
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Birmingham parallel genetic algorithm and its application to the direct DFT global optimisation of IrN (N = 10–20) clusters†
Jack B. A.
Davis
*a,
Armin
Shayeghi
b,
Sarah L.
Horswell
a and
Roy L.
Johnston
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: jbad90@gmail.com; r.l.johnston@bham.ac.uk; Tel: +44 (0)1214 147477
bEduard-Zintl-Institut, Technische Universität Darmstadt, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany. E-mail: shayeghi@cluster.pc.chemie.tu-darmstadt.de
First published on 22nd July 2015
Abstract
A new open-source parallel genetic algorithm, the Birmingham parallel genetic algorithm, is introduced for the direct density functional theory global optimisation of metallic nanoparticles. The program utilises a pool genetic algorithm methodology for the efficient use of massively parallel computational resources. The scaling capability of the Birmingham parallel genetic algorithm is demonstrated through its application to the global optimisation of iridium clusters with 10 to 20 atoms, a catalytically important system with interesting size-specific effects. This is the first study of its type on Iridium clusters of this size and the parallel algorithm is shown to be capable of scaling beyond previous size restrictions and accurately characterising the structures of these larger system sizes. By globally optimising the system directly at the density functional level of theory, the code captures the cubic structures commonly found in sub-nanometre sized Ir clusters.
1. Introduction
Nanosized materials are currently being investigated for potential use in a variety of applications. This is because the nanosizing effects seen in such materials result in properties different from those of the bulk material. These properties can also be tuned, normally through altering the size and shape of the cluster.Metallic nanoparticles are such materials, with potential optical, magnetic and catalytic applications.1 Small Ir nanoparticles, in particular, are currently used as catalysts for a range of organic reactions, including olefin hydrogenation, oligomerisation, and ring-opening of cycloalkanes.2 Ir has been shown both experimentally3 and theoretically4 to exhibit significant nanosize-induced hydrogen adsorption capacity. Larger Ir nanoparticles have been shown to be active in C–C bond hydrogenolysis.5 Selective molecular recognition has also been seen in supported Ir cluster-based catalysts.6
A key step in rationalising properties, such as the catalytic activity of nanoparticles, is their structural characterisation. To achieve this it is necessary to sample comprehensively the potential energy landscape (PES) of the nanoparticle. A wide variety of methods is available for the exploration of the PES. These methods include statistical mechanical methods, such as the CBEV/FCEM method,7,8 basin-hopping methods,9 such as GMIN,10 and genetic algorithms, such as the Birmingham cluster genetic algorithm (BCGA).11 The choice of method largely depends on the size and complexity of the system.
It is necessary to decide the level of theory required to replicate accurately a particular PES of a system. For example, the electronic structure of larger nanoparticles is thought to resemble closely that of the bulk material. This means the use of empirical potentials, such as the Gupta potential,12 is suitable for the accurate representation of the PES. Statistical mechanical methods, such as CBEV/FCEM, may be best suited to these larger systems.8 However, for smaller, sub-nanometre clusters a much more computationally demanding quantum mechanical description of the cluster PES is necessary as quantum-size effects, such as spin–orbit coupling, tend to dominate.13–15 A variety of methods has been developed to achieve this, many of which have been outlined by Heiles and Johnston.16
The cubic structures adopted by sub-nanometre Ir clusters have been previously shown up to the CCSD(T) level of theory,17–24 differing from the fcc structure of the bulk material. It is therefore vital that any global optimisation of IrN (N = 10–20) structures is performed directly at the density functional theory (DFT) level of theory at least.
A quantum description of the PES greatly increases the cost of exploring it comprehensively, limiting the size of the cluster it is possible to investigate.16 It is therefore necessary that efficient parallel methodologies, with the ability to utilise greater computational resources, are developed. There have been several implementations of parallel schemes within genetic algorithms for both atomic and molecular clusters but few combine this parallelism with direct DFT global optimisation.25–29
This work presents the global optimisation of IrN (N = 10–20) clusters directly at the DFT level of theory. This is achieved using the Birmingham parallel genetic algorithm (BPGA), a new open-source genetic algorithm available via Bitbucket.30 The BPGA utilises a pool genetic algorithm methodology combined with the evaluation of potential cluster geometries in parallel.25 This combination ensures highly efficient scaling when compared with generation based genetic algorithms and allows the structural characterisation of larger and more complex systems. The pool methodology has been recently applied to metallic clusters31 and was benchmarked and applied successfully to the global optimisation of the much studied Au10 and Au20 clusters.31 Its predictions for Ag10+ have also been shown to be accurate when compared with spectra from molecular beam experiments.32 The BPGA incorporates this highly efficient algorithm within a flexible Python framework.
Due to the 5d76s2 ground state and other low lying states originating from its 5d86s1 configuration,33 the spin of the IrN clusters must be considered in the calculations.33 To account for this, spin-polarised DFT global optimisations are performed. The use of spin-polarised local minimisations effectively doubles the computational cost and can only now be performed due to the parallelism of the BPGA.
The BPGA is also capable of globally optimising bimetallic nanoalloys, whose PES is complicated by the presence of homotops.34 It is hoped that this work demonstrates that scaling capability of the BPGA and its ability to utilise massively parallel architectures, which enable the program to predict accurately the geometries of metallic nanoparticles.
2. Methodology
2.1. Birmingham parallel genetic algorithm
The BPGA is a parallel genetic algorithm for the structural characterisation of nanoparticles. The program is written in object-oriented Python. This allows greater flexibility and the ability to utilise the large existing libraries of Python code, such as the atomic simulation environment.35 Python is well suited to job submission, required by the DFT interface, on a large shared HPC resource, such as ARCHER.36 The program is open-source and available via BitBucket.30The BPGA utilises a pool methodology, shown in Fig. 1.25,31 This differs from a generation-based code, where structures belong to and are evaluated generation by generation. When executed in parallel, multiple instances of the BPGA are started sequentially within a run. Each instance is a separate BPGA run with its own set of processes. The BPGA initially generates a fixed-size pool of n random geometries and places them in a central database file which is available to the other instances of the program. In the present study the pool is set to n = 15 random geometries. These initial geometries are fixed so that no two atoms are overlapping.
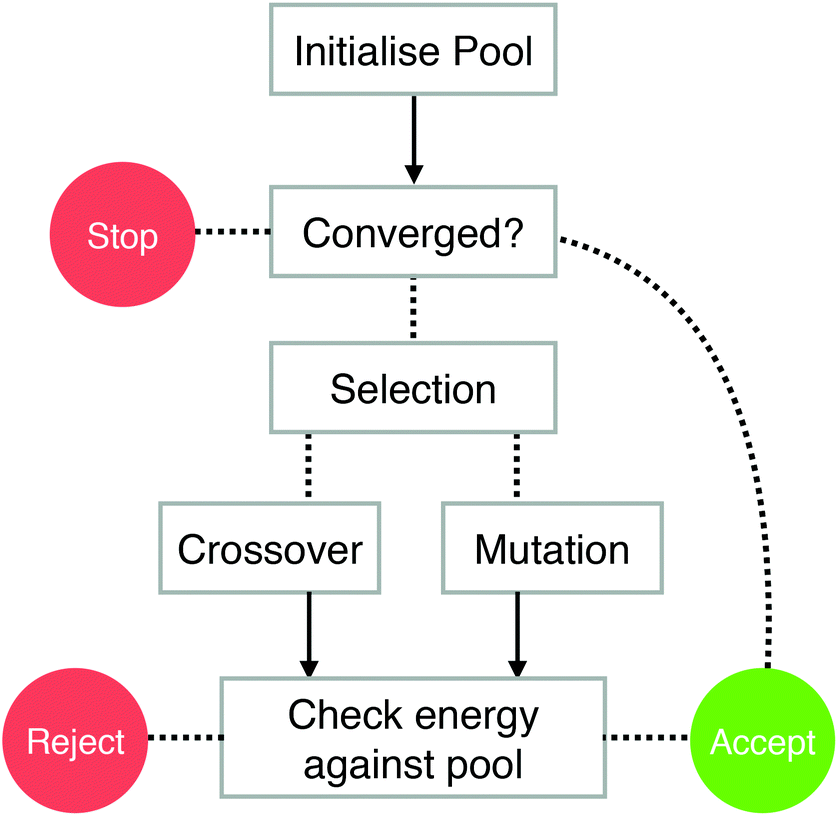 | ||
| Fig. 1 The pool scheme used by the BPGA. Arrows represent DFT local minimisations. | ||
In the local minimisation the energy of a structure is minimised with respect to its coordinates. This transforms the PES into a simpler stepped surface, greatly reducing the search space. If an instance becomes free and all structures in the pool are being or have been minimised the instance will continue to evaluate further random structures. If one of these new structures is lower in energy than the highest energy cluster in the pool, the new lower energy structure will replace it.
Once the initial pool of structures has been minimised, offspring and mutants are produced through crossover and mutation. The choice of producing either an offspring or mutant is based on the mutation rate, which is set to anywhere between 0–100% of the fixed pool size. In the present work the mutation rate is set to 10%.
Mutation is defined as the selection of a cluster at random from the pool and the displacement of two of its atoms by up to 1 Å. Other mutations schemes are available in the code, including generating a new random geometry or, for bimetallic systems, swapping unlike atoms.
Selection for crossover is carried out using the tournament method. Once selected, clusters undergo crossover according to the Deaven and Ho cut and splice method.37 The cutting plane is weighted based on the fitness of each of the clusters selected. A higher fitness represents a lower energy.
A local minimisation of the offspring is performed and its energy is checked against those of the other structures in the pool. If the offspring's energy is lower than that of the highest energy structure in the pool, the offspring structure replaces it. Convergence is achieved when the energies of the structures in the pool differ by no less than 10−3 eV. For larger clusters, convergence may not be achievable. In this case the lowest energy structure in the pool after a run of around 500 separate minimisations is taken as the putative global minimum.
The BPGA also has the ability to perform a DFT-level global optimisation of a cluster supported on a surface. This method, within a generation based code, has been demonstrated previously.38
2.2. DFT
Gamma-point, spin-polarised DFT calculations were performed with VASP.39–43 Projected-augmented wave (PAW) pseudopotentials were used with the PBE exchange correlation functional.44,45 A plane-wave basis set with a cut-off of 400 eV was used. Methfessel–Paxton smearing, with a sigma value of 0.01 eV, was utilised to improve metallic convergence.462.3. Energetics
The binding energies per atom were calculated using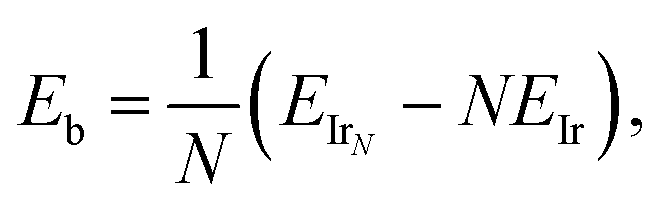 | (1) |
The stability of the clusters, relative to their N + 1 and N − 1 neighbours, is given by their second-order differences Δ2E, calculated using
| Δ2E = 2EIrN − (EIrN+1 + EIrN−1). | (2) |
3. Results
The BPGA calculations were performed on the UK's national supercomputer ARCHER.36 Each was run in parallel with eight instances of the code operating on the pool. The theoretical scaling of this parallel pool methodology has been shown previously.31 Around 500 structures were evaluated for each cluster size. Due to the high computational cost of the calculations multiple runs are not possible for each system size.The parallelism within the code, and the scaling capabilities of VASP on ARCHER, allows for spin-polarised calculations to be carried out during the global optimisation. The binding energies Eb, point groups and spin multiplicities of the putative global minimum are given in Table 1. The coordinates of the global minima and the additional minima discussed are supplied in the ESI.†
| Cluster | Point group | E b/eV | (2S + 1) |
|---|---|---|---|
| Ir10 | C s | −4.914 | 3 |
| Ir11 | C 1 | −4.932 | 4 |
| Ir12 | D 4h | −5.172 | 3 |
| Ir13 | C s | −5.139 | 4 |
| Ir14 | C s | −5.220 | 3 |
| Ir15 | C 2v | −5.206 | 2 |
| Ir16 | C s | −5.301 | 3 |
| Ir17 | C s | −5.348 | 4 |
| Ir18 | D 4h | −5.452 | 7 |
| Ir19 | C 1 | −5.416 | 2 |
| Ir20 | C 1 | −5.436 | 3 |
Overall, the putative global minima from the BPGA searches are in good agreement with structures suggested in previous work on Ir clusters.18,21–23 Some structures have been previously characterised and give a good indication of the ability of the BPGA to find the putative global minimum at a given level of theory. Other structures are reported here for the first time.
The BPGA successfully finds the C2v dimer-capped (“house”) structure, shown in Fig. 2, as the putative global minimum for Ir10 and scales successfully well beyond this previous 10-atom limit. The putative global minimum structures of IrN (N = 11–20) clusters are shown in Fig. 3.
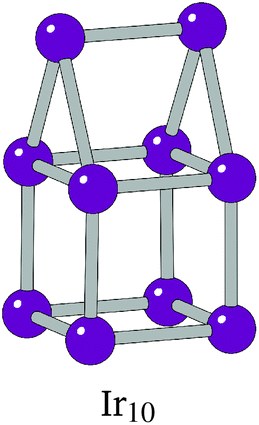 | ||
| Fig. 2 The dimer-capped (“house”) structure of Ir10. | ||
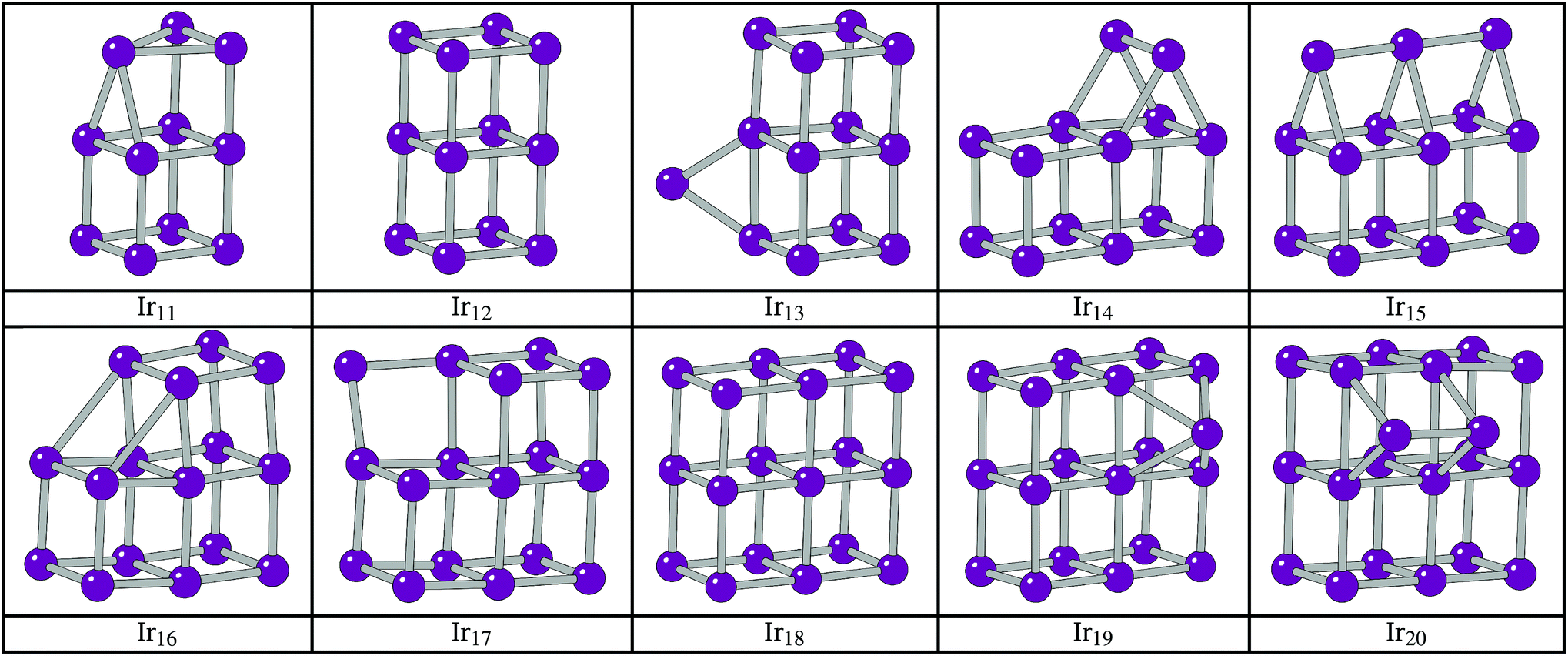 | ||
| Fig. 3 Putative global minimum structures for IrN (N = 10–20) from the BPGA searches. | ||
The overall global minimum structure for Ir11 is a triangle-capped cube. This structure, together with a second highly competitive low lying minimum, an edge-bridged structure based on the Ir10 “house”, are shown in Fig. 4. The two structures differ by 0.05 eV. The global minimum structure is a high spin structure with a spin multiplicity of 4, compared with the competitive minimum's multiplicity of 2. The spin polarised DFT global optimisation has allowed this lower energy putative global minimum to be reported for the first time.
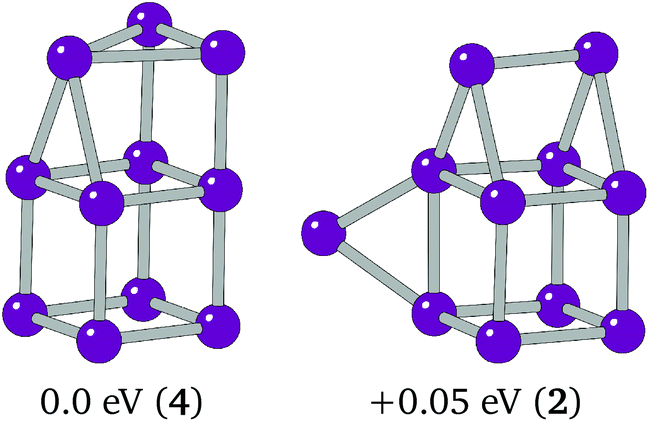 | ||
| Fig. 4 Putative global minimum (left) and competitive low-lying minima (right). Relative energies and spin multiplicities (in brackets) are shown below. | ||
The additional Ir in Ir12 now makes it possible to complete a third cubic face and the 3 × 2 × 2 D2h cuboid structure becomes the global minimum. For Ir13 the extra Ir bridges an edge on one of the cubes.
It was thought that the global minimum structure for Ir13 may be a C4v structure, with an Ir atom capping an end of the cuboid, and that the cubic bounding cell used to generate the initial random geometries may have biased the search against any elongated structures. Local minimisations were carried out on C2v centre edge-bridged, Cs top edge-bridged and C4v top-capped cuboid structures, shown in Fig. 5. The C2v centre edge-bridged structure locally minimises into a face-capped Cs structure. The structures were found to lie 0.26, 0.33 and 0.97 eV higher in energy than the putative global minimum structure from the BPGA search, respectively. This structural preference is also confirmed by previous work on Ir13.19,20
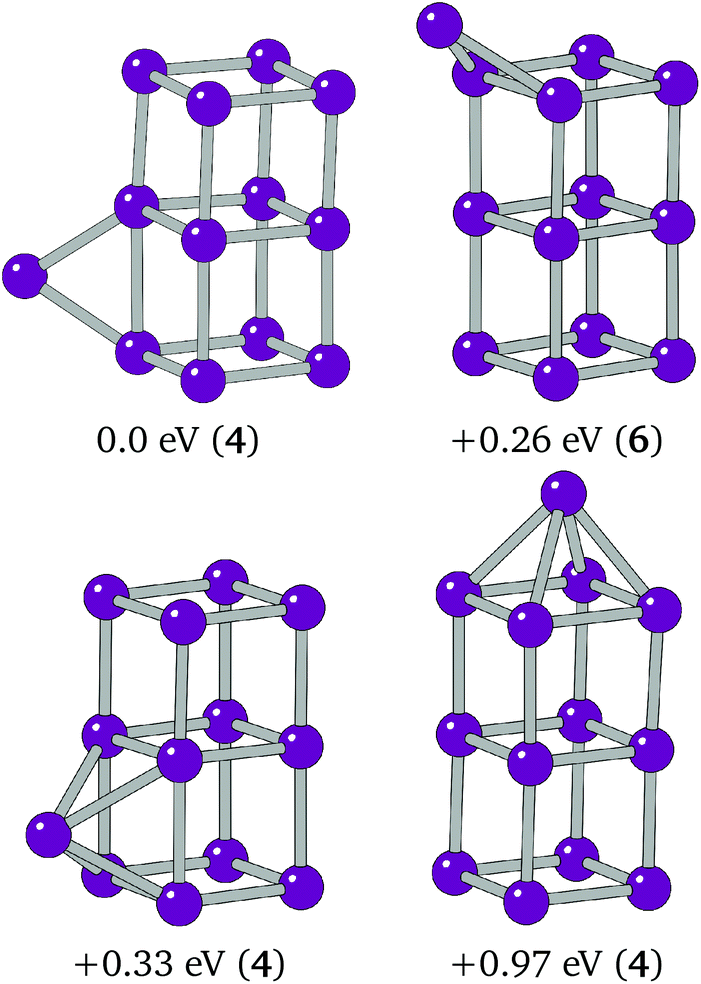 | ||
| Fig. 5 Global minimum (top-left), top edge-bridged (top-right), centre edge-bridged (bottom-left) and top-capped (bottom-right) cubic structures assessed for Ir13. The centre edge-bridged structure is shown after local minimisation to the face-capped cuboid structure. | ||
The structure of Ir14 is a dimer-capped 3 × 2 × 2 cuboid, analogous to the Ir10 structure with the dimer lying perpendicular to the long edges of the Ir12 cuboid structure. This structure has been previously shown.21 Upon addition of an extra Ir, the structure of Ir15 becomes a trimer-capped cuboid, with the Ir3 trimer lying parallel to the long sides of the rectangular face.
The preference of Ir16 is shown to be a slightly deformed L-shaped cubic structure, so that two elongated 3.1 Å bonds can form between two cubes of the structure. Cubic bounding may also have affected Ir16, as it follows from the structure of Ir8, a cube, and Ir12 that the global minimum could be a 4 × 1 × 1 cuboid. This structure was assessed alongside two other low-lying minima, T-capped and square-capped cuboid structures. The structures and relative energies of these minima are shown in Fig. 6, with the 4 × 1 × 1 cuboid found to lie 0.88 eV higher in energy than the BPGA global minimum.
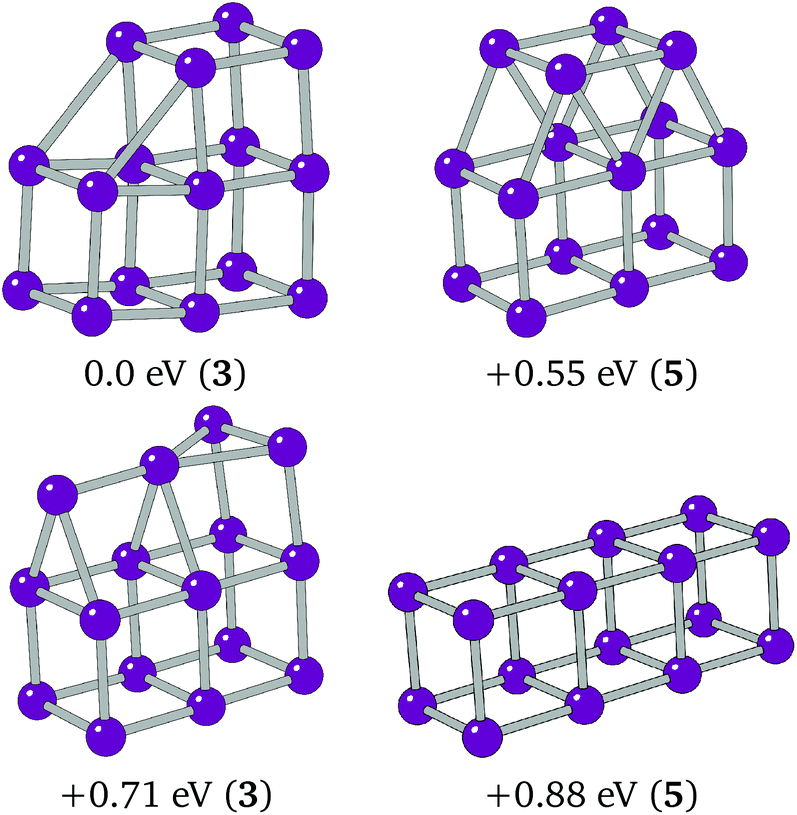 | ||
| Fig. 6 Putative global minimum (top-left), 4 × 1 × 1 cuboid, T-capped and square capped 3 × 1 × 1 cubic structures assessed for Ir16. | ||
The structure of Ir17 shows the additional Ir in between two cubes of the Ir16 cuboid, the start of a complete 3 × 3 × 2 cuboid. The additional Ir of Ir18 sits between two cubes of the L-shaped Ir17 cluster and forms the complete 3 × 3 × 2 cuboid.
The extra Ir in Ir19 caps a cubic face on the 3 × 3 × 2 cuboid. This putative global minimum was tested against an edge-bridged structure, which was believed to be the more likely global minimum structure following the trend seen in smaller clusters. The C1 edge-bridged structure, shown in Fig. 7, was found to have a spin multiplicity of 6 and to be just 0.008 eV lower in energy than the BPGA global minimum.
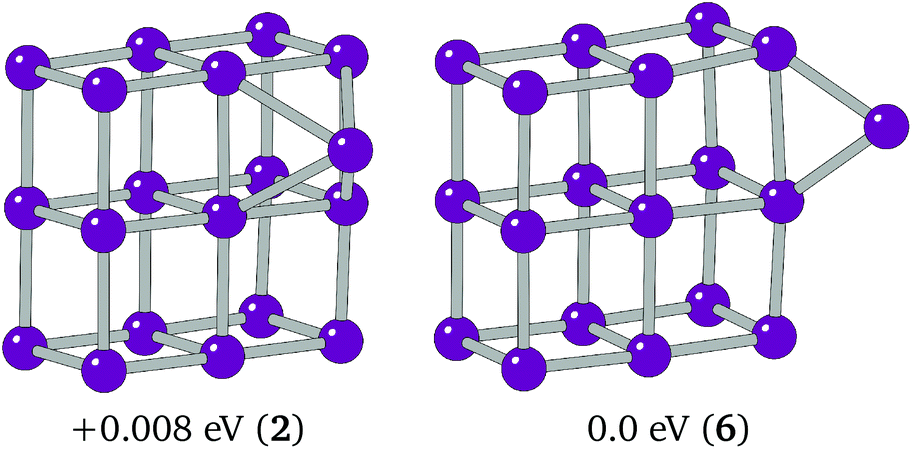 | ||
| Fig. 7 Putative global minimum (left) and edge-bridged cuboid structure for Ir19. | ||
The energetic differences between the low lying minima of Ir11 and Ir19 are far smaller than that seen for the various Ir13 minima and smaller than the error in the current DFT calculations. To determine accurately the global minimum, higher level calculations, coupled with experiment, will be required in the future.
The structure of Ir20 is analogous to that of Ir13 with an Ir2 dimer lying perpendicular to the long edges of the 3 × 3 × 2 cuboid structure.
The binding energies of the clusters give an indication of the relative stability of the clusters, with more negative values indicating greater stability. Eb values are shown in Fig. 8. Overall, Eb tends to decrease as N increases, with Ir18 having the lowest Eb.
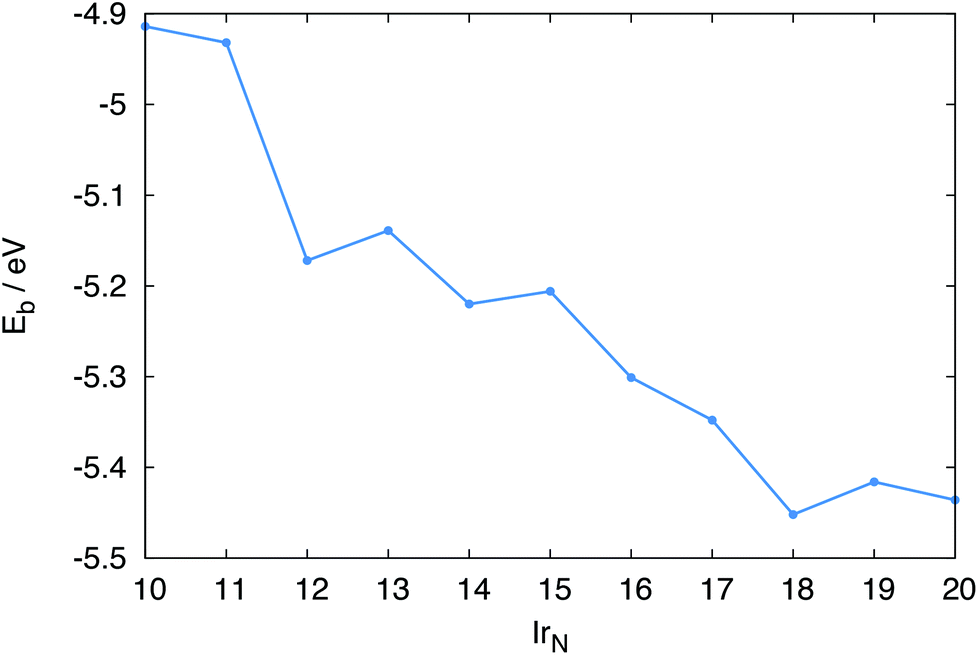 | ||
| Fig. 8 Binding energies Eb, for IrN (N = 10–20) clusters. | ||
For several N-atom clusters Eb is higher (less stable) than for the N − 1 cluster: in particular Ir13, Ir15 and Ir19. Ir20, despite Ir forming a dimer capping a face, has a higher Eb than Ir18. The increased stability displayed by the even numbered clusters is more clearly shown by their second-order difference plot shown in Fig. 9. Each odd numbered cluster clearly shows decreased stability, indicated by a positive Δ2E, relative to its even numbered neighbours.
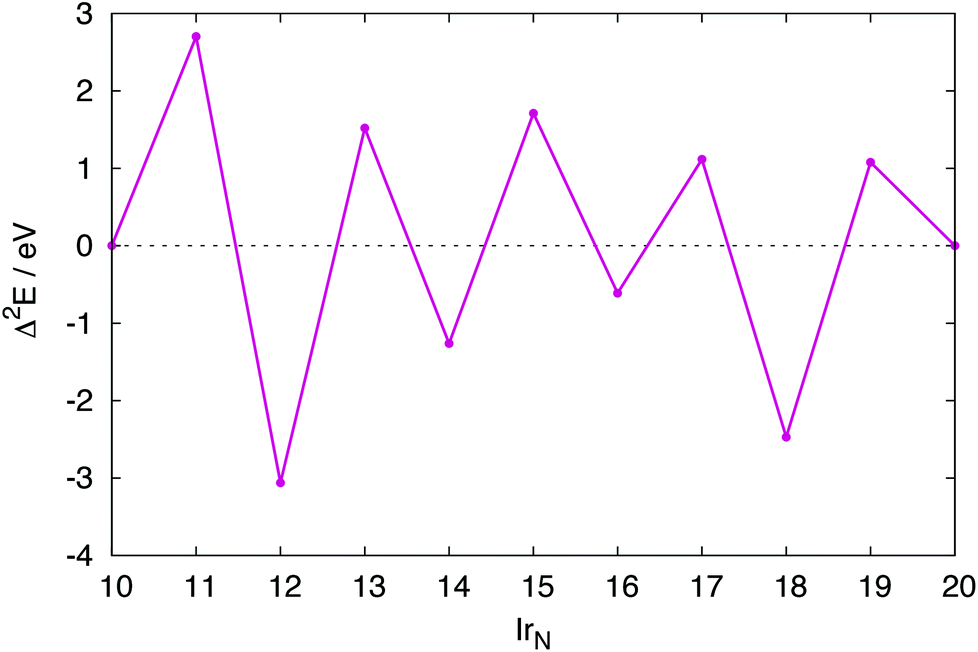 | ||
| Fig. 9 Second-order differences Δ2E, for IrN (N = 10–20) clusters. | ||
The spin multiplicities of the clusters are given in Table 1. All global minima are found to have multiplicities of between 2 and 7. This is a clear indication of the importance of magnetic effects in these clusters. In particular, high spin structures are shown to be the global minimum structures of Ir11 and Ir19. It is likely that these and other low energy minima would have been excluded from the searches if non-spin-polarized calculations had been carried out.
4. Conclusions
The BPGA has successfully coupled the computation resources of ARCHER36 with the scaling capability of a pool genetic algorithm methodology. This has allowed the direct DFT global optimisation of IrN (N = 10–20) clusters, with some global minimum structures being reported here for the first time.The code has captured the cubic nature of the sub-nanometre Ir clusters with the putative global minima evaluated and compared with previous results. For Ir11 and Ir19 their spin has been shown to determine their global minimum structures, which would have otherwise been missed in a low-spin search. The use of spin polarized calculations has been made possible because of the BPGA's ability to utilise greater computational resources. The structural characterisation of a system is a vital first step in exploring its catalytic properties. The structures of the IrN clusters will form the basis of future studies of the catalytic properties of the system, including modelling their interaction with small molecules.
The cubic structures found here are in agreement with higher level CCSD(T) and CASSCF calculations on Ir8, reported by Dixon et al.18 In future work, we will investigate the effect of changing the functional in DFT calculations on Ir clusters; in particular we will explore the use of meta-GGA functionals.47,48
The development of the BPGA will continue. This will include the implementation of new features, such as the global optimisation of a system in the presence of a ligand or directly on a variety of surfaces. Further improvements to the efficiency of the parallel scheme will also be made. The code will be applied to a variety of new supported and ligated mono- and bimetallic cluster systems. The number of interfaces to common quantum chemistry programs within the BPGA will be expanded and applied to the global optimisation of systems at theory levels beyond DFT.
Acknowledgements
J.B.A.D. and R.L.J. acknowledge the Engineering and Physical Sciences Research Council, U.K. (EPSRC) for funding under Critical Mass Grant EP/J010804/1 “TOUCAN: Towards an Understanding of Catalysis on Nanoalloys”.A.S. acknowledges financial support by the DFG (grant SCHA885/10-2) and the Merck'sche Gesellschaft für Kunst und Wissenschaft e.V.
Calculations were performed via membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work made use of the facilities of ARCHER, the UK's national high-performance computing service, which is funded by the Office of Science and Technology through EPSRC's High End Computing Programme.
References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- A. Uzun, D. A. Dixon and B. C. Gates, ChemCatChem, 2011, 3, 95–107 CrossRef CAS PubMed.
- H. Kobayashi, M. Yamauchi and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 6893–6895 CrossRef CAS PubMed.
- J. B. A. Davis, S. L. Horswell, L. Piccolo and R. L. Johnston, J. Organomet. Chem., 2015 DOI:10.1016/j.jorganchem.2015.04.03.
- L. Piccolo, S. Nassreddine, G. Toussaint and C. Geantet, ChemSusChem, 2012, 5, 1717–1723 CrossRef CAS PubMed.
- A. Okrut, R. C. Runnebaum, X. Ouyang, J. Lu, C. Aydin, S.-J. Hwang, S. Zhang, O. A. Olatunji-Ojo, K. A. Durkin, D. A. Dixon, B. C. Gates and A. Katz, Nat. Nanotechnol., 2014, 9, 459–465 CrossRef CAS PubMed.
- M. Polak and L. Rubinovich, Surf. Sci., 2005, 584, 41–48 CrossRef CAS PubMed.
- J. B. A. Davis, R. L. Johnston, L. Rubinovich and M. Polak, J. Chem. Phys., 2014, 141, 224307 CrossRef PubMed.
- D. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- http://www-wales.ch.cam.ac.uk/GMIN/ .
- R. L. Johnston, Dalton Trans., 2003, 4193–4207 RSC.
- F. Cleri and V. Rosato, Phys. Rev. B: Condens. Matter, 1993, 48, 22–33 CrossRef CAS.
- S. Heiles, A. J. Logsdail, R. Schäfer and R. L. Johnston, Nanoscale, 2012, 4, 1109–1115 RSC.
- C. J. Heard and R. L. Johnston, Eur. Phys. J. D, 2013, 67, 34 CrossRef PubMed.
- P. C. Jennings and R. L. Johnston, Comput. Theor. Chem., 2013, 1021, 91–100 CrossRef CAS PubMed.
- S. Heiles and R. L. Johnston, Int. J. Quantum Chem., 2013, 113, 2091–2109 CrossRef CAS PubMed.
- Y. Chen, M. Huo, T. Chen, Q. Li, Z. Sun and L. Song, Phys. Chem. Chem. Phys., 2015, 17, 1680–1687 RSC.
- M. Chen and D. A. Dixon, J. Phys. Chem. A, 2013, 117, 3676–3688 CrossRef CAS PubMed.
- M. J. Piotrowski, P. Piquini, M. M. Odashima and J. L. Da Silva, J. Chem. Phys., 2011, 134, 134105 CrossRef PubMed.
- M. Zhang and R. Fournier, Phys. Rev. A, 2009, 79, 043203 CrossRef.
- J. Du, X. Sun, J. Chen and G. Jiang, J. Phys. Chem. A, 2010, 114, 12825–12833 CrossRef CAS PubMed.
- T. Pawluk, Y. Hirata and L. Wang, J. Phys. Chem. B, 2005, 109, 20817–20823 CrossRef CAS PubMed.
- W. Zhang, L. Xiao, Y. Hirata, T. Pawluk and L. Wang, Chem. Phys. Lett., 2004, 383, 67–71 CrossRef CAS PubMed.
- G. Ping, Z. Ji-Ming, Z. Pei, Z. Lin-Lin and R. Zhao-Yu, Chin. Phys. B, 2010, 19, 083601 CrossRef.
- B. Bandow and B. Hartke, J. Phys. Chem. A, 2006, 110, 5809–5822 CrossRef CAS PubMed.
- L. B. Vilhelmsen and B. Hammer, J. Chem. Phys., 2014, 141, 044711 CrossRef PubMed.
- Y. Ge and J. D. Head, J. Phys. Chem. B, 2004, 108, 6025–6034 CrossRef CAS.
- J. M. Dieterich and B. Hartke, Mol. Phys., 2010, 108, 279–291 CrossRef CAS PubMed.
- F. Weigend, J. Chem. Phys., 2014, 141, 134103 CrossRef PubMed.
- https://bitbucket.org/JBADavis/bpga/ .
- A. Shayeghi, D. Götz, J. B. A. Davis, R. Schäfer and R. L. Johnston, Phys. Chem. Chem. Phys., 2015, 17, 2104–2112 RSC.
- A. Shayeghi, R. L. Johnston and R. Schäfer, J. Chem. Phys., 2014, 141, 181104 CrossRef CAS PubMed.
- J. E. Sansonetti and W. C. Martin, J. Phys. Chem. Ref. Data, 2005, 34, 1777–1781 CrossRef PubMed.
- J. Jellinek and E. B. Krissinel, Chem. Phys. Lett., 1996, 258, 283–292 CrossRef CAS.
- https://wiki.fysik.dtu.dk/ase/ .
- http://www.archer.ac.uk .
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS.
- C. J. Heard, S. Heiles, S. Vajda and R. L. Johnston, Nanoscale, 2014, 6, 11777–11788 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter, 1996, 54, 533–539 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758–1775 CrossRef CAS.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter, 1989, 40, 3616–3621 CrossRef CAS.
- L. A. Constantin, E. Fabiano and F. Della Sala, J. Chem. Theory Comput., 2013, 9, 2256–2263 CrossRef CAS.
- P. Hao, J. Sun, B. Xiao, A. Ruzsinszky, G. I. Csonka, J. Tao, S. Glindmeyer and J. P. Perdew, J. Chem. Theory Comput., 2013, 9, 355–363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C5NR03774C |
| This journal is © The Royal Society of Chemistry 2015 |