
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the relaxation rates of dual-mode T1/T2 nanoparticle contrast agents: a study into the ideal system†
Natasha A.
Keasberry
a,
Manuel
Bañobre-López
b,
Christopher
Wood
a,
Graeme. J.
Stasiuk
ac,
Juan
Gallo
*abd and
Nicholas. J.
Long
*ad
aDepartment of Chemistry, Imperial College London, South Kensington, London, SW7 2AZ, UK. E-mail: n.long@imperial.ac.uk; juan.gallo@inl.int
bInternational Iberian Nanotechnology Laboratory (INL), 4715-330 Braga, Portugal
cSchool of Biological, Biomedical and Environmental Sciences, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
dComprehensive Cancer Imaging Centre, Department of Surgery & Cancer, Hammersmith Campus, Imperial College London, Du Cane Road, London W12 0NN, UK
First published on 9th September 2015
Abstract
Magnetic resonance imaging (MRI) is an excellent imaging modality. However the low sensitivity of the technique poses a challenge to achieving an accurate image of function at the molecular level. To overcome this, contrast agents are used; typically gadolinium based agents for T1 weighted imaging, or iron oxide based agents for T2 imaging. Traditionally, only one imaging mode is used per diagnosis although several physiological situations are known to interfere with the signal induced by the contrast agents in each individual imaging mode acquisition. Recently, the combination of both T1 and T2 imaging capabilities into a single platform has emerged as a tool to reduce uncertainties in MR image analysis. To date, contradicting reports on the effect on the contrast of the coupling of a T1 and T2 agent have hampered the application of these specialised probes. Herein, we present a systematic experimental study on a range of gadolinium-labelled magnetite nanoparticles envisioned to bring some light into the mechanism of interaction between T1 and T2 components, and advance towards the design of efficient (dual) T1 and T2 MRI probes. Unexpected behaviours observed in some of the constructs will be discussed. In this study, we demonstrate that the relaxivity of such multimodal probes can be rationally tuned to obtain unmatched potentials in MR imaging, exemplified by preparation of the magnetite-based nanoparticle with the highest T2 relaxivity described to date.
Introduction
MRI is a powerful technique perfectly suited for biological imaging.1,2 Properties like non-invasiveness, high penetration, high spatial resolution and absence of ionising radiations,2 make it one of the first choices not only for human clinical diagnosis, but also for the monitoring of the progression of diseases/treatments, and for drug design. MRI images water protons, measuring the relaxation time of their nuclear magnetic spins once they have been excited with a radio-frequency pulse under a static magnetic field.3 The contrast in MR images arises from the different chemical environment of these water protons, which induces differences in the water proton relaxation times (T). This relaxation time can be greatly affected by the presence of paramagnetic or superparamagnetic compounds in the surroundings of the water molecules, leading to an enhanced contrast on the MR images. There are two major MRI modalities in use in hospitals when it comes to the use of contrast agents; T1- and T2-weighted imaging. Gadolinium-containing T1 contrast agents are the preferred contrast agent in the clinic as the enhanced signal produced is more favourable and easier to distinguish (although adipose tissue is a well-known interference).4T2 agents such as iron oxide nanoparticles (IONPs), on the other hand produce a decrease in signal intensity which can be misinterpreted as bleeding, calcification or other abnomalies.5,6 Nanoparticles are a versatile platform to which targeting molecules and drugs can be coupled.7,8 Thus, having a single multifunctional probe integrating both T1 and T2 contrast capabilities can overcome the drawbacks of both MRI modalities, with compatible T1 and T2 acquisitions, allowing for artefacts to be reduced and diagnostics to be doubly verified.The earliest example of dual contrast T1/T2 MR imaging was carried out by Weissleder et al. to image liver tumours in rats. When Gd-DTPA and ferrite nanoparticles were co-administered, an enhanced tumour signal from the Gd-DTPA was observed while the liver showed a negative signal intensity from the accumulation of ferrite.9 Their results showed an increase in r1 but no effect on r2 when both agents were present at the same time. The same effect was observed when the contrast agents were administered sequentially in humans, in studies carried out by Semelka et al. and Kubaska et al.10,11 Theoretically, integration of the T1 and T2 contrast as a single entity as opposed to having them separately, would result in T1 spin alignment in the same direction as the magnetic field induced by the T2 material, enhancing the T1 effect while maintaining the T2 signal.4,12 This theory was in part confirmed by experimental evidence from Gao et al.12 Gd2O3-embedded Fe3O4 nanoparticles (GdIO) were synthesised and presented a synergistic enhancement of r1 and r2. The GdIO showed higher r2 than Fe3O4 of similar size, as well as higher r1 than Gd2O3 of similar size. In addition to this, the Gd2O3 nanoparticles showed no enhanced T2 contrast, while Fe3O4 nanoparticles showed limited enhanced T1 contrast. Other inorganic hybrid systems combining T1 and T2 nanoparticles such as those synthesised by Im et al.13 (Fe3O4/MnO) and Kim et al.14 (Gd-doped iron oxide nanoparticles) have also shown enhanced T1 contrast while retaining the T2 effects.
Although inorganic nanoparticle hybrids have shown some promising results, simple conjugation of a paramagnetic chelate to the surface of iron oxide nanoparticles is an even more attractive method to produce dual-mode contrast agents according to the reports published so far.15–18 The theoretical assumption is that these simpler combined agents will present an enhanced r1 without the r2 being significantly affected. Actual results however, are much more complex. Yang et al.15 and Bae et al.16 showed independently with different T1/T2 systems, that r2 strongly decreases when iron oxide nanoparticles are conjugated to Gd chelates, accompanied by an enhancement in the r1. Choi et al.17 also observed the same trend with a possible dependence on the distance between the Gd and the magnetic core. Finally, Huang et al.18 showed the opposite trend where both r1 and r2 increased with Gd concentration when the T1 and T2 moieties were coupled. In all these cases, r1 increases when Gd is incorporated. However, the r2 trends are not consistent between different publications/systems.
These contradicting results demonstrate that further studies are required to understand the relationship between the final relaxation rates and the design and structure of the T1/T2 probes. Thus, in this research, a systematic series of iron oxide nanoparticles functionalised with Gd chelates were prepared to better understand the effect of this interaction on the final relaxivity of the system, as well as to search for the ideal (dual) MRI probe design. A rational screening of different parameters arising from the combination of these T1 and T2 moieties was performed i.e. nature of the organic coating of the magnetic nanoparticles, distance between the magnetic and paramagnetic components, magnetic properties, and nature and presentation of the paramagnetic component.
Materials and methods
General
All reagents except DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) (CheMatech) and PEG molecules (Quanta Biodesign and Iris Biotech) were purchased from Sigma-Aldrich and used as supplied. 6 nm Fe3O4 nanoparticles were prepared according to Sun et al.19 by thermal decomposition. Fourier transform infrared (FT-IR) spectra were recorded using a Perkin Elmer Spectra 100 FT-IR Spectrometer. UV-Vis spectra were recorded using a Perkin Elmer Lambda 25 UV/Vis Spectrometer. A JEOL 2010 transmission electron microscope working at 200 keV was used to image the nanoparticles. Hydrodynamic size studies were performed either/both on a Malvern Zetasizer Nano ZS instrument or a Perkin Elmer Delsa NanoS. T1 and T2 relaxation times were measured with a Minispec mq60 relaxometer working at 1.47 T and selected samples were measured also in a DRX400 Bruker NMR spectroscope at 9.4 T. Imaging was performed in a 3 T horizontal bore MR Solutions Benchtop MRI system (Guildford, UK) equipped with 48 G cm−1 actively-shielded gradients. For imaging the sample, a 56 mm diameter quadrature birdcage coil was used in transmit/receive mode.Ligand exchange on 6 nm Fe3O4 nanoparticles with 11-aminoundecanoic acid
Prior to the ligand exchange, 2 mL (3 mg mL−1 Fe) of the original nanoparticle solution were washed extensively three times with methanol, and twice with each ethanol and acetone (5 mL each), followed by centrifugation for 5 minutes at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g. Finally the sample was resuspended in THF (4 mL) and mixed with water (4 mL) containing 11-aminoundecanoic acid (50 mg, 0.24 mmol). This solution was stirred for 48 h and then was directly centrifuged for 5 minutes at 17000g, the supernatant discarded and the pellet washed in the same way with a 1:1 mixture of THF:water (5 mL total). Finally the pellet was resuspended in water (2 mL) and stored in the fridge until further use.
000g. Finally the sample was resuspended in THF (4 mL) and mixed with water (4 mL) containing 11-aminoundecanoic acid (50 mg, 0.24 mmol). This solution was stirred for 48 h and then was directly centrifuged for 5 minutes at 17000g, the supernatant discarded and the pellet washed in the same way with a 1:1 mixture of THF:water (5 mL total). Finally the pellet was resuspended in water (2 mL) and stored in the fridge until further use.
Ligand exchange on 6 nm Fe3O4 nanoparticles with alendronic acid
Bulk oleic acid-capped nanoparticles (2 mL, 3 mg mL−1 Fe) were precipitated out of hexane and resuspended in THF (2 mL). Sodium alendronate trihydrate (50 mg) was dissolved in water (5 mL) and the pH adjusted to 9 with 1 M KOH. The THF solution of the nanoparticles was added to the ligand solution, and stirred for 2 days at room temperature. Then, the stirrer bar was removed and the nanoparticles isolated with a magnet. The supernatant was decanted and the black solid suspended in water (2 mL). Acetone (10 mL) was added to wash out any remaining ligand and this was repeated a further two times. Finally, the nanoparticles were resuspended in water (5 mL) and the pH checked to ensure it was at pH 8 (adjusted accordingly with KOH solution).Fmoc-PEGx-CO2H coupling
Bifunctional carboxylic/Fmoc PEG molecules of different molecular weights were used for the fuctionalisation of the nanoparticles. In general, Fe3O4 nanoparticles (1 mg, 1.6 × 10−9 mol) were mixed in DMSO (2 mL) with PEG (1.6 × 10−6 mol), and EDC (10 mg, excess) was added to the solution. The reaction was allowed to take place overnight. Next day the sample was centrifuged (17000g, 5 minutes), the supernatant discarded, and the pellet washed three times with water in the same way. Then, to deprotect the amine, the pellet was resuspended with ultrasonication in a 80/20 mixture of DMF/piperidine (5 mL) and shaken for 15 min. After centrifugation under the same conditions, this deprotection step was repeated two more times and finally the pellet was resuspended in water and stored in the fridge until further use.
DOTA coupling
DOTA (194.1 μg, 4.8 × 10−7 mol) was activated in water with EDC (100.8 μg, 5.28 × 10−7 mol) and NHS (93.23 μg, 8.1 × 10−7 mol). After 90 minutes the product of this reaction was mixed with PEG functionalised nanoparticles (1 mg, 1.6 × 10−9 mol), and stirred overnight. To purify the final product, acetone (1 mL) was added and the particles were centrifuged (17000g, 5 minutes). The supernatant was kept to calculate the yield of the reaction and the pellet was washed two more times in the same way (acetone addition followed by centrifugation). Finally the nanoparticles were resuspended in water (2 mL), aliquoted, and stored in the freezer until further use.
The yield of the DOTA coupling was measured by incubating the washings from the coupling reaction with a known amount of GdCl3 overnight and subsequently measuring the amount of free non-chelated Gd(III) using xylenol orange and UV-Vis spectroscopy (ESI, S3†).20
Gd(III) chelation
In a model reaction, DOTA-functionalised nanoparticles (1.3 × 10−9 mol) were mixed in acetate buffer (2 mL, 0.2 M, pH 6.5) with a solution of GdCl3·6H2O (12.36 μg, 7.86 × 10−8 mol). The mixture was shaken overnight. After this time, the solution was destabilised by the addition of acetone (1 mL) and centrifuged for 5 minutes at 17000g. The supernatant was discarded, and the pellet was resuspended in water (2 mL) and stored until further use.
The amount of Gd(III) was calculated from three independent ICP-OES measurements.
Relaxivity measurements
Four aqueous dilutions of different nanoparticle concentrations (between 0 and 0.6 mM Fe and 0 and 0.02 mM Gd, when present) were prepared of each sample. For measurements in the Minispec (1.47 T) 200 μL of samples were prepared in relaxometer tubes in water. For measurements at 9.4 T, the samples (600 μL) were placed in standard NMR tubes along with a capillary tube filled with deuterated water to facilitate the locking. Standard CPMG and saturation recovery sequences were used for T2 and T1 measurements respectively. All experiments were performed at 37 °C. The relaxivity constants (both r1 and r2) were calculated as the slope of the curve fitting T1−1 or T2−1 values versus the metal of interest concentration in mM.TEM sample preparation
The nanoparticles were deposited from their solutions (7 μL) onto holey carbon TEM grids with 300 mesh (Agar Scientific, UK) and dried at room temperature before the imaging.Magnetic measurements
The magnetic properties of the hybrid systems were measured in a superconducting quantum interference device (SQUID) from Quantum Design. For the sample preparation, 20 μL of the solution sample were introduced in a PTFE sample holder also provided by Quantum Design. Then, the solvent was evaporated under vacuum until the sample was perfectly dry. Afterwards, the sample holder was closed and placed in a brass row. Magnetization curves as a function of the magnetic field (hysteresis loops) were measured at room temperature and 2 K under an applied magnetic field up to 2 T.MR imaging
For the phantom measurements, the samples at different concentrations (between 25 and 100 μM in Fe/Gd) were dissolved in 200 μL of water in 300 μL tubes. All MR images of the phantoms were acquired with an image matrix 256 × 252, FOV 60 × 60 mm, 3 slices with a slice thickness of 1 mm and 1 mm slice gap. For T2-weighted imaging a fast spin echo (FSE) sequence with the following parameters was used: TE = 11 ms, TR = 12000 ms, NA = 32. For T1-weighted imaging a fast spin echo based (FSE) sequence with the following parameters was used: TE = 11 ms, TR = 720 ms, NA = 32.
Results
The most straightforward idea to combine T2 and T1 effects into a single probe is the direct coupling of both species currently approved for human diagnostics, namely Gd(III) chelates and iron oxide nanoparticles. This strategy, in principle simpler than inorganic approaches,12,17,21,22 seems also more interesting as, according to previous reports,15,16,18,23 unexpected effects arising from this coupling can be observed in the final performance of the contrast agents. In this sense, this approach allows us to finely tune several design parameters that affect the relaxivity properties of the nanoprobes, independently from each other, and so establish a cause-effect relationship on their relaxivity properties and their suitability as T1 and/or T2 MRI probes. In our search for the ideal MRI contrast agent we decided to systematically explore this approach through the evaluation of fully characterised iron oxide nanoparticles functionalised with DOTA-Gd(III) molecules. This study was divided into several sample subsets to evaluate the effect of different parameters into the final relaxivity performance (Scheme 1). First, different molecules were studied as protecting/stabilising ligands on the nanoparticles (set 1). Once this first step was optimised, different T1 moieties were evaluated (set 2), and finally, the effect of the distance between the T1 and T2 components was also examined (set 3). | ||
| Scheme 1 Overview of the different modifications to the T1/T2 systems, with the particular modification of each set highlighted by the dashed lines. | ||
To evaluate the potential interactions between T2 and T1 moieties in a single probe, iron oxide magnetic nanoparticles were covalently functionalised with Gd chelates. A thermal decomposition protocol was adopted for the preparation of iron oxide nanoparticles as this methodology provides highly crystalline products and the reaction conditions can be closely controlled to obtain samples with different particle size and narrow size distribution.24 Highly monodisperse magnetite (Fe3O4) nanoparticles of 6 nm of particle size were prepared using iron acetylacetonate as starting material, as shown by TEM (Fig. 1A, ESI, Fig. S1†).19 Both the phase formation and high crystallinity of the nanoparticles were evidenced by selected area electron diffraction (SAED) and X-ray diffraction (XRD). The presence of secondary phases due to impurities was completely discarded as no extra reflection peaks were identified (Fig. 1).
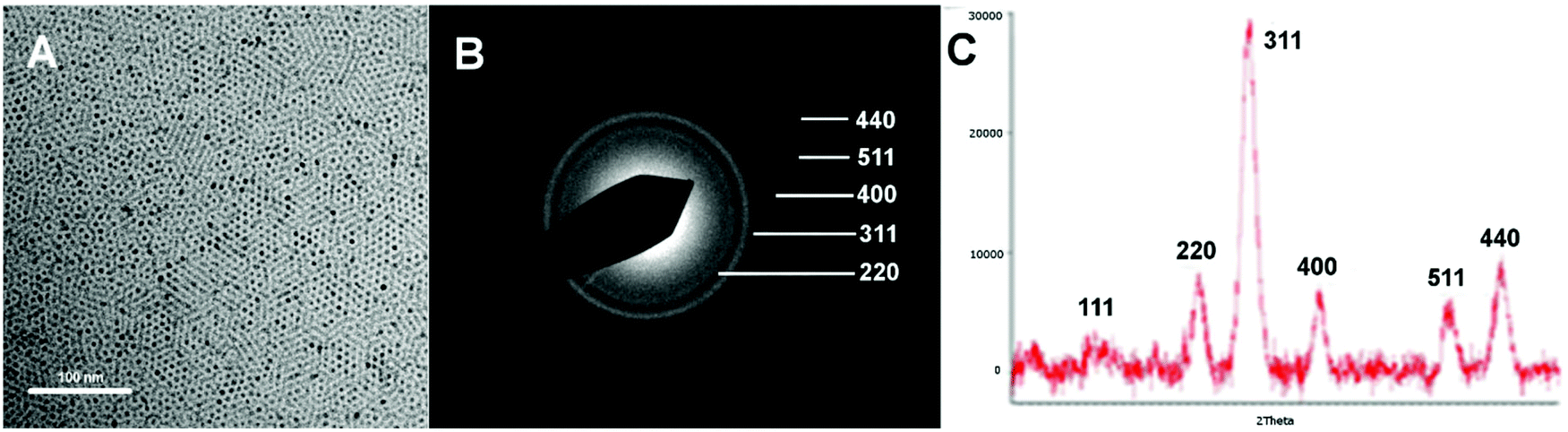 | ||
| Fig. 1 A, TEM micrograph of as-prepared Fe3O4 nanoparticles (oleic acid capped). B, SAED showing Fe3O4 lattice ring patterns. C, XRD diffractogram with indexed peak positions. Scale bar, 100 nm. | ||
As-prepared nanoparticles were only soluble in apolar solvents and thus not compatible with biological applications. A ligand exchange strategy was chosen to transfer the nanoparticles from organic to aqueous solution using more suitable bifunctional water soluble molecules. For this purpose, and to investigate the effect of ligands protecting/stabilising the nanoparticles on the final relaxivity, the nanoparticle surface was first functionalised with one of the following ligands (Scheme 2): 11-aminoundecanoic acid (AUA, a classic bifunctional carboxylic/amine small molecule, NP1), O-(2-phosphonoethyl)-O′-(2-aminoethyl)pentaethylene glycol (P-PEG6-NH2, a bifunctional phosphate/amine ligand, NP2), or alendronic acid, (ALA, a bifunctional, bisphosphonate/amine molecule, NP3). Phophates/bisphosphonates have not been explored as much as carboxylates as iron oxide ligands, but they have also been shown to render water soluble nanoparticles with good stability and magnetic properties.6,24–27 To evaluate the efficiency of the ligand exchange, the nanoparticles were characterised by infrared spectroscopy, (ESI, Fig. S2 and S3†). Functional groups with a strong stretching frequency, such as C–O in the carboxylic group (around 1500 cm−1), and the phosphonate resonance (strong P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch around 1100 cm−1), can be used to follow the nanoparticle surface functionalization.
O stretch around 1100 cm−1), can be used to follow the nanoparticle surface functionalization.
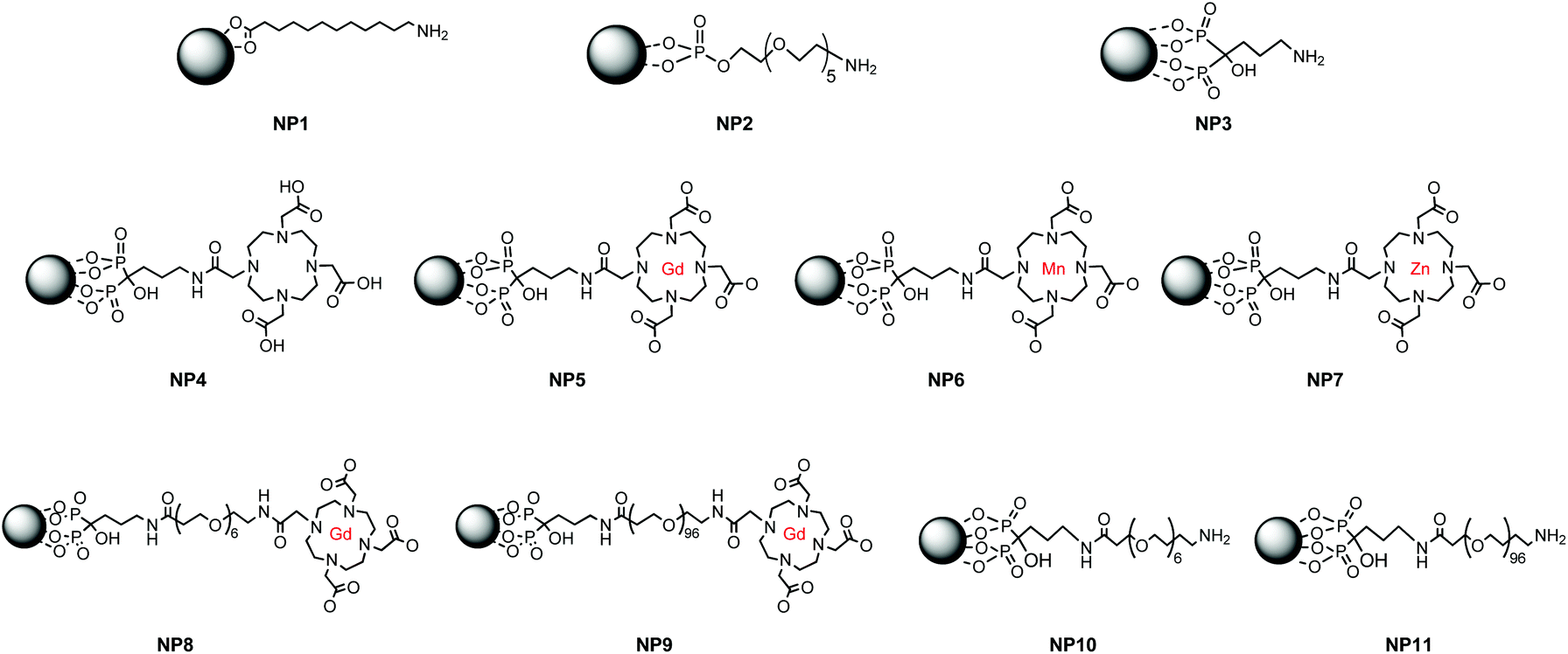 | ||
| Scheme 2 Structures of the series of nanoparticles used in this study. | ||
The transversal relaxivity value, r2, for NP3 (360.0 mM−1 s−1 at 1.47 T and 37 °C, Table 1) was found to be higher than most of the other iron oxide nanoparticles (IONPs) coated with carboxylate-based ligands28–30 including NP1 (153.8 mM−1 s−1). It was found to be also higher than the r2 of phosphate protected particles, NP2 (178.7 mM−1 s−1). On the basis of the quantum mechanical outer sphere theory, the T2 relaxivity is highly dependent, among other factors, on the saturation magnetisation of the nanoparticles.31 The preparation method of the nanoparticles, together with the nature of the coating ligands (depending on the functional group anchoring the ligand to the particle) have been found to greatly affect the saturation magnetisation, resulting in differing relaxation rates. Surface effects drastically increase when the particle size decreases. In the particular case of magnetite, core–shell structures are generally assumed in which the bulk spin arrangement of the core contrasts with the spin canting effects present in the atomic surface layers, which are supposed to be the reason of a decreasing saturation magnetisation as the particle size is reduced. This phenomena has been shown for both carboxylate- and phosphate/phosphonate-coated nanoparticles.32 However, advances in chemical surface functionalization strategies have re-opened discussions about the effect of coating agents on the magnetic ‘dead’ outer layer in magnetic nanoparticles.33,34 In this sense, although previous works reported atomic disorder and spin-glass behaviour at the surface of magnetite nanoparticles,32,35 more recent studies show evidence of a highly crystallized nanoparticle surface with long range atomic order induced by the effect of the anchoring groups of coating ligands.36,37 Therefore, different anchoring functional groups affect differently the net saturation magnetisation via spin disorder at the nanoparticle surface.32 Spin canting is less significant in phosphate-based ligands than in carboxylate-based ones,32 and thus phosphate-coated nanoparticles were expected to present a higher magnetisation and therefore higher relaxation rates (Fig. 2 and ESI, Fig. S4†). Additionally, the use of high temperature thermal decomposition synthesis technology is known to lead to IONPs with superior crystallinity, which is also correlated to higher nanoparticle magnetisation.38,39 These two factors, the lack of spin canting, and the high crystallinity of the core, justify the exceptionally high relaxation rate of NP3, and its better performance when compared to NP1 and NP2.
 | ||
| Fig. 2 Magnetic characterisation of the NPs. A, Room temperature magnetic curves as a function of the applied magnetic field for NPs with different ligands (NP1, NP2, NP3). B, Room temperature magnetic curves as a function of the applied magnetic field for NPs with different metals (NP3, NP5, NP7). C, Room temperature magnetic curves as a function of the applied magnetic field for NPs with different functionalisations (NP3, NP5, NP8, NP9, NP10 and NP11)). Results not corrected for diamagnetic component from the sample holder. | ||
| Modification | NP | r 2 ([Fe]) | r 1 ([Fe + Gd]) | r 1 ([Gd]) |
|---|---|---|---|---|
| a r 1 values calculated with respect to Fe concentration. b r 1 value calculated with respect to Mn concentration. c r 1 value calculated with respect to Zn concentration. | ||||
| Ligand | NP1 | 153.8 ± 25.4 | 15.0 ± 2.5a | — |
| NP2 | 178.7 ± 15.2 | 3.9 ± 1.2a | — | |
| NP3 | 360.0 ± 30.5 | 29.4 ± 3.2a | — | |
| Initial functionalization | NP3 | 360.0 ± 30.5 | 29.4 ± 3.2a | — |
| NP4 | 358.6 ± 28.3 | 17.3 ± 2.4a | — | |
| Metal | NP4 | 358.6 ± 28.3 | 17.3 ± 2.4 | — |
| NP5 | 836.7 ± 51.1 | 31.6 ± 2.6 | 451.5 ± 34.4 | |
| NP6 | 324.5 ± 24.6 | 14.2 ± 2.1 | 248.6 ± 20.0b | |
| NP7 | 209.9 ± 19.9 | 15.8 ± 2.6 | 192.7 ± 19.8c | |
| Distance NP-Gd | NP5 | 836.7 ± 51.1 | 31.6 ± 2.6 | 451.5 ± 34.4 |
| NP8 | 272.7 ± 16.5 | 7.0 ± 1.7 | 191.0 ± 23.6 | |
| NP9 | 212.7 ± 21.7 | 6.1 ± 2.1 | 404.0 ± 29.8 | |
| PEGylation | NP3 | 360.0 | 29.4 ± 3.2a | — |
| NP10 | 197.6 | 4.1 ± 1.1a | — | |
| NP11 | 530.8 | 19.1 ± 2.8a | — | |
The next step of this study was to optimise the nature of the paramagnetic moiety. Gd(III) is the most effective paramagnetic metal for use as a T1 contrast agent due to its seven unpaired electrons and suitable magnetic moment.40 The main concern with Gd(III) comes from its toxicity. Other paramagnetic ions, such as Mn(II) (five unpaired electrons in high spin configuration) have also been used as T1 contrast agents to some extent. Zn(II), a diamagnetic ion was used as a blank in these studies as no magnetic effects are expected from it. In the clinic, Gd(III) has to be used in combination with an appropriate chelator to avoid its interaction with biological processes.41 The thermodynamic stability and dissociation constant of the resulting complexes are crucial parameters that will determine the level of toxicity at the final application. The most widely used of these chelates is DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) due to its biocompatibility and good stability,42 and therefore, it was selected as the chelator of choice for this study. The conjugation of DOTA to the nanoparticles was carried out stoichiometrically, using standard peptidic chemistry to couple one of the carboxylic acids on the chelate to one of the amines on the surface of the nanoparticles. EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) and NHS (N-hydroxysuccinimide) were used as coupling reagents. To determine the stoichiometry of reagents required, the density of ligands on the nanoparticles was first determined (ESI, Fig. S5, Table S1†) by TGA to be 1.02 ligands per nm2. The yield of the DOTA coupling reactions was calculated measuring the amount of non-reacted chelate through an indirect colorimetric method (ESI, Fig. S6 and S7†).20 DOTA coupling yields were >80%. Finally, Gd(III) (or Mn(II)/Zn(II)) was incorporated into the nanoparticles via an overnight incubation with GdCl3 (MnCl2/ZnCl2) salts in pH 6.5 acetate buffer. Any unbound Gd(III), (Mn(II) or Zn(II)) was removed by centrifugation. The yield of Gd(III), (Mn(II), Zn(II)) incorporation was calculated (from ICP measurements) to be above 95% (with respect to DOTA) for all the samples.
The incorporation of a paramagnetic moiety on the nanoparticles brings with it a conceptual problem in the calculation of the relaxivity rates. The calculation of the r1/2 of a contrast agent is achieved by plotting the inverse of the relaxation time versus the concentration of the element responsible for the change in the relaxation. While Gd(III) is known to have a negligible effect on the transversal relaxation of protons, iron oxide nanoparticles present a significant effect on the longitudinal relaxation and therefore could/should be considered for the calculation of r1. In these nanoparticulate systems, the concentration of Fe is around one order of magnitude higher than that of Gd, which means that when considered together for the calculation of the relaxivity, only a small difference is observed from Gd contribution. This is why some authors decide to obviate Fe and calculate r1 only in respect to the concentration of Gd.12,22
Prior to comparing the effects of paramagnetic elements on the global relaxivity, r1/2 of NP3 were compared to those of NP4. As expected, there were no significant changes on the r2 before and after the coupling of DOTA (360.0 vs. 358.6 mM−1 s−1). On the other hand, when comparing the longitudinal relaxivity, a significant decrease was observed (29.4 vs. 17.3 mM−1 s−1). Unlike T2 effects where no contact is needed between the proton and the magnetic component (spin–spin relaxation) to effectively modify the longitudinal relaxation, a T1 relaxation mechanism implies a direct contact between the water molecules and the contrast agent. Thus, any modification in the outer shell of the nanoparticles has the potential to change the r1.
The introduction of paramagnetic species in the probe is assumed to have a bigger impact on the r1. When considering only the newly introduced element, the obtained r1 values for NP5, 6 and 7 followed the expected trend; the most paramagnetic ion, Gd(III) presented the highest value, followed by Mn(II) (weaker paramagnetic), and then by Zn(II) (451.5 > 248.6 > 192.7 mM−1 s−1). DOTA-Gd(III) on its own presents a much lower relaxation rate of around 4 mM−1 s−1 at 1.47 T. This difference in relaxivity (from 31.6 to 4 mM−1 s−1) results from the combination of a multimeric effect on the nanoparticle and changes in the tumbling rate at this field. When r1 values were calculated considering the combined amount of Fe + Gd (Mn/Zn), the results were not that easy to explain. Gd also produced an increase in the r1 (31.6 vs. 17.3 mM−1 s−1). Surprisingly, both Mn and Zn brought a non-significant decrease in the relaxivity (14.2 and 15.8 vs. 17.3 mM−1 s−1). Due to the low X/Fe ratio, only a significant increase is observed in the case of X = Gd when the sum of Fe + X is taken as parameter of normalization, because of Gd strong effect (7 unpaired electrons) on T1. In the case of Zn, where no magnetic effects are foreseen, the relaxivity value showed no change compared NP4. NP6 (Mn2+) does not show any significant difference compared to NP4, the behaviour being very similar to Zn. Mn(H2DOTA) is 6 coordinate with a distorted octahedral geometry (through solid state crystal structure). In solution however, the uncoordinated carboxylates are expected to ionise (due to low pKa values).43 Deprotonation of uncoordinated pendant arms could lead to binding of the pendant arm to the metal centre, resulting in a net increase in the metal coordination number. Mn2+ ion has a maximum coordination number of 8, and when this deprotonation and subsequent binding of the pendant arm occurs in solution, the Mn2+ for these particles would reach coordinative saturation and only the outer sphere water exchange will contribute to relaxation rates.43
A similar theme is found when analysing the r2 values for the same series of particles. Similarly to r1 (Fe + Gd) values, r2 is anomalously enhanced by the incorporation of Gd(III) (836.7 vs. 358.6 mM−1 s−1), in agreement with the higher saturation magnetisation of the hysteresis loop (Fig. 2 and ESI, Fig. S4†). To the best of our knowledge, this r2 value is the highest reported to date for magnetite nanoparticles of 6 nm size. To double-check this extraordinary value, the r2 of this nanoparticle was also measured at 9.4 T. The value obtained, 414.61 ± 35 mM−1 s−1, is still the highest relaxivity reported to date for this kind of particle at high field (the relaxivity decreases strongly at high fields). MR images acquired at 3 T of phantoms of this samples at different concentrations, further confirmed the outstanding properties of NP5, both in T2-weighted and T1-weighted mode (ESI, Fig. S8 and S9†). Under the imaging conditions used (see Materials and methods section), NP5 produced a clear change in the contrast in T2-weighted mode at any of the concentrations tested. NP3 also produced a significant change at 100 and 50 μM, while the nanoparticle with the classic carboxylate ligand, NP1, did not change the contrast even at the highest concentration tested (100 μM). In T1-weighted mode, again only NP5 produced a clear signal. In this case the strongest signal change was observed, as expected, from the commercial agent Dotarem® at a concentration of 100 μM in Gd.
Mn and Zn incorporation brought a decrease in the r2, which was statistically significant in the case of Zn. As mentioned above, both a decrease and an increase in the r2 caused by the coupling of a paramagnetic moiety has been reported before.15,16,18 In our system, only Gd shows an effect on r2 which suggests that Mn2+ paramagnetism is not strong enough to produce a change in the relaxivity of the final probe. In T1/T2 constructs, the increase in r2 has been attributed to an alignment of the electronic spins of the paramagnetic ion by the induced magnetic field generated by the superparamagnetic particles.12
In a different set of experiments, the influence of the distance between the superparamagnetic and the paramagnetic units was explored. In order to investigate this potential distance dependency, a bifunctional carboxy/amine polyethylene glycol (PEG) spacer was included between DOTA and the bisphosphonate molecule (ESI, Scheme S1†). Two different PEG sizes were tested, a short PEG comprising 12 units (600 Da, NP8), and a long PEG of 96 units (5000 Da, NP9). The construction of the probes was achieved in three steps. First, PEG molecules were coupled to the nanoparticles in water following standard peptidic chemistry. Then, the amine terminal side of the PEGs was deprotected in DMF:piperidine (80:20), and finally DOTA was coupled stoichiometrically using peptidic chemistry again. To have a better control over the system, these complex probes were fully characterised by TGA to determine the average number of PEG molecules per nanoparticles (71 PEG600 and 57 PEG5000 per NP, ESI, Table S2†).
Relaxivity results for this set of samples showed a clear dependency of the r2 with the distance. When the paramagnetic and superparamagnetic components are close by, the final r2 of the particles is greatly enhanced (NP4 358.6 vs.NP5 836.7 mM−1 s−1). From there, with increasing Mw of the PEG, the r2 of the probes decreases to values below that of the original particles (NP8 272.7 and NP9 212.7 mM−1 s−1). This r2 distance dependency has already been observed by other authors in inorganic systems,17 and was simply attributed to a decrease of the magnetic field generated by the superparamagnetic particles with the distance (1/r3, r being the distance from the particle). In our system, the hydrodynamic size also increases with PEG size (NP5 31.87 nm, NP8 37.70 nm and NP9 40.70 nm, ESI, Tables S3 and S4†). This decrease in r2 could then be the consequence of a weaker magnetic coupling between the superparamagnetic iron oxide nanoparticle and the paramagnetic Gd ion as the PEG molecular weight increases.
r 1 (Fe + Gd) values follow the same trend as those of r2 (NP4 17.3, NP5 31.6, NP8 7.0, NP9 6.1 mM−1 s−1), with an initial increase when Gd is closest to the nanoparticles, followed by a significant decrease as the distance between the two moieties is increased. The presence of a PEG spacer may account for part of this decrease as the tumbling rate of the chelates coupled to these flexible molecules will be faster than that of the chelates directly attached to the more rigid alendronic acid (the longer the PEG, the faster the tumbling rate). Furthermore, a magnetic interaction between the superparamagnetic magnetite core and the paramagnetic Gd complex at that minimal distance could be responsible for the observed r1 enhancement. As the length of the PEG chain becomes longer and the packing of the PEG chains around the magnetic core is more favoured, this magnetic interaction would disappear and only the Gd ion would contribute primarily to the r1, bringing a significant decrease. This is also supported by the magnetic results (ESI, Fig. S4†). The hysteresis loops for the samples NP4 and NP5 (minimum distance between Fe3O4 and Gd) show the highest saturation magnetization, so that the possibility of a magnetic interaction between the magnetic core and the Gd complex must be considered. On the other hand, the longer the PEG spacer, the lower the saturation magnetisation. Therefore, in addition to a longer separation distance, a lower intensity of the magnetic field coming from the magnetite core, would justify for samples NP8 and NP9 a negligible magnetic coupling with the Gd complex and the corresponding significant decrease of r1 compared to NP5. When only the concentration of Gd(III) is considered for the calculation of the longitudinal relaxivity, there is an initial strong decrease in r1 from NP5 to NP8 (451.5 vs. 191.0 mM−1 s−1) followed by a recovery of the r1 from NP8 to NP9 (191.0 vs. 404.0 mM−1 s−1). These values are more difficult to rationalise and require a more detailed study of these particles and the intermediate functionalised probes. Comparison of the r2 values of NP3, NP10 and NP11 (360.0, 197.6 and 530.8 mM−1 s−1) give initially the same trend as the one observed when Gd is present; there is an initial decrease in r2 with the short PEG chain. The introduction of a longer PEG chain, surprisingly increases the relaxivity of the probes. r1 values follow a similar trend with a decrease with the short PEG and a partial recovery of the r1 with the longer PEG. To try to explain these unexpected changes in MR performance, the conformation of the PEG molecules on the surface of the particles was studied. PEG molecules can adopt two different conformations on a surface, brush (extended), or mushroom (coiled). The space that each PEG molecule occupies on the surface of the nanoparticle (D) can be calculated and compared to the Flory radius (Rf) of that PEG. If Rf is bigger than D then the PEGs adopt a mushroom conformation.44 Calculation of these parameters (ESI, Table S5†) showed that the most probable conformation for both PEGs was brush, so the conformation does not help to explain the differences in relaxivity. Although this theory has been shown very helpful in many cases, for this particular application a more suitable approach that takes into account the curvature of the particles might be needed to explain effects such as those observed here. In our case the coupling of a much heavier PEG molecule is only accompanied by a modest increase in hydrodynamic size. This might suggest an intermediate PEG conformation between brush and mushroom not contemplated in planar models.
A detailed study of the M vs. H curves for samples NP3, NP10 and NP11 shows a decrease of the saturation magnetisation as the PEG chain is introduced, compared to the situation in which only alendronic acid is attached to the particle surface (ESI, Fig. S4†). However, and in agreement with the observed trend in relaxivity, the saturation magnetisation recovers as the number of ethylene units in the PEG increases (NP11). According to the outer sphere relaxation approach, an increase of r2 from NP10 to NP11 would also be expected from the observed partial recovery of the saturation magnetisation. However, although the magnetic and relaxivity data are in agreement, we believe that more detailed experiments need to be conducted in order to assure that the observed relaxivity values are directly coupled exclusively to the magnetic data and no other factors i.e. the degree of freedom of the PEG chains.
Conclusion
The low sensitivity of MRI requires the use of contrast agents, especially in fields like oncology where detection of small features (early detection of tumours) is crucial for patient survival. Current contrast agents are far from ideal, with severe limitations both in performance (particularly at high fields) and ease of diagnosis. The combination of T1 and T2 imaging capabilities into a single probe is one of the ways forward, at least in respect to trustworthy diagnosis. This field of dual-modal MRI agents is only in its early development and results so far lack uniformity. Systematic studies such as this one are required to evaluate the effect of the coupling of a paramagnetic moiety to a superparamagnetic one. In this report, several parameters involved in the design of these dual probes, such as nature of the ligands, spacing between the two active components, and nature of the paramagnetic element, have been studied in detail. The importance of exerting close control over some of these parameters has been highlighted by this work. The distance between the two components is shown to be key to maximise the r1/2 of the probes. A close proximity between the paramagnetic and superparamagnetic moieties is needed to maximise the potential of the probes. Also, Gd(III) seems to be irreplaceable as the paramagnetic moiety, as weaker paramagnetic ions like Mn2+, do not exert a sufficiently strong influence on the final probe.Even though other parameters (apart from r1 and r2) have to be taken into consideration to evaluate the potential of a probe as dual T1/T2 contrast agent (like the r2/r1 ratio), some of the nanoparticles reported in this work are incredibly promising for diagnostic applications. For example, NP5 shows the highest r2 reported to date for a 6 nm Fe3O4 based particle, and NP11 presents a very high r2 together with the stealth properties coming from PEG functionalisation.
Acknowledgements
This work was partially supported by POCTEP (Operational Programme for Cross-border Cooperation Spain-Portugal), by the ERDF (European Regional Development Fund) under grant InveNNta Project and by the P.O Norte CCDR-N/ON.2 programme. JG acknowledges the financial support from the Marie Curie COFUND Programme (NanoTRAINforGrowth). We also wish to thank the Ministry of Education of the Government of Brunei for funding a studentship (to NK).References
- R. Weissleder, Science, 2006, 312, 1168–1171 CrossRef CAS PubMed.
- D. E. Sosnovik and R. Weissleder, Curr. Opin. Biotechnol., 2007, 18, 4–10 CrossRef CAS PubMed.
- D. Weishaupt, V. D. Kochli and B. Marincek, How Does MRI Work?: An introduction to the Physics and Function of Magnetic Resonance Imaging, Springer, 2nd edn, 2006 Search PubMed.
- F. Hu and Y. S. Zhao, Nanoscale, 2012, 4, 6235–6243 RSC; Z. Hashim, M. Green, P. H. Chung, K. Suhling, A. Protti, A. Phinikaridou, R. Botnar, R. A. Khanbeigi, M. Thanou, L. A. Dailey, N. J. Commander, E. Rowland, J. Scott and D. Jenner, Nanoscale, 2014, 6, 8376–8386 RSC.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484–499 CrossRef CAS PubMed; L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. J. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Bottnar, P. J. Blower, M. A. Green and R. T. M. de Rosales, ACS Nano, 2013, 7, 500–512 CrossRef PubMed.
- G. R. Reddy, M. S. Bhojani, P. McConville, J. Moody, B. A. Moffat, D. E. Hall, G. Kim, Y.-E. L. Koo, M. J. Woolliscroft, J. V. Sugai, T. D. Johnson, M. A. Philbert, R. Kopelman, A. Rehemtulla and B. D. Ross, Clin. Cancer Res., 2006, 12, 6677–6686 CrossRef CAS PubMed; N. E. Blanco, M. Jauregui-Osoro, M. Cobaleda-Silesim, C. R. Maldonado, M. Henriksen-Lacey, D. Padro, S. Clark and J. C. Mareque-Rivas, Chem. Commun., 2012, 48, 4211–4213 RSC.
- J. R. McCarthy, F. A. Jaffer and R. Weissleder, Small, 2006, 2, 983–987 CrossRef CAS PubMed.
- R. Weissleder, S. Saini, D. D. Stark, J. Wittenberg and J. T. Ferrucci, Am. J. Roentgenol., 1988, 150, 561–566 CrossRef CAS PubMed.
- R. C. Semelka, J. K. T. Lee, S. Worawattanakul, T. C. Noone, R. H. Patt and S. M. Ascher, J. Magn. Reson. Imaging, 1998, 8, 670–674 CrossRef CAS PubMed.
- S. Kubaska, D. V. Sahani, S. Saini, P. F. Hahn and E. Halpern, Clin. Radiol., 2001, 56, 410–415 CrossRef CAS PubMed.
- Z. Zhou, D. Huang, J. Bao, Q. Chen, G. Liu, Z. Chen, X. Chen and J. Gao, Adv. Mater., 2012, 24, 6223–6228 CrossRef CAS PubMed.
- G. H. Im, S. M. Kim, D.-G. Lee, W. J. Lee, J. H. Lee and I. S. Lee, Biomaterials, 2013, 34, 2069–2076 CrossRef CAS PubMed.
- J. Kim, C. Lee and S. Lee, Bull. Korean Chem. Soc., 2009, 30, 6–9 Search PubMed.
- H. Yang, Y. Zhuang, Y. Sun, A. Dai, X. Shi, D. Wu, F. Li, H. Hu and S. Yang, Biomaterials, 2011, 32, 4584–4593 CrossRef CAS PubMed.
- K. H. Bae, Y. B. Kim, Y. Lee, J. Hwang, H. Park and T. G. Park, Bioconjugate Chem., 2010, 21, 505–512 CrossRef CAS PubMed.
- J.-S. Choi, J.-H. Lee, T.-H. Shin, H.-T. Song, E. Y. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015–11017 CrossRef CAS PubMed.
- C.-C. Huang, C.-Y. Tsai, H.-S. Sheu, K.-Y. Chuang, C.-H. Su, U.-S. Jeng, F.-Y. Cheng, C.-H. Su, H.-Y. Lei and C.-S. Yeh, ACS Nano, 2011, 5, 3905–3916 CrossRef CAS PubMed.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS PubMed.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, Contrast Media Mol. Imaging, 2006, 1, 184–188 CrossRef PubMed.
- N. Tresilwised, P. Pithayanukul, P. S. Holm, U. Schillinger, C. Plank and O. Mykhaylyk, Biomaterials, 2012, 33, 256–269 CrossRef CAS PubMed.
- T.-H. Shin, J. Choi, S. Yun, I.-S. Kim, H.-T. Song, Y. Kim, K. I. Park and J. Cheon, ACS Nano, 2014, 8, 3393–3401 CrossRef CAS PubMed.
- A. Szpak, S. Fiejdasz, W. Prendota, T. Strączek, C. Kapusta, J. Szmyd, M. Nowakowska and S. Zapotoczny, J. Nanopart. Res., 2014, 16, 1–11 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- T. J. Daou, G. Pourroy, J. M. Greneche, A. Bertin, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2009, 4442–4449 RSC.
- T. Daou, S. Begin-Colin, J. M. Greneche, F. Thomas, F. Derory, P. Bernhardt, P. Legare and G. Pourroy, Chem. Mater., 2007, 19, 4494–4505 CrossRef CAS.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS PubMed.
- E. D. Smolensky, H.-Y. E. Park, T. S. Berquó and V. C. Pierre, Contrast Media Mol. Imaging, 2011, 6, 189–199 CAS.
- S. Tong, S. Hou, Z. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607–4613 CrossRef CAS PubMed.
- U. Tromsdorf, O. Bruns and S. Salmen, Nano Lett., 2009, 9, 4434–4440 CrossRef CAS PubMed.
- Y. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS PubMed.
- T. J. Daou, J. M. Grenèche, G. Pourroy, S. Buathong, A. Derory, C. Ulhaq-Bouillet, B. Donnio, D. Guillon and S. Begin-Colin, Chem. Mater., 2008, 20, 5869–5875 CrossRef CAS.
- C. Rodriguez, M. Banobre-Lopez, Y. V. Kolen'ko, B. Rodriguez, P. Freitas and J. Rivas, IEEE Trans. Magn., 2012, 48, 3307–3310 CrossRef CAS.
- C. Pereira, A. M. Pereira, C. Fernandes, M. Rocha, R. Mendes, M. P. Fernández-García, A. Guedes, P. B. Tavares, J.-M. Grenèche, J. P. Araújo and C. Freire, Chem. Mater., 2012, 24, 1496–1504 CrossRef CAS.
- B. Martínez, X. Obradors, L. Balcells, A. Rouanet and C. Monty, Phys. Rev. Lett., 1998, 80, 181–184 CrossRef.
- P. Guardia, B. Batlle-Brugal, A. G. Roca, O. Iglesias, M. P. Morales, C. J. Serna, A. Labarta and X. Batlle, J. Magn. Magn. Mater., 2007, 316, e756–e759 CrossRef CAS PubMed.
- A. G. Roca, D. Niznansky, J. Poltierova-Vejpravova, B. Bittova, M. A. González-Fernández, C. J. Serna and M. P. Morales, J. Appl. Phys., 2009, 105, 114309 CrossRef PubMed.
- D. Margulies, F. Parker, F. Spada, R. Goldman, J. Li, R. Sinclair and A. Berkowitz, Phys. Rev. B: Condens. Matter, 1996, 53, 9175–9187 CrossRef CAS.
- D. Margulies, F. Parker, M. Rudee, F. Spada, J. Chapman, P. Aitchison and A. Berkowitz, Phys. Rev. Lett., 1997, 79, 5162–5165 CrossRef CAS.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- J. G. Penfield and R. F. Reilly, Nat. Clin. Pract. Nephrol., 2007, 3, 654–668 Search PubMed.
- G. Stasiuk and N. Long, Chem. Commun., 2013, 49, 2732 RSC.
- S. Wang and T. D. Westmoreland, Inorg. Chem., 2009, 48, 719–727 CrossRef CAS PubMed.
- C. Allen, N. Dos Santos, R. Gallagher, G. N. C. Chiu, Y. Shu, W. M. Li, S. A. Johnstone, A. S. Fanoff, L. D. Mayer, M. S. Webb and M. B. Bally, Biosci. Rep., 2002, 22, 225–250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr04400f |
| This journal is © The Royal Society of Chemistry 2015 |