
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Core–shell polymer nanoparticles for prevention of GSH drug detoxification and cisplatin delivery to breast cancer cells†
Bapurao
Surnar
,
Kavita
Sharma
and
Manickam
Jayakannan
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER)-Pune, Dr. HomiBhabha Road, Pune 411008, Maharashtra, India. E-mail: jayakannan@iiserpune.ac.in
First published on 30th September 2015
Abstract
Platinum drug delivery against the detoxification of cytoplasmic thiols is urgently required for achieving efficacy in breast cancer treatment that is over expressed by glutathione (GSH, thiol-oligopeptide). GSH-resistant polymer–cisplatin core–shell nanoparticles were custom designed based on biodegradable carboxylic functional polycaprolactone (PCL)-block-poly(ethylene glycol) diblock copolymers. The core of the nanoparticle was fixed as 100 carboxylic units and the shell part was varied using various molecular weight poly(ethylene glycol) monomethyl ethers (MW of PEGs = 100–5000 g mol−1) as initiator in the ring-opening polymerization. The complexation of cisplatin aquo species with the diblocks produced core–shell nanoparticles of 75 nm core with precise size control the particles up to 190 nm. The core–shell nanoparticles were found to be stable in saline solution and PBS and they exhibited enhanced stability with increase in the PEG shell thickness at the periphery. The hydrophobic PCL layer on the periphery of the cisplatin core behaved as a protecting layer against the cytoplasmic thiol residues (GSH and cysteine) and exhibited <5% of drug detoxification. In vitro drug-release studies revealed that the core–shell nanoparticles were ruptured upon exposure to lysosomal enzymes like esterase at the intracellular compartments. Cytotoxicity studies were performed both in normal wild-type mouse embryonic fibroblast cells (Wt-MEFs), and breast cancer (MCF-7) and cervical cancer (HeLa) cell lines. Free cisplatin and polymer drug core–shell nanoparticles showed similar cytotoxicity effects in the HeLa cells. In MCF-7 cells, the free cisplatin drug exhibited 50% cell death whereas complete cell death (100%) was accomplished by the polymer–cisplatin core–shell nanoparticles. Confocal microscopic images confirmed that the core–shell nanoparticles were taken up by the MCF-7 and HeLa cells and they were accumulated both at the cytoplasm as well at peri-nuclear environments. The present investigation lays a new foundation for the polymer-based core–shell nanoparticles approach for overcoming detoxification in platinum drugs for the treatment of GSH over-expressed breast cancer cells.
1 Introduction
cis-Diamminedichloridoplatinum(II) (CDDP, cisplatin) is one of the widely employed anticancer drugs as a first-level chemotherapeutic agent for breast, testicular, ovarian, head and neck, and lung cancers.1,2 To overcome the complaints from patients such as nausea-vomiting, fatigue and nephrotoxicity, several other platinum derivatives such as oxaliplatin, picoplatin, carboplatin and satraplatin have also been developed and approved by the FDA for clinical trials.3,4 One of the major obstacles in delivering platinum drugs to cancer tissues has been identified as detoxification by cytoplasmic thiol species such glutathione (GSH, an oligopeptide) at the intracellular compartments.5–10 Recent in vitro cell line studies have confirmed the over-expression of GSH in breast cancer cells and its influence on cisplatin detoxification during drug administration in cancers.8–10 Further, platinum drugs are also sensitive to ions (cations/anions) and proteins in blood plasma and they require additional stability for intravenous administration.11 Polymer-based drug delivery approaches have emerged as new trend to overcome the above limitations in cisplatin drug administration. Polymer–cisplatin drug conjugates also have additional advantages of their passive selective accumulation at cancer tissues through an enhanced permeability and retention (EPR) effect.12 PEGylated cisplatin (or oxaliplatin) liposomal formulations are currently being tested for phase II level trials.13 PEGylated block copolymers of poly(isobutylene-maleic acid)14 and polyacrylates15–20 are some of the important polymer systems that have been explored for cisplatin conjugation. In these cases, the aquo cisplatin complex was conjugated with a carboxylic acid functionality in the polymer backbone to preserve the drug in its active form prior to delivery. Kataoka and co-workers used PEGylated-poly(L-glutamic acid) copolymers for cisplatin conjugation and the resultant micellar drug conjugates have been found to be effective in suppressing the growth of solid tumours.21–23 Conjugated polyelectrolyte–cisplatin nanoparticles have been developed for simultaneous imaging and drug tracking.24 Shell cross-linked Knedel-like nanoparticles have been employed for cisplatin delivery and these nanoparticles were found to exhibit significant antitumor activity.25 Folic acid-conjugated platinum nanoparticles,26 PEGylated mesoporous silica nanoparticles,27 functionalized carbon nanotubes28 and graphenes,29,30 lipids31,32 and amphiphilic oligomer-based micelles,33 cyclic tripeptides,34 and gold nanoparticles35,36 are some of the other approaches developed for cisplatin delivery. Despite the above examples observed the importance of platinum drug delivery, the stability of cisplatin drugs in the polymer scaffolds against detoxification by cytoplasmic thiol species such as GHS has not been addressed. Further, most of the above polymer scaffolds are non-biodegradable; thus, the polymer–drug cleavage mechanism at the intracellular compartments is not clearly understood. Hence, new efforts are required to develop polymer–cisplatin conjugates that are resistant to detoxification against cytoplasmic thiols (like GSH) for accomplishing efficient treatment against breast cancer that are found to be over-expressed by GSH.Enzyme-cleavable polymer scaffolds are emerging as an important approach for drug administration exclusively at the intracellular compartments.37 We and other research groups have earlier reported polysaccharide vesicles,38–40 amphiphilic dendrons41,42 and block copolymer assemblies43 having enzyme-responsiveness for delivering anticancer drug molecules such as doxorubicin and camptothecin. Polycaprolactone (PCL) is an important biodegradable aliphatic polyester that could be ruptured by lysosomal enzymes like esterase at the intracellular compartments for drug delivery.44 Recently, we have reported new PEG-b-carboxylic PCL as a pH-responsive vesicular scaffold for oral delivery of anticancer drugs under the gastrointestinal tract.45 Further, the preliminary studies on this system have revealed that these copolymers are capable of cisplatin drug chemical conjugation.46 This has provided a new opportunity for us to develop biodegradable diblock copolymer assemblies for cisplatin delivery to accomplish 100% cell killing in breast cancer cells. The current design has three important components: (i) PEG chains for hydrophilic shell size-control and also for enhancing the solubility of the nanoparticles in aqueous medium for drug administration; (ii) carboxylic acid units anchored in the PCL backbone for conjugating the cisplatin aquo complex which constitutes the drug core; and (iii) the hydrophobic PCL backbone acting as a protecting layer between the shell and core against platinum drug detoxification by cytoplasmic thiols such as GSH. Further, this design also has an in-built enzyme-responsive PCL layer; thus, the drug–polymer conjugate can be cleaved by lysosomal enzymes at the intracellular compartments. This scaffold design for cisplatin delivery is shown in Fig. 1.
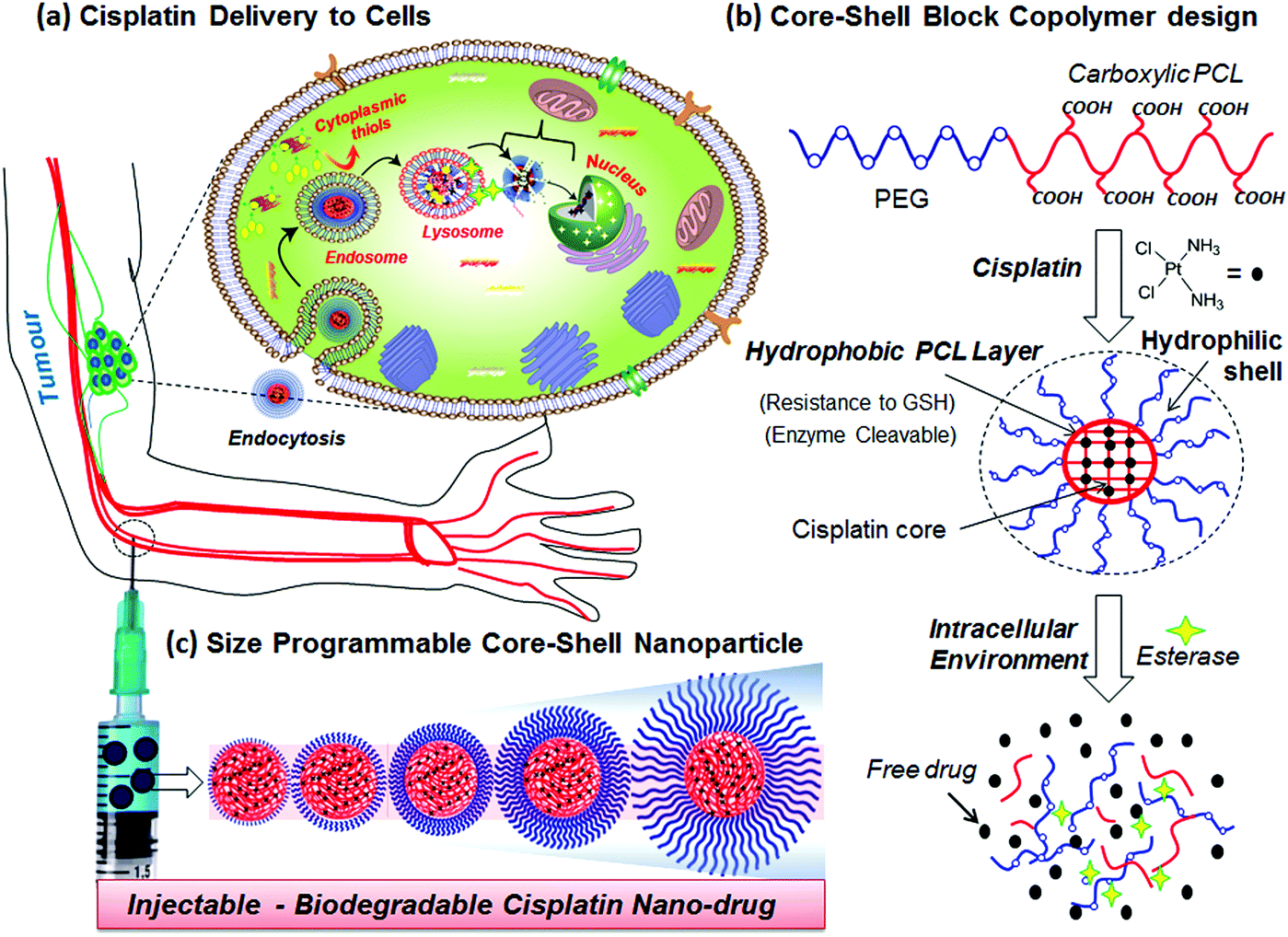 | ||
| Fig. 1 Core–shell polymer–cisplatin nanoparticle delivery to cancer cells (a). The delivery of the drugs at the intracellular compartments and their resistance to detoxification by cytoplasmic thiol species GSH (b). Diblock copolymer nano-drug design with variable shell size and fixed drug core (c). | ||
The present investigation aims to develop new, size-controllable and biodegradable diblock copolymer nanoparticle assemblies that are capable of producing cisplatin complexation that also exhibit shielding against the drug core from the detoxification of cytoplasmic thiols (GSH) in breast cancer cells (see Fig. 1). The PEGx-b-CPCL diblocks were designed in such a way so that the numbers of carboxylic functional units are maintained the same, as ∼100 units in all the blocks, and the PEG chains –(CH2CH2O)x– are systematically varied, from x = 3, 7, 17, 45 and 113, to achieve size-controllable core–shell prodrug nanoparticles (see Fig. 1c). The cisplatin drug was chemically conjugated at the central core through Pt-OOR-PCL linkages and is also protected at the periphery by the PEG-shells. This design enabled us to retain the central core as 75.0 ± 5 nm and vary the size of the shell precisely up to 190 nm. The role of the PEG-shell on the drug stability [in saline, PBS and fetal bovine serum (FBS)] and action against cytoplasmic thiol species like cysteine (amino acid residue) and glutathione (GSH) are investigated in detail. Within the intracellular environment, the core–shell particle is cleaved by the esterase enzyme (present in the lysosome) to release the active Pt-drug for cell death. Thus, the new core–shell polymer–cisplatin nanoparticle design is both stable against detoxification thiol-residues as well as cleavable at the intracellular compartments by esterase to deliver cisplatin drugs. Cellular uptake and cytotoxicity of the core–shell nanoparticles were studied in normal (Wt-MEFs) and cancer (HeLa and MCF-7) cell lines. It was found that prodrugs showed selective and enhanced cytotoxicity exclusively in the breast cancer cells compared to other cell lines.
2 Experimental methods
2.1 Materials
1,4-Cyclohexanediol, t-butyl acrylate, potassium t-butoxide, pyridinium chlorochromate (PCC), meta-chloroperbenzoic acid (mCPBA), tin(II) 2-ethylhexanoate (Sn(Oct)2), triethylene glycol monomethyl ether (TEG), poly(ethylene glycol) monomethyl ether (here after referred to as PEG) with molecular weights of 350, 750, 2000 and 5000, cisplatin, silver nitrate, ortho-phenylenediammine (OPD), glutathione (GSH) and esterase were purchased from Aldrich chemicals. TEG and PEGs were dried under vacuum prior to use. Wild-type mouse embryonic fibroblasts (Wt-MEFs), cervical cancer (HeLa cells) and human breast cancer cells (MCF-7) were maintained in DMEM (phenol red-free medium: Gibco) containing 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin–streptomycin at 37 °C under a 5% CO2 humidified atmosphere. Cells were washed with 40% DPBS (Gibco), trypsinised using 0.05% trypsin (Gibco) and seeded in 96- or 6-well (as per experiment) flat-bottomed plastic plates (Costar) for all assays. Tetrazolium salt, 3,4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide (MTT), DMSO, Hoechst and 4% paraformaldehyde were obtained from Sigma. Fluoromount was obtained from Southern Biotech. All solvents such as tetrahydrofuran (THF), dichloromethane (DCM) and trifluoroacetic acid (TFA) were purified and distilled prior to use.2.2 Methods
NMR spectra were recorded using a 400 MHz JEOL NMR spectrophotometer. All spectra were recorded in CDCl3 containing TMS as an internal standard. Analysis by gel permeation chromatography (GPC) was performed using a Viscotek VE 1122 pump, Viscotek VE 3580 RI detector, and Viscotek VE 3210 UV–Vis detector in tetrahydrofuran (THF) using polystyrene as the standard. The thermal stability of the polymers was determined using a Perkin-Elmer thermal analyser STA 6000 model at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Absorption spectra were recorded using a Perkin-Elmer Lambda 45 UV–Vis spectrophotometer. Dynamic light scattering (DLS) was carried out using a Nano ZS-90 apparatus utilizing a 633 nm red laser (at a 90° angle) from Malvern Instruments. At 90° scattering, fluctuations were detected to generate the correlation function [g2(t)]; from this function the diffusion coefficient (D) was calculated using the cumulant method. By applying the Stokes–Einstein equation, the particle diameter was calculated. The reproducibility of the data was checked at least three times using independent polymer solutions. FE-SEM images were recorded using a Zeiss Ultra Plus scanning electron microscope. For FE-SEM analysis, the samples were prepared by drop-casting on to silicon wafers. Atomic force microscope images were recorded for drop-cast samples using Agilent instruments. The reproducibility of the data was checked for at least three independent amphiphilic solutions. TEM images were recorded using a Technai-300 instrument by drop-casting the sample on to a Formvar®-coated copper grid. Fluorescent micrographs were collected using a Carl Zeiss Axiovert 200 microscope. Confocal micrographs were collected using an LSM710 microscope.2.3 Synthetic procedure for monomer
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v) as eluent. Yield: 39 g (74.1%). 1H-NMR (400 MHz, CDCl3) δ ppm: 3.63 (m, 3H, O–CH2– and O–CH), 3.27–3.39 (m, 1H, CH–OH), 2.38 (t, 2H, –CH2CO–), 1.96–1.81 (m, 4H, OCH(CH2)2), 1.64–1.32 (m, 4H, CO(CH2)2), 1.44 (s, 9H, –C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ ppm: 171.01, 80.42, 69.49, 63.84, 63.60, 32.54, 30.32, 29.20 and 27.48. FT-IR (cm−1): 3422, 2979, 2937, 2863, 1731, 1462, 1393, 1366, 1215, 1158, 1116 and 1134. HR-MS (ESI+): m/z [M + Na+] calcd for C13H24O4 [M+]: 267.1572; found: 267.1588.
:10 v/v) as eluent. Yield: 39 g (74.1%). 1H-NMR (400 MHz, CDCl3) δ ppm: 3.63 (m, 3H, O–CH2– and O–CH), 3.27–3.39 (m, 1H, CH–OH), 2.38 (t, 2H, –CH2CO–), 1.96–1.81 (m, 4H, OCH(CH2)2), 1.64–1.32 (m, 4H, CO(CH2)2), 1.44 (s, 9H, –C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ ppm: 171.01, 80.42, 69.49, 63.84, 63.60, 32.54, 30.32, 29.20 and 27.48. FT-IR (cm−1): 3422, 2979, 2937, 2863, 1731, 1462, 1393, 1366, 1215, 1158, 1116 and 1134. HR-MS (ESI+): m/z [M + Na+] calcd for C13H24O4 [M+]: 267.1572; found: 267.1588.
2.4 Synthesis of tert-butyl 3-((4-oxocyclohexyl)oxy)propanoate (B)
PCC (46.3 g, 214.8 mmol) was added to a solution of compound A (35.0 g, 143.2 mmol) in dry DCM (400 mL) under inert conditions and the reaction mixture was stirred at 25 °C for 4 h. A catalytic amount of molecular sieves was added to absorb moisture. The filtrate was condensed and the resultant liquid was purified by passing through a silica gel column by eluting with petroleum ether/EtOAc (1:4 v/v). The product was obtained as a colourless liquid. Yield: 33.0 g (95%). 1H-NMR (400 MHz, CDCl3) δ ppm: 3.55 (m, 3H, O–CH2 and O–CH), 2.57 (t, 2H, –CH2–CO), 2.63 (m, 2H, –(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH2–), 2.25 (m, 2H, –(CO)CH2–) 2.10 (m, 2H, –(CO)CH2–) 1.94 (m, 2H, –(CO)CH2–), 1.45 (s, 9H, –C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ ppm: 211.40, 170.99, 80.56, 72.74, 64.02, 37.02, 36.56, 30.40 and 20.04. FT-IR (cm−1): 2974, 2874, 2360, 1716, 1456, 1419, 1393, 1368, 1316, 1239, 1212 and 1109. HRMS (ESI+): m/z [M + Na+] calcd for C13H22O4 [M+]: 265.1415; found: 265.1401.
O)CH2–), 2.25 (m, 2H, –(CO)CH2–) 2.10 (m, 2H, –(CO)CH2–) 1.94 (m, 2H, –(CO)CH2–), 1.45 (s, 9H, –C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ ppm: 211.40, 170.99, 80.56, 72.74, 64.02, 37.02, 36.56, 30.40 and 20.04. FT-IR (cm−1): 2974, 2874, 2360, 1716, 1456, 1419, 1393, 1368, 1316, 1239, 1212 and 1109. HRMS (ESI+): m/z [M + Na+] calcd for C13H22O4 [M+]: 265.1415; found: 265.1401.
2.5 Synthesis of tert-butyl 3-((7-oxooxepan-4-yl)oxy)propanoate (monomer-CCL)
m-Chloroperbenzoic acid (42.7 g, 247.6 mmol) was added slowly to a stirred solution of B (30.0 g, 123.8 mmol) in dry DCM (600 mL) under nitrogen atmosphere. To the above reaction mixture, anhydrous NaHCO3 (31.2 g, 371.4 mmol) was added and the reaction was continued at 25 °C for 12 h. The solvent was removed and the residue was neutralized with saturated aqueous NaHCO3 solution (20 mL) and saturated aqueous Na2S2O3 solution (20 mL). It was extracted with ethyl acetate and the organic layer was dried over anhydrous Na2SO4. After solvent evaporation, the crude product was purified by passing through a silica gel column using ethyl acetate and petroleum ether (4:6 v/v). Yield = 28 g (87.5%). 1H-NMR (400 MHz, CDCl3) δ ppm: 4.41 (dd, 1H, COOCH), 4.04 (dd, 1H, COOCH) 3.60 (m, 4H, OCH2, OCH and COCH), 2.99 (dd, 1H, COCH), 2.48 (t, 2H, COCH2), 2.45–1.85 (m, 4H, OCH–(CH2)2), 1.44 (s, 9H, C(CH3)3). 13C-NMR (100 MHz, CDCl3) δ ppm: 176.35, 171.19, 81.01, 75.24, 64.24, 63.66, 36.80, 34.15, 28.40, 27.84 and 27.61. FT-IR (cm−1): 2928, 1727, 1446, 1393, 1367, 1253, 1155, 1110 and 1058. HRMS (ESI+): m/z [M + K+] calcd for C13H32O5 [M+]: 299.3187; found: 299.3163.
2.6 Synthesis of PEGx-b-BPCL diblock polymers
The typical synthetic procedure was elucidated for PEG5000-b-BPCL, where initial monomer-to-initiator ratio ([M0]/[I0]) is kept as 100. The initiator PEG5000 (193.8 mg, 0.0387 mmol), catalyst Sn(Oct)2 (7.8 mg, 0.0193 mmol) and the substituted caprolactone monomer (1.0 g, 3.87 mmol) were taken in a flame-dried Schlenk tube under nitrogen atmosphere. A high vacuum was applied to this reaction mixture for 45 min with stirring at room temperature. After achieving inert conditions inside the tube, it was immersed in a preheated oil bath at 130 °C. The polymerization was continued for 6 h with constant stirring. The polymer mixture was cooled to room temperature and precipitated in cold MeOH. The polymer was redissolved in THF and precipitated again in cold-methanol, the process being repeated at least twice to obtain pure polymer. Yield: 700 mg (70%). 1H-NMR (400 MHz, CDCl3) δ ppm: 4.13 (s, 2H), 3.63 (s, 6.51 H), 3.43 (s, 1H), 3.38 (s, 1H), 2.44 (t, 2H), 2.35 (t, 2H), 1.93–1.81 (m, 2H), 1.81–1.67 (m, 4H), 1.44 (s, 9H, t-butyl). 13C-NMR (100 MHz, CDCl3): 173.69, 170.81, 80.77, 75.62, 70.66, 65.13, 61.47, 36.60, 33.04, 29.81, 28.86, and 28.22. FT-IR (cm−1): 2973, 2931, 1727 (CO ester), 1457, 1364, 1251, 1156, 1156, 1100, 1062, 959, 900, 845, and 757. GPC molecular weights: Mn = 18400, Mw = 24900, and Mw/Mn = 1.35.
A similar procedure was followed for the synthesis of other block copolymers PEGx-b-BPCL where x = 3, 7, 17 and 45 (details are given in the ESI†).
2.7 Synthesis of PEGx-b-CPCL diblock polymers
Trifluoroacetic acid (0.2 mL) was added slowly into PEG5000-b-BPCL (200 mg) in dry DCM (5.0 mL), and the polymer solution was stirred at 25 °C for 30 min. The solvent was evaporated and the polymer was redissolved in THF and precipitated in cold methanol. The purification was repeated at least twice to obtain pure polymer. 1H-NMR (400 MHz, CDCl3) δ: 4.14 (t, 2H, CH2OH), 3.84–3.64 (m, 3.7 H, PEG and OCH2), 3.57 (m, 1H, OCH), 2.56 (t, 2H, CH2COOH), 2.38 (t, 2H, COCH2), 1.99–1.67 (m, 4H, –OCH(CH2)2). 13C-NMR (100 MHz, CDCl3): 173.68, 170.86, 80.72, 75.58, 70.72, 65.13, 61.50, 36.60, 33.14, 29.81, and 28.22. FT-IR (cm−1): 3447, 2932, 2450, 1711 (CO), 1365, 1257, 1175, 1099, 1063, and 960.
2.8 Preparation of aquated cisplatin [Pt(NH3)2(OH2)2]2+
Cisplatin (CP; 50 mg, 166 mmol, 1 equiv.) was dispersed in H2O (50.0 mL) with constant stirring at 37 °C. To this mixture, silver nitrate (56.4 mg, 332 mmol, 2 equiv.) was added and the resulting reaction mixture was stirred for 24 h in dark conditions. Formation of the aquated cisplatin complex was confirmed by milky-white silver chloride precipitation. Silver chloride was removed by centrifuging at 10000 rpm for 1 h. Finally, the aquated cisplatin was obtained by filtration through a 0.2 μm filter. The sample was lyophilized and stored at 4 °C.
2.9 Synthesis of the polymer–cisplatin conjugate
A typical procedure for preparation of PEG5000-b-CPCL-CP is explained in detail. PEG5000-b-CPCL diblock polymer (20.0 mg) was dissolved in NaOH (2 mL, 1 mg mL−1) and the solution was stirred at 37 °C for 30 min. Aquated cisplatin (16.4 mg, 55 mmol, the lyophilized sample) was added to the above polymer solution and the complex was stirred for 24 h at 37 °C. The solution was transferred to a dialysis bag of molecular weight cut-off (MWCO = 1000) and dialyzed against a large amount of distilled water for 2 days. The distilled water was replaced periodically to ensure the removal of un-encapsulated molecules from the dialysis tube. The solution recovered from the dialysis tube was filtered through a 0.45 μm filter, lyophilized and stored at 4 °C. FT-IR (cm−1): 3300, 2920, 2880, 1660, 1560, 1395, 1360, 1090, 1050, 930, 830 and 548.A similar procedure was used for making cisplatin complexes of copolymers and the details are given in the ESI.†
2.10 In vitro drug-release studies
Cisplatin-loaded nanoparticles were taken in a dialysis bag (in 3 mL), and they were immersed in a 100 mL beaker and dialyzed at 37 °C with constant stirring. Various dialysis conditions like saline, PBS and FBS were employed for these studies. At specific time intervals, 3.0 mL of the dialysate was withdrawn and replaced with an equal volume of fresh buffer (or FBS). The amount of molecule (or drug) released in each aliquot was measured using absorption spectroscopy using ortho-phenylenediammine (OPD) assay to quantify the percentage of cumulative release. For esterase-aided release studies, 10 units of enzyme were used following the above-mentioned procedure.Cumulative release (%) = Cn × V0/m × 100, where Cn is the amount of loaded cargo in the nth sample, V0 is the total volume, and m is the total amount loaded on the nanoparticles.
2.11 ortho-Phenylenediamine (OPD) colorimetric assay
Samples with unknown cisplatin (Pt) content were added to 0.5 mL of OPD solution in N,N-dimethylformamide (DMF) (1.2 mg mL−1) and heated for 2 h at 100 °C. The amount of Pt present in the sample was determined by measuring the absorbance at 706 nm (the absorbance maximum of the OPD–Pt complex). The molar extinction coefficient was calculated for OPD–Pt as 24310 L mol−1 cm−1. The concentration of Pt released from the drug–conjugate was expressed as a ratio of the amount of platinum in the releasing solution from the polymer backbone.
The drug loading efficiency (DLE) and drug loading content (DLC) were determined by absorption spectroscopy using the OPD colorimetric assay from the following respective equation:39–41
| DLE (%) = {weight of drug in NPs/weight of drug in feed} × 100% |
| DLC (%) = {weight of drug in NPs/weight of drug-loaded NPs} × 100%. |
2.12 Preparation of Nile red-encapsulated Pt-coordinated complex
Nile red (NR) was loaded (co-encapsulated) with cisplatin into diblock polymers by adding an acetone solution of Nile red (20 μL, 1 mM) and an aqua solution of cisplatin (13 mM) to the polymer solution in water (5 mL, 1 mg mL−1), which was then stirred overnight. In order to completely remove the acetone, free Nile red and free cisplatin, the solution was dialyzed against water for 16 h at room temperature with an MWCO of 1000. After dialysis the solution was filtered through a 0.4 μm filter, lyophilized and stored at 4 °C.2.13 Cell-viability assay (MTT assay)
To perceive the effect of cisplatin (CP), drug-loaded scaffolds and polymers alone, a cell-viability assay was performed using the WT-MEF cell line, HeLa cell line and MCF-7 cell line using the tetrazolium salt, 3,4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide (MTT). In a 96-well plate (Corning, USA), 1000 cells were seeded per well in 100 μL of DMEM with 10% FBS (fetal bovine serum) and allowed to adhere for 16 h. Prior to drug treatment, media from the cells was aspirated and various concentrations of CP and scaffold with encapsulated CP were added as feed. A blank control, DMEM with FBS in the absence of compound, was used in each experiment. All control and treated experiment wells were run in triplicate. Cells were incubated for 72 h without a change in medium. After 72 h, the drug-containing medium was aspirated. A freshly prepared stock solution of MTT in sterile PBS (5 mg mL−1) was diluted to 50 μg mL−1 in DMEM. 100 μL of this solution was added to each well. Cells were then incubated with MTT for 4 h at 37 °C. Medium with MTT was then aspirated from the wells and the purple formazan crystals that formed as a result of reduction of MTT by mitochondrial dehydrogenase enzyme from the cells were dissolved in 100 μL of 100% DMSO (added per well). The absorbance from the formazan crystals was immediately measured using a micro plate reader at 570 nm (Varioskan Flash) and was representative of the number of viable cells per well. Values from triplicates run for each control and treated set were noted and their mean value was used for calculations. The values thus obtained for the untreated control samples were equated to 100% and the relative percentage values for CP, the scaffold alone and CP-loaded nanoparticles were calculated accordingly. To mimic in vivo conditions a short time MTT assay was studied, wherein MCF-7 cells were seeded in 96-well plates with 1 × 103 cells per well for 16 h. Later, the medium was removed and treated with various concentrations of cisplatin and polymer–cisplatin conjugates. The compounds were incubated with cells for different time intervals of 1, 2, 3 and 4 h separately. After incubation, the media was aspirated and fresh media was added. These cells were then incubated for additional 72 h. After termination of the experiment, the cell viability was determined using the MTT assay.2.14 Cellular uptake of PEG-b-BPCL-CP-NR by confocal microscopy
HeLa cells were seeded at a density of 1 × 105 cells on flame-dried cover slips placed in 6 well-plates containing DMEM medium with 10% FBS and incubated at 37 °C for 16 h. The cells were then exposed to the required concentration of cisplatin alone, the polymer scaffold alone, and NR-loaded cisplatin–polymer nanoparticles for 4 h in a CO2 incubator at 37 °C. After incubation, the drug-containing medium was aspirated from each well, and cells were washed twice with PBS (2 × 1 mL) and fixed with 4% paraformaldehyde solution in PBS for 10 min at room temperature. The cells were washed twice with PBS (2 × 1 mL) and stained with Hoechst solution in PBS. After incubation for 2 min, at room temperature in the dark, the excess dye was washed from the plate and the cells were again gently rinsed with PBS for 1 min. The cover slips were mounted on slides using Fluoromount™ mounting medium (Southern Biotech) and dried overnight at room temperature in the dark. The cells were imaged using a confocal microscope using the λ = 420 nm (blue channel) and λ = 560 nm (red channel) lasers. Images thus obtained were opened using ImageJ analysis software and the image for each channel was separated.3 Results and discussion
3.1 Synthesis of block copolymer cisplatin prodrug
The t-butyl carboxylic ester-substituted caprolactone monomer (named CCL) was prepared from 1,4-cyclohexanediol through multi-step reactions (see Fig. SF1, in the ESI†). Diblock copolymers having a variable poly(ethylene glycol) chain length with fixed carboxylic PCL units were synthesized as shown in Scheme 1. The PEG content in each block was varied using monomethyl ether end-capped oligo (ethylene glycol) CH3(OCH2CH2)xOH (x = 3, 7, 17, 45 and 113) as initiator for the ring-opening polymerization (ROP). For this purpose, monomethyl ethers of triethylene glycol, PEG-350, PEG-750, PEG-2000 and PEG-5000 were employed as initiators along with Sn(Oct)2 as a catalyst. The monomer CCL to initiator ratio was maintained as [M]/[I] = 100 and a solvent-free bulk (or melt) ROP process was developed to produce these block copolymers in high purity for biomedical applications. The resultant diblock copolymers were named as PEGx-b-BPCL, where x represents PEG chains (MW = 350, 750, 2000 and 5000), and where B represents the t-butyl ester. The triethylene glycol monomethyl ether (TEG-OMe)-initiated polymers are referred as BPCL. | ||
| Scheme 1 Synthetic scheme for diblock polymers and their complexation with cisplatin. | ||
The 1H-NMR spectra of a few of the representative diblocks PEGx-b-BPCL (B = butyl-protected) are shown in Fig. 2a (see ESI SF2† for other blocks). The chemical structure of the diblock copolymer is given and the different protons are assigned by using letters a–h (see Fig. 2a). With an increase in the PEG content in the diblocks, the peak at 3.64 ppm corresponding to (OCH2CH2)x increased (see proton ‘a’ in Fig. 2a) in the diblock copolymers (compare the spectra of BPCL and PEG5000-b-BPCL). The protons ‘b’ in the carboxylic CCL unit merged with protons ‘a’ in the PEG. The intensities of protons ‘c’ in the PCL backbone appeared at 4.15 ppm. Thus, the subtraction of peak intensities [(a + b) − c] provided the actual number of protons ‘a’. Comparison of peak integrals of ‘a’ (protons at 3.65–3.68 ppm) with protons ‘c’ (at 4.15 ppm) or t-butyl protons ‘g’ (at 1.44 ppm) provided the number average degree of polymerization (n) for the PCL backbone in the diblock structure. A similar approach was adopted and the “n” values were determined for all the diblock polymers. The number average molecular weights (Mn) were estimated (Mn = n × repeating unit mass) and these values are summarized in Table ST1 (in ESI†).
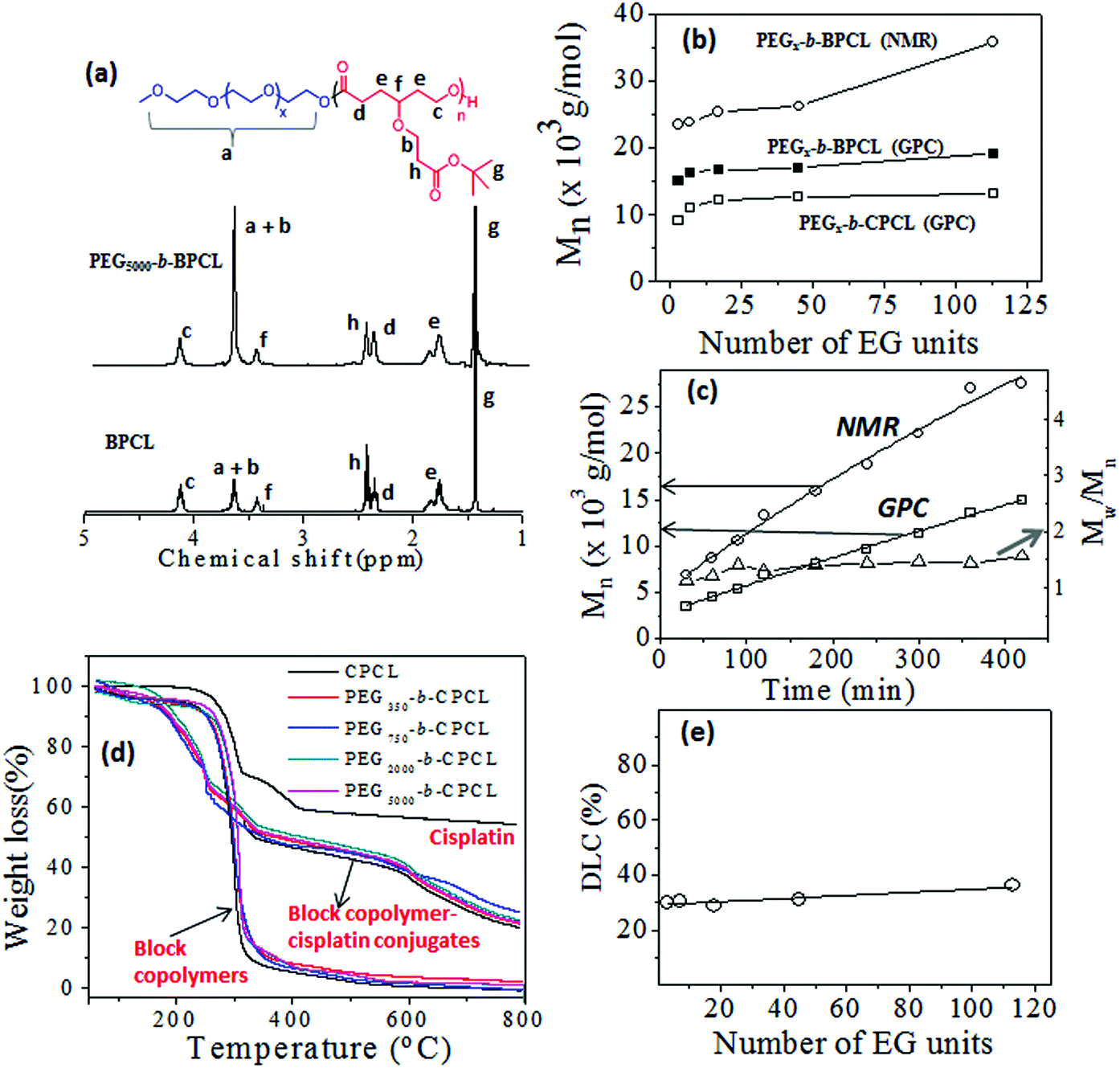 | ||
| Fig. 2 1H-NMR of PEG-b-BPCL block polymers (a). Plots of Mn against number of EG units (b). Plots of Mn and PDI against polymerization time (c). TGA plots of free cisplatin, diblock polymers and their cisplatin conjugates (d). Plot of DLC against number of EG units (e). | ||
The molecular weights of the polymers were determined by GPC in THF. The GPC chromatograms (see Fig. SF3†), the Mn and Mw values, and the polydispersity (PDI) of the polymers (see Table ST1†) are given in the ESI. The plots of Mn obtained from 1H-NMR and GPC techniques were plotted and are shown in Fig. 2b. The molecular weights of the polymers increased with the PEGx chain length in the feed. The GPC technique underestimated the molecular weights of the diblocks compared to 1H-NMR. This trend was attributed to the use of polystyrene standards for the GPC calibration. To study the control (or livingness) of the ROP of the CCL monomer, kinetic polymerization experiments were performed for the PEG2000-b-BPCL diblock copolymer and aliquots were collected at regular intervals. The molecular weights of these aliquots were determined by GPC (see SF3†) and 1H-NMR. The plots of Mn (from NMR and GPC) and polydispersity (from GPC) versus the reaction time are shown in Fig. 2c. The plots for Mn followed a linear trend over the reaction time, confirming the occurrence of the controlled process with respected to a living ROP process. The polydispersity (PDI = Mw/Mn) values of the samples were obtained as ≈1.4, indicating the formation of narrow molecular weight diblock copolymers. The t-butyl ester of the carboxylic functional group was hydrolysed by trifluoroacetic acid to yield the corresponding carboxylic acid diblock copolymer PEGx-b-CPCL, where C represents carboxylic acid (see Scheme 1). The hydrolysis of the t-butyl units was confirmed by 1H-NMR and their spectra are given in (SF4†). The molecular weights of these de-protected diblocks were determined by GPC and their details are given in Fig. SF3 and Table ST1.† The plots of Mnversus the PEG chain length in PEGx-b-CPCL diblocks (see Fig. 2b) revealed that the molecular weights were not affected by the de-protection step. Based on the above analysis, we can conclude that the diblock copolymers with a variable PEG chain length with fixed carboxylic PCL units were produced through the solvent-free ROP process of the new CCL monomer.
The cis-diamminediaquoplatinum(II) complex was formed by cisplatin and AgNO3 in Milli-Q water and the resultant AgCl precipitate was removed by filtration.17 The cisplatin aquo complex was reacted with sodium salts of PEGx-b-CPCL diblocks in deionized water for 24 h in the dark (see Scheme 1). The cisplatin aquo complex to diblock copolymer ratio was maintained as 1.0:1.1 (in molar ratio) to achieve complete chelation in the polymer. The resultant polymer–cisplatin complexes were filtered through 0.45 μm filters and dialyzed for 48 h to remove un-reacted cisplatin aquo complex. The dialyzed solutions were lyophilized to yield dark brown-coloured polymer–cisplatin drug conjugates. Formation of the polymer–cisplatin conjugate was confirmed by FT-IR (see Fig. SF5†). The carbonyl (–CO) stretching frequency appeared as a distinct band around 1720 cm−1 in the nascent polymer which disappeared upon complexation. A new band appeared around 1560 cm−1 with respect to the (Pt–O–CO) stretching frequency of the metal carboxylate functional group.47,48 Additionally, a distinct peak at around 550 cm−1 corresponding to the Pt–O (metal alkoxide) bond stretching was clearly visible in the drug–polymer conjugate.47,48 These cisplatin-conjugated diblock polymers are referred to as PEGx-b-CPCL-CP (CPCL-CP in the case for TEG). Thermal gravimetric analysis (TGA) was used to estimate the drug-loading content of cisplatin in the polymer–drug conjugates. The TGA plots for the polymer–drug conjugates, diblock polymers and free cisplatin are shown in Fig. 2d. Briefly, these calculations were elucidated for PEG5000-b-CPCL-CP. The decomposition of the polymer started at 260 °C and was completely degraded at 500 °C. Cisplatin (alone) underwent stepwise decomposition and showed a 60% weight loss below 400 °C and the platinum content remain unchanged up to 800 °C. The diblock copolymers exhibited a single-step decomposition at 300 °C. In contrast, the cisplatin–polymer conjugates showed different decomposition profiles: (i) below 380 °C with respect to the ligands attached to cisplatin; (ii) from 380 to 580 °C with respect to the degradation of the PEG-b-CPCL block; and (iii) the residual platinum metal above at 600 °C. The drug conjugation efficiency (DCE) was estimated by following the procedure reported by Xu et al.49
| DCE = mPtexp/mPttheo × 100% = (WPt/MPt)/(Wacid/2Macid) × 100% |
3.2 Size and shape of polymer–cisplatin prodrug
To study the self-assembled structures of the polymer–drug conjugates, the samples were dispersed in water and subjected to dynamic light scattering (DLS) analysis. The DLS histograms of the polymer–drug conjugates for various PEGx-b-CPCL diblocks are shown in Fig. 3a–e. All histograms showed a uniform monomodal distribution and the sizes of the self-assembled objects were found to increase with PEG content in the diblocks. The smallest conjugate CPCL-CP (with the triethylene glycol hydrophilic unit) showed the formation of 70 ± 5 nm size assemblies. With increase in the EG unit, the size of the aggregates in DLS increased up to 190 ± 5 nm (see Fig. 3k). Field emission scanning electron microscopy (FE-SEM) images of these polymer–cisplatin conjugates are shown in Fig. 3f–j. The FE-SEM images show the formation of the spherical nano-particular morphologies, and the sizes of the nanoparticles in the images are in very good agreement with the DLS sizes of the samples in water (see Fig. 3k).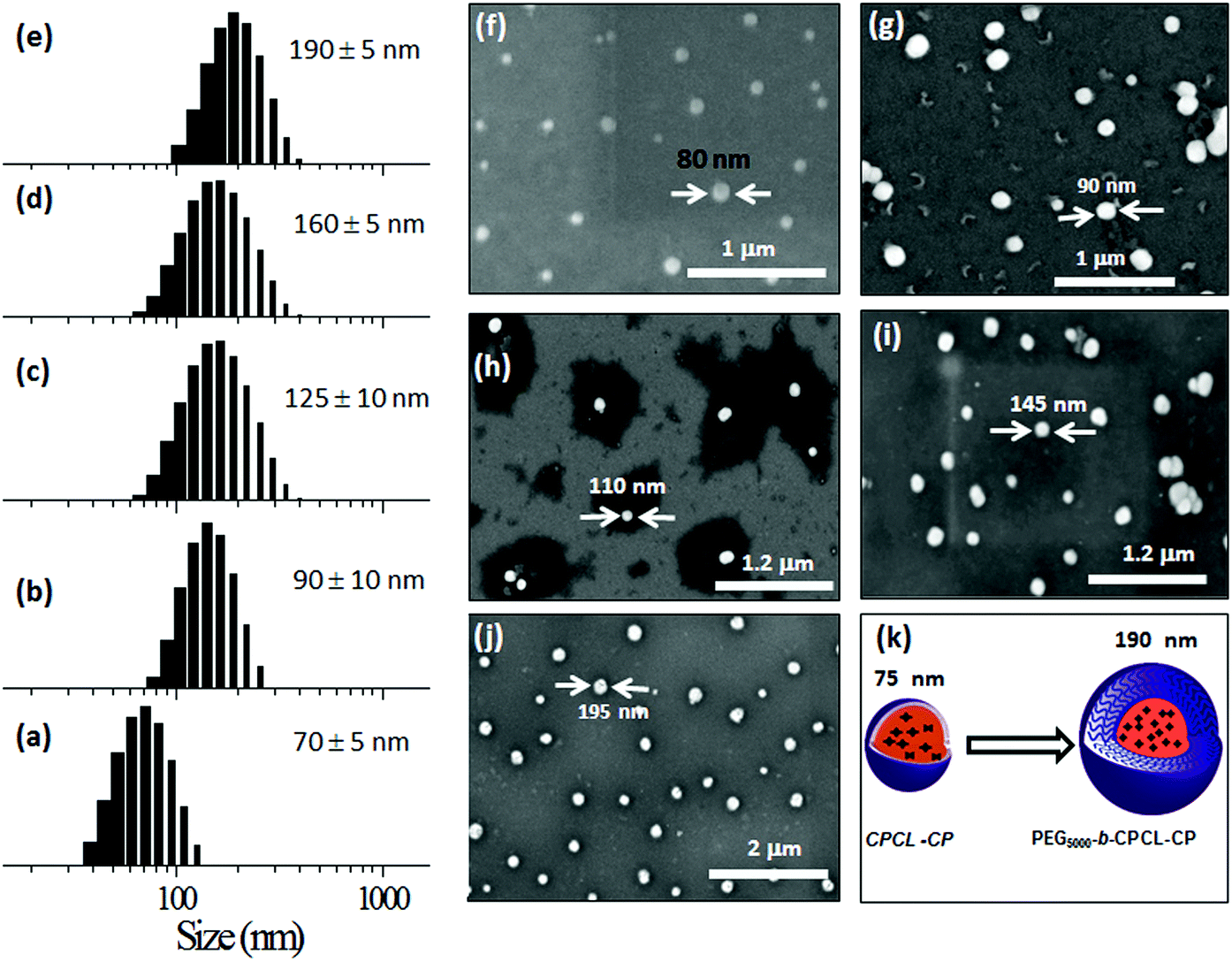 | ||
| Fig. 3 DLS histograms of CPCL-CP (a), PEG350-b-CPCL-CP (b), PEG750-b-CPCL-CP (c), PEG2000-b-CPCL-CP (d) and PEG5000-b-CPCL-CP (e). FE-SEM images of CPCL-CP (f), PEG350-b-CPCL-CP (g), PEG750-b-CPCL-CP (h), PEG2000-b-CPCL-CP (i) and PEG5000-b-CPCL-CP (j). Pictorial representation of core–shell nanoparticles (k). | ||
A core–shell nanoparticle model is proposed for the increase in the polymer–drug-conjugated nanoparticle size with number of ethylene glycol units in the diblocks as represented in Fig. 4a. The core of the nanoparticles is retained as the same and the hydrophilic shell varied by the increase in the PEG chains. This was accomplished since all the diblocks in PEGx-b-CPCL have the same number of carboxylic units (100 units and they have identical DLC, see Fig. 2e) for cisplatin conjugation. This model is further validated by plotting the size of the nanoparticles obtained from DLS and FE-SEM against the number of ethylene glycol units on the periphery (see Fig. 4b and 4c). These plots showed a linear trend with an intercept of 70 nm and slopes of 1.06 and 1.01, for DLS and FE-SEM images respectively. The intercept value of 70 nm corresponds to the core of the nanoparticles in CPCL-CP. Thus, 70 nm is assigned to the core of all the nanoparticles since they have same number of COOH units and cisplatin content (as evident from Fig. 2e). The increase in the nanoparticle size to more than 70 nm was attributed to the increase in the ethylene glycol units on the periphery of the nanoparticles. The slopes of the plots in Fig. 4b and 4c were obtained as ∼1.0, which further supported the linear increase in overall core–shell nanoparticle size with increase in the (OCH2CH2) units at the periphery. The core–shell nanoparticles were subjected to atomic force microscopy (AFM) and high-resolution transmission electron microscope (HR-TEM) analysis. The HR-TEM images of PEG-b-CPCL-CP are shown in Fig. 5a and 5b. The nanoparticles appeared as spherical objects having a dark contrast at the hydrophobic core filled with platinum metal (see more HR-TEM images in Fig. SF6†). In Fig. 5b, the lattice fringes were observed and the space between two subsequent fringes was calculated as 0.23 nm, as reported in the literature.25 AFM analysis was carried out for two conjugates (CPCL-CP and PEG2000-b-CPCL-CP) and their images are shown in Fig. 5c and 5d respectively. The AFM image of CPCL-CP confirmed the existence of nanoparticles of size 75 ± 10 nm and PEG2000-b-CPCL-CP showed the formation of 150 ± 10 nm nanoparticles. Thus, both AFM and HR-TEM analysis support the observation of the spherical core–shell nanoparticle morphology by FE-SEM (also DLS).
 | ||
| Fig. 4 Schematic representation of nanoparticles (a). Plots of nanoparticle size from DLS (b) and FE-SEM (c) against number of ethylene glycol units in the diblocks. | ||
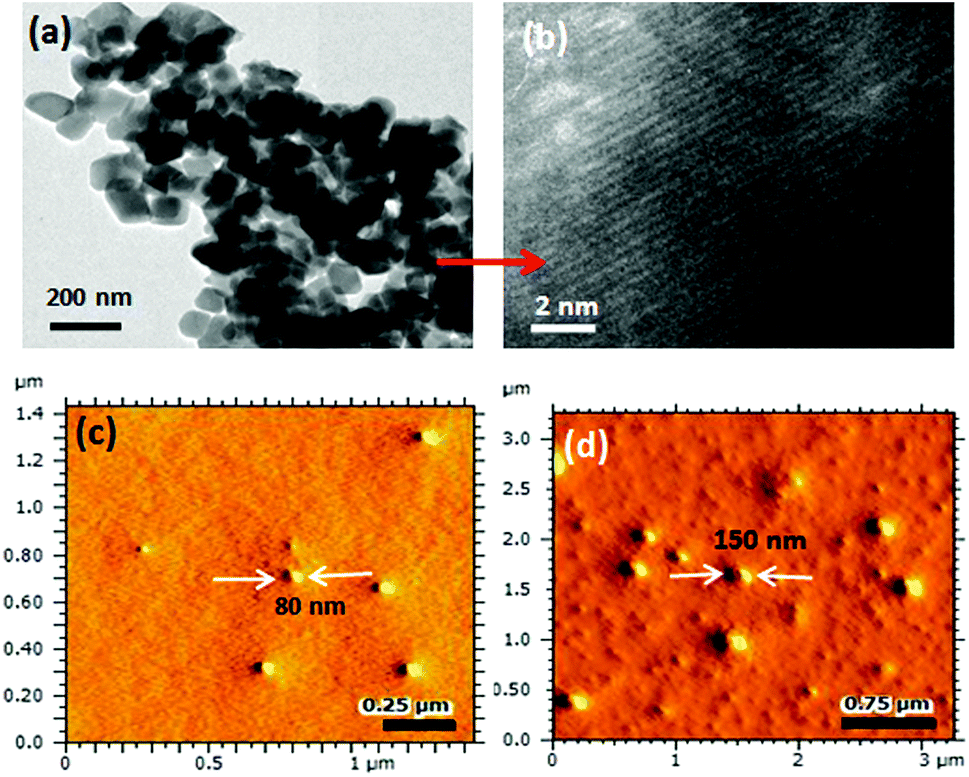 | ||
| Fig. 5 High-resolution TEM image (a) and lattice fringes (b) of PEG2000-b-CPCL-CP. AFM images of CPCL-CP (c) and PEG2000-b-CPCL-CP (d) in tapping mode. | ||
3.3 Core–shell nanoparticle stability in saline, PBS and FBS
To study the effect of the PEG shell on the stability of nanoparticle and cisplatin release from the nanoparticle core, the release studies were carried out under various in vitro conditions. The stability of the core–shell nanoparticles was investigated in Milli-Q water (pH = 6.8), saline (pH = 7.2), PBS (pH = 7.4) and FBS (pH = 7.2) and the results are shown in Fig. 6. Earlier it had been reported that cisplatin prodrugs were susceptible to cleaving in the presence of chloride ions. To test the role of the PEG-shell on the de-chelation, two polymer–cisplatin drug conjugates CPCL-CP and PEG5000-b-CPCL-CP were chosen and subjected to release studies in saline solution and water. For this purpose, typically, the polymer–cisplatin conjugates were dialyzed at 37 °C in the respective media (saline or water) in a semi-permeable membrane having the MWCO = 1000. The amount of cisplatin released in the medium was quantified using the OPD colorimetric assay and the details are given in the ESI (see Fig. SF7†). The cumulative release was calculated as follows:| Cumulative release (%) = Cn × Vo/m × 100% |
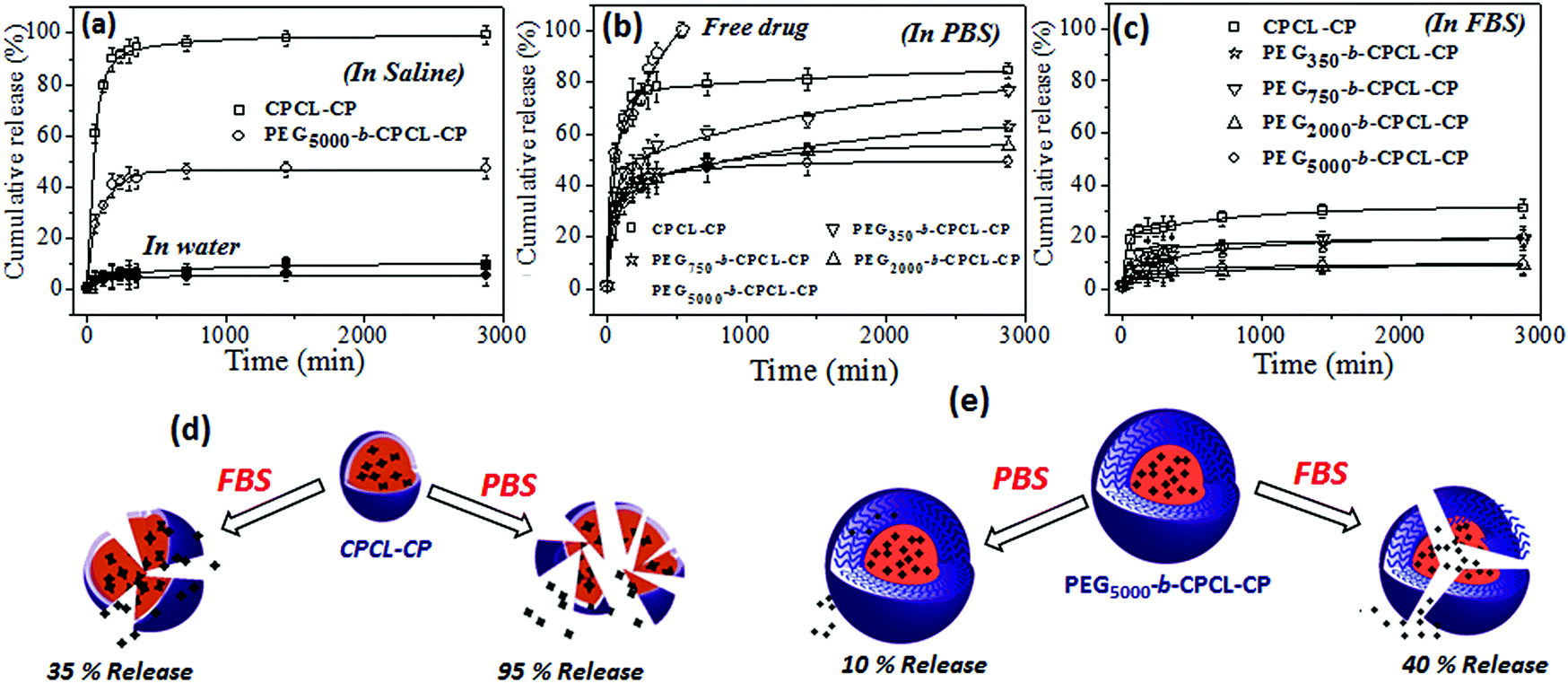 | ||
| Fig. 6 Cumulative release of cisplatin from nanoparticles in saline and water (a), in PBS (b) and in FBS (c) at 37 °C. Possible cleavage mechanism for the drug release from CPCL-CP (d) and PEG5000-b-CPCL-CP (e). | ||
The stability of the polymer–cisplatin nanoparticles in phosphate buffered saline (PBS) is shown in Fig. 6b. The phosphate ions PO42− in the PBS were capable of de-chelating the polymer–cisplatin drug.50 In the CPCL-CP nanoparticles (without any PEG shell protection) these showed >85% of cisplatin leaching. The PEG shell protection in the PEGx-b-CPCL-CP nanoparticles avoided the leaching in saline (Fig. 6a). The drug-release trend in PBS was found to be almost similar to that in saline solution. Interestingly, in FBS (fetal bovine serum, pH = 7.2) the polymer–cisplatin nanoparticles showed significant enhancement in stability compared to PBS (also in saline). The nanoparticles without a PEG shell (CPCL-CP) showed <20% leaching, whereas the PEG shell-protected nanoparticles (PEG5000-b-CPCL-CP) were found to be very stable (<10% leaching in FBS). The schematic representations of breakage of the polymer core–shell nanoparticle in saline, PBS and FBS with respect to PEG shell protection are shown in Fig. 6d and 6e. The PEG shell protection was found to be a crucial factor for stabilizing the cisplatin drug conjugates for intravenous administration. The PEG shell enhanced the solubility of the core–shell nanoparticles against anions such as Cl− and PO42−etc. Further, the enhanced stability in FBS was an added advantage for the intravenous delivery of these nanoparticle systems. These results support the proposal that once the polymer–drug conjugate enters the blood stream, they could be very stable against proteins and other biological species present in the serum (as shown in the hand model Fig. 1a for intravenous administration). Thus, based on the above results the polymer–cisplatin nanoparticles can be stored in Milli-Q water and can be used for direct administration into the blood stream for maximizing their efficacy in cancer treatment.
3.4 GSH resistance and enzyme-responsive cleavage
Sulfur-containing biological species such as cysteine (amino acid residues) and oligopeptides like glutathione (GSH) have been reported to be detoxifying for cisplatin drugs in cancer therapy.6 Reactions of GSH and cysteine with cisplatin are known to produce the S–Pt bond that could be monitored by absorption spectroscopy.36 To study the stability of the core–shell nanoparticles against cytoplasmic thiol residues, several model release studies were performed. The reactions were carried out in the dark at 37 °C in 0.1 mM Tris-HCl buffer containing 4.6 mM NaCl at pH 7.4. The UV–Vis absorbance spectra of the reaction product of free cisplatin with GSH are shown in Fig. 7a. Cisplatin reaction with GSH produced a new absorbance peak at 260 nm, which was assigned to the formation of the Pt–S bond (see Fig. 7a).36 Interestingly, the reaction product of GSH with PEGx-b-CPCL-CP core–shell nanoparticles did not show any new peak for the Pt–S bond (see Fig. 7b). Similar results were also observed for the reaction with cysteine (details are given in Fig. SF9†). The absorbance was plotted against time for GSH and cysteine action on free cisplatin and two polymer nanoparticles CPCL-CP and PEG2000-b-CPCL-CP. From Fig. 7c, it is very clear that the free cisplatin reacted with these thiol species and produced Pt–S bonds. This provides direct evidence that the cisplatin underwent detoxification in the cytoplasm due to the S-rich biological species as pictorially represented in Fig. 7e. On the other hand, the polymer–cisplatin core–shell nanoparticles were found to be resistant to these cytoplasmic thiol species. There was no increase in the absorbance peak corresponding to Pt–S bond formation (see Fig. 7d). This observation supports that the newly designed core–shell nanoparticles are able to stabilise the platinum drug against cytoplasmic thiol species for efficient delivery to cancer cells, as shown schematically in Fig. 7f. | ||
| Fig. 7 Absorbance spectra of free cisplatin drug (a) and core–shell nanoparticles (b) in reaction with GSH. Monitoring of the extent of reaction (at the absorbance maximum of 260 nm) against time of the reaction for free cisplatin (c) and core–shell cisplatin nanoparticles (d) upon exposure to cysteine and GSH. Schematic representation of free cisplatin (e) and cisplatin polymer nanoparticles (f) entry into the cytoplasm and their possible interaction with GSH or cysteine in Pt–S bond formation. | ||
To track the intracellular fate of the nanoparticles in the presence of enzyme-rich lysosomes and cytoplasmic thiols, two different types of experiments were performed using the PEG2000-b-CPCL-CP conjugate (see Fig. 8). In the type-1 experiment, the samples were subjected to lysosomal compartment conditions wherein esterase leads to cleavage of the polymer–cisplatin conjugate made up of biodegradable PCL units. To address this concept, the cumulative release profiles of the polymer–drug conjugates were studied in the presence of 10 U of esterase.39–41 The amount of cisplatin released was monitored using the OPD assay. This esterase enzyme was added in two ways: (i) at the initial stage at 37 °C, and (ii) after incubation of the nanoparticles for 24 h at 37 °C. The cumulative release profiles for the addition of esterase at the initial stage (see Fig. 8a) revealed that the PCL backbone was cleaved by the enzyme and cisplatin release occurred at a fast rate. In the second experiment, incubation of the nanoparticles showed an initial 40% leaching (as observed in Fig. 6b). The administration of esterase after 24 h facilitated the instantaneous rupturing of the PCL core and release of the remaining drug (see Fig. 8a). Hence, it can be concluded that the core–shell nanoparticles preserved the drug and were only ruptured in the presence of lysosomal enzymes to release their cargo at the intracellular compartments. The type-2 experiment was performed to understand the nature of the interaction between GSH and the polymer–cisplatin conjugate. Here, GSH was administrated first to mimic entry of the cisplatin conjugate and immediate exposure to GSH in the cytoplasm upon being taken up by cells. The data in Fig. 8b confirm that the cisplatin–polymer conjugate is stable against GSH for a period of more than 6 h. Upon subsequent administration of esterase (after 6 h of GSH exposure), the polymer backbone immediately disassociated to release the cisplatin drug. At this stage, the drug has exposure to GSH already present in the medium to produce Pt–S bonds. In the actual in vitro cellular administration, the drug would have equal chance to reach the nucleus to promote cell death. This control experiment attempts to mimic the intracellular environment, and it is clearly evident that the polymer–cisplatin conjugate is stable against GSH and that it can be cleaved by the esterase enzyme present in the lysosomal compartments. Cleavage mechanism of the polymer–cisplatin nanoparticles in the presence of esterase and GSH in our type-1 and type-2 model experiments is shown schematically in Fig. 8c. The results clearly demonstrate the polymer nanoparticles resistance to drug detoxification against GSH and ensure efficient cisplatin administration to the nucleus of the cell to enhance cell death.
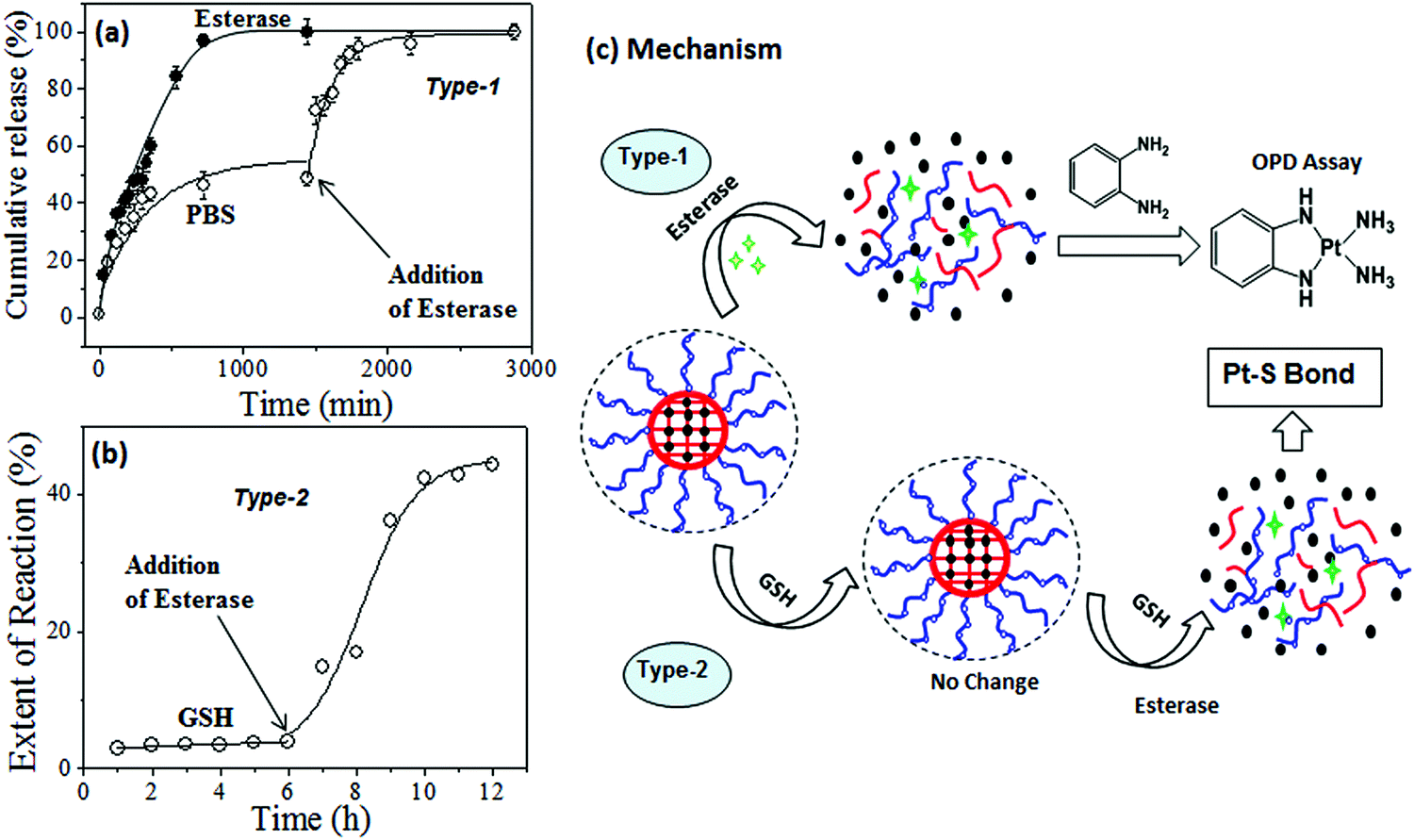 | ||
| Fig. 8 (a) Cumulative drug release of cisplatin from PEG2000-b-CPCL-CP with respect to the administration of esterase at either the beginning or after incubating in PBS for 24 h. (b) Extent of GSH attack (monitored at 260 nm absorbance) on PEG2000-b-CPCL-CP for initial 6 h and after administration esterase enzyme in Tris-buffer. (c) Schematic representation of nanoparticle dissociation by esterase enzyme alone (type-1) and its resistance to GSH and subsequent cleavage followed by the addition of esterase enzyme (type-2). | ||
3.5 Cellular uptake and cytotoxicity
The cytotoxicity of the diblock copolymers and their cisplatin conjugates was investigated in wild-type mouse embryonic fibroblasts (WT-MEFs), breast cancer (MCF-7) and cervical cancer (HeLa) cell lines. Breast cancer cells (MCF-7) are inherently 10 times over-expressed with cytoplasmic thiol residues such as GSH (in mM) compared to other cancer cells (HeLa).7 The cytotoxicity of the nascent diblock copolymers was first tested in HeLa cell lines by varying their concentration up to 400 μg mL−1. The data shown in Fig. 9a indicate a more than 80% cell viability for 10 μg mL−1 concentration.19 At higher concentrations the cell viability gradually decreased with increase in the polymer content (see Fig. SF10†). These results confirmed that the custom-designed diblock copolymers possess high biocompatibility up to 10 μg mL−1 which is a concentration typically employed for synthetic polymers in drug delivery.17 The cytotoxicity of free cisplatin drug and polymer–drug conjugates without protection (CPCL-CP) and with PEG shell protection (PEG2000-b-CPCL-CP) were tested in normal (WT-MEFs) and cancer (MCF-7 and HeLa) cell lines. The free cisplatin drug concentration was maintained as equivalent to drug conjugates in the polymer (DLC = 33%). The concentration of cisplatin drug was varied from 0.1 to 8.0 μg mL−1 and their cytotoxicity data are summarized for WT-MEFs, HeLa and MCF-7 in Fig. 9b–d, respectively. In Fig. 9b, the data support that the free cisplatin is toxic to normal cells; however, the cisplatin polymer nanoparticles are non-toxic. In the HeLa cell lines, both free cisplatin drug and polymer–drug conjugates show 50% killing of cells at 1.0 μg mL−1 concentrations which is in accordance with earlier observations.51 At higher drug concentrations of >4.0 μg mL−1, less than 10% cells were viable (see Fig. 9c). Interestingly, the breast cancer cells showed a dramatic difference for free cisplatin and polymer-conjugated drugs. Free cisplatin drug showed 50% killing in cells at 1.0–2.0 μg mL−1 which is similar to that of earlier observation.52 The cell viability for free cisplatin drug did not change much at higher concentrations and 40% of cells were viable even at larger drug concentrations of 10 μg mL−1. On the other hand, the polymer–cisplatin drug conjugate showed significant improvement in the cell killing. The % cell death increased significantly with increase in the concentration of polymer–cisplatin drug conjugates and only <10% of the cells were viable at 4.0 μg mL−1 (see Fig. 9d). The free cisplatin drug was ineffective for achieving complete cell killing in MCF-7 cells, which was overcome by conjugating the drug in the diblock structures. The ineffective cell death for the free drug can be attributed to the over-expression of GSH in the MCF-7 cells and the detoxification of free cisplatin drugs. On the other hand, the cisplatin in the core of the polymer assembly provided shielding against detoxification (see Fig. 7 and 8b) and enhanced the drug stability for increasing the cell death. Further, the polymer–cisplatin conjugates could also be cleaved in a controlled manner by intracellular enzymes like esterase so that the drug is released in a controlled manner for a longer period of time to achieve complete cell killing (evident in Fig. 8a). Thus, the polymer–drug conjugates carry these additional advantages to accomplish complete cell death which is not possible in free cisplatin drugs. The present in vitro studies provide direct evidence for the need to conjugate cisplatin drugs in the polymer scaffold to achieve complete cell death in the presence of GSH which is over-expressed in breast cancer tissues.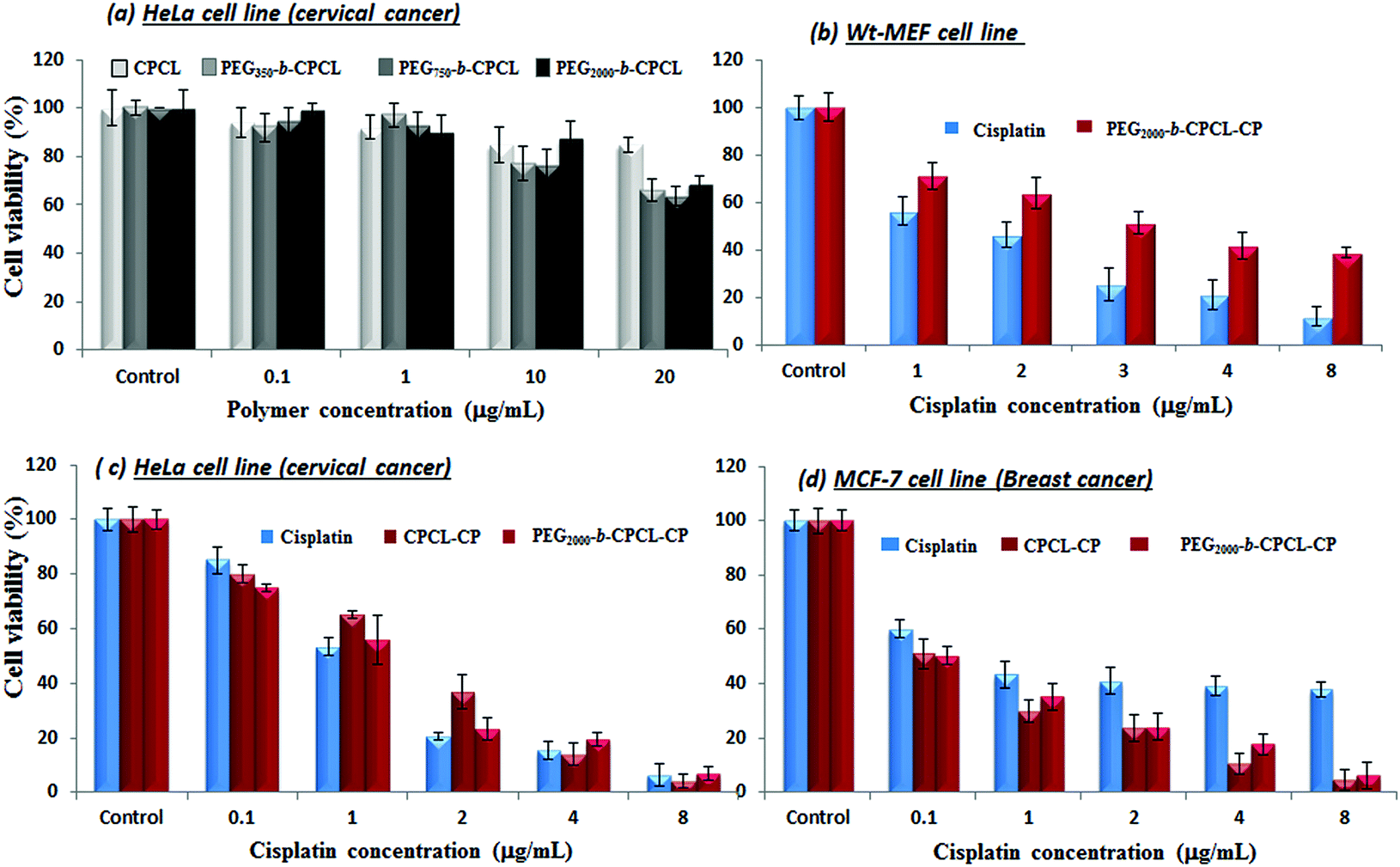 | ||
| Fig. 9 Histogram depicting cytotoxicity of diblock polymers in HeLa cells at various concentrations (a). Cytotoxicity of cisplatin and PEG2000-b-CPCL in WT-MEF (b) at various concentrations. Cytotoxicity of cisplatin, CPCL-CP and PEG2000-b-CPCL in HeLa (c) and MCF cells (d) at various concentrations. | ||
Peer and co-workers developed in vitro cell line experiments that could mimic in vivo type conditions for nanoparticle treatment in cancer cells.53,54 As per this experimental protocol, free and polymer-bound drugs were administered and incubated for a short period (typically 1–4 hours). The excess drug removed by washing and cells were left for a longer time (up to 72 h) for cell proliferation. In this process, the drugs were initially taken up by the cells (prior to the washing) to control the cell proliferation and their growth was similar to that of in vivo conditions. In the present investigation, a similar experiment was carried out for the free drug and cisplatin–polymer nanoparticles in MCF-7 cells and the details are given in Fig. 10. The cells were initially administered and incubated for 1, 2, 3 and 4 hours with free cisplatin and in the polymer-conjugated form. The drug concentrations in the above experiment were varied as 1.0 and 4.0 μg mL−1 (based on the MCF-7 data in Fig. 9d) and the cells were incubated for 72 h. According to Fig. 10a and 10b, it is clear that at a lower drug concentration (1.0 μg mL−1) both free cisplatin and the drug–polymer nanoparticle did not exhibit significant cell killing. Thus at the lower drug concentration, the in vitro experiments in which the cells were allowed to proliferate continuously (see Fig. 9d) behave similar to those of the in vivo-mimicking experiment. Interestingly, at a higher drug concentration (at 4.0 μg mL−1), the free drug and cisplatin–polymer nanoparticle exhibited a significant difference in the cell killing. For example, the free drug showed only 50% cell death whereas the cisplatin–polymer nanoparticle accomplished almost 90% cell death. This in vivo-mimicking experiment confirmed the efficient cell killing by the cisplatin–polymer nanoparticles, similar to that observed in the in vitro data (see Fig. 9d). Hence, it may be concluded that the custom-designed cisplatin–polymer nanoparticles are very efficient prodrugs for breast cancer treatment. Further, in vivo experiments in mice models would provide more insight into the drug action which will be done in future studies.
 | ||
| Fig. 10 Histograms showing the in vivo-mimicking cytotoxicity experiment. Cisplatin and PEG2000-b-CPCL-CP in MCF-7 cells at various drug concentration levels (1 and 4 μg mL−1) and incubation time (1 to 4 h). The cells were washed after the said incubation time and allowed to proliferate for 72 h post-washing. | ||
The cellular uptake of polymer–cisplatin nanoparticles into the cytoplasm was scrutinised in HeLa and MCF-7 cells using confocal laser scanning microscopy (CLSM) and fluorescence microscopy. Since, cisplatin is non-luminescent in nature, it needs to be co-encapsulated with a fluorescent molecule. Nile red (NR) was chosen as a probe which was encapsulated in the hydrophobic PCL core. Further cisplatin aquo complex and NR were stirred together with PEG2000-b-CPCL100-CP at 37 °C for 24 h. The unencapsulated dyes were removed by dialysis (MWCO = 1000). Structural characterization of these NR-loaded polymer–cisplatin nanoparticles was carried out and the details are shown in Fig. SF11.† NR-loaded nanoparticles (PEG2000-b-CPCL-CP-NR) were incubated with HeLa and MCF-7 cells for 4 h at 37 °C. The nanoparticle concentration used for treatment was 10 μg mL−1 with 3.3 μg of cisplatin and 0.17 μg of NR. The red fluorescence from NR at ∼560 nm was monitored through the red channel (λ = 568 nm). The blue fluorescence produced by the cell nuclei after Hoechst staining was observed through the blue channel (λ = 461 nm). The images corresponding to PEG2000-b-CPCL-CP and Hoechst fluorescence in HeLa and MCF-7 cell along with the merged image are shown in Fig. 11. As shown in the images, strong NR fluorescence is observed at the intracellular compartment in the cytoplasm and at the peri-nuclear region (more images are shown in Fig. SF12†). At present, it is rather difficult to make any conclusion about the difference in the uptake of the nanoparticles based on the fluorescence intensity of the cell images. The cellular uptake of the nanoparticles was also studied by using the fluorescent microscopy technique in HeLa cells (see Fig. SF13†). Fluorescence microscopy images also supported the peri-nuclear localization of cisplatin-loaded nanoparticles inside the cells. The images revealed that the polymers are capable of delivering the cargoes both at the cytoplasm and the peri-nuclear environment. Based on these studies, it may be concluded that these nanoparticles accumulate in the cytoplasm and also in the peri-nuclear region. Further, in vivo studies are required to check the administration of these nanoparticles in animal models. Nevertheless, the present investigation has provided proof of the need to conjugate cisplatin drugs for achieving resistance to detoxification by GSH in breast cancer cells and for enhancing their efficacy. Though, the approach has been demonstrated only for cisplatin, in principle, it can be expanded to other platinum-based drugs for resistance against cytoplasmic thiol residues. Further, the core–shell strategy can also, in principle, be expanded to other metal nanoparticles for increasing their stabilization in aqueous medium. Currently, efforts are being taken to proceed in these directions to expand the diblock polymer core–shell approach for various biomedical applications.
 | ||
| Fig. 11 CLSM images of HeLa cells and MCF-7 cells incubated with PEG2000-b-CPCL-NR nanoparticles. The nucleus was counter-stained with Hoechst (blue). The cells were observed through the red channel to locate NR fluorescence (red). | ||
4 Conclusion
In summary, new classes of biodegradable diblock copolymer core–shell nanoparticle assemblies has been designed and developed for cisplatin delivery against detoxification of cytoplasmic thiol residues in breast cancer cells. The complexation of a cisplatin aquo complex with the above-mentioned diblock copolymers produced core–shell nanoparticles. In this process, the core of the particles was fixed at 75 nm and the hydrophilic PEG shell was varied by varying the PEG chain length in the ROP process. The core–shell nanoparticles were found to be very stable in FBS and water. In PBS and saline solution, the drug stability increased with increase in the PEG shell protection layer. In vitro drug-release studies revealed that the PEG shell protected the cisplatin drug against attack by the cytoplasmic thiol residues like GSH and thus the drug was free from detoxification. Further, the biodegradable aliphatic PCL ester backbone was found to shield the cisplatin core against GSH action, with rupture only being accomplished upon exposure to esterase enzyme at conditions identical to those of the intracellular compartments. The cytotoxicity of the polymer and polymer–cisplatin conjugates was tested in MCF-7 and HeLa cell lines. The nascent polymers were found to be biocompatible and non-toxic to cells. In the HeLa cell line, both the free cisplatin drug and polymer–cisplatin core–shell nanoparticles showed almost identical cytotoxicity. The free cisplatin drug failed to kill all of the cells in the MCF-7 study and the cells were more than 50% viable even at very high drug concentrations. Over-expression of GSH in MCF-7 was attributed to poor cell killing by the free cisplatin drug. The polymer–cisplatin nanoparticles showed enhanced cell killing for MCF-7 and the cell viability is found to be <10% at the 4 μg mL−1 drug concentration. This selective and enhanced cell killing in MCF-7 cells by the polymer nanoparticles was attributed to their resistance to drug detoxification by GSH. Cellular uptake of the nanoparticles into the cytoplasm and peri-nuclear environment assemblies was confirmed by confocal and fluorescence microscopic analysis using Nile red as fluorophore. The present polymer–cisplatin core–shell approach provides a new platform for platinum-based drugs against cytoplasmic thiol detoxification which is very useful for enhancing the killing of breast cancer cells that have over-expression of GSH to 100%.Author information
All the authors contributed to the work. The authors declare no financial interest.Acknowledgements
The authors thank research grant from Department of Science and Technology (DST) for the project SB/S1/OC-37/2013. B.S. thanks UGC, New Delhi for Ph.D. research fellowship. The authors thank National Chemical Laboratory, Pune, India for HR-TEM facility. AFM facility is sponsored by the DST FIST Project. Authors thank Dr Harinath Chakrapani, IISER-Pune, for providing cell culture facilities and for scientific discussions.References
- A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS PubMed.
- M. A. Foertes, A. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645–662 CrossRef PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- J. C. Dabrowiak, J. Goodisman and A. K. Souid, Drug Metab. Dispos., 2002, 30, 1738–1744 CrossRef.
- G. K. Balendiran, R. Dabur and D. Fraser, Cell Biochem. Funct., 2004, 22, 343–352 CrossRef CAS PubMed.
- R. R. Perry, J. Mazzeta, M. Levin and S. C. Barranco, Cancer, 1993, 72, 783–787 CrossRef CAS.
- Y. Kasherman, S. Sturup and D. Gibson, J. Med. Chem., 2009, 52, 4319–4438 CrossRef CAS PubMed.
- C. Syng-ai, A. L. Kumari and A. Khar, Mol. Cancer Ther., 2004, 3, 1101–1107 CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- D. Nowotnik and E. Cvitkovic, Adv. Drug Delivery Rev., 2009, 61, 1214–1219 CrossRef CAS PubMed.
- J. M. Rademaker-Lakhai, C. Terret, S. B. Howell, C. B. Baud, R. F. D. B. Boer, D. Pluim, J. H. Beijnen, J. H. M. Schellens and J.-P. Droz, Clin. Cancer Res., 2004, 10, 3386–3395 CrossRef CAS PubMed.
- L. Nuhn, S. Hartmann, B. Palitzsch, B. Gerlitzki, E. Schmitt, R. Zentel and H. Kunz, Angew. Chem., Int. Ed., 2013, 52, 10652–10656 CrossRef CAS PubMed.
- H. T. T. Doung, V. T. Huynh, P. De Souza and M. H. Stenzel, Biomacromolecules, 2010, 11, 2290–2299 CrossRef PubMed.
- V. T. Huynh, G. Chen, P. De Souza and M. H. Stenzel, Biomacromolecules, 2011, 12, 1738–1751 CrossRef CAS PubMed.
- S.-M. Lee, T. V. O'Halloran and S. B. T. Nguyen, J. Am. Chem. Soc., 2010, 132, 17130–17138 CrossRef CAS PubMed.
- V. T. Huynh, P. De souza and M. H. Stenzel, Macromolecules, 2011, 44, 7888–7900 CrossRef CAS.
- K. J. A. Karim, S. Binauld, W. Scarano and M. H. Stenzel, Polym. Chem., 2013, 4, 5542 RSC.
- S. Kaida, H. Cabral, M. Kumagai, A. Kishimura, Y. Tereda, M. Sekino, I. Aoki, N. Nishiyama, T. Tani and K. Kataoka, Cancer Res., 2010, 70, 7031–7041 CrossRef CAS PubMed.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamato, Y. Sugiayama, K. Nishio, Y. Mastumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS.
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172–13175 CrossRef CAS PubMed.
- D. Ding, K. Li, Z. Zhu, K.-Y. Pu, Y. Hu, X. Jiang and B. Liu, Nanoscale, 2011, 3, 1997–2002 RSC.
- F. Zhang, M. Elsabahy, S. Zhang, L. Y. Lin, J. Zou and K. L. Wooley, Nanoscale, 2013, 5, 3220–3225 RSC.
- Y. Teow and S. Valiyaveettil, Nanoscale, 2010, 2, 2607–2613 RSC.
- C. Morelli, P. Maris, D. Sisci, E. Perrotta, E. Brunelli, I. Perrotta, M. L. Panno, A. Tagarelli, C. Versace, M. F. Casula, F. Testa, S. Ando, J. B. Nagy and L. D. Pascqua, Nanoscale, 2011, 3, 3198–3207 RSC.
- L. Muzi, C. Menard-Moyon, J. Russier, J. Li, C. F. Chin, W. H. Ang, G. Pastorin, G. Risuleo and A. Beanco, Nanoscale, 2015, 7, 5383–5394 RSC.
- F. Wang and J. Liu, Nanoscale, 2014, 6, 7079–7084 RSC.
- A. R. Maity, A. Chakraborty, A. Mondal and N. R. Jana, Nanoscale, 2014, 6, 2752–2758 RSC.
- S. Khiati, D. Luvino, K. Oumzil, B. Chauffert, M. Campio and P. Barthelemy, ACS Nano, 2011, 5, 8649–8655 CrossRef CAS PubMed.
- S. Guo, Y. Wang, L. Miao, Z. Xu, C. M. Lin, Y. Zhang and L. Huang, ACS Nano, 2013, 7, 9896–9904 CrossRef CAS PubMed.
- X. Qi, N. Li, H. Gu, Y. Xu, Y. Xu, Y. Jiao, Q. Xu, H. Li and J. Lu, Nanoscale, 2013, 5, 8925–8929 RSC.
- Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2254 CrossRef CAS PubMed.
- S. Setua, M. Ouberai, S. G. Piccirillo, C. Watts and M. Welland, Nanoscale, 2014, 6, 10865–10873 RSC.
- Y. Min, C.-Q. Mao, S. Chen, G. Ma, J. Wang and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS PubMed.
- Q. Hu, P. S. Katti and Z. Gu, J. Am. Chem. Soc., 2014, 6, 12273–12286 CAS.
- J. Zhuang, M. R. Gordon, J. Venture, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- P. S. Pramod, K. Takamura, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Biomacromolecules, 2012, 13, 3627–3640 CrossRef CAS PubMed.
- P. S. Pramod, R. Shaw and M. Jayakannan, Nanoscale, 2015, 7, 6636–6652 RSC.
- P. S. Pramod, R. Shaw, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Nanoscale, 2014, 6, 11841–11855 RSC.
- A. J. Harnoy, I. Rosenboum, E. Tirosh, Y. Ebenstein, R. Saharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- S. Samarajeeva, R. Shrestha, Y. Li and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 1235–1242 CrossRef PubMed.
- E. A. Rainbolt, K. E. Washington, M. C. Biewer and M. C. Stefan, Polym. Chem., 2015, 6, 2369–2381 RSC.
- B. Surnar and M. Jayakannan, Biomacromolecules, 2013, 14, 4377–4387 CrossRef CAS PubMed.
- B. Surnar, P. S. Pramod and M. Jayakannan, Z. Anorg. Allg. Chem., 2014, 640, 1119–1126 CrossRef CAS PubMed.
- R. W. Ski, J. Kuduk-Jaworska and D. Michalska, J. Mol. Struct. (THEOCHEM), 2006, 758, 169–179 CrossRef PubMed.
- A. D. Allen and T. Theophanindes, Can. J. Chem., 1964, 42, 1551–1554 CrossRef CAS.
- J. Xu, Q. Fu, J.-M. Ren, G. Bryant and G. G. Qiao, Chem. Commun., 2013, 49, 33–35 RSC.
- R. C. Todd, K. S. Lovejoy and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 6370–6371 CrossRef CAS PubMed.
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172–13175 CrossRef CAS PubMed.
- T. C. Johnstone, N. Kulak, E. C. Pridgen, O. C. Farokhzad, R. Langer and S. J. Lippard, ACS Nano, 2013, 7, 5675–5683 CrossRef CAS PubMed.
- K. Kohen, R. Emmanuel, E. Kishin-Finfer, D. Shabath and D. Peer, ACS Nano, 2014, 8, 2183–2195 CrossRef PubMed.
- G. Bachar, K. Kohen, R. Hod, R. Feinmesser, A. Mizrachi, T. Shpitzer, O. Katz and D. Peer, Biomaterials, 2011, 32, 4840–4848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA profile and DSC thermogram of all polymers, DLS data, AFM image, 1H-NMR, 13C-NMR, and MALDI spectra of all polymers and monomers. See DOI: 10.1039/c5nr04963f |
| This journal is © The Royal Society of Chemistry 2015 |