
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D assembly of silica encapsulated semiconductor nanocrystals†
Christin
Rengers
a,
Sergei V.
Voitekhovich
b,
Susann
Kittler
a,
André
Wolf
a,
Marion
Adam
c,
Nikolai
Gaponik
*a,
Stefan
Kaskel
c and
Alexander
Eychmüller
a
aPhysical Chemistry, TU Dresden, Bergstr. 66b, 01062, Germany. E-mail: nikolai.gaponik@chemie.tu-dresden.de
bResearch Institute for Physical Chemical Problems, Belarusian State University, Leningradskaya Str. 14, 220030 Minsk, Belarus
cInorganic Chemistry I, TU Dresden, Bergstr. 66, 01062, Germany
First published on 19th June 2015
Abstract
Non-ordered porous networks, so-called aerogels, can be achieved by the 3D assembly of quantum dots (QDs). These materials are well suited for photonic applications, however a certain quenching of the photoluminescence (PL) intensity is observed in these structures. This PL quenching is mainly attributed to the energy transfer mechanisms that result from the close contact of the nanoparticles in the network. Here, we demonstrate the formation of a novel aerogel material with non-quenching PL behaviour by non-classical, reversible gel formation from tetrazole capped silica encapsulated QDs. Monitoring of the gelation/degelation by optical spectroscopy showed that the optical properties of the nanocrystals could be preserved in the 3D network since no spectral shifts and lifetime shortening, which can be attributed to the coupling between QDs, are observed in the gels as compared to the original colloidal solutions. In comparison with other QD-silica monoliths, QDs in our gels are homogeneously distributed with a distinct and controllable distance. In addition we show that the silica shell is porous and allows metal ions to pass through the shell and interact with the QD core causing detectable changes of the emission properties. We further show the applicability of this gelation method to other QD materials which sets the stage for facile preparation of a variety of mixed gel structures.
Introduction
Non-ordered porous networks, so-called aerogels, are fine inorganic superstructures with an enormously high porosity and inner surface area, resulting in densities as low as 3 times that of air.1 These materials offer a wide variety of applications, e.g. in the field of catalysis,1 due to their attractive catalytic, thermo resistant, piezoelectric, antiseptic, and many other properties, originating from the combination of the specific properties of nanoparticles by macroscale self-assembly.In the early 1930s, the first synthesis of aerogels made from metal oxides was reported by Kistler2 and was further developed starting from the late 1960s.1,3 So far, the most investigated aerogel materials are silica or metal oxides as well as their mixtures. But recently, creating aerogels from noble metal nanoparticles, colloidal quantum dots (QDs) or mixtures of both has attracted interest, since these materials may open opportunities in areas such as semiconductor technology, photocatalysis, optoelectronics, and photonics.4–12 In 2005, Brock et al. first reported on CdS and CdSe nanocrystal aerogels.13 In order to perform gel formation, they had to transfer the non-polar organic stabilized QDs into polar organic or aqueous media. Later in 2008, our group developed a photochemical treatment system to create aerogels from thiol-capped CdTe nanocrystals, directly synthesized in water12,14 and a reversible gelation procedure for tetrazole-capped CdTe nanocrystals in 2010.15 Although the typical emission properties of the initial quantum confined nanocrystal building blocks could be preserved, usually in these structures a certain quenching of the photoluminescence (PL) intensity is observed compared to the colloidal solutions of the nanoparticles. This PL quenching is mainly attributed to the energy transfer mechanisms that result from the close contact among the nanoparticles in the network. Introducing a spacer into the network and thus increasing the distance between the nanoparticles to a certain value may be a possibility to avoid this quenching behaviour.5,15 For this purpose, applying a dielectric shell of several nm thickness on fluorescent QDs, consisting of silica or a polymer (e.g. polystyrene), might be an option to overcome this problem.
Thus far, silica coating of inorganic nanocrystals has been under investigation16–21 and the preparation methods can be classified into two approaches, i.e. the Stöber19,22 method and the reverse microemulsion technique.23–25 Both approaches usually cause a drop in the PL of the QDs. Only recently, modifications of the Stöber method allowing the retention or even an enhancement of the PL were reported.26,27 However, aiming for shell thicknesses of only several nanometers, it seems to be more promising to use the reverse microemulsion approach since it allows for better control of the particle size distribution for small sizes (from 20 nm to 150 nm) in comparison with the Stöber method.28,29
So far, in order to produce assembled structures, QDs were incorporated into a silica matrix as a film or a monolithic item. Thereby, QD composites were derived by surface exchange using organofunctional alkoxysilanes followed by a sol–gel process.26–31
In this article, we show the formation of hydrogels and aerogels from silica encapsulated QDs, following a procedure reported earlier by our group.15 In comparison with other QD-silica monoliths, our gel is not derived via a sol–gel process but through the interconnection of silica-QD nanoparticles leading to a homogeneous distribution of the QDs in the whole structure with a distinct and controllable distance to each other. Therefore, the silica coated QDs were functionalized with a tetrazole ligand specially designed to bind to the silica surface that allows for a controlled gel formation by interconnection of the negatively charged tetrazole rings through complexation with metal ions. This process is reversible and dissolution of the hydrogel can be achieved by the addition of a stronger complexant, e.g. EDTA, which removes the metal ions from the network. Furthermore, we show monitoring of the gelation/degelation by optical spectroscopy, i.e. absorption and PL spectroscopy as well as PL lifetime and PL quantum yield (QY) measurement. These studies show that we were able to create a novel aerogel material with non-quenching PL behaviour, since no spectral shifts and lifetime shortening, which can be attributed to the coupling between QDs, are observed in gels as compared to the original colloidal solutions. Furthermore we show that the silica shell is not densely packed but rather porous and allows metal ions to pass through the shell and interact with the QD core.
