
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High binding yet accelerated guest rotation within a cucurbit[7]uril complex. Toward paramagnetic gyroscopes and rolling nanomachines†
G.
Casano
a,
F.
Poulhès
a,
T. K.
Tran
a,
M. M.
Ayhan
ab,
H.
Karoui
a,
D.
Siri
a,
A.
Gaudel-Siri
a,
A.
Rockenbauer
c,
G.
Jeschke
d,
D.
Bardelang
*a,
P.
Tordo
*a and
O.
Ouari
*a
aAix-Marseille Université, CNRS, Institut de Chimie Radicalaire, UMR 7273, 13013 Marseille, France. E-mail: david.bardelang@univ-amu.fr; paul.tordo@univ-amu.fr; olivier.ouari@univ-amu.fr; Fax: +33 4 91 28 8758; Tel: +33 4 91 28 8610
bDepartment of Chemistry, Gebze Technical University, P.K.:141, 41400 Gebze, Kocaeli, Turkey
cInstitute of Materials and Environmental Chemistry, Hungarian Academy of Sciences, 1519 Budapest, P.O. Box. 286, and Department of Physics, Budapest University of Technology and Economics 1111 Budapest, Hungary
dETH Zurich, Laboratory of Physical Chemistry, Vladimir-Prelog-Weg 2, CH-8093 Zurich, Switzerland
First published on 12th June 2015
Abstract
The (15-oxo-3,7,11-triazadispiro[5.1.5.3]hexadec-7-yl)oxidanyl, a bis-spiropiperidinium nitroxide derived from TEMPONE, can be included in cucurbit[7]uril to form a strong (Ka ∼ 2 × 105 M−1) CB[7]@bPTO complex. EPR and MS spectra, DFT calculations, and unparalleled increased resistance (a factor of ∼103) toward ascorbic acid reduction show evidence of deep inclusion of bPTO inside CB[7]. The unusual shape of the CB[7]@bPTO EPR spectrum can be explained by an anisotropic Brownian rotational diffusion, the global tumbling of the complex being slower than rotation of bPTO around its “long molecular axis” inside CB[7]. The CB[7] (stator) with the encapsulated bPTO (rotator) behaves as a supramolecular paramagnetic rotor with increased rotational speed of the rotator that has great potential for advanced nanoscale machines requiring wheels such as cucurbiturils with virtually no friction between the wheel and the axle for optimum wheel rotation (i.e. nanopulleys and nanocars).
Introduction
Molecular machines are increasingly being considered as promising architectures for advanced machineries proceeding at the nanoscale. Among them, nanocars1 are outstanding examples of such nanomachines where increased degrees of control are progressively gained over their design2 and their movements.3 However, all reports of such nanocars show the wheels covalently linked to the chassis implying friction problems upon movement. Among the macrocycles that could be used as wheels in a non-covalent strategy, cucurbiturils are rigid symmetrical round-shaped molecules4–6 with binding constants up to 7.2 × 1017 M−1. Here we show that suitably designed guests with high affinity can efficiently rotate in cucurbiturils with low friction. During the last two decades, the host–guest chemistry of CB[n] has been studied extensively7,8 using a combination of electronic absorption and NMR spectroscopy, mass spectrometry, and X-ray crystallography. In the past few years, EPR spectroscopy has also been used as an additional tool to explore the binding properties of CB[n] with paramagnetic molecules, containing one or several nitroxide moieties as probes.9–20 Lucarini first showed that TEMPO can be complexed by CB[7] (Ka ∼ 25 ± 2 × 103 M−1),15 the free and complexed radical exchanging slowly on the EPR time scale, and the latter showing smaller nitrogen hyperfine splitting and larger g factor values (ΔaN = 0.11 mT, Δg = 0.0008). Kaifer et al.16 showed that the TEMPO moiety of 4-amido-2,2,6,6-tetramethylpiperidine-1-oxyl)cobaltocenium is engulfed in CB[8] to form a very stable inclusion compound (Ka = 2.1 ± 1 × 108 M−1). The binding of one and two CB[8] macrocycles has been used to allosterically regulate the extent of spin exchange coupling in paramagnetic molecules bearing several nitroxide moieties.17 At concentrations above 10−3 M, an interesting selective aggregation of three supramolecules of nitroxide@CB[8] could be detected by EPR with various nitroxides.18–20 The three supramolecules are arranged in a triangular geometry that leads to spin exchange between the three radical centers. No such aggregation was evident in the case of CB[7] complexes. Nitroxide probes are widely used to investigate biological systems,9,21–24 however their use in vivo is often limited by their rapid reduction to EPR silent compounds.25–28 Various approaches have been developed to obtain nitroxide probes with increased resistance to bioreduction.29–31 One strategy to protect nitroxides from bioreductants is to include them into macrocycles such as cyclodextrins (CD).32–36 We37–40 and others41–44 have shown that the half-lives of various stable nitroxides or persistent nitroxide spin adducts37 can indeed be enhanced in the presence of CDs. However, because of the relatively weak binding constants of CD@nitroxide complexes, reductants, such as glutathione (GSH) or ascorbate, still remain active. Recently, we reported that CB[7] is a promising candidate in protecting the TEMPO nitroxide in the presence of an excess of ascorbate.18 However, limitations still remain, due to the inherent dynamic inclusion complex equilibrium that leaves a fraction of the nitroxides exposed to the reductants. Recently different authors45–47 reported that a high degree of size and shape complementarity, and the presence on the guest of two positive charges, both positioned to interact with the CB[7]'s ring carbonyl oxygens through ion–dipole interactions, can lead to unprecedented CB[7]–guest affinity, with values (up to Ka = 7.2 × 1017 M−1) higher than that of the avidin–biotin pair.Based on these results, we designed nitroxides (2,2-dimethyl-4-oxo-1,9-diazaspiro[5.5]undec-1-yl)oxidanyl PTO, and bPTO, having in water one or two protonated amine functions prone to position near the two carbonyl laced portals, and to force the N–O˙ group to stand near the center of the CB[7] or CB[8] cavity (Scheme 1). Compared to TEMPONE we found that the binding affinities of bPTO for CB[7] and CB[8] are significantly increased, and once complexed bPTO becomes particularly resistant to reduction with ascorbate. Moreover, the EPR spectra of CB[7]@bPTO and CB[8]@bPTO complexes have a rather unusual shape. The high field line of the 14N triplet is not broadened, as predicted due to the expected longer correlation time of the complexes compared to free bPTO. This behaviour can be explained by an anisotropic Brownian rotational diffusion, the global tumbling of the complexes being slower than the rotation of bPTO along its “long molecular axis” inside CB, the CB (stator) with the encapsulated bPTO (rotator) behaving as a supramolecular paramagnetic molecular rotor. Our results are presented and discussed hereafter.
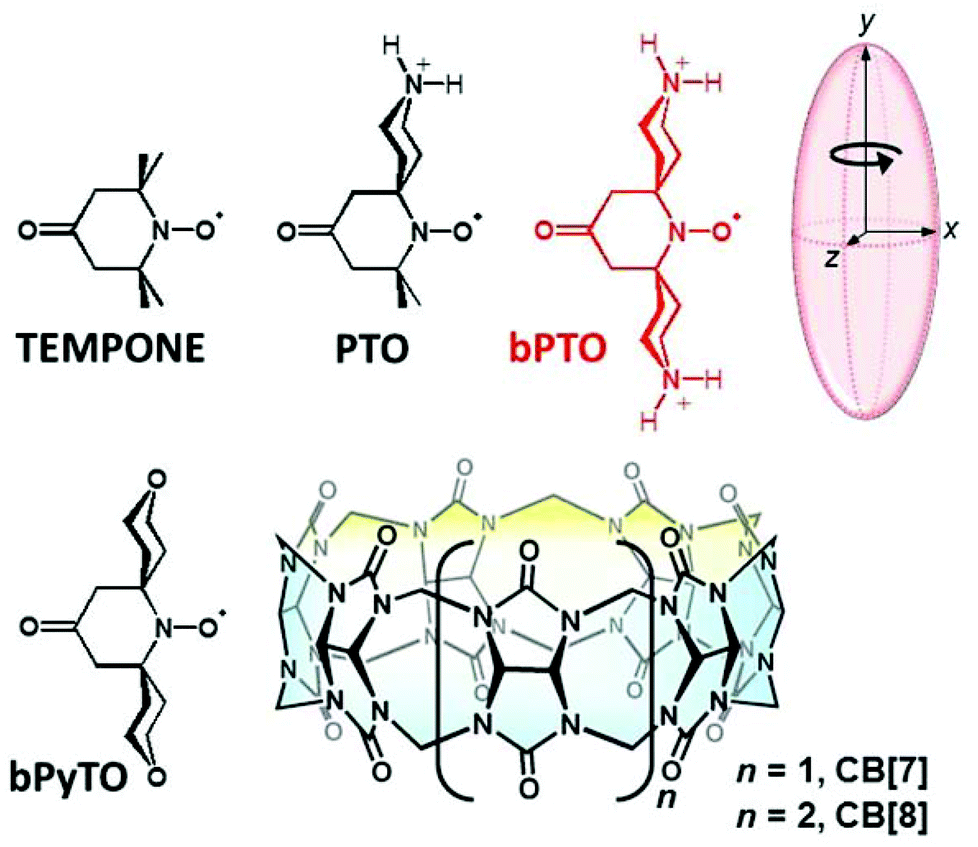 | ||
| Scheme 1 Structures of TEMPONE derivatives and of cucurbit[n]urils. | ||
Results and discussion
PTO and bPTO were prepared in a three-step sequence; experimental details for reaction procedures and characterization are given in the ESI.† All the experiments were performed in water, and with a pKa of piperidine around 11.2, we will consider for the following discussion that PTO and bPTO are protonated at the amine sites.Mass spectrometry
High resolution mass spectra of equimolar solutions of bPTO and CB[7] (1 mM) in water showed one peak at m/z 708.2644 corresponding to a doubly charged cation of the formula C55H66N31O162+ which is in agreement with the composition [(CB7)(bPTO˙)]2+. Similarly with CB[8], the detection of a cation at m/z 791.2893 corresponding to the formula C61H72N35O182+ is in agreement with a complex of the composition [(CB8)(bPTO˙)]2+.EPR characterization
EPR spectra of PTO and bPTO show a typical three line pattern with a width at half height of 0.26 mT and 0.33 mT respectively and nitrogen coupling constants aN of 1.57 and 1.53 mT respectively (gPTO = 2.0058, gbPTO = 2.0061, Fig. 1).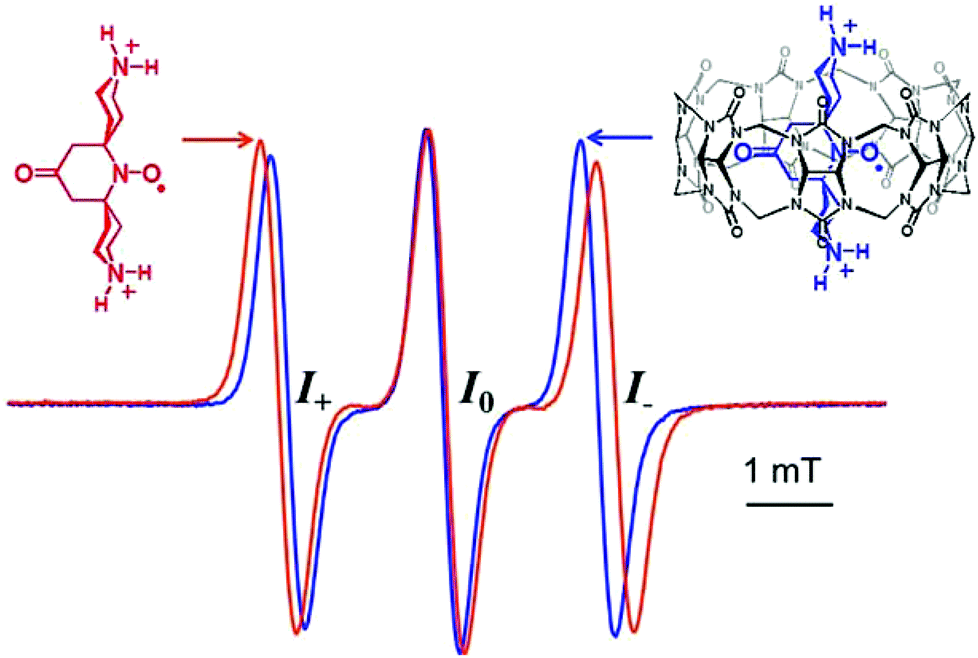 | ||
| Fig. 1 EPR spectra in water of bPTO alone (0.2 mM, red line) and in the presence of CB[7] (1.4 eq., blue line), highlighting the reduced aN coupling constant and the increased intensity of the high field line (I−) upon binding. | ||
With regard to TEMPONE (0.08 mT),48 the larger linewidth observed for bPTO is mainly due to additional long range hyperfine couplings with γ- and δ-hydrogens (see the ESI†). For bPTO, in the presence of CB[7], the nitrogen hyperfine coupling constant decreases significantly (ΔaN = 0.12 mT) and the g factor increases (Δg = 0.0006), in agreement with the formation of a CB[7]@bPTO inclusion complex, which is accompanied by the N–O˙ group localization in the less polar surrounding of the CB[7] cavity. Usually, together with changes in aN and g values, the formation of a nitroxide inclusion complex is accompanied by a broadening of the EPR high field line (I−), resulting from an increase of the correlation time.15,16 This broadening was not observed with CB[7]@bPTO (Fig. 1), and as discussed below this result can be accounted for by an anisotropic rotational diffusion tensor for the included bPTO.
EPR titration experiments were performed recording a series of EPR spectra obtained by gradually increasing the CB[7] or CB[8] concentrations. Using a 2D simulation program,49 binding constants Ka ∼ 9 × 103 M−1 and 2.8 × 105 M−1 were determined (Table 1) for the complexation of PTO with CB[7] and CB[8] respectively. The significantly smaller Ka value obtained for CB[7] is presumably due to steric hindrance at the PTO carbonyl–nitroxide region (O–O˙ distance ≈7.7 Å) with van der Waals radii with respect to the cavity of CB[7] (entrance ≈5.8 Å, inner part ≈7.8 Å). For bPTO, due to the presence of two piperidinium rings the affinity for CB[7] and CB[8] is expected to be higher. It reached 1.8 × 105 M−1 for CB[7] and was estimated (because we are close to the limit of reliable quantitative estimation of binding using EPR) to be above 106 M−1 for CB[8]. The best fit between experimental and calculated EPR spectra was obtained assuming the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes. Reduction experiments of the N–O˙ group in the presence of ascorbic acid were performed (i) as an indication of the accessibility of the nitroxide function and (ii) as a way to determine the shielding effect, i.e. the efficacy of cucurbiturils to enhance the lifetime of nitroxides in biologically relevant media. Ascorbic acid was selected because it is known to be one of the most powerful reductants of nitroxides in biological fluids or cells, leading to very fast decay of their EPR signals in biological systems.26,50 We first monitored the EPR signals of the included bPTO (0.1 mM) in CB[7] and CB[8] (0.35 mM) after addition of ascorbic acid (2 mM). Over 90 minutes, the signal decay was very slow while at the same ascorbic acid concentration, the nitroxide alone is instantaneously reduced (Fig. 2).
:1 complexes. Reduction experiments of the N–O˙ group in the presence of ascorbic acid were performed (i) as an indication of the accessibility of the nitroxide function and (ii) as a way to determine the shielding effect, i.e. the efficacy of cucurbiturils to enhance the lifetime of nitroxides in biologically relevant media. Ascorbic acid was selected because it is known to be one of the most powerful reductants of nitroxides in biological fluids or cells, leading to very fast decay of their EPR signals in biological systems.26,50 We first monitored the EPR signals of the included bPTO (0.1 mM) in CB[7] and CB[8] (0.35 mM) after addition of ascorbic acid (2 mM). Over 90 minutes, the signal decay was very slow while at the same ascorbic acid concentration, the nitroxide alone is instantaneously reduced (Fig. 2).
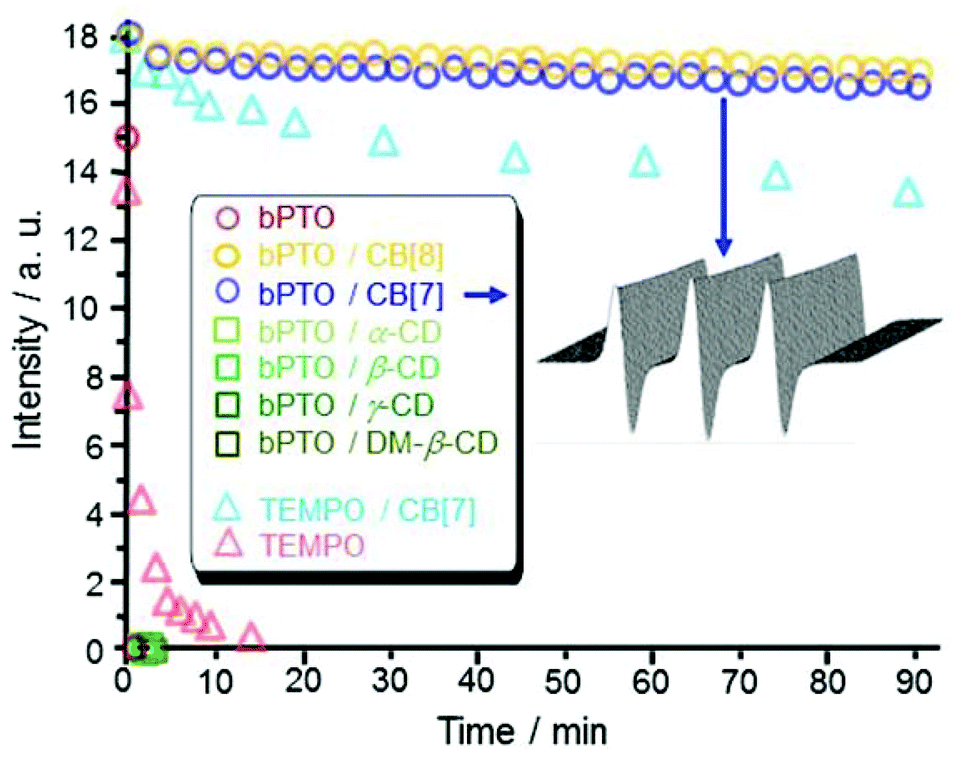 | ||
Fig. 2 Reduction experiments (decay of the EPR signal) of bPTO (0.2 mM  ) and TEMPO (0.1 mM ) and TEMPO (0.1 mM  ) and in the presence of CB[7] (12.75 mM for TEMPO ( ) and in the presence of CB[7] (12.75 mM for TEMPO ( ) and 0.35 mM for bPTO ) and 0.35 mM for bPTO ), CB[8] (0.35 mM ), CB[8] (0.35 mM  ), α-CD (50 mM ), α-CD (50 mM  ), β-CD (10 mM ), β-CD (10 mM  ), γ-CD (100 mM ), γ-CD (100 mM  ) and DM-β-CD (200 mM ) and DM-β-CD (200 mM  ) by ascorbic acid (2 mM, and sodium ascorbate: 2 mM for TEMPO and TEMPO/CB[7]). ) by ascorbic acid (2 mM, and sodium ascorbate: 2 mM for TEMPO and TEMPO/CB[7]). | ||
| a N/mT | K a/M−1 | |
|---|---|---|
| TEMPONE | 1.61 | — |
| TEMPONE/CB[7] | 1.54 | ∼103 |
| TEMPONE/CB[8] | 1.53 | 40 |
| PTO | 1.57 | — |
| PTO/CB[7] | 1.50 | 9.0 × 103 |
| PTO/CB[8] | 1.45 | 2.8 × 105 |
| bPTO | 1.53 | — |
| bPTO/CB[7] | 1.41 | 1.8 × 105 |
| bPTO/CB[8] | 1.40 | >106 |
Interestingly, α-cyclodextrin (α-CD 50 mM), β-cyclodextrin (β-CD 10 mM), γ-cyclodextrin (γ-CD 100 mM) and 2,6-di-O-methyl-β-cyclodextrin (DM-β-CD 200 mM) that also show signs of inclusion of bPTO (Fig. 3a) afforded no protection, and no EPR signal could be detected 45 seconds after the addition of the reductant. These results show that CB[7] and CB[8] behave as effective shields around bPTO, and indicate that the N–O˙ group is deeply immersed in their cavity. We previously showed that CB[7] (12.75 mM, 100-fold excess) improved the protection of TEMPO (0.1 mM) regarding ascorbate reduction (2 mM), increasing its half-life to 254 minutes.18 The protection is much more efficient for bPTO (0.2 mM) with CB[7] (0.35 mM). The intensity of the CB[7]@bPTO EPR lines is reduced by only ∼23% after 16 hours which corresponds to an approximate t1/2 of ∼17 h. Because under the same experimental conditions, the half-life time of bPTO alone is <1 min, complexation with CB[7] affords a ca. 103 fold enhancement in the protection of the N–O˙ group. The results obtained using CB[8] (t1/2 ∼ 21 h, ESI†) are very similar to those found with CB[7].
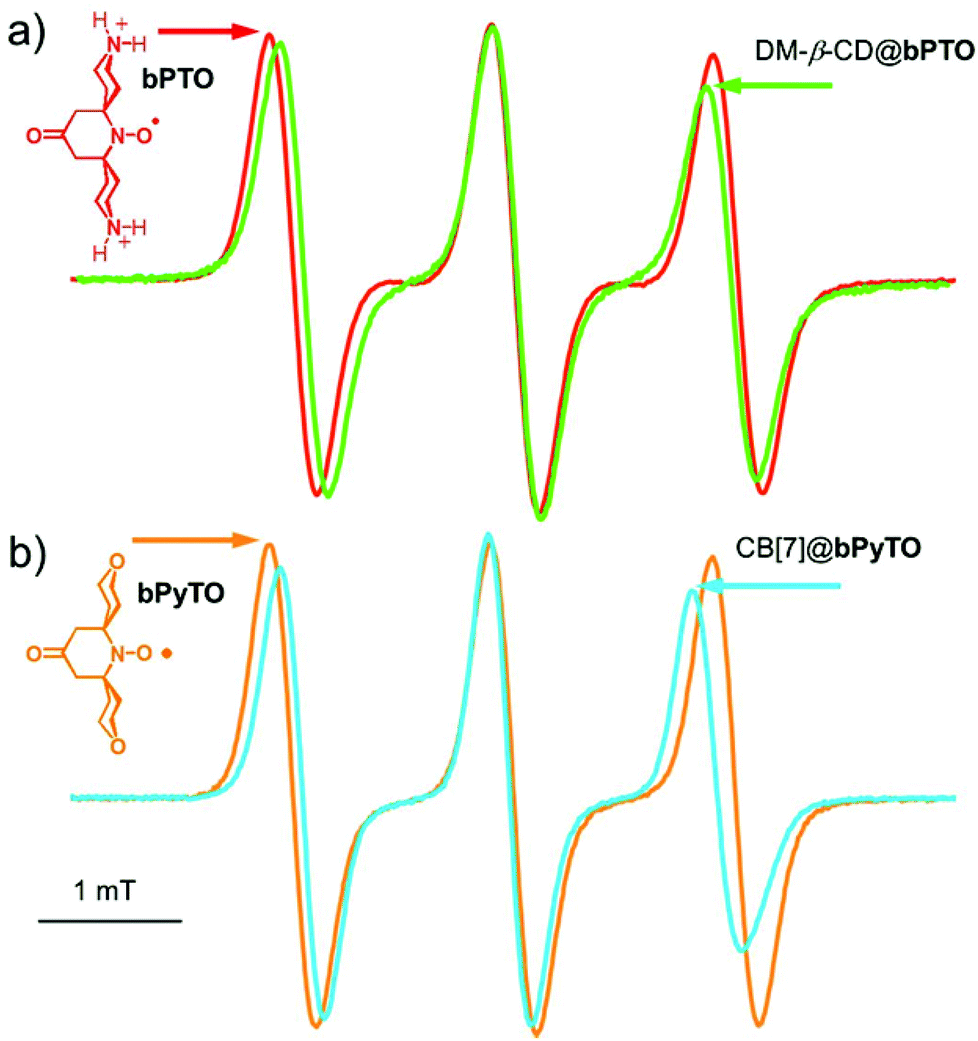 | ||
| Fig. 3 EPR spectra of (a) bPTO (0.2 mM) alone (red line) and in the presence of DM-β-CD (200 mM, green line). (b) bPyTO (0.2 mM, orange line) and with CB[7] (8 mM, blue line). | ||
Rotational dynamics
Our results show that for CB[7]@bPTO the host and the guest have no dynamic cohesion. As previously mentioned by Mock,55 the reason for the absence of mechanical coupling is the nearly cylindrical symmetry of cucurbiturils which allows guests to keep an axis of rotational freedom. However, usually the rotational motion of the complexed guest becomes restricted due to steric constraints.45,55 For bPTO, slowdown of rotational diffusion about the x and z axes on inclusion into CB is expected because of the larger effective radius of the particle undergoing rotational diffusion. The fact that rotational diffusion about the y axis remains fast and actually appears to speed up is unexpected. The data strongly suggest that bPTO rotates about its long axis after inclusion into CB with less friction than in pure water. This increase of rotational motion around the y axis could be reasonably accounted for by the successive formation of the same set of hydrogen bonds, between ammonium hydrogen atoms and the CB portal carbonyl groups, requiring a small energy barrier as in molecular ball bearings. In order to get more evidence on the rotational dynamics, we also examined the changes (Fig. 3) in the EPR spectra of bPTO and bPyTO, in the presence of a large excess of 2,6-di-O-methyl-β-cyclodextrin (DM-β-CD) and CB[7] respectively.
In both cases, the observed decrease of aN and the broadening of the high field line are in agreement with the formation of an inclusion complex (ΔaN = 0.07 mT for DM-β-CD@bPTO, and ΔaN = 0.1 mT for CB[7]@bPyTO). Spectral calculations using the first type of fit indicated again the absence of dynamic cohesion. Interestingly, in the absence of either the CB[7] carbonyl groups for DM-β-CD@bPTO or the ammonium ions for CB[7]@bPyTO (Fig. 3b), calculations predicted a slowing down (Table S1 and Fig. S23†) of the rotational dynamics after complexation.
We want to point out that the actual CB[7]@bPTO motion may be more complex, and the relative line intensities in the fast motion regime may not provide enough information for fully characterizing it. However, both types of EPR fits indicate a speed-up of rotational motion around the y axis, and we believe that the values from the first type of fit are more realistic than the extreme speed-up found with the second type of fit.
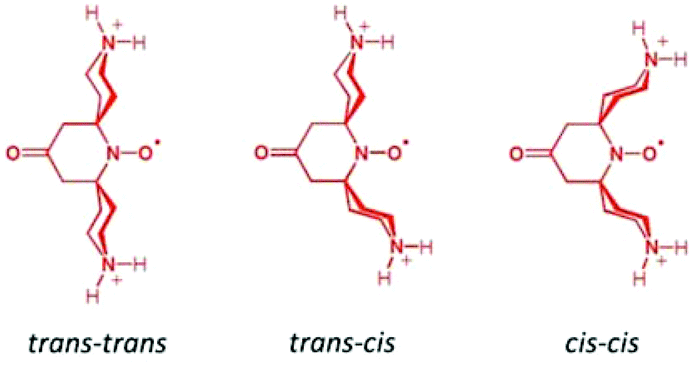 | ||
| Scheme 2 Three favored conformers of bPTO which are very close in energy (within 0.3 kcal mol−1). | ||
As shown in Fig. 4, the N–O˙ group is strongly shielded, positioned near the geometric center of the macrocycle. For CB[7]@bPTO, the distances between the CB[7] geometric center and the two ammonium nitrogens are 4.33 and 4.15 Å. The trans–trans conformer (Fig. 4a) corresponds to the major conformer, the two others being at least 6 kcal mol−1 higher in energy (see the ESI†). For this major conformer, the bPTO moiety is tightly bound, and the axis connecting the two nitrogen atoms of the piperidinium rings is colinear with the CB[7] C7 axis. For CB[8]@bPTO, all three conformers have closer energies (within 2.3 kcal mol−1), reflecting higher degrees of freedom inside the larger macrocycle, and the axis connecting the two atoms of the piperidinium rings is tilted up to ≈24° from that of the CB[8] C8 axis (ESI†).
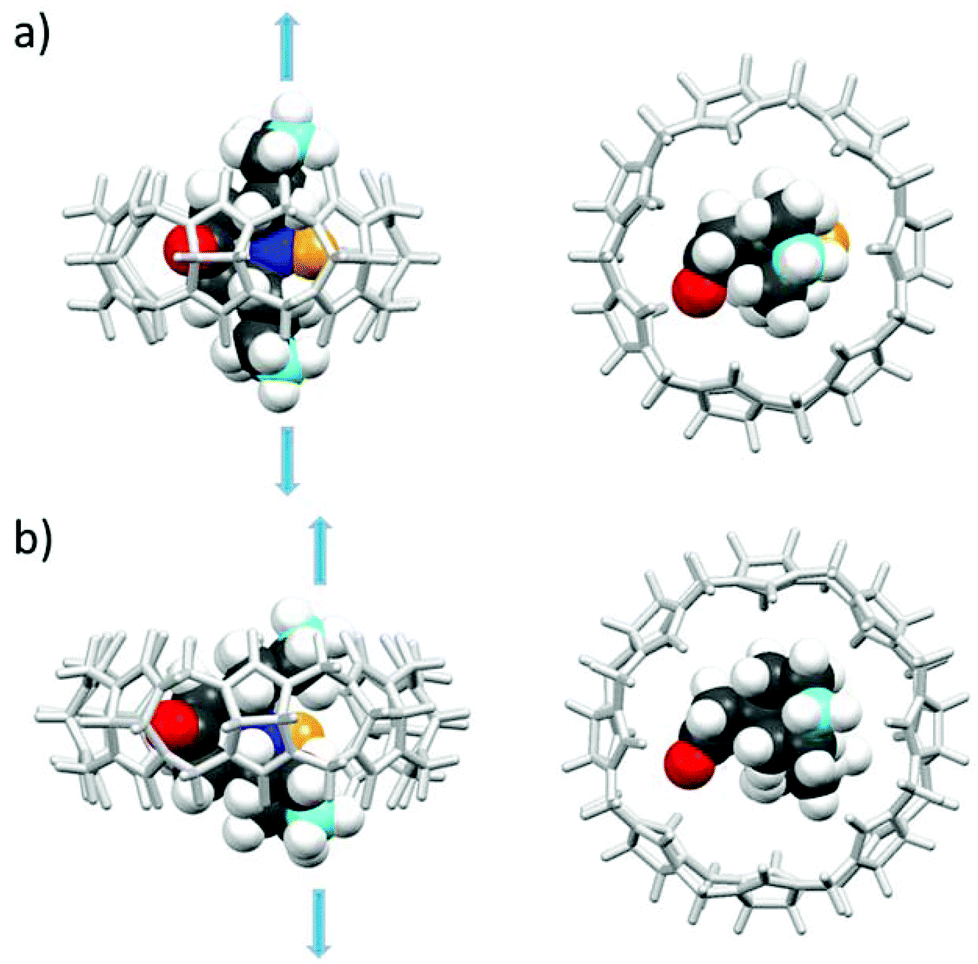 | ||
| Fig. 4 Side and top views of the inclusion complex of bPTO in CB[7] (a, trans–trans conformer) and in CB[8] (b, cis–cis conformer) as found after DFT minimization (lowest energy structures) with water continuum (for other conformers, see the ESI;† the colours of the N and O atoms of the N–O˙ group are dark blue and yellow respectively). | ||
 | ||
| Fig. 5 (a) Distribution of distances between the guest atom H20 and the host atom O1 (b) for a 100 ns trajectory in water. (c) Distance between the geometric center of CB[7] and the nitrogen atom carrying H20. (d) Vectors V1 and V2 defined as collinear with respect to the N–O˙ bond and a C–H bond of the cucurbituril. (e) Distribution of θ values over two 100 ns trajectories, starting from θ = 0° and θ = 180° respectively. | ||
There are few studies reporting guest rotational dynamics in molecular containers.63–67 Because guests were reported to have slower dynamics when included in cucurbiturils,45,55,68 the present acceleration of guest rotation upon binding was unexpected and represents an alternative solution to the oligoketone guest proposed by Keinan69 for a “lowered-friction” molecular rotary motor. We think that the present jumping model, where the hydrogen bonding ammonium function moves almost freely by increments of nearly 50°, is due to preorganization of the CB[7] carbonyl crown where the ketone oxygen atoms are ready to hydrogen bond (on both sides of the CB) thus lowering the barrier to jump from one ketone to the next. In this view, the multiply hydrogen bonded network of bulk water (solvent shell) certainly plays a role because the two ammoniums of bPTO are less solvated when included in CB[7]. Such a solvent vs. preorganized macrocycle effect has already been reported for related systems70,71 such as in lubricated molecular shuttles,72 ring rotations within catenanes73 or in simple N-arylimide molecules.74 Still the present results will prove to be useful in the design of advanced CB[n] based molecular machines75–78 like supramolecular gyroscopes79–83 and molecular ball-bearings.84 More generally, the present guest design offers new perspectives for any application requiring fast-spinning wheels where cucurbiturils can be used, and also critical spinning information can be obtained from the free radical labelled guest and EPR spectroscopy.
Conclusion
Reduction of nitroxides is recognized to be one of the main limitations for their use in biology. We have shown that sequestration with high affinity in CB[7] or CB[8] of suitably-designed nitroxides can dramatically improve their resistance to reduction (lifetime of several hours with minor decay in the presence of 20 fold excess of ascorbate). Our results could open new perspectives to the use of nitroxides in biological milieu. Additionally, our results highlight the advantages of cucurbiturils as stators offering restricted friction for optimized rotational motion in tailored molecular rotors. Such non-covalent molecular rotors can open new avenues toward nanoscale molecular machines on which one could exert control over the rotator for fast spinning movements such as nanopulleys and all nanoscale machines where pulleys are involved or for the construction of small motor vehicle chassis such as nanomotorcycles or nanocars. In particular, the present study highlights the great potential of cucurbiturils for the construction of CB-wheeled nanocars. Work along this line is in progress and will be reported in due course.Acknowledgements
The CNRS, Aix-Marseille Université, Renard RPE TGE and Région PACA (project “Masked Spins”) are acknowledged for financial and technical support. We also thank Laszlo Jicsinszky (Cyclolab), Sébastien Combes and the CRCMM, ‘Centre Régional de Compétences en Modélisation Moléculaire de Marseille’ for computing facilities.Notes and references
- G. Vives and J. M. Tour, Acc. Chem. Res., 2009, 42, 473–487 CrossRef CAS PubMed.
- C. Joachim and G. Rapenne, ACS Nano, 2013, 7, 11–14 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Macia, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- L. Cao, M. Sekutor, P. Y. Zavalij, K. Mlinaric-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC.
- D. Bardelang, M. Hardy, O. Ouari and P. Tordo, Spin Labels and Spin Probes, in Handbook of Free Radical Chemistry and Biology, John Wiley & sons, 2012, vol. 4, pp. 1965–2015 Search PubMed.
- S. Yi, B. Captain and A. E. Kaifer, Chem. Commun., 2011, 47, 5500–5502 RSC.
- M. Porel, S. Jockusch, M. F. Ottaviani, N. J. Turro and V. Ramamurthy, Langmuir, 2011, 27, 10548–10555 CrossRef CAS PubMed.
- E. Mileo, C. Casati, P. Franchi, E. Mezzina and M. Lucarini, Org. Biomol. Chem., 2011, 9, 2920–2924 CAS.
- M. Spulber, S. Schlick and F. A. Villamena, J. Phys. Chem. A, 2012, 116, 8475–8483 CrossRef CAS PubMed.
- I. Kirilyuk, D. Polovyanenko, S. Semenov, I. Grigorev, O. Gerasko, V. Fedin and E. Bagryanskaya, J. Phys. Chem. B, 2010, 114, 1719–1728 CrossRef CAS PubMed.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2007, 13, 7223–7233 CrossRef CAS PubMed.
- S. Yi, B. Captain, M. F. Ottaviani and A. E. Kaifer, Langmuir, 2011, 27, 5624–5632 CrossRef CAS PubMed.
- D. Bardelang, G. Casano, F. Poulhès, H. Karoui, J. Filippini, A. Rockenbauer, R. Rosas, V. Monnier, D. Siri, A. Gaudel-Siri, O. Ouari and P. Tordo, J. Am. Chem. Soc., 2014, 136, 17570–17577 CrossRef CAS PubMed.
- D. Bardelang, K. Banaszak, H. Karoui, A. Rockenbauer, M. Waite, K. Udachin, J. A. Ripmeester, C. I. Ratcliffe, O. Ouari and P. Tordo, J. Am. Chem. Soc., 2009, 131, 5402–5404 CrossRef CAS PubMed.
- E. Mileo, E. Mezzina, F. Grepioni, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2009, 15, 7859–7862 CrossRef CAS PubMed.
- N. Jayaraj, M. Porel, M. F. Ottaviani, M. V. S. N. Maddipatla, A. Modelli, J. P. Da Silva, B. R. Bhogala, B. Captain, S. Jockusch, N. J. Turro and V. Ramamurthy, Langmuir, 2009, 25, 13820–13832 CrossRef CAS PubMed.
- L. J. Berliner, Spin Labeling: A Modern Perspective, in Stable Radicals, John Wiley & sons, 2010, pp. 521–534 Search PubMed.
- V. V. Khramtsov and J. L. Zweier, Functional in vivo EPR Spectroscopy and Imaging Using Nitroxide and Trityl Radicals, in Stable Radicals, John Wiley & sons, 2010, pp. 537–563 Search PubMed.
- M. A. Sowers, J. R. McCombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 5460 CrossRef PubMed.
- A. Rajca, Y. Wang, M. Boska, J. T. Paletta, A. Olankitwanit, M. A. Swanson, D. G. Mitchell, S. S. Eaton, G. R. Eaton and S. Rajca, J. Am. Chem. Soc., 2012, 134, 15724–15727 CrossRef CAS PubMed.
- Y. Wang, J. T. Paletta, K. Berg, E. Reinhart, S. Rajca and A. Rajca, Org. Lett., 2014, 16, 5298–5300 CrossRef CAS PubMed.
- A. A. Bobko, I. A. Kirilyuk, I. A. Grigorev, J. L. Zweier and V. V. Khramtsov, Free Radicals Biol. Med., 2007, 42, 404–412 CrossRef CAS PubMed.
- K. J. Liu, M. W. Grinstaff, J. Jiang, K. S. Suslick, H. M. Swartz and W. Wang, Biophys. J., 1994, 67, 896–901 CrossRef CAS.
- Y. Y. Woldman, S. V. Semenov, A. A. Bobko, I. A. Kirilyuk, J. F. Polienko, M. A. Voinov, E. G. Bagryanskaya and V. V. Khramtsov, Analyst, 2009, 134, 904–910 RSC.
- J. T. Paletta, M. Pink, B. Foley, S. Rajca and A. Rajca, Org. Lett., 2012, 14, 5322–5325 CrossRef CAS PubMed.
- I. A. Kirilyuk, Y. F. Polienko, O. A. Krumkacheva, R. K. Strizhakov, Y. V. Gatilov, I. A. Grigorev and E. G. Bagryanskaya, J. Org. Chem., 2012, 77, 8016–8027 CrossRef CAS PubMed.
- A. A. Bobko, A. Ivanov and V. V. Khramtsov, Free Radical Res., 2013, 47, 74–81 CrossRef CAS PubMed.
- P. Franchi, M. Lucarini, E. Mezzina and G. F. Pedulli, J. Am. Chem. Soc., 2004, 126, 4343–4354 CrossRef CAS PubMed.
- G. Ionita, A. Caragheorgheopol, H. Caldararu, L. Jones and V. Chechik, Org. Biomol. Chem., 2009, 7, 598–602 CAS.
- G. Ionita, V. Meltzer, E. Pincu and V. Chechik, Org. Biomol. Chem., 2007, 5, 1910–1914 CAS.
- J. Martinie, J. Michon and A. Rassat, J. Am. Chem. Soc., 1975, 97, 1818–1823 CrossRef CAS.
- Y. Kotake and E. G. Janzen, J. Am. Chem. Soc., 1989, 111, 5138–5140 CrossRef CAS.
- H. Karoui, A. Rockenbauer, S. Pietri and P. Tordo, Chem. Commun., 2002, 3030–3031 RSC.
- D. Bardelang, A. Rockenbauer, H. Karoui, J. P. Finet, I. Biskupska, K. Banaszak and P. Tordo, Org. Biomol. Chem., 2006, 4, 2874–2882 CAS.
- D. Bardelang, L. Charles, J. P. Finet, L. Jicsinszky, H. Karoui, S. R. A. Marque, V. Monnier, A. Rockenbauer, R. Rosas and P. Tordo, Chem. – Eur. J., 2007, 13, 9344–9354 CrossRef CAS PubMed.
- M. Hardy, D. Bardelang, H. Karoui, A. Rockenbauer, J.-P. Finet, L. Jicsinszky, R. Rosas, O. Ouari and P. Tordo, Chem. – Eur. J., 2009, 15, 11114–11118 CrossRef CAS PubMed.
- P. Franchi, M. Fani, E. Mezzina and M. Lucarini, Org. Lett., 2008, 10, 1901–1904 CrossRef CAS PubMed.
- M. Okazaki and K. Kuwata, J. Phys. Chem., 1985, 89, 4437–4440 CrossRef CAS.
- C. Ebel, K. U. Ingold, J. Michon and A. Rassat, New J. Chem., 1985, 9, 479–485 CAS.
- C. Ebel, K. U. Ingold, J. Michon and A. Rassat, Tetrahedron Lett., 1985, 26, 741–744 CrossRef CAS.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737–20742 CrossRef CAS PubMed.
- L. Cao, M. Sekutor, P. Y. Zavalij, K. Mlinaric-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed.
- S. R. Burks, M. A. Makowsky, Z. A. Yaffe, C. Hoggle, P. Tsai, S. Muralidharan, M. K. Bowman, J. P. Y. Kao and G. M. Rosen, J. Org. Chem., 2010, 75, 4737–4741 CrossRef CAS PubMed.
- A. Rockenbauer, T. Szabo-Planka, S. Arkosi and L. Korecz, J. Am. Chem. Soc., 2001, 123, 7646–7654 CrossRef CAS PubMed.
- G. I. Roshchupkina, A. A. Bobko, A. Bratasz, V. A. Reznikov, P. Kuppusamy and V. V. Khramtsov, Free Radicals Biol. Med., 2008, 45, 312–320 CrossRef CAS PubMed.
- D. Kivelson, J. Chem. Phys., 1960, 33, 1094–1106 CrossRef CAS PubMed.
- A. K. Vorobiev, V. S. Gurman and T. A. Klimenko, Phys. Chem. Chem. Phys., 2000, 2, 379–385 RSC.
- M. G. Santangelo, M. Levantino, A. Cupane and G. Jeschke, J. Phys. Chem. B, 2008, 112, 15546–15553 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1989, 111, 2697–2699 CrossRef CAS.
- J. Heo, S.-Y. Kim, D. Whang and K. Kim, Angew. Chem., Int. Ed., 1999, 38, 641–643 CrossRef CAS.
- S. Lim, H. Kim, N. Selvapalam, K.-J. Kim, S. J. Cho, G. Seo and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 3352–3355 CrossRef CAS PubMed.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed.
- P. Thuéry, Cryst. Growth Des., 2008, 8, 4132–4143 Search PubMed.
- D. Bardelang, K. A. Udachin, D. M. Leek, J. Margeson, G. Chan, C. I. Ratcliffe and J. A. Ripmeester, Cryst. Growth Des., 2011, 11, 5598–5614 CAS.
- D. Bardelang, K. A. Udachin, R. Anedda, I. Moudrakovski, D. M. Leek, J. A. Ripmeester and C. I. Ratcliffe, Chem. Commun., 2008, 4927–4929 RSC.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- A. Scarso, H. Onagi and J. Rebek Jr., J. Am. Chem. Soc., 2004, 126, 12728–12729 CrossRef CAS PubMed.
- P. D. Kirchhoff, J.-P. Dutasta, A. Collet and J. A. McCammon, J. Am. Chem. Soc., 1999, 121, 381–390 CrossRef CAS.
- J. S. Mugridge, G. Szigethy, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 16256–16264 CrossRef CAS PubMed.
- R. Kulasekharan, N. Jayaraj, M. Porel, R. Choudhury, A. K. Sundaresan, A. Parthasarathy, M. F. Ottaviani, S. Jockusch, N. J. Turro and V. Ramamurthy, Langmuir, 2010, 26, 6943–6953 CrossRef CAS PubMed.
- H. Kitagawa, Y. Kobori, M. Yamanaka, K. Yoza and K. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10444–10448 CrossRef CAS PubMed.
- D. Banik, J. Kuchlyan, A. Roy, N. Kundu and N. Sarkar, J. Phys. Chem. B, 2015, 119, 2310–2322 CrossRef CAS PubMed.
- I. B. Shir, S. Sasmal, T. Mejuch, M. K. Sinha, M. Kapon and E. Keinan, J. Org. Chem., 2008, 73, 8772–8779 CrossRef PubMed.
- F. G. Gatti, S. León, J. K. Y. Wong, G. Bottari, A. Altieri, M. Angeles Farran Morales, S. J. Teat, C. Frochot, D. A. Leigh, A. M. Brouwer and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10–14 CrossRef CAS PubMed.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS PubMed.
- M. R. Panman, B. H. Bakker, D. den Uyl, E. R. Kay, D. A. Leigh, W. J. Buma, A. M. Brouwer, J. A. J. Geenevasen and S. Woutersen, Nat. Chem., 2013, 5, 929–934 CrossRef CAS PubMed.
- D. A. Leigh, A. Murphy, J. P. Smart, M. S. Deleuze and F. Zerbetto, J. Am. Chem. Soc., 1998, 120, 6458–6467 CrossRef CAS.
- B. E. Dial, P. J. Pellechia, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2012, 134, 3675–3678 CrossRef CAS PubMed.
- Y. H. Ko, I. Hwang, H. Kim, Y. Kim and K. Kim, Chem. – Asian J., 2015, 10, 154–159 CrossRef CAS PubMed.
- W. S. Jeon, E. Kim, Y. H. Ko, I. Hwang, J. W. Lee, S.-Y. Kim, H.-J. Kim and K. Kim, Angew. Chem., Int. Ed., 2004, 44, 87–91 CrossRef PubMed.
- S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 12912–12914 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275–277 CrossRef CAS.
- R. Pievo, C. Casati, P. Franchi, E. Mezzina, M. Bennati and M. Lucarini, ChemPhysChem, 2012, 13, 2659–2661 CrossRef CAS PubMed.
- E. Mezzina, M. Fani, F. Ferroni, P. Franchi, M. Menna and M. Lucarini, J. Org. Chem., 2006, 71, 3773–3777 CrossRef CAS PubMed.
- K. Skopek, M. C. Hershberger and J. A. Gladysz, Coord. Chem. Rev., 2007, 251, 1723–1733 CrossRef CAS PubMed.
- C. A. Schalley, Angew. Chem., Int. Ed., 2002, 41, 1513–1515 CrossRef CAS.
- R. J. Blanch, A. J. Sleeman, T. J. White, A. P. Arnold and A. I. Day, Nano Lett., 2002, 2, 147–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, syntheses, DFT and experimental and calculated EPR spectra. See DOI: 10.1039/c5nr03288a |
| This journal is © The Royal Society of Chemistry 2015 |