
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light driven water splitting over BiFeO3 photoanodes grown via the LPCVD reaction of [Bi(OtBu)3] and [Fe(OtBu)3]2 and enhanced with a surface nickel oxygen evolution catalyst†
Savio J. A.
Moniz
*a,
Christopher S.
Blackman
b,
Paul
Southern
c,
Paul M.
Weaver
 d,
Junwang
Tang
a and
Claire J.
Carmalt
b
d,
Junwang
Tang
a and
Claire J.
Carmalt
b
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: s.moniz@ucl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
cUCL Healthcare Biomagnetics Laboratories, 21 Albemarle Street, London, W1S 4BS, UK
dNational Physical Laboratory, Hampton Road, Teddington, Middlesex TW11 0LW, UK
First published on 11th September 2015
Abstract
Phase-pure BiFeO3 films were grown directly via dual-source low-pressure CVD (LPCVD) from the ligand-matched precursors [Bi(OtBu)3] and [Fe(OtBu)3]2, without the requirement for oxidising gas or post deposition annealing. Photocatalytic testing for water oxidation revealed extremely high activity for PEC water splitting and photocatalytic water oxidation under visible light irradiation (λ > 420 nm) with a benchmark IPCE for BiFeO3 of 23% at 400 nm. The high activity is ascribed to the ultrafine morphology achieved via the LPCVD process. The performance was enhanced by over four times when the BiFeO3 photoanode is coupled to a Ni–B surface OEC.
Introduction
Solar generation of fuels using wide spectrum responsive photocatalysts is a potential solution to meet the rapid increase in energy demands of a growing global population.1,2 The most commonly used materials for photocatalytic water splitting are transition metal oxides but the band-gaps of these materials (over 3.0 eV) are too high to serve as efficient photocatalysts under visible light irradiation.3 A number of steps have been taken to reduce their band-gap and increase their visible response, mainly through doping (e.g. with nitrogen, sulfur) or through coupling with visible light responsive molecular catalysts or dyes. The search for efficient, stable, low cost photocatalysts for the kinetically slow four electron process of water oxidation is regarded to be a significant hurdle for the introduction of large-scale water splitting devices.4 Recombination of electron–hole pairs is an additional limiting factor over efficiency; however a viable solution is the use of a photoelectrochemical (PEC) cell, where an external voltage can efficiently separate photo-generated charge carriers to drive electron migration toward the conducting back-contact to reach the counter-electrode via an external wire, where proton reduction to hydrogen gas may occur. In addition, the loading of co-catalysts that can act as either electron or hole acceptors for improved charge separation is a promising strategy, leading to the adaptation of a junction architecture.5Perovskite bismuth ferrite (BiFeO3, “BFO”) exhibits a direct band-gap of approximately 2.2 eV and is a promising multifunctional material that also exhibits simultaneous ferroelectric and ferromagnetic ordering in addition to photovoltaic6 and photocatalytic properties;7–9 Indeed, the recent successful application of inorganic and hybrid perovskite structured materials such as BiFeO3, CsSnI3, and CH3NH3PbI3 for solar energy conversion emphasises that polar semiconductors can be used in conventional photovoltaic architectures and has led to renewed interest in these “photoferroic” materials.10 BiFeO3 has been demonstrated to be a promising oxygen evolution photocatalyst exhibiting high efficiencies under both UV- and visible light irradiation,11,12 and recently, we have demonstrated the material to be an excellent wide spectrum photocatalyst for water oxidation, showing that the material has sufficient overpotential for water oxidation in the absence of an electrical or chemical bias.13 A further advantage of BiFeO3 is that its spontaneous polarization in the ferroelectric domains leads to band bending, that transports photo-generated electrons and holes in opposite directions, resulting in spatially resolved reactivity and improved charge separation.14
A major hurdle for water splitting devices is the difficulty in scale up of production, which typically requires high surface area catalysts to be grown uniformly under relatively mild conditions to be cost effective.15 Thin film growth via Chemical Vapour Deposition (CVD) has many potential advantages including excellent substrate coverage, low-cost, ease of scale-up, control over thickness and morphology and high throughput capabilities. However, growth of stoichiometrically pure BiFeO3 films using chemical deposition techniques is challenging due to the prevalence of parasitic co-phases and evaporation of bismuth at elevated temperatures;16–18 and in all previous examples the use of oxidising gases or post-growth heat treatment have been required for fully oxygenated films.19 Typical bismuth precursors for CVD also suffer from drawbacks such as poor volatilities {[BiI3],20 [Bi(C6H5)3],21 [Bi(O2CNPri2)3]},22 decomposition characteristics {[Bi(thd)3]},23 or sensitivity to air and moisture – {[Bi(mmp)3],24 [Bi(CH3)3]},25 which makes the growth of pure BiFeO3 particularly challenging.
Here, we report the growth of phase-pure BiFeO3 films via a dual-source LPCVD reaction utilizing volatile [Bi(OtBu)3] and [Fe(OtBu)3]2 precursors. The use of these ligand-matched precursors decreases the likelihood of unwanted side-reactions occurring in the gas-phase and also eliminates the need for an oxidizing gas during deposition or a post-growth annealing step. The effect of substrate temperature and system pressure on composition and phase was also investigated. Films were fully characterized and surprisingly, were found to be highly active photocatalysts for water oxidation under both full arc and visible light irradiation showing excellent stability and respectable efficiency. A nickel surface OEC further improves the water splitting capability by increasing charge separation efficiency and improving kinetics for the process, resulting in stable H2 and O2 evolution under an applied bias.
Experimental
Precursor synthesis
Standard Schlenk line procedures were used. Bismuth(III) tert-butoxide, [Bi(OtBu)3], was synthesised according to the literature via the metathesis reaction of BiCl3 and three molar equivalents of NaOtBu.26 Iron(III) tert-butoxide, [Fe(OtBu)3]2, synthesised using a modified literature preparation via the metathesis reaction between anhydrous FeCl3 and three molar equivalents of NaOtBu, was obtained as dark green crystals via sublimation under reduced pressure (10−1 mbar, 120 °C).27 Both products were stored in a nitrogen filled MBraun Unilab glovebox. TGA studies were conducted using a Netzsch 449 C Instrument under a flow of helium gas (50 sccm) with a heating rate of 10 °C min−1. TGA samples were sealed in a glovebox into aluminium pans and pierced with a hole in the lid prior to analysis. Vapour pressure measurement for [Fe(OtBu)3]2 was obtained from SAFC Ltd. The decomposition characteristics of [Bi(OtBu)3] and [Fe(OtBu)3]2 were analysed via TGA and their vapour pressures were plotted to identify suitable evaporation conditions (Fig. S1, ESI†).Film growth
A schematic diagram of the LPCVD apparatus is shown in Scheme 1 in the ESI.† A base pressure of 2 × 10−3 mbar was achieved before use, to ensure as far as possible that no air and moisture remained in the system. The total reactor pressure was varied via the use of a mechanical throttle valve. Each deposition was carried out for 1 hour. Films were deposited on borosilicate glass microscope slides (3 cm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×2 cm), FTO-coated glass and platinised silicon wafers. For deposition of BiFeO3 films, a 2:1 molar ratio of Bi:Fe precursor was utilised {[Fe(OtBu)3]2 exists as a dimer}. By information obtained from the vapour pressure and TGA curves, 230 mg (0.54 mmol) of [Bi(OtBu)3] was used for each deposition and evaporated at 110 °C (Vp = 1.1 mmHg). 150 mg of [Fe(OtBu)3]2 (0.27 mmol) was evaporated at 100 °C (Vp = 2.4 mmHg). Nitrogen (BOC, 99.96%, 50 sccm) was used as the carrier gas for each precursor with a plain line flow of nitrogen (50 sccm). Photo-assisted electrodeposition of Nickel–borate (Ni–B) was carried out in a solution of 1 mM nickel(II) nitrate in 0.1 M aqueous potassium borate, using an applied bias of 0.5 V for a period of 15 minutes under 100 mW cm−2 irradiation, then rinsed with de-ionised water and dried at room temperature. A current density of 25 μA cm−2 was observed during the deposition.
×2 cm), FTO-coated glass and platinised silicon wafers. For deposition of BiFeO3 films, a 2:1 molar ratio of Bi:Fe precursor was utilised {[Fe(OtBu)3]2 exists as a dimer}. By information obtained from the vapour pressure and TGA curves, 230 mg (0.54 mmol) of [Bi(OtBu)3] was used for each deposition and evaporated at 110 °C (Vp = 1.1 mmHg). 150 mg of [Fe(OtBu)3]2 (0.27 mmol) was evaporated at 100 °C (Vp = 2.4 mmHg). Nitrogen (BOC, 99.96%, 50 sccm) was used as the carrier gas for each precursor with a plain line flow of nitrogen (50 sccm). Photo-assisted electrodeposition of Nickel–borate (Ni–B) was carried out in a solution of 1 mM nickel(II) nitrate in 0.1 M aqueous potassium borate, using an applied bias of 0.5 V for a period of 15 minutes under 100 mW cm−2 irradiation, then rinsed with de-ionised water and dried at room temperature. A current density of 25 μA cm−2 was observed during the deposition.
Film analysis
X-ray diffraction was carried out using a Bruker-AXS D4 powder diffractometer in reflection geometry using Cu-Kα radiation (λ = 1.54056 Å) on a rotating sample holder in the range 10–70° 2θ, 0.05° step size, with 2 s per step. Phase information was obtained from the Diffracplus EVA program suite (version 2) and ICSD. Scanning electron microscopy (SEM) was used in order to examine surface morphology and film thickness. Images were obtained on a Jeol JSM-6301F Field Emission Microscope at 5 kV, after coating samples with an ultrathin layer of gold to prevent charging. Quantitative analyses of bismuth and iron were carried out via WDX using a Philips XL30ESEM Machine operating at 10 kV, equipped with an Oxford Instruments INCA detector. Films were carbon coated prior to analysis to prevent charging. XPS analysis was performed using a Kratos AXIS Ultra machine with a delay line detector under a pressure of 10−9 torr. A monochromated Al-Kα X-ray source producing a full width at half maximum (FWHM) on the Ag 3d5/2 peak of 0.48 eV was used. Raman spectra were acquired using a Renishaw Raman 1000 System using a helium-neon laser wavelength of 514.5 nm at liquid nitrogen temperature (−195 °C) using a cold stage and temperature controller equipped with a cryo pump. AFM analysis was performed using a Veeco Dimension 3100 machine in intermittent contact mode. UV-Vis spectra were recorded in transmission mode over the range 300–2500 nm using a Perkin Elmer Lambda 950 photospectrometer. Magnetism measurements (M–H hysteresis and ZFC-FC magnetisation) were conducted using a Quantum Design SQUID Vibrating Sample Magnetometer (VSM) with a maximum field setting of 7 T (70000 Oe). Films were mounted on a 4 mm diameter quartz rod using a vinyl phenolic adhesive (code GE7031, stable up to 400 K) and suspended parallel to the magnetic field (in-plane). The magnetic moment was corrected by subtracting the diamagnetic contribution from the uncoated substrate.
Photocatalytic testing
| E(RHE) = E(Ag/AgCl) + (0.059 × pH) + 0.197 V |
The light source was a 150 W Xenon lamp (Newport, USA) equipped with a standard AM 1.5 G filter; the light intensity was calibrated to 1 sun (100 mW cm−2), measured using a silicon photodiode and Newport hand-held Optical Meter (Model 1918-R). Visible light experiments were conducted using a long-pass filter (λ > 420 nm, Newport). The scan rate was 10 mV s−1, a mechanical chopper was used to chop the light and the scanned range was 0 to +1.5 V (vs. Ag/AgCl). The illuminated area was 0.5 cm × 0.5 cm. Faradaic efficiency (gas evolution) measurements were carried out using a gas-tight custom-made reactor PEC cell (Adams and Schittenden Co., USA) which was thoroughly purged with argon for 1 hour prior to irradiation. The solution was stirred vigorously (1000 rpm) during testing to ensure as much dissolved oxygen as possible is transferred to the headspace. Gas concentration analysis was performed using a GC (Varian 430-GC, TCD, 5 Å molecular sieve column, argon carrier gas 99.999%) by taking 250 μl samples of the gas in the headspace of the PEC reactor at regular intervals. An external bias of 1.2 V vs. RHE was used.
Incident photon to current conversion efficiency (IPCE) was measured with the aid of a monochromator, and calculated using the following equation:30
| IPCE (%) = [1240 × Photocurrent density]/[Wavelength × photon flux] × 100% |
Results and discussion
The LPCVD reaction of [Fe(OtBu)3]2 and [Bi(OtBu)3] resulted in adherent films at a temperatures between 475 and 550 °C. All films displayed good coverage, were red-orange in colour and could not be removed via tissue or scotch tape, but were scratched with a steel stylus. The deposition conditions are summarised in Table 1.| Substrate temp/°C | Reactor pressure/mbar | Phase(s) obtained via XRD | Thickness/nm | Deposition rate/nm min−1 | Band-gap/eV | At. % Bi:Fe (from WDX) |
|---|---|---|---|---|---|---|
| 475 | 8 | Bi2Fe4O9 + Bi25FeO40 | 470 | 8 | — | 16:84 |
| 500 | 8 | BiFeO3 + Bi2Fe4O9 + Bi25FeO40 | 600 | 10 | 2.2 | 34:66 |
| 525 | 8 | BiFeO3 + Bi25FeO40 | 640 | 11 | 2.3 | 39:61 |
| 550 | 8 | BiFeO3 | 880 | 15 | 2.4 | 48:52 |
| 575 | 8 | BiFeO3 + Bi25FeO40 | 210 | 4 | 2.2 | 45:54 |
| 550 | 15 | BiFeO3 + Bi2Fe4O9 | 940 | 16 | 2.2 | 55:45 |
| 550 | 30 | BiFeO3 + Bi2Fe4O9 | 1200 | 20 | 2.4 | 59:41 |
| 550 | 45 | BiFeO3 + Bi2Fe4O9 | 1450 | 24 | 2.3 | 37:63 |
BiFeO3 was only detected above 500 °C (Fig. 1) with films deposited below this temperature comprised of Bi2Fe4O9 and Bi25FeO40/Bi24Fe2O39 from XRD analysis (ESI Fig. S2†); surprisingly WDX measurements showed the low temperature films to be compositionally rich in iron rather than bismuth, with bismuth incorporation increasing as a function of deposition temperature. At 550 °C there were virtually no impurity phases observed (Fig. 1) and a near 1:1 Bi:Fe ratio was observed as expected for BiFeO3. Hexagonal unit cell parameters of a = b = 5.58 (2) Å, c = 13.90 (1) Å, (α = β = 90°, γ = 120°, space group R3c) were obtained via unit cell refinement and are in agreement with literature values for bulk rhombohedral BiFeO3 (a = b = 5.57414 (4) Å, c = 13.85882 (6) Å, JCPDS = 71-2494);31 a relatively small shift in 2 theta value was noticed (less than 1°) but is likely due to experimental error. Film growth rates as function of temperature are shown in Table 1 and plotted as a function of temperature in the ESI, Fig. S3.† The activation energy for the process was calculated to be approximately 360 kJ mol−1 (ca. 90 kcal mol−1). Increasing the total system pressure whilst maintaining a growth temperature of 550 °C led to formation of Bi2Fe4O9 impurity and iron rich films (ESI Fig. S4†), likely due to the complex kinetics of the Fe2O3–Bi2O3 system which can easily lead to co-existence of Bi2Fe4O9 and BiFeO3. Bi2Fe4O9 is a kinetically stable phase32–35 and the activation energies for formation of BiFeO3 and Bi2Fe4O9 are very similar (96.6 ± 9 kcal mol−1 and 99.4 ± 9 kcal mol−1, respectively),36 contributing to the difficulties in growing phase-pure BiFeO3. A dramatic increase in film thickness was also observed at increased deposition pressure (Table 1).
 | ||
| Fig. 1 X-ray diffraction patterns of the BiFeO3 films deposited at 500, 525, 550 and 575 °C at 8 mbar pressure. Peaks marked with # and * correspond to Bi25FeO40 and Bi2Fe4O9 phases respectively. | ||
The morphologies of the films were investigated using SEM (Fig. 2), showing films with dense and uniform packing of the particles, particularly at 575 °C where films appeared flatter with fewer voids but more coalescence between particles. Individual particles were estimated to be 50 nm in diameter. Upon increasing the substrate temperature the morphology appeared more uniform and dense, whereas increasing the system pressure appeared to have little effect upon the morphology of BiFeO3. Estimation of the average particle size using the Scherrer equation gave particle diameters of 35 nm and 65 nm for films deposited at 15 mbar and 30 mbar, suggesting that the increase in deposition rate results in larger particle sizes. At 30 mbar, distinct crescent-shaped structures were apparent, in contrast to those obtained at 8 mbar, where films were composed of globular particles approximately 50 nm in diameter.
 | ||
| Fig. 2 Top-down SEM images of BiFeO3 films deposited at 550 and 575 °C, and at 30 and 45 mbar (550 °C). | ||
AFM analysis of the BiFeO3 film deposited at 550 °C, 8 mbar (Fig. 3) showed globular and spherical particles of approximately 50 nm diameter clustered together to form larger aggregates. Although the globular morphology is similar to that observed in the top-down SEM images, the morphology appears much rougher with more features. The coverage nevertheless is continuous over the substrate and there are no voids. The root mean square (rms) roughness of the sample was calculated at 60 nm which is high for typical LPCVD films, but is similar to the value obtained for AACVD-grown BiFeO3 films reported recently (62 nm).13 This high surface roughness (area) is likely to be beneficial for surface area applications such as photocatalysis.
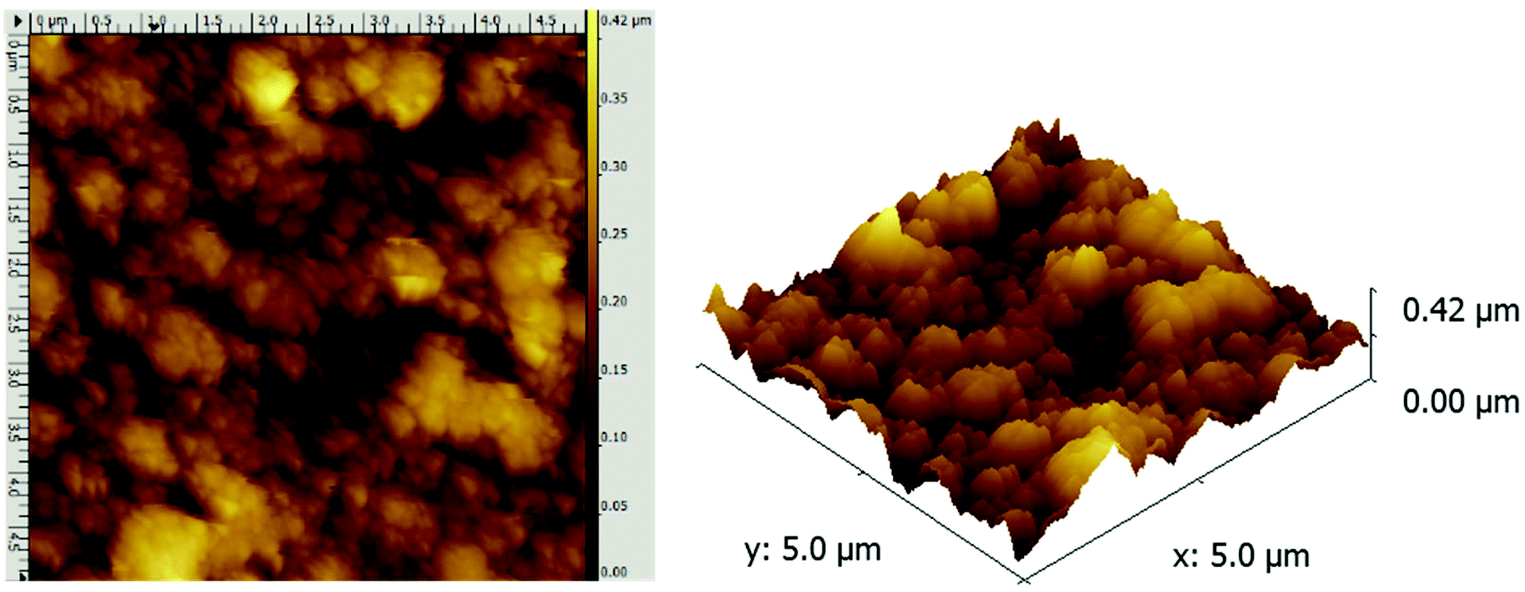 | ||
| Fig. 3 A 5 μm field size (left) and the corresponding 3D AFM image of the BiFeO3 film formed via LPCVD of [Bi(OtBu)3] and [Fe(OtBu)3]2 at 550 °C, 8 mbar. | ||
Low temperature Raman spectroscopy was used to verify phase purity of a BiFeO3 films deposited at 550 °C, 8 mbar (Fig. 4). For single crystal BiFeO3, one would expect to observe a total of 13 peaks in the Raman spectrum due to the 4A1 and 9E phonon modes.37 Peaks assigned to the A1 phonon mode were observed at 147, 175, 223 and 410 cm−1, whilst peaks assigned to the E phonon mode were observed at 134, 265, 280, 351, 374, 473 and 526 cm−1 and is consistent with those reported in the literature.38,39 Encouragingly, peaks for BixFeyOz or FeOx impurity phases were not observed in our spectra.
 | ||
| Fig. 4 Raman spectra of a BiFeO3 film deposited at 550 °C, 8 mbar, recorded at −195 °C. | ||
The chemical environment of a BiFeO3 film deposited at 550 °C (8 mbar) was analysed via XPS. The Fe 2p core level (Fig. 5) is split with the Fe 2p1/2 peak at 724.8 eV and a broad asymmetric Fe 2p3/2 peak at 711 eV revealing the predominant presence of iron in the +3 oxidation state, and typical of iron in BiFeO3 species. As reported in the literature18 the Fe3+ satellite peak is usually broad and here it was observed at 717 eV as a broad shoulder to the main Fe 2p3/2 peak, characteristic of the presence of pure Fe3+ in a sample. The spectra is largely flat in this region so it is difficult to assign satellites with any certainty. Furthermore, it is unlikely that the Fe ionisation originates from impurity Fe3O4 species as an additional broad shoulder (at 707 eV) to the peak at 711 eV is expected.40 Our spectrum is almost identical to that reported in the seminal text by Eerenstein et al. for BiFeO3 films grown via PLD.41 In addition, the smaller peak at 730 eV corresponds to the second Fe 2p1/2 satellite peak, in agreement with XPS spectra of pure Fe3+ in α-Fe2O3,42 therefore a mixed valence state of Fe2+ and Fe3+ is unlikely in these films.
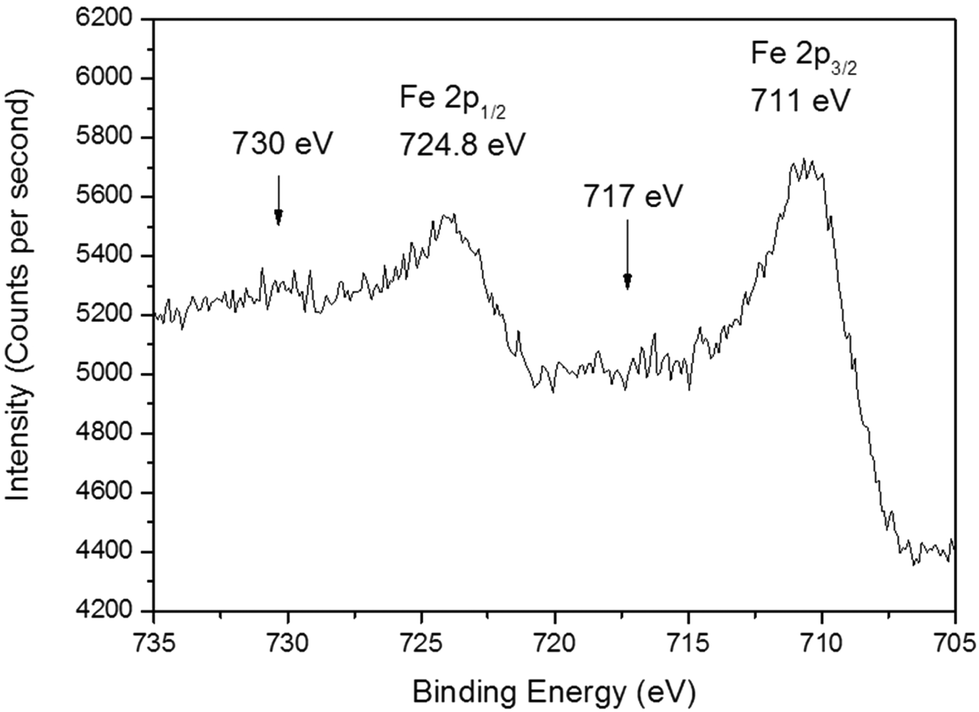 | ||
| Fig. 5 High resolution XPS spectrum of the iron 2p region. | ||
XPS analysis of the bismuth 4f region showed two distinct peaks; the Bi4f7/2 peak at 159.3 eV and the Bi 4f5/2 peak at 164.6 eV (ESI Fig. S5†). Both these peaks and their positions are characteristic of the presence of bismuth in the +3 oxidation state.43 XPS of the oxygen 1s region (ESI Fig. S6†) revealed two broad, asymmetric peaks at 532.4 eV (surface OH) and 529.7 eV (metal oxide). For ferroelectric characterisation, BiFeO3 films were grown directly on to 1 cm2 Pt/Si wafers. The room temperature P-E loop measured at 1 kHz (ESI Fig. S7†) revealed a remnant polarisation of 2.4 μC cm−2 and saturated polarisation of 3.9 μC cm−2 with coercivity of 45 kV cm−1. The remnant polarisation is much higher than those reported by Tasaki et al.44 for their CVD-grown BiFeO3 films (Pr ∼ 0.2 μC cm−2, Ps ∼ 1.75 μC cm−2) but lower than that reported for AACVD-grown BiFeO3 (8.7 μC cm−2).13 However, given the shape of the loop and the associated large dielectric loss, the polarisation at zero electric field from these loops cannot be accurately ascribed to the electrical remnant polarisation. Magnetism measurements were performed on a BiFeO3 film deposited at 550 °C on glass at both 5 K and 300 K (Fig. 6, Inset). At 5 K, the film displayed weak ferromagnetic behavior, with a saturation magnetization of 17.5 emu cm−3, slightly larger than that expected for BiFeO3 from DFT calculations.18,45 At 300 K, the saturation magnetisation decreased to 4.9 emu cm−3, more consistent with the expected value for BiFeO3.46,47 and a decrease in coercivity (∼80 Oe) was also observed. For clarity, enlarged plots of the magnetic coercivity at both temperatures may be found in the ESI, Fig. S8 and S9.† The temperature dependence of the magnetisation (field cooled (FC) and zero field cooled (ZFC) curves under a 200 Oe applied field is shown in Fig. 6.
 | ||
| Fig. 6 ZFC and FC curves for the 880 nm thick BiFeO3 film formed via LPCVD at an applied field of 200 Oe. | ||
The sharp cusp at ∼48 K in the ZFC data has been observed previously and is characteristic of spin-glass behaviour (TB, blocking temperature).48–50 Above 50 K, the ZFC data reveals an increase in magnetisation due to spin re-alignment, however at elevated temperatures thermal motion predominates and the spins randomly orientate themselves resulting in a decrease in magnetisation, leading to a disordered state with no net magnetisation. Our findings are in agreement with those recorded by Scott et al.51 for BiFeO3 single crystals – the splitting of the curves below 250 K is characteristic of spin-glass behaviour. The spins orientate themselves in a preferred direction, however if given sufficient thermal excitation can flip direction. If the measurement time of the magnetometer (τM) is smaller than the time taken for the spins to change direction (Neel relaxation time τN) the magnetisation will not change during the measurement and the particle can be viewed in a “blocked” state unless the temperature is increased. It is important to establish what type of magnetism is present and hence we demonstrate that CVD-grown BiFeO3 films still possess good ferroelectric and ferromagnetic ordering and display spin-glass behaviour at low temperature.
The UV-vis absorption spectra of BiFeO3 films deposited at 525 °C and 550 °C at (8 mbar) are shown in Fig. 7(a). The band-gap (Eg) of a semiconductor could be inferred from its UV-Vis spectra using the following equation:
| (αhv)n = A(hv − Eg) |
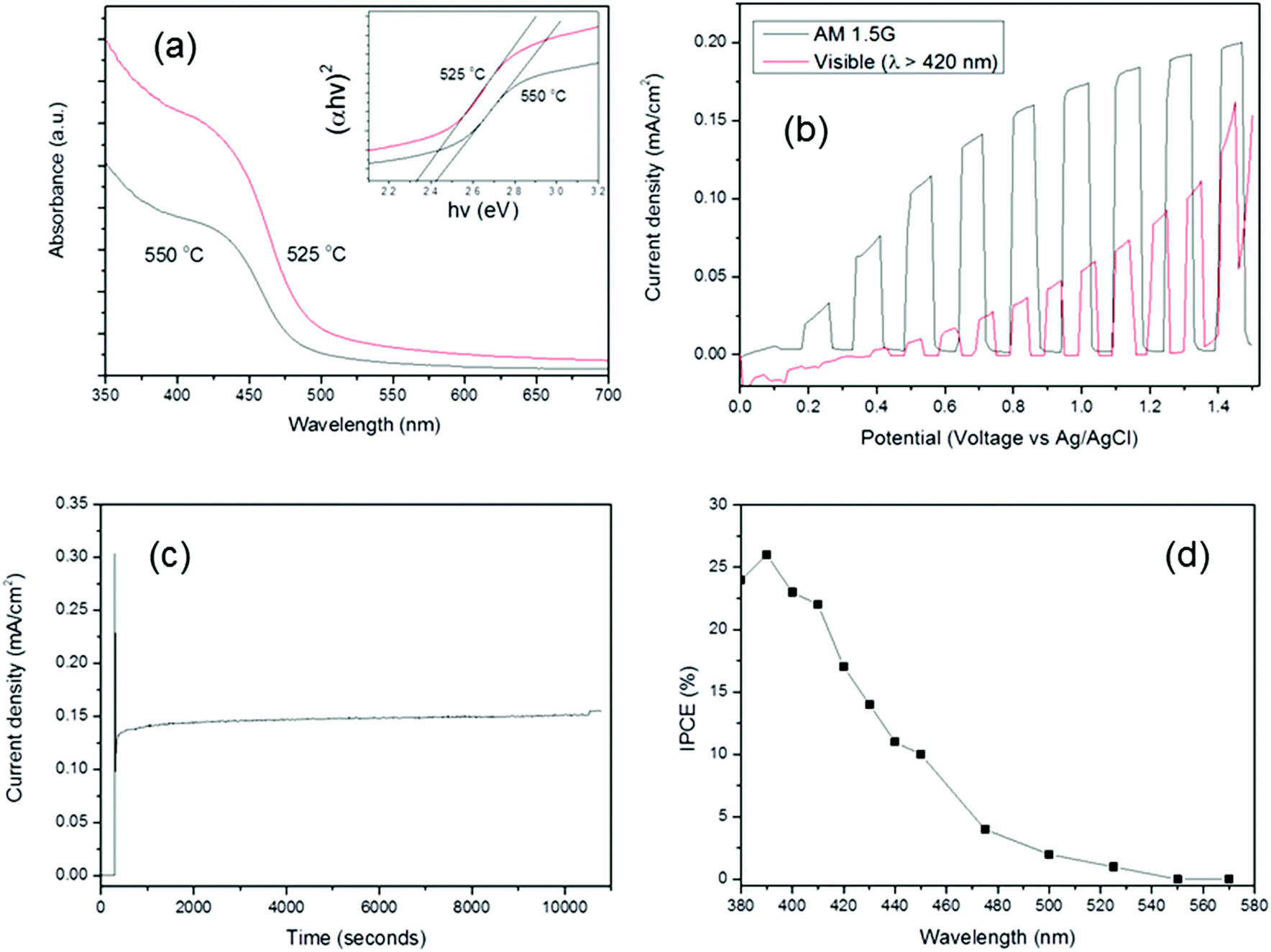 | ||
| Fig. 7 (a) UV-vis absorption spectra of BiFeO3 films grown at 525 and 550 °C via LPCVD. Inset figure shows the plot of (αhν)2versus energy for band-gap calculations; (b) I–V curve for BiFeO3 under both 100 mW cm−2 AM 1.5 G and visible light irradiation in Na2SO4 electrolyte (pH 6.5); (c) I–T curve for BiFeO3 measured under full arc AM 1.5 G illumination, at an applied potential of 1 V (vs. Ag/AgCl) for 3 hours; (d) IPCE spectrum for BiFeO3 photoelectrode measured at 1.2 V (vs. RHE). | ||
where α corresponds to the absorption coefficient, hυ is the photon energy, and n represents the index which depends on the electronic transition of the semiconductor (for direct band-gap semiconductors, n = 2).52 In addition, A is a proportionality constant related to the material. The band-gap energy was obtained from the intercept of the tangent line in the plot of (αhυ)2versus energy (inset Fig. 7), and the value was determined to be between 2.3–2.5 eV for BiFeO3 for both samples in good agreement with literature reports.53,54 A band-gap value of 2.18 eV was recently elucidated for BiFeO3 nanoparticles7 and values of between 2.2–2.76 eV were found for thin films of BiFeO3 grown via sputtering; values in the higher part of this range were attributed to band-gap defect states and oxygen vacancies.55 In our case the presence of additional Bi25FeO40 phase at 525 °C is likely to have an effect on the band-gap energy estimated, leading to an overall decrease compared to pure BiFeO3 which is in agreement with literature reports for Bi25FeO40.56
The photocurrent–voltage (I–V) and current–time (I–T) curves for a BiFeO3 film deposited directly onto a FTO-coated glass substrate at 550 °C and illuminated using full arc AM 1.5 G and visible light (λ > 420 nm) are displayed in Fig. 7(b). As previously demonstrated,13 we observe that the onset potential for BiFeO3 is 0.1 V (vs. Ag/AgCl), corresponding to a potential of ∼0.6 V vs. RHE. This is similar to the measured onset potential of α-Fe2O3 (haematite) electrodes, as expected given that both BiFeO3 and α-Fe2O3 possess similar flat-band potentials.57 It also suggests that because the onset potential is considerably more positive than the flat-band potential (by approximately 0.3 V for BiFeO3), there are a number of serious limiting factors governing the photocurrent produced by BiFeO3 (ca. 170 μA cm−2 at 1 V vs. Ag/AgCl), the reasons of which are likely to be similar to α-Fe2O3.58 The photocurrent generated under visible light is much lower, (ca. 60 μA cm−2 at 1 V vs. Ag/AgCl); this shows that the majority (ca. 60%) of photocurrent generated by BFO is due to the small UV portion of the AM 1.5 G spectrum. Importantly, the onset potential under visible irradiation also shifts positively, suggesting that the majority of photocurrent produced by BiFeO3 is driven by higher energy UV photons. This is rationalised by the sharp increase in the absorption spectra below 400 nm (Fig. 7a); a similar phenomenon has recently been observed more dramatically on Au/BiFeO3, where under UV-Vis excitation a near fivefold increase in oxygen evolution was measured compared to that under visible irradiation (λ > 420 nm).59 A similar increase in absorption below 400 nm was observed. This is significant particularly in the case of BiFeO3, which is widely reported to be a visible-light driven photocatalyst, and currently we are investigating the reasons behind these phenomena. Nevertheless, this clearly demonstrates visible-light driven activity for BiFeO3 photoanodes. It is also higher than that reported recently for epitaxial BFO/SRO/STO films grown by RF sputtering.60 Strikingly, the transient photocurrent, recorded at 1 V vs. Ag/AgCl for a period of three hours under full arc irradiation (Fig. 7c), revealed very good stability for BiFeO3, with no obvious loss of activity during the test. In order to assess the light harvesting efficiency of the BiFeO3 photoelectrode, IPCE (Fig. 7(d)) was measured using a monochromator under an applied voltage of 1.2 V vs. RHE (0.6 V vs. Ag/AgCl). The IPCE at 420 nm was recorded as 17%, a marked improvement over the ca. 4% recorded at a similar voltage (0.5 V vs. Ag/AgCl) by Chen et al.61 for PLD-grown BiFeO3 electrodes and is in good agreement with the band-gap absorption edge of approximately 480 nm exhibited in the UV-vis absorption spectra (Fig. 7(a)). This demonstrates that these CVD-grown BiFeO3 films possess superior light harvesting and charge transfer characteristics and compare favourably to those recorded for spray-deposited mesoscopic α-Fe2O3 electrodes.62 There are a variety of reasons for the observed enhancement in PEC behaviour. The LPCVD process is able to deposit phase-pure, highly conformal, nanostructured films in comparison to other methods of film deposition, thus enabling a high surface area and more reaction sites. This is evidenced by the higher photocurrent exhibited for these LPCVD-grown BiFeO3 films compared to films grown via AACVD, which exhibited no obvious well-defined nanostructuring.13 Furthermore, ferroelectric measurements confirmed room temperature polarisation in these films; the internal dipolar field within a ferroelectric creates charged surfaces that cause photogenerated carriers to move in opposite directions, which separates electrons and holes and causes oxidation and reduction products to be generated at different locations.14 This has already been demonstrated in BiFeO3 photocatalysts and is akin to an internal p–n junction.63,64
In order to verify that BiFeO3 can produce oxygen from photocatalytic water splitting under visible light, films deposited at 525 °C and 550 °C at (8 mbar) were investigated for photo-oxidation of water using persulphate ions as a sacrificial electron scavenger under simulated visible light irradiation (λ > 420 nm) in the absence of external bias and co-catalysts (see Experimental section).65 An oxygen electrode (Clark cell) was used to measure the voltage as a function of time, which is proportional to the dissolved oxygen concentration in the solution, and when an uncoated substrate was tested, no oxygen evolution was detected as expected. The Clark cell is extremely accurate for immediately detecting low concentrations of oxygen dissolved in aqueous electrolytes. Each film was suspended in the solution and irradiated with the light source for approximately 70 minutes and graphs plotting voltage against time for the two samples are shown in Fig. 8. Both films displayed appreciable photo-activities over the duration of the experiment. The small spike in the data recorded for the 640 nm thick film is due to the very high sensitivity of the oxygen electrode resulting in the detection of a brief, sharp increase in voltage; however this does not affect the overall gradient of the slope. As a control experiment, no increase in voltage was detected under dark conditions or in the absence of photocatalyst.
 | ||
| Fig. 8 Plots of voltage against time measured during the photo-oxidation of water (without applied bias) for two BiFeO3 films of 880 nm and 640 nm thicknesses under visible light illumination (λ > 420 nm). The gradient of the slope was used to calculate the rate of oxygen produced. | ||
The rate of oxygen production observed under visible irradiation (Table 2) were 1–2 orders of magnitude higher than those obtained from the use of anatase films deposited on glass via APCVD (360–2820 μmol h−1 m−2) using a 365 nm UV-light source,66 and are higher than those recently reported for BiFeO3 grown via AACVD.13 Joshi et al. have investigated the potential of BiFeO3 nanocubes to serve as visible-light water oxidation catalysts and found low activities in the region of 2.5 μmol O2 after 12 hours, at a rate of 0.5 μmol h−1.67 However, very high surface area mesoporous BiFeO3 was recently reported to exhibit an oxygen evolution rate of ca. 66 μmol g−1 h−1.59 Clearly, quantum yield is not the optimal method to measure the efficiency of thin film photoelectrodes; thus we place emphasis of Faradaic efficiency and IPCE (see later). However, on a per gram basis, taking the density of BiFeO3 to be ∼9 g cm−3 with the film volume of ca. 6 × 10−10 m3, the actual mass of BiFeO3 present would be ∼6 mg.68 On a per gram basis, the oxygen evolution would be between 250–520 μmol h−1 g−1, much higher than the recently aforementioned mesoporous BiFeO3. There are several reasons why the photocatalytic activity is comparable to, or much higher than those previously observed for BiFeO3. Firstly, our films possess a high surface area as demonstrated by the relatively high roughness values found using AFM analysis. Secondly, the likelihood of introducing charge trapping sites from parasitic phases is much lower due to the high purity of the films deposited at 550 °C in our system, as evidenced in the lower performance of the film deposited at 525 °C. In agreement with the transient photocurrent test, there were no distinct changes observed via XRD for both samples after being submerged in the basic electrolyte and irradiated with the solar simulator, thus demonstrating excellent stability for BiFeO3 photocatalysts grown via this CVD method.
| Phase(s) obtained | Film thickness/nm | Band-gap/eV | Rate of O2 production/μmol h−1 | Rate of O2 production/μmol h−1 m−2 |
|---|---|---|---|---|
| BiFeO3 + Bi25FeO40 | 640 | 2.3 | 1.49 | 7400 |
| BiFeO3 | 880 | 2.4 | 3.14 | 15700 |
In order to improve the kinetics for the sluggish process of water oxidation on BiFeO3, a Nickel borate (Ni–B) oxygen evolution catalyst (OEC) was introduced via photo-assisted electrodeposition to the BiFeO3 grown on FTO glass.69,70 The OEC catalyst functions through collection of photoholes from the light absorbing semiconductor, thus aiding charge separation, and also lowers the activation energy for water oxidation, which occurs solely on its surface. The mechanism of its formation has recently been explained by Park et al. for their BiVO4 photoanodes.70 In addition, we recently demonstrated that the Ni–B OEC possesses further functionality as an inert, earth abundant passivation layer for unstable photoelectrodes.71 The high resolution XPS spectrum of the Ni 2p region is shown in the ESI Fig. S10,† revealing the presence of nickel in the OEC as Ni2+. The I–V curve of a Ni–B/BiFeO3 photoelectrode under full arc conditions tested in aqueous 0.1 M potassium borate electrolyte is shown in Fig. 9a. The photocurrent reaches a maximum of 0.72 mA cm−2 at 1 V vs. Ag/AgCl, which is over four times higher than that exhibited by the bare BiFeO3 electrode (Fig. 7b). Furthermore, the onset potential has cathodically shifted by almost 0.3 V, similar to our recent result with ZnO photoanodes,71 and shows that the OEC efficiently traps photoholes on the surface for the oxygen evolution reaction. This is crucial for widening the operating window of a PEC device in order to operate under minimum external bias. The increase in dark current above 1 V (vs. Ag/AgCl) is likely due to the electrocatalytic function of the OEC or due to the Ni2+/Ni3+ redox peak.70 The stability of the junction photoelectrode was confirmed through prolonged testing (almost 3 hours) with no loss of current detected (Fig. 9b), thus confirming the Ni–B/BiFeO3 junction is stable under the operating conditions. Importantly, Faradaic efficiency was determined to provide further verification of the water splitting reaction on Ni–B/BiFeO3 photoanodes. As can be seen from Fig. 9c, stoichiometric gas evolution, close to 70% Faradaic efficiency is observed for oxygen and over 85% for hydrogen evolution over a 3 hour test period, demonstrating that BiFeO3 could serve as an alternative, stable photoanode material for water splitting reactions because most of the photogenerated charges were consumed for water splitting and H2/O2 production in the current system. It is worth mentioning that O2 dissolved in the electrolyte is not measured by the GC which can result in an underestimation of Faradaic efficiency. Moreover, the lack of separation between evolved gases in the single compartment PEC cell and systematic error in gas measurement using gas syringe and GC (which are not contained together within a closed system) yields efficiencies that can deviate from unity. In spite of these limitations, these results clearly demonstrate the potential of BiFeO3 thin films to act as powerful visible-light photocatalysts for the kinetically slow four electron process of water oxidation and could be promising photocatalysts for environmental remediation applications.
 | ||
| Fig. 9 (a) I–V curve for a Ni–B/BiFeO3 photoanode under 100 mW cm−2 AM 1.5 G irradiation in 0.1 M potassium borate electrolyte (pH 9.2); (b) corresponding transient photocurrent for Ni–B/BiFeO3 measured at an applied potential of 1 V (vs. Ag/AgCl) for 3 hours; (c) inset figure shows PEC gas evolution over Ni–B/BiFeO3 photoelectrode compared with the theoretical gas evolution deduced from photocurrent generation. | ||
Conclusions
For the first time, we demonstrate the growth of high quality, pure phase multiferroic BiFeO3 films via dual-source LPCVD using the ligand-matched solid organometallic precursors [Fe(OtBu)3]2 and [Bi(OtBu)3] without the need for an additional oxygen source. The effect on the phase(s) of BFO obtained whilst varying the CVD system pressure and growth temperature was investigated, and it was found that a temperature of 550 °C was required to obtain phase-pure BiFeO3, and that an increase in system pressure brought about a variation in morphology to crescent-shaped nanoparticulate clusters. Films displayed ferromagnetic magnetisation and spin-glass behaviour, together with weak ferroelectric polarisation due to the absence of epitaxial strain caused by lattice mismatch, thus confirming multiferroic properties at room temperature. The photocatalytic activity of the films was assessed via PEC measurements and water oxidation using persulfate ions as an electron scavenger, which revealed very high activity for these BFO films. The initial enhanced activity was ascribed to the high surface area, nanostructured morphology brought about by the LPCVD process, coupled with an improved charge separation efficiency induced by the internal polarisation present within perovskite BiFeO3. For the first time we observe a visible light driven PEC response from BiFeO3 in the absence of any scavengers with concomitant hydrogen and oxygen gas evolution, which is promising not only for photocatalytic water splitting but also for environmental remediation applications. In addition, we have shown that a surface Ni OEC can enhance the oxygen production on BiFeO3, by improving the reaction kinetics and lowering the overpotential for water oxidation. Further enhancement could be possible with the incorporation of noble metal particles to improve charge separation or through doping.30 Furthermore our results complement those recently reported for Au-BFO nanowires which recently exhibited a dramatic oxygen evolution rate under visible light irradiation and demonstrate the potential of BiFeO3 as a viable replacement for hematite photoanodes once further studies concerning carrier dynamics are undertaken.12Acknowledgements
EPSRC are acknowledged for funding a PhD studentship. SAFC Hitech Ltd are thanked for vapour pressure measurement of [Fe(OtBu)3]2. SM and JT acknowledge financial support from the EU FP7 Project 4G-PHOTOCAT – 309636.Notes and references
- A. Bard and M. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- D. Martin, G. Liu, S. Moniz, Y. Bi, A. Beale, J. Ye and J. Tang, Chem. Soc. Rev., 2015 10.1039/c5cs00380f.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. Martin, Z. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 Search PubMed.
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C.-H. Yang, M. D. Rossell, P. Yu, Y.-H. Chu, J. F. Scott, J. W. Ager, L. W. Martin and R. Ramesh, Nat. Nanotechnol., 2010, 5, 143–147 CrossRef CAS PubMed.
- F. Gao, X. Y. Chen, K. B. Yin, S. Dong, Z. F. Ren, F. Yuan, T. Yu, Z. G. Zou and J.-M. Liu, Adv. Mater., 2007, 19, 2889–2892 CrossRef CAS.
- S. Li, Y. Lin, B. Zhang and Y. Wang, J. Phys. Chem. C, 2010, 114, 2903–2908 CrossRef CAS.
- X. Xu, Y.-H. Lin, P. Li, L. Shu and C.-W. Nan, J. Am. Ceram. Soc., 2011, 94, 2296–2299 CrossRef CAS.
- K. Butler, J. M. Frost and A. Walsh, Energy Environ. Sci., 2014, 8, 838–848 Search PubMed.
- F. Gao, Y. Yuan, K. F. Wang, X. Y. Chen, F. Chen, J.-M. Liu and Z. F. Ren, Appl. Phys. Lett., 2006, 89, 102506 CrossRef.
- S. Li, J. Zhang, M. G. Kibria, Z. Mi, M. Chaker, D. Ma, R. Nechache and F. Rosei, Chem. Commun., 2013, 49, 5856–5858 RSC.
- S. J. A. Moniz, R. Quesada-Cabrera, C. S. Blackman, J. Tang, P. Southern, P. M. Weaver and C. J. Carmalt, J. Mater. Chem. A, 2014, 2, 2922–2927 RSC.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
- S. Y. Yang, F. Zavaliche, L. Mohaddes-Ardabili, V. Vaithyanathan, D. G. Schlom, Y. J. Lee, Y. H. Chu, M. P. Cruz, Q. Zhan, T. Zhao and R. Ramesh, Appl. Phys. Lett., 2005, 87, 102903 CrossRef.
- M. Kartavtseva, O. Gorbenko and A. Kaul, Surf. Coat. Technol., 2007, 201, 9149–9153 CrossRef CAS.
- J. Thery, C. Dubourdieu, T. Baron, C. Ternon, H. Roussel and F. Pierre, Chem. Vap. Deposition, 2007, 13, 232–238 CrossRef CAS.
- S. J. A. Moniz, C. J. Carmalt, L. G. Bloor and D. C. Pugh, Comprehensive Inorganic Chemistry II, Elsevier, 2013, 2, 1021–1052 Search PubMed.
- M. Schuisky and A. Hårsta, Chem. Vap. Deposition, 1996, 2, 235–238 CrossRef CAS.
- G. Bandoli, D. Barreca, E. Brescacin, G. A. Rizzi and E. Tondello, Chem. Vap. Deposition, 1996, 2, 238–242 CrossRef CAS.
- S. D. Cosham, M. S. Hill, G. A. Horley, A. L. Johnson, L. Jordan, K. C. Molloy and D. C. Stanton, Inorg. Chem., 2014, 53, 503–511 CrossRef CAS PubMed.
- S. Kang and S. W. Rhee, Thin Solid Films, 2004, 468, 79–83 CrossRef CAS.
- P. A. Williams, A. C. Jones, M. J. Crosbie, P. J. Wright, J. F. Bickley, A. Steiner, H. O. Davies, T. J. Leedham and G. W. Critchlow, Chem. Vap. Deposition, 2001, 7, 205–209 CrossRef CAS.
- H. Kim, J. Myung and S. Shim, Solid State Commun., 2006, 137, 196–198 CrossRef CAS.
- M.-C. Massiani, R. Papiernik, L. G. Hubert-Pfalzgraf and J.-C. Daran, Polyhedron, 1991, 10, 437–445 CrossRef CAS.
- S. Mathur, M. Veith, V. Sivakov, H. Shen, V. Huch, U. Hartmann and H.-B. Gao, Chem. Vap. Deposition, 2002, 8, 277–283 CrossRef CAS.
- S. J. A. Moniz, C. S. Blackman, C. J. Carmalt and G. Hyett, J. Mater. Chem., 2010, 20, 7881–7886 RSC.
- A. Mills and M. A. Valenzuela, J. Photochem. Photobiol., A, 2004, 165, 25–34 CrossRef CAS.
- S. J. A. Moniz, J. Zhu and J. Tang, Adv. Energy Mater., 2014, 4, 201301590 Search PubMed.
- A. Reyes, C. Delavega, M. Fuentes and L. Fuentes, J. Eur. Ceram. Soc., 2007, 27, 3709–3711 CrossRef CAS.
- J.-T. Han, Y.-H. Huang, X.-J. Wu, C.-L. Wu, W. Wei, B. Peng, W. Huang and J. B. Goodenough, Adv. Mater., 2006, 18, 2145–2148 CrossRef CAS.
- Y. Li, Y. Zhang, W. Ye, J. Yu, C. Lu and L. Xia, New J. Chem., 2012, 36, 1297–1300 RSC.
- F. Tyholdt, S. Jørgensen, H. Fjellvåg and A. E. Gunnaes, J. Mater. Res., 2005, 20, 2127–2139 CrossRef CAS.
- F. Tyholdt, H. Fjellvag, A. E. Gunnæs and A. Olsen, J. Appl. Phys., 2007, 102, 074108 CrossRef.
- L. Mukherjee and F. Y. Yang, J. Am. Ceram. Soc., 1971, 51, 31–34 CrossRef.
- H. Fukumura, H. Harima, K. Kisoda, M. Tamada, Y. Noguchi and M. Miyayama, J. Magn. Magn. Mater., 2007, 310, e367–e369 CrossRef CAS.
- D. Rout, K.-S. Moon and S.-J. L. Kang, J. Raman Spectrosc., 2009, 40, 618–626 CrossRef CAS.
- A. Perejón, N. Murafa, P. E. Sánchez-Jiménez, J. M. Criado, J. Subrt, M. J. Diánez and L. A. Pérez-Maqueda, J. Mater. Chem. C, 2013, 1, 3551 RSC.
- L. Pan, G. Zhang, C. Fan, H. Qiu, P. Wu, F. Wang and Y. Zhang, Thin Solid Films, 2005, 473, 63–67 CrossRef CAS.
- W. Eerenstein, F. D. Morrison, J. Dho, M. G. Blamire, J. F. Scott and N. D. Mathur, Science, 2005, 307, 1203a CrossRef PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- V. S. Dharmadhikari, S. Sainkar, S. Badrinarayan and A. Goswami, J. Electron Spectrosc. Relat. Phenom., 1982, 25, 181–189 CrossRef CAS.
- Y. Tasaki, T. Kanoko, M. Kabeya, N. Chifu and S. Yoshizawa, Integr. Ferroelectr., 2006, 81, 281–288 CrossRef CAS.
- C. Ederer and N. Spaldin, Phys. Rev. B: Condens. Matter, 2005, 71, 1–4 Search PubMed.
- T. Zhao, A. Scholl, F. Zavaliche, K. Lee, M. Barry, A. Doran, M. P. Cruz, Y. H. Chu, C. Ederer, N. A. Spaldin, R. R. Das, D. M. Kim, S. H. Baek, C. B. Eom and R. Ramesh, Nat. Mater., 2006, 5, 823–829 CrossRef CAS PubMed.
- J. Wang, J. B. Neaton, H. Zheng, S. Scholl, V. Nagarajan, S. B. Ogale, B. Liu, D. Viehland, V. Vaithyanathan, D. G. Schlom, L. Mohaddes-Ardabili, U. V. Waghmare, N. A. Spaldin, K. M. Rabe, T. Zhao, M. Wuttig and R. Ramesh, Science, 2005, 307, 1203b CrossRef.
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463–2485 CrossRef CAS.
- B. Liu, B. Hu and Z. Du, Chem. Commun., 2011, 47, 8166–8168 RSC.
- T.-J. Park, G. C. Papaefthymiou, A. J. Viescas, A. R. Moodenbaugh and S. S. Wong, Nano Lett., 2007, 7, 766–772 CrossRef CAS PubMed.
- M. Singh, W. Prellier, M. Singh, R. Katiyar and J. Scott, Phys. Rev. B: Condens. Matter, 2008, 77, 1–5 Search PubMed.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- S. Li, Y.-H. Lin, B.-P. Zhang, C.-W. Nan and Y. Wang, J. Appl. Phys., 2009, 105, 056105 CrossRef.
- J. Luo and P. A. Maggard, Adv. Mater., 2006, 18, 514–517 CrossRef CAS.
- A. J. Hauser, J. Zhang, L. Mier, R. A. Ricciardo, P. M. Woodward, T. L. Gustafson, L. J. Brillson and F. Y. Yang, Appl. Phys. Lett., 2008, 92, 222901 CrossRef.
- R. Köferstein, T. Buttlar and S. G. Ebbinghaus, J. Solid State Chem., 2014, 217, 50–56 CrossRef.
- J. Y. Kim, G. Magesh, D. H. Youn, J.-W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 2681 CrossRef PubMed.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- I. Papadas, J. A. Christodoulides, G. Kioseoglou and G. S. Armatas, J. Mater. Chem. A, 2015, 3, 1587–1593 RSC.
- W. Ji, K. Yao, Y.-F. Lim, Y. C. Liang and A. Suwardi, Appl. Phys. Lett., 2013, 103, 062901 CrossRef.
- X. Y. Chen, T. Yu, F. Gao, H. T. Zhang, L. F. Liu, Y. M. Wang, Z. S. Li, Z. G. Zou and J.-M. Liu, Appl. Phys. Lett., 2007, 91, 022114 CrossRef.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184–17191 CrossRef CAS PubMed.
- A. M. Schultz, Y. Zhang, P. A. Salvador and G. S. Rohrer, ACS Appl. Mater. Interfaces, 2011, 3, 1562–1567 Search PubMed.
- Y. Zhang, A. M. Schultz, P. A. Salvador and G. S. Rohrer, J. Mater. Chem., 2011, 21, 4168–4174 RSC.
- S. Elouali, A. Mills, I. P. Parkin, E. Bailey, P. F. McMillan and J. A. Darr, J. Photochem. Photobiol., A, 2010, 216, 110–114 CrossRef CAS.
- G. Hyett, J. A. Darr, A. Mills and I. P. Parkin, Chem. – Eur. J., 2010, 16, 10546–10552 CrossRef CAS PubMed.
- U. A. Joshi, J. S. Jang, P. H. Borse and J. S. Lee, Appl. Phys. Lett., 2008, 92, 242106 CrossRef.
- J. A. Schneeloch, Z. Xu, J. Wen, P. M. Gehring, C. Stock, M. Matsuda, B. Winn, G. Gu, S. M. Shapiro, R. J. Birgeneau, T. Ushiyama, Y. Yanagisawa, Y. Tomioka, T. Ito and G. Xu, Phys. Rev. B: Condens. Matter, 2015, 91, 064301 CrossRef.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801–6809 CrossRef CAS PubMed.
- S. K. Choi, W. Choi and H. Park, Phys. Chem. Chem. Phys., 2013, 15, 6499–6507 RSC.
- C. Jiang, S. J. A. Moniz, M. Khraisheh and J. Tang, Chem. – Eur. J., 2014, 20, 12954–12961 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr04804d |
| This journal is © The Royal Society of Chemistry 2015 |