
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ optical sequencing and structure analysis of a trinucleotide repeat genome region by localization microscopy after specific COMBO-FISH nano-probing
M.
Stuhlmüller
a,
J.
Schwarz-Finsterle
a,
E.
Fey
b,
J.
Lux†
a,
M.
Bach
a,
C.
Cremer
ac,
K.
Hinderhofer
b,
M.
Hausmann
*a and
G.
Hildenbrand
ad
aKirchhoff-Institute for Physics, Heidelberg University, Im Neuenheimer Feld 227, 69120 Heidelberg, Germany. E-mail: hausmann@kip.uni-heidelberg.de; Tel: +49-6221-549824; Fax: +49-6221-549112
bInstitute for Human Genetics, Heidelberg University, Im Neuenheimer Feld 366, 69120 Heidelberg, Germany
cInstitute for Molecular Biology, Ackermannweg 4, 55128 Mainz, Germany
dDept. Radiooncology, University Medical Centre Mannheim, Theodor-Kutzer-Ufer 1-3, 68135 Mannheim, Germany
First published on 2nd October 2015
Abstract
Trinucleotide repeat expansions (like (CGG)n) of chromatin in the genome of cell nuclei can cause neurological disorders such as for example the Fragile-X syndrome. Until now the mechanisms are not clearly understood as to how these expansions develop during cell proliferation. Therefore in situ investigations of chromatin structures on the nanoscale are required to better understand supra-molecular mechanisms on the single cell level. By super-resolution localization microscopy (Spectral Position Determination Microscopy; SPDM) in combination with nano-probing using COMBO-FISH (COMBinatorial Oligonucleotide FISH), novel insights into the nano-architecture of the genome will become possible. The native spatial structure of trinucleotide repeat expansion genome regions was analysed and optical sequencing of repetitive units was performed within 3D-conserved nuclei using SPDM after COMBO-FISH. We analysed a (CGG)n-expansion region inside the 5′ untranslated region of the FMR1 gene. The number of CGG repeats for a full mutation causing the Fragile-X syndrome was found and also verified by Southern blot. The FMR1 promotor region was similarly condensed like a centromeric region whereas the arrangement of the probes labelling the expansion region seemed to indicate a loop-like nano-structure. These results for the first time demonstrate that in situ chromatin structure measurements on the nanoscale are feasible. Due to further methodological progress it will become possible to estimate the state of trinucleotide repeat mutations in detail and to determine the associated chromatin strand structural changes on the single cell level. In general, the application of the described approach to any genome region will lead to new insights into genome nano-architecture and open new avenues for understanding mechanisms and their relevance in the development of heredity diseases.
Introduction
Trinucleotide repeat expansion in the DNA of chromatin has been identified as the reason for more than 40 neurological disorders such as Fragile-X syndrome, Huntington disease, inherited ataxias and muscular dystrophy. Until now it is not clearly understood how the expansions take place and develop during cell proliferation or what in general could be the reason behind them.1 Under the assumption that the genome architecture is important for genome function,2,3 investigations of structural changes of chromatin in intact, 3D-conserved cell nuclei may help to better understand mechanisms behind disease induction and development.Microscopic nano-techniques have offered new insights into cell biology especially of cell membranes and are bridging the gap of the mechanistic understanding of cell function, e.g. immunological response, and medical diagnostics.4 Basic structural features of cell nuclei and their time dependent changes have been elucidated and their profound functional correlation has been demonstrated.5,6 However, so far comparable investigations failed because of probe resolution and specificity referring to very small chromatin regions (nano-architecture of the cell nucleus) even if modern optical super-resolution microscopy or electron microscopy were applied. The use of localization microscopy (e.g. PALM, STORM or as applied here Spectral Position Determination Microscopy [SPDM])7,8 in combination with COMBO-FISH (COMBinatorial Oligonucleotide Fluorescence In Situ Hybridization)9,10 allows the investigation on the one hand of the native spatial structure of chromatin for instance trinucleotide repeat expansions and on the other hand of the optical sequencing of repetitive sequences by counting the number of oligonucleotide probes bound to the target area within the genome of 3D-conserved cell nuclei cells.
Fragile-X syndrome (FXS) or Martin-Bell-Syndrome is one main cause of inherited mental retardation.1 FXS belongs to the group of the so called ‘trinucleotide repeat expansion disorders’ consisting of the expansion of a trinucleotide frequency [(CGG)n-expansion] in the 5′ untranslated region of the Fragile-X Mental Retardation 1 gene (FMR1) on the X-chromosome.11,12 The enlargement of the CGG triplet-repeat results in a hypermethylisation13 within the promoter-region of the FMR1-gene causing a change of the spatial structure of the chromatin.11,14 This leads to a deactivation of the gene and so to a lack of FMR1-protein, which is meant to be required for synaptic plasticity.15 Normally approx. 30 triplets are found. If the number of triplet repeats exceeds 200, (CGG)>200, this is named full mutation. Repeats in the range between (CGG)≥45 and (CGG)≤54 are known as intermediates or grey zone alleles and repeats from (CGG)≥55 to (CGG)≤200 are called pre-mutations.16,17
In current medical diagnostics, the length of the trinuclueotide repeat expansions is usually estimated by Southern blotting and PCR amplification.11 This lengthy method has not only limits in the resolution of the Southern blot; it also averages the result of a certain amount of cells. Therefore, it is hard to differentiate between pre-mutation and full mutation if the expansion rate on average approximates 200 (CGG) repeats.
Lukáš et al. published an article18 about sequestration of muscleblind-like protein 1 (MBNL1) in tissues of patients with myotonic dystrophy type 2, which is known to be caused by a (CCTG)n tetraplet repeat expansion in intron 1 of the zinc finger 9 (ZFN9 gene) on chromosome 3q 21.3.19 They used RNA-FISH and immunofluorescence-FISH to demonstrate a different frequency of nuclei containing the foci of the CCUG transcript, a different expression pattern of MBNL1 protein and a different sequestration of MBNL1 by (CCUG)n repeats in different tissues.18
Here, for the first time we present microscopic structural and sequence measurements of very small chromatin regions (down to the order of one kb) in 3D-conserved cell nuclei on the nano-scale using localization microscopy. With the design and application of specifically labelled oligonucleotides it is shown that COMBO-FISH (COMBinatorial Oligonucleotide FISH) allows counting of the number of CGG-repeats in an expansion region inside the 5′ untranslated region of the FMR1 gene. The number of repeats verified by Southern blotting indicated a full mutation of the Fragile-X syndrome case analysed here. With appropriate sets of oligonucleotides for labelling, we found in this case that the FMR1 promotor region was similarly condensed as the centromeric region whereas the arrangement of the probes labelling the expansion region seemed to indicate a loop-like structure.
Results and discussion
In order to get a more precise estimate about the length of the expansion region we developed a procedure for microscopy based repeat counting on the single cell level. We used combinatorial oligonucleotide fluorescence in situ hybridization (COMBO-FISH)9 in combination with super-resolution single molecule localization microscopy (SPDM).7,10COMBO-FISH uses a combination of computer based designed oligonucleotide probes with a typical stretch length of 15-30 nucleotides. By data bank based analysis for accessory binding sites within the whole genome, the design of the probes is optimized so that the specificity of the used probe combination is increased by co-localization within the target region.20 We extracted multiplets of 6 trinucleotide units [(CGG)6 or (CCG)6 probes] referred to as expansion-probes, showing the highest specificity to the (CGG)-repeat expansion region within the 5′ untranslated region of the FMR1 gene with a minimum of accessory binding sites within the whole genome. Three accessory binding sites with a maximum of six probes within a 250 kb-range were identified. Considering a probe length of six trinucleotide units with a linked fluorochrome at one end, simulations of the probes binding to the target region let us expect 45 ± 2 linearly bound probes in an expansion region with 477 (CGG)-repeats (∼1.4 kb) occurring in the cell line used here. In addition, a probe set of different oligonucleotides specifically co-localizing in the FMR1 promotor region, further referred to as promotor-set or promotor-probes, was designed. The design of this promotor-set is based on following conditions: a minimum probe length of fifteen nucleotides, a melting-point difference between promotor-probes and expansion-probes less than 10 K, less than three binding sites within the remaining genome and a distance between the binding sites of the probes of about 490 nucleotides. This search resulted in 50 suitable sequences. Out of this pool the 20 shortest probes were chosen as the promotor-set. Finally, a repetitive probe, unique and thus highly specific for centromere 9 was designed for control experiments.