Experimental
Chemicals and apparatus
All chemicals used were of analytical grade or of the highest purity available. All aqueous solutions were prepared from Milli-Q water (Millipore).Synthesis of tetrazole capped silica encapsulated QDs
Silica encapsulation procedure was adopted from the literature.32 In detail, in a 500 mL round bottom flask, 2.4 mL of IGEPAL® CO-520 were added to 40 mL of anhydrous cyclohexane, followed by vigorous stirring for 15 min. Meanwhile, organic capped QDs (2 × 10−9 mol) were precipitated by addition of 1 mL of acetone and centrifugation at 4000 RPM for 3 min. The precipitate was dissolved in 1 mL of cyclohexane and then added to the stirred IGEPAL solution. After 15 min of stirring, TEOS was added. In order to obtain 20 nm sized silica particles, 15 μL of TEOS were used. Higher amounts of TEOS resulted in larger particles. Finally, after 30 min of stirring, 280 μL of ammonium hydroxide (28% in water) were slowly added and the mixture was stirred for 48 h at RT in a sealed flask. Purification was done by mixing with 50 mL of ethanol and centrifuging at 5100 RPM for 30 min at 10 °C. The supernatant was discarded and the process was repeated twice using pure ethanol. The particles were then redispersed in 1 mL of 4 mM NaOH aqueous solution. The solution was sonicated for 1 min and centrifuged at 2500 RPM for 3 min in order to remove any aggregates. The supernatant containing QDs encapsulated by silica shells was stored at 4 °C. Tetrazole capping was performed as follows. In 200 μL of aqueous silica-QD solution a spatula tip of solid (MeO)3Si–(CH2)3–NH–CH2–CN4H was dissolved. Under these conditions, the ligand will immediately grow onto the silica surface resulting in flocculation. The addition of a few drops of 1 M NaOH led to a clear, stable solution, since tetrazole rings were deprotonated which allows charge stabilization of the silica particles.Preparation of hydrogels from tetrazole capped silica encapsulated QDs and their degelation
Hydrogels were obtained by the stepwise addition of Zn(OAc)2 aqueous solution (10−2 M) to the tetrazole capped silica-QD colloidal solution up to complete gelation of the nanoparticles. The hydrogels were redissolved back into their colloidal form by the addition of an aliquot of 10−2 M ethylenediaminetetraacetic acid (EDTA) aqueous solution (adjusted to pH 12 by the addition of 1 M NaOH) equal to the Zn2+ concentration in the gelated sample. The gelation process was monitored directly in a quartz cuvette with a light path of 10 mm by the stepwise addition of Zn(OAc)2 solution into the diluted silica-QD colloid and subsequently measurements of the absorbance, PL and PL lifetime were obtained.Preparation of aerogels from hydrogels
A critical point drier (13200J-AB from Spi Supplies) was used for supercritical CO2 drying to prevent the fine nanostructures from collapsing and to obtain self-supporting aerogel monoliths. This drying technique has been described previously.12 In brief, acetone was added in small portions to the hydrogel and the resulting water/acetone mixture was removed a few times to ensure that most of the water was exchanged with acetone. Subsequently, the sample was placed in a vacuum desiccator with 100% acetone containing some anhydrous CaCl2. The desiccator was evacuated until the acetone boiled, was sealed under low pressure, and left under these conditions for ca. 20 h. The procedure provides a very efficient but yet gentle exchange of the remaining water with acetone.Characterization
UV/Vis absorption spectra were obtained using a Cary 50 spectrophotometer (Varian Inc.) and a Cary 5000 spectrophotometer (Varian Inc.) with an integrating sphere. PL measurements were recorded using a FluoroMax-4 spectrofluorimeter (HORIBA Jobin Yvon Inc.). PL life time measurements were recorded on a Fluorolog-3 spectrofluorimeter (HORIBA Jobin Yvon Inc.) equipped with a 200 ps pulsed LED diode emitting at 403 nm and a TCSPC module. All measurements were performed at room temperature. PL QY of colloidal solutions was determined in reference to rhodamine 6G and rhodamine 101. For scattering samples, absolute PL QY were recorded using a Fluorolog-3 spectrofluorimeter (HORIBA Jobin Yvon Inc.) equipped with a Quanta-ϕ integrating sphere.Transmission electron microscopy imaging was done on a LIBRA microscope operating at 200 kV (Carl Zeiss AG). The samples were prepared beforehand by dropping diluted nanoparticle solutions onto a carbon coated copper grid and solvent evaporation. In the case of hydrogels, the sample was sonicated a few seconds before dropcasting.
Scanning electron microscopy (SEM) was performed by using a Zeiss DSM 982 Gemini instrument. The samples for SEM characterisation were prepared by dropping diluted aerogel dispersions in acetone onto silicon slides and subsequent solvent evaporation.
Nitrogen and carbon dioxide physisorption measurements were performed on an Autosorp 1C (Quantachrome Instruments) using nitrogen (99.999%) and carbon dioxide (99.9995%). Specific surface area (SSA) was calculated using the equation from Brunauer, Emmett and Teller (BET) in the relative pressure range of 0.05–0.20 p/p0, and total pore volume was determined at 0.99 p/p0. Nonlocal density functional theory (NLDFT) was used to obtain the pore size distribution. Prior to gas physisorption experiments, the sample was activated at 298 K for 48 h under vacuum.
Results and discussion
The gelation mechanism shown here is based on previous results of our group as reported by Lesnyak et al. in 2010.15 Using this technique, gels are formed by metal ion assisted complexation of tetrazole capped nanoparticles. This is a fast, controllable and reversible method to subsequently fabricate 3D networks. The difference here in comparison with the former work, lies in the usage of silica encapsulated QDs instead of bare QDs. The silica shell acts as a spacer between the fluorescent nanocrystals in the three-dimensional network and thus prevents them from quenching, which has been observed before when no spacer was present. Additionally, it provides an option to form hydrogels and aerogels from a broad spectrum of QD materials since our newly developed tetrazole ligand can be applied to any silica coated QD material. The schematic pathway of the 3D assembly of silica encapsulated QDs is shown in Fig. 1. In the first step, the QDs are coated with a dielectric shell, in this case silica. Following this, non-classical gel formation is performed by surface capping with tetrazoles and subsequently metal ion addition.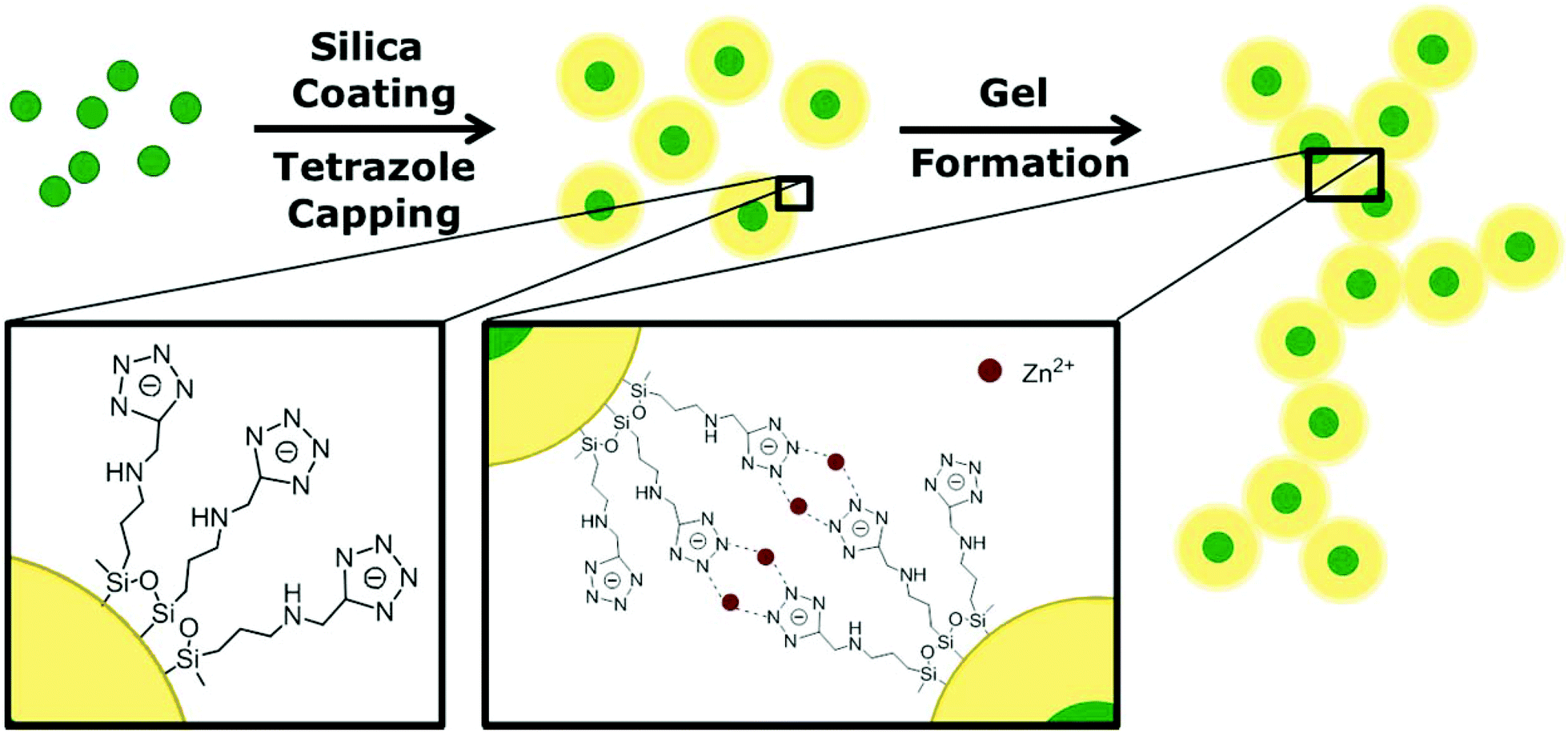 | ||
| Fig. 1 Schematic pathway of luminescent silica gel formation. In the first step the synthesized CdSe/CdS core/shell QDs are covered with a silica shell using a reversed microemulsion technique. After coating with a specially designed tetrazole ligand, the hydrogel is formed by metal ion assisted complexation in the second step. Subsequently, the aerogel can be achieved by supercritical point drying. | ||
The QDs were encapsulated with silica using a reversed microemulsion technique.32 Details are described in the Experimental section. Here we used CdSe/CdS core/shell nanocrystals as a model system to show gel formation and characterization, but we also proved the applicability of other QD materials as will be explained later. The PL QY of the employed QDs was in the range of 30–40%, whereas the PL QY of silica coated QDs was typically between 15 and 20%. PL QY data for a representative sample are given in Table 1.
| Sample | PL QY [%] |
|---|---|
| CdSe/CdS | 31.5 |
| CdSe/CdS@silica | 14.1 |
| CdSe/CdS@silica hydrogel | 16.8 |
The silica shell thickness was varied from 10 to 20 nm in order to efficiently disable coupling between the QDs while preserving a high PL intensity. More information on the influence of the silica shell on the PL of the QDs can be found elsewhere.33,34 An example of silica nanospheres with a CdSe/CdS nanocrystal core and an average diameter of 30 nm is depicted in the TEM image in Fig. 2A. According to the image shown, not all silica particles contain QDs. The appearance of silica spheres without QD core is common for most of the reported silanisation procedures. In our case, based on the analysis of several representative TEM images it did not exceed 5%. After silica coating, the spheres were functionalized with a specially designed tetrazole ligand, 5-(3-(trimethoxysilyl)propylaminomethyl)tetrazole which has the ability to couple to the silica surface via a silane group. Under basic conditions the tetrazole moieties attached to the surface of the silica spheres undergo deprotonation generating negatively charged tetrazolate anions. The latter displays a high nucleophilicity and a good coordination potential towards transition metals.35,36
 | ||
| Fig. 2 (A) TEM image of ∼30 nm sized silica nanoparticles with CdSe/CdS QD core. (B) TEM image of silica hydrogel formed out of 20 nm sized silica nanoparticles. After supercritical point drying an aerogel is obtained. (C) SEM image; (D) and (E) are photographs of the solid aerogel under daylight and UV light. | ||
In particular, tetrazolato ligands are known to form stable complexes with Zn2+ cations in water. In these complexes, the heteroring exhibits bridging modes through several N atoms coordinated to the metal cations which predetermines the networking of nanoparticles by the complexes formed.37,38 This gives an opportunity to interconnect the particles with the aid of metal ions leading to a 3D structure.39 This non-classical gelation is a controllable and reversible method and has not been applied for silica gels so far. The resulting hydrogel is a very porous non-ordered network as can be seen in the TEM image (Fig. 2B). The network is built up from silica nanospheres which have aligned during the gelation and have formed a wire-like 3D assembly. Due to the silica shell, the QDs are well separated from each other in the resulting gel, which has a positive effect on the emission properties as will be discussed below.
In order to obtain a solid material, the hydrogel can be converted into an aerogel by supercritical CO2 drying. This is a very gentle method to exchange the solvent in the fine pores of the 3D network with air, while maintaining the porous structure and avoiding shrinking of the gel and thus fissures in the material as much as possible. The SEM image in Fig. 2C shows the characteristic sponge-like structure of a silica-QD aerogel produced by this drying procedure. The porous gel structure is preserved during the washing and solvent exchange procedure and the aerogel structure is also robust against moisture due to the high stability of the tetrazole–metal ion complex.
The resulting silica-QD aerogel exhibits the typical properties of a silica aerogel, e.g. white transparent colour, and those of a QD aerogel. This can be easily seen in the photographs in Fig. 2D and E. Under normal daylight, the aerogel seems to appear as a plain silica gel, still slightly displaying the absorption colour of the QDs, whereas the bright emission colour of the QDs becomes visible when the illumination is changed to UV light.
For the comparison of the silica-QD gel with the pure QD gels, the optical properties of the silica encapsulated QDs have been monitored during the gel formation process by absorption and fluorescence spectroscopy as well as fluorescence lifetime measurements (see Fig. 3). Lesnyak et al.15 studied the development of absorption and fluorescence spectra during the gelation and degelation of tetrazole capped CdTe nanocrystals. They reported an increase of absorbance upon gel formation due to increased scattering. The same trend can be observed for the silica-QD gel (see Fig. 3A), meaning that only diffuse reflectance occurs but the absorption properties remain unchanged (see absorption spectrum in Fig. S1 in the ESI,† recorded using an integrating sphere setup). Furthermore, they described a decrease of PL intensity accompanied by a redshift of the PL maximum and a much faster decay of the PL in the hydrogel due to energy transfer mechanisms. At this point, regarding the PL development in Fig. 3B the advantage of using a spacer to create a defined distance between the nanoparticles becomes very clear. In hydrogels formed from silica encapsulated QDs, the optical properties of the nanoparticles are not changed in comparison with the colloidal solution. The absorption remains unchanged and the PL spectrum does not display a shift of the emission maximum and the PL decay does not change from the colloidal silica-QD solution to the hydrogel, meaning that the applied silica coating successfully avoids energy transfers. Additionally, we observe an apparent increase in the PL intensity upon gel formation. However, this increase is not confirmed by PL QY measurements as can be seen from Table 1. This observation is a result solely of the increased emitter concentration induced by the volume contraction during colloid-to-gel transition.
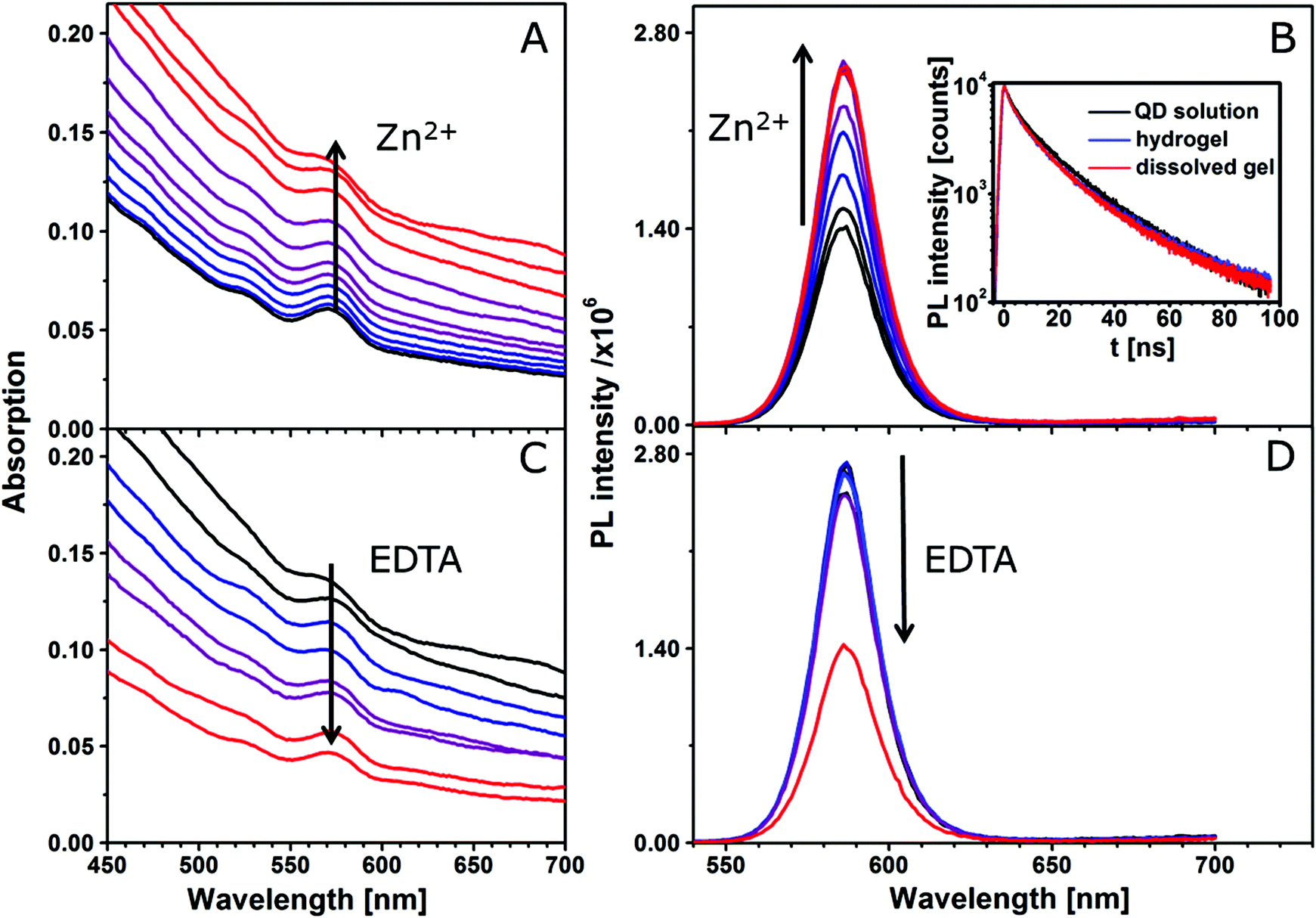 | ||
| Fig. 3 Reversible gel formation monitored by absorption and fluorescence spectroscopy. (A) Absorption spectra during gel formation. The stepwise addition of Zn2+ ions to the tetrazole capped silica-QDs leads to successive gel formation indicated by the steadily increasing absorption caused by scattering. The corresponding fluorescence spectra (B) meanwhile show an increase of the PL intensity. Since the position of the 1st absorption and the emission maximum do not change during gel formation, it is suggested that the nanocrystals are well separated because of the silica shell and thus do not interact with each other. This also corresponds to the PL decays, inset in (B), where no difference between the QD solution and the hydrogel is observed. (C) and (D) show the absorption and fluorescence spectra during the dissolution of the gel by stepwise addition of EDTA solution. The opposite progress of these spectra compared to (A) and (B) shows the reversibility of this process. | ||
The reversibility of this gelation process was demonstrated by the addition of a strong complexing agent, i.e. EDTA, which removes the metal ions from the network and thus initiates degelation. The addition of an equimolar amount of EDTA with respect to the amount of Zn2+ leads to complete dissolution of the gel as can be seen from Fig. 3C as decreased scattering. The resulting clear, colloidal solution can restore 100% of the PL intensity of the initial solution.
Monitoring the gelation of different batches of silica-QDs analogous to the experiment, as shown in Fig. 3, we observed a different behaviour from that explained above when particle batches of lower QY were used. For the gelation of the low QY silica-QDs, Zn2+ ions were added to the colloidal solution (for spectroscopy data see Fig. S2 in the ESI†). The results of gelation monitoring are in good agreement with those of Fig. 3 except for one main difference. Upon stepwise Zn2+ ion addition to the colloidal silica-QD solution, a healing process is observed before the gelation takes place. The healing is observable as an increase of the PL intensity and PL lifetime while the absorption remains unchanged (see Fig. 4). When healing is completed, further Zn2+ ion addition leads to gel formation with the same characteristics as shown in Fig. 3, i.e. increase of the PL intensity and absorption due to scattering and unchanged PL lifetime. Subsequently, the addition of an excess amount of EDTA with respect to the amount of Zn2+ added previously leads to complete gel dissolution. Hence the absorption decreases to the value of the initial colloidal solution. The PL intensity also decreases but only to the value that was reached after healing and the PL lifetime remains unchanged. In short, a permanent healing of the silica-QDs takes place due to the influence of Zn2+ ions. From earlier studies we know that the addition of metal ions to QD solutions may lead to surface healing and thus an increase of QY.40,41 However, performing the same experiment using Pb2+ instead of Zn2+ results in PL quenching (see Fig. 4). While absorption is retained, the PL intensity and the PL lifetime of the silica-QDs are reduced in the presence of Pb2+ ions. Gels formed with the usage of Pb2+ ions show a reduced PL intensity and a faster PL decay compared to the initial solution (for spectroscopy data see Fig. S3 in the ESI†). Furthermore, this quenching seems to be irreversible since the addition of an excess amount of EDTA does not lead to a recovery of the emission properties.
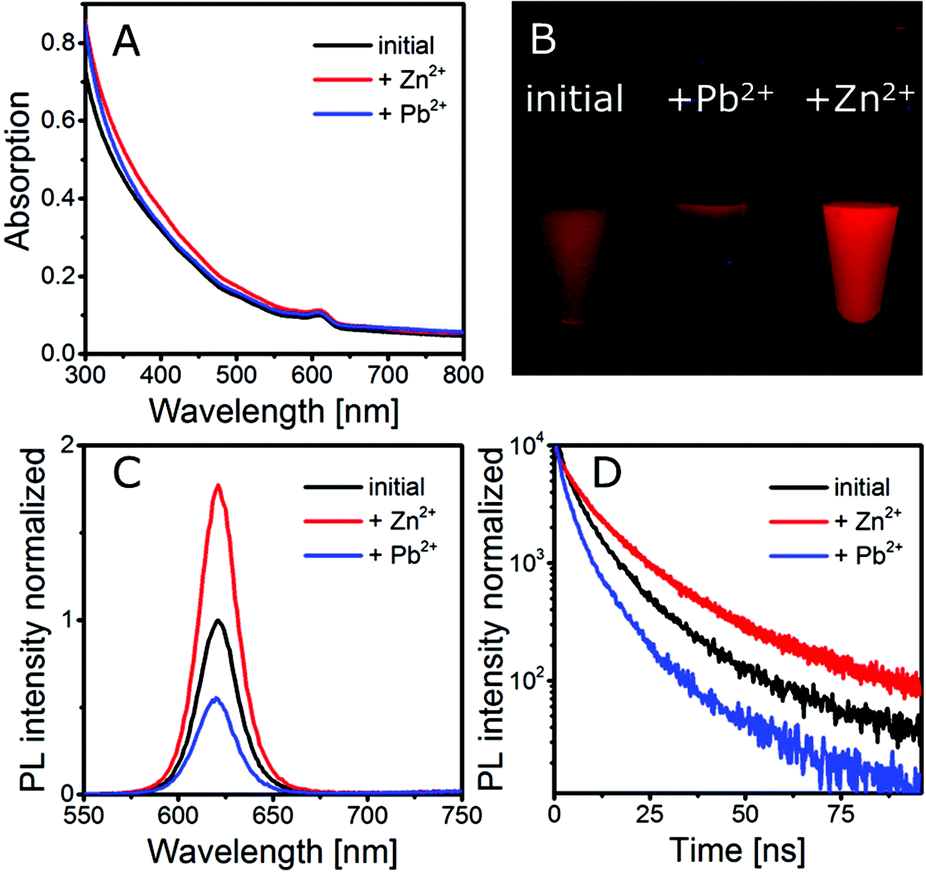 | ||
| Fig. 4 (A) Absorption, (C) fluorescence spectra and (D) PL decay of a silica-QDs exposed to Zn2+ and Pb2+. A photograph of the corresponding solutions is shown in (B). | ||
From this we conclude that the silica shell coated on our QDs is not densely packed but rather porous and allows small species, such as metal ions, to pass through the silica shell to the QD core and hence enables interaction with the QD surface. In our case, this interaction is observed as a permanent enhancement of the PL intensity and lifetime in the case of Zn2+ and a decrease of the PL intensity and lifetime in the case of Pb2+.
According to the literature, silica shells are expected to act as a protecting layer to prevent the QDs from photodegradation and impede the release of heavy metal ions.42,43
However, several studies confirmed the existence of a certain porosity in the examined silica shells. Although no indication of porosity is given by high-resolution TEM, the existence of micro- and mesoporous cavities in colloidal silica particles was observed in BET gas adsorption isotherms.44,45 Liz-Marzán and Mulvaney showed, for glass coated Ag nanoparticles grown with sodium silicate, that the shells are porous and the core can undergo a variety of chemical reactions.46 The reaction rate thereby is reduced with the increasing shell thickness, but even for the largest shell thickness of 23 nm, the reaction was not negligible. The porous nature has also been reported for sol–gel derived silica spheres.47 However, it should be mentioned that the porous structure strongly depends on the preparation and cleaning conditions as well as on the shell thickness48 and might therefore vary between different preparation techniques.
To prove the porosity of our silica particles derived by the microemulsion approach, we determined the specific surface area using nitrogen physisorption measurement at 77 K. The adsorption isotherm is presented in Fig. 5. In the range of 10−6 up to 0.8 p/p0, the isotherm matches well with the type I isotherm that is specific for microporous materials according to the IUPAC classification. The strong increase at higher relative pressure (>0.8 p/p0) is attributed to the filling of interparticle pores with nitrogen molecules. The BET specific surface area (SSA) was determined to be 102 m2 g−1, which is high compared to the theoretical geometric surface area of the silica nanoparticles of 75.2 m2 g−1, supporting the existence of a certain porosity. The geometric surface area was determined using the average external diameter of the spheres from a TEM of 42 nm and a bulk density of 1.9 g cm−3.47 The pore size distribution from the NLDFT model exhibits pore widths within the range of micro- and mesopores (see Fig. S4, ESI†). The measurement of carbon dioxide adsorption at 273 K allows a more detailed statement about the microporosity of the material. In Fig. S4C† the CO2 isotherm in the range of 10−4 up to 3 × 10−2p/p0 is shown, which presents the adsorption in micropores. A micropore volume of 0.11 cm3 g−1 was determined, indicating good porosity of the silica material.
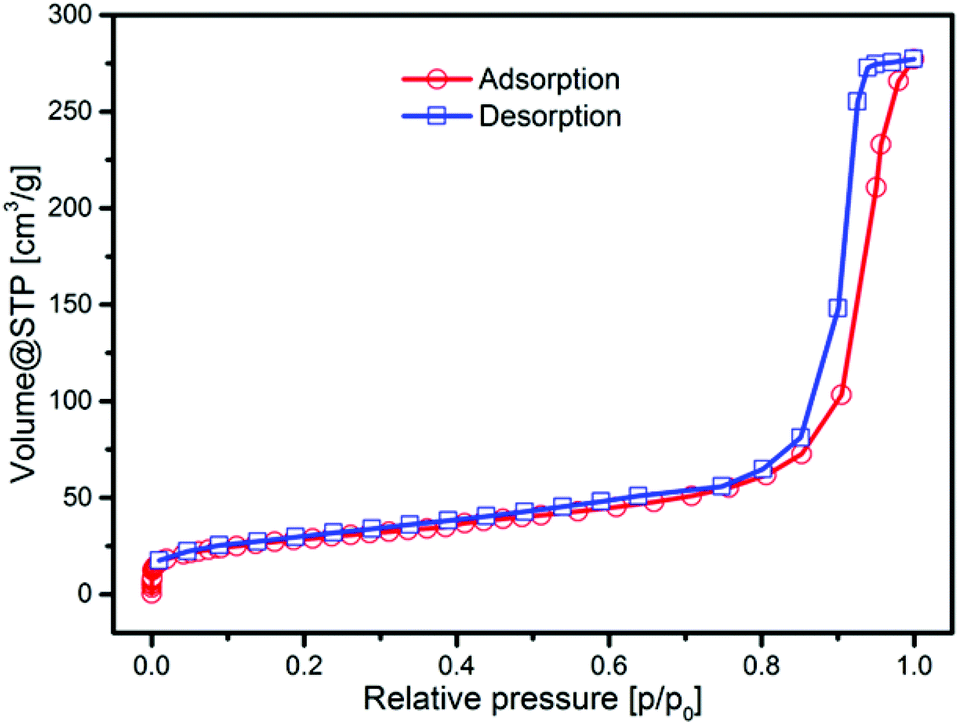 | ||
| Fig. 5 Nitrogen physisorption (at 77 K) isotherm of microemulsion derived silica nanoparticles with an average diameter of 42 nm. | ||
This porosity might be useful with respect to future sensing applications of the material. As shown before, the PL properties are sensitive towards metal ions. This concentration dependent influence of metal ions (see Fig. S2 and S3†) makes them a promising ion sensing material. The porosity of the material is therefore a necessary requirement to provide the transport of the analyte from the outer media to the QD core.
The gelation method shown here is applicable to any silica coated nanoparticle material. We performed the same procedure not only with CdSe/CdS QDs but also with three other QD materials, namely In(Zn)P/GaP/ZnS, CdSe/CdS/ZnS and a CdSeZnS alloy. Fig. 6 shows SEM images and photographs under UV light of the aerogels produced from these materials. As can be seen from the photographs, the PL intensities of the aerogels made from different materials differ significantly. This is caused by two main reasons: (i) the differing QY from the different QD materials and (ii) PL losses during silica coating. In particular the second aspect is highly disadvantageous. We assume that additional trap states are formed on the QD surface during the silica coating procedure, e.g. caused by ligand exchange of the organic ligands with partially hydrolysed TEOS.17,18,34 The extent of PL loss was observed to be lower for core/shell QDs with several monolayers of the shell material compared to bare QD cores, but can differ between different QD batches.
 | ||
| Fig. 6 SEM images and photographs (insets) of silica aerogels with different QD cores. The QD cores are (A) In(Zn)P/GaP/ZnS, (B) CdSe/CdS/ZnS and (C) a CdSeZnS alloy. | ||
The possibility to produce 3D networks from silica coated QDs with different QD core materials simultaneously opens the opportunity to produce a variety of mixed structures, since similar surface chemistry is provided by the silica shell and the attached tetrazole ligand. Mixing, for example, two differently emitting silica-QD species makes it very easily possible to tune the colour of the resulting gel. Since energy transfer is disabled in this architecture due to the large distance between the emitting species, no spectral shifts have to be considered when mixing the desired emission colour. This can be demonstrated in the following experiment. Fig. 7A shows a photograph of two differently emitting silica-QD species, i.e. QD1 and QD2 and the mixtures of both species, i.e. Mix1–Mix3, in different ratios in colloidal solution and as hydrogels. As expected, the emission colour in solution and in the gel is identical. This observation is supported by the fluorescence spectra of the solutions and the corresponding gels (see Fig. 7B). No spectral shifts or significant changes of the peak area ratios are observed for all the three mixtures in solution and in the gel. Thanks to this, the colour of the mixed gel can be easily tuned. Fig. 7C demonstrates that subsequent increase of the red component equivalently shifts the colour of the mixture providing the possibility to create different shades of yellow.
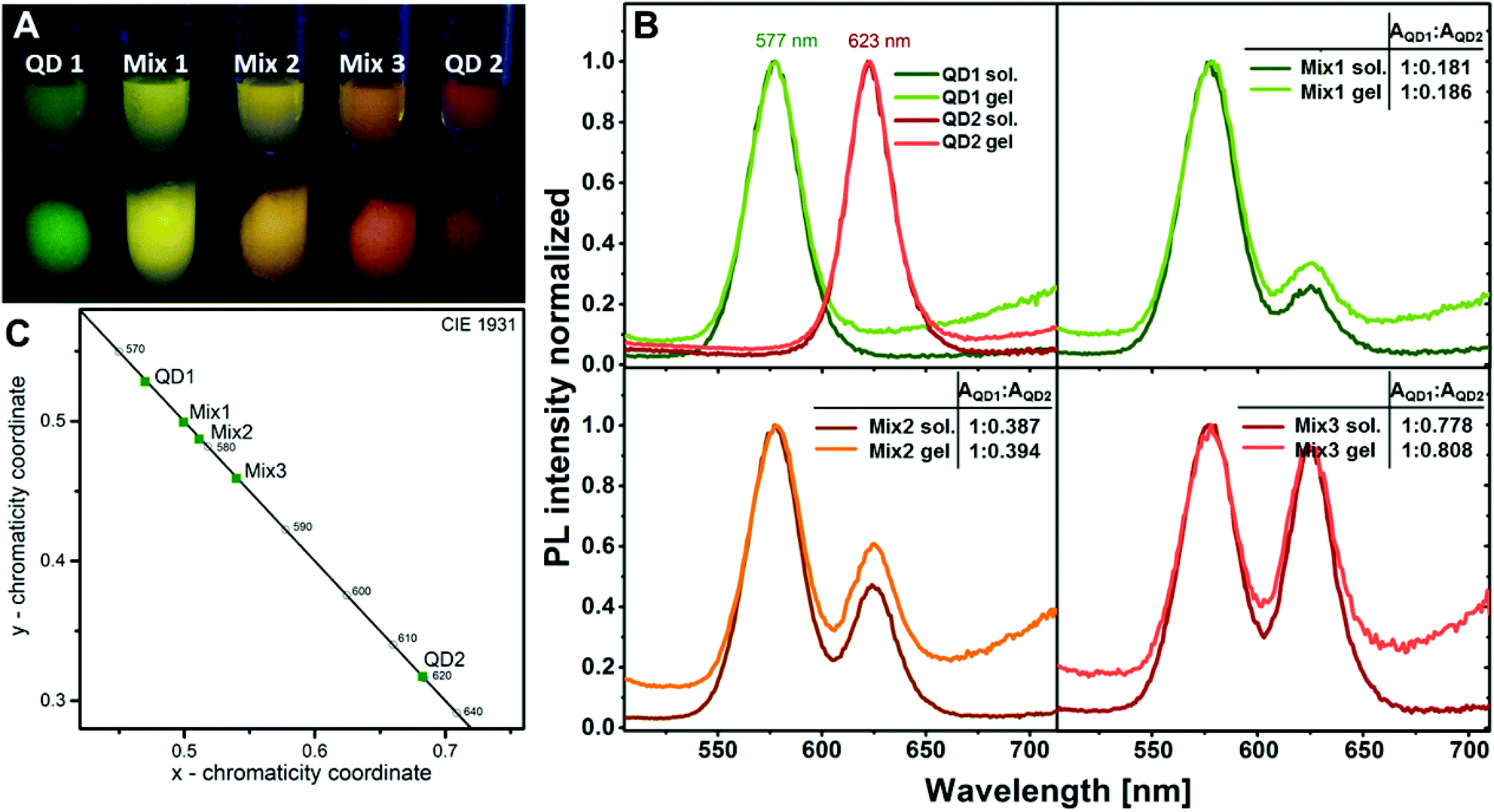 | ||
Fig. 7 (A) Photograph of two differently emitting silica-QDs and mixtures of both species in differing ratios in solution (upper row) and as hydrogels (lower row). The emission colours are not changed in the hydrogel compared to the solution, since energy transfer from donor particles to acceptor particles is disabled due to the large distance between the species. (B) Fluorescence spectra of two differently emitting silica-QDs and mixtures of the species in different ratios in solution and as hydrogels. The optical properties of the initial solutions are preserved in the gel as can be seen from the comparison of the peak area ratios AQD1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AQD2 of the species QD1 and QD2 in the three different mixtures. (C) CIE (Commission internationale de l’éclairage) coordinates of the two differently emitting silica-QDs and their mixtures. :AQD2 of the species QD1 and QD2 in the three different mixtures. (C) CIE (Commission internationale de l’éclairage) coordinates of the two differently emitting silica-QDs and their mixtures. | ||
Conclusions
We have developed a new aerogel material with non-quenching PL behaviour from silica encapsulated semiconductor QDs, based on a method reported for colloidal solutions of semiconductor and metal nanoparticles. In comparison with other monolithic silica-QD structures, we provide a defined and controllable distance between the QDs in our gel, providing control over energy transfer processes. Silica encapsulation has been done by using a well-known reverse microemulsion approach that allows uniform and controllable shell formation. In order to perform reversible gel formation, we developed a new tetrazole ligand that was intentionally designed to specifically bind to the silica surface. A facile 3D assembly of silica-QDs capped with this ligand was achieved by metal ion complexation of the negatively charged tetrazole rings. By monitoring the gel formation process using optical spectroscopy, we were able to show that the optical properties of the nanocrystals could be preserved in the 3D network. No spectral shifts, lifetime shortening, PL quenching or decrease of PL QY is observed for this novel gel material, as has all along been the case for these kinds of gels prepared from non-coated QDs, until now. Using nitrogen and carbon dioxide physisorption measurements, we showed that the silica shell is porous and allows metal ions, such as Zn2+ and Pb2+ to pass through the shell and interact with the QD core causing detectable changes of the emission properties. This makes them a promising material for metal ion sensing application. We further showed the applicability of this gelation method to other QD materials which sets the stage for facile preparation of a variety of mixed gel structures. With respect to future applications in the field of solid state lighting, we showed the advantage of the architecture of our aerogel for colour tuning. As an example, we demonstrated on the basis of two differently emitting silica-QD species that colour tuning of the resulting mixed gel is very straightforward, because no energy transfer and thus no spectral shifts require to be considered.Acknowledgements
This work was supported by DFG project EY16/10-2, the European Research Council (ERC-2013-AdG project AEROCAT) and the EU FP7 Network of Excellence “Nanophotonics for Energy Efficiency”. S.V. gratefully acknowledges the European Science Foundation for the exchange grant within ESF activity “New Approaches to Biochemical Sensing with Plasmonic Nanobiophotonics (PLASMON-BIONANOSENSE)”. Special thanks to Zoran Popoviç for imparting information on silica coating.Notes and references
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- S. S. Kistler, Nature, 1931, 127, 741–741 CrossRef CAS PubMed.
- H. D. Gesser and P. C. Goswami, Chem. Rev., 1989, 89, 765–788 CrossRef CAS.
- N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- V. Lesnyak, A. Wolf, A. Dubavik, L. Borchardt, S. V. Voitekhovich, N. Gaponik, S. Kaskel and A. Eychmüller, J. Am. Chem. Soc., 2011, 133, 13413–13420 CrossRef CAS PubMed.
- S. Bag, I. U. Arachchige and M. G. Kanatzidis, J. Mater. Chem., 2008, 18, 3628–3632 RSC.
- S. Bag, P. N. