
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Weighing the surface charge of an ionic liquid†
Nicklas
Hjalmarsson
a,
Daniel
Wallinder
b,
Sergei
Glavatskih
cd,
Rob
Atkin
e,
Teodor
Aastrup
b and
Mark W.
Rutland
*af
aSurface and Corrosion Science, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
bAttana AB, SE-11419, Stockholm, Sweden
cSystem and Component Design, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
dDepartment of Mechanical Construction and Production, Ghent University, B-9052 Zwijnaarde, Belgium
eDiscipline of Chemistry, The University of Newcastle, Callaghan, NSW 2308, Australia
fChemistry, Materials and Surfaces, SP Technical Research Institute Sweden, SE-11428 Stockholm, Sweden. E-mail: mark@kth.se
First published on 9th September 2015
Abstract
Electrochemical quartz crystal microbalance has been used to measure changes in the composition of the capacitive electrical double layer for 1-ethyl-3-methylimidazolium tris(pentafluoroethyl)-trifluorophosphate, an ionic liquid, in contact with a gold electrode surface as a function of potential. The mass difference between the cation and anion means that the technique can effectively “weigh” the surface charge accurately with high temporal resolution. This reveals quantitatively how changing the potential alters the ratio of cations and anions associated with the electrode surface, and thus the charge per unit area, as well as the kinetics associated with these interfacial processes. The measurements reveal that it is diffusion of co-ions into the interfacial region rather than expulsion of counterions that controls the relaxation. The measured potential dependent double layer capacitance experimentally validates recent theoretical predictions for counterion overscreening (low potentials) and crowding (high potentials) at electrode surfaces. This new capacity to quantitatively measure ion composition is critical for ionic liquid applications ranging from batteries, capacitors and electrodeposition through to boundary layer structure in tribology, and more broadly provides new insight into interfacial processes in concentrated electrolyte solutions.
Introduction
The behaviour of Ionic Liquids (ILs) at interfaces is of decisive importance for their application in fields ranging from electrochemistry to tribology.1,2 Their charged nature provides an additional control parameter compared with conventional liquids,2 of controlling interfacial structure through application of surface potentials.3–7 Despite this importance and recent theoretical advances,1,8,9 the nature of the so called electrical double layer (EDL) remains experimentally intransigent.1,2,10 ILs have several attractive properties that have sparked a recent surge in interest; amongst other things, they can be thermally and electrochemically stable, have a low vapour pressure and can dissolve both polar and apolar moieties. These properties render ILs relevant for electrochemical applications such as electrodiscs,11 electrowetting,12 and molecular gating.13 For electrochemistry the nature of the EDL, (the arrangement of ions at a charged interface) is of particular importance; however, the traditional description of the EDL structure for aqueous electrolyte solutions is not applicable for ILs1 where electrostatic interactions are only manifested over the distance of an ion-pair.14,15 Furthermore, the cations and anions comprising an IL are usually bulky and asymmetric, with a delocalised charge, which is not commensurate with assumptions of point charges: there has thus been a need to find new descriptions for the EDL structure of ILs.1,8–10,16–19Bazant et al. developed a simple Landau–Ginzburg-type continuum theory to predict the structure of ILs on charged interfaces.8 The calculations showed that at low to moderate voltages, i.e. when the surface charge is relatively low compared to the charge of a monolayer of counterions, that the surface is “overscreened” by a layer of counterions which is compensated for by an excess of co-ions in the next layer (illustrated in Fig. 1a). This behaviour changes at high voltages since the charge of a monolayer of counterions is insufficient to screen the surface charge. The counterions will thus also be in excess in the second monolayer, “crowding”, and the co-ion dominated layer will not be manifested until the third layer (Fig. 1b). Experimental studies of the EDL for ILs have used a plethora of techniques such as X-ray reflectivity (XRR),4,5,20–23 Neutron Reflectivity,24 Atomic Force Microscopy (AFM),15,18,25–29 Surface Force Apparatus,14,30–33 Neutral Impact Collision Ion Scattering Spectroscopy,34–41 Angle Resolved X-ray Photoelectron Spectroscopy,37,42–44 Rutherford Backscattering,45 and Vibrational Sum Frequency Spectroscopy34,46–49 however few have investigated the charged solid–liquid interface, particularly as a function of surface charge. In a landmark paper, Mezger et al. reported on the distribution of anion and cations for three different ILs (where the anion remained the same) on a charged sapphire surface using high energy XRR. The results showed extensive molecular layering of the different ILs on the surface but in that system it was not possible to vary the voltage on the surface and thus not study the EDL as a function of surface charge.5 They followed up this paper by reporting on the behaviour of several other ILs (where the cation remained the same) with similar results.4 Yamamoto et al. took their XRR-study one step further and were able to independently apply a voltage (up to ±3 V) to a gold surface. They clearly demonstrated the effects of overscreening at low voltages but were unable to induce a crowding effect even at ±3 V. Fedorov and Kornyshev point out in their recent review1 that it is most likely an effect of the neutral tails which, being sufficiently long, can shift the onset of lattice saturation to larger voltages. This has been shown in several papers where the effect of the neutral tail has been simulated.16,50,51 Another possible reason is that the applied voltages do not exert a large enough electric field to completely turn on crowding. In another work, using AFM, Hayes et al. studied the layering of two different ILs by applying a potential to a gold surface.15 The force measurements revealed 4 to 7 layers which demonstrates the EDL structure, with the distance of the observed “steps” being correlated to the ion-pair diameter. They found that structural forces were stronger for negative polarisations and ascribed this to the different molecular structure of cation and anion. Cations with the more localised charge groups enhance layering, which is a simple demonstration of the tunability of ILs. It is worth noting that in the experimental work done on the EDL structure it is the voltage which has been controlled, and independent verification of the surface charge is often challenging.
 | ||
| Fig. 1 A schematic representation of the difference between overscreening (a) and crowding (b) for ions at a charged interface at low (a) and high (b) surface charge, respectively. The figure is redrawn from Bazant et al.8 | ||
The structure of the EDL for ILs has important ramifications for the sliding of ionic layers, and thus the friction. Tribotronics,52 the use of applied potentials to influence the friction of a lubricating contact, has interesting implications for the development of novel systems where the amount of friction, and even its mechanism, can be controlled in situ. Recent work, using AFM3 and Molecular Dynamics simulations,6 have shown that it is possible to control the friction of a system in situ using ILs and also achieve very low friction by applying an electrical potential. Li et al. recently applied potentials to different imidazolium ILs on gold and measured the friction using a colloidal probe AFM.53 They showed that the magnitude of the friction depended on which ions were sliding against each other and lowest friction was achieved with a high negative potential – cations sliding against each other (as long as the cation was smaller than the anion). In a similar study, Li et al. used a comparable system and achieved superlubricity (arbitrarily defined as a friction coefficient below 0.01) with an IL confined between a highly ordered pyrolytic graphite interface and an AFM tip.54 In the latter case, the lowest friction was achieved with a high positive potential (suggesting anions at the sliding interface). Clearly the surfaces must affect the local structuring of the ILs, but there is thus a need for an increased understanding of the interfacial behaviour of ILs and how this relates to friction.
In this study, the magnitude and sign of the surface potential applied to a gold surface immersed in 1-ethyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate [EMIm][FAP] was varied to investigate surface response using an electrochemical Quartz Crystal Microbalance (QCM). The mass difference between the ions allows the change in charge in the EDL to be directly measured, while a measure of the surface charge can be independently obtained from monitoring the capacitance of the system.
Experimental details
1-Ethyl-3-methylimidazolium tris(pentafluoroethyl)trifluorophosphate [EMIm][FAP] was acquired from Merck KGaA in ultrapure quality and used as received. An A200 QCM-instrument from Attana AB (Stockholm, Sweden) was used with a custom-built experimental setup for electrochemical measurements that was powered by a 9 V battery and included several resistors and operational amplifiers to minimise the current passing through (as this could potentially disrupt the QCM-crystal and/or facilitate electrochemical reactions). The current was measured over that last resistor and it was determined to be smaller than 0.2 μA (detection limit of the multimeter used). The chip (a self-contained insertable liquid cell made of PTFE with a QCM-crystal inside) was modified so that an electrode could be fitted inside. This quasi-reference electrode used in this paper was a 0.25 mm thick Pt-wire placed in the middle of the cell, 25–50 μm above the 10 MHz gold coated quartz crystal (working electrode) which was prepared by sputtering and has an area of 15.9 mm2 and thickness of 150 nm. The cell volume is approximately 1.5 μl and is completely sealed by a PTFE O-ring and screws to prevent both leakage and contamination of the liquid. The modest dimensions of the cell preclude a third electrode largely due to the substantially increased risk of leakage. A two-electrode system can be used in an electrochemical cell whenever the current is small,55 which is the case here. The chip (with accessories) was cleaned according to manufacturer's standard protocol (ESI†) and the gold surface was rinsed with ethanol before being assembled. The system was put in a desiccator for at least 12 h to allow for minimal water ingression before introducing [EMIm][FAP]. Karl–Fischer Titration (Metrohm) was used to monitor the water content and the measured values were always lower than 0.03 wt% H2O (which translates to less than one water molecule per 100 ion-pairs). The system was left to stabilise overnight in the QCM for every experiment to reduce drift (which was never higher than 0.2 Hz min−1 and usually considerably lower). Drift was corrected for by extrapolating a linear fit.56 The setup was always left to stabilise for at least 20 min before changing the potential to allow the open-circuit potential (OCP) as well as the surfaces changes in the QCM to stabilise. The OCP between the gold surface and the Pt quasi-reference electrode was recorded each time the potential was switched off and converged to a value of −0.018 V ± 0.01 V. The Sauerbrey equation57 was used to convert measured frequency changes to mass changes in ng cm−2 with an instrument value of C = 4.4 Hz cm2 ng−1.Results and discussion
Mass change as a function of potential
Fig. 2 shows a QCM-experiment where a negative potential of varying magnitude was applied to a gold surface immersed in [EMIm][FAP]. In this figure the potential is ramped from 0 V to −2 V and the mass difference per area on the gold surface is plotted against applied potential. The mass associated with QCM crystal (extracted from vibration frequency) clearly changes as a function of the magnitude of the potentials applied. Notably, positive potentials always increased the sensed mass whereas negative potentials resulted in a reduction. Assuming that the mass changes reflect primarily a change in the composition of the ion layers in close proximity to the surface, then this result can be rationalised in terms of surface charge induced ion exchange. This figure proves the hypothesis that QCM can be used to monitor the surface charge via changes in mass. In this IL the anion is significantly heavier; the molecular weights (Mw) of the cation and anion are 111 and 445 g mol−1, respectively. Thus the application of a negative potential leads to a reduction in the number of anions and an increase in the number of cations in the surface region, with a net mass loss of 334 g mol−1 of charge. Conversely, a positive potential enriches anions at the expense of cations, and the mass increases. The initial rapid change in mass we interpret as being associated with local “flipping” as co-ions and counterions change places in the closest layers. During the second regime the mass changes more slowly towards its plateau value over the order of a minute which reflects migration of ions to and from the surface layers over larger distances (inset Fig. 2). Before changing the potential to its next value, or changing the polarity, the voltage was disconnected and a recovery period was allowed during which the system relaxed. | ||
| Fig. 2 Dependence of mass change on applied potential between 0 V and −2 V. The inset shows a representative graph of mass difference as a function of time. | ||
The mass was also monitored during this relaxation and some of the results are shown in Fig. 3. The relaxation time is much longer than the time taken to reach the plateau (Fig. 2). The mass difference (with respect to its value at time zero – the point at which the voltage was switched off) is plotted against t1/2. The resulting linear relationship demonstrates a diffusion dependent change in the Sauerbrey mass and this is in agreement with Fick's law of diffusion at short time scales.58 Remarkably, the gradient is steeper for the extinguishing of the positive potential (a factor 2.0 ± 0.2) when comparing potentials of the same magnitude but different polarity. It is known59 that the self-diffusion coefficients of [FAP]− and [EMIm]+ are 1 × 10−11 and 2 × 10−11 m2 s−1, respectively, reflecting their different sizes. Thus the ratio of factor 2.0 between the gradients of the relaxation data for different polarities is entirely consistent with the literature ratio of the diffusion coefficients. More importantly, it indicates that the relaxation of the charge is associated primarily with movement of only one of the ions in each case. The relative diffusion rates indicate that the equilibration of the concentration gradient is limited not by diffusion of counterions away from the surface, but by diffusion of the co-ions towards the electrode surface. The demands of local electroneutrality lead to an equal number of co-ions (to the surface charge) being essentially repelled to “infinite distance”. For electroneutrality to be restored, the co-ions are required to diffuse over larger distances. This is further elaborated in the discussion of Fig. 4. The continuous data from a full experiment, where the voltage is varied multiple times at both polarities and over a wider range of values than in Fig. 2, is shown as mass change versus time in the ESI.†
 | ||
| Fig. 3 The two lines represent the diffusion controlled mass (or charge) relaxation for +1 V (thick line) and −1 V (thin line) when the potential is switched off. The ratio of the gradients reflects the ratio of the diffusion coefficients of the anion and cation. (Fig. 2 of the ESI† shows this data on a linear scale.) | ||
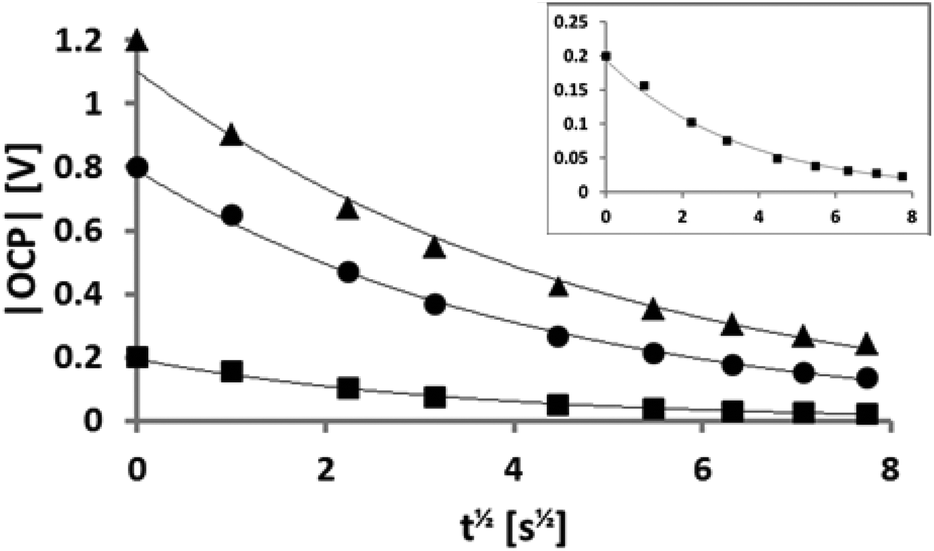 | ||
| Fig. 4 OCP recorded as a function of t1/2. The symbols correspond to −0.2 V (squares), −0.8 V (spheres), and −1.2 V (triangles). The inset is a zoomed version for −0.2 V. All fits are exponential as a function of t1/2 with excellent agreement (R2 > 0.98). | ||
Capacitance effect as a function of potential
To investigate the capacitive effect of the system and to ensure that the system relaxed to a consistent open-circuit potential (OCP), the OCP was also recorded when the potential was switched off, using a separate voltmeter. In Fig. 4 the OCP is plotted as a function of t1/2 for three different voltages of the same polarity: −0.2 V (squares), −0.8 V (spheres), and −1.2 V (triangles). There is an excellent agreement with the exponential fit indicated by the line in each case. The y-intercept at t = 0 corresponds to the respective, independently measured, applied potential and these results can thus be applied to a self-discharge equation: | (1) |
| C = C0·t1/2 | (2) |
 | (3) |
Mass charge as a function of capacitance charge
Data of the kind shown in Fig. 3 and 4 provide independent measures of the surface charge (in the latter case convoluted with the resistance of the system which is assumed to be constant for each experiment, reflecting the distance between the electrodes). Comparison of such values is made in Fig. 5. Here we have chosen to distinguish the measure of the surface charge extracted from eqn (3) as the “capacitance charge” to indicate its experimental origin., In this experiment the negative potential was systematically ramped up to investigate whether evidence could be obtained for transitions from overscreening to crowding in the double layer, as recently predicted.8 | ||
| Fig. 5 Calculated mass charge as a function of the capacitance charge. The data points for the two different regions were fitted separately to straight lines. The capacitance charge was normalised so that the slope of the fitted curve to the squared data points equaled 1. | ||
The mass change in Fig. 5 reflects the enrichment of cations at the electrode surface. The experiment was conducted in such a way that the potential was always switched off between each measurement to enable to record both the mass change and the corresponding exponential decay of the OCP. In Fig. 5 the “mass charge” (measured from the QCM) is plotted against the capacitance charge derived earlier by using the exponential constant and the value of the applied potential (the capacitance charge is convoluted by a constant which is characteristic of each experiment, so is plotted in arbitrary units such that the gradient is constrained to 1 for low charges in the figure). The mass charge is obtained by assuming that the mass change in the system is due to an anion–cation-exchange which reflects the surface charge. The QCM measures a change in resonance frequency (which provides a measure of the adsorbed mass change using Sauerbrey's equation) rather than the total mass. We here assumed that as a first approximation the mass change is due to an exchange of co-ions for counterions. It is known that the gold surface can reconstruct in the present of ILs which conceivably could lead to a mass change but for the IL used here it has been demonstrated that no such reconstruction occurs.60 If there were to be orientational changes of layers or significant changes in viscosity or density in the fluid adjacent to the crystal, this would also change the resonance frequency. We consider that the assumption is entirely reasonable for the following reasons. (1) The sign of the mass loss follows the sign of the applied potential. (2) As Fig. 5 shows there is a linear relationship between the mass change and the independently measured charge via the capacitance. (3) The mass (charge) relaxation is diffusion limited which is unlikely to be the case if the frequency change is due to changes in orientation or density/viscosity.
To relate the mass to that corresponding to a monolayer of cations it is necessary to have an estimate of the cation size. Perkin et al. estimated the [EMIm]+ cation size using two independent methods, molecular mechanics (CHARMM code) and a semi-empirical package (MOPAC, PM3 method, restricted) and both methods agreed to the dimensions: (0.76 ± 0.03 nm) × (0.36 ± 0.08 nm) × (0.22 ± 0.04 nm).61 Furthermore, together with several other publications,46,48,49 they propose that an imidazolium cation adsorbs with the aromatic ring parallel to the surface. Baldelli points out however that this can change if the water content is high.49 Since our measured water content is low, and assuming that the ring is parallel to the surface, then the charge density equivalent to one monolayer of cations is equal to 4.8 μC cm−2 for [EMIm]+. In Fig. 5 there are two distinct linear regions, which display a significant difference in slope. We speculate that these two regions reflect the conditions of overscreening and crowding.8 In the case of overscreening (lower charge density, squares, depicted in Fig. 1a) the double layer can be thought of as a charge sandwich where a layer of counterions is flanked on either side by a layer of surface charge and a layer of co-ions. Thus, as the surface charge approaches the limiting density which can be accommodated via an overscreening configuration, the surface charge should be approximately half the value of the charge of the full monolayer. According to the calculation above, the transition from overscreening to crowding should thus occur at about 2.4 μC cm−2.
The transition point appears at approximately 2 μC cm−2 mass charge which is highly comparable with the calculated value of 2.4 μC cm−2. This difference in slope is the key evidence that we are sensitive to the transition from overscreening to crowding. If the QCM detected only the change in mass of the ion exchange in the sensed volume then this curve would remain linear and no transition would be observed. The change in slope reflects changes in the ordering of the IL in several different ways. Firstly the mass sensitivity decreases with distance from the surface in an exponential fashion.62,63 Secondly, the reorganisation of the IL in response to the surface charge by definition must cause a local change in both density and viscosity. Both of these parameters should affect the QCM response,64 though not in a transparent fashion. Simulations, albeit of equally sized spheres, clearly show that such density changes are expected to occur in association with the transition. Specifically, the average density of the first few molecular layers should increase significantly (of the order of 10 to 20%, see Fig. 3 in ESI,† data from ref. 65). The increased gradient observed in Fig. 5 is strongly indicative of such an effect. A full, quantitative explanation of the difference in slopes of the two regions would require knowledge of the molecular orientations, the local viscosity and density changes associated with the charge rearrangements and the implications of the size mismatch between the ions in the third layer, which are well beyond the scope of this work. The observed trend however is in full, and almost quantitative, agreement with the proposed model of Bazant et al.8 that predicts a transition from overscreening to crowding.
Conclusion
A new method of investigating the surface structure of ILs using electrical QCM has been developed. The results corroborate previous simulation studies on ILs at charged interfaces. The significant differences in mass between the anion and cation can be leveraged to “weigh” the surface charge since it causes an imbalance in the relative numbers of anions and cations in the surface region sensed by the QCM. For ions of equal mass the technique is unlikely to be very sensitive, but based on the resolution found here, a significantly smaller mass difference could be viable. The effective experimental limits will be explored in future work by systematically varying the relative sizes of the ions. The observation that measurements of both mass and OCP relaxation were dependent on t1/2 allowed two conclusions to be established – firstly that the relaxation is limited by diffusion of counterions back to the surface region, and secondly that independent measures of the surface charge could be established. The ratio of the diffusion coefficients of the anion and cation determined here are in quantitative agreement with literature values, indicating that the QCM can also be used to obtain measures of diffusion parameters. Comparison of the two measures of the charge allows the issues of relating charge to potential to be circumvented and reveals a clear transition at values predicted to be associated with a transition from overscreening to crowding. In addition to providing valuable information on surface structure, which is key for understanding the tribotronics of ILs the results provide strong experimental support for current theoretical predictions.Acknowledgements
The Swedish Research Council and the Knut and Alice Wallenberg Foundation are gratefully acknowledged for their financial support. R.A. thanks the Australian Research Council Future Fellowship (FT120100313) and Discovery Project (DP120102708).References
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- J. Sweeney, F. Hausen, R. Hayes, G. B. Webber, F. Endres, M. W. Rutland, R. Bennewitz and R. Atkin, Phys. Rev. Lett., 2012, 109, 155502 CrossRef.
- M. Mezger, S. Schramm, H. Schroder, H. Reichert, M. Deutsch, E. J. De Souza, J. S. Okasinski, B. M. Ocko, V. Honkimaki and H. Dosch, J. Chem. Phys., 2009, 131 Search PubMed.
- M. Mezger, H. Schroder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schoder, V. Honkimaki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- O. Y. Fajardo, F. Bresme, A. A. Kornyshev and M. Urbakh, Sci. Rep., 2015, 5 Search PubMed.
- R. Capozza, A. Vanossi, A. Benassi and E. Tosatti, J. Chem. Phys., 2015, 142, 064707 CrossRef CAS PubMed.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106 Search PubMed.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
- A. A. Kornyshev and R. Qiao, J. Phys. Chem. C, 2014, 118, 18285–18290 CAS.
- A. A. Kornyshev, M. A. Vorotyntsev and E. Spohr, Electrochemical interfaces: at the border line, in Encyclopedia of electrochemistry, volume 1, thermodynamics and electrified interfaces, Wiley-VCH, 2002, pp. 33–132 Search PubMed.
- C. W. Monroe, L. I. Daikhin, M. Urbakh and A. A. Kornyshev, Phys. Rev. Lett., 2006, 97 Search PubMed.
- T. Albrecht, K. Moth-Poulsen, J. B. Christensen, J. Hjelm, T. Bjørnholm and J. Ulstrup, J. Am. Chem. Soc., 2006, 128, 6574–6575 CrossRef CAS PubMed.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CAS.
- M. V. Fedorov, N. Georgi and A. A. Kornyshev, Electrochem. Commun., 2010, 12, 296–299 CrossRef CAS PubMed.
- V. Lockett, M. Horne, R. Sedev, T. Rodopoulos and J. Ralston, Phys. Chem. Chem. Phys., 2010, 12, 12499–12512 RSC.
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954–5960 CrossRef CAS PubMed.
- C. Merlet, D. T. Limmer, M. Salanne, R. van Roij, P. A. Madden, D. Chandler and B. Rotenberg, J. Phys. Chem. C, 2014, 118, 18291–18298 CAS.
- R. Yamamoto, H. Morisaki, O. Sakata, H. Shimotani, H. Yuan, Y. Iwasa, T. Kimura and Y. Wakabayashi, Appl. Phys. Lett., 2012, 101, 053122 CrossRef PubMed.
- M. Mezger, B. M. Ocko, H. Reichert and M. Deutsch, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3733–3737 CrossRef CAS PubMed.
- A. Uysal, H. Zhou, G. Feng, S. S. Lee, S. Li, P. Fenter, P. T. Cummings, P. F. Fulvio, S. Dai, J. K. McDonough and Y. Gogotsi, J. Phys. Chem. C, 2014, 118, 569–574 CAS.
- H. Zhou, M. Rouha, G. Feng, S. S. Lee, H. Docherty, P. Fenter, P. T. Cummings, P. F. Fulvio, S. Dai, J. McDonough, V. Presser and Y. Gogotsi, ACS Nano, 2012, 6, 9818–9827 CrossRef CAS PubMed.
- Y. Lauw, M. D. Horne, T. Rodopoulos, V. Lockett, B. Akgun, W. A. Hamilton and A. R. J. Nelson, Langmuir, 2012, 28, 7374–7381 CrossRef CAS PubMed.
- J. M. Black, M. Baris Okatan, G. Feng, P. T. Cummings, S. V. Kalinin and N. Balke, Nano Energy, 2015, 15, 737–745 CrossRef CAS PubMed.
- R. Atkin, N. Borisenko, M. Druschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849–6857 RSC.
- R. Atkin, N. Borisenko, M. Drüschler, F. Endres, R. Hayes, B. Huber and B. Roling, J. Mol. Liq., 2014, 192, 44–54 CrossRef CAS PubMed.
- H. Li, R. J. Wood, F. Endres and R. Atkin, J. Phys.: Condens. Matter, 2014, 26, 284115 CrossRef PubMed.
- H.-W. Cheng, P. Stock, B. Moeremans, T. Baimpos, X. Banquy, F. U. Renner and M. Valtiner, Adv. Mater. Interfaces, 2015, 2, 1500159 Search PubMed.
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. C, 2014, 118, 6491–6503 CAS.
- M. A. Gebbie, M. Valtiner, X. Banquy, E. T. Fox, W. A. Henderson and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9674–9679 CrossRef CAS PubMed.
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572–6574 RSC.
- R. G. Horn, D. F. Evans and B. W. Ninham, J. Phys. Chem., 1988, 92, 3531–3537 CrossRef CAS.
- D. Wakeham, P. Niga, C. Ridings, G. Andersson, A. Nelson, G. G. Warr, S. Baldelli, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2012, 14, 5106–5114 RSC.
- C. Ridings, V. Lockett and G. Andersson, Phys. Chem. Chem. Phys., 2011, 13, 17177–17184 RSC.
- C. Ridings, V. Lockett and G. Andersson, Phys. Chem. Chem. Phys., 2011, 13, 21301–21307 RSC.
- T. Hammer, M. Reichelt and H. Morgner, Phys. Chem. Chem. Phys., 2010, 12, 11070–11080 RSC.
- C. Ridings, V. Lockett and G. Andersson, Colloids Surf., A, 2012, 413, 149–153 CrossRef CAS PubMed.
- M. Reichelt, T. Hammer and H. Morgner, Surf. Sci., 2011, 605, 1402–1411 CrossRef CAS PubMed.
- I. J. Villar-Garcia, S. Fearn, N. L. Ismail, A. J. S. McIntosh and K. R. J. Lovelock, Chem. Commun., 2015, 51, 5367–5370 RSC.
- I. J. Villar-Garcia, S. Fearn, G. F. De Gregorio, N. L. Ismail, F. J. V. Gschwend, A. J. S. McIntosh and K. R. J. Lovelock, Chem. Sci., 2014, 5, 4404–4418 RSC.
- V. Lockett, R. Sedev, S. Harmer, J. Ralston, M. Horne and T. Rodopoulos, Phys. Chem. Chem. Phys., 2010, 12, 13816–13827 RSC.
- C. Kolbeck, T. Cremer, K. R. J. Lovelock, N. Paape, P. S. Schulz, P. Wasserscheid, F. Maier and H. P. Steinrück, J. Phys. Chem. B, 2009, 113, 8682–8688 CrossRef CAS PubMed.
- T. Cremer, M. Stark, A. Deyko, H. P. Steinrück and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS PubMed.
- K. Nakajima, S. Oshima, M. Suzuki and K. Kimura, Surf. Sci., 2012, 606, 1693–1699 CrossRef CAS PubMed.
- C. Romero, H. J. Moore, T. R. Lee and S. Baldelli, J. Phys. Chem. C, 2007, 111, 240–247 CAS.
- C. S. Santos and S. Baldelli, J. Phys. Chem. B, 2007, 111, 4715–4723 CrossRef CAS PubMed.
- C. Romero and S. Baldelli, J. Phys. Chem. B, 2006, 110, 6213–6223 CrossRef CAS PubMed.
- S. Baldelli, J. Phys. Chem. B, 2003, 107, 6148–6152 CrossRef.
- N. Georgi, A. A. Kornyshev and M. V. Fedorov, J. Electroanal. Chem., 2010, 649, 261–267 CrossRef CAS PubMed.
- J. Vatamanu, O. Borodin and G. D. Smith, J. Am. Chem. Soc., 2010, 132, 14825–14833 CrossRef CAS PubMed.
- S. Glavatskih and E. Hoglund, Tribol. Int., 2008, 41, 934–939 CrossRef PubMed.
- H. Li, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14616–14623 RSC.
- H. Li, R. J. Wood, M. W. Rutland and R. Atkin, Chem. Commun., 2014, 50, 4368–4370 RSC.
- D. S. Silvester, E. I. Rogers, R. G. Compton, K. J. McKenzie, K. S. Ryder, F. Endres, D. Macfarlane and A. P. Abbott, in Electrodeposition from Ionic Liquids, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, ch. 11, pp. 287–351, DOI:10.1002/9783527622917.
- N. Nordgren and M. W. Rutland, Nano Lett., 2009, 9, 2984–2990 CrossRef CAS PubMed.
- G. Sauerbrey, Z. Physik, 1959, 155, 206–222 CrossRef CAS.
- J. Crank, The Mathematics of Diffusion, Clarendon Press, Oxford, Eng, 2nd edn, 1975 Search PubMed.
- S. Seki, N. Serizawa, K. Hayamizu, S. Tsuzuki, Y. Umebayashi, K. Takei and H. Miyashiro, J. Electrochem. Soc., 2012, 159, A967–A971 CrossRef CAS PubMed.
- N. Borisenko, R. Atkin and F. Endres, Electrochem. Soc Interface, 2014, 23(1), 59–63 CAS.
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243–1247 RSC.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS PubMed.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- W. P. Mason, J. Colloid Sci., 1948, 3, 147–162 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Electrochim. Acta, 2008, 53, 6835–6840 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr03965g |
| This journal is © The Royal Society of Chemistry 2015 |