After hybridization of the expansion- and promotor-sets, fluorescence signals (spots) within more than 80% of the confocal microscopy images of the examined cells were detected. By simultaneous co-localization of two expansion probe sets (same oligonucleotide stretches with different dyes) in a two colour experiment, we proved the specificity of the designed expansion-probes. The observed co-localization efficiency was over 70%. The same results were obtained in co-localization tests of the FMR1-promotor-set and the (CGG)-probes bound to the expansion region (Fig. 1).
 | ||
| Fig. 1 Nucleus with DNA staining (a), specific labelling for the (CGG)-repeat expansion region (b) and for the FMR1-promotor region (c) (arrows), scale bar 2 μm. (a) Confocal overview image of an examined cell, counterstained with TO-PRO(R)-3; (b) confocal image of Alexa 488 stained expansion-probes; (c) confocal image of Alexa 568 stained promotor-probes; (d) overlay of (a) with thresholded images (b) coloured in green and (c) coloured in red; (e) overlay of TO-PRO(R)-3 counterstained nuclei with thresholded confocal images of expansion-probes (green) and promotor-probes (red). | ||
For further examinations, such as the number of expansion-probes bound to the target area and their binding behaviour, dyes undergoing reversible photo-bleaching were used21 in order to acquire high resolution images by SPDM.6,7,22 Image time stacks were processed by specially developed MATLAB-based programs to detect molecular blinking events that represent the position of the individual dye molecules linked to the probes. As a result of this blinking detection a matrix is created containing the exact position and the localization precision of each blinking event. This matrix is used to create the localization images and for further cluster analysis.
One subscript of these programs additionally gives the opportunity to crop the localization images and the underlying matrix to a region of interest, which contains the COMBO-FISH signals in a wide field image. Thereby, the signal-to-noise-ratio is improved and the local background within the region of interest is reduced.
A cluster analysis that is applied to the data-matrix detects ‘cluster-points’ within the localization data according to user-defined parameters. These parameters are the minimum number of fluorescence signals within a defined circular region around each blinking event and the radius of this region. The mode of operation is schematically shown in Fig. 2. An event is identified as a ‘cluster-point’ if it has at least as many neighbours within the determined radius as preset by the user (see Fig. 2d and e). An event is not counted to be in a cluster if the preset number of neighbours within this area is not reached (see Fig. 2b and c). After this analysis has been done for all events within the data-matrix the final cluster in turn is set as the incessant union of ‘cluster-points’ (Fig. 2f and g). For visualisation of the clusters as seen in Fig. 4c and g the area of each single cluster gets coloured. By this process not only the localization events get coloured but also the corresponding regions around each event. In addition the regions around events identified not to be inside a cluster are excluded.
 | ||
| Fig. 2 Mode of operation of the cluster-analysis-script, schematic localization images. (a) Theoretical localization image; (b) calculation of the number of neighbouring points within the circular area with radius r for the first point; (c) if the number is under the designated parameter the point is set as ‘not in a cluster’ (blue point); (d) calculation of the number of neighbouring points within the circular area with radius r for the second point; (e) if the number of neighbours is higher than this parameter the point is identified as ‘cluster-point’ (red point); (f) image representing the results of the cluster-point identification for all points within the localization data; (g) contiguous areas of cluster-points are combined to clusters. | ||
Additionally, the script provides different information about these identified clusters such as the number of signals within the clusters, the cluster diameter and the distance distributions of signals inside and outside of clusters. Comparing and validating the position and size of clusters recorded by conventional fluorescence microscopy vs. SPDM, we achieved the parameters representing clusters optimally. This way the number of probes bound to the examined region is given by the number of signals within the specific clusters.
To verify this newly developed analysis procedure and optical sequencing strategy, we used human B-lymphocytes (Coriell GM06897) which were sampled from a male donor with an exhibition of 477 (CGG)-repeats according to the provider. Analysing these cells with the common method based on Southern blotting revealed an expansion length of 490 ± 100 (CGG)-repeats (Fig. 3). This indicates a certain inhomogeneity of the cells and further motivates our single cell approach.
 | ||
| Fig. 3 Southern blot of the examined cell lines. (a) Female pre-mutation control: normal and expanded fragment in the pre-mutation range (both fragments methylated and non-methylated); (b) female wild type control: normal fragment (methylated and non-methylated); (c) Coriell GM06897, EBV B-lymphoblastoid cell line of a male patient with Fragile-X syndrome: smear of bands (approx. 490 (+/−100) (CGG)-repeats) in the full mutation range (non-methylated); kb: kilobase; PM: pre-mutation; WT: wild type; FM: full mutation. | ||
The application of our new method based on COMBO-FISH and SPDM resulted in 65 ± 5 signals within the clusters that were identified as bound probes (Fig. 4). Assuming that a target region with 490 ± 100 (CGG)-repeats should lead to about 45 ± 10 linear bound probes extending our simulations of 477 repeats in a worst case. According to the above mentioned simulation, the maximum amount of probes bound linearly to a target area of 590 (CGG)-repeats, which is about the maximum length of the expansion region based on the Southern blot analysis, should therefore be about 60 occurring with a probability of 4% only.
 | ||
| Fig. 4 SPDM image analysis. (a) Confocal overview image of an examined cell after hybridization with an Alexa 568-linked expansion-probe; (b) localization image resulting from the analysis of the image stacks taken by a localization microscope; (c) cluster image resulting from the localization data (cluster parameters: minimum 10 neighbours within a circular region with radius 100 nm); (d) overlay of (a) and (b); (e) overlay of (a) and (c); (f) localization image resulting from the analysis of the image stacks taken by a localization microscope cropped to the ROI (green 2 μm × 2 μm square) in (a); (g) cluster image resulting from the cropped localization data (cluster parameters: minimum 10 neighbours within a circular region with radius 100 nm); [scale in (a)–(e): green square/scale bar upper left 2 μm; scale in (f) and (g): whole image 2 μm × 2 μm]. | ||
Measurements comparing the distances of the expansion-probes with those of the promotor-probes and centromere 9 probes (for probe design see ref. 10) indicated a significant difference in the signal and thus probe densities (Fig. 5). While the non-repetitive promotor- and centromere-probes typically showed a mean distance of 23.3 ± 3.7 nm (assuming linear 0.34 nm/nucleotide: 69 ± 11 nucleotides) and 26.1 ± 4.3 nm (77 ± 13 nucleotides), respectively, between neighbouring fluorophores, the repetitive expansion-probes showed a mean distance of only 12.3 ± 3.3 nm (36 ± 10 nucleotides). The only difference between these probes was the character (repetitive or non-repetitive units) of their sequences and the structure of their binding sites. The difference in the probe distance between theoretical (490 nucleotides) and measured values (69 ± 11 nucleotides) in the case of the promotor-probes hints to an inactivated, i.e., dense DNA superstructure. This explanation is proved by densely packed centromere-probes. One can assume that centromere regions and similarly densely packed chromatin regions are the highest compaction form of chromatin. Therefore, the apparently “more densely packed” expansion-probe signals may hint on the contrary to an extended, loop-like and therefore well accessible chromatin conformation that allows increasing the real number of bound probes so that the detected distances of the fluorochromes are apparently shortened. The difference between the measured number of probes bound to the target areas and the simulated average number indicates DNA superstructures since the simulation assumes a linear binding behaviour only. However, a possible non-linear probe binding behaviour could not be strictly excluded for repetitive probe units (Fig. 6). For stoichiometric reasons, these binding formations would also require appropriate free volumes around the target strand so that again a loop-like conformation seems to be preferred.
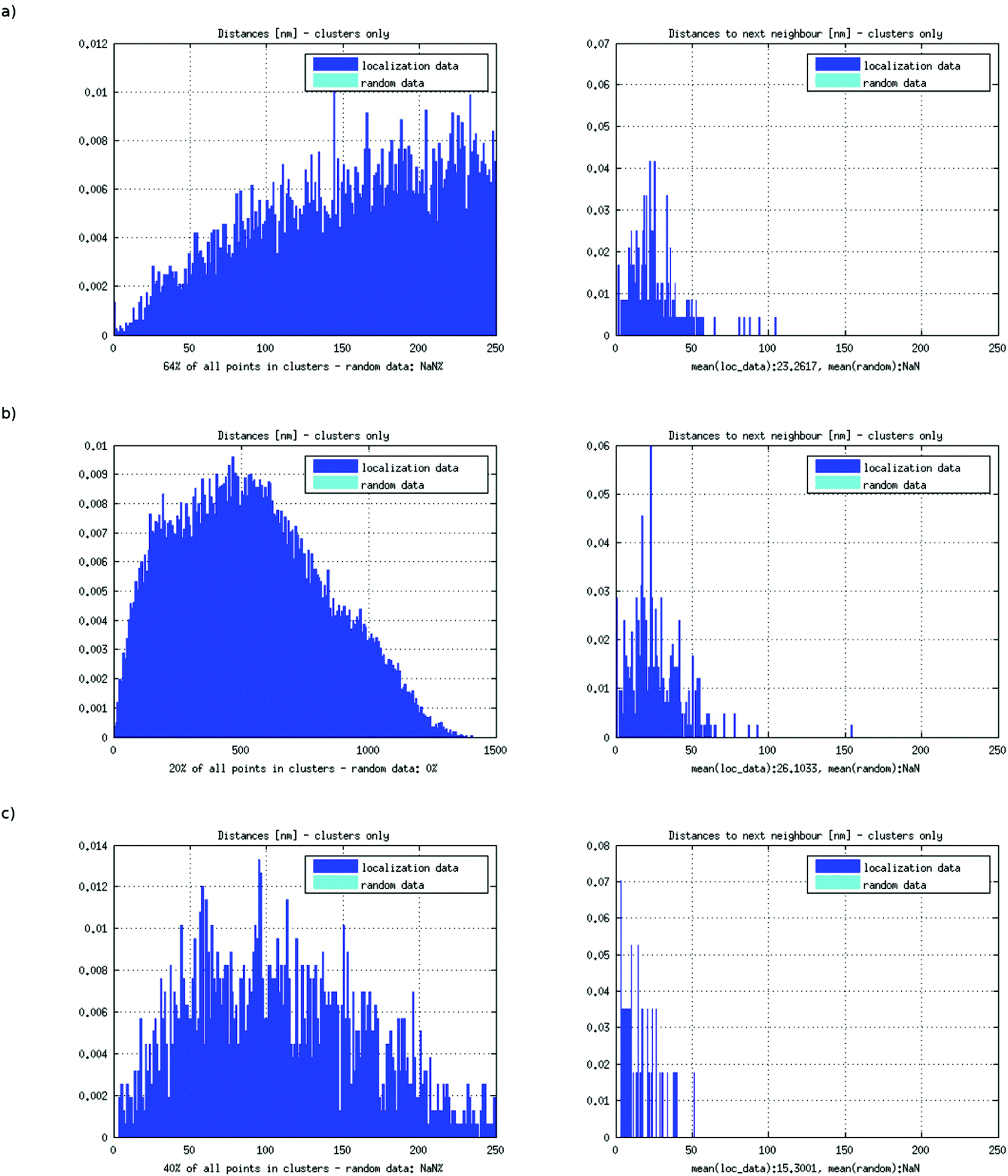 | ||
| Fig. 5 Results of the cluster analysis for the different probes. (a) Distance frequency distribution resulting from the cluster analysis for expansion probes; (b) distance frequency distribution resulting from the cluster analysis for promotor-probes; (c) distance frequency distribution resulting from the cluster analysis for centromere 9-probes. | ||
 | ||
| Fig. 6 Possible non-linear binding behaviours of the repetitive expansion-probes. Both mentioned binding behaviours result in shortening the distances between two neighbouring fluorophores and so result in an increased number of signals within the target area. | ||
The combination of COMBO-FISH9,10 and SPDM,7,23,24i.e. super-resolution microscopy, enables analyses of extremely small target areas of the genome and represents an important step towards the optical sequencing of trinucleotide region expansions or any other genome target region, for which appropriate probes are available. Besides aspects of probe binding one should, however, also consider resolution limits in practical SPDM. Physical constraints have an influence on localization accuracy and precision especially for dye molecule distances in the range of 10 nm.25 Thus nowadays the usually applied detection and recording systems of SPDM may run into registration problems under the labelling conditions applied here, so that small probe numbers densely arranged have to be carefully identified. Thus, our experimental requirements might be a technological challenge towards front-line localization microscopy. However, novel developments in optics and detector sensitivity as well as sophisticated informatics will overcome such shortcomings.
Experimental
Cell culture
GM06897 an Epstein–Barr-virus (EBV)-transformed B-lymphoblastoid cell line derived from a patient with Fragile-X syndrome was obtained from Coriell Institute for Medical Research, Camden, NJ, USA. Cells were grown in RPMI 1640 supplemented with L-glutamine, non-essential amino acids, antibiotics/antimycotics, gentamycin (Gibco, Life Technologies GmbH, Darmstadt, Germany), and fetal calf serum (Biochrom GmbH, Berlin, Germany) under a 5% carbon dioxide atmosphere. For the preparation of acetic acid/methanol suspensions cell cultures were treated with hypotonic solutions (75 mM KCl, pH 7.0 and 37.5 mM KCl, pH 7.0) and fixed with acetic acid/methanol (1/3). DNA was extracted by the salting out method according to Miller et al. (1988).26 Southern blot analysis was carried out using the restriction endonucleases EagI and HindIII and the 32P-labelled probe pP2 to re-evaluate the length of CGG repeats in the FMR1 gene as described by Meijer et al. (1994).27COMBO-FISH
While conventional fluorescence in situ hybridization (FISH) often uses BAC-probes with a length of about 104 nucleotides, combinatorial oligonucleotide fluorescence in situ hybridization (COMBO-FISH), first published by Hausmann et al. (2003),9 uses short probes with a length of 15–30 nucleotides. The design of these probes is based on a computer search for appropriate oligonucleotides for a highly specific labelling of the genome region of interest.10 Through the small size of the probes the specificity of the single probes drops down. This is compensated by a unique highly specific co-localization of many probes within the target region.Probe design
Within this work we had to find probes which are highly specific for the Fragile-X trinucleotide repeat expansion region, which only contains nucleotide triplets. This requires an optimized probe design of multiplets of these triplets so that the specificity rises by co-localization within the target region.By simulating the binding of probes with a different number of (CGG)-repeats on targets of different lengths and a simultaneous search for accessory binding sites of these probes within the human genome we ascertained that the optimal length for the COMBO-FISH probes is (CGG)6. At this length we expected 45 ± 10 probes to bind linearly within an assumed target region of the used cells. Additionally, the data bank search let us expect three accessory binding sites in which three or six probes can bind within a range of 250 kb.28 These results for the (CGG)-probes can be transferred directly to the complimentary (CCG)-probes.
According to these results three oligonucleotide probes with one fluorochrome molecule on the 5′ end were commercially synthesized. Two of these probe types are (CCG)6-probes, one labelled with Alexa 488 and one labelled with Alexa 568. The third kind are (CCG)6-probes labelled with Alexa 568.
Slide preparation
Cell nuclei suspensions of lymphocytes were dropped on slides. For permeabilisation of the nuclear membrane, the slides were incubated in 0.7% Triton-X 100 in 2× SSC at room temperature for 30 min. After rinsing twice in 2× SSC for 5 min, the specimens were incubated in 100 μl RNase A (10 mg ml−1 in water with 10 mM TrisHCl, pH 8.0) at 37 °C for 0.5 h. After washing three times in 2× SSC for 3 min each, the slides were equilibrated in 1× PBS for 5 min followed by treatment with 0.25 μl proteinase K (a stock solution consisting of 20 mg ml−1 in H2O) diluted in 100 mL 20 mmol l−1 Tris-b and 2 mmol l−1 CaCl2 in H2O at 37 °C for 10 min. After dehydration by an ethanol series (70%, 80%, 95%) for 3 min each of the slides were denatured at 72 °C for 5 min in 70% Formamide/2× SSC at pH 7. This was followed by dehydration with an ice cold ethanol series (70%, 80%, 95%) 3 min each. After air-drying 20 μl hybridization mixture consisting of 200 ng probe DNA (0.36 μl), 3 M NaCl, 0.6 M MgCl2, 1 M NaOAc was dropped on the specimen slides, which were covered with a coverslip and incubated in a humidified chamber at 37 °C for 24 h. The specimens were washed twice in 2× SSC, pH 7.0, at room temperature for 5 min. For counterstaining the slides were incubated in 50 μl DAPI (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20000 in 1× PBS) and 50 μl TO-PRO-3 (1:1000 in 1× PBS) for 5 min. Finally, the slides were mounted in 20 μl ProlongGold antifade solution and covered with a coverslip.
:20000 in 1× PBS) and 50 μl TO-PRO-3 (1:1000 in 1× PBS) for 5 min. Finally, the slides were mounted in 20 μl ProlongGold antifade solution and covered with a coverslip.
Localization microscopy (SPDM)
A single molecule localization microscope equipped with a Plan APO 100×/NA 1.4 oil immersion objective lens, DPSS lasers at 405 nm (Bfi Optilas) and 671 nm (Beijing Viasho Technology), a Sapphire HP 488 nm and a Sapphire HP 568 nm laser (both Coherent Inc.) was used. The precise setup of the used experimental microscope setup has been described in former articles.22The appropriate excitation wavelength was selected by Python-based software. The fluorescence was detected by using a Sensicam QE with 1376 × 1040 pixels and a pixel size of 6.45 μm (PCO). Typically, 2000 frames were acquired with an integration time of 50 ms each.
Digital image analysis
The image stacks acquired by the SPDM were processed by specially developed MATLAB-based programs. For further details on the localization algorithm, please refer to Kaufmann et al.23,29 and Lemmer et al.24Additionally, some newly developed programs were used to select specified regions within the widefield images of the data stacks and crop the raw data onto these regions. This leads to a much better noise-to-signal-ratio within the region of interest so that it became much more likely to detect the COMBO-FISH-signals within the data. Furthermore, an appropriate cluster detection algorithm was developed.
Conclusions
We were able to show that with the application of nano-probing using the COMBO-FISH labelling technique in combination with localization microscopy like SPDM, it is feasible to get an estimate about the state (number of sequence units) of the trinucleotide repeat expansion (“optical sequencing”) and the associated chromatin nano-structure changes behind it. For the first time we present microscopic structural and sequence measurements of very small chromatin regions (down to the order of one kb) in 3D-conserved cell nuclei on the nano-scale. With appropriate sets of oligonucleotides for labelling, we found in the case analysed that the FMR1 promotor region was similarly condensed as a centromeric region whereas the arrangement of the probes labelling the expansion region seemed to indicate a loop-like structure.These results demonstrate that in situ chromatin nano-structure measurements are feasible in intact cell nuclei. Changes in these nano-structures will provide new information about mechanisms behind genomic aberration inductions. In general the applied methodological approach will lead to new quantifiable insights into mechanisms correlated with the genome nano-architecture and functional consequences especially with aspects relating to the relevance of the development of heredity diseases.
Competing interests
The authors declare that they have no competing interests.Authors’ contributions
M.S. collected, processed and refined data, developed MATLAB-scripts and wrote the paper; J.S.-F. coordinated the project and was the scientific editor; E.F. did the cell culture and Southern blot; J.L. designed the expansion- and promotor-probes and did first measurements; M.B. did first data analysis; C.C. cooperated in localization microscopy; K.H. and M.H. designed the study and were coordinating and applicant investigators; G.H. was the scientific councillor and was involved in study design. All authors discussed the results and commented on the manuscript.Acknowledgements
We thank Nikon Imaging Center at the University of Heidelberg for access to their ERS-6 spinning disk confocal microscope. This work was supported by the Innovation Fond “Frontier” of the Heidelberg University within the excellence initiative of the Deutsche Forschungsgemeinschaft (DFG) granted to M. Hausmann and K. Hinderhofer.References
- H. T. Orr and H. Y. Zoghbi, Annu. Rev. Neurosc., 2007, 30.1, 575 CrossRef PubMed.
- M. Nicodemi and A. Pombo, Curr. Opin. Cell Biol., 2014, 28, 90 CrossRef CAS PubMed.
- T. Cremer, Cell News, 2013, 39, 6 Search PubMed.
- J. Pi, H. Jin, F. Yang, Z. W. Chen and J. Cai, Nanoscale, 2014, 6, 12229 RSC.
- D. Smeets, Y. Markaki, V. J. Schmid, F. Kraus, A. Tattermusch, A. Cerase, M. Sterr, S. Fiedler, J. Demmerle, J. Popken, H. Leonhardt, N. Brockdorff, T. Cremer, L. Schermelleh and M. Cremer, Epigenet. Chromatin, 2014, 7, 8 CrossRef PubMed.
- Y. Zhang, G. Máté, P. Müller, S. Hillebrandt, M. Krufczik, M. Bach, R. Kaufmann, M. Hausmann and D. W. Heermann, PLoS One, 2015, 10, e0128555 Search PubMed.
- C. Cremer, R. Kaufmann, M. Gunkel, S. Pres, Y. Weiland, P. Müller, T. Ruckelshausen, P. Lemmer, F. Geiger, M. Degenhard, C. Wege, N. Lemmermann, R. Holtappels, H. Strickfaden and M. Hausmann, Biotechnol. J., 2011, 6.9, 1037 CrossRef PubMed.
- C. Cremer and B. R. Masters, Eur. Phys. J. H, 2013, 38, 281 CrossRef PubMed.
- M. Hausmann, R. Winkler, G. Hildenbrand, J. Finsterle, A. Weisel, A. Rapp, E. Schmitt, S. Janz and C. Cremer, BioTechniques, 2003, 35.3, 564 Search PubMed.
- P. Müller, Y. Weiland, R. Kaufmann, M. Gunkel, S. Hillebrandt, C. Cremer and M. Hausmann, Int. J. Mol. Sci., 2010, 11, 4094 CrossRef PubMed.
- C. D'Hulst and R. F. Kooy, J. Med. Genet., 2009, 46.9, 577 CrossRef PubMed.
- H. A. Lubs, Am. J. Hum. Genet., 1969, 21, 231 CAS.
- D. Kumari and K. Usdin, J. Biol. Chem., 2009, 284, 7413 CrossRef CAS PubMed.
- M. Zemánek, J. Kypr and M. Vorlíčková, Int. J. Biol. Macromol., 2005, 36, 23 CrossRef PubMed.
- S. J. Martin, P. D. Grimwood and R. G. M. Morris, Annu. Rev. Neurosc., 2000, 23.1, 649 CrossRef PubMed.
- M. D. Wittenberger, R. J. Hagermann, S. L. Sherman, A. McConkie-Rosell, C. K. Welt, R. W. Rebar, E. C. Corrigan, J. L. Simpson and L. M. Nelson, Fertil. Steril., 2007, 87.3, 456 CrossRef PubMed.
- V. Biancalana, D. Glaeser, S. McQuaid and P. Steinbach, Eur. J. Hum. Genet., 2014, 23, 417 Search PubMed.
- Z. Lukáš, M. Falk, J. Feit, O. Souček, I. Falková, L. Štefančíková, E. Janoušová, L. Fajkusová, J. Zaorálková and R. Hrabálková, Neuromuscular Disord., 2012, 22.7, 604 CrossRef PubMed.
- G. Meola and R. T. Moxley, J. Neurol., 2004, 251.10, 1173 CrossRef PubMed.
- E. Schmitt, J. Wagner and M. Hausmann, J. Comput. Sci., 2012, 3.5, 328 CrossRef PubMed.
- J. N. Henderson, A. Hui-wang, R. E. Campbell and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6672 CrossRef CAS PubMed.
- S. Rossberger, G. Best, D. Baddeley, R. Heintzmann, U. Birk, S. Dithmar and C. Cremer, J. Opt., 2013, 15.9, 094003 CrossRef.
- R. Kaufmann, P. Lemmer, M. Gunkel, Y. Weiland, P. Müller, M. Hausmann, D. Baddeley, R. Amberger and C. Cremer, Proc. SPIE, 2009, 7185, 71850J1 CrossRef PubMed.
- P. Lemmer, M. Gunkel, Y. Weiland, P. Müller, D. Baddeley, R. Kaufmann, A. Urich, H. Eipel, R. Amberger, M. Hausmann and C. Cremer, J. Microsc., 2009, 235.2, 927 Search PubMed.
- S. Ram, E. S. Ward and R. J. Ober, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4457 CrossRef CAS PubMed.
- A. Miller, D. D. Dykes and H. F. Polesky, Nucleic Acids Res., 1988, 16.3, 1215 CrossRef PubMed.
- H. Meijer, E. de Graaff, D. M. Merckx, R. J. Jongbloed, C. E. de Die-Smulders, J. J. Engelen, J. P. Fryns, P. M. Curfs and B. Oostra, Hum. Mol. Genet., 1994, 3.4, 615 Search PubMed.
- J. Lux, Diploma thesis, University of Heidelberg, 2012.
- R. Kaufmann, P. Müller, G. Hildenbrand, M. Hausmann and C. Cremer, J. Microsc., 2011, 242.1, 46 CrossRef PubMed.
Footnote |
| † Present address: Institute of Environmental Physics, Heidelberg University, Im Neuenheimer Feld 229, 69120 Heidelberg, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |