
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of nanomaterials for water treatment
Renyuan
Li
,
Lianbin
Zhang
and
Peng
Wang
*
Water Desalination and Reuse Center, Division of Biological and Environmental Science and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: peng.wang@kaust.edu.sa
First published on 26th August 2015
Abstract
The ever-increasing human demand for safe and clean water is gradually pushing conventional water treatment technologies to their limits. It is now a popular perception that the solutions to the existing and future water challenges will hinge upon further developments in nanomaterial sciences. The concept of rational design emphasizes on ‘design-for-purpose’ and it necessitates a scientifically clear problem definition to initiate the nanomaterial design. The field of rational design of nanomaterials for water treatment has experienced a significant growth in the past decade and is poised to make its contribution in creating advanced next-generation water treatment technologies in the years to come. Within the water treatment context, this review offers a comprehensive and in-depth overview of the latest progress in rational design, synthesis and applications of nanomaterials in adsorption, chemical oxidation and reduction reactions, membrane-based separation, oil–water separation, and synergistic multifunctional all-in-one nanomaterials/nanodevices. Special attention is paid to the chemical concepts related to nanomaterial design throughout the review.
 Renyuan Li | Renyuan Li is currently an MS/PhD student at King Abdullah University of Science and Technology (KAUST) under the supervision of Professor Peng Wang. He received his B.S. degree in material science and engineering at Beijing University of Science and Technology in 2014. His research interest is in functionalized nanomaterials and surfaces for water purification. |
 Lianbin Zhang | Lianbin Zhang is currently a research scientist in Professor Peng Wang's group at KAUST. He received his B.Sc. degree in polymer material and engineering in 2005 and Ph.D. degree in polymer chemistry and physics in 2010, both from Jilin University, China. He then conducted postdoctoral research studies in Hong Kong University of Science and Technology in 2010 and at KAUST from 2010 to 2012. His scientific interests are focused on functionalized interfacial materials, stimuli-responsive nanomaterials, and their applications in water and environment. |
 Peng Wang | Professor Peng Wang joined KAUST in September 2009 as a founding faculty member and he is currently an associate professor and program chair of Environmental Science and Engineering program at KAUST. He received his Ph.D. degree from the University of California, Santa Barbara (UCSB) in 2008. His research focuses on rational design, synthesis, and application of nanomaterials for clean water and clean energy production. |
1. Introduction
Water pollution and water scarcity are among the most challenging problems facing mankind nowadays.1 With rapid population growth, steadily improving life standards, fast industrialization and modernization of developing countries, these challenges will persist, if not worsen, in the years to come.2 Conventional water treatment technologies, including adsorption,3–5 chemical treatment,6–9 membrane-based separation,10,11 and biological treatment,12–14 are generally designed on the basis of bulk water chemistry and without any doubt, these technologies have made critical contributions to sustaining human society in the past century. However, the ever-increasing demand for safe and clean water is gradually pushing them to their limits.On the other hand, ever since 1959, when the term “nanotechnology” was first used by Richard Feynman in his famous lecture entitled “there's plenty of room at the bottom”,15 the field of nanomaterials/nanotechnology has been experiencing literally explosive growth, especially in the last two decades.16–21 In a traditional sense, nanomaterials have two primary advantages over conventional bulk materials: (1) very small size and huge specific surface area, which are beneficial to many interface-related applications;16,22–27 (2) distinctively tunable chemical, physical, optical, electronic and mechanical properties, which can be rationally adjusted by controlling their size, surface morphology, shape and crystal orientation.28–34 As a result, the concept of going to nanoscale has opened up numerous new avenues that would otherwise be impossible with conventional bulk materials.16,27
Moreover, the application of nanomaterials/nanotechnology for water treatment and more broadly for environmental remediation has steadily grown into a distinct field with the expected growth rate on an exponential rise.35–45 In the early stages, the concept of nanomaterials first attracted attention from researchers in environmental science and it was regarded as having unconfirmed potential in environmental remediation. At this stage, major efforts have been made in searching for available nanomaterials developed by materials scientists in a trial-and-error manner. Occasionally, materials scientists were also involved in the water treatment research in a bid to find some applications for their newly developed nanomaterials. Although a high level of disconnection between the researchers who synthesized the nanomaterials and those who tested their performances was previously not uncommon, this pioneering work demonstrated the great potential of nanomaterials in the water treatment field35 and therefore naturally pushed it to the next stage, which can be described as the rational design of nanomaterials for water treatment. By this stage, researchers realized that the chemistry and ultimately the functions of nanomaterials could be deliberately pre-designed for a desired purpose before embarking on nanomaterial synthesis. Within the scheme of rational design, less attention is paid to the inherent properties of the currently available nanomaterials and more focus is on designing and calculatedly imparting synergistic multi-functionalities to the as-developed nanomaterials or in some cases nanodevices to target a clearly defined problem.46–51 In this rational design stage, the nanomaterials are generally made of multi-components, and the material design, synthesis and application have been seamlessly integrated within one entity to ensure effective and iterative communication/feedback between the nanomaterial design and the final performance toward well-defined purposes, which leads to a high likelihood of achieving the final goals.
The focus of this review is on the rational design of nanomaterials for water treatment applications. However, the review is not intended to be exhaustive and instead it aims to give a comprehensive overview of this exciting field using a limited number of examples. It is for this purpose that some topics, for example, nano-assisted bio-remediation, nano-assisted ion-exchange, nanomaterial-based water pollutant sensors, nano-assisted microbial fuel cells and design of nanoscale zero-valent iron (NZVI)-based treatment systems, although interesting, are not included in the review. It can be noted that the review defines nanomaterials as substances with controllable features at the nanoscale, instead of on a particle size basis, which makes many materials and devices eligible for this review. The review is divided into the following sub-topics: (1) rational design of nanoporous materials and their pore structure controls, (2) rational design of nano-adsorbents by surface chemistry engineering; (3) rational design of nano-assisted oxidation and reduction processes; (4) rational design of nano-assisted membrane-based separation; (5) rational design of superwetting surfaces for oil–water separation; (6) multifunctional all-in-one nanomaterials and nanodevices for designed purposes, and (7) concluding remarks.
2. Rational design of nanoporous materials and their pore structure controls
As the size of a particle decreases, its specific surface area (i.e., the surface area per unit mass) increases drastically (Fig. 1a)52 and it was the ultra-high specific surface area of nano-sized materials that first attracted attention from the water treatment field because numerous water treatment processes rely on interface-related processes (e.g., adsorption, chemical reaction, and catalysis),5–7,9 whose performance is positively dependent on the material surface area. Exciting published results have demonstrated the effectiveness of the strategy of going to nano-size for an enhanced performance by fabricating smaller and smaller materials.22,53–55 As an important example, research groups all around the world demonstrated the remarkably higher degradation rate of trichloroethylene (TCE) by nanoscale zero-valent iron (NZVI) (generally sized 10–100 nm) than by bulk iron filings (with size >2 mm) in conventional permeable reactive barriers (PRBs).56–63 NZVI for TCE degradation heavily dominated research into the application of nanomaterials for water treatment, especially groundwater treatment, in the early stages.37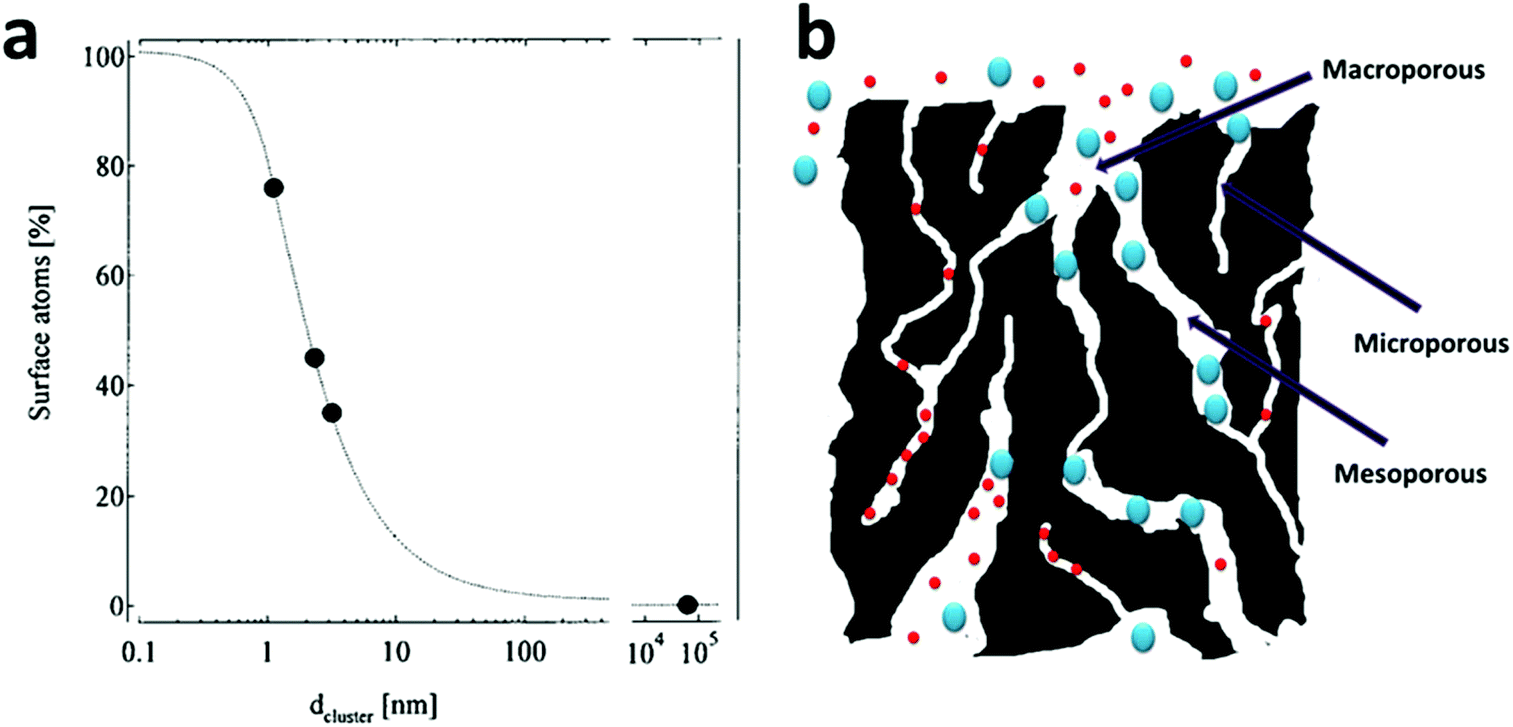 | ||
| Fig. 1 (a) The average percentage of surface atoms as a function of the nanoparticle diameter. Reprinted with permission from ref. 52. Copyright Springer 2000. (b) A structural scheme for a typical activated carbon, which contains highly disordered macropores (>50 nm), mesopores (2–50 nm) and micropores (<2 nm). The micropores contribute to a large part of its surface area, but are only available for the adsorption of small molecules and may be easily jammed by large molecules. | ||
However, an inevitable drawback of having nano-sized particles is the separation and recycling problems after their use, which diverts some research into making numerous nano-sized pores in larger materials. With a well-designed nanoporous material, the material simultaneously possesses both a high surface area and a reasonably large particle size, which alleviate separation and recycling concerns to a large extent. Thus, nanoporous materials have found themselves numerous applications in water treatment as nanoporous adsorbents,64–66 nanoporous catalysts67 and nanoporous hosting substrates for nano-sized catalysts.68,69 Historically, activated carbon, whose surface area is generally between 500 and 1500 m2 g−1, is a conventional nanoporous material and has been widely utilized for water treatment in the past century.70 Its ultra-high surface area, along with its high stability in aqueous environments, makes it an important and versatile adsorbent for a variety of water pollutants. However, the major shortcomings of activated carbon are as follows: (1) its pore structure is disordered and (2) the majority of its pores is within the range of micropores (pores with size ≤2 nm) (Fig. 1b), which leads to its low adsorption capacity for large molecules, its sluggish mass transfer kinetics and long equilibration time. It is common for many activated carbons to take days or even weeks to reach their adsorption mass transfer equilibrium.70–76
Significant efforts were made in finding ordered porous silica-based nanostructures with a high surface area in the 1990s. In 1991, Dr Whitesides from Harvard University highlighted the concept of self-assembly, which is the key strategy for the synthesis of various nanostructures.77 In 1992, the Mobil company in the USA highlighted a liquid-crystal template mechanism, which further facilitated pore structure control and led to the creation of the well-known ordered mesoporous silica materials, MCM-41, MCM-48 and MCM-50 (commonly known as the MCM-41s), with well-ordered and uniform pore sizes of 2–8 nm.78 The creation of MCM-41s demonstrates a meaningful route in preparing ordered nanoporous structures and in controlling the pore diameters using different surfactants, adopting different hydrothermal/calcination treatment temperatures and adding micelle swelling agents. Compared to conventional activated carbon, MCM-41s have ordered pore structures, controllable pore sizes and accordant pore diameters. These advantages made controllable nano-synthesis a very attractive method for the further fabrication of new porous materials at that time. The field of pore size and pore structure control was fast moving. In 1993, the concept of cooperative self-assembly was put forward by Dr Stucky79 (Fig. 2).80 Five years later, a new branch of well-ordered mesoporous materials with larger pore diameters (7.5 to 32.0 nm) and thicker walls (3.1 to 6.4 nm) was fabricated at the University of California, Santa Barbara (UCSB), named SBA-15, using block-copolymers with large molecular weights as the structure-directing agents.81 The increase in the wall thickness increases the stability of mesoporous silica and the larger pores make the adsorption of large molecules feasible and at the same time dramatically increase the mass transfer kinetics within the pores.
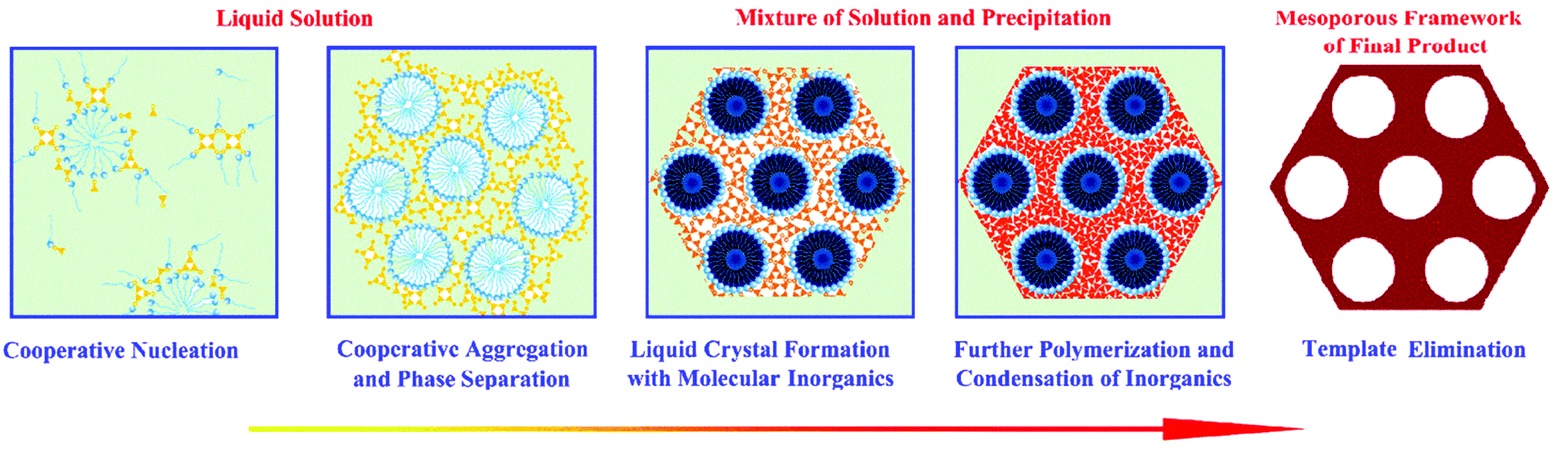 | ||
| Fig. 2 The formation of mesoporous material via a cooperative self-assembly strategy. In the first step, surfactants and inorganic species are attracted together due to interactions arising from hydrogen bonds or electrostatic attractions; in the second step, continuous hydrolysis and crosslinking of the inorganic species cause further cooperative aggregation and eventually lead to phase separation from the solution; in the third step, the surfactant-inorganic species form an ordered mesostructure to decrease the total interfacial energy; the fourth step involves further polymerization and condensation of inorganic species. Upon surfactant removal, the final mesoporous product is obtained. Reprinted with permission from ref. 80. Copyright American Chemical Society 2007. | ||
In 1999, Ryoo et al. first demonstrated the fabrication of a mesoporous carbon material, CMK-1, with an ordered and uniform mesoporosity by a nanocasting method.82 Later, a family of mesoporous carbon, named CMK-X (X = 1–9), was similarly fabricated using different mesoporous silica as hard templates and different precursors as the carbon source.83 The mesopores of these materials come from the removal of the silica templates and therefore correspond to the wall thickness of their mother templates, which is in the range of 1–4 nm.83 As expected, compared with conventional activated carbon, the mesoporous carbon showed dramatically enhanced adsorption kinetics and capacities.75 In one example, ordered mesoporous carbonaceous phenol–formaldehyde resins prepared by a nanocasting method exhibited adsorption capacities of 317.5 mg g−1 and 134.2 mg g−1 for fuchsin and aniline, respectively, with the adsorption equilibration time being less than 1 hour.84
However, the nanocasting synthesis of mesoporous carbon uses mesoporous silica as a template, which makes it a multi-step and material-intensive procedure. In 2004, Dai et al. reported a direct synthesis of ordered mesoporous carbon using a block-copolymer as a structure directing agent and resorcinol as a carbon precursor via a soft template method.85 Soon afterwards, Zhao et al. reported an improved direct synthesis strategy for mesoporous carbon, FDU-14, FDU-15 and FDU-16, using low molecular phenolic resin as a precursor and a Poloxamer-type block-copolymer as a structure directing agent (Fig. 3a).86–88 This method was then quickly extended to the synthesis of mesoporous carbon via an aqueous solution, which makes it suitable for large scale production.89 To date, the pore size of the FDU family of mesoporous carbon can be readily tuned from 12 to 37 nm, which opens up a lot of applications for them.90–93
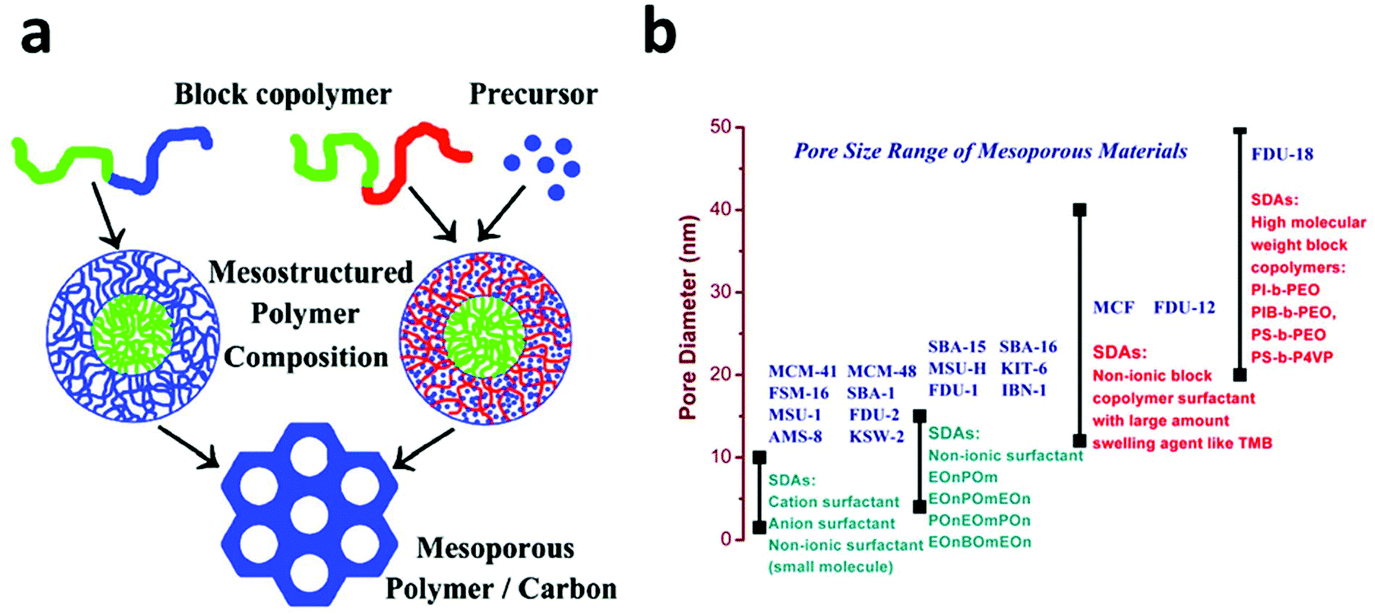 | ||
| Fig. 3 (a) Two strategies for the direct synthesis of mesoporous carbon using a block copolymer as a structure directing agent. In the first strategy (left), one block of the block copolymer is used as the carbon source and the other block is selectively removed to form uniform mesopores. In the second strategy (right), the carbon precursor is added separately and the entire block copolymer is removed after the carbonization process. Reprinted with permission from ref. 88. Copyright American Chemical Society 2008. (b) Classification of Mesoporous materials according to their pore size distribution and the surfactant used in their synthesis. | ||
The excellent progress with respect to the synthesis of mesoporous silica and carbon with controllable, uniform and large pore sizes (Fig. 3b) provides these materials with plenty of opportunities in water treatment. The removal of microcystins from water by adsorption is an insightful example. It is well known that microcystins are extremely toxic with a large molecular weight of about 1000 Da and a large size of 1–2 nm; they are usually produced in cyanobacterial blooms occurring in many eutrophic waters.94 Activated carbon has been adopted for the adsorption of microcystins and it is found that the small micropores of the activated carbon do not contribute to the adsorption due to the molecular sieve effect.95 In 2007, Zhao et al. employed a mesoporous silica with a pore size of 2.3 nm to adsorb microcystins and found that over 95.4% of microcystins was removed and adsorbed onto silica within 60 seconds, which dramatically contrasted with the very slow adsorption of microcystins on activated carbon.96 In 2013, the same group compared microcystins’ adsorption on a series of mesoporous silica with different pore sizes and pore structures and found that the mesoporous silica with larger mesopores (>6 nm) had a much higher adsorption capacity than those with small mesopores (2–3 nm). In one case, SBA-15 with a pore size of 8.7 nm, despite its smaller specific surface area (800 m2 g−1) exhibited a microcystin adsorption capacity six times that of MCM-41 with a pore size of 2.8 nm and a surface area of 1180 m2 g−1.97 This result convincingly demonstrates the necessity and effectiveness of pore size engineering. However, due to the weak surface interaction between microcystins and silica, the microcystins’ adsorption capacity on mesoporous silica is far from satisfactory. Recently, ordered mesoporous carbon was utilized and showed an improved performance for microcystin adsorption. An ordered mesoporous carbon with bimodal mesopores (2.8 and 5.8 nm), a surface area of 1680 m2 g−1 and a pore volume of 1.67 cm3 g−1 exhibited an unprecedented microcystin adsorption capacity of 526 mg g−1, which was 30 times that of the powder activated carbon tested under the same conditions.75
In the past decade, ultra-thin two dimensional (2D) solids, defined as crystals with very high aspect ratios and with thicknesses corresponding to a single or a few atomic layers, have attracted tremendous research attention98 due to their extremely high surface atom ratio along with other attractive features. In water treatment, the 2D materials are particularly appealing, mainly for the following reasons: (1) the ultra-thin layer structure of the 2D materials endows them with a high specific surface area and stable structures. For example, the surface area of monolayer graphene is around 2630 m2 g−1;99 (2) the edges and defect sites on the layers of the 2D materials can be conveniently functionalized; (3) when stacked together, they form a tight structure but with a controllable interlayer space, which is desirable for many design purposes;100,101 (4) in practice, few-layer-stacked 2D structure-based materials can be facilely synthesized and can be used to make various types of bulk materials such as membranes, powders and fibers.100–102 Among these, graphene, an atomic monolayer consisting of densely packed carbon atoms, along with its derivatives (especially graphene oxide (GO) and reduced graphene oxide (rGO)), are by far the most studied 2D materials.98 Due to its ultra-thin wall thickness, a graphene-based ultra-porosity sponge with an extremely high pore volume has proven very effective as an oil sorbent, removing as much as 69 times that of its own weight.102–104
In 2011, a new and exciting group of 2D materials composed of early transition metal carbides and/or carbonitrides, now known as MXenes, was introduced by Gogotsi's group105 and its family has been growing henceforth, along with its prospective applications.98 MXenes are produced by etching out the A layers from Mn+1AXn phases, where M is an early transition metal, A is mainly a group IIIA or IVA (i.e., group 13 or 14) element, X is C and/or N, and n = 1, 2, or 3.98,106 Their natural tendency to adsorb cations onto their surfaces and their high surface area endow MXenes with highly desirable properties in energy storage and water treatment areas.
3. Rational design of nano-adsorbents by surface chemistry engineering
Nanoporous, especially well-ordered mesoporous materials, are cut out to be excellent adsorbents, given their high surface areas, large and regularly ordered mesoscale channels, and fast mass transfer kinetics.75,96,97,107 However, a high and fast adsorption relies not only on a large surface area to provide active adsorption sites and unobstructed pathways for the adsorbates to quickly reach their adsorption sites, but also on the interaction between the active sites and the targeted adsorbates, which controls the strength and the selectivity of the adsorption.108,109 While the surfaces of carbon-based nanoporous materials are largely chemically inert and thus difficult to functionalize, the surfaces of silica materials are covered by silanol groups, which only provide weak interactions with polar adsorbates, leading to a limited adsorption capacity and unsatisfactory selectivity. However, given their well-known rich chemistry, silica surfaces and silanol groups are susceptible to a variety of chemical modifications, which is also due to the wide availability of various organosilane coupling reagents.108–110 Therefore, in parallel with pore size and pore structure engineering, surface chemical functionalization, especially of nanoporous silica based materials, is also a basis of many rational designs of nano-adsorbents for water pollutant removal.3.1 Charged species removal
Most of the functional ligand groups can be easily linked to the surface of mesoporous silica by grafting organosilanes with the corresponding terminal groups via –Si–O–Si– covalent bonds (Fig. 4a).108,113 The surface grafting can be achieved either by a post-grafting strategy or by a one-pot co-condensation strategy and both of these have been well developed in the last decades.114–116 One successful example is the mesoporous silica materials functionalized with thiol-based ligand groups (thiol, thiourea, and thioether) on their pore surfaces for the removal of Hg2+ by adsorption.109,112,117 These rationally designed adsorbents show extremely high capacity, fast kinetics, and high selectivity over common competing metal cations due to the specific complex chemistry between Hg2+ and thiol-based groups, in which the Hg to –SH molar ratio can be as high as 1.0. In 1997, Liu et al. synthesized a mesoporous silica material and grafted its pore surface with (methoxy)mercaptopropylsilane to achieve 76% coverage of the pore surface with the –SH groups (Fig. 4b), and the synthesized material exhibited a high Hg2+ adsorption capacity of 505 mg g−1.117 In addition to the post grafting method, also feasible is the introduction of the –SH group onto the pore surface of mesoporous silica by a one-pot co-condensation strategy with an even higher –SH group loading (4.1 mmol g−1), which led to an Hg2+ adsorption capacity of more than 800 mg g−1 in one case.118
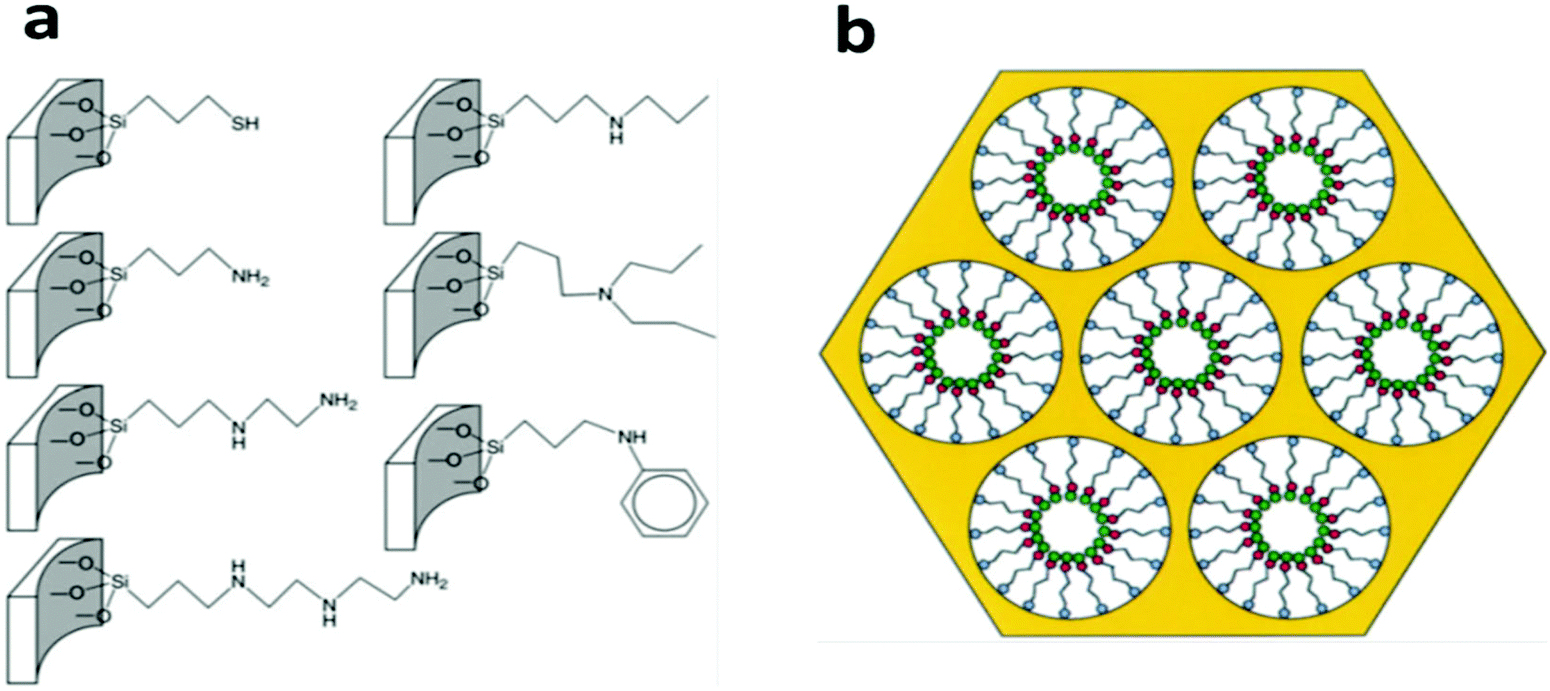 | ||
| Fig. 4 (a) Several commonly-used organo-functional groups grafted on the pore surface via Si–O–Si covalent bonds for the preparation of mesoporous silica-based adsorbents. Reprinted with permission from ref. 108. Copyright Royal Society of Chemistry 2010. (b) The scheme illustrates the structural model of self-assembled monolayers on a mesoporous supports (SAMMS) adsorbent with a monolayer functional group (red dots) on its mesopore surface, which shows efficient adsorption of the target metal ion pollutants (green dots). Reprinted with permission from ref. 117. Copyright American Association for the Advancement of Science 1997. | ||
In 2000, Kawi et al. compared thiol- and amino-functionalized SBA-15 silica for the adsorption of heavy metal ions and found that the thiol-functionalized SBA-15 showed high removal efficiency for Hg2+, but was much less successful for Cu2+, Zn2+, Cr3+ and Ni2+ due to the mismatch of coordination chemistry.119 On the other hand, amino-functionalized SBA-15 showed good removal efficiency for all ions except Hg2+. However, because the binding strength between amino-groups and these metal cations (Cu2+, Zn2+, Cr3+, Ni2+ and Hg2+) is much weaker than that between the thiol-group and Hg2+, amino-functionalized adsorbents only lead to a low capacity and poor selectivity in comparison to thiol-functionalized ones for Hg2+ removal. For example, the maximum adsorption capacities of Ni(II), Cd(II) and Pb(II) ions with a –NH2 group-functionalized mesoporous silica MCM-41 were only 12.4, 18.3 and 57.7 mg g−1, respectively.120 In an attempt to further increase the binding strength between the amino group and the metal ions, melamine-based dendrimer amines were utilized as the functional groups to modify SBA-15 and the functionalized SBA-15 exhibited adsorption capacities of 130, 126 and 98 mg g−1 for Pb(II), Cu(II) and Cd(II), respectively.121
In 2010, Tao et al. functionalized the surface of a mesoporous silica with a complex amino group (–CH2–CH2–O–CH2–CH(OH)–CH2–NH2), which was further reacted with Rhodamine B to form a complicated ligand, as shown in Fig. 5b.122 Interestingly, unlike the general amino-functionalized mesoporous silica, this material exhibited a high selectivity for Hg2+ from an aqueous matrix containing Na+, Mg2+, Mn2+, Co2+, Ni2+, Zn2+, Cd2+, Ag+, Pb2+, and Cu2+. Although the mechanism behind the high selectivity in this case is not fully understood, the results hint that beyond the simple groups as discussed above, the design of more complicated ligands may provide some unprecedented opportunity to achieve exclusive binding to target heavy metal cations of interest with high selectivity.
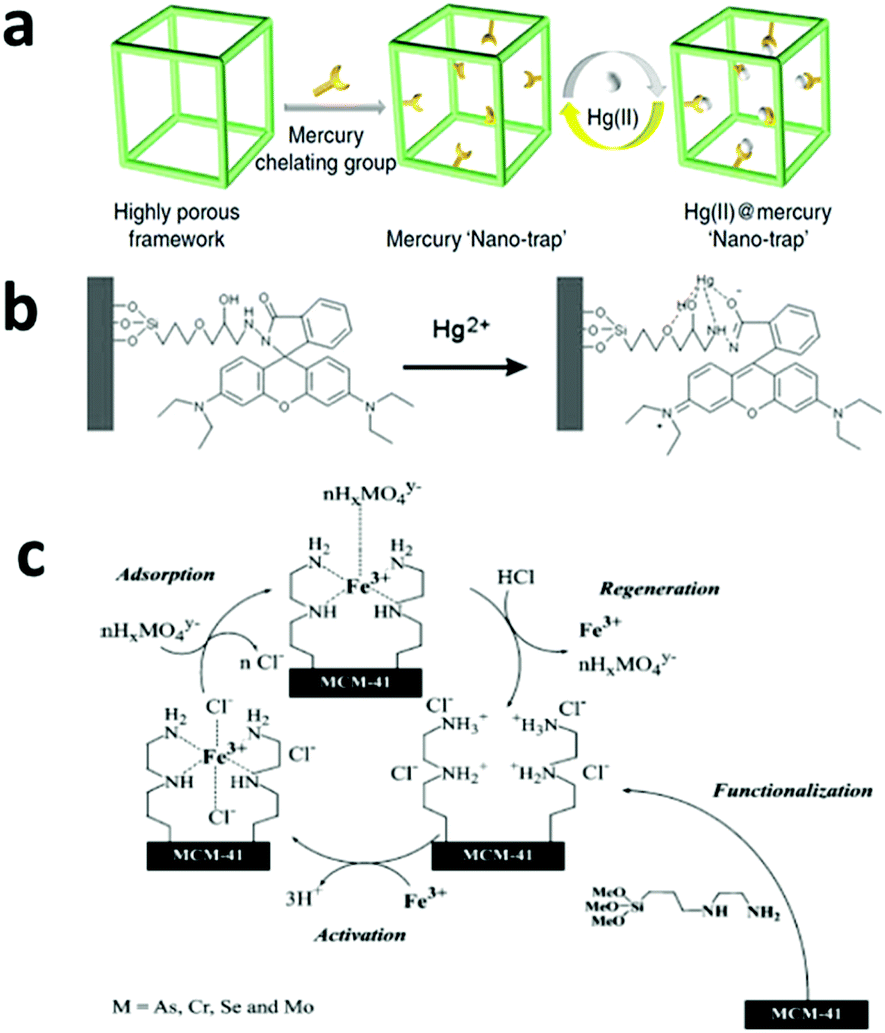 | ||
| Fig. 5 (a) A highly efficient adsorbent with a mercury nano trap is fabricated by linking the mercury chelating group –SH to a highly porous aromatic framework PAF-1, which has a Langmuir surface area of 7100 m2 g−1. The product shows a high adsorption capacity and can be easily regenerated for reuse. Reprinted with permission from ref. 123. Copyright Nature Publishing Group 2014. (b) A proposed model for the selective binding of Hg2+ with a specially designed ligand with a fluorescence group, which makes it an Hg2+ sensor as well as a selective adsorbent. Reprinted with permission from ref. 122. Copyright Royal Society of Chemistry 2010. (c) A mesoporous silica-based adsorbent with amino groups on its pore surface. After it chelates with Fe3+, the adsorbent shows high affinity with oxyanionic heavy metal pollutants and can be regenerated by HCl washing. Reprinted with permission from ref. 142. Copyright Elsevier Inc. 2004. | ||
One shortcoming of the silica-based materials is the poor stability of the –Si–O–Si– bond in basic conditions, which may cause leaching of the surface-grafted functional groups.123 Very recently, Li et al. reported a thiol-functionalized, porous, organic polymer-based nano-trap for selective Hg2+ removal and the material achieved an Hg2+ adsorption capacity of over 1000 mg g−1 along with a high selectivity and fast adsorption kinetics (Fig. 5a).123 More importantly, this material showed high stability in water under a wide pH range, which was attributed to its stable C–C bond, and it remained stable at high temperatures up to 270 °C. Other abundant and inexpensive polymers, such as polystyrene and polysaccharide, have also been tested in adsorption applications and showed good performances in the removal of heavy metals, organic dyes and other organic compounds.124,125
Metal oxides besides SiO2 were also examined for their potentials in cationic pollutant adsorption. For example, Dubey et al. reported manganese oxide nanoparticles as a good adsorbent for Hg2+.126 Afkhami et al. reported 2,4-dinitrophenylhydrazine-modified aluminum oxide nanoparticles, which showed adsorption capacities for Pb(II) at 100 mg g−1, Cd(II) at 83 mg g−1, and Co(II) at 41 mg g−1.127 Their adsorption ability can be ascribed to the electrostatic attraction between these metal oxide adsorbents and cationic heavy metal ions.128
Recently, 2D materials have been employed in adsorption. For example, Xie et al. reported a graphene oxide aerogel with a Cu2+ adsorption capacity of 19 mg g−1.129 Sampath et al. investigated the dye adsorption and found that cationic dyes showed higher adsorption onto exfoliated graphene oxide, whereas rGO favored anionic dyes.130 In 2014, Zhang et al. developed a large scale preparation method of MXenes and the produced MXenes were able to selectively reduce an aqueous Pb2+ concentration to lower than 2 ug L−1. In this study, the strong selectivity was attributed to the strong metal–ligand interaction between [Ti–O]−H+ groups and Pb(II), the large surface area of MXenes led by HF exfoliation and the lower hydration energy of Pb(II) compared to other cations such as Ca(II) and Mg(II), which leads to Pb(II) being more easily adsorbed onto an OH-rich surface.131 While the exact adsorption mechanism remains to be further explored and confirmed, MXenes have shown good adsorption performances for Cr and dyes.132,133
In 1992, Ramana et al. found that copper-chelated pyridyl and tertiary ammonium polymers exhibited a high affinity toward arsenate (AsO33−), due to the ultralow solubility of cupric arsenate.139 In 1999, Liu et al. designed a strategy for the adsorption of arsenate and chromate (CrO42−) using similar metal–chelated ligands as active sites,140 in which an ethylenediamine group was firstly grafted onto the surface of mesoporous silica and then chelated with Cu2+. This metalized adsorbent exhibited a high adsorption capacity of 142 mg g−1 for arsenate and 132 mg g−1 for chromate. It was proposed that in the course of adsorption, the target oxyanions, arsenate or chromate, would directly bind to the Cu2+ ions by releasing one of three ethylenediamine ligands that previously chelated with Cu2+ and oxyanions would thus be locked in the complex trap formed by Cu2+ and ethylenediamine ligands.
In 2003, Tatsumi's group investigated these types of metalized adsorbents in more detail using three different amino groups as the ligands, namely, –NH2 (1N), –NH–CH2–CH2–NH2 (2N) and –NH–CH2–CH2–NH–CH2–CH2–NH2 (3N), and Fe3+, Co2+, Ni2+, and Cu2 as the metal additives.141 It was found that Fe3+ exhibited the best performance due to the strong interaction between iron and arsenic (Fig. 5c). In case of MCM-48 as the matrix, the 2N group as the ligand and Fe3+ as the metal additive, one Fe3+ bound to about 2.7 arsenate anions and the final arsenic adsorption capacity was as high as 353 mg g−1. In 2004, the same group further demonstrated the high adsorption capacities of the same materials for chromate (115 mg g−1), selenate (116 mg g−1) and molybdate (206 mg g−1).142
As mentioned above, iron ions form very strong binding interactions with arsenate and chromate and therefore all types of iron-containing materials, such as zero-valent iron, iron oxides, iron oxyhydroxides and iron hydroxides, have been utilized for the removal of arsenic and chromium in the last two decades,134 and they are regarded as promising adsorbents due to their natural abundance, low cost and non-toxicity. In 2000, Lackovic et al. demonstrated that both As(III) and As(V) could be effectively removed from an aqueous solution by zero-valent iron due to surface precipitation or complexation of arsenic with iron.143 By making smaller and smaller iron-containing nanoparticles, the adsorption capacity with respect to arsenic has been greatly improved. In 2006, Yavuz et al. synthesized monodispersed Fe3O4 nanoparticles with diameters of 12 nm, which exhibited a high arsenic adsorption capacity of 200 mg g−1.22 Such a high capacity is mainly attributed to the high fraction of exposed iron on the surface of these ultra-small particles. However, the ultra-small particles have a strong tendency to agglomerate, which reduces their specific surface area and thus adsorption capacity.144 Several strategies have recently been developed to overcome this challenge. In 2009, Lo and Wang synthesized a mesoporous γ-Fe2O3 with a self-supported porous framework with a particle size larger than 200 nm and a pore wall thickness smaller than 10 nm, which exhibited a chromate adsorption capacity of 15.6 mg g−1.145 In 2014, a mesoporous cerium iron mixed oxide material was similarly synthesized and exhibited adsorption capacities of ∼106.2 and ∼75.4 mg g−1 for arsenate and chromate, respectively.146 In addition to making self-supported mesoporous frameworks, an alternative strategy was recently explored, which involved loading ultra-small iron nanoparticles onto a high surface nanoporous substrate to avoid particle aggregation. Zhao et al. loaded Fe2O3 nanoparticles with a size less than 10 nm on a mesoporous carbon matrix with a high loading capacity of 52 wt%, and the arsenic adsorption capacity of this composite material was 29.4 mg g−1.147 Yu et al. designed a structure in which ultra-small γ-Fe2O3 nanoparticles (6 nm) were well dispersed within a mesoporous silica foam with a pore size of around 100 nm and a pore volume of 1.6 cm3 g−1.148 The non-agglomeration of γ-Fe2O3 in the structure maintained a high level of active adsorption sites, leading to high adsorption capacities for As(III) at 320 mg g−1 and As(V) at 248 mg g−1. In addition, the large particle size and large pore size of the hosting silica foams ensured fast adsorption kinetics and made possible a direct packing of the materials into a filter cartridge for household drinking water treatment at an ambient pressure.148
Besides the strong interaction of Fe species with As and Cr oxyanions, some other strong interactions between metal oxide and anionic ions were also discovered and utilized.128 For example, it is now known that iron oxide, magnesium oxide, zirconium oxide and alumina all form strong interactions with fluoride, and the nanostructures of these metal oxides have been utilized for fluoride adsorption.128 Ahn et al. compared mesoporous alumina and activated alumina and concluded that a large surface area and mesopore size of the mesoporous alumina were desirable for a high fluoride adsorption capacity and fast adsorption kinetics.149 Chen et al. employed zirconium oxide nanoparticles for fluoride removal and achieved a capacity of 78 mg g−1.150 It has also been reported that zirconium phosphate (ZrP) can effectively remove trace fluoride from contaminated water.150 Zhang et al. designed a porous polystyrene-encapsulated zirconium phosphate nanocomposite and the material showed high selectivity towards fluoride in a matrix with high concentrations of SO42−, NO3−, and Cl−.151
3.2 Charge-neutral species removal
Hydrophobic organic compounds (HOCs), including polychlorinated biphenyls (PCBs), polycyclic aromatic hydrocarbons (PAHs) and hydrophobic pesticides, are one important category of water pollutants.152–154 Their primary removal strategy is based on the principle of like dissolves like by extraction with a hydrophobic medium. Activated carbons are the most widely applied adsorbents for HOC removal, but they suffer from a large fraction of micropores. Surfactant micelles have a capability of extracting HOCs from contaminated water by solubilizing HOCs within their very hydrophobic cores, but they cannot be directly separated from water.155–157In 1992, mesoporous silica was first synthesized via cooperative self-assembly between surfactants and silica79 and the as-made materials before calcination can be regarded as unconfined micelle arrays that are solidified by silica matrices via weak electrostatic interactions. In the late 1990s, Denoyel et al. demonstrated that these hybrid materials exhibit a high efficiency in removing various chlorophenols from an aqueous solution.158,159 However, the problem of surfactant leaching out of these hybrid materials during the adsorption and regeneration process inhibited their practical applications. In 2008, a study by UCSB rationally designed magnetic permanently confined micelle arrays (Mag-PCMAs) to overcome the surfactant leaching problem (Fig. 6a).160 In their design, a special surfactant, 3-(trimethoxysily) propyl-octadecyldimethylammonium chloride (TPODAC), which is able to form covalent bonds with silica frameworks, was utilized in a cooperative self-assembly and therefore the micelle arrays, once formed, were permanently solidified within the silica frameworks by covalent bonds. Due to the permanent confinement of the micelles, Mag-PCMAs can be easily regenerated by simple solvent washing without losing their HOC adsorption capacity and due to the magnetic core of this core–shell structured nanomaterial, Mag-PCMAs work well in ex situ soil washing as well. Some co-workers later applied a micelle-swelling strategy during the material synthesis (Fig. 6b) and created extra space inside the permanently confined micelles, which led to an increase in the HOC adsorption capacity by as much as 3.5 times.161 Cai et al. synthesized a similar core–shell magnetic mesoporous silica adsorbent by a two-step method and the final product exhibited a good performance with respect to PAH removal.162
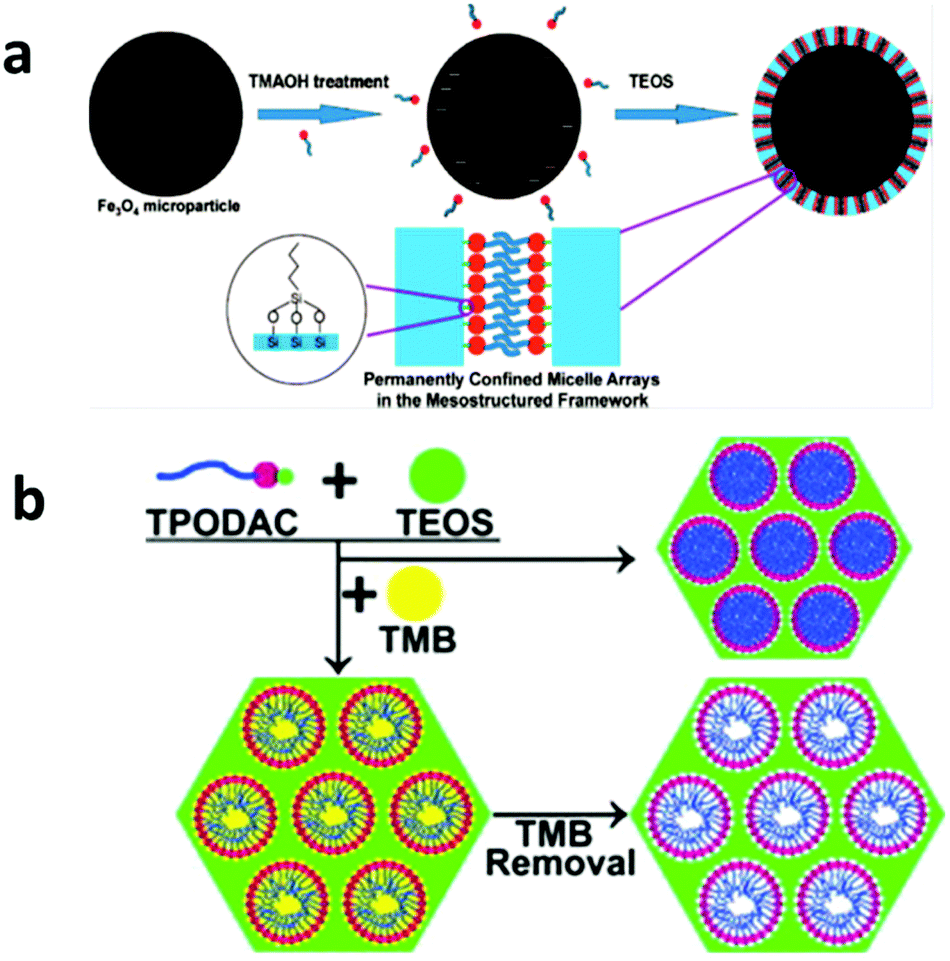 | ||
| Fig. 6 (a) The synthesis procedure for Mag-PCMAs, which is designed for the removal of HOCs. The mesostructured hybrid shell layer is constructed of a mesoporous silica framework with the micelle arrays formed by special surfactants with an active –Si(OCH3)3 end group inside the mesopores. In this material, the surfactant micelles are permanently anchored in the silica framework via a Si–O–Si bond, which solves the surfactant leaching problem. Reprinted with permission from ref. 160. Copyright American Chemical Society 2008. (b) By using the micelle swelling agent trimethylbenzene (TMB) in the synthesis of PCMAs and removing TMB after the synthesis, additional cavities can be created inside the confined micelle arrays, thus significantly increasing the HOC removal efficiency. Reprinted with permission from ref. 161. Copyright Elsevier Inc. 2012. | ||
Compared with many good material designs for HOC removal, the adsorption of charge-neutral hydrophilic pollutants with environmental significance, such as many of pharmaceuticals and personal care products (PPCPs) and dissolved natural organic matter (NOM), is always challenging and problematic. A few attempts were made using carbon-based materials, such as activated carbon and ordered mesoporous carbon, and some of these materials showed a good performance towards PPCP adsorption.163–165 However, these carbon materials are nonselective and versatile adsorbents and thus their application to this end is not within the rational design domain. The sluggish progress in hydrophilic water pollutant adsorption is not due to lack of effort, but the intrinsic hydrophilicity of these compounds makes them reluctant to leave water. Based on the principle of like dissolves like, good adsorbents for charge-neutral hydrophilic pollutants can be materials whose hydrophilicity is such that it maximizes its differences with water but at the same time minimizes its difference with the pollutants of interest, which however has not yet been confirmed experimentally. Molecular recognition-based adsorption has shown some promise with respect to the removal of this group of water pollutants, but it is still too early to draw any definite conclusions.
3.3 Molecular recognition based adsorption
Adsorption based on molecular recognition has gained some attention in the past decade, with aptamer and molecular imprinting approaches being two good examples.166–168 The commonality of all molecular recognition approaches lies in the use of the target pollutant species as a template to select or create the adsorbent that has the capability to precisely recognize and, more importantly, select the target from a wide range of species even where similar structures are present.Aptamer is a recently developed type of new ligand that can exhibit highly specific and strong affinity to the target molecule with multiple interaction points from three-dimensional directions.169,170 For a specific target molecule, a special aptamer can be selected exclusively for the target from a huge library of DNA molecules containing randomly created sequences via the systematic evolution of ligands by exponential enrichment (SELEX), also known as in vitro selection.171,172 This strategy has been adopted in various research areas, such as nanomaterials synthesis and sensor development, and has recently been utilized in water treatment. In 2009, Kim et al. selected eight aptamers from a random DNA library for arsenic (As(V) and As(III)) binding via SELEX171,172 and the selected aptamers showed extremely high affinity to both As(V) and As(III) with nanomolar scale dissociation constants (Kd) of 4.95 and 7.05 nM, respectively.173 When immobilized on streptavidin agarose resin, the aptamers removed almost all the arsenic from contaminated natural water from Vietnam with an excellent selectivity to arsenic. In 2011, Zhou et al. utilized the same strategy for the removal of trace (ng L−1) quantities of cocaine and diclofenac from drinking water and obtained a removal efficiency as high as 88%–95% (Fig. 7a).174
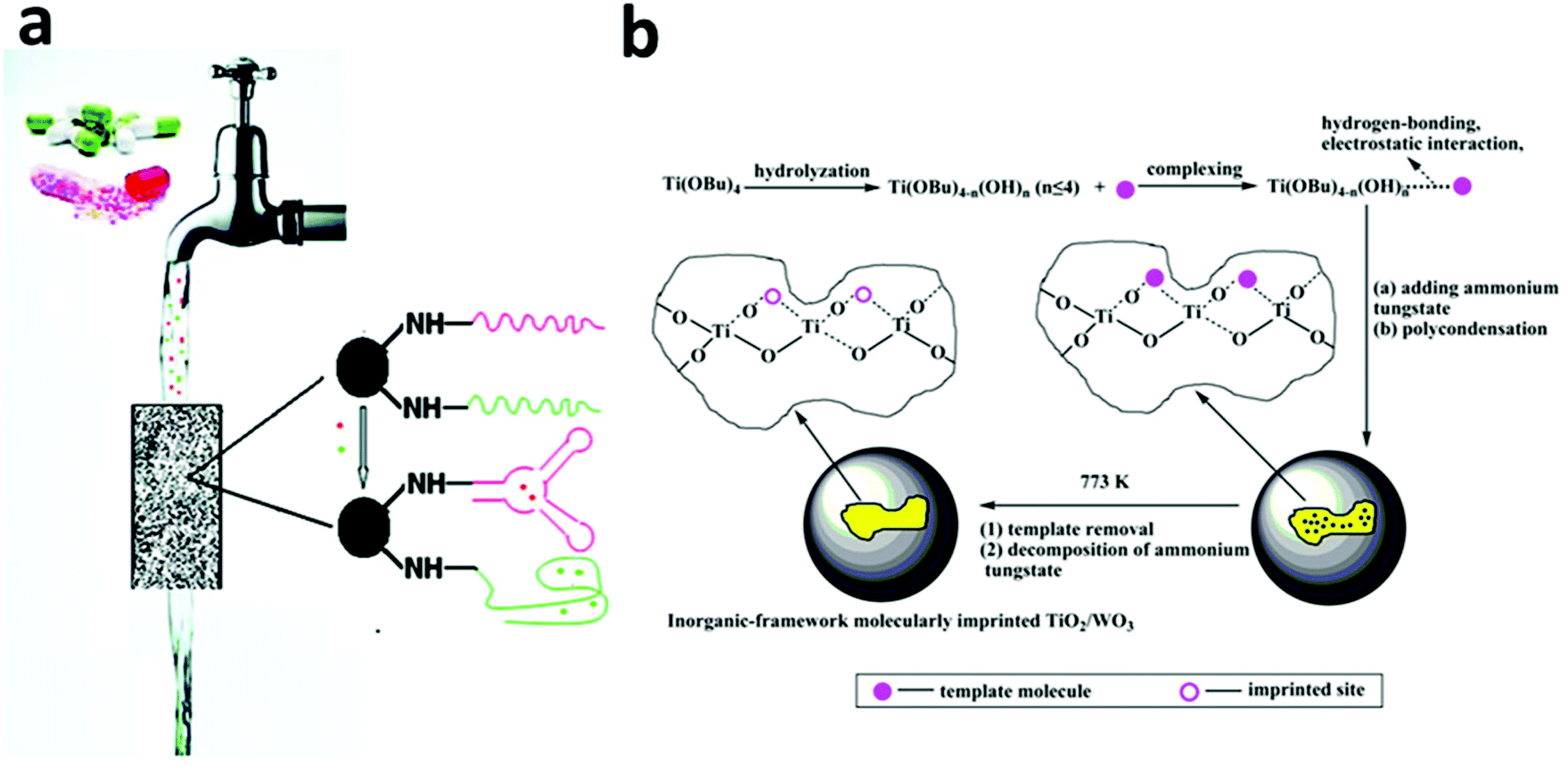 | ||
| Fig. 7 (a) A column packed with adsorbents functionalized with a selected aptamer showed the ability to removal trace amounts (ng L−1) of pharmaceuticals (e.g. cocaine and diclofenac) in drinking water with 88%–95% removal efficiency. Reprinted with permission from ref. 174. Copyright American Chemical Society 2011. (b) The synthesis procedure of an inorganic-framework molecular imprinted TiO2/WO3 nanocomposite. The synthesized product showed a selective and high photo-degradation rate for 2-nitrophenol (2NP) or 4-nitrophenol (4NP), depending on the molecular template used in the synthesis process. Reprinted with permission from ref. 110. Copyright American Chemical Society 2013. | ||
Based on the lock-and-key mechanism used by enzymes for substrate recognition, molecular imprinting is concerned with employing a target molecule as a template to create template-shaped cavities in a matrix, generally polymeric and in some cases inorganic, with a memory of the template molecule.166–168 Similar to the aptamer-based strategy, the molecular imprinting strategy has high selectivity and affinity towards the template molecule even in the presence of interfering substances that may be thousands to millions of times more abundant than the target.166,168 One significant study on molecular imprinting for water treatment concerns the development of core–shell structured nanocomposites, consisting of a magnetic-nanoparticle core and a molecular-imprinted-polymer shell, for selective adsorption of water pollutants. Li et al. reported a synthesis of a core–shell magnetic molecular imprinted polymer by surface RAFT polymerization for the fast and selective detection and removal of endocrine-disrupting chemicals, such as Bisphenol A, from aqueous solutions.175,176 Other nanocomposites with similar magnetic core–shell structures were developed for the selective removal of herbicides from water,177 creatinine, albumin, and lysozyme from urine;178 4-chlorophenol from water;179 and methyl parathion from a soil solution.180
Recently, the concept of molecular imprinting has been extended to the field of photocatalysis to overcome a general non-selectivity issue, especially in TiO2 based photocatalysis. In 2007, Tang and Zhu coated a molecular imprinted polymer on the surface of a TiO2-type photocatalyst, P25, and identified the selective decomposition of target pollutants in the presence of high-level interfering substances.181 For example, in the case of 4-chlorophenol (4CP) being used as the template, the molecular-imprinted-polymer-coated-TiO2 showed a much faster rate of decomposition for 4CP than that of phenol (k4CP/kphenol = 20.6), whereas for the original P25, k4CP/kphenol was only 8.68. The same group also extended this method to 2-nitrophenol, 4-nitrophenol and salicylic acid and achieved high activity and selectivity toward the photo-degradation of the targets.182,183 The effectiveness of this strategy has also been proven by other groups on the selective decomposition of 2-nitrophenol and 4-nitrophenol using inorganic-framework molecularly imprinted TiO2/WO3 nanocomposites as photocatalysts (Fig. 7b).110
4. Rational design of nano-assisted oxidation and reduction processes
4.1 Nano-assisted advanced oxidation processes
Chemical processes account for a significant fraction of conventional water treatment technologies, among which advanced oxidation processes (AOPs) have been playing an important role in wastewater treatment especially in developed countries and particularly for organic pollutant decomposition since its inception in the late 19th century.6–9,184,185 In a Fenton reaction, a typical AOP, peroxides (usually H2O2) react with iron ions to form highly reactive hydroxyl radicals (OH˙), as described in eqn (1) and (2).186 OH˙ is one of the most powerful oxidants known to us and its oxidation potential (E = 2.80 V) is even higher than those of atomic oxygen (2.42 V) and ozone (2.07 V),187 thus it is capable of oxidizing most organic pollutants present in wastewater.184| Fe2+ + H2O2–Fe3+ + OH˙ + OH− k = 63 M−1 s−1 | (1) |
| Fe3+ + H2O2–Fe2+ + HO2˙ + H+ k = 0.002–0.01 M−1 s−1 | (2) |
A conventional Fenton reaction is carried out in a homogeneous catalysis system whose advantages include readily available iron ions, negligible mass transfer limitations, and thus high reaction efficiency.186,188–190 However, the conventional homogeneous Fenton reaction has to take place in an acidic aqueous solution with a pH that is generally lower than 3 to optimize the performance and to avoid losing iron ions by precipitation186 (Fig. 8a and b). The acidification of the entire bulk water before the reaction and its adjustment back to neutral pH after the reaction adds to the operation cost of the conventional Fenton system.
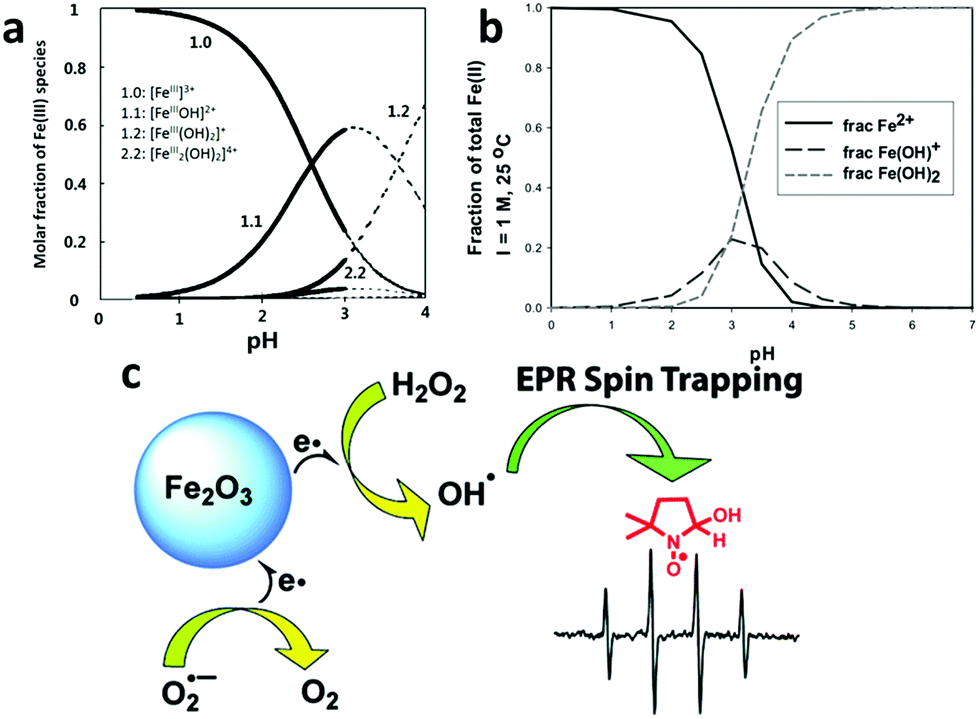 | ||
| Fig. 8 (a,191 b192) The speciation of different Fe3+ and Fe2+ species in an acidic aqueous solution at different pHs, showing that precipitation occurs when the pH is higher than 4. Reprinted with permission from ref. 191, Copyright Elsevier Inc. 1999 and reprinted with permission from ref. 192, Copyright Royal Society of Chemistry 1968, respectively. (c) A newly proposed mechanism for the heterogeneous Fenton reaction. The comparative spin-trapping EPR experiments on a γ-Fe2O3 nanoparticle catalyst showed that the free radical production should mainly be attributed to the surface iron ions rather than those dissolved metal ions released by the nanoparticles as previously thought. Reprinted with permission from ref. 198 Copyright American Chemical Society 2010. | ||
Beginning in the mid-1990s, researchers started to develop heterogeneous catalysts for the Fenton reaction using solid iron-containing compounds as the catalysts.193–195 The advantages of the heterogeneous systems are obvious: (1) drastic alleviation of the problem of Fe(OH)3 precipitation as few iron ions are present in the aqueous phase, (2) easy separation of the catalyst after application; (3) a broadened pH range wherein the Fenton reaction can take place.196,197 Moreover, Smirnova et al. investigated the free radical generation rate using a spin-trapping electron paramagnetic resonance (EPR) technique and found that at least 50-fold more OH˙ free radicals were generated on the iron-oxide surface than by dissolved Fe3+ in a homogeneous Fenton system (Fig. 8c).198
The past decade has experienced great progress in the rational design of nanoparticle-based Fenton catalysts (e.g., Fe3O4, α-FeOOH, α-Fe2O3, and γ-Fe2O3), assisted by a steadily deepening understanding of the Fenton reaction mechanisms. For example, it is known that in a homogeneous Fenton reaction, the Fe2+ ion produces hydroxyl radicals much faster than the Fe3+ species.186 Guided by this knowledge, in 2008, Guimarães et al. applied hydrogen thermal reduction treatment to α-FeOOH to induce Fe(II) on the material surface, which was found to significantly increase the degradation rate of quinolone.199 Compared to other iron-containing compounds, magnetite (i.e., Fe3O4) has recently attracted a considerable amount of interest because it is one of the most abundant iron oxides with Fe(II) in its crystal structure and it is still quite stable in air.200 In addition, Fe(II) in Fe3O4 is located in the octahedral sites of the spinel phase crystal and therefore exhibits high surface exposure, which is expected to be an advantage for the Fenton reaction. Many published results have proven that magnetite gives a better performance than other iron-containing substances.200
In efforts to break the conventional Fenton reaction efficiency limit, many other nano-assisted processes have been rationally combined with the Fenton reaction and have resulted in a number of successful Fenton variants (e.g., photo–Fenton reaction,201–205 electro–Fenton reaction,206–208 and “Fenton-like” reaction196,209,210). However, due to the space limitation, these topics are not covered in this review and interested readers should direct their attention to AOP or Fenton-focused review articles.184,186,206,211,212
4.2 Nano-assisted TiO2 based photocatalysis
Photocatalysis-based water treatment processes, based on oxidization much more than reduction, have long been studied. Among all semiconductor catalysts, TiO2 has distinguished itself majorly due to the fact that TiO2 is by far the most photostable photocatalyst in an aqueous environment.213 However, its performance under sunlight is limited by two key bottlenecks. The first one comes from its wide band gap nature (3.0 eV for rutile and 3.2 eV for anatase) and thus it is only responsive to ultraviolet (UV) light, which represents only 5% of the total solar spectrum.19,214 The second one is the ultra-fast recombination rate of photo-generated electron–hole pairs within this catalyst, which decreases the quantum efficiency.19,214,215 Research efforts concerning TiO2 based water treatments have been mainly concentrated on overcoming these two bottlenecks within the last two decades.In an attempt to expand the TiO2 light responsive range, hetero-element doping (e.g. N, F and C) has been widely employed and has been successful in increasing TiO2's adsorption of visible light and in inducing TiO2 to show certain levels of photocatalytic activity within the visible-light range.19,216 However, the stability of hetero-element-doped TiO2 is generally decreased and its performance under the entire solar spectrum, including UV and visible light, does not usually show a significant improvement in most cases. This is mainly ascribed to the fact that the incorporation of these foreign impurities (e.g. N, F and C) is inevitably accompanied by a significantly increased number of electron–hole recombination centers in the material, which thus decreases the quantum efficiency.19,216
Recently, the hetero-element doping strategy has been gradually giving way to so-called self-doping by Ti3+via various in situ reduction methods (e.g. H2 reduction, NaBH4 and electrochemical reduction).217–221 In 2011, Mao et al. demonstrated that reducing the TiO2 surface layer by H2 treatment could significantly shift the band gap of TiO2 from 3.3 eV in their case to 1.54 eV, which led to the final TiO2 product having a black color (Fig. 9a).222 Black TiO2 was proven to be an effective photocatalyst for decomposing organic water pollutants. Recently, many types of “colorful” TiO2 materials, which indicated their visible light responsiveness, were fabricated by various reduction approaches and their enhanced photocatalytic activities were widely reported.223,224 For example, using an aluminum-mediated reduction method, Xie et al. synthesized gray TiO2, which exhibited visible-light and even IR absorption with high photocatalytic activity toward organic pollutant degradation.225
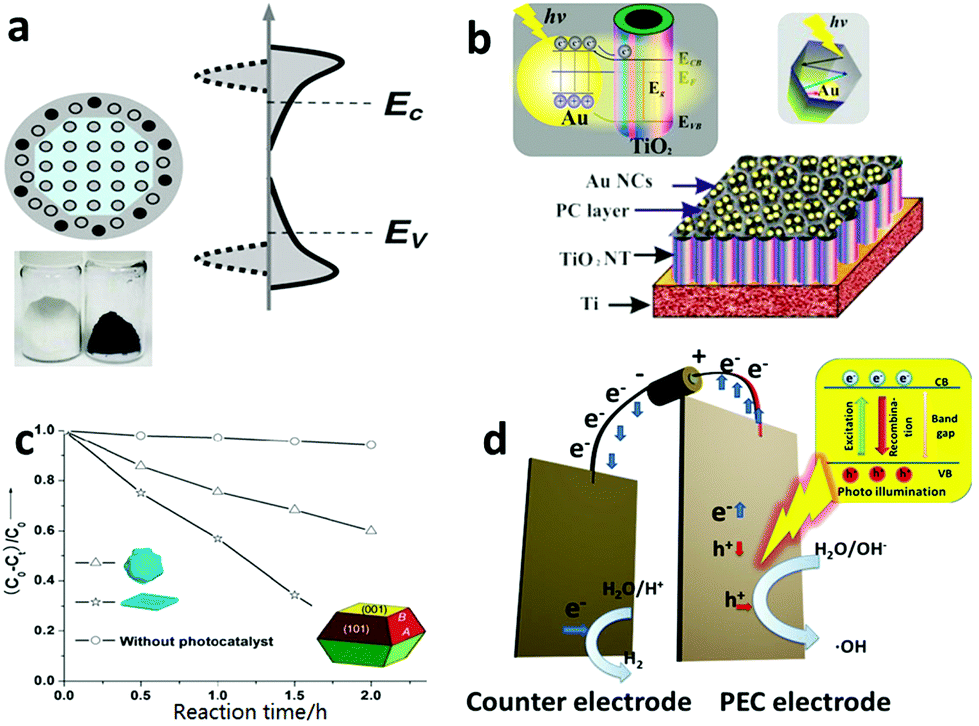 | ||
| Fig. 9 (a) The crystal structure, photo image and electronic density of states (DOS) of hydrogen reduced black TiO2. Its band gap is greatly narrowed because the crystal structure disorder leads to broadened tails of states, extending into the forbidden band gap. Reprinted with permission from ref. 222. Copyright American Association for the Advancement of Science 2011. (b) The structure scheme and energy band structure of a plasmonic gold nanocrystal-decorated TiO2 nanotube array. The gold nanocrystals can capture visible light to produce hot electrons via an SPR process, inject them into a TiO2 nanotube and thus endow TiO2 with a visible light spectrum response. Reprinted with permission from ref. 227. Copyright American Chemistry Society 2013. (c) A unique TiO2 single crystal material with controllable exposure of high active {001} crystal planes. The TiO2 catalyst with a higher percentage of {001} planes showed higher catalytic activity in the photodecomposition of 4-chlorophenol. Reprinted with permission from ref. 241. Copyright Royal Society of Chemistry 2009. (d) Schematic structural view of a PCE cell, in which an external bias can be applied to the electrode to vary the energy level of the electrons to meet the demands of a specific reaction. | ||
In addition to the doping strategy, to rationally engineer the band structure of TiO2, TiO2 hetero-junctions (e.g. P–N junctions, noble-metal–TiO2 junctions and Schottky junctions) with other visible light photoactive nanomaterials are also a popular options in the same line.226–228 In 2013, Wang et al. designed and fabricated a gold (Au) nanocrystal–TiO2 nanotube array nanocomposite material, in which the Au nanocrystals were responsive to visible light via surface plasmonic resonance (SPR) and injected the photo-generated hot-electrons into the conduction band of the adjacent TiO2, thus letting visible light contribute to the photocatalysis reaction (Fig. 9b). The shape and size of the Au nanocrystals could be rationally designed and synthesized to engineer their SPR wavelength to make an optimal match with the TiO2 substrate to maximize the photocatalytic performance of the nanocomposite material.227 Besides acting as an antenna to capture visible light energy, the second phase material of these hetero-junctions may benefit fast electron–hole separation by design and thus increase the quantum efficiency. Wang et al. designed a palladium (Pd) and TiO2 Schottky junction, which considerably reduced the recombination of photogenerated electrons and holes, promoted electron transfer and ultimately led to an improved performance with respect to photocatalytic oxidation of organic water pollutants.229
Because of a general mismatch between the optical path length required for light absorption (at least 1 μm for 90% light absorption) and the optimal charge diffusion length (usually 70 nm for minority carrier: hole) in TiO2, it has inherently fast photoelectron–hole recombination in its particle form.230–232 As it allows for optimization of the optical path length and the charge diffusion length relatively independently, one-dimensional nanostructures of TiO2, such as nanotubes (NTs), nanorods and nanowire, are rational solutions to the mismatch issue in the TiO2 particle form.19,206 Over the past decade, some reliable and well-established synthesis methods for one-dimensional TiO2 have been developed, including hydrothermal233,234 and solvothermal syntheses235,236 and anodization.237,238 Wang et al. reported a facile two-step anodization method that was able to produce a hierarchical TiO2 nanotube array with a well-controlled surface morphology, which achieved a record-high photoactivity in the category of a pure and unmodified TiO2 material.215
A highly reactive crystal plane is another essential factor for enhancing the photocatalytic performance. In 2005, Selloni et al. studied the crystal plane catalytic reactivity through investigation of methanol adsorption using density functional theory (DFT) calculations and first principles molecular dynamics (MD) simulations.239 Their results theoretically proved the high reactivity of the TiO2 {001} crystal plane.240 However, the unsettling reality is that the {101}, rather than the {001}, crystal plane is usually the most dominant one in natural and regular TiO2 materials. This is so because compared with the {101} crystal plane whose average surface energy is only 0.44 J m−2, the high average surface energy of the {001} crystal plane (0.9 J m−2) makes it thermodynamically unfavorable and thus during the crystal growing process, the {001} crystal plane diminishes quickly to lower the total system energy.241 In 2008, Lu and coworkers first reported a uniformly large percentage of {001} crystalline (47%) formation using hydrofluoric acid as a morphology control agent.242 In 2009, Yu et al. reported a microwave-assisted method of synthesizing micro-sheet anatase TiO2 with an 80% level of the reactive {001} plane, which showed a significantly higher 4-chlorophenol degradation performance than the {101} crystal plane dominated TiO2 (Fig. 9c).241
Although powder-TiO2-based photocatalysis has been mainstream in water treatment,243–245 TiO2-based photoelectrocatalysis (PEC), in which the photocatalyst is made/deposited on an electrode, has gained tremendous attention. Powder TiO2 generally has a higher specific surface area and thus a higher level of interaction with the target pollutant in water than the PEC based system. However, the advantages of the PEC-based electrode system over the powder-based one should not be underplayed.41,246–249 As in a TiO2 PEC system, the photogenerated electrons go through an external circuit before being ultimately accepted by electron acceptors on the cathode side. Within the external circuit, an external bias can be applied to vary the energy level of the electrons so to induce some reactions that may not be possible within the TiO2 powder based system (Fig. 9d).250,251 One example is that pure TiO2 powder, i.e., unmodified with co-catalyst, is not able to reduce water to produce H2 gas due to the high activation energy barrier of hydrogen evolution, even though the TiO2's photoelectron energy level lies below the hydrogen evolution level. With a PEC system, an applied external bias can easily induce the electrons to jump over the activation energy barrier of the same reaction to split water to generate both H2 and O2.215,227,252–254
4.3 Nano-assisted reduction processes
Despite the dominance of oxidation processes in water treatment, there are places wherein reduction-based water treatment is essential. For example, the reductive dechlorination of legacy chlorinated groundwater pollutants, especially TCE, by NZVI is an important process for eliminating the environmental impact of these human carcinogens from water resources. NZVI in the reaction acts as a reductant.56–63,255–257Given the high water solubility and low adsorptive capacity of such water pollutants as oxyanions (e.g. NO3−, ClO4− and BO3−), nitrosamines and PPCPs, conventional water treatment approaches suffer from their own limitations. The development of highly efficient heterogeneous chemical catalysts for the reduction of these compounds has now become a more popular strategy.258 Nanoscale Pd-based catalysts possess high activities in lots of reduction reactions and have found wide applications in the reductive transformation of many water pollutants. In the mid-1990s, Abu-Omar and Espenson found that a rhenium(V) complex could serve as an efficient catalyst for the reduction of perchlorate to chlorate, using hypophosphorous acid (H3PO2) as a reducing agent.239,259 Although their method is being continuously improved,260 such a homogeneous catalysis system with soluble reducing agents is not suitable for water purification systems. In 2007, Shapley et al. developed the first oxorhenium(VII)-based heterogeneous catalyst for perchlorate reduction using carbon as a support along with Pd metal particles (Re–Pd/C),261 which promoted the complete reduction of perchlorate to chloride using hydrogen as the reducing agent under acidic conditions (pH < 3). In 2009, Shapley et al. further reported that the presence of substituted pyridine ligands was able to greatly improve the activity and stability of this Re–Pd/C catalyst.262 However, in this system, the rhenium species were immobilized in the activated carbon support only by electrostatic interactions, which led to a rhenium leaching problem.263 In 2013, Choi et al. reported a Pd on N-doped activated carbon (Pd/N-AC) system for perchlorate reduction, in which the N-doped carbon surface provided adsorption sites for perchlorate due to the basic nitrogen functional groups on the surface and the supported Pd clusters acted as the catalyst for perchlorate reduction in a hydrogen atmosphere (Fig. 10a).264 The adsorption of perchlorate on this N-doped carbon support (3.67 mg g−1) was believed to be a key process for this method. However, the low adsorption capacity and low ion selectivity of the activated carbon necessitates frequent regeneration of the adsorbents. In 2014, Strathmann and Werth along with coworkers designed ultra-small Pd clusters (<2 nm) within ion-exchange resin as an adsorption/catalysis bi-functional material for perchlorate reduction. In this system, a ClO4− selective ion-exchange resin was adopted to replace the activated carbon support due to its high adsorption capacity (∼200 mg g−1) and selectivity, which significantly improved the ClO4− reduction efficiency and the catalyst reusability.265
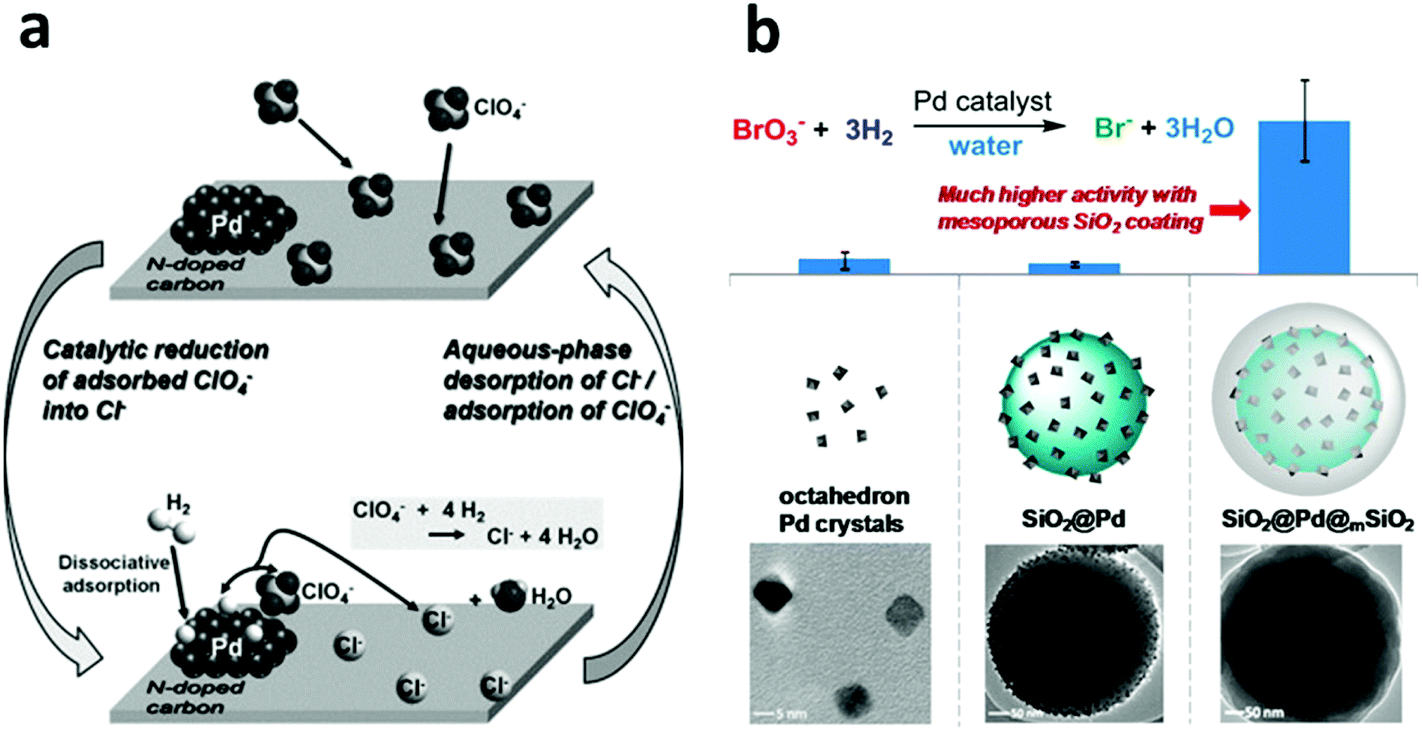 | ||
| Fig. 10 (a) The perchlorate adsorption–reduction cycle on a Pd/N-doped activated carbon (Pd/N-AC) catalyst. The N-doped carbon provides ample adsorption sites for perchlorate and thus benefits the subsequent catalytic reduction by Pd clusters in a hydrogen atmosphere. Reprinted with permission from ref. 264. Copyright Elsevier Inc. 2013. (b) A core–shell structured catalyst, composed of a Pd-nanoparticle-decorated SiO2 nanosphere core and a mesoporous silica shell, exhibits much higher activity in the reduction of bromate. Reprinted with permission from ref. 69. Copyright American Chemical Society 2014. | ||
Furthermore, In 2011, Reinhard et al. used bimetallic palladium–indium (Pd–In) nanoparticles supported on alumina for the reduction of N-nitrosodimethylamine (NDMA) and found that indium served as a promoter metal.266 In 2013, Werth et al. examined the activity of Pd nanoparticles in the catalytic reduction of nitrite (NO2−), NDMA, and diatrizoate as a function of the Pd crystal plane.267 However, the ultra-small size of the Pd nanoparticles is not conducible to their practical application due to the particle aggregation problem and the difficulties associated with catalyst separation and recovery. In 2014, Strathmann and Werth, along with coworkers, reported a core–shell-structured catalyst with encapsulated Pd nanoparticles for the reduction of bromate (BrO3−) using H2 as a reducing agent at room temperature.69 The Pd nanoparticles with a size of 6 nm were firstly attached to the surface of the solid silica nanospheres, which were then further encapsulated with an ordered mesoporous silica shell with a 2.3 nm pore size. The mesoporous shell provided a physical barrier to prevent Pd from leaching and aggregation and at the same time, due to its ordered porous structure, it ensured the accessibility of the Pd nanoparticles by the reactants. It was, although surprising, quite interesting that the mesoporous silica shell could promote BrO3− adsorption near the Pd active sites and thus greatly enhance the catalytic activity by a factor of 10, compared to a catalyst that was otherwise the same but without the mesoporous silica shell (Fig. 10b). The dual functions of the mesoporous shell, enhancing the Pd catalyst activity and preventing aggregation of the active nanoparticles, suggest a general and promising strategy of using metal nanoparticle catalysts for water treatment and/or other relevant aqueous-phase applications.
5. Rational design of nano-assisted membrane based separation
Over the past decade, nanomaterials have set foot in almost all areas of conventional membrane based separations, including microfiltration (MF), nanofiltration (NF), ultrafiltration (UF), membrane distillation (MD), forward osmosis (FO) and reverse osmosis (RO), and many nano-assisted membrane processes have recorded a significant progress.11,268–278 This section discusses the rational design of nano-assisted membrane processes by focusing majorly on RO and FO, along with emerging next-generation inorganic membranes and active membrane filtration.5.1 Nano-assisted RO and FO performance enhancement
In general, the modern FO and RO membranes share commonality in structural configuration, both consisting of a thin, dense active layer, supported on a thick microporous support layer.270,271,279–281 The support layer does not contribute to the salt rejection and it is the active layer wherein the separation occurs. The dense, active-layer backbone material in the modern RO and FO membranes is dominated by polyamide due to its high salt rejection.271,279,281,282 The major concern in RO nowadays is with its energy consumption caused by high operation pressure, which is in turn forced by the need for high water flux.270,272,274 Consequently, research into nano-assisted RO membranes has been focused majorly on increasing the water flux by blending selected nanomaterials (e.g. zeolite, silica nanoparticles, CNTs and aquaporin protein) in polyamide-based active layers.283–287 The essence of the nanomaterial blending strategy lies in the preferential water channels created by placing the selected nanomaterials in a polyamide matrix.11,272Inspired by the superior molecular sieving effect in zeolite, in 2007, Hoek et al. fabricated a zeolite blended polyamide active layer on top of a polysulfone support (Fig. 11a), which exhibited a water flux twice that of the non-modified membrane under an optimized zeolite particle loading without decreasing the salt rejection rate.286 In 2001, Hummer et al. reported a MD simulation result that showed water molecules were able to rapidly move through carbon nanotubes with a pulse-like transmission behavior288 and in 2003, their MD simulation result further showed that water molecules may flow through membranes of open-ended CNTs under an osmotic gradient in an almost friction-less manner and the resultant water flow rate was comparable to those measured for biological water channels such as aquaporin.289 In 2011, Zhang et al. synthesized a functionalized multi-walled carbon nanotubes (MWCNTs)-incorporated polyamide-based active layer in a RO membrane, which exhibited a significantly increased water flux, from 26 L m−2 h−1 (LMH) with the original membrane to 71 LMH with the MWCNTs-blended membrane.290 Due to the hydrophilicity and thermal stability of silica, in 2008, Singh et al. blended silica nanoparticles (16 nm and 3 nm) into a polyamide active layer in a RO membrane and reported a water flux increase of 200% with the same level of salt rejection under an optimal nanoparticle loading (21.3 vs. 9.0 LMH with and without the silica blending).291 Later, Deng et al. incorporated mesoporous silica MCM-41 into a polyamide active layer and recorded a water flux of 46.6 LMH in comparison to 28.5 LMH without MCM-41. They suggested that the internal pores of MCM-41 contributed significantly to the increase in water permeability.292
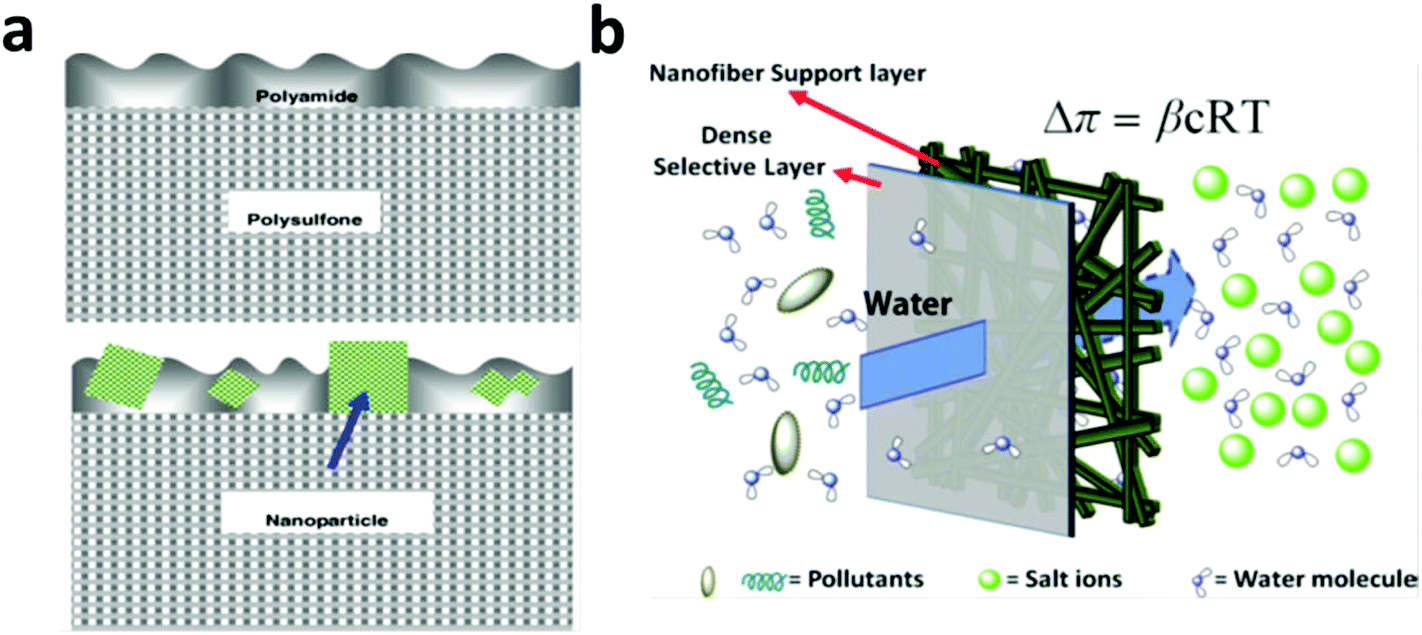 | ||
| Fig. 11 (a) Schematic of a typical RO or FO membrane configuration with a top thin dense active layer (made of polyamide) supported on a thick microporous support layer (made of polysulfone) (top figure) and a membrane with zeolite nanoparticles blended in the top thin active layer (bottom figure). From ref. 286 Copyright © 2007 Elsevier B.V. All rights reserved. (b) Schematic of an FO membrane with a thin, dense active layer and a highly porous support layer made of electrospun fibers to alleviate the adverse effect from ICP. From ref. 302 Copyright © 2007 American Institute of Chemical Engineers (AIChE). | ||
Biological membranes have evolved an effective way for water transportation via aquaporin proteins and this has been inspirational with respect to RO and FO membrane modification.272,293 In 2007, Zilles and Clark et al. fabricated amphiphilic triblock-polymer vesicles containing aquaporin Z, which showed complete salt rejection and water productivity 800 times that of the pure polymer without aquaporin Z.294 Recently, Tang et al. directly incorporated aquaporin Z into the active layer of a RO membrane via the interfacial polymerization method and the prepared membrane exhibited water permeability an order of magnitude higher than a typical seawater RO membrane.295
Concentration polarization is a serious issue in both RO and FO, as it causes a significant decrease in water flux and therefore in operation efficiency.296–298 FO relies on natural osmotic pressure to drive water selectively through a FO membrane, and internal concentration polarization (ICP) is a phenomenon in which the water permeating through the porous support layer concentrates/dilutes the salts inside the porous support layer. ICP leads to reduced osmotic pressure in FO and thus is very problematic as it would cost a drastic loss of driving force in the process.299,300 In early times, when RO membranes were directly taken for FO processes, more than 90% of the driving force was lost due to the ICP problem. Even with the latest progress in FO membrane fabrication, ICP still resulted in a loss of more than 50% of the driving force.296,301 Therefore, at this point, in the FO membrane, it is not the active layer, but the support layer that is the bottleneck of the FO process. As expected, a nano-assisted and rationally engineered FO support layer has been making its contribution to reduce the adverse effect of ICP.
From a rational design point of view, it has been proposed that an ideal FO support layer should have a large porosity, a thin layer structure and low tortuosity, and at the same time, it should provide enough mechanical strength to support the active layer.302,303 Following this idea, in 2011, Sun et al. fabricated a FO membrane with a support layer made of electrospun nanofibers (Fig. 11b), which showed a water flux 3.5 times that of a commercial FO membrane tested under otherwise the same conditions.302 Tang et al. proposed that the incorporation of porous nanomaterials in a FO support layer would alleviate the adverse ICP effect, and they demonstrated the effectiveness of this proposal by blending zeolite and nanoporous silica gels into polysulfone support layers.274 Their results showed that with these rationally selected nanoparticles in the support layers, a significantly improved water flux (2.5 times) was obtained compared with the original unmodified FO membrane and their work identified an optimal 10 nm silica gel pore size for the best FO performance.
5.2 Nano-assisted RO and FO anti-fouling
Membrane fouling occurs when suspended solids, microbes, and organic materials are deposited on the surface of RO and FO membranes.304–307 Membrane fouling significantly decreases the membrane lifespan and increases operation costs, but is seemingly an inevitable byproduct of selective water permeation in RO and FO membranes. With the recent progress in RO and FO water flux enhancement, research into membrane anti-fouling becomes more and more pressing as membrane fouling worsens along with increasing water flux. Two common strategies toward membrane anti-fouling are developed, which can be described as (1) anti-adhesion modification and308–310 (2) anti-microbial modification.306,311,312The majority of research activity on the anti-adhesion modification front focuses on surface hydrophilic modification in light of the fact that most membrane foulants, such as protein, bacteria and large organic compounds, are largely hydrophobic;313 this was first uncovered by Belfort et al. in 1997.314 With a hydrophilic surface modified RO or FO membrane, a thin water layer would form on the membrane surface, preventing the hydrophobic fouling substances from adhering to the membrane. Based on this idea, in 2007, Cao et al. reported a method for surface grafting a poly(ethylene glycol) (PEG) layer during active layer fabrication and the PEG grafted RO membrane showed an improved antifouling performance.315 Takahara et al. modified membrane surfaces with several polymer brushes with different surface tensions to investigate the relationship between surface wettability and antifouling behaviors and their results proved that the hydrophilic surface had much better antifouling properties towards hydrophobic foulants than the hydrophobic ones.316
Given the ubiquitous presence of microorganisms, fouling by microorganisms, especially bacteria-induced fouling, is not uncommon in many RO and FO membranes and therefore a biocide-induced anti-biofouling strategy is a rational solution in these cases.11,276,313 The centuries-old knowledge of the antimicrobial properties of silver metal has led scientists to use silver, especially silver nanoparticles, for antifouling purposes due to their high antibacterial activity and simple synthesis. In 2009, Yang et al. prepared a silver nanoparticle-coated commercial polyamide RO membrane and tested its anti-biofouling properties in a seawater desalination process. Their results showed an obvious decrease in the microbial concentration on the membrane surface.317 In 2014, Elimelech et al. modified a polyamide RO membrane with silver nanoparticles via a layer-by-layer (LbL) assembly, followed by further modification with a polymer brush of poly(sulfobetaine) or PDMS. All modified membrane surfaces exhibited a significant reduction in irreversible bacterial cell adhesion as well as strong anti-bacterial activity (Fig. 12).318
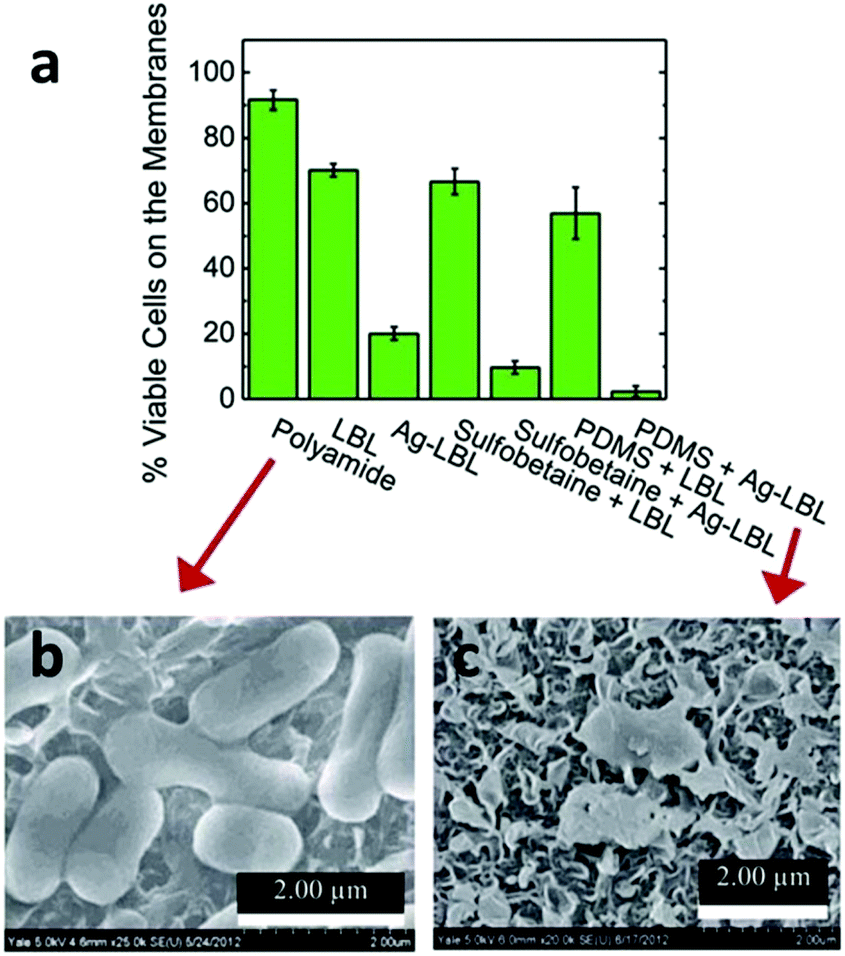 | ||
| Fig. 12 (a) The histogram of residual live cells (E. coli K12) on different modified membranes through live/dead assay. The SEM images of the cells on the surface of (b) polyamide and (c) PDMS/Ag modified membranes, respectively, which represented the worst and best antibacterial performance among these materials. Reprinted with permission from ref. 318 Copyright Royal Society of Chemistry 2014. | ||
In 2007, Elimelech et al. first reported the antibacterial activity of single-walled CNTs (SWCNTs)319 and in 2012 the same group prepared an antimicrobial film through LbL assembly of SWCNTs with polypeptides.320 In 2013, it was further revealed that bundled SWCNTs had much faster bacterial inactivation kinetics than unbundled ones.321 In 2014, it was found that graphene oxide (GO) possessed a broad spectrum of antimicrobial activities.322 Very recently, Elimelech's group investigated the GO's antimicrobial activity as a function of its size323 and showed that the GO's antimicrobial activity increased sharply as its size decreased due to the higher defect density of smaller GO sheets. The same group prepared a GO nanosheet-modified polyamide active layer on a RO membrane via surface amide coupling and reported high-level and fast antibacterial activity on the membrane.324
5.3 Emerging next-generation membranes
Unbounded by the currently available membranes, the development of nanomaterials in the past 10 years, constantly aided by MD simulation as a design guide, has led to many brand-new membranes, especially inorganic membranes, that are completely beyond the limits of conventional membrane materials.288,289,325–328 The recent developments in this regard include membranes wholly made of CNTs,329 graphene,330 graphene-oxide (GO),331 and reduced graphene-oxide (rGO).332 Among them, graphene based membranes, inclusive of GO and rGO, have shown unprecedented performances and thus represented a very promising direction in next-generation water treatment, especially in nanofiltration and seawater RO desalination, which will be the focus of the next paragraph.In 2010, using MD simulation, Aluru and coworkers first investigated and compared the water penetration process within a graphene membrane327 and pointed out the potential of a graphene-based membrane for water filtration. In 2012, a detailed MD study by Grossman and Cohen-Tanugi revealed that nanometer-scale pores in single-layer freestanding graphene could effectively filter NaCl salt from water and they predicted that its water permeability would be several orders of magnitude higher than conventional RO membranes.326 In the same year, Geim, the 2010 Nobel Prize laureate for his work with graphene, and collaborators prepared a semimicrometer-thick GO membrane by spin coating and their permeation experiments showed that the GO membrane was impermeable to the tested liquids, gases and vapors, but allowed unimpeded permeation of water with a flow rate 10 orders of magnitude faster than helium (Fig. 13a).331 In 2014, Geim et al. further synthesized a 5.0 um thick GO membrane by a simple vacuum filtration method and found that the GO membrane, if immersed in water, acted as a molecular sieve, blocking all solutes with hydrated radii larger than 4.5 angstroms. Moreover, it was found that smaller ions permeated through the membranes at rates thousands of times faster than what is expected for simple diffusion.101 They attributed the anomalously fast permeation of the small ions to a capillary–like high pressure acting on ions inside the graphene interlayer spaces. The fact that the major ionic species in seawater, including Na+, K+, Mg2+ and Cl−, can easily permeate through the GO membrane makes it impossible for RO and FO to be used for water desalination. However, the ultrafast ion transport and precise molecular sieving size cutoff promise the GO membrane numerous applications in the nanofiltration category. In 2014, Park et al. synthesized a double-layered graphene membrane and perforated it using focused ion beam milling to produce plenty of pores ranging from 10 nm to 1 micrometer and the perforated graphene membrane showed water permeation far in excess of those shown by finite-thickness membranes.333 In 2015, a single layered graphene membrane with nanoscale pores was created by a team from Oak Ridge National Laboratory using a plasma etching process (Fig. 13b) and the resulting membrane exhibited a salt (e.g. LiCl, NaCl and KCl) rejection rate of nearly 100% and rapid water transport, making it a promising next-generation RO and FO membrane for water desalination.330 Other interesting work is briefly described in this section: using vacuum filtration followed by HI reduction, Zhang et al. synthesized a freestanding ultrathin (less than 20 nm) rGO membrane, which showed an outstanding performance as a FO membrane.332 Mi et al. prepared a GO membrane via LbL assembly and subsequent crosslinking and the membrane showed water flux 4–10 times higher than most commercial NF membranes.334 MXenes, due to their atomic layered structures similar to graphene-based materials, highly controllable chemical compositions and high aspect ratios, promise a bright prospective in membrane-based filtration, but unfortunately there have been no published results on this so far.
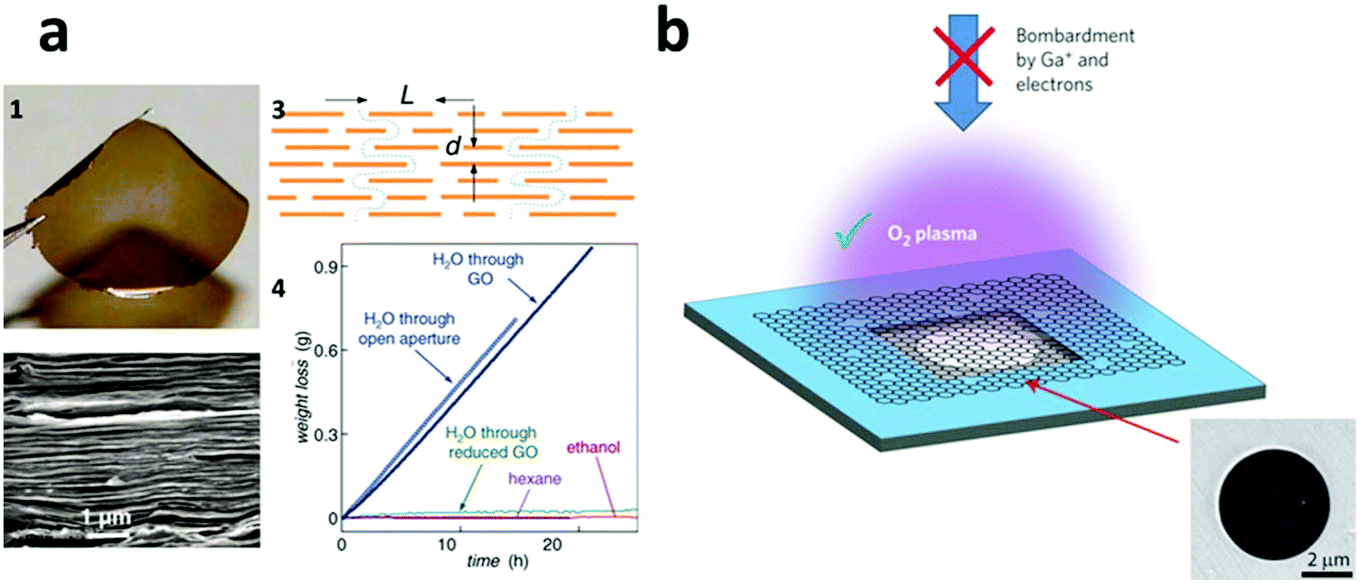 | ||
| Fig. 13 (a) (1) The optical and (2) SEM image of a 1 um-thick GO membrane prepared by a spin coating method; (3) a possible penetration mechanism through the GO membrane; (4) the permeation rate results showed that the membrane was impermeable to ethanol and hexane, whereas water could quickly pass through it. Reprinted with permission from ref. 331. Copyright American Association for the Advancement of Science 2012. (b) Ultra-small nanoholes were fabricated on a single-layered graphene membrane supported by a copper foil via O2 plasma treatment, which showed a much better performance in water desalination than those treated by electrons or gallium ions. Reprinted with permission from ref. 330. Copyright Nature Publishing Group 2015. | ||
5.4 Active filtration membranes
In comparison to the traditional concept of membranes being physical and permeable barriers for the physical separation of two bulk phases, such as in RO and FO processes, a trend is arising wherein adsorption and/or more importantly chemical processes (e.g. catalysis, reduction and oxidization) are being combined with membrane filtration to achieve active filtration for a more energy-efficient water treatment.335–340 When operated under a pressure-driven convective flow, the active membranes provide reactants with rapid access to active sites, thereby minimizing the mass transfer limitations associated with other high surface area-to-volume materials and leading to enhanced treatment performance.337 Examples of active filtration membranes include TiO2 or other semiconductor-based photo(electrochemical)-catalytically active membrane filters,338 CNT-based electrochemically active filters,136 noble-metal (e.g. Au and Pd) based catalytically active membrane filters,336 and Fenton-reaction reactive membrane filters.337,341 In a typical electrochemically or photoelectrochemically active membrane filtration system, the membrane serves as a working electrode (either cathode or anode depending on the targeted reactions), which is connected to a counter electrode to provide the required potential. Therefore, it is required that the membrane material has to be or be made electro-conductive.136 Among suitable candidate materials, CNT-based electrochemically active filters have been studied intensively. For instance, CNT-based electrochemically active filters have been demonstrated to be effective in removing aqueous organic pollutants such as salt, proteins, viruses, azo dyes, PPCPs, perfluorinated chemicals, and phenol.137,342–349 However, the potential of other conductive materials, especially graphene, rGO, MXenes and conductive polymers in this regard should not be underestimated.6. Rational design of superwetting surfaces for oil–water separation
With fossil fuel, especially gasoline and diesel, being a dominant energy source in personal transportation, the deliberate and accidental release of oil into the aqueous environment takes place in every step of the lifecycle of petroleum and this is nowadays a serious environmental concern. Efficient oil–water separation technologies are highly sought after as environmental response measures. The conventional oil–water separation technologies, such as physical skimmers, hydrocyclone-based separation and membrane-based separation, generally involve external energy sources to drive the separation.350–352 Recently, the rapid development of interface science along with bionics has helped in evolving a brand-new concept of using superwetting materials,353,354 generally in the form of two-dimensional membranes, for oil–water separation in the absence of an external energy input.350–352,355–361 The superwetting capability of materials, which is a result of a proper combination of their surface micro-nano hierarchical structure and surface chemistry, refers to extreme wetting behaviors such as superhydrophobicity, superhydrophilicity, superoleophobicity, superoleophilicity, superamphiphilicity, and superamphiphobicity. The inspirations of the superwetting materials usually have their roots in nature, and bio-inspired materials with superwetting capability have shown tremendous advantages over conventional methods in the field of oil–water separation as they allow for gravity-driven separation.In 2002, Jiang et al. revealed that a combination of spatial micro- and nanometer-scale hierarchical surface structures and proper chemical composition resulted in the superhydrophobic self-cleaning effect of the lotus leaf,362 which led them to develop the first example of a superwetting (superhydrophobic/superoleophilic in their case) membrane for oil–water separation in 2004.363 In their study, a low-surface-energy material of polytetrafluoroethylene (PTFE) coated stainless steel mesh, which had a water contact angle greater than 150° and a diesel contact angle of ∼0°, was used to effectively separate oil from water.
Later, researchers came to realize that because the superhydrophobic and superoleophilic materials removed only oil, these oil-removing types of materials could be easily fouled or even blocked by oils because of their intrinsic oleophilic properties, which seriously impact the lifetime of the materials.350–352 In addition, the oil adhered to or adsorbed on the materials is hard to remove, resulting in secondary pollution during the post-treatment process.350–352 In an effort to solve this problem, Jiang and coworkers, inspired by the oil-repellent capability of fish scale (Fig. 14a),364 recently fabricated a novel superhydrophilic and underwater superoleophobic hydrogel coated mesh for oil–water mixture separation (Fig. 14b).365 This water-removing type of material has the opposite wettability to traditional hydrophobic and oleophilic materials and thus overcomes a propensity to fouling and recycling problems because they (1) selectively allow water, instead of oil, to pass and thus prevent oil from making contact with the materials, which effectively avoids or reduces the possibility of membrane clogging caused by a viscous oil phase; (2) allow for a true gravity-driven separation of oil/water phases due to the fact that water is generally heavier than the oil phase (Fig. 14).366,367 Similarly, Jin and coworkers recently reported the fabrication of a novel poly-(acrylic acid)-grafted PVDF filtration membrane using a salt-induced phase-inversion approach. A hierarchical micro/nanoscale structure was constructed on the membrane surface, which endowed the membrane with a superhydrophilic and underwater superoleophobic property and thus allowed for effective separation of oil-in-water emulsions.368
 | ||
| Fig. 14 (a) Surface structures of fish scale. Inset in (a): shape of an oil droplet on fish scales in water, showing the superoleophobicity of the fish scales. Reprinted with permission from ref. 364. Copyright WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 2009. (b) Oil/water separation by the hydrogel-coated mesh. The coated mesh was fixed between two glass tubes and a mixture of crude oil and water was put into the upper glass tube. Water selectively permeated through the coated mesh, whereas oil was repelled and remained in the upper glass tube. Reprinted with permission from ref. 365. Copyright WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 2011. (c) Schematic for the preparation of a self-cleaning underwater superoleophobic mesh for oil–water separation. Reprinted with permission from ref. 371. Copyright Nature Publishing Group 2013. (d) Preparation and characterization of a surface with switchable superoleophilicity and superoleophobicity on a non-woven textile substrate. (e) Switch of wettability between underwater superoleophilicity and superoleophobicity. Reprinted with permission from ref. 372. Copyright Nature Publishing Group 2012. | ||
Although (super)hydrophilic and underwater superoleophobic membranes are effective for the separation of oil–water mixtures, in practical applications, the hydrophilic or superhydrophilic surfaces of the separation materials are still prone to contamination by low-surface-energy substances present in the mixture due to their intrinsically high surface energy.369,370 These low-surface-energy contaminants, once adsorbed, are difficult to remove and often diminish the surface wetting behavior, thus impairing the separation performance. It is for this reason that the frequent washing-based maintenance for the separation membranes is indispensable, which adds to the high operational cost of the separation. To this end, Wang et al. reported a self-cleaning underwater superoleophobic mesh for oil–water separation, which was prepared by LbL assembly of sodium silicate and TiO2 nanoparticles on a stainless steel mesh.371 Compared with an organic separation membrane, which suffers from poor stability and may become unstable under harsh conditions, the all-inorganic silicate/TiO2 coating provided improved stability. Furthermore, the integration of the self-cleaning property of TiO2 into the all-inorganic separation mesh enables convenient removal of the fouling contaminants by ultraviolet (UV) illumination and allows for easy recovery of the separation ability of the mesh once contaminated (Fig. 14c).
In view of the diversity of oil–water mixtures and complexity of oil-spill incidents, a controllable oil–water separation is highly desirable, i.e. the separation material allows either oil or water to pass through on demand.372–377 Wang et al. for the first time demonstrated a smart surface with switchable superoleophilicity and superoleophobicity in aqueous media for controllable oil–water separation,372 as illustrated in Fig. 14d and e. This surface is the first one of its type that can switch between superoleophilicity and superoleophobicity at room temperature without any organic solvent being involved. To obtain a smart surface with switchable oil wettability in aqueous media, especially between superoleophobicity and superoleophilicity, the chemistry on the surface should be delicately designed such that it comprises both hydrophilic and oleophilic/hydrophobic characteristics, with either characteristic becoming dominant over the other in response to environmental conditions. In this study, they grafted a block copolymer comprising pH-responsive poly(2-vinylpyridine) and oleophilic/hydrophobic polydimethylsiloxane blocks (i.e., P2VP-b-PDMS) to functionalize inexpensive and easily available materials, including non-woven textiles and polyurethane sponges, and the functionalized materials possessed switchable superoleophilicity and superoleophobicity in aqueous media (Fig. 14d), which made them highly efficient in controllable oil–water separation. The P2VP block on the grafted block copolymer can alter its wettability and its conformation via protonation and deprotonation in response to the pH of the aqueous media, which in turn provides controllable and switchable access of oil by the oleophilic PDMS block on the surface (Fig. 14e).
7. Multifunctional all-in-one nanomaterials and nanodevices for designed purposes
Given the inherent complexity of natural water and the contrasting application scenarios in reality, an ideal design of nanomaterial for water treatment is expected to be proactively complex, necessitating multi-functions on individual nanomaterials working hand-in-hand to achieve a designed goal. In the past few years, some enlightening synergistically multi-functional all-in-one nanomaterials and in many cases, integrated nano-devices, have been proposed, prepared and successfully tested, many of which represent proof-of-concepts of some ground-breaking and next-generation concepts in water treatment and more broadly in clean water production. The following are three examples we selected among many interesting and inspirational cases.7.1 An all-in-one, on-demand Fenton-active filtration device
In 2007, Bhattacharyya et al. rationally designed and fabricated an all-in-one Fenton-reaction-active filtration system for advanced oxidation toward water treatment applications, integrating nanostructured materials, enzymatic catalysis, and iron-catalyzed free radical reactions within pore-functionalized synthetic membrane platforms (Fig. 15a).337 In this study, within a two-layered membrane, glucose oxidase was immobilized in the top membrane layer to in situ generate H2O2 by reacting with deliberately added glucose in the raw water, which allowed for the flexibility of on-demand initiation of the Fenton reaction. Moreover, once H2O2 was generated, it was flushed down to the second membrane layer wherein it reacted with the polymer-immobilized Fe2+/Fe3+ or iron oxide nanoparticles to kick off the Fenton reaction for the oxidation of pollutants in the raw water within the confined membrane pore space. The rational design of this active filtration system represents a great stride in on-demand initiated chemical reactions and in moving the chemical reaction within confined spaces to break the conventional reaction efficiency limit. | ||
| Fig. 15 (a) Schematic of a Fenton reactive membrane-based filtration system with a stacked configuration. The bioactive (top) membrane contains immobilized enzyme for the catalytic production of H2O2 from glucose. The bottom membrane contains either immobilized iron ions or ferrihydrite/iron oxide nanoparticles for the decomposition of hydrogen peroxide to form powerful free radical oxidants for AOP.337 Reprinted with permission from ref. 337. Copyright National Academy of Sciences, USA 2011. (b) (1) The fabrication process of a solar-light-to-heat conversion membrane by coating a polypyrrole layer on a stainless steel mesh followed by fluoroalkylsilane modification. (2) The infrared (IR) thermal images demonstrated the high efficiency of the interfacial heating produced by the fabricated membrane under solar illumination. Reprinted with permission from ref. 384. Copyright WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim 2015. (c) Schematic of a micro/nanofluidic desalination system designed by utilizing the ion concentration polarization effect, which involves using bifurcated channels to separate desalted and salted streams and is able to obtain continuous desalination flow. Reprinted with permission from ref. 385. Copyright Nature Publishing Group 2011. | ||
7.2 An all-in-one, self-floating and self-healing solar-driven desalination device
Solar evaporation is an important approach to the large-scale production of clean water. However, conventional solar evaporation experiences a high level of energy loss and thus a low evaporation rate due to its bulk water heating nature.378,379 Recently, heating based on inorganic photothermal nanomaterial, especially carbon black, CNTs and gold nanoparticles,380–383 has become a promising strategy for improving the energy efficiency of solar-driven water evaporation. In 2015, Wang et al. rationally designed and fabricated an interfacial heating membrane, which spontaneously stayed at the water–air interface due to its hydrophobicity, collected and converted solar light into heat with high efficiency, and locally heated only water near the air/water interface.384 In this study, polypyrrole (PPy) was chosen as the polymeric photothermal material because of its high adsorption of solar light, photostability, and easy processing, and it was coated on the surface of stainless steel by electropolymerization. The PPy-coated mesh was modified to be hydrophobic with Wenzel's wetting behavior for high heating efficiency (Fig. 15b). The rationally-designed membrane possessed a significantly enhanced water evaporation rate with a solar energy to heat conversion efficiency of 58% in comparison to the natural solar bulk heating efficiency of only 24%. Moreover, given the likelihood of losing its hydrophobicity as a result of UV irradiation during application, the photothermal mesh in this study was made capable of recovering its hydrophobicity once lost. Based on this concept, an all-in-one and point-of-use solar desalination device was fabricated and could produce ca. 750 g m−2 fresh water from seawater or wastewater in less than 5 hours under natural solar irradiation.3847.3 An all-in-one, point-of-use water desalination cell
In 2010, Han et al. employed the concept of ion concentration polarization within nanofluidics channels and created an external pressure free, fouling-free, all-in-one direct seawater desalination device385 (Fig. 15c). In this device, a continuous seawater flow was divided into desalted and concentrated flows by an ion concentration polarization effect. Since the salts and larger particles were pushed away from the channels during permeation, the possibility of membrane fouling and salt accumulation were both greatly reduced. Although the electrical efficiency was miscalculated in the original paper,386 the unconventional desalination concept from this work is enlightening and represents a great effort in next generation desalination technologies, especially in the point-of-use front.It is generally true that the complexity in the synergistically multi-functionalized nanomaterial design and subsequently synthesis is paid off in the application stage as these nanomaterials lessen the requirements for applications or even open new applications that were impossible with conventional water treatment systems.
8. Concluding remarks
From the discussion throughout this review, one can see that the rational design emphasizes ‘design-for-purpose’. Unlike the trial-and-error approach, on the basis of an in-depth understanding of water treatment processes, a rational design process always starts with scientifically, generally chemically, defining the problem to be solved in detail, such as what the barrier is, what the key to the solution is and the conditions required to solve the problem. Based on the clear problem definition, a conceptual design of a nanomaterial-based solution is proposed, which is then fed back to the problem definition to be scientifically tested. The communication is iterated until both the problem definition and nanomaterial design agree well with each other. Next, the conceptually designed nanomaterial, which just passes the scientific check, is checked with the currently available synthesis capability and can then be synthesized if possible. Otherwise, the iteration back to the nanomaterial design will take place until the designed nanomaterial can be successfully synthesized. The performance of the synthesized nanomaterial is then assessed with respect to its design purpose, which has been unambiguously defined in the problem definition step and the iteration back to the nanomaterial design will take place again in the event of an unsatisfactory performance of the nanomaterial (Fig. 16b).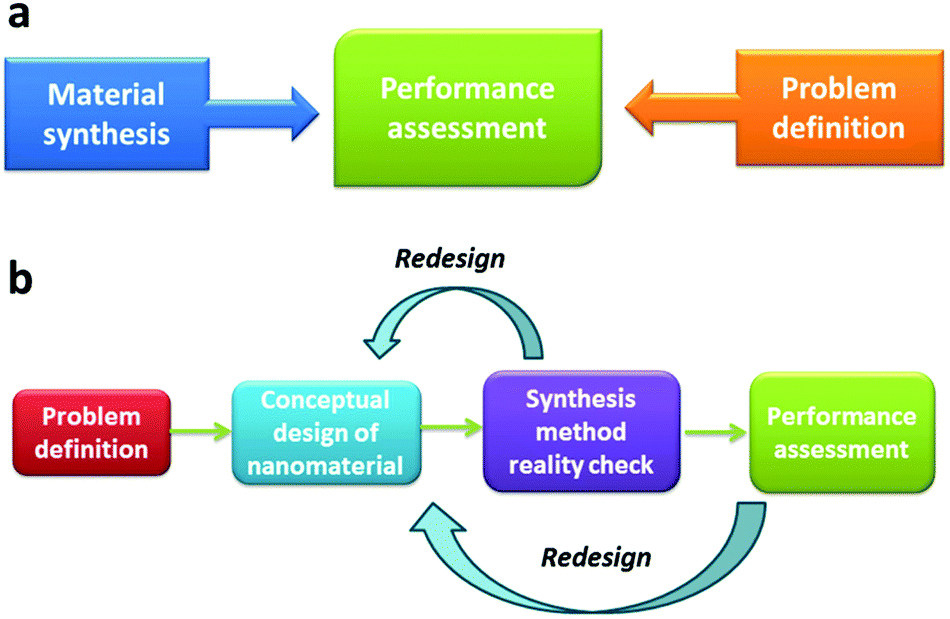 | ||
| Fig. 16 (a) Trial-and-error approach versus (b) rational design of nanomaterials for purpose. | ||
Rational design, involving “thinking-outside-the-box”, is not bounded by the available nanomaterials, and thus has a high potential of creating next-generation and ground-breaking solutions to the water challenges of our times. In theory, any material developed within the scheme of rational design is new and thus contributes to the already vast library of nanomaterials. However, it is not highly likely that the nanomaterials designed and produced based on the rational design concept find themselves as effective in processes other than those they have been designed for.
In light of the exciting progress in the field in the past decade, we truly believe that the rational design of nanomaterials will continue to evolve and offer us even more unprecedented opportunities to solve the water challenges in sustainable ways. Looking at the near future, the following are some of our humble expectations: (1) the molecular dynamics and other simulation tools would extend their presence in the field and would gradually be taken as indispensable guiding tools in both the problem definition and the conceptual design of the nanomaterial steps (Fig. 16b); (2) the next few years would see more multi-functional and all-in-one nanomaterial designs for groundbreaking water applications; (3) smart and intelligent nanomaterials, nano-devices, and nano-systems, which are capable of autonomously adjusting their function(s) in response to ambient conditions for the purpose of achieving the best performance with respect to their designed goals, would most likely emerge in the water treatment field and would gain popularity thereafter.
Acknowledgements
The authors are grateful to KAUST for very generous funding support. We would like to thank Professor Chuyang Tang from the University of Hong Kong and Dr Zhenyu Li from the WDRC at KAUST for their suggestions and comments on the membrane section of the review.References
- R. P. Schwarzenbach, T. Egli, T. B. Hofstetter, U. von Gunten and B. Wehrli, Annu. Rev. Environ. Resour., 2010, 35, 109–136 CrossRef.
- WHO, Guidelines for Drinking-water Quality, 4th edn, 2011 Search PubMed.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, J. Hazard. Mater., 2010, 177, 70–80 CrossRef CAS PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- S. Babel and T. A. Kurniawan, J. Hazard. Mater., 2003, 97, 219–243 CrossRef CAS.
- V. K. Gupta, I. Ali, T. A. Saleh, A. Nayak and S. Agarwal, RSC Adv., 2012, 2, 6380–6388 RSC.
- B. A. Lyon, R. Y. Milsk, A. B. DeAngelo, J. E. Simmons, M. P. Moyer and H. S. Weinberg, Environ. Sci. Technol., 2014, 48, 6743–6753 CrossRef CAS PubMed.
- D. Ghernaout, B. Ghernaout and M. W. Naceur, Desalination, 2011, 271, 1–10 CrossRef CAS PubMed.
- M. Catalá, N. Domínguez-Morueco, A. Migens, R. Molina, F. Martínez, Y. Valcárcel, N. Mastroianni, M. López de Alda, D. Barceló and Y. Segura, Sci. Total Environ., 2015, 520, 198–205 CrossRef PubMed.
- B. Van Der Bruggen, C. Vandecasteele, T. Van Gestel, W. Doyen and R. Leysen, Environ. Prog., 2003, 22, 46–56 CrossRef CAS PubMed.
- M. M. Pendergast and E. M. V. Hoek, Energy Environ. Sci., 2011, 4, 1946–1971 CAS.
- T.-w. Hao, P.-y. Xiang, H. R. Mackey, K. Chi, H. Lu, H.-k. Chui, M. C. M. van Loosdrecht and G.-H. Chen, Water Res., 2014, 65, 1–21 CrossRef CAS PubMed.
- J. P. Scott and D. F. Ollis, Environ. Prog., 1995, 14, 88–103 CrossRef CAS PubMed.
- X.-D. Hao, Q.-L. Wang, J.-Y. Zhu and M. C. M. Van Loosdrecht, Crit. Rev. Environ. Sci. Technol., 2010, 40, 239–265 CrossRef CAS PubMed.
- R. P. Feynman, J. Microelectromech. Syst., 1992, 1, 60–66 CrossRef.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- E. Ozbay, Science, 2006, 311, 189–193 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- Z. Luo, M. Ibáñez, A. M. Antolín, A. Genç, A. Shavel, S. Contreras, F. Medina, J. Arbiol and A. Cabot, Langmuir, 2015, 31, 3952–3957 CrossRef CAS PubMed.
- Y. Wang, D. Chen, Y. Wang, F. Huang, Q. Hu and Z. Lin, Nanoscale, 2012, 4, 3665–3668 RSC.
- L.-F. Chen, H.-W. Liang, Y. Lu, C.-H. Cui and S.-H. Yu, Langmuir, 2011, 27, 8998–9004 CrossRef CAS PubMed.
- Z. Li, D. V. Potapenko and R. M. Osgood, ACS Nano, 2015, 9, 82–87 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- K. Zhou, X. Wang, X. Sun, Q. Peng and Y. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS PubMed.
- M. L. Mastronardi, K. K. Chen, K. Liao, G. Casillas and G. A. Ozin, J.Phys. Chem. C, 2015, 119, 826–834 CAS.
- A. L. Swindle, A. S. E. Madden, I. M. Cozzarelli and M. Benamara, Environ. Sci. Technol., 2014, 48, 11413–11420 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhou, E. Villarreal, Y. Lin, S. Zou and H. Wang, Nano Lett., 2015, 15, 4161–4169 CrossRef CAS PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- X. Qu, J. Brame, Q. Li and P. J. J. Alvarez, Acc. Chem. Res., 2013, 46, 834–843 CrossRef CAS PubMed.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS PubMed.
- W.-x. Zhang, J. Nanopart. Res., 2003, 5, 323–332 CrossRef CAS.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS.
- P. G. Tratnyek and R. L. Johnson, Nano Today, 2006, 1, 44–48 CrossRef.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- X. Quan, S. Yang, X. Ruan and H. Zhao, Environ. Sci. Technol., 2005, 39, 3770–3775 CrossRef CAS.
- S. J. Tesh and T. B. Scott, Adv. Mater., 2014, 26, 6056–6068 CrossRef CAS PubMed.
- J. Li, T. Zhao, T. Chen, Y. Liu, C. N. Ong and J. Xie, Nanoscale, 2015, 7, 7502–7519 RSC.
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugan and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075–8109 CAS.
- Y. Huang and A. A. Keller, Water Res., 2015, 80, 159–168 CrossRef CAS PubMed.
- S. Tao, Y. Wang and Y. An, J. Mater. Chem., 2011, 21, 11901–11907 RSC.
- T. Wang, L. Zhang, C. Li, W. Yang, T. Song, C. Tang, Y. Meng, S. Dai, H. Wang, L. Chai and J. Luo, Environ. Sci. Technol., 2015, 49, 5654–5662 CrossRef CAS PubMed.
- Y. Liu, S. Yu, R. Feng, A. Bernard, Y. Liu, Y. Zhang, H. Duan, W. Shang, P. Tao, C. Song and T. Deng, Adv. Mater., 2015, 27, 2768–2774 CrossRef CAS PubMed.
- C. Lu, X. Liu, Y. Li, F. Yu, L. Tang, Y. Hu and Y. Ying, ACS Appl. Mater. Interfaces, 2015, 7, 15395–15402 CAS.
- K. N. Knust, D. Hlushkou, R. K. Anand, U. Tallarek and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 8107–8110 CrossRef CAS PubMed.
- Z. Liu, H. Bai and D. D. Sun, New J. Chem., 2011, 35, 137–140 RSC.
- C. Nützenadel, A. Züttel, D. Chartouni, G. Schmid and L. Schlapbach, Eur. Phys. J. D, 2000, 8, 245–250 CrossRef.
- J. Gómez-Pastora, E. Bringas and I. Ortiz, Chem. Eng. J., 2014, 256, 187–204 CrossRef PubMed.
- J. T. Mayo, C. Yavuz, S. Yean, L. Cong, H. Shipley, W. Yu, J. Falkner, A. Kan, M. Tomson and V. L. Colvin, Sci. Technol. Adv. Mater., 2007, 8, 71–75 CrossRef CAS PubMed.
- J. Hu, G. Chen and I. M. C. Lo, Water Res., 2005, 39, 4528–4536 CrossRef CAS PubMed.
- X.-q. Li, D. W. Elliott and W.-x. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111–122 CrossRef CAS PubMed.
- Z.-m. Xiu, Z.-h. Jin, T.-l. Li, S. Mahendra, G. V. Lowry and P. J. J. Alvarez, Bioresour. Technol., 2010, 101, 1141–1146 CrossRef CAS PubMed.
- Y. Liu, S. A. Majetich, R. D. Tilton, D. S. Sholl and G. V. Lowry, Environ. Sci. Technol., 2005, 39, 1338–1345 CrossRef CAS.
- J. Gotpagar, E. Grulke, T. Tsang and D. Bhattacharyya, Environ. Prog., 1997, 16, 137–143 CrossRef CAS PubMed.
- A. Taghavy, J. Costanza, K. D. Pennell and L. M. Abriola, J. Contam. Hydrol., 2010, 118, 128–142 CrossRef CAS PubMed.
- J. Zhan, T. Zheng, G. Piringer, C. Day, G. L. McPherson, Y. Lu, K. Papadopoulos and V. T. John, Environ. Sci. Technol., 2008, 42, 8871–8876 CrossRef CAS.
- T. Zheng, J. Zhan, J. He, C. Day, Y. Lu, G. L. McPherson, G. Piringer and V. T. John, Environ. Sci. Technol., 2008, 42, 4494–4499 CrossRef CAS.
- B. Sunkara, J. Zhan, J. He, G. L. McPherson, G. Piringer and V. T. John, ACS Appl. Mater. Interfaces, 2010, 2, 2854–2862 CAS.
- Z. Yue and J. Economy, J. Nanopart. Res., 2005, 7, 477–487 CrossRef CAS.
- Y. Zhang, S. Wei, F. Liu, Y. Du, S. Liu, Y. Ji, T. Yokoi, T. Tatsumi and F.-S. Xiao, Nano Today, 2009, 4, 135–142 CrossRef CAS PubMed.
- S. Singh, V. C. Srivastava and I. D. Mall, Colloids Surf., A, 2009, 332, 50–56 CrossRef CAS PubMed.
- H. Li, Z. Bian, J. Zhu, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 4538–4539 CrossRef CAS PubMed.
- J.-J. Li, X.-Y. Xu, Z. Jiang, Z.-P. Hao and C. Hu, Environ. Sci. Technol., 2005, 39, 1319–1323 CrossRef CAS.
- Y. Wang, J. Liu, P. Wang, C. J. Werth and T. J. Strathmann, ACS Catal., 2014, 4, 3551–3559 CrossRef CAS.
- D. Mohan and C. U. Pittman Jr., J. Hazard. Mater., 2006, 137, 762–811 CrossRef CAS PubMed.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, Chem. Eng. J., 2013, 219, 499–511 CrossRef CAS PubMed.
- C. Stoquart, P. Servais, P. R. Bérubé and B. Barbeau, J. Membr. Sci., 2012, 411–412, 1–12 CrossRef CAS PubMed.
- R. Toor and M. Mohseni, Chemosphere, 2007, 66, 2087–2095 CrossRef CAS PubMed.
- C. Namasivayam and D. Kavitha, Dyes Pigm., 2002, 54, 47–58 CrossRef CAS.
- W. Teng, Z. Wu, J. Fan, H. Chen, D. Feng, Y. Lv, J. Wang, A. M. Asiri and D. Zhao, Energy Environ. Sci., 2013, 6, 2765–2776 CAS.
- W.-J. Huang, B.-L. Cheng and Y.-L. Cheng, J. Hazard. Mater., 2007, 141, 115–122 CrossRef CAS PubMed.
- G. Whitesides, J. Mathias and C. Seto, Science, 1991, 254, 1312–1319 CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS PubMed.
- A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. F. Chmelka, Science, 1993, 261, 1299–1303 CAS.
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- A. H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS PubMed.
- Z. Wu, Y. Meng and D. Zhao, Microporous Mesoporous Mater., 2010, 128, 165–179 CrossRef CAS PubMed.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- Y. Wan, Y. Shi and D. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef CAS.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS PubMed.
- Z. Wu, P. A. Webley and D. Zhao, Langmuir, 2010, 26, 10277–10286 CrossRef CAS PubMed.
- K. Wang, H. Yang, L. Zhu, Z. Ma, S. Xing, Q. Lv, J. Liao, C. Liu and W. Xing, Electrochim. Acta, 2009, 54, 4626–4630 CrossRef CAS PubMed.
- L. C. C. d. Silva, L. B. O. d. Santos, G. Abate, I. C. Cosentino, M. C. A. Fantini, J. C. Masini and J. R. Matos, Microporous Mesoporous Mater., 2008, 110, 250–259 CrossRef PubMed.
- Z. Wu, Y. Yang, B. Tu, P. Webley and D. Zhao, Adsorption, 2009, 15, 123–132 CrossRef CAS.
- E. M. Jochimsen, W. W. Carmichael, J. An, D. M. Cardo, S. T. Cookson, C. E. M. Holmes, M. B. Antunes, D. A. de Melo Filho, T. M. Lyra, V. S. T. Barreto, S. M. F. O. Azevedo and W. R. Jarvis, N. Engl. J. Med., 1998, 338, 873–878 CrossRef CAS PubMed.
- P. Pendleton, R. Schumann and S. H. Wong, J. Colloid Interface Sci., 2001, 240, 1–8 CrossRef CAS PubMed.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- W. Teng, Z. Wu, D. Feng, J. Fan, J. Wang, H. Wei, M. Song and D. Zhao, Environ. Sci. Technol., 2013, 47, 8633–8641 CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- Q. Liu, J. Shi, J. Sun, T. Wang, L. Zeng and G. Jiang, Angew. Chem., Int. Ed., 2011, 50, 5913–5917 CrossRef CAS PubMed.
- B. Mi, Science, 2014, 343, 740–742 CrossRef CAS PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- X. Dong, J. Chen, Y. Ma, J. Wang, M. B. Chan-Park, X. Liu, L. Wang, W. Huang and P. Chen, Chem. Commun., 2012, 48, 10660–10662 RSC.
- D. N. H. Tran, S. Kabiri, T. R. Sim and D. Losic, Environ. Sci.: Water Res. Technol., 2015, 1, 298–305 CAS.
- H. Bi, X. Xie, K. Yin, Y. Zhou, S. Wan, L. He, F. Xu, F. Banhart, L. Sun and R. S. Ruoff, Adv. Funct. Mater., 2012, 22, 4421–4425 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- L. Mercier and T. J. Pinnavaia, Adv. Mater., 1997, 9, 500–503 CrossRef CAS PubMed.
- A. Walcarius and L. Mercier, J. Mater. Chem., 2010, 20, 4478–4511 RSC.
- L. Mercier and T. J. Pinnavaia, Environ. Sci. Technol., 1998, 32, 2749–2754 CrossRef CAS.
- X. Luo, F. Deng, L. Min, S. Luo, B. Guo, G. Zeng and C. Au, Environ. Sci. Technol., 2013, 47, 7404–7412 CAS.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- A. Bibby and L. Mercier, Chem. Mater., 2002, 14, 1591–1597 CrossRef CAS.
- G. Li, Z. Zhao, J. Liu and G. Jiang, J. Hazard. Mater., 2011, 192, 277–283 CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- P. K. Jal, S. Patel and B. K. Mishra, Talanta, 2004, 62, 1005–1028 CrossRef CAS PubMed.
- A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403–1419 CrossRef CAS.
- X. Feng, G. E. Fryxell, L.-Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923–926 CrossRef CAS.
- J. Aguado, J. M. Arsuaga and A. Arencibia, Microporous Mesoporous Mater., 2008, 109, 513–524 CrossRef CAS PubMed.
- A. M. Liu, K. Hidajat, S. Kawi and D. Y. Zhao, Chem. Commun., 2000, 1145–1146 RSC.
- A. Heidari, H. Younesi and Z. Mehraban, Chem. Eng. J., 2009, 153, 70–79 CrossRef CAS PubMed.
- A. Shahbazi, H. Younesi and A. Badiei, Chem. Eng. J., 2011, 168, 505–518 CrossRef CAS PubMed.
- C. Wang, S. Tao, W. Wei, C. Meng, F. Liu and M. Han, J. Mater. Chem., 2010, 20, 4635–4641 RSC.
- B. Li, Y. Zhang, D. Ma, Z. Shi and S. Ma, Nat. Commun., 2014, 5, 5537 CrossRef CAS PubMed.
- I. G. B. Kaya, D. Duranoglu, U. Beker and B. F. Senkal, CLEAN – Soil, Air, Water, 2011, 39, 980–988 CrossRef CAS PubMed.
- B. Pan, B. Pan, W. Zhang, L. Lv, Q. Zhang and S. Zheng, Chem. Eng. J., 2009, 151, 19–29 CrossRef CAS PubMed.
- S. P. Mishra, S. S. Dubey and D. Tiwari, J. Colloid Interface Sci., 2004, 279, 61–67 CAS.
- A. Afkhami, M. Saber-Tehrani and H. Bagheri, J. Hazard. Mater., 2010, 181, 836–844 CrossRef CAS PubMed.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- X. Mi, G. Huang, W. Xie, W. Wang, Y. Liu and J. Gao, Carbon, 2012, 50, 4856–4864 CrossRef CAS PubMed.
- G. K. Ramesha, A. Vijaya Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS PubMed.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef CAS PubMed.
- Y. Ying, Y. Liu, X. Wang, Y. Mao, W. Cao, P. Hu and X. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 1795–1803 CAS.
- O. Mashtalir, K. M. Cook, V. N. Mochalin, M. Crowe, M. W. Barsoum and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 14334–14338 CAS.
- D. Mohan and C. U. Pittman Jr., J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- S. Chakravarty, V. Dureja, G. Bhattacharyya, S. Maity and S. Bhattacharjee, Water Res., 2002, 36, 625–632 CrossRef CAS.
- B. Daus, R. Wennrich and H. Weiss, Water Res., 2004, 38, 2948–2954 CrossRef CAS PubMed.
- H. Liu, K. Zuo and C. D. Vecitis, Environ. Sci. Technol., 2014, 48, 13871–13879 CrossRef CAS PubMed.
- M. Dakiky, M. Khamis, A. Manassra and M. Mer'eb, Adv. Environ. Res., 2002, 6, 533–540 CrossRef CAS.
- A. Ramana and A. K. Sengupta, J. Environ. Eng., 1992, 118, 755–775 CrossRef CAS.
- G. E. Fryxell, J. Liu, T. A. Hauser, Z. Nie, K. F. Ferris, S. Mattigod, M. Gong and R. T. Hallen, Chem. Mater., 1999, 11, 2148–2154 CrossRef CAS.
- H. Yoshitake, T. Yokoi and T. Tatsumi, Chem. Mater., 2003, 15, 1713–1721 CrossRef CAS.
- T. Yokoi, T. Tatsumi and H. Yoshitake, J. Colloid Interface Sci., 2004, 274, 451–457 CrossRef CAS PubMed.
- J. A. Lackovic, N. P. Nikolaidis and G. M. Dobbs, Environ. Eng. Sci., 2000, 17, 29–39 CrossRef CAS.
- Y.-F. Lin and J.-L. Chen, RSC Adv., 2013, 3, 15344–15349 RSC.
- P. Wang and I. M. C. Lo, Water Res., 2009, 43, 3727–3734 CrossRef CAS PubMed.
- B. Chen, Z. Zhu, J. Hong, Z. Wen, J. Ma, Y. Qiu and J. Chen, Dalton Trans., 2014, 43, 10767–10777 RSC.
- Z. Wu, W. Li, P. A. Webley and D. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- J. Yang, H. Zhang, M. Yu, I. Emmanuelawati, J. Zou, Z. Yuan and C. Yu, Adv. Funct. Mater., 2014, 24, 1354–1363 CrossRef CAS PubMed.
- G. Lee, C. Chen, S.-T. Yang and W.-S. Ahn, Microporous Mesoporous Mater., 2010, 127, 152–156 CrossRef CAS PubMed.
- J. He and J. P. Chen, J. Colloid Interface Sci., 2014, 416, 227–234 CrossRef CAS PubMed.
- Q. Zhang, Q. Du, T. Jiao, Z. Zhang, S. Wang, Q. Sun and F. Gao, Sci. Rep., 2013, 3, 2551 Search PubMed.
- J. M. Blais, D. W. Schindler, D. C. G. Muir, L. E. Kimpe, D. B. Donald and B. Rosenberg, Nature, 1998, 395, 585–588 CrossRef CAS.
- M. T. O. Jonker, S. A. van der Heijden, J. P. Kreitinger and S. B. Hawthorne, Environ. Sci. Technol., 2007, 41, 7472–7478 CrossRef CAS.
- A. G. Carr, R. Mammucari and N. R. Foster, Chem. Eng. J., 2011, 172, 1–17 CrossRef CAS PubMed.
- P. Wang and A. A. Keller, Water Res., 2008, 42, 2093–2101 CrossRef CAS PubMed.
- P. Wang and A. A. Keller, Water Res., 2009, 43, 706–714 CrossRef CAS PubMed.
- C. T. Jafvert, Environ. Sci. Technol., 1991, 25, 1039–1045 CrossRef CAS.
- R. Denoyel and E. Sabio Rey, Langmuir, 1998, 14, 7321–7323 CrossRef CAS.
- K. Hanna, I. Beurroies, R. Denoyel, D. Desplantier-Giscard, A. Galarneau and F. Di Renzo, J. Colloid Interface Sci., 2002, 252, 276–283 CrossRef CAS PubMed.
- P. Wang, Q. Shi, Y. Shi, K. K. Clark, G. D. Stucky and A. A. Keller, J. Am. Chem. Soc., 2009, 131, 182–188 CrossRef CAS PubMed.
- Y. Shi, B. Li, P. Wang, R. Dua and D. Zhao, Microporous Mesoporous Mater., 2012, 155, 252–257 CrossRef CAS PubMed.
- X.-l. Zhang, H.-y. Niu, W.-h. Li, Y.-l. Shi and Y.-q. Cai, Chem. Commun., 2011, 47, 4454–4456 RSC.
- B. Pan, P. Ning and B. Xing, Environ. Sci. Pollut. Res., 2009, 16, 106–116 CrossRef CAS PubMed.
- J. Xu, L. Wu and A. C. Chang, Chemosphere, 2009, 77, 1299–1305 CrossRef CAS PubMed.
- S. Suárez, M. Carballa, F. Omil and J. Lema, Rev. Environ. Sci. Biotechnol., 2008, 7, 125–138 CrossRef.
- X. Shen, L. Zhu, N. Wang, L. Ye and H. Tang, Chem. Commun., 2012, 48, 788–798 RSC.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- T. Takeuchi and J. Haginaka, J. Chromatogr., B: Biomed. Sci. Appl., 1999, 728, 1–20 CrossRef CAS.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biomol. Eng., 2007, 24, 191–200 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- M. Kim, H.-J. Um, S. Bang, S.-H. Lee, S.-J. Oh, J.-H. Han, K.-W. Kim, J. Min and Y.-H. Kim, Environ. Sci. Technol., 2009, 43, 9335–9340 CrossRef CAS PubMed.
- X. Hu, L. Mu, Q. Zhou, J. Wen and J. Pawliszyn, Environ. Sci. Technol., 2011, 45, 4890–4895 CrossRef CAS PubMed.
- Y. Li, X. Li, J. Chu, C. Dong, J. Qi and Y. Yuan, Environ. Pollut., 2010, 158, 2317–2323 CrossRef CAS PubMed.
- Y. Li, C. Dong, J. Chu, J. Qi and X. Li, Nanoscale, 2011, 3, 280–287 RSC.
- B. Liu, M. Han, G. Guan, S. Wang, R. Liu and Z. Zhang, J. Phys. Chem. C, 2011, 115, 17320–17327 CAS.
- M.-H. Lee, J. L. Thomas, M.-H. Ho, C. Yuan and H.-Y. Lin, ACS Appl. Mater. Interfaces, 2010, 2, 1729–1736 CAS.
- L. Chang, S. Chen and X. Li, Appl. Surf. Sci., 2012, 258, 6660–6664 CrossRef CAS PubMed.
- S. Xu, C. Guo, Y. Li, Z. Yu, C. Wei and Y. Tang, J. Hazard. Mater., 2014, 264, 34–41 CrossRef CAS PubMed.
- X. Shen, L. Zhu, J. Li and H. Tang, Chem. Commun., 2007, 1163–1165 RSC.
- X. Shen, L. Zhu, G. Liu, H. Yu and H. Tang, Environ. Sci. Technol., 2008, 42, 1687–1692 CrossRef CAS.
- X. Shen, L. Zhu, H. Yu, H. Tang, S. Liu and W. Li, New J. Chem., 2009, 33, 1673–1679 RSC.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS PubMed.
- E. Erdim, A. R. Badireddy and M. R. Wiesner, J. Hazard. Mater., 2015, 283, 80–88 CrossRef CAS PubMed.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS.
- M. C. Pereira, L. C. A. Oliveira and E. Murad, Clay Miner., 2012, 47, 285–302 CrossRef CAS PubMed.
- K. Rusevova, F.-D. Kopinke and A. Georgi, J. Hazard. Mater., 2012, 241–242, 433–440 CrossRef CAS PubMed.
- W. G. Kuo, Water Res., 1992, 26, 881–886 CrossRef CAS.
- E. Lipczynska-Kochany, G. Sprah and S. Harms, Chemosphere, 1995, 30, 9–20 CrossRef CAS.
- H. Gallard, J. De Laat and B. Legube, Water Res., 1999, 33, 2929–2936 CrossRef CAS.
- C. F. Wells and M. A. Salam, J. Chem. Soc. A, 1968, 24–29 RSC.
- K. Fajerwerg and H. Debellefontaine, Appl. Catal., B, 1996, 10, L229–L235 CrossRef CAS.
- W. Z. Tang and R. Z. Chen, Chemosphere, 1996, 32, 947–958 CrossRef CAS.
- F. Lücking, H. Köser, M. Jank and A. Ritter, Water Res., 1998, 32, 2607–2614 CrossRef.
- E. G. Garrido-Ramírez, B. K. G. Theng and M. L. Mora, Appl. Clay Sci., 2010, 47, 182–192 CrossRef PubMed.
- A. Dhakshinamoorthy, S. Navalon, M. Alvaro and H. Garcia, ChemSusChem, 2012, 5, 46–64 CrossRef CAS PubMed.
- M. A. Voinov, J. O. S. Pagán, E. Morrison, T. I. Smirnova and A. I. Smirnov, J. Am. Chem. Soc., 2011, 133, 35–41 CrossRef CAS PubMed.
- I. R. Guimarães, L. C. A. Oliveira, P. F. Queiroz, T. C. Ramalho, M. Pereira, J. D. Fabris and J. D. Ardisson, Appl. Catal., A, 2008, 347, 89–93 CrossRef PubMed.
- S. Rahim Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef CAS PubMed.
- R. G. Zepp, B. C. Faust and J. Hoigne, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef CAS.
- R. F. P. Nogueira, M. C. Oliveira and W. C. Paterlini, Talanta, 2005, 66, 86–91 CrossRef CAS PubMed.
- P. L. Huston and J. J. Pignatello, Water Res., 1999, 33, 1238–1246 CrossRef CAS.
- Z. Miao, S. Tao, Y. Wang, Y. Yu, C. Meng and Y. An, Microporous Mesoporous Mater., 2013, 176, 178–185 CrossRef CAS PubMed.
- S. Esplugas, J. Giménez, S. Contreras, E. Pascual and M. Rodríguez, Water Res., 2002, 36, 1034–1042 CrossRef CAS.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- S. Khoufi, F. Aloui and S. Sayadi, Water Res., 2006, 40, 2007–2016 CrossRef CAS PubMed.
- H. Liu, C. Wang, X. Li, X. Xuan, C. Jiang and H. n. Cui, Environ. Sci. Technol., 2007, 41, 2937–2942 CrossRef CAS.
- J. H. Ramirez, F. J. Maldonado-Hódar, A. F. Pérez-Cadenas, C. Moreno-Castilla, C. A. Costa and L. M. Madeira, Appl. Catal., B, 2007, 75, 312–323 CrossRef CAS PubMed.
- K. Dutta, S. Mukhopadhyay, S. Bhattacharjee and B. Chaudhuri, J. Hazard. Mater., 2001, 84, 57–71 CrossRef CAS.
- R. Andreozzi, V. Caprio, A. Insola and R. Marotta, Catal. Today, 1999, 53, 51–59 CrossRef CAS.
- M. M. Huber, S. Canonica, G.-Y. Park and U. von Gunten, Environ. Sci. Technol., 2003, 37, 1016–1024 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- Z. Zhang and P. Wang, Energy Environ. Sci., 2012, 5, 6506–6512 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- I. Justicia, P. Ordejón, G. Canto, J. L. Mozos, J. Fraxedas, G. A. Battiston, R. Gerbasi and A. Figueras, Adv. Mater., 2002, 14, 1399–1402 CrossRef CAS.
- Z. Zhang, M. N. Hedhili, H. Zhu and P. Wang, Phys. Chem. Chem. Phys., 2013, 15, 15637–15644 RSC.
- Z. Zhang, X. Yang, M. N. Hedhili, E. Ahmed, L. Shi and P. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 691–696 CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Müller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef CAS PubMed.
- H. Yin, T. Lin, C. Yang, Z. Wang, G. Zhu, T. Xu, X. Xie, F. Huang and M. Jiang, Chem. – Eur. J., 2013, 19, 13313–13316 CrossRef CAS PubMed.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, M. N. Hedhili, H. Zhang and P. Wang, Nano Lett., 2013, 13, 14–20 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- Z. Zhang, Y. Yu and P. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 990–996 CAS.
- M. Liu, N. de Leon Snapp and H. Park, Chem. Sci., 2011, 2, 80–87 RSC.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS PubMed.
- M. Takahashi, K. Tsukigi, T. Uchino and T. Yoko, Thin Solid Films, 2001, 388, 231–236 CrossRef CAS.
- L. C. Kao, C. J. Lin, C. L. Dong, C. L. Chen and S. Y. H. Liou, Chem. Commun., 2015, 51, 6361–6364 RSC.
- X. Zhang, J. H. Pan, A. J. Du, W. Fu, D. D. Sun and J. O. Leckie, Water Res., 2009, 43, 1179–1186 CrossRef CAS PubMed.
- Z. Bian, J. Zhu, F. Cao, Y. Huo, Y. Lu and H. Li, Chem. Commun., 2010, 46, 8451–8453 RSC.
- P. Si, S. Ding, J. Yuan, X. W. Lou and D.-H. Kim, ACS Nano, 2011, 5, 7617–7626 CrossRef CAS PubMed.
- Z. Zhang, Y. Yuan, G. Shi, Y. Fang, L. Liang, H. Ding and L. Jin, Environ. Sci. Technol., 2007, 41, 6259–6263 CrossRef CAS.
- J. M. Macák, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2005, 44, 2100–2102 CrossRef PubMed.
- M. M. Abu-Omar and J. H. Espenson, Inorg. Chem., 1995, 34, 6239–6240 CrossRef CAS.
- X.-Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS PubMed.
- D. Zhang, G. Li, X. Yang and J. C. Yu, Chem. Commun., 2009, 4381–4383 RSC.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- J. A. Byrne, B. R. Eggins, N. M. D. Brown, B. McKinney and M. Rouse, Appl. Catal., B, 1998, 17, 25–36 CrossRef CAS.
- S. Malato, J. Blanco, J. Cáceres, A. R. Fernández-Alba, A. Agüera and A. Rodríguez, Catal. Today, 2002, 76, 209–220 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS PubMed.
- X. Z. Li, H. L. Liu, P. T. Yue and Y. P. Sun, Environ. Sci. Technol., 2000, 34, 4401–4406 CrossRef CAS.
- D. H. Kim and M. A. Anderson, Environ. Sci. Technol., 1994, 28, 479–483 CrossRef CAS PubMed.
- X. Z. Li and H. S. Liu, Environ. Sci. Technol., 2005, 39, 4614–4620 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol. C, 2010, 11, 179–209 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Z. Liu, X. Zhang, S. Nishimoto, M. Jin, D. A. Tryk, T. Murakami and A. Fujishima, J. Phys. Chem. C, 2008, 112, 253–259 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Gong, Y. Lai and C. Lin, Electrochim. Acta, 2010, 55, 4776–4782 CrossRef CAS PubMed.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- B.-Z. Wu, H.-Y. Chen, S. J. Wang, C. M. Wai, W. Liao and K. Chiu, Chemosphere, 2012, 88, 757–768 CrossRef CAS PubMed.
- W. S. Orth and R. W. Gillham, Environ. Sci. Technol., 1996, 30, 66–71 CrossRef CAS.
- C.-B. Wang and W.-x. Zhang, Environ. Sci. Technol., 1997, 31, 2154–2156 CrossRef CAS.
- J. Liu, J. K. Choe, Y. Wang, J. R. Shapley, C. J. Werth and T. J. Strathmann, ACS Catal., 2015, 5, 511–522 CrossRef CAS.
- M. M. Abu-Omar, E. H. Appelman and J. H. Espenson, Inorg. Chem., 1996, 35, 7751–7757 CrossRef CAS.
- M. M. Abu-Omar, L. D. McPherson, J. Arias and V. M. Béreau, Angew. Chem., Int. Ed., 2000, 39, 4310–4313 CrossRef CAS.
- K. D. Hurley and J. R. Shapley, Environ. Sci. Technol., 2007, 41, 2044–2049 CrossRef CAS.
- K. D. Hurley, Y. Zhang and J. R. Shapley, J. Am. Chem. Soc., 2009, 131, 14172–14173 CrossRef CAS PubMed.
- J. K. Choe, J. R. Shapley, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2010, 44, 4716–4721 CrossRef CAS PubMed.
- Y.-N. Kim, Y.-C. Lee and M. Choi, Carbon, 2013, 65, 315–323 CrossRef CAS PubMed.
- J. K. Choe, M. H. Mehnert, J. S. Guest, T. J. Strathmann and C. J. Werth, Environ. Sci. Technol., 2013, 47, 4644–4652 CrossRef CAS PubMed.
- M. G. Davie, K. Shih, F. A. Pacheco, J. O. Leckie and M. Reinhard, Environ. Sci. Technol., 2008, 42, 3040–3046 CrossRef CAS.
- D. Shuai, D. C. McCalman, J. K. Choe, J. R. Shapley, W. F. Schneider and C. J. Werth, ACS Catal., 2013, 3, 453–463 CrossRef CAS.
- J. Yin and B. Deng, J. Membr. Sci., 2015, 479, 256–275 CrossRef CAS PubMed.
- A. Subramani and J. G. Jacangelo, Water Res., 2015, 75, 164–187 CrossRef CAS PubMed.
- D. Li and H. Wang, J. Mater. Chem., 2010, 20, 4551–4566 RSC.
- K. P. Lee, T. C. Arnot and D. Mattia, J. Membr. Sci., 2011, 370, 1–22 CrossRef CAS PubMed.
- C. Tang, Z. Wang, I. Petrinić, A. G. Fane and C. Hélix-Nielsen, Desalination, 2015, 368, 89–105 CrossRef CAS PubMed.
- H. M. Hegab and L. Zou, J. Membr. Sci., 2015, 484, 95–106 CrossRef CAS PubMed.
- N. Ma, J. Wei, S. Qi, Y. Zhao, Y. Gao and C. Y. Tang, J. Membr. Sci., 2013, 441, 54–62 CrossRef CAS PubMed.
- L.-x. Dong, H.-w. Yang, S.-t. Liu, X.-m. Wang and Y. F. Xie, Desalination, 2015, 365, 70–78 CrossRef CAS PubMed.
- K. Lutchmiah, A. R. D. Verliefde, K. Roest, L. C. Rietveld and E. R. Cornelissen, Water Res., 2014, 58, 179–197 CrossRef CAS PubMed.
- C. A. Crock, A. R. Rogensues, W. Shan and V. V. Tarabara, Water Res., 2013, 47, 3984–3996 CrossRef CAS PubMed.
- P. P. Mane, P.-K. Park, H. Hyung, J. C. Brown and J.-H. Kim, J. Membr. Sci., 2009, 338, 119–127 CrossRef CAS PubMed.
- W. J. Lau, A. F. Ismail, N. Misdan and M. A. Kassim, Desalination, 2012, 287, 190–199 CrossRef CAS PubMed.
- J. Wei, C. Qiu, C. Y. Tang, R. Wang and A. G. Fane, J. Membr. Sci., 2011, 372, 292–302 CrossRef CAS PubMed.
- N. Y. Yip, A. Tiraferri, W. A. Phillip, J. D. Schiffman and M. Elimelech, Environ. Sci. Technol., 2010, 44, 3812–3818 CrossRef CAS PubMed.
- J. E. Cadotte, R. J. Petersen, R. E. Larson and E. E. Erickson, Desalination, 1980, 32, 25–31 CrossRef.
- M. L. Lind, A. K. Ghosh, A. Jawor, X. Huang, W. Hou, Y. Yang and E. M. V. Hoek, Langmuir, 2009, 25, 10139–10145 CrossRef CAS PubMed.
- H. S. Lee, S. J. Im, J. H. Kim, H. J. Kim, J. P. Kim and B. R. Min, Desalination, 2008, 219, 48–56 CrossRef CAS PubMed.
- M. L. Lind, B.-H. Jeong, A. Subramani, X. Huang and E. M. V. Hoek, J. Mater. Res., 2009, 24, 1624–1631 CrossRef CAS.
- B.-H. Jeong, E. M. V. Hoek, Y. Yan, A. Subramani, X. Huang, G. Hurwitz, A. K. Ghosh and A. Jawor, J. Membr. Sci., 2007, 294, 1–7 CrossRef CAS PubMed.
- M. Kazemimoghadam, Desalination, 2010, 251, 176–180 CrossRef CAS PubMed.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS PubMed.
- A. Kalra, S. Garde and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10175–10180 CrossRef CAS PubMed.
- L. Zhang, G.-Z. Shi, S. Qiu, L.-H. Cheng and H.-L. Chen, Desalin. Water Treat., 2011, 34, 19–24 CrossRef CAS.
- G. L. Jadav and P. S. Singh, J. Membr. Sci., 2009, 328, 257–267 CrossRef CAS PubMed.
- J. Yin, E.-S. Kim, J. Yang and B. Deng, J. Membr. Sci., 2012, 423–424, 238–246 CrossRef CAS PubMed.
- P. Agre, M. Bonhivers and M. J. Borgnia, J. Biol. Chem., 1998, 273, 14659–14662 CrossRef CAS PubMed.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- Y. Zhao, C. Qiu, X. Li, A. Vararattanavech, W. Shen, J. Torres, C. Hélix-Nielsen, R. Wang, X. Hu, A. G. Fane and C. Y. Tang, J. Membr. Sci., 2012, 423–424, 422–428 CrossRef CAS PubMed.
- J. R. McCutcheon and M. Elimelech, J. Membr. Sci., 2006, 284, 237–247 CrossRef CAS PubMed.
- S. S. Sablani, M. F. A. Goosen, R. Al-Belushi and M. Wilf, Desalination, 2001, 141, 269–289 CrossRef CAS.
- E. Matthiasson and B. Sivik, Desalination, 1980, 35, 59–103 CrossRef CAS.
- G. T. Gray, J. R. McCutcheon and M. Elimelech, Desalination, 2006, 197, 1–8 CrossRef CAS PubMed.
- S. Zhang, K. Y. Wang, T.-S. Chung, H. Chen, Y. C. Jean and G. Amy, J. Membr. Sci., 2010, 360, 522–535 CrossRef CAS PubMed.
- T. Y. Cath, A. E. Childress and M. Elimelech, J. Membr. Sci., 2006, 281, 70–87 CrossRef CAS PubMed.
- X. Song, Z. Liu and D. D. Sun, Adv. Mater., 2011, 23, 3256–3260 CrossRef CAS PubMed.
- J. R. McCutcheon and M. Elimelech, AIChE J., 2007, 53, 1736–1744 CrossRef CAS PubMed.
- X. Lu, L. H. Arias Chavez, S. Romero-Vargas Castrillón, J. Ma and M. Elimelech, Environ. Sci. Technol., 2015, 49, 1436–1444 CrossRef CAS PubMed.
- K. R. Zodrow, E. Bar-Zeev, M. J. Giannetto and M. Elimelech, Environ. Sci. Technol., 2014, 48, 13155–13164 CrossRef CAS PubMed.
- M. Ben-Sasson, X. Lu, E. Bar-Zeev, K. R. Zodrow, S. Nejati, G. Qi, E. P. Giannelis and M. Elimelech, Water Res., 2014, 62, 260–270 CrossRef CAS PubMed.
- D. Rana, Y. Kim, T. Matsuura and H. A. Arafat, J. Membr. Sci., 2011, 367, 110–118 CrossRef CAS PubMed.
- E. M. Vrijenhoek, S. Hong and M. Elimelech, J. Membr. Sci., 2001, 188, 115–128 CrossRef CAS.
- D. L. Shaffer, H. Jaramillo, S. Romero-Vargas Castrillón, X. Lu and M. Elimelech, J. Membr. Sci., 2015, 490, 209–219 CrossRef CAS PubMed.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed.
- X. Lu, S. Romero-Vargas Castrillón, D. L. Shaffer, J. Ma and M. Elimelech, Environ. Sci. Technol., 2013, 47, 12219–12228 CrossRef CAS PubMed.
- T.-H. Bae, I.-C. Kim and T.-M. Tak, J. Membr. Sci., 2006, 275, 1–5 CrossRef CAS PubMed.
- G.-d. Kang and Y.-m. Cao, Water Res., 2012, 46, 584–600 CrossRef CAS PubMed.
- A. Nabe, E. Staude and G. Belfort, J. Membr. Sci., 1997, 133, 57–72 CrossRef.
- G. Kang, M. Liu, B. Lin, Y. Cao and Q. Yuan, Polymer, 2007, 48, 1165–1170 CrossRef CAS PubMed.
- M. Kobayashi, Y. Terayama, H. Yamaguchi, M. Terada, D. Murakami, K. Ishihara and A. Takahara, Langmuir, 2012, 28, 7212–7222 CrossRef CAS PubMed.
- H.-L. Yang, J. C.-T. Lin and C. Huang, Water Res., 2009, 43, 3777–3786 CrossRef CAS PubMed.
- M. S. Rahaman, H. Therien-Aubin, M. Ben-Sasson, C. K. Ober, M. Nielsen and M. Elimelech, J. Mater. Chem. B, 2014, 2, 1724–1732 RSC.
- S. Kang, M. Pinault, L. D. Pfefferle and M. Elimelech, Langmuir, 2007, 23, 8670–8673 CrossRef CAS PubMed.
- S. Aslan, M. Deneufchatel, S. Hashmi, N. Li, L. D. Pfefferle, M. Elimelech, E. Pauthe and P. R. Van Tassel, J. Colloid Interface Sci., 2012, 388, 268–273 CrossRef CAS PubMed.
- S. Aslan, J. Maatta, B. Z. Haznedaroglu, J. P. M. Goodman, L. D. Pfefferle, M. Elimelech, E. Pauthe, M. Sammalkorpi and P. R. Van Tassel, Soft Matter, 2013, 9, 2136–2144 RSC.
- J. Chen, H. Peng, X. Wang, F. Shao, Z. Yuan and H. Han, Nanoscale, 2014, 6, 1879–1889 RSC.
- F. Perreault, A. F. de Faria, S. Nejati and M. Elimelech, ACS Nano, 2015, 9, 7226–7236 CrossRef CAS PubMed.
- F. Perreault, M. E. Tousley and M. Elimelech, Environ. Sci. Technol. Lett., 2014, 1, 71–76 CrossRef CAS.
- D. Konatham, J. Yu, T. A. Ho and A. Striolo, Langmuir, 2013, 29, 11884–11897 CrossRef CAS PubMed.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed.
- M. E. Suk and N. R. Aluru, J. Phys. Chem. Lett., 2010, 1, 1590–1594 CrossRef CAS.
- K. Zhao and H. Wu, Nano Lett., 2015, 15, 3664–3668 CrossRef CAS PubMed.
- H. Y. Yang, Z. J. Han, S. F. Yu, K. L. Pey, K. Ostrikov and R. Karnik, Nat. Commun., 2013, 4, 2220 Search PubMed.
- S. P. Surwade, S. N. Smirnov, I. V. Vlassiouk, R. R. Unocic, G. M. Veith, S. Dai and S. M. Mahurin, Nat. Nanotecchnol., 2015, 10, 459–464 CrossRef CAS PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- H. Liu, H. Wang and X. Zhang, Adv. Mater., 2015, 27, 249–254 CrossRef CAS PubMed.
- K. Celebi, J. Buchheim, R. M. Wyss, A. Droudian, P. Gasser, I. Shorubalko, J.-I. Kye, C. Lee and H. G. Park, Science, 2014, 344, 289–292 CrossRef CAS PubMed.
- M. Hu and B. Mi, Environ. Sci. Technol., 2013, 47, 3715–3723 CrossRef CAS PubMed.
- S. Mozia, Sep. Purif. Technol., 2010, 73, 71–91 CrossRef CAS PubMed.
- H. Wang, Z. Dong and C. Na, ACS Sustainable Chem. Eng., 2013, 1, 746–752 CAS.
- S. R. Lewis, S. Datta, M. Gui, E. L. Coker, F. E. Huggins, S. Daunert, L. Bachas and D. Bhattacharyya, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8577–8582 CrossRef PubMed.
- S. P. Albu, A. Ghicov, J. M. Macak, R. Hahn and P. Schmuki, Nano Lett., 2007, 7, 1286–1289 CrossRef CAS PubMed.
- B. Sensale-Rodriguez, R. Yan, S. Rafique, M. Zhu, W. Li, X. Liang, D. Gundlach, V. Protasenko, M. M. Kelly, D. Jena, L. Liu and H. G. Xing, Nano Lett., 2012, 12, 4518–4522 CrossRef CAS PubMed.
- J. Liu, M. Beals, A. Pomerene, S. Bernardis, R. Sun, J. Cheng, L. C. Kimerling and J. Michel, Nat. Photonics, 2008, 2, 433–437 CrossRef CAS PubMed.
- A. Julbe, D. Farrusseng and C. Guizard, J. Membr. Sci., 2001, 181, 3–20 CrossRef CAS.
- B. Guo, E. V. Pasco, I. Xagoraraki and V. V. Tarabara, Sep. Purif. Technol., 2015, 149, 245–254 CrossRef CAS PubMed.
- H. A. Shawky, S.-R. Chae, S. Lin and M. R. Wiesner, Desalination, 2011, 272, 46–50 CrossRef CAS PubMed.
- A. V. Dudchenko, J. Rolf, K. Russell, W. Duan and D. Jassby, J. Membr. Sci., 2014, 468, 1–10 CrossRef CAS PubMed.
- C. D. Vecitis, M. H. Schnoor, M. S. Rahaman, J. D. Schiffman and M. Elimelech, Environ. Sci. Technol., 2011, 45, 3672–3679 CrossRef CAS PubMed.
- W. Duan, A. Dudchenko, E. Mende, C. Flyer, X. Zhu and D. Jassby, Environ. Sci.: Processes Impacts, 2014, 16, 1300–1308 CAS.
- M. S. Rahaman, C. D. Vecitis and M. Elimelech, Environ. Sci. Technol., 2012, 46, 1556–1564 CrossRef CAS PubMed.
- G. Gao and C. D. Vecitis, ACS Appl. Mater. Interfaces, 2012, 4, 1478–1489 CAS.
- C. F. de Lannoy, D. Jassby, D. D. Davis and M. R. Wiesner, J. Membr. Sci., 2012, 415–416, 718–724 CrossRef CAS PubMed.
- Z. Chu, Y. Feng and S. Seeger, Angew. Chem., Int. Ed., 2015, 54, 2328–2338 CrossRef CAS PubMed.
- Y. Zhu, D. Wang, L. Jiang and J. Jin, NPG Asia Mater., 2014, 6, e101 CrossRef CAS PubMed.
- B. Wang, W. Liang, Z. Guo and W. Liu, Chem. Soc. Rev., 2015, 44, 336–361 RSC.
- X. Deng, L. Mammen, H.-J. Butt and D. Vollmer, Science, 2012, 335, 67–70 CrossRef CAS PubMed.
- Z. Shi, W. Zhang, F. Zhang, X. Liu, D. Wang, J. Jin and L. Jiang, Adv. Mater., 2013, 25, 2422–2427 CrossRef CAS PubMed.
- B. Su, W. Guo and L. Jiang, Small, 2015, 11, 1072–1096 CrossRef CAS PubMed.
- S. Wang and L. Jiang, Adv. Mater., 2007, 19, 3423–3424 CrossRef CAS PubMed.
- A. Lafuma and D. Quere, Nat. Mater., 2003, 2, 457–460 CrossRef CAS PubMed.
- T. Sun, L. Feng, X. Gao and L. Jiang, Acc. Chem. Res., 2005, 38, 644–652 CrossRef CAS PubMed.
- X.-M. Li, D. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 1350–1368 RSC.
- X. Zhang, F. Shi, J. Niu, Y. Jiang and Z. Wang, J. Mater. Chem., 2008, 18, 621–633 RSC.
- Q. Li, P. Xu, W. Gao, S. Ma, G. Zhang, R. Cao, J. Cho, H.-L. Wang and G. Wu, Adv. Mater., 2014, 26, 1378–1386 CrossRef CAS PubMed.
- L. Feng, S. Li, Y. Li, H. Li, L. Zhang, J. Zhai, Y. Song, B. Liu, L. Jiang and D. Zhu, Adv. Mater., 2002, 14, 1857–1860 CrossRef CAS PubMed.
- L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 116, 2046–2048 CrossRef PubMed.
- M. Liu, S. Wang, Z. Wei, Y. Song and L. Jiang, Adv. Mater., 2009, 21, 665–669 CrossRef CAS PubMed.
- Z. Xue, S. Wang, L. Lin, L. Chen, M. Liu, L. Feng and L. Jiang, Adv. Mater., 2011, 23, 4270–4273 CrossRef CAS PubMed.
- Q. Wen, J. Di, L. Jiang, J. Yu and R. Xu, Chem. Sci., 2013, 4, 591–595 RSC.
- F. Zhang, W. B. Zhang, Z. Shi, D. Wang, J. Jin and L. Jiang, Adv. Mater., 2013, 25, 4192–4198 CrossRef CAS PubMed.
- W. Zhang, Y. Zhu, X. Liu, D. Wang, J. Li, L. Jiang and J. Jin, Angew. Chem., Int. Ed., 2014, 53, 856–860 CrossRef CAS PubMed.
- J. A. Howarter and J. P. Youngblood, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS PubMed.
- J. A. Howarter and J. P. Youngblood, Macromol. Rapid Commun., 2008, 29, 455–466 CrossRef CAS PubMed.
- L. Zhang, Y. Zhong, D. Cha and P. Wang, Sci. Rep., 2013, 3, 2326 Search PubMed.
- L. Zhang, Z. Zhang and P. Wang, NPG Asia Mater., 2012, 4, e8 CrossRef PubMed.
- B. Wang and Z. Guo, Chem. Commun., 2013, 49, 9416–9418 RSC.
- D. Tian, X. Zhang, Y. Tian, Y. Wu, X. Wang, J. Zhai and L. Jiang, J. Mater. Chem., 2012, 22, 19652–19657 RSC.
- B. Xue, L. Gao, Y. Hou, Z. Liu and L. Jiang, Adv. Mater., 2013, 25, 273–277 CrossRef CAS PubMed.
- Z. Xu, Y. Zhao, H. Wang, X. Wang and T. Lin, Angew. Chem., Int. Ed., 2015, 127, 4610–4613 CrossRef PubMed.
- L. Zhang, J. Wu, M. N. Hedhili, X. Yang and P. Wang, J. Mater. Chem. A, 2015, 3, 2844–2852 CAS.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- T. Oki and S. Kanae, Science, 2006, 313, 1068–1072 CrossRef CAS PubMed.
- Z. Fang, Y.-R. Zhen, O. Neumann, A. Polman, F. J. García de Abajo, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 1736–1742 CAS.
- Z. Wang, Y. Liu, P. Tao, Q. Shen, N. Yi, F. Zhang, Q. Liu, C. Song, D. Zhang, W. Shang and T. Deng, Small, 2014, 10, 3234–3239 CrossRef CAS PubMed.
- R. W. Wood, Philos. Mag. Ser. 6, 1902, 4, 396–402 CrossRef PubMed.
- O. Neumann, A. S. Urban, J. Day, S. Lal, P. Nordlander and N. J. Halas, ACS Nano, 2013, 7, 42–49 CrossRef CAS PubMed.
- L. Zhang, B. Tang, J. Wu, R. Li and P. Wang, Adv. Mater., 2015, 27, 4889–4894 CrossRef CAS PubMed.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef CAS PubMed.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2013, 8, 609–609 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |