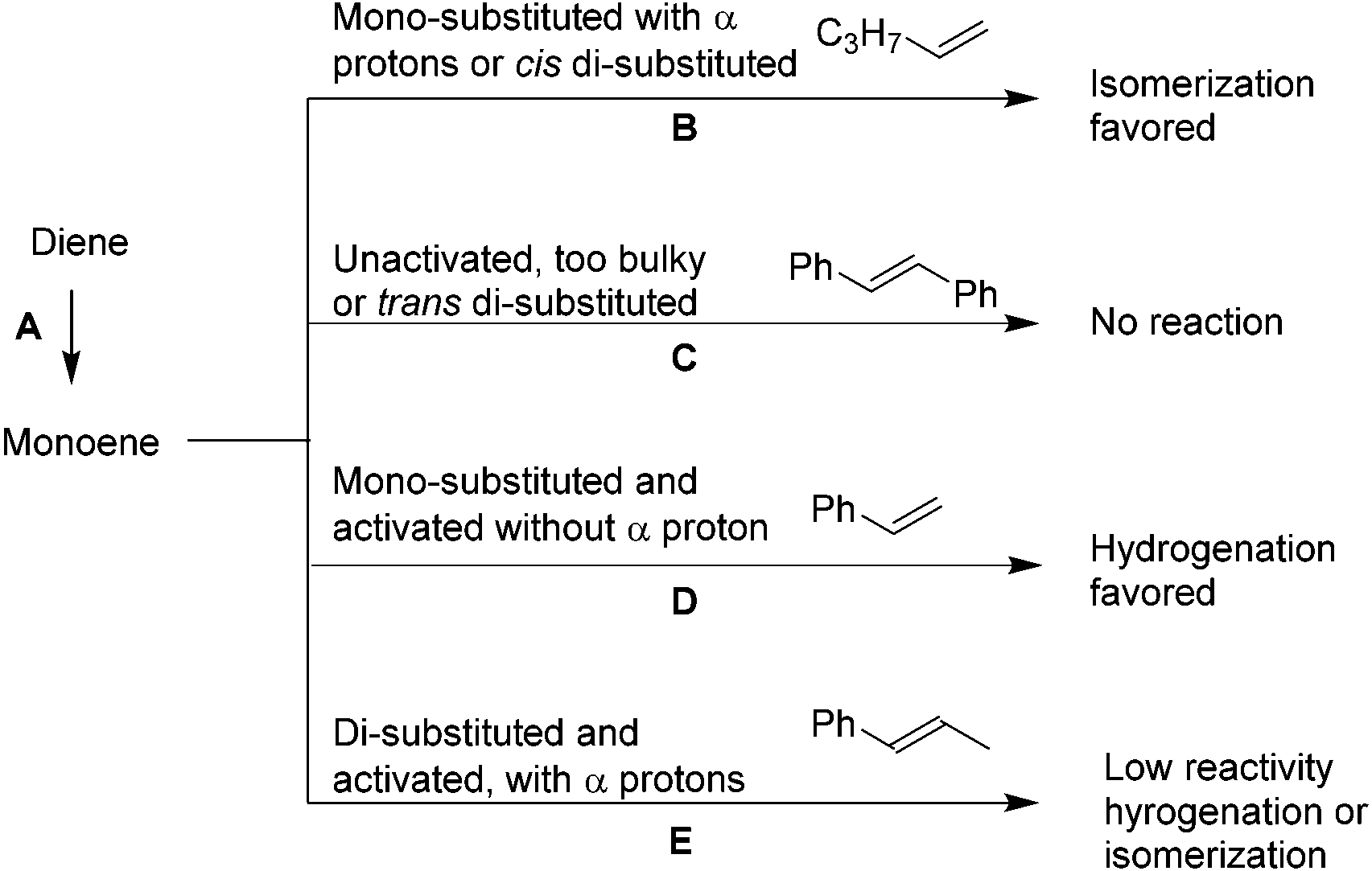 | ||
| Scheme 2 Expected reactivity of alkenes on PdNP. | ||
To test these predictions, the reactivity of 1-phenyl-trans-1,3-butadiene (24) was examined. This is an interesting diene due to the presence of two activated alkenes capable of being hydrogenated. Based on the generalizations, 24 will undergo one hydrogenation event, forming 25 with small amounts of 23. Mono-substituted 23 with αCH2 will then isomerize to 26, which can further isomerize to 25. As a result, the monoene major product expected is di-substituted monoene 25. Indeed, GC/MS analysis after 24 h reaction reveals that the reaction was 72% selective for 25, with a trans/cis ratio of 4.2. Its isomer 26 was produced in 12% with a trans/cis ratio of 4.7 (Scheme 3). 24 underwent 15% conversion to the complete hydrogenation product 27via both 25 (major) and 26 (minor) as intermediates. In comparison, 21 underwent only 8% conversion to its corresponding hydrogenation product. The increased hydrogenation activity for 24 compared to 21 can be accounted for by the better p orbital overlap of 24, which allows it to be adsorbed closer to the surface of PdNP initially.
 | ||
| Scheme 3 Isomerization and hydrogenation products of 24. | ||
Conclusions
We have taken advantage of the specific di-σ bond that must form with Pd surface before hydrogenation takes place in higher alkenes. Thiolate ligands prevent sterically hindered substrates from forming these di-σ bonds unless the substrate has good p orbital overlap capable of forming multiple Pd–π bond interactions. After a single hydrogenation in these substrates, only π interactions can be made with PdNP due to the loss of p orbital overlap, discouraging subsequent hydrogenation from occurring. Even though isomerization can still take place via mono-σ bonded intermediates, this can be exploited in order to selectively synthesize di-substituted internal alkenes.Acknowledgements
This study was supported in part by National Institute of General Medical Science (#SC3GM089562) and the Undergraduate Education Grant Program of the W. M. Keck Foundation. NMR instrumentation was provided for by the National Science Foundation (MRI CHE-1337559).Notes and references
- Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185 CrossRef.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687 CrossRef CAS PubMed.
- T. Shibata, Y. Tahara, K. Tamura and K. Endo, J. Am. Chem. Soc., 2008, 130, 3451 CrossRef CAS PubMed.
- B. M. Fraga, R. Guillermo, M. G. Hernandéz, M. C. Chamy and J. A. Garbarino, J. Nat. Prod., 2009, 72, 87 CrossRef CAS PubMed.
- T. J. A. Graham, T. H. Poole, C. N. Reese and B. C. Goess, J. Org. Chem., 2011, 76, 4132 CrossRef CAS PubMed.
- S. Mahapatra and R. G. Carter, J. Am. Chem. Soc., 2013, 135, 10792 CrossRef CAS PubMed.
- E. Osawa, Y. Tahara, A. Togashi, T. Iizuka, N. Tanaka, T. Kan, D. Farcasiu, G. J. Kent, E. M. Engler and P. v. R. Schleyer, J. Org. Chem., 1982, 47, 1923 CrossRef CAS.
- K. M. Nicholas, J. Am. Chem. Soc., 1975, 97, 3254 CrossRef CAS.
- J. Goodman, V. V. Grushin, R. B. Larichev, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2010, 132, 12013 CrossRef CAS PubMed.
- J. A. Osborn, F. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC.
- J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130 CrossRef CAS PubMed.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS.
- Q.-A. Chen, Z.-S. Ye, Y. Duan and Y.-G. Zhou, Chem. Soc. Rev., 2013, 42, 497 RSC.
- W. Long, H. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700 CrossRef CAS.
- M. P. Conley, C. Coperet and C. Thieuleux, ACS Catal., 2014, 4, 1458 CrossRef CAS.
- V. L. Budarin, P. S. Shuttleworth, J. H. Clark and R. Luque, Curr. Org. Synth., 2010, 7, 614 CrossRef CAS.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS PubMed.
- A. M. Buchbinder, N. A. Ray, J. Lu, R. P. Van Duyne, P. C. Stair, E. Weitz and F. M. Geiger, J. Am. Chem. Soc., 2011, 133, 17816 CrossRef CAS PubMed.
- L. Huang, T. P. Ang, Z. Wang, J. Tan, J. Chen and P. K. Wong, Inorg. Chem., 2011, 50, 2094 CrossRef CAS PubMed.
- A. Sarkany, Appl. Catal., A, 1997, 165, 87 CrossRef CAS.
- P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- D. J. Gavia and Y.-S. Shon, ChemCatChem, 2015, 7, 892 CrossRef CAS PubMed.
- D. J. Gavia and Y.-S. Shon, Langmuir, 2012, 28, 14502 CrossRef CAS PubMed.
- E. Sadeghmoghaddam, C. Lam, D. Choi and Y.-S. Shon, J. Mater. Chem., 2011, 21, 307 RSC.
- E. Sadeghmoghaddam, K. Gaïeb and Y.-S. Shon, App. Catal., A, 2011, 405, 137 CrossRef CAS PubMed.
- D. J. Gavia, M. S. Maung and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2013, 5, 12432 CAS.
- E. Sadeghmoghaddam, H. Gu and Y.-S. Shon, ACS Catal., 2012, 2, 1838 CrossRef CAS.
- A. S. Canning, S. D. Jackson, A. Monaghan and T. Wright, Catal. Today, 2006, 116, 22 CrossRef CAS PubMed.
- D. Teschner, Z. Révay, J. Borsodi, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Milroy, S. D. Jackson, D. Torres and P. Sautet, Angew. Chem., Int. Ed., 2008, 47, 9274 CrossRef CAS PubMed.
- J. Adeleke and B. J. Booth, Organomet. Chem., 1988, 339, 223 CrossRef CAS.
- Y. Wang, A. V. Biradar and T. Asefa, ChemSusChem, 2012, 5, 132 CrossRef CAS PubMed.
- P. Santra and P. Sagar, J. Mol. Catal. A: Chem., 2003, 197, 37 CrossRef CAS.
- A. M. Doyle, S. K. Shaikhutdinov and H.-J. Freund, Angew. Chem., Int. Ed., 2005, 44, 629 CrossRef CAS PubMed.
- A. M. Doyle, S. K. Shaikhutdinov, S. D. Jackson and H. J. Freund, Angew. Chem., Int. Ed., 2003, 42, 5240 CrossRef CAS PubMed.
- C. Vericat, M. E. Vela, G. Corthey, E. Pensa, E. Cortés, M. H. Fonticelli, F. Ibañez, G. E. Benitez, D. Carro and R. C. Salvarezza, RSC Adv., 2014, 4, 27730 RSC.
- The earlier reports from our group demonstrated the high recyclability of alkanethiolate-capped Pd nanoparticles in a similar catalytic reaction condition.26–28.
- B. Natarajan, S. Gupta, N. Jayaraj, V. Ramamurthy and N. Jayaraman, J. Org. Chem., 2012, 77, 2219 CrossRef CAS PubMed.
- D. Riedel, M. Cranney, M. Martin, R. Guillory, G. Dujardin, M. Dubois and P. Sonnet, J. Am. Chem. Soc., 2009, 131, 5414 CrossRef CAS PubMed.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, methods, materials, and characterization data. See DOI: 10.1039/c5nr05090a |
| This journal is © The Royal Society of Chemistry 2015 |