
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards 4th generation biomaterials: a covalent hybrid polymer–ormoglass architecture†
N.
Sachot
ab,
M. A.
Mateos-Timoneda
abc,
J. A.
Planell
ab,
A. H.
Velders
d,
M.
Lewandowska
e,
E.
Engel
abc and
O.
Castaño
*abf
aBiomaterials for Regenerative Therapies, Institute for Bioengineering of Catalonia (IBEC), Barcelona, Spain. E-mail: ocastano@ibecbarcelona.eu
bCIBER en Bioingeniería, Biomateriales y Nanomedicina, CIBER-BBN, Zaragoza, Spain
cMaterials Science and Metallurgical Engineering, Universitat Politècnica de Catalunya (UPC), Barcelona, Spain
dBioNanoTechnology, Wageningen University (WU), Wageningen, The Netherlands
eFaculty of Materials Science and Engineering, Warsaw University of Technology, Warsaw, Poland
fMaterials Science and Metallurgical Engineering, Universitat de Barcelona (UB), Barcelona, Spain
First published on 19th August 2015
Abstract
Hybrid materials are being extensively investigated with the aim of mimicking the ECM microenvironment to develop effective solutions for bone tissue engineering. However, the common drawbacks of a hybrid material are the lack of interactions between the scaffold's constituents and the masking of its bioactive phase. Conventional hybrids often degrade in a non-homogeneous manner and the biological response is far from optimal. We have developed a novel material with strong interactions between constituents. The bioactive phase is directly exposed on its surface mimicking the structure of the ECM of bone. Here, polylactic acid electrospun fibers have been successfully and reproducibly coated with a bioactive organically modified glass (ormoglass, Si–Ca–P2 system) covalently. In comparison with the pure polymeric mats, the fibers obtained showed improved hydrophilicity and mechanical properties, bioactive ion release, exhibited a nanoroughness and enabled good cell adhesion and spreading after just one day of culture (rMSCs and rEPCs). The fibers were coated with different ormoglass compositions to tailor their surface properties (roughness, stiffness, and morphology) by modifying the experimental parameters. Knowing that cells modulate their behavior according to the exposed physical and chemical signals, the development of this instructive material is a valuable advance in the design of functional regenerative biomaterials.
Introduction
Various stimuli from tissue architecture and the cell microenvironment, known as “the stem cell niche”, affect the proliferation and fate1 of stem cells. This microenvironment is composed of biochemical (O2, cations, growth factors, cytokines) and biophysical cues (extracellular matrix (ECM) properties), and cells also make a contribution. Adhesion motifs and binding sites comprise the biochemical information contained in the ECM, while mechanical information contributes in the form of substrate stiffness and deformability.In the last decade, the classic tissue engineering strategy has been validated. However, it involves extensive and time-consuming cell expansion processes before implantation. A new approach has been introduced to bypass these ex vivo steps in order to obtain a suitable microenvironment to repair structural and functional tissues:2in situ tissue regeneration. It consists of the development of a target-specific scaffold that can efficiently control the host microenvironment and recruit host stem or tissue-specific progenitor cells to the injury site, using the body's own capacity for regeneration.
Since the middle of the last century, biomaterials have evolved exponentially. The 1st generation (inert materials) gave way to bioabsorbable or bioactive materials (2nd generation) and then to the combination of both resorbable and bioactive ones (3rd generation),3 and in the last decade this has led to a so-called 4th generation of biomaterials. These try to mimic the ECM of natural tissues, recreating the molecular architecture and biochemical environment to surround cells with proper stimuli. However, we are not yet close to accomplishing this feat. There are plenty of studies using sophisticated materials that involve complicated up-scalability due to difficult chemistries and fabrication methods. Some of these studies have become high-impact papers, but translation to the clinic does not seem to be feasible, at least in the medium term. The reasons for this are the lack of reproducibility from batch to batch and high costs for production, storing and application, as well as the lack of standardization and regulation.
Nowadays an extensive range of biomaterials and processing techniques allows the fabrication of scaffolds for numerous applications in regenerative medicine.4 Currently, hybrid materials are being extensively investigated with the aim of mimicking the ECM microenvironment to be able to develop tough, cost-efficient, bioactive scaffolds for bone tissue engineering.5–8 This approach is of great interest for scientists working in the field, as it enables the fabrication of materials which are composed of the combined and synergistic properties of the different constituents. For example, a synthetic polymer and a glass can be associated with a unique material in which the glass constitutes the bioactive phase of the matrix and the polymer supports its mechanical properties.9 However, these hybrids present two major drawbacks. The first one is their non-homogeneous degradation in body fluids. Although it has been shown that these materials possess chemical interactions at the nanoscale,10 these interactions are usually weak (i.e. van der Waals, ionic, hydrogen bonding) and easy to break. As a consequence, this leads to non-homogeneous and rapid detachment of the different phases. But, to be recognized as a successful temporary implant, scaffolds should degrade at a rate that matches the formation of the new tissue. Therefore, a strong chemical interaction between the glass and the polymer is required to have better control of the material degradation rate, as well as an improvement in the global mechanical properties of the whole matrix.11,12 The second problem is the non-homogeneous dispersion of the bioactive compound inside the polymer. This is a common issue in hybrids because both compounds are usually mixed with each other without proper control of their respective dispersed positions.13,14 In this case, the polymer often masks the glass, and cells cannot detect it. Generally, synthetic biocompatible and biodegradable polymers do not have an intrinsic bioactivity and cells prefer to attach to the bioactive inorganic constituent.15,16 It is thus essential to produce materials that present a better surface exposure of the bioactive compound in order to be detected by the cells, thus improving the cell–material interactions. In such a case, no prior degradation of the polymer would be needed to uncover the glass, inducing an enhancement of the adhesion efficiency and spread of the cells.17 This is due to the fact that all the ions released from the glass during its degradation could be immediately perceived by the biological entities, promoting their functions.18 There is clear evidence that high Ca2+ concentrations promote cell homing, migration, and differentiation, as well as tubulogenesis.19–21 Furthermore, we hypothesized and validated in prior studies that angiogenesis can be triggered through two signaling synergistic pathways: mechanical and biochemical.16In vitro results using a composite scaffold made of a calcium phosphate glass and polylactic acid (PLA) demonstrated that the Ca2+ released by the scaffold induced a higher expression of vascular endothelial growth factor (VEGF), through the calcium-sensing receptor (CaSR) in endothelial rat progenitor cells (rEPCs) and rat mesenchymal stem cells (rMSCs). On the other hand, the mechanical properties of the scaffold induced the expression and synthesis of the VEGF receptor 2 (VEGFR2). We also evidenced that the optimal cell response was maximum in the range of [Ca2+]∼10 mM.22 There is, therefore, a need to design hybrid scaffolds using novel material fabrication strategies to overcome these challenges.
Here we present a novel coating protocol to produce hybrid materials for bone tissue engineering applications that, in contrast with other approaches, possess a bioactive phase (organically modified glass: ormoglass) fully exposed on their surface and covalently linked to the organic domain. This approach, based on the sol–gel method and subsequent surface treatments,23 enables the fabrication of materials with tailorable surface properties, topography or ion release by modifications in composition or hydrolysis ratio. The results are electrospun mats with flexible properties, the lack of a potential wedge effect that can lead to stress concentration during mechanical loading, and the versatility of the method to be transferred to other processing methods (film, rapid prototyping, solvent casting–particle leaching, freeze-drying, etc.), as it depends on the existence of carboxylic groups at the surface rather than the scaffolding processing manner. It was not the usual bioactivity through the precipitation of an apatite layer on the surface that was the main goal for the bioactivity of these nanostructured scaffold materials, but rather a controlled calcium release.
The ormoglass system used (Si–Ca–P2) was selected according to its well-documented osteointegrative and osteogenic properties.24–26 PLA was chosen as a polymeric compound because of its biodegradability, biocompatibility and excellent features as a bone graft substitute.27–29 Moreover, it can be easily processed to produce materials with different shapes.30,31 Taking into account that the material structure influences cellular activity (adhesion and migration, for example), the choice of a proper implant architecture is a key point for the design of fully functional scaffolds. Fibers produced by the electrospinning technique have been shown to mimic the fibrous structure of the extracellular matrix (ECM) of natural bone32 and exhibit a high surface-to-volume ratio when considered as a highly 3D porous mat.33 Therefore, this technique has been used to shape PLA as nanofibers in order to obtain a biomimicked substrate. Together with the coating modulation (i.e. change of the surface properties), this aims to provide a suitable environment for cells to promote their adhesion, proliferation, and subsequent differentiation.
Results and discussion
Surface treatments
PLA fibers were successfully coated with two Si–Ca–P2 ormoglass compositions, S60 and S40, following a new and promising protocol recently developed by our group.23 As described in more detail in the Experimental section and seen in Fig. 1, this protocol consists of the bonding of ormoglass nanoparticles onto the surface of polymeric fibers by means of a coupling agent (APTES). It involves the hydrolysis of the fibers’ surface, several pre-functionalization treatments and a condensation process that enables the particles to be linked to the intermediate coupling agent.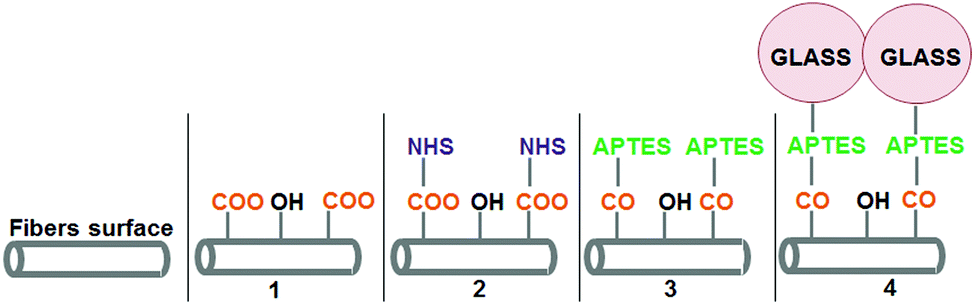 | ||
| Fig. 1 Chemical modifications performed on the fibers’ surface to coat polymeric fibers with ormoglass particles. (1) After NaOH hydrolysis; (2) after EDC/NHS treatment; (3) after immersion in APTES solution; (4) after ormoglass coating. | ||
To ensure that each treatment was efficiently performed, changes in the surface electrostatic potential were evaluated by measuring the zeta potential (ZP) – the potential at the solid–liquid interface – of the fibrous layer obtained. Table 1 summarizes the ZP values after each treatment step for pH = 7 (indicative value selected for comparison) and isoelectric point values (IEP, pH value at ZP = 0). The curves associated with Table 1 can be seen in the ESI (Fig. S1†). Except for the last treatment, significant changes in the electrical potential attest that modifications at the surface of the fibers occurred. Pristine PLA fibers produced by electrospinning showed an IEP equal to 3.06. After hydrolysis, this value decreased to 2.73 due to the formation of reactive negatively charged carboxylic groups (COO−) at the fibers’ surface. The fibrous layer became much less electronegative after activation with a value of 6.07. This is attributed to the positive surface charges mainly related to the presence of imide groups. Then, a significant drop in the IEP value was observed once APTES was grafted on the surface (IEP = 3.68). This can be explained by the introduction of the silane group on the surface through the bonding of the APTES molecule. After the addition of the ormoglass (both compositions), IEP values remained approximately unchanged compared to the value obtained after APTES functionalization. Therefore, the efficiency of this final step was not clear. In order to verify if the ormoglass particles had indeed been linked to the polymer, EDS measurements were performed. As shown in Fig. S2,† calcium and phosphate were detected on the spectra related to the coated fibers, while no traces of these elements were observed just after APTES functionalization. Also, the peak associated with the silicon was more intense after coating due to the higher amount of silicon atoms present on the fibers’ surface (ormoglass network based on Si–O–Si bonds). These complementary observations confirmed that the final step was successfully achieved.
| Treatment | ZP (mV) | IEP (pH) |
|---|---|---|
| Raw fibers (PLA) | −68 | 3.06 |
| Hydrolysis | −85 | 2.73 |
| Activation | −18 | 6.07 |
| APTES functionalization | −73 | 3.68 |
| Coated S60 fibers | −74 | 3.43 |
| Coated S40 fibers | −76 | 3.24 |
Scaffold composition and surface morphology
The exact composition of the coating was determined by EDS measurements (Table 2). The first composition of the coating (molar percentage ratio) was 60Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32Ca:8P2 (S60 fibers) and the second one 39Si:46Ca:15P2 (S40 fibers). PLA fibers were coated with two different ormoglass compositions in order to demonstrate the versatility and potential of this approach to produce materials with different chemical features. As chemical cues received by cells from the material are essential for the triggering of specific cellular responses, this ability to change the coating composition simply by adapting the protocol to a selected glass reflects the great potential of this method for tissue engineering applications. In the case of the present ormoglass coatings, silicon, calcium and phosphate ions are expected to promote a better integration of the scaffold in the host tissue and to stimulate cell differentiation towards an osteoblastic lineage.
:32Ca:8P2 (S60 fibers) and the second one 39Si:46Ca:15P2 (S40 fibers). PLA fibers were coated with two different ormoglass compositions in order to demonstrate the versatility and potential of this approach to produce materials with different chemical features. As chemical cues received by cells from the material are essential for the triggering of specific cellular responses, this ability to change the coating composition simply by adapting the protocol to a selected glass reflects the great potential of this method for tissue engineering applications. In the case of the present ormoglass coatings, silicon, calcium and phosphate ions are expected to promote a better integration of the scaffold in the host tissue and to stimulate cell differentiation towards an osteoblastic lineage.
| PLA | S60 fibers | S40 fibers | |
|---|---|---|---|
| Si (%) | — | 59.7 ± 8.9 | 38.7 ± 2.8 |
| Ca (%) | — | 32.1 ± 6.2 | 45.9 ± 4.2 |
| P2 (%) | — | 8.2 ± 4.9 | 15.5 ± 2.0 |
| Water contact angle (°) | 122.1 ± 3.2 | 29.1 ± 3.0 | 28.6 ± 3.2 |
Differences in the surface morphology of the fibers were observed before and after coating. Coated fibers showed a rough nanostructured topography, whereas PLA fibers exhibited a smoother surface. A change in the ormoglass composition seemed also to lead to a modification of the surface topography. As seen in Fig. 2, the morphology of S60 coated fibers appears to be rougher than the S40 ones. However, according to water contact angle measurements, both scaffolds showed excellent hydrophilic properties in comparison with the pure PLA ones (Table 2).
 | ||
| Fig. 2 Surface morphology of (a) pure PLA fibers, (b) S40 fibers and (c) S60 fibers (FESEM images: the scale is equal to 500 nm). | ||
Infrared characterization
Fig. 3 shows the magnified relevant areas of FTIR spectra of the coated fibers acquired by the ATR method (completed spectra can be observed in the ESI in Fig. S3†) and Table 3 summarizes the assignment of the peaks according to different previous studies.34–44 Spectra of both coating compositions were similar to each other. However, differences were observed between the spectra of the PLA fibers, APTES functionalized fibers and coated fibers. Most of these changes occurred in the fingerprint region. Only one difference can be noticed between 4000 cm−1 and 1600 cm−1: a small peak located at 3446 cm−1 appeared after APTES functionalization. This peak was assigned to the secondary amine of PLA-linked APTES (N–H stretching) which also resulted in an increase of the IR signal at 756 cm−1 (N–H wagging) after APTES grafting. The peaks characteristic for Si-OCH2CH3 groups from APTES were also observed at 1213 and 1183 cm−1, inferring that APTES has been successfully attached to the fibers. Another peak, located at 919 cm−1, also confirmed the presence of APTES molecules on the fibers’ surface. This peak resulted from silanol groups (Si–OH) of some APTES molecules already partially hydrolyzed. It is assumed that these silanol groups were formed during the APTES functionalization step because of the ambient humidity or during the IR measurements.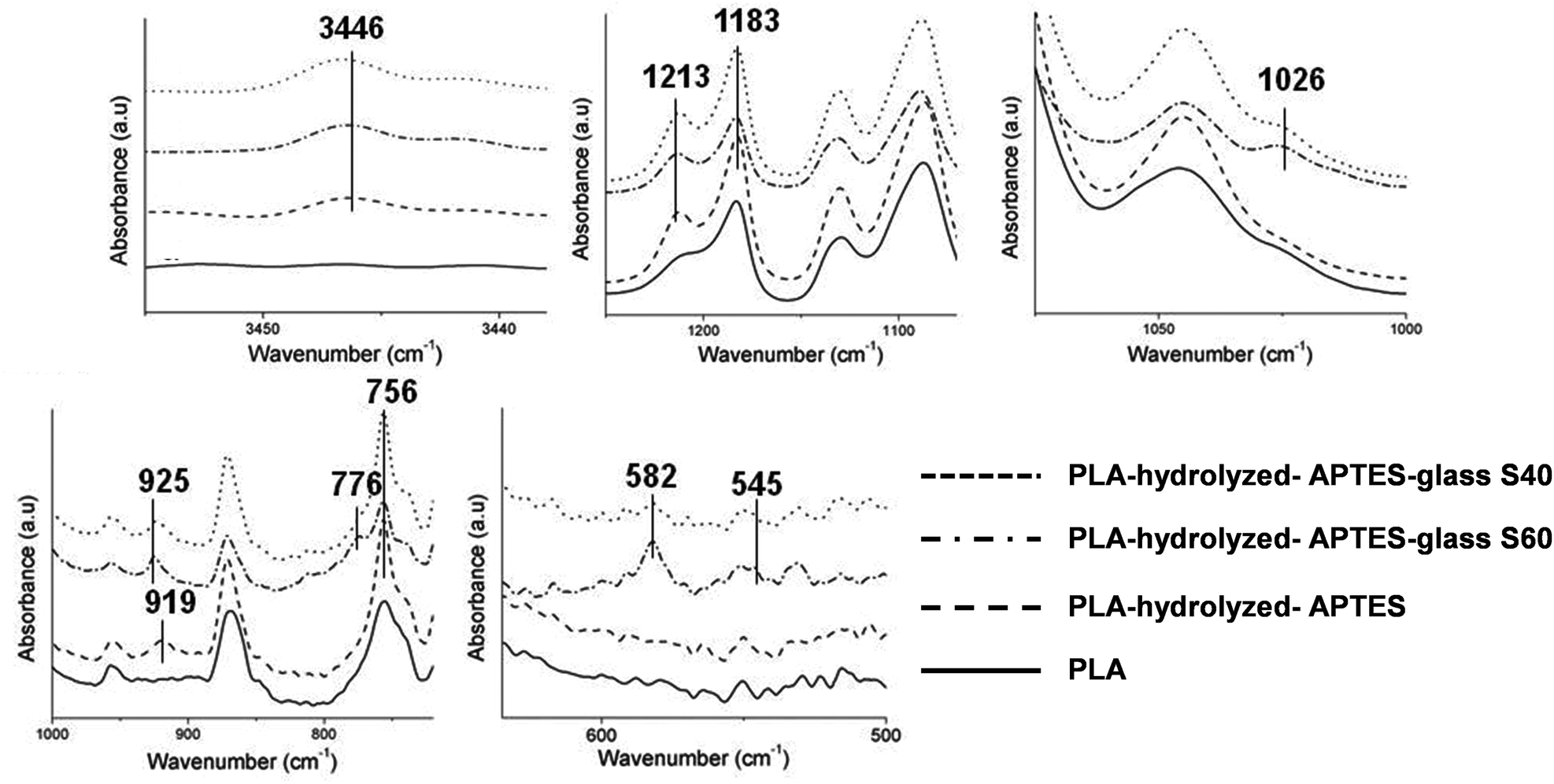 | ||
| Fig. 3 Relevant areas of the FTIR spectra obtained for the PLA fibers, hydrolyzed and APTES functionalized fibers, and coated fibers (S40 and S60). | ||
| Wavenumber (cm−1) | Assignment | Ref. | Spectra |
|---|---|---|---|
| 3446 | N–H stretching | 34 | APTES functionalized fibers and coated fibers |
| 1213, 1183 | Si-OCH2CH3 Si–O–Si | 35–37 | APTES functionalized fibers and coated fibers Coated fibers |
| 1026 | Si–O–Si asymmetric stretching | 34,37–39 | Coated fibers |
| 919, 925 | Si–OH stretching | 37,40,41 | APTES functionalized fibers Coated fibers |
| 776 | Si–O–Si symmetric stretching | 38,41 | Coated fibers |
| 756 | N–H bending | 42 | APTES functionalized fibers and coated fibers |
| 582, 545 | P–O vibrations | 43,44 | Coated fibers |
After the coating, novel peaks associated with phosphate and silicon species were observed. Siloxane bond (Si–O–Si) signals appear at 1026 and 776 cm−1. It can be moreover noticed that the peak located at 1213 cm−1, attributed to ethoxy groups of previously grafted APTES, is still observed after the coating with no apparent intensity decrease. This can be surprising considering that APTES molecules should be hydrolyzed to create the bonding, and a decrease in the peak intensity would be expected. But signals related to Si–O–Si groups can also coincide with the same wavenumber range of the ethoxy groups of APTES. Therefore, the fact that no changes were observed before and after the coating with the ormoglass for this peak (1213 cm−1) can be explained by two possibilities: the first one implies that the whole signal is associated with a full substitution of the Si-OCH2CH3 groups from APTES by Si–O–Si groups from the ormoglass coating network; the second one implies that the observed intensity is an accumulation of the intensity from some non-hydrolyzed Si-OCH2CH3 groups from APTES plus Si–O–Si groups from the ormoglass network. Notice that the Si–O–Si signal can be associated with Siglass–O–Siglass or SiAPTES–O–Siglass, which can be assigned to a covalent bond between the polymeric fiber surface and the ormoglass coating network. The peak located at 925 cm−1 in the spectra of the coated fibers is typically due to silanol groups and is assigned to the silanol groups from the ormoglass network. On the APTES functionalized fibers, these groups (silanols) gave signals at 919 cm−1. This shift in the wavenumber is attributed to the difference in the chemical environment of the silanols. Our interpretation is that the 919 cm−1 peak might also appear in the spectra of the coated fibers but it is difficult to see because of the diminution of its intensity due to the formation of a SiAPTES–O–Siglass bond and the presence of the 925 cm−1 peak. The other peaks observed at lower wavenumbers were assigned to a phosphate complex.
It can be noticed that signals corresponding to the ormoglass were not very intense, especially for the S40 coating, probably because of the thinness of the grafted ormoglass. As seen in the FESEM pictures, S40 particles seemed to be much smaller than the S60 ones. This was in fact confirmed by dynamic light scattering (DSL) measurements (ESI, Fig. S4†). Small particles thus lead to a thinner coating than large particles. This is particularly true if the coating is constituted by a monolayer of ormoglass particles and if bilayers, trilayers, etc. are energetically penalized. Otherwise, this would result in the random growth of the coating layer. But this is not the case here. Observations under FESEM of the remaining inorganic phase obtained after the thermogravimetry analysis indeed revealed that the coating is made of a mono-layer of particles (see the Ormoglass coating thickness and weight percentage section) and that the coating thickness seems to be directly controlled by the particle size.
Scaffold mechanical properties and roughness
An important aspect to be considered when designing hybrid materials is the mechanical properties of the scaffold. Even when the combination of a polymer with a glass (usually an extremely brittle compound) enabled the fabrication of tough materials, other problems related to mechanical imperfections started to be pointed out in the literature. The most recurrent is probably the non-union between the two phases. It results, in fact, in the non-homogeneous and fast degradation of the scaffold. More control of the phase interactions should thus be achieved in order to better tailor the degradation rate and global mechanical properties of the scaffold. As reported previously,23 high silicon content scaffolds showed important increases in Young's modulus and yield strength values (tensile-strain assays), while remaining fully flexible. AFM investigation also revealed that these scaffolds had a higher stiffness than the pure polymeric ones. According to the results obtained for the S40 fibers, the expected results consisting of enhancing the tensile properties and stiffness of pristine PLA fibers by coating them with an ormoglass were confirmed (Fig. 4). Young's modulus and yield strength (tensile tests) of S40 fibers were also significantly increased, as well as the DMT modulus that revealed these fibers to be the stiffest substrate of the study. The scaffold was also easy to bend; a great advantage in terms of material handling for further in vitro and in vivo assays and possible clinical trials. Our hypothesis is: considering that Si is the element that forms the backbone of the network formed during the condensation process (note that Si binds four other elements through oxygen bonding and calcium only two), the higher content of silicon, the higher the strength of the network, leading to stronger mechanical features. In contrast, we expect in further analysis that flexibility and fatigue resistance will be lower as the Si content decreases. We have to also take into account that the thickness of S60 is higher than that of S40, which also contributes to the higher mechanical resistance and stiffness.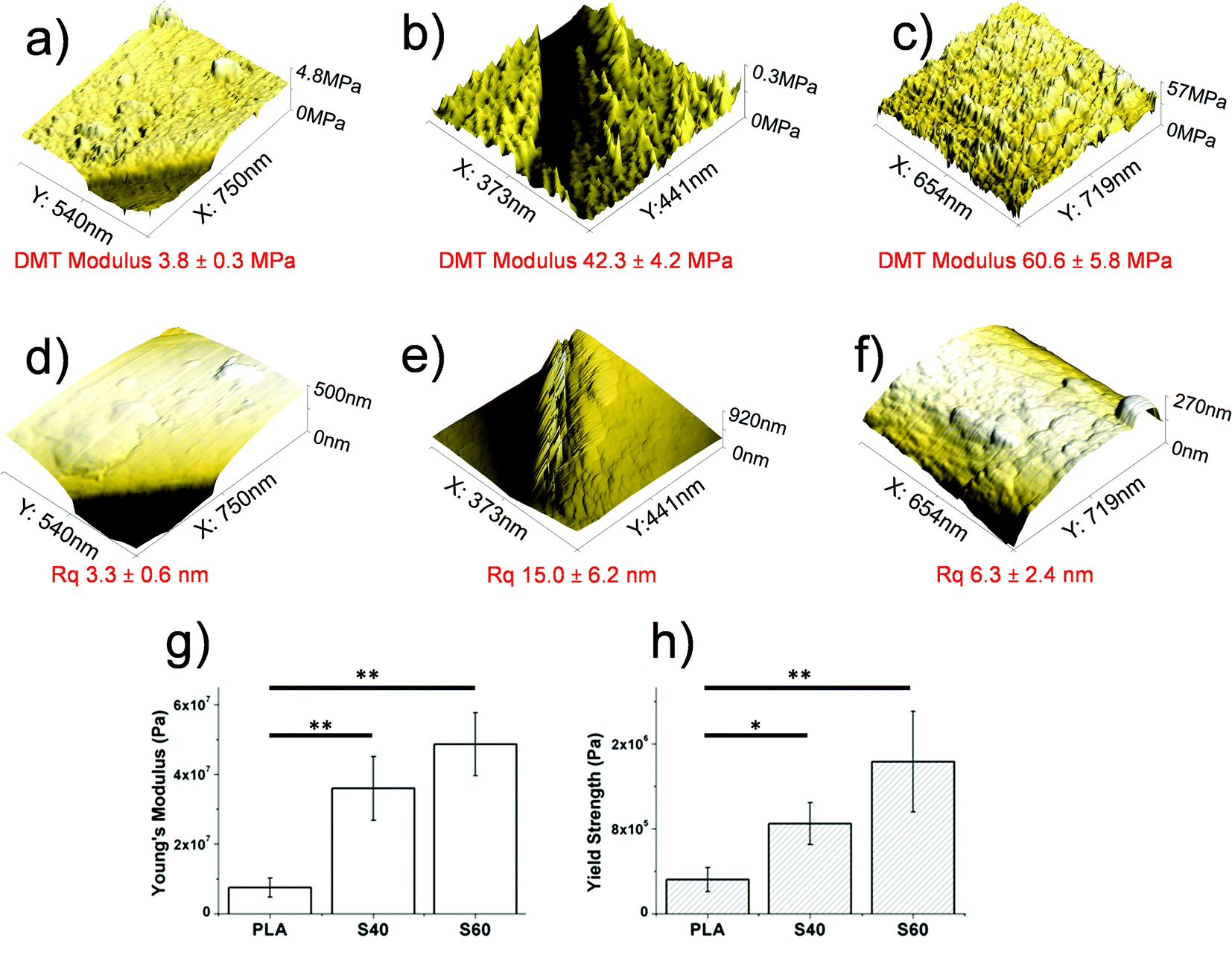 | ||
| Fig. 4 Top: DMT modulus (a–c) and roughness (d–f) of (a, d) PLA, (b, e) S40 and (c, f) S60 fibers (AFM analysis). Bottom: mechanical properties of PLA and coated fibers obtained by tensile tests: (g) the Young's modulus approach; (h) the yield strength. | ||
In addition, AFM measurements also enabled the determination of the roughness of the fibers’ surface. As noticed in the FESEM pictures, S60 fibers seemed to be rougher than the S40 ones. This has been assessed by the roughness mean square (Rq) values associated with each fiber type. Software analysis revealed that the Rq of PLA fibers was doubled after being coated with the S40 ormoglass composition, while it increased five-fold after being coated with the S60 ormoglass composition. Thus it can be concluded that a change in the ormoglass composition indeed leads to modifications in the fibers’ topography. Two reasons can be given to explain these observations. As reported in the ESI (Fig. S4†), the differences in composition of the ormoglass influenced the particle formation. The S40 ormoglass had fewer silicon atoms available for condensation and this could have minimized the particles size. Even if a bigger amount of water was used to hydrolyze S40 ormoglass (molar ratio of Si:H2O of 1:2) than to hydrolyze the S60 one (Si:H2O of 1:1), this did not promote the formation of bigger S40 particles. Another factor that might have also influenced the thickness of the coating can be the dilution of the ormoglass solution with ethanol which was higher for the S40 ormoglass than for the S60 one. It might have limited the possible diameter growth of the particles during the last step of treatment. Note that ethanol dilutes dispersion of the nanoparticles to avoid agglomeration and to slow the hydrolysis of the precursor, therefore slowing condensation and particle growth. The result is that the time and amount of hydrolysis can be selected to stop reactions and bind the particle to the polymer. In this way, the roughness control is excellent. If ethanol is not added, the particles would be bigger and would probably form aggregates, provoking an uncontrolled increase in roughness.
Measurements of the fibers’ diameters, before and after coating, were made using the pictures obtained from FESEM and ImageJ software to determine the thickness of the coated ormoglass. In comparison with the PLA fibers’ diameter (686 ± 48 nm), coated fibers showed an increase in thickness: 976 ± 84 nm for the S60 fibers and 770 ± 66 nm for the S40 fibers. This corresponds to thicknesses of ∼150 nm for the S60 coating and ∼45 nm for the S40 one. This attests that the lower the silicon content, the thinner the coating. In order to quantify this difference, a thermogravimetric analysis was performed. This technique is widely used to evaluate the percentage of inorganic phase contained in hybrid organic–inorganic materials and to determine their thermal stability.45,46
Ormoglass coating thickness and weight percentage
As organic compounds degrade at relatively low temperatures, it is possible to quantify the inorganic part of the ormoglass that was grafted on the polymeric fibers when applying proper heat treatment. TGA results and morphology of the sample after thermal treatment are presented in Fig. 5. The remaining mass percentage of the inorganic part of the ormoglass (non-degraded phase) related to the S60 fibers is higher than that of the S40 fibers (5.6 and 1.7% respectively). Correlated to the fibers’ thickness measurements, this confirms that less ormoglass has been coated for the low silicon content composition than for the one with higher silicon content. To visualize the remaining inorganic part of the ormoglass and to measure the thickness of the inorganic shell, FESEM pictures of the samples were taken after thermal degradation. For the S60 fibers, the thickness of the fibers is easily measured because the tubular structure of the fibers is well maintained. On the contrary, for the S40 fibers, it was not possible to measure the thickness of the inorganic shell because the sample collapsed resulting in a sintered powder when trying to transfer it to the FESEM sample holder (see ESI, Fig. S5†). This suggests that the coating was in fact thinner and mechanically weaker for this ormoglass composition. From this assay, it can be also noticed that the thickness of the remaining inorganic shell of the S60 fibers differs from the measurements of the fiber diameter. Indeed, the thickness of the S60 coatings appears smaller after the thermal treatment (72 ± 14 nm for S60 fibers) than the one determined on the coated fibers (∼150 nm). This difference can be explained by the loss of the organics contained in the ormoglass particles during the heating. The ormoglass, which was prepared by the partial hydrolysis of a mix containing alkoxides, possessed with high probability a significant amount of organic groups in the network (i.e. nonhydrolyzed organic ligands from alkoxides). Therefore, when these groups were degrading, a shrinkage of the ormoglass could have occurred, resulting thus in a decrease of the layer thickness. On the other hand, FESEM pictures additionally seemed to show that fibers were coated with a monolayer of particles, as already mentioned previously.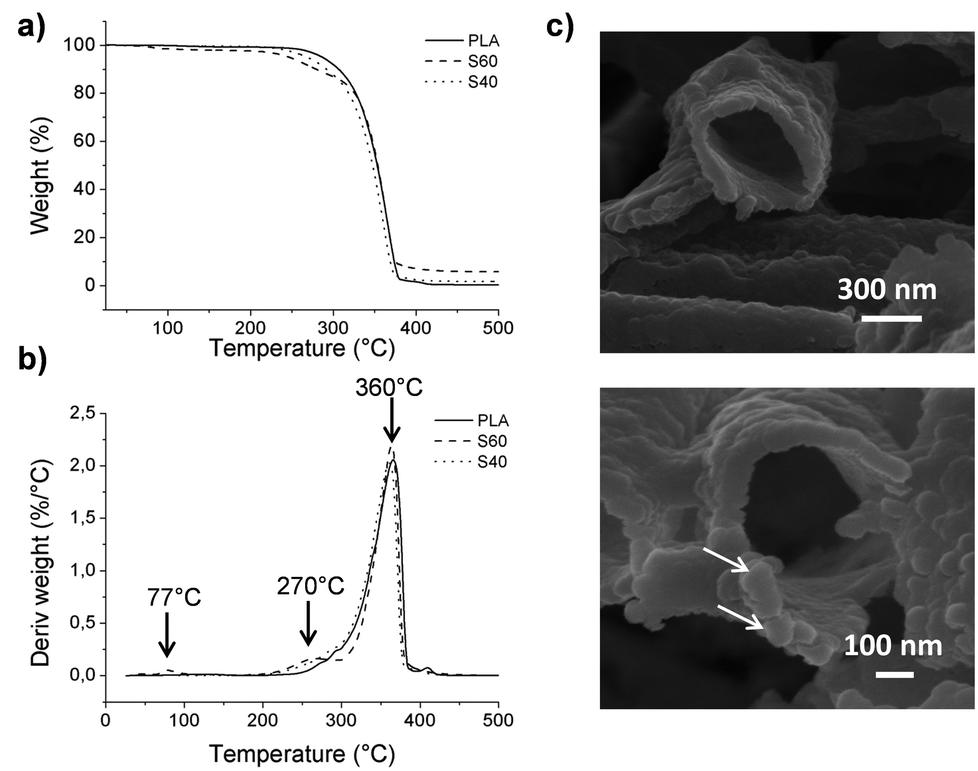 | ||
| Fig. 5 (a) TGA and (b) TGA derivative curves obtained for PLA and coated fibers. (c) Morphology of the inorganic shell remaining after thermal treatment of the S60 fibers (FESEM images). | ||
Information about the degradation byproducts was obtained from the TGA assays. Differences in the graphics of the TGA derivative curves are observed: peaks at ∼77 and ∼270 °C that were not observed in the pure PLA curve are clearly noticed in the S60 fibers curve. The first peak is not observed for the S40 fibers, but a very small shoulder can be seen for the second peak. To determine to which compounds these changes were related, measurements with TGA coupled with FTIR were performed. The peak at the lowest temperature was attributed to the presence of triethoxysilane molecules and the second one to the degradation of PLA, as well as the third peak common to all samples (see ESI, Fig. S6† for spectra). This suggests that the coating influences the degradation of the polymer and favors the decomposition of some polymeric fragments before the usual PLA degradation temperature (characteristic peak at 360 °C). This can be explained by the degradation of short polymeric fragments created by the hydrolysis of the fibers during the first surface treatment (chain scission), promoting their earlier decomposition. The peak corresponding to the triethoxysilane compound can be related to the presence of non-reacted molecules that could have been trapped in the ormoglass network during its formation. As this peak is not observed for the S40 fibers, it is assumed that this coating possessed only residual silane molecules, whose concentration was lower than the detection limit of the device. Gas chromatography assay in fact revealed that silanes were indeed found in the S40 fibers (ESI, Fig. S7†). These silane molecules might remain in the coatings because of the fast formation of the colloidal suspension, which can lead to the quick encapsulation of non-reacted molecules. Assigned to the ormoglass, the mass loss related to this peak (77 °C) should thus be taken into account for the quantification of ormoglass coated. This is especially relevant for the S60 fibers for which a significant mass loss of 2.2% was detected by TGA at this temperature. Finally, the ormoglass mass percentage of the coating of these fibers is a little bit higher (7.8%) than the final inorganic mass percentage that was defined at the end of the heat treatment (5.6%). Although it is clear that the S40 fibers contained a little amount of non-reacted silanes in the coating according to gas chromatography, the final inorganic mass percentage (1.7%) was however considered as the representative amount of ormoglass coated, because no significant mass loss was measured by TGA at this temperature. Moreover, it is assumed that these percentages are approximate values of the exact amount of coated ormoglass as they do not include the organic fragments that are in the ormoglass but whose mass could not be determined by the assays carried out.
Polymer thermal calorimetric characterization
To determine whether changes in the polymer occur after coating, DSC measurements were performed. All data summarizing the different characteristics of the thermal properties of the fibers are presented in Table 4 (see the ESI for the curves, Fig. S8†). Curve analysis showed that the crystalline onset (Tonsetc) and the melting onset (Tonsetm) temperatures of both coating compositions are shifted towards higher values than those associated with pure PLA fibers (presenting the typical thermal curve of a semicrystalline polymer). The increase of ∼12–13 °C in the Tonsetc, and therefore the need of more energy to crystallize, reflects the mobility limitations of the polymer chains after coating. This means that the bonding between the ormoglass particles and the polymer prevents the rearrangement of some polymer chains in a well-ordered phase by restricting its mobility. This also suggests that strong interactions between the polymer and the ormoglass might have been efficiently created. Another observation considering the Tonsetc is the change in the intensity of the peak: the DSC curve of PLA shows a more intense and well defined exothermal peak than the one observed for the coated fibers. This possibly means that less amorphous phase can be ordered after the coating protocol, as reflected by the ΔHc values. The small increase of ∼2 °C in the Tm demonstrates a slight improvement in the thermal stability of the crystalline phase of the hybrids. On the other hand, the crystallinity of the coated fibers (around 44%) significantly increased in comparison with one of the PLA fibers, which initially had a low crystalline percentage (10%). X-ray data confirmed that after coating, a crystalline phase attributed to the PLA compound was formed (ESI, Fig. S9†). Complementary DSC results additionally showed that the increase in crystallinity was not due to the hydrolysis of the fibers (first treatment) and a possible chain rearrangement because of the shortening of the chains (ESI, Fig. S10†). It is thus believed that the coating acts as a nucleating agent that promotes PLA crystallization.47,48 This could be an additional explanation for the shift in the crystallization temperatures associated with the coated fibers. These already ordered parts might hinder the organization of the potentially structurable chains. According to this crystallinity level, it can also be concluded that the mechanical properties assessed under tensile-strain assays are not only due to the presence of the ormoglass but also to this crystallinity gain. Both aspects may contribute to the modification of the mechanical properties of the scaffold.| Sample | T onsetm (°C) | T peakm (°C) | ΔHm (J g−1) | T onsetc (°C) | T peakc (°C) | ΔHc (J g−1) | χ (%) |
|---|---|---|---|---|---|---|---|
| PLA | 153.4 | 159.1 | 34.5 | 72.4 | 82.2 | 24.6 | 10.7 |
| S60 | 155.3 | 161.4 | 39.8 | 86.5 | 92.5 | 1.8 | 44.2 |
| S40 | 155.4 | 161.3 | 43.9 | 85.1 | 91.9 | 4.0 | 43.6 |
Calcium release and pH analysis
The lower the silicon content, the higher the calcium release. In Fig. 6, the release of the calcium ion from the two different coatings can be observed. Release concentration was normalized to the coating thickness, assuming the formation of a monolayer of particles. S40 showed higher and more sustained release of calcium ions, even when S60 had a stronger earlier burst. In both cases, calcium release stabilizes for longer periods. However, and in contrast to what was observed in previous studies with similar compounds,29 pH in a SBF solution remains practically unmodified, and we observed a fast tendency to recover the original pH of 7.4, indicating the importance of the low amount of bioactive material in coatings. These results seem to confirm previous conclusions, as S40 was the sample with a higher normalized release rate of calcium and the most constant sustained decrease at higher time points. In the case of the S60 coating, the remaining calcium was lower, especially after the early burst. S40 continued to be the composition with the highest calcium and most sustained release in longer time periods. Note, however, that without the normalization, the absolute calcium released in S60 would be higher due to the higher amount of deposited material.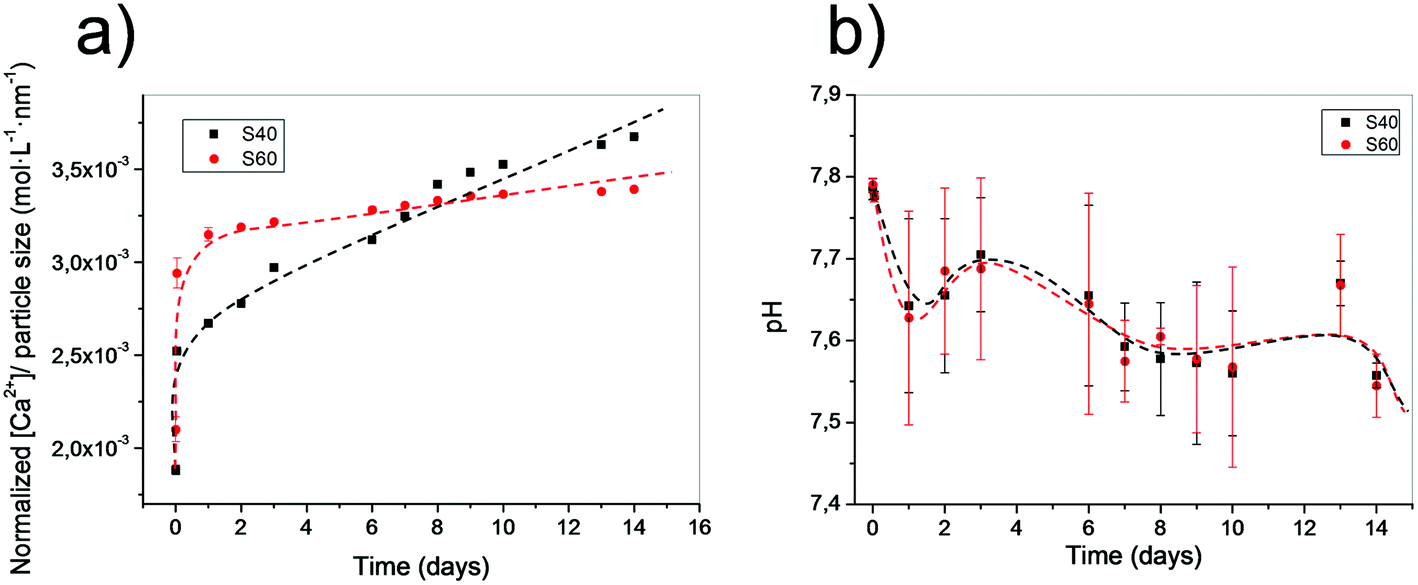 | ||
| Fig. 6 (a) Particle size normalized calcium release of both samples in HEPES buffered solution at different time points. (b) pH of the media for both immersed samples at different time points. | ||
In vitro assays
As described previously, the materials developed in this study aim to improve interactions between cells and materials. To verify that the exposure of the bioactive phase directly at the material surface had a positive effect on cellular response, preliminary in vitro assays were performed. rMSCs and rEPCs were seeded on the fibers separately. These two types of cells were chosen according to published results that demonstrated the potential of this glass system to promote mesenchymal stem cell differentiation into osteoblastic cell lineage and induce vascularization.49–51 After 1 day of culture on the materials, cells were fixed and stained with specific fluorophores to assess their morphology. Pictures obtained from confocal microscope imaging revealed that both cell types restricted their spreading on PLA fibers, whereas they spread significantly on the coated fibers for both coating compositions (Fig. 7). Cells on polymeric scaffolds presented a round shape with only a few filopodia, while on the coated ones cells show filopodia following the direction of the fibers and extending their cytoskeleton. Stress fibers are even observed inside the cells, corroborating the good adhesion of cells on the scaffold.52,53 Quantification of cell adhesion efficiency also showed that S40 shows a slightly better spread cell behavior and better adhesion than S60, although not significant differences were found for both cell lineages. However, significant differences were found when compared with pure PLA confirming morphology observations. Further cultures should be performed to specifically study the difference in occupied area percentages between cell lineages.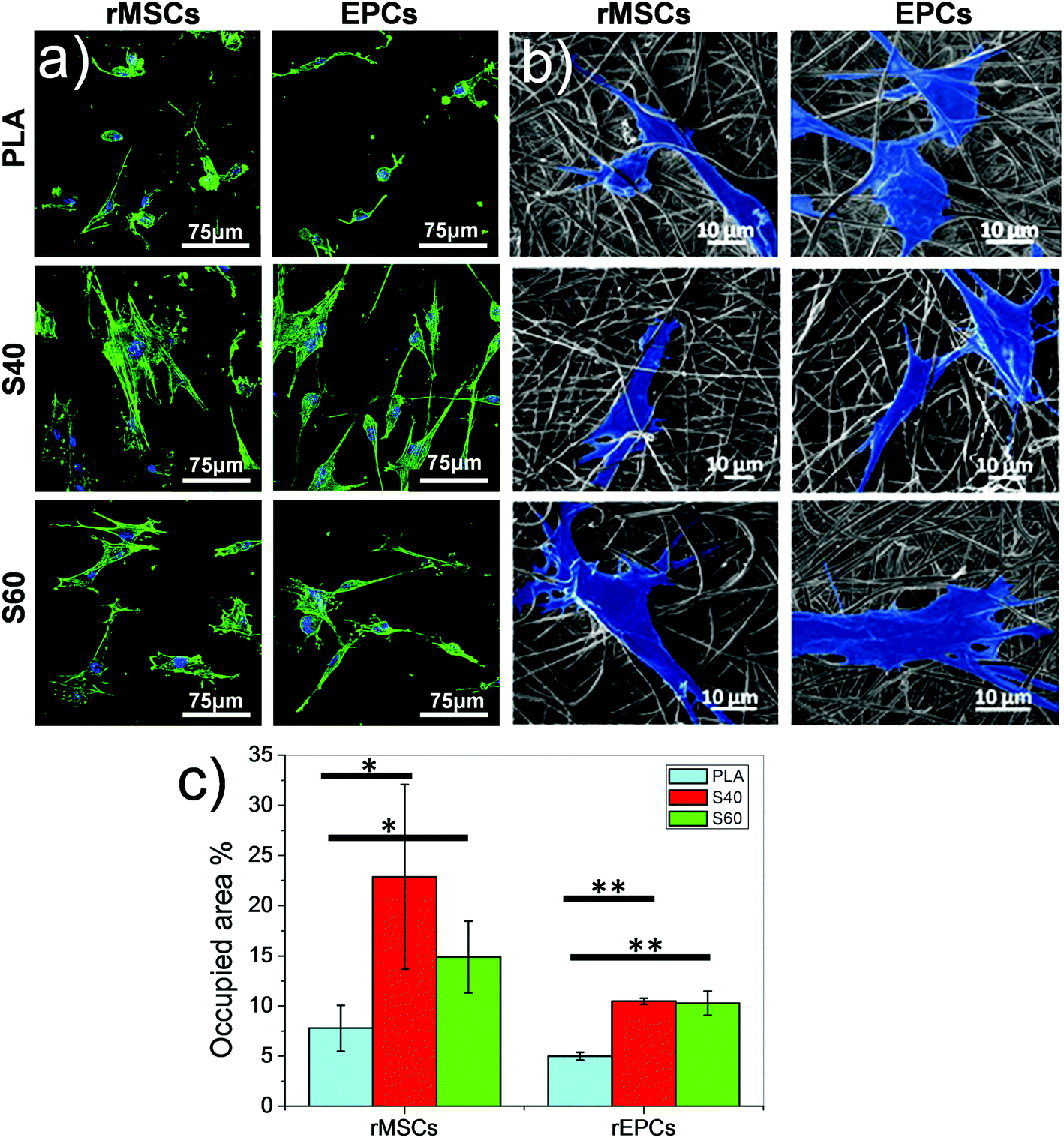 | ||
| Fig. 7 Morphology of rat mesenchymal stem cells (rMSCs) and endothelial progenitor cells (rEPCs) after 1 day of culture on PLA and coated fibers: (a) fluorescent micrographs. (b) FESEM micrographs where cells were artificially highlighted. (c) Cell adhesion efficiency quantification by cell spread surface measurement. | ||
This assay thus demonstrates that even after a short time, cells immediately modulate their spreading according to the fibers’ surface properties of the materials. The rapid and consequent spreading of the cells on the coated fibers can be explained by several factors. The first one is the direct exposure of the bioactive ormoglass at the material surface which can be immediately perceived by biological entities; the second one is the focal adhesions that are promoted by the existence of the nanoroughness of the coating54,55 and, finally, the significant hydrophilicity of the scaffold.56 Although further investigations such as proliferation and differentiation assays are needed to confirm these scaffolds as promising materials for bone tissue engineering, these first cellular results show that the coating enables an immediate positive interaction of the cells with the material. The nanoscale and nanofeatures of these scaffolds represent moreover significant advantages over macro or microstructured materials as they play an important key role in controlling sub-cellular events and in instructing cell response.57
Experimental
Fiber fabrication
Poly-l/DL-lactic acid 95/5 (PLA95/5) (Purasorb PLDL 9562, inherent viscosity midpoint 6.2 dl g−1, Mw ≈ 125000 g mol−1) was dissolved in 2,2,2-trifluoroethanol (TFE, Sigma-Aldrich ≥99%) (3% w/w) and 15 mL of the obtained polymeric solution was electrospun. The experimental setup conditions were set as: flow rate of 0.5 mL h−1, voltage of 7.5 kV and a tip–collector distance of 12 cm. Fibers were collected on a flat metallic aluminum foil.
Si–Ca–P2 ormoglass precursors’ preparation
The ormoglass was prepared by applying the sol–gel method to a solution containing silicon, calcium and phosphorus. For that purpose, tetraethylorthosilicate (TEOS), calcium 2-methoxyethoxide and phosphorus pentoxide precursor solutions were mixed under a nitrogen atmosphere. Calcium and phosphorus precursor solutions were prepared in our laboratory, whereas TEOS was commercially purchased (Sigma-Aldrich ≥99%). Calcium metallic (Sigma-Aldrich, 98%) and P2O5 powder (Sigma-Aldrich, 99.99%) were refluxed in 2-methoxyethanol (Sigma-Aldrich, anhydrous, 99.8%) and absolute ethanol (Panreac, 99.98%) respectively, following already published protocols.58,59 The mix was done to obtain the ormoglass precursors with two different compositions: 60Si:30Ca:10P2 (labeled as S60) and 40Si:45Ca:15P2 (labeled as S40) molar ratios. The final ormoglass precursors were kept under an inert atmosphere until hydrolysis was performed.
Surface treatments
The coating process is based on a 4-step surface treatment: hydrolysis of the fiber surface to create carboxylic groups, activation of these groups, coupling agent functionalization (3-aminopropyltriethoxysilane, APTES coupling molecule, Acros Organics, 99%) and a condensation process enabling the bonding of ormoglass particles to the PLA by means of APTES.23 Once removed from the aluminum foil, fiber sheets were thus hydrolyzed in a NaOH 0.1 M solution for 150 s. Fibers were then immersed in a 0.1 M ethyl(dimethylaminopropyl)carbodiimide/0.2 M N-hydroxysuccinimide (EDC, Aldrich, 97%/NHS, Aldrich, 98%) ethanolic solution for 1 hour and placed afterwards in an APTES solution (10% v/v in absolute ethanol) for 2 h. At this point, ormoglass precursor solution was removed from the inert atmosphere and immediately partially hydrolyzed to obtain a suspension of ormoglass nanoparticles (Si–Ca–P network).23 Precursors were both hydrolyzed for 150 s but with different silicon:water molar ratios: 1 to 1 for the S60 one and 1 to 2 for the S40 one. The chosen ratios were determined to be the maximum quantity of water that can be introduced into each ormoglass precursor solution system without inducing a full gelation over 2 hours. The particles were kept well dispersed, minimizing aggregates. This was essential for the final coating stage during which single particles were aimed to be linked to the fibers with a homogeneous distribution. After hydrolysis, ethanol was added to dilute the suspensions in order to avoid the dissolution of the fibers that can occur when they are placed in contact with a so concentrated solution of alkaline compounds such as alkoxides. The dilution factor was fixed at 1 mL ethanol for 1 mL of S60 ormoglass suspension, and 4 mL ethanol for 1 mL of S40 ormoglass suspension. The diluted ormoglass suspensions were then added to the pre-APTES functionalized fibers to initiate the condensation process, creating a siloxane bond between APTES molecules and the ormoglass nanoparticles. To ensure that the ormoglass particles were strongly attached to the fiber's surface, fibers were finally placed in an 80 W ultrasonic bath (VWR, Mollet del Vallès, Spain) for 5 minutes. All residual particles were thus washed. Fibers were left on a petri dish to dry at room temperature.
Note that nanofibrous PLA can be hydrophobic enough to avoid the entry of a NaOH aqueous solution within the fibers in the bulk of the mat. To ensure the entire coating of the fibers, the mats were immersed in absolute ethanol to increase wettability of the PLA fibers.60,61 After this, NaOH aqueous solution wetted and etched the inner fibers. The rest of the treatments are performed in ethanol and the whole fibrous mat is perfectly wet.
Material characterization
Zeta potential measurements were performed in a 1 mM KCl electrolyte solution using the “adjustable gap cell” set-up (electrokinetic analyzer SurPASS, Anton Paar Ltd).Fiber morphology and fibers with fixed cultured cells were assessed using a Field Emission Scanning Electron Microscope (FESEM, Nova™-Nano SEM-230; FEI Co.) and fiber composition was assessed using an Energy Dispersive X-ray Spectrometer (EDS, Quanta 200 XTE 325/D8395; FEI Co.). In both cases, fibers were coated with a thin layer of carbon before analysis.
Infrared measurements (Fourier Transform Infrared Spectroscopy, FTIR) were performed in attenuated total reflectance (ATR) mode (Nicolet 8700 Thermo Scientific), placing the sample directly in contact with the ATR crystal without a preliminary special preparation. FTIR spectra were averaged from 64 scans at a resolution of 4 cm−1 and collected in the 4000–400 cm−1 wavenumber range.
Tensile-strain assays (Adamel Lhomargy DY-34) were performed with 5 × 1 cm samples cut from the coated fibrous mats, whose thicknesses were preliminarily determined using FESEM cross section pictures. Young's modulus was attained considering the slope of the lineal elastic area of the stress–strain curves and yield strength was considered as the intersection between the measured stress and a line with the origin at 0.2% of the strain (parallel to the elastic area).
Atomic Force Microscopy (AFM, MultiMode 8, Bruker) was used to assess the stiffness of the fibers, previously fixed on an adhesive borosilicate substrate. Measurements were performed in tapping Peakforce mode. DMT modulus was assessed to determine the stiffness of the fibers and the root mean square roughness (Rq) to evaluate their roughness (NanoScope Analysis V1.20 software).
To evaluate the content of ormoglass in the hybrid materials, thermogravimetry analysis was used (TGA, Q5000 TA). Samples were heated at 10 °C min−1 at up to 700 °C in air. In order to identify the gaseous products that degrade during this thermal treatment, an additional TGA analysis coupled FTIR was carried out under nitrogen flow (10 ml min−1) on the fibers coated with the ormoglass S60. On the other hand, Differential Scanning Calorimetry (DSC) analysis was performed to check if changes in the organic phase occur after the coating. A DSC Q2000 TA device and 5 mg samples confined in hermetic aluminum pans were used. Samples were heated at a rate of 10 °C min−1 starting from 25 °C up to 180 °C. The software used to analyze TGA and DSC results was Universal Analysis 2000 v4.7A and the degree of crystallinity was calculated using eqn (1):62
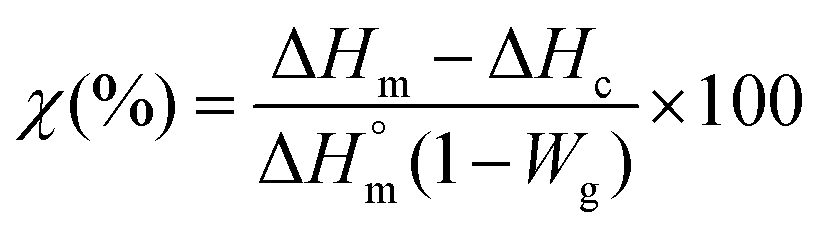 | (1) |
pH and Ca2+ releases of the samples were evaluated by immersion in c-SBF64 (pH evaluation) and in a pH ∼7.4 ± 0.1 buffered solution (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, HEPES, 0.1% to avoid CaHPO4·H2O precipitation and 0.02 M KCl for a constant ionic strength in the Ca2+ release evaluation). Discrete measurements for different time points were collected, renewing the liquid at each time point. They were performed using a Crison GLP22+ pH-meter (Crison Spain), a Crison pH microelectrode, a Crison Ca2+ selective electrode and an Ag/AgCl reference electrode. The released calcium concentration was normalized to the coating thickness to allow sample comparison without the influence of particle size and coating weight.
In vitro assays were conducted as preliminary biological tests. This study involved the use of rat endothelial progenitor cells and rat mesenchymal stem cells. All experiments were performed in compliance with the Spanish regulations and institutional guidelines, and also the institutional committee that approved the experiments. rMSCs and rEPCs cells from the rat bone marrow were isolated and cultured following a method described before22 and detailed in the ESI.† Cells were expanded in flasks and trypsinized before reaching confluence. Before being seeded on the materials at a density of 10000 cells per well (culture plates of 24 wells), fibers were rinsed twice with phosphate buffered saline (PBS) and incubated in culture medium for 2 hours at 37 °C. After seeding, plates were incubated for 1 day at 37 °C under a CO2 atmosphere. Cells were then fixed with a PFA solution, stained with green phalloidin (cytoskeleton) and DAPI (nucleus) and observed under a confocal microscope. Image stacks were reconstructed using the ImageJ software.65 Adhesion efficiency quantification based on the surface of spread cells was performed by ImageJ using 5 images for each condition to quantify the efficiency of cell adhesion on the ormoglass coating.
FE-SEM images for cell adhesion were prepared as follows: samples were seeded for 1 day and then fixed for 10 min at room temperature in 2.5% glutaraldehyde in PBS. The fixed samples were then dehydrated for 5 min by immersion in different diluted ethanol aqueous solutions (40%, 60%, 80%, 95% and 100%) and critical-point dried. Then, samples were coated with a thin layer of gold and observed by the previously described FESEM device. Images were artificially colored using blue for the cell surface.
Results are shown as the means ± standard deviation and analyzed via one-way ANOVA. A value of p < 0.05 was considered statistically significant (*) and p < 0.005 highly statistically significant (**).
Conclusion
A novel coating protocol that allows the fabrication of materials with tailorable surface properties (topography, chemical composition and stiffness) has been successfully developed. This approach, involving the sol–gel method, is an efficient, cost-effective and versatile option to produce hybrid materials which possess the bioactive phase fully exposed at their surface and strong interactions between their constituents. The materials produced showed excellent fast cellular response, Ca2+ release and improved mechanical properties. Moreover, the ability to easily modify their surface properties by changing the ormoglass composition or the ormoglass hydrolysis ratio, for example, highlights the potential of this promising protocol. As materials’ surface properties influence cell functionalities, a wide range of biomaterials that could possibly trigger different cellular responses could be produced.The protocol can be also transferred to other polymer structures prepared by different processing methods and other bioactive glasses, offering the possibility of expanding the applications to additional tissue types depending on the scaffolds’ architecture and composition: angiogenic coating for muscle regeneration, tube coating for arterial replacement, or antimicrobial glass coating for wound healing, for example. This functionalization method therefore represents an essential improvement towards the design of functional materials for the regenerative medicine field.
Acknowledgements
The authors thank the European Commission (European ERANET project PI11/03030, NANGIOFRAC), the Spanish Ministry of Economy and Competitiveness (project MAT2011-29778-C02-01) and the National Center for Research and Development (contract no. 04/Euronanomed/2012) for funding. O. Castaño also acknowledges the MINECO for the “Ramon y Cajal” and I3 programe. N. Sachot thanks the AGAUR (travel grant fellowship) for financial support. We also thank Tomasz Jaroszewicz, Ewa Kijeńska and Emilia Choińska for their help with DSC, FTIR and TGA devices respectively. Dr Jungyou Wang is also acknowledged for his advice on the DLS measurements.References
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Sci., 2009, 324, 1673–1677 CrossRef CAS PubMed.
- I. K. Ko, S. J. Lee, A. Atala and J. J. Yoo, Exp. Mol. Med., 2013, 45, e57 CrossRef PubMed.
- M. Navarro, A. Michiardi, O. Castaño and J. A. Planell, J. R. Soc., Interface, 2008, 5, 1137–1158 CrossRef CAS PubMed.
- D. W. Hutmacher and S. Cool, J. Cell. Mol. Med., 2007, 11, 654–669 CrossRef CAS PubMed.
- M. Wang, Biomaterials, 2003, 24, 2133–2151 CrossRef CAS.
- Q. Chen, J. A. Roether and A. R. Boccaccini, in Tissue engineering, 2008, vol. 4 Search PubMed.
- R. A. Martin, S. Yue, J. V. Hanna, P. D. Lee, R. J. Newport, M. E. Smith and J. R. Jones, Philos. Trans.: Math., Phys. Eng. Sci., 2012, 370, 1422–1443 CrossRef CAS PubMed.
- N. Sachot, E. Engel and O. Castaño, Curr. Org. Chem., 2014, 18(18), 2299–2314 CrossRef CAS.
- J. R. Jones, Acta Biomater., 2013, 9, 4457–4486 CrossRef CAS PubMed.
- G. Poologasundarampillai, C. Ionescu, O. Tsigkou, M. Murugesan, R. G. Hill, M. M. Stevens, V. Hanna, M. E. Smith and J. R. Jones, J. Mater. Chem., 2010, 20, 8952–8961 RSC.
- O. Mahony, O. Tsigkou, C. Ionescu, C. Minelli, L. Ling, R. Hanly, M. E. Smith, M. M. Stevens and J. R. Jones, Adv. Funct. Mater., 2010, 20, 3835–3845 CrossRef CAS PubMed.
- G. Poologasundarampillai, B. Yu, O. Tsigkou, E. Valliant, S. Yue, P. D. Lee, R. W. Hamilton, M. M. Stevens, T. Kasuga and J. R. Jones, Soft Matter, 2012, 8, 4822–4832 RSC.
- T. Niemela, H. Niiranen, M. Kellomaki and P. Tormala, Acta Biomater., 2005, 1, 235–242 CrossRef CAS PubMed.
- H.-W. Tong, M. Wang, Z.-Y. Li and W. W. Lu, Biomed. Mater., 2010, 5, 054111 CrossRef PubMed.
- M. Charles-Harris, M. A. Koch, M. Navarro, D. Lacroix, E. Engel and J. A. Planell, J. Mater. Sci. Mater. Med., 2008, 19, 1503–1513 CrossRef CAS PubMed.
- A. Aguirre, A. González, M. Navarro, O. Castaño, J. A. Planell and E. Engel, Eur. Cells Mater., 2012, 24, 90–106 CAS.
- M. Charles-Harris, M. A. Koch, M. Navarro, D. Lacroix, E. Engel and J. A. Planell, J. Mater. Sci. Mater. Med., 2008, 19, 1503–1513 CrossRef CAS PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757–2774 CrossRef CAS PubMed.
- A. Aguirre, A. González, J. A. Planell and E. Engel, Biochem. Biophys. Res. Commun., 2010, 393, 156–161 CrossRef CAS PubMed.
- A. Aguirre, J. A. Planell and E. Engel, Biochem. Biophys. Res. Commun., 2010, 400, 284–291 CrossRef CAS PubMed.
- A. González-Vázquez, J. A. Planell and E. Engel, Acta Biomater., 2014, 10, 2824–2833 CrossRef PubMed.
- A. Aguirre, A. González, J. A. Planell and E. Engel, Biochem. Biophys. Res. Commun., 2010, 393, 156–161 CrossRef CAS PubMed.
- N. Sachot, O. Castaño, M. A. Mateos-timoneda, E. Engel and J. A. Planell, J. R. Soc., Interface, 2013, 10, 1–5 CrossRef PubMed.
- B. Blencke, H. Brömer, K. Deutscher and W. Hennig, Biomed. Tech. Band, 1978, 23, 360–361 Search PubMed.
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res., 1971, 5, 117–141 CrossRef PubMed.
- I. D. Xynos, A. J. Edgar, L. D. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151–157 CrossRef CAS.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- H. Zhou, J. G. Lawrence and S. B. Bhaduri, Acta Biomater., 2012, 8, 1999–2016 CrossRef CAS PubMed.
- O. Castano, N. Sachot, E. Xuriguera, E. Engel, J. A. Planell, J.-H. Park, G.-Z. Jin, T.-H. Kim, J.-H. Kim and H.-W. Kim, ACS Appl. Mater. Interfaces, 2014, 6(10), 7512–7522 CAS.
- M. Charles-Harris, M. Navarro, E. Engel, C. Aparicio, M. P. Ginebra and J. A. Planell, J. Mater. Sci. Mater. Med., 2005, 16, 1125–1130 CrossRef CAS PubMed.
- T. Serra, J. A. Planell and M. Navarro, Acta Biomater., 2013, 9, 5521–5530 CrossRef CAS PubMed.
- Z. Ma, M. Kotaki, R. Inai and S. Ramakrishna, Tissue Eng., 2005, 11, 101–109 CrossRef CAS PubMed.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- J. Coates and R. A. M. Ed, in Encyclopedia of analytical chemistry, 2000, pp. 10815–10837 Search PubMed.
- P. J. Launer, in Silicone Coumpounds Register and Review, 1987, pp. 100–103 Search PubMed.
- E. T. Vandenberg, L. Bertilsson, B. Liedberg, K. Uvdal, R. Erlandsson, H. Elwing and I. Lundström, J. Colloid Interface Sci., 1991, 147, 103–118 CrossRef CAS.
- G. E. A. Swann and S. V. Patwardhan, Clim. Past, 2011, 7, 65–74 CrossRef.
- M. Manzano, A. J. Salinas, F. J. Gil and M. Vallet-Regí, J. Mater. Sci. Mater. Med., 2009, 20, 1795–1801 CrossRef CAS PubMed.
- R. Ravarian, H. Wei and F. Dehghani, Eng. Med. Biol. Soc., 2011, 3593–3596 CAS , 2011 Annu. Int. Conf. IEEE.
- R. Peña-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2006, 42, 595–603 CrossRef.
- V. C. Costa, H. S. Costa, W. L. Vasconcelos, M. D. M. Pereira, R. L. Oréfice and H. S. Mansur, Mater. Res., 2007, 10, 21–26 CrossRef CAS.
- J. E. Stewart, J. Chem. Phys., 1959, 30, 1259–1265 CrossRef CAS PubMed.
- M. Vallet-Regi, A. J. Salinas, J. Ramirez-Castellanos and J. M. Gonzalez-Calbet, Chem. Mater., 2005, 17, 1874–1879 CrossRef CAS.
- C. C. Ribeiro, I. Gibson and M. A. Barbosa, Biomaterials, 2006, 27, 1749–1761 CrossRef CAS PubMed.
- M. Pluta, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3392–3405 CrossRef CAS PubMed.
- A. R. McLauchlin and N. L. Thomas, Polym. Degrad. Stab., 2009, 94, 868–872 CrossRef CAS PubMed.
- W. S. Chow and S. K. Lok, J. Therm. Anal. Calorim., 2009, 95, 627–632 CrossRef CAS.
- H. Pan and Z. Qiu, Macromolecules, 2010, 43, 1499–1506 CrossRef CAS.
- L. L. Hench, J. Eur. Ceram. Soc., 2009, 29, 1257–1265 CrossRef CAS PubMed.
- A. Leu, S. M. Stieger, P. Dayton, K. W. Ferrara and J. K. Leach, Tissue Eng., Part A, 2009, 15, 877–885 CrossRef CAS PubMed.
- L.-C. Gerhardt, K. L. Widdows, M. M. Erol, C. W. Burch, J. A. Sanz-Herrera, I. Ochoa, R. Stämpfli, I. S. Roqan, S. Gabe, T. Ansari and A. R. Boccaccini, Biomaterials, 2011, 32, 4096–4108 CrossRef CAS PubMed.
- I. K. Buckley and K. R. Porter, Protoplasma, 1967, 64, 349–380 CrossRef CAS.
- P. W. Oakes, Y. Beckham, J. Stricker and M. L. Gardel, J. Cell Biol., 2012, 196, 363–374 CAS.
- A. M. Ross, Z. Jiang, M. Bastmeyer and J. Lahann, Small, 2012, 8, 336–355 CrossRef CAS PubMed.
- M. Arnold, E. A. Cavalcanti-Adam, R. Glass, J. Blümmel, W. Eck, M. Kantlehner, H. Kessler and J. P. Spatz, ChemPhysChem Eur. J. Chem. Phys. Phys. Chem., 2004, 5, 383–388 CAS.
- B. A. Dalton, C. D. Mcfarland, R. G. Thomas, H. J. Griesser and J. G. Steele, J. Biomater. Sci., Polym. Ed., 1998, 9, 781–799 CrossRef CAS PubMed.
- L. Zhang and T. J. Webster, Nano Today, 2009, 4, 66–80 CrossRef CAS PubMed.
- D. M. Pickup, S. P. Valappil, R. M. Moss, H. L. Twyman, P. Guerry, M. E. Smith, M. Wilson, J. C. Knowles and R. J. Newport, J. Mater. Sci., 2009, 44, 1858–1867 CrossRef CAS.
- A. F. Ali, P. Mustarelli, E. Quartarone and A. Magistris, J. Mater. Res., 1999, 14, 327–329 CrossRef CAS.
- M. Lundgren, N. L. Allan, T. Cosgrove and N. George, Langmuir, 2002, 18, 10462–10466 CrossRef CAS.
- A. K. Metya, S. Khan and J. K. Singh, J. Phys. Chem. C, 2014, 118, 4113–4121 CAS.
- T. H. Lee, F. Y. C. Boey and K. A. Khor, Compos. Sci. Technol., 1995, 53, 259–274 CrossRef CAS.
- D. E. Henton, P. Gruber, J. Lunt and J. Randall, in Natural Fibers, Biopolymers and Biocomposites, 2005, pp. 527–578 Search PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- W. S. Rasband, US Natl. Inst. Heal, Bethesda, USA, http//imagej.nih.gov/ij/, 1997, 2014.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr04275e |
| This journal is © The Royal Society of Chemistry 2015 |