
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of diphenylalanine backbone homologues and their combination with functionalized carbon nanotubes†
Bhimareddy
Dinesh
a,
Marco A.
Squillaci
b,
Cécilia
Ménard-Moyon
a,
Paolo
Samorì
b and
Alberto
Bianco
*a
aCNRS, Institut de Biologie Moléculaire et Cellulaire, Laboratoire d'Immunopathologie et Chimie Thérapeutique, 67000 Strasbourg, France. E-mail: a.bianco@ibmc-cnrs.unistra.fr
bISIS & icFRC, Université de Strasbourg & CNRS, 8 allée Gaspard Monge, 67000 Strasbourg, France
First published on 7th September 2015
Abstract
The integration of carbon nanotubes (CNTs) into organized nanostructures is of great interest for applications in materials science and biomedicine. In this work we studied the self-assembly of β and γ homologues of diphenylalanine peptides under different solvent and pH conditions. We aimed to investigate the role of peptide backbone in tuning the formation of different types of nanostructures alone or in combination with carbon nanotubes. In spite of having the same side chain, β and γ peptides formed distinctively different nanofibers, a clear indication of the role played by the backbone homologation on the self-assembly. The variation of the pH allowed to transform the nanofibers into spherical structures. Moreover, the co-assembly of β and γ peptides with carbon nanotubes covalently functionalized with the same peptide generated unique dendritic assemblies. This comparative study on self-assembly using diphenylalanine backbone homologues and of the co-assembly with CNT covalent conjugates is the first example exploring the capacity of β and γ peptides to adopt precise nanostructures, particularly in combination with carbon nanotubes. The dendritic organization obtained by mixing carbon nanotubes and peptides might find interesting applications in tissue engineering and neuronal interfacing.
Introduction
In natural systems, biomolecules often assemble spontaneously and reversibly through the combination of noncovalent interactions to generate highly ordered and functional nanostructures.1–3 Such biomolecular systems in form of nanofibrils, nanospheres and hydrogels have been exceptionally useful in the field of biomedical applications, including drug delivery, tissue engineering and biosensors.2,4,5 Among the different types of biomolecules, peptides are extremely attractive tools due to their chemical properties, structural diversity and biocompatibility.6,7 Diphenylalanine (FF) – the core recognition motif of the Alzheimer β-amyloid polypeptide – is considered one of the simplest and most studied building block.7,8 The ease of synthesis and modification of this small peptide facilitated the design of various nanostructures such as nanotubes,9 nanofibers,10 nanospheres,11 nanovesicles,12 and nanowires.13The structure and the function of peptides are the resultant of their amino acid composition. They can be altered by applying strategic modifications at the level of each amino acid residues. These modifications may include alterations made on the backbone or on the side chain of the residues.14,15 The peptides with such modified parts display enhanced biological stability toward proteolysis.16,17 Besides this, the use of backbone homologated amino acids bearing proteinogenic side chains has emerged as a successful approach to generate secondary structural motifs in peptidomimetics, called foldamers.14,18,19 The additional methylene groups on the backbone of the peptide provides several degrees of conformational space resulting in various secondary structures. The hybrids of phenylalanine dipeptides have shown excellent self-assembling properties leading to the formation of nanospheres and nanotubes.20,21 However, the understanding of the structural diversity of the assemblies formed by these hybrid peptides is still limited due to the prevalent use of only natural residues. Further investigations using exact analogues of backbone homologated diphenylalanines will allow to better explore the characteristics of the assemblies originated by the backbone extensions, alone or in combination with nanomaterials, and to expand their applications.
Over the past two decades, carbon nanotubes (CNTs) have gained enormous attention due to their peculiar chemical, electrical and mechanical properties and found potential uses in electronics as well as in biomedical engineering.22,23 In spite of their unique properties, the major stumbling drawback was their solubility in aqueous and non-aqueous media. In this context, surface functionalization of CNTs with biologically relevant molecules such as nucleobases or oligonucleotides and peptides allowed to obtain hybrid materials much easier to manipulate with properties that were exploited for sensing and biomedical applications.22,24,25 Such nanomaterials could also lead to well-defined structures driven by hydrogen bonding and π–π interactions.26 Among all conjugates, covalent CNT–peptide conjugates have been proved to be very promising as biosensors and biomedical tools.27–29 However, the field of the self-assembly of CNTs in combination with amino acids or peptides remains still little explored considering the wide range of possibilities using all α-amino acid homologues and mimetics. Extended studies in this direction would be helpful for obtaining a better understanding on the interaction between these two classes of molecules.
Herein, we present a study on the self-assembly of the backbone-homologated analogues of diphenylalanine motif constituted by β3(R)Phe and γ4(R)Phe. These amino acids are derived by homologation of the naturally occurring α(S)Phe. The structural variations induced by solvent and pH changes in both peptides have been also explored. Moreover, we studied the co-assembly of β and γ dipeptides and functionalized CNTs (f-CNTs). The self-assemblies were analyzed by using complementary electron microscopy and spectroscopy techniques.
Results and discussion
Self-assembly of β and γ peptides
The N-terminal tert-butyloxycarbonyl (Boc)-protected FF peptide has been extensively studied and it was found to form tubular structures in water and spherical assemblies in ethanol.11 In this regard we have been initially interested to investigate if these assemblies persist with the increase in the number of carbon atoms within the backbone of the peptide. Hence, we selectively choose to synthesize two peptides, corresponding to Boc-β3(R)Phe-β3(R)Phe-OH (1) and Boc-γ4(R)Phe-γ4(R)Phe-OH (2) (Fig. 1). These two peptides are backbone homologues of Boc-protected diphenylalanine (Boc-α(S)Phe-α(S)Phe-OH) formed by inserting two and four CH2 groups in β (1) and γ (2) analogues, respectively. | ||
| Fig. 1 Chemical structures of Boc-β3(R)Phe-β3(R)Phe-OH (1) and Boc-γ4(R)Phe-γ4(R)Phe-OH (2). The additional methylene groups within the peptide backbone are highlighted in red. | ||
Taking into consideration the different experimental conditions reported for the assembly of Boc-Phe-Phe-OH,11,30 we started our observations using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion ethanol/water solution (50% ethanol solution). To assemble the peptides 1 and 2, the stock solution of each peptide in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 100 mg mL−1 was diluted by adding 50% ethanol solution yielding the desired concentrations of peptide (see Experimental section in ESI†). The vortexed solutions were allowed to self-assemble over 12 hours.
:1 proportion ethanol/water solution (50% ethanol solution). To assemble the peptides 1 and 2, the stock solution of each peptide in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 100 mg mL−1 was diluted by adding 50% ethanol solution yielding the desired concentrations of peptide (see Experimental section in ESI†). The vortexed solutions were allowed to self-assemble over 12 hours.
The peptide self-assembly at different concentrations starting from 10 down to 1 mg mL−1 was observed using transmission electron microscopy (TEM). At all concentrations, both β and γ peptides were found to form huge aggregated nanofibers (not shown). To gain better insights into the organization of these fibers, the peptides were observed in more diluted solutions (25–100 μg mL−1 range). At the concentration of 25 μg mL−1, peptide 1 was able to form nanofiber bundles originating from a central nucleating point (Fig. 2a and b). The diameter of the fibers was measured in the range of 20 to 80 nm, with several microns in length. The nanofibers formed by peptide 2 (at 40 μg mL−1) were instead shorter, more compact and aligned in parallel-like arrangement creating a sort of film (Fig. 2c and d). Highly oriented nanofibers of both peptides at the same concentrations of TEM were also observed by atomic force microscopy (AFM) (Fig. 3). The line profile (Fig. 3b) of peptide 1 shows that the nanofibers formed by depositing 100 μL solution of the 25 μg mL−1 concentration measure about 20 nm in height and 60 nm in width. Furthermore, to assess the stability and the lifetime of these assemblies, the peptide solutions were stored at room temperature for 30 days and their assemblies were found unaltered as shown by scanning electron microscopic (SEM) analysis (Fig. S1†), thus revealing their long time stability towards ageing.
 | ||
| Fig. 2 TEM images of the assemblies of peptide 1 (at 25 μg mL−1) (a), (b) and 2 (at 40 μg mL−1) (c), (d). The assembly of both peptides leads to distinctively different fibrillar structures. | ||
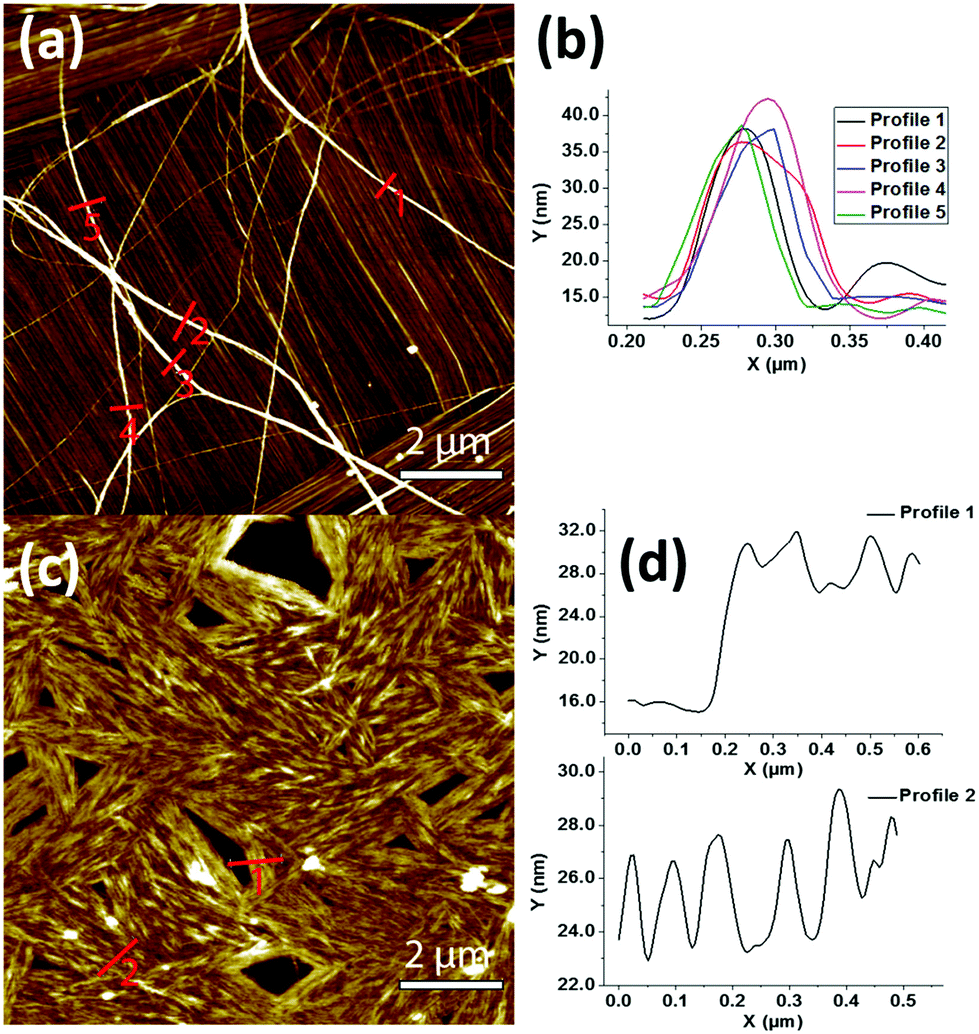 | ||
| Fig. 3 AFM images of peptides 1 (a) and 2 (c) nanofibers formed from 25 and 40 μg mL−1 solutions, respectively, and corresponding topographical profiles of peptide 1 (b) and 2 (d). (Image Z scale (a) = 30 nm, (b) = 45.5 nm). | ||
Next, to check the nature of these assemblies in similar protic solvents and to evaluate the role of water and time course to form the nanostructures, the dilutions were carried out in pure methanol from a stock solution of the peptides in pure methanol (without HFIP). A well-mixed peptide solution was immediately drop cast and after the solvent evaporation, the TEM images (Fig. S2†) allowed to observe nanofibers with almost similar morphology to those obtained in 50% ethanol solution (Fig. 2). The dendritic assemblies of peptide 1 are formed almost instantaneously, whereas the assembly of peptide 2 led to a uniform film rather than to defined nanofibers. This indicates that the self-assembled structures were formed in a very short time with no effects in changing the type of protic solvent. Then we decided to observe the assembly of both peptides in water. We diluted the peptide stock solutions in HFIP with distilled water to a concentration of 1 mg mL−1, and allowed the peptides to assemble before observation by TEM (Fig. S3†). The morphology still remains nanofibrillar unlike Boc-αPhe-αPhe-OH, for which it was found that the structure changed from spherical to tubular structures upon changing the solvent from ethanol to water.11 The observations of peptides 1 and 2 revealed that our nanostructures are stable at different concentrations and solvents. A recent study on N-acetyl capped β3 peptide hetero-oligomers reported that the N-terminal capping was a critical parameter in promoting fiber growth.31 Here, in order to verify this parameter, both C- and N-terminal free β peptide [β3(R)Phe-β3(R)Phe] was probed for self-assembly. TEM analysis showed the complete absence of regular structures (Fig. S4†) supporting the key role of N-capping in forming the fibers. The highly hygroscopic oily nature of the free γ peptide [γ4(R)Phe-γ4(R)Phe] prevented us to conduct the control self-assembly experiments. Further spectroscopic observations were then performed to assess in more details the secondary structure of these systems.
The assembly of respective β and γ peptides 1 and 2 obtained from 50% ethanol solution was analyzed by solid-state Fourier-transform infrared (FT-IR) spectroscopy.32 The region 3500–3200 cm−1 is important for assessing N–H stretching vibrations, which provide hydrogen bonding information in peptides and proteins.33–35 The region 1800–1500 cm−1 corresponds to the stretching band of amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and the bending peak of amide II (C–N stretching and N–H bending).33,36Fig. 4a shows two defined N–H stretching bands at 3357 and 3324 cm−1 for peptide 1, and a band at 3351 with a shoulder at 3324 cm−1 for peptide 2, respectively. It is well-established that the non-hydrogen bonded N–H corresponds to a higher energy bands, while the lower energy bands are due to intramolecularly hydrogen bonded N–H.36,37 In this context, the relatively higher band proportion at 3324 cm−1 of 1 in comparison to 2 may be due to a stronger intramolecular hydrogen bonding. This could be attributed to the observed highly ordered fibers in 1.
O stretching) and the bending peak of amide II (C–N stretching and N–H bending).33,36Fig. 4a shows two defined N–H stretching bands at 3357 and 3324 cm−1 for peptide 1, and a band at 3351 with a shoulder at 3324 cm−1 for peptide 2, respectively. It is well-established that the non-hydrogen bonded N–H corresponds to a higher energy bands, while the lower energy bands are due to intramolecularly hydrogen bonded N–H.36,37 In this context, the relatively higher band proportion at 3324 cm−1 of 1 in comparison to 2 may be due to a stronger intramolecular hydrogen bonding. This could be attributed to the observed highly ordered fibers in 1.
 | ||
| Fig. 4 FT-IR spectra of peptides 1 and 2 in the amide A region (a) and amide I and amide II regions (b). | ||
The amide I peaks at 1656 cm−1 in 1 and at 1645 cm−1 in 2 evidence the presence of helical conformations in both peptides within the fibers.38 This is in agreement with the reported C14-helical structures of homooligomeric γ-peptides.39 The lower frequency band at 1645 cm−1 in 2 may be due to the higher flexibility in the backbone of this peptide combined to a higher solvent exposure, resulting in different stretching frequency than that of peptide 1.38 The frequency of amide I bands at ∼1685/1689 cm−1 is generally assigned to the β-sheet structure, similarly to other polypeptides and protein secondary structures.33,38 In contrast to this assumption, a recent study suggests that the band around this frequency may also originate from the carbamate functionality,40 as it is likely the case of our Boc-protected peptides. Moreover, it is interesting to bring to the attention the aromatic CC stretching in the region 1500–1400 cm−1, which is due to the contribution of the side chains of the peptides. In Fig. 4b, the peak at 1446 cm−1 in 1 is less intense than that in 2 at 1453 cm−1. This is likely due to significant π–π interactions in peptide 1.
The diphenylalanine based self-assembled nanostructures can be modulated by varying the experimental conditions such as solvents, concentrations, pH and temperature. For example, in the case of diphenylalanine the reversible transition between nanotubes and nanovesicles was demonstrated by adjusting the concentration,41 and from nanospheres to nanotubes by changing the solvents from ethanol to water.11 In this regard, since the peptides 1 and 2 form nanofibers in both tested solvents (i.e. 50% ethanol and water), we decided to study the effect of pH.
To probe the influence of pH, 30 μL of 0.1 M HCl solution was added to the peptide solutions (1 mL) at 25 μg mL−1 of 1 and 40 μg mL−1 of 2 in 50% ethanol. The solutions were carefully mixed and allowed to self-assemble overnight. The TEM analysis revealed the transformation of the fibers into partly coalesced spherical structures in the case of peptide 1 (Fig. 5a and S5a†) and led to the disassembly with formation of an irregular film in the case of peptide 2 (Fig. 5b and S5b†). The size distribution of the nanospheres of peptide 1 measured from ∼2000 non-coalesced spheres from TEM images varied from 25 to 140 nm with an average of ∼95 nm (Fig. S6†). The coalesced spheres formed here are similar to the microspheres observed in aromatic tripeptide assemblies42 and metal-mediated modified Phe–Phe dipeptide-based soft spherical structures.43 The structural differences between the spherical and fibrilar assemblies could be due to π–π interactions43 and difference in the hydrogen bond strength as already observed in the transition between spherical to tubular assemblies.44 The spherical structures of 1 at low pH may be due to stronger hydrogen bonding that could withstand the pH variation. The disassembly of 2 at low pH may be due instead to the weaker hydrogen bonding as observed by FT-IR analysis (Fig. 4a). Overall, the dendritic fibrillar assembly of beta peptide 1 and parallel-like arranged nanofibers of gamma peptide 2 were stable upon changing the solvent systems from alcohol to water. The parent analogue Boc-Phe-Phe-OH modifies instead its assembly from spherical to tubular nanostructures upon changing the solvent system (from ethanol to water).11 The structural stability of diphenylalanine homologues in different solvents implies that their assembly is driven mostly by π–π interactions rather than hydrogen bonding. In contrast, the structures of Boc-Phe-Phe-OH were altered by a simple change in solvent, accounting for an assembly stabilized by weaker hydrogen bonding.
 | ||
| Fig. 5 TEM images after changing the pH by adding 0.1 M HCl. Coalesced nanospheres of peptide 1 (a). Disassembled morphology of the peptide 2 (b). | ||
Self-assembly of peptide-functionalized carbon nanotubes
In the recent years, we and others have demonstrated the utility of f-CNTs in different domains, covering for example cellular uptake by a wide range of cells,45 formation of catalytic silver nanoparticles,46 and assembly into precise nanostructures.47,48 In all these reports the binding affinity between specific moieties and CNTs played an important role. For example, adenine covalently functionalized to CNTs was responsible to trigger the formation of catalytic nanoparticles with controlled size.46 In another study, uracil covalently conjugated to CNTs self-assembled into nanorings.47 In these previous works, the binding affinity between nucleobases and CNTs due to aromaticity influenced the formation of different molecular architectures. Similarly, other studies also draw conclusions that peptides containing aromatic residues have greater interactions with CNTs.49–52 More recently, the diphenylalanine nanotubes combined with CNTs were used in amperometric biosensors to detect NADH and phenol.53,54 In all these works, the aromatic moiety bound on the surface of CNTs either through covalent or noncovalent interactions resulted efficient in dispersing CNTs for various electronic and biomedical applications. On the basis of all these studies, we have focused our interest to the investigation of the precise self-assembled structures generated by combining aromatic peptides 1 and 2 with CNTs, either modified with the same peptide or just simply using the CNT precursors.Based on our long term experience in CNTs, here we decided to study the CNT–peptide assembly using multi-walled carbon nanotubes (MWCNTs). Peptide 1 was covalently conjugated to MWCNTs from both N- and C-terminal parts (Scheme 1) and peptide 2 from C-terminus. The peptides were conjugated to CNTs by inserting a diaminotriethylene glycol (TEG) linker (MWCNT-TEG-NH3+) to provide a certain flexibility for the self-assembly of the peptide. The different conjugates were characterized by thermogravimetric analysis (TGA) and TEM. The loading of the N-free peptide on MWCNT-TEG-β3(R)Phe-β3(R)Phe-NH2 (3) and MWCNT-TEG-γ4(R)Phe-γ4(R)Phe-NH2 (5) was 126 μmol g−1 (Fig. S7a†) and 84 μmol g−1 (Fig. S7c†), respectively, while the amount of C-free peptide on MWCNT-TEG-β3(R)Phe-β3(R)Phe-OH (4) was 57 μmol g−1 (Fig. S7b†).
 | ||
| Scheme 1 Synthesis of MWCNT–peptide conjugates. (i) (a) Neat (COCl)2; (b) Boc-NH-(CH2CH2O)2-CH2CH2-NH2, dry tetrahydrofuran; (c) 4 N HCl in dioxane. (ii) (a) Boc-β3(R)Phe-β3(R)Phe-OH, EDC, DIPEA, HOBt; (b) 4 N HCl in dioxane; (c) neutralized with 2 N NaOH. (iii) Succinic anhydride, DIPEA. (iv) (a) EDC, DIPEA, HOBt, H2N-β3(R)Phe-β3(R)Phe-OMe; (b) alkaline hydrolysis; (c) 1 N HCl. (v) (a) Boc-γ4(R)Phe-γ4(R)Phe-OH, EDC, DIPEA, HOBt; (b) 4 N HCl in dioxane; (c) neutralized with 2 N NaOH. | ||
We first decided to focus our attention on the self-assembly study of the C-free terminus CNT–peptide conjugate 4. The stock solution of the CNT–peptide conjugates was directly prepared in 1:1 proportion ethanol/water by sonicating for a few minutes until the dispersion became homogeneous. Then, the conjugates, diluted with 50% ethanol to a final concentration of 50 μg mL−1, were allowed to self-assemble overnight. The supernatant solution analyzed by TEM shows disorganized structures while there are no changes in the morphology of the peptide-functionalized CNTs in comparison to the CNT precursors (Fig. S8†). On the basis of these findings, we wanted to explore the behavior of the N-free CNT–peptide conjugate 3 in the same conditions. The high resolution scanning electron microscopy (HR-SEM) and TEM analyses revealed the presence of big aggregated CNTs forming interesting rope-like morphologies of several hundred microns in length (Fig. S9a and S10a†). The formation of these aggregates may be attributed to the strong interaction between the unreacted carboxylic acid functions of CNTs and the amine functions of the peptide through strong hydrogen bonding, in addition to the intertube aromatic interactions of the phenyl groups of the peptide. To assess this assumption, the solution of conjugate 3 was protonated by adding 50 μL of 0.1 M HCl solution. The HR-SEM observation showed that the CNT assembly was clearly disrupted, likely by breaking the hydrogen bonds following the protonation of the peptide N-terminal amines (Fig. S9b†). Hence, these morphological differences observed for conjugates 3 and 4 could be due to the strong hydrogen bonding capacity of the terminal NH of the peptide in conjugates 3 and the carboxylic groups of CNTs, leading to formation of rope-like structures. Whereas weaker interactions between the terminal OH of the peptide in conjugates 4 and CO of CNTs might not be enough to retain a similar kind of morphology. To explore further the network properties of the conjugate 3, we studied its co-assembly with the peptide 1.
Based on previous observations, we decided to use a concentration of the peptide 1 at 25 μg mL−1 and 50 μg mL−1 of conjugate 3 (previously sonicated to obtain a homogeneous dispersion). We mixed the solutions of CNT–peptide conjugate and peptide in the ratios 1:1, 1:2 and 1:5, vortexed for one minute, and allowed to self-assemble overnight. Interestingly, the TEM observation of the 1:5 ratio revealed the presence of regular dendritic assemblies, where CNTs were at the core of the ramifications likely nucleating their formation (Fig. 6a).
 | ||
| Fig. 6 TEM images of co-assembled peptide 1 and CNT–peptide conjugate 3 (a), and co-assembled peptide 2 and CNT–peptide conjugate 5 (b) showing similar dendritic assemblies with CNTs nucleating the structures. | ||
Intrigued by the behavior of CNT–peptide conjugate 3 and its co-assembly with β peptide 1, we decided to study the assembled nanostructures of CNT–peptide conjugate 5 in similar conditions. The TEM analysis of incubated CNT–peptide conjugate 5 (50 μg mL−1) showed the formation of aggregated clusters (Fig. S10b†), morphologically different from that of CNT–peptide conjugate 3. We then co-assembled the CNT–peptide conjugate 5 and peptide 2 (40 μg mL−1) by mixing them homogeneously in the ratios 1:1, 1:3 and 1:5. Surprisingly, the TEM analysis of the 1:1 ratio revealed the complete absence of assembly (Fig. S11†), but the 1:5 ratio displayed the formation of the dendritic structures similar to those of the co-assembled peptide 1 and CNT–peptide conjugate 3 (Fig. 6b).
These dendritic assemblies are clearly influenced by the formation of a network between the peptide and the CNT–peptide conjugate. The CNT conjugates outside the nucleation point are showing the tendency towards the formation of fibers (Fig. S12a†). It was then interesting to check whether the amino group on CNTs without peptide would have similar kind of interactions leading again to dendritic assembly. Hence, we examined the co-assembly of peptide 1 with the MWCNT-TEG-NH2, the precursor of conjugate 3. The TEM analysis showed no dendritic assembly with varied mix proportions of the peptide 1 and MWCNT-TEG-NH2 (Fig. S13†). This shows that the peptide conjugated to CNTs is responsible of forming the dendritic network. Next, in order to check if the variation of the pH, leading to the formation of the nanospheres of 1, has an effect on the dendritic morphology of co-assembled CNT–peptide conjugates, we mixed the solution of CNT–peptide conjugate 3 and peptide 1 at 1:5 ratio (0.6 mL) and added 50 μL of 0.1 M HCl. The TEM image shows no dendritic assembly but a random distribution of peptide nanospheres and dispersed CNT–peptide conjugates. However, some interactions between the peptide nanospheres and the CNT–peptide conjugates can be still seen (Fig. S14†).
The known stability of unnatural amino acids and the dendritic interactions between the backbone extended peptides and CNTs, found by our investigations, can open the possibility to explore their use in electronic and biomedical applications. Altogether, the observations from the present study suggest that the peptides containing higher homologues of α-amino acids with proteinogenic side chains can be the promising building blocks to design various types of nanostructures.
Conclusions
In summary, we have demonstrated the supramolecular structural differences between the backbone homologated β and γ diphenylalanine analogues. We have found that β peptide Boc-β3(R)Phe-β3(R)Phe-OH and γ peptide Boc-γ4(R)Phe-γ4(R)Phe-OH self-assemble to generate fibers of different dimensions and shapes, which undergo morphological changes at acidic pH. This pH sensitivity of the systems could provide the basis for developing novel pH sensitive β and γ peptide-based materials including controlled drug release systems. On the other hand, we have found that the N- and C-terminally free β peptide (β3(R)Phe-β3(R)Phe-OH) failed to form any regular structures. Furthermore, co-assembly of β and γ peptides and CNT–peptide covalent conjugates have shown an excellent mutual affinity by forming regular dendritic morphologies. The stable fibrillar nature of β and γ peptides in water as well as 50% ethanol suggests the idea to use these systems as scaffolds for designing stable hydrogels.55,56 Future work will focus on using these self-assembled systems for developing hybrid hydrogels. Using these peptides and CNTs with suitable modifications it would be possible to incorporate and deliver therapeutic agents or envisage their use also as support for neuronal interfacing.Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique (CNRS), by the Agence Nationale de la Recherche (ANR) through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC), the EC-ERC project SUPRAFUNCTION (GA-257305) and by the International Center for Frontier Research in Chemistry (icFRC). BD wishes to thank support by the ANR project Nanomultisens (ANR-13-BS10-0003-02). MS wishes to acknowledge for support by the EC project MSCA-SYNCHRONICS (GA-643238). The authors wish to acknowledge Cathy Royer and Valérie Demais for TEM and SEM analyses at the “Plateforme Imagerie in Vitro” at the Center of Neurochemistry (Strasbourg, France). Rajendra Kurapati is acknowledged for fruitful discussions during the manuscript preparation.References
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- Y. Loo, S. Zhang and C. A. E. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS PubMed.
- A. Bianco, M. Venanzi and C. Aleman, J. Pept. Sci., 2011, 17, 73–74 CrossRef CAS PubMed.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- E. Gazit, Nat. Chem., 2015, 7, 14–15 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- R. Huang, R. Su, W. Qi, J. Zhao and Z. He, Nanotechnology, 2011, 22, 245609 CrossRef PubMed.
- L. Adler-Abramovich, N. Kol, I. Yanai, D. Barlam, R. Z. Shneck, E. Gazit and I. Rousso, Angew. Chem., Int. Ed., 2010, 49, 9939–9942 CrossRef CAS PubMed.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- J. Ryu, S.-W. Kim, K. Kang and C. B. Park, ACS Nano, 2010, 4, 159–164 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- S. Hecht and I. Huc, Foldamers: structure, properties and applications, John Wiley & Sons, 2007 Search PubMed.
- E. A. Harker and A. Schepartz, ChemBioChem, 2009, 10, 990–993 CrossRef CAS PubMed.
- R. W. Cheloha, A. Maeda, T. Dean, T. J. Gardella and S. H. Gellman, Nat. Biotechnol., 2014, 32, 653–655 CrossRef CAS PubMed.
- D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CAS.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- S. Maity, S. Nir and M. Reches, J. Mater. Chem. B, 2014, 2, 2583–2591 RSC.
- S. Parween, A. Misra, S. Ramakumar and V. S. Chauhan, J. Mater. Chem. B, 2014, 2, 3096–3106 RSC.
- S. Kruss, A. J. Hilmer, J. Zhang, N. F. Reuel, B. Mu and M. S. Strano, Adv. Drug Delivery Rev., 2013, 65, 1933–1950 CrossRef CAS PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015 DOI:10.1021/acs.chemrev.5b00008.
- A. Battigelli, C. Ménard-Moyon, T. Da Ros, M. Prato and A. Bianco, Adv. Drug Delivery Rev., 2013, 65, 1899–1920 CrossRef CAS PubMed.
- S. Marchesan, K. Kostarelos, A. Bianco and M. Prato, Mater. Today, 2015, 18, 12–19 CrossRef CAS.
- W. j. Jeong and Y. b. Lim, Macromol. Biosci., 2012, 12, 49–54 CrossRef CAS PubMed.
- C. Klumpp, K. Kostarelos, M. Prato and A. Bianco, Biochim. Biophys. Acta, 2006, 1758, 404–412 CrossRef CAS PubMed.
- C. Gaillard, M. Duval, H. Dumortier and A. Bianco, J. Pept. Sci., 2011, 17, 139–142 CrossRef CAS PubMed.
- C. Fabbro, H. Ali-Boucetta, T. Da Ros, K. Kostarelos, A. Bianco and M. Prato, Chem. Commun., 2012, 48, 3911–3926 RSC.
- S. Yuran, Y. Razvag and M. Reches, ACS Nano, 2012, 6, 9559–9566 CrossRef CAS PubMed.
- M. P. Del Borgo, A. I. Mechler, D. Traore, C. Forsyth, J. A. Wilce, M. C. J. Wilce, M.-I. Aguilar and P. Perlmutter, Angew. Chem., Int. Ed., 2013, 52, 8266–8270 CrossRef CAS PubMed.
- S. Bera, P. Jana, S. K. Maity and D. Haldar, Cryst. Growth Des., 2014, 14, 1032–1038 CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin, 2007, 39, 549–559 CrossRef CAS PubMed.
- V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- C. Toniolo and M. Palumbo, Biopolymers, 1977, 16, 219–224 CrossRef CAS PubMed.
- G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 1054–1062 CrossRef CAS.
- B. Dinesh, V. Vinaya, S. Raghothama and P. Balaram, Eur. J. Org. Chem., 2013, 3590–3596 CrossRef CAS.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- K. Basuroy, B. Dinesh, M. B. M. Reddy, S. Chandrappa, S. Raghothama, N. Shamala and P. Balaram, Org. Lett., 2013, 15, 4866–4869 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- X. Yan, Y. Cui, Q. He, K. Wang, J. Li, W. Mu, B. Wang and Z.-c. Ou-yang, Chem. – Eur. J., 2008, 14, 5974–5980 CrossRef CAS PubMed.
- S. Maity, P. Jana, S. K. Maity and D. Haldar, Langmuir, 2011, 27, 3835–3841 CrossRef CAS PubMed.
- G. Kaur, L. A. Abramovich, E. Gazit and S. Verma, RSC Adv., 2014, 4, 64457–64465 RSC.
- H. Matsui and C. Holtman, Nano Lett., 2002, 2, 887–889 CrossRef CAS.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, W. Sebastien, J. Luangsivilay, S. Godefroy, D. Pantarotto, J.-P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- P. Singh, G. Lamanna, C. Ménard-Moyon, F. M. Toma, E. Magnano, F. Bondino, M. Prato, S. Verma and A. Bianco, Angew. Chem., Int. Ed., 2011, 50, 9893–9897 CrossRef CAS PubMed.
- P. Singh, F. M. Toma, J. Kumar, V. Venkatesh, J. Raya, M. Prato, S. Verma and A. Bianco, Chem. – Eur. J., 2011, 17, 6772–6780 CrossRef CAS PubMed.
- P. Singh, J. Kumar, F. M. Toma, J. Raya, M. Prato, B. Fabre, S. Verma and A. Bianco, J. Am. Chem. Soc., 2009, 131, 13555–13562 CrossRef CAS PubMed.
- S. Wang, E. S. Humphreys, S.-Y. Chung, D. F. Delduco, S. R. Lustig, H. Wang, K. N. Parker, N. W. Rizzo, S. Subramoney, Y.-M. Chiang and A. Jagota, Nat. Mater., 2003, 2, 196–200 CrossRef CAS PubMed.
- H. Xie, E. J. Becraft, R. H. Baughman, A. B. Dalton and G. R. Dieckmann, J. Pept. Sci., 2008, 14, 139–151 CrossRef CAS PubMed.
- C. G. Salzmann, M. A. H. Ward, R. M. J. Jacobs, G. Tobias and M. L. H. Green, J. Phys. Chem. C, 2007, 111, 18520–18524 CAS.
- S. M. Tomásio and T. R. Walsh, J. Phys. Chem. C, 2009, 113, 8778–8785 Search PubMed.
- L. Adler-Abramovich, M. Badihi-Mossberg, E. Gazit and J. Rishpon, Small, 2010, 6, 825–831 CrossRef CAS PubMed.
- J. Yuan, J. Chen, X. Wu, K. Fang and L. Niu, J. Electroanal. Chem., 2011, 656, 120–124 CrossRef CAS.
- J. Nanda and A. Banerjee, Soft Matter, 2012, 8, 3380–3386 RSC.
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr04665c |
| This journal is © The Royal Society of Chemistry 2015 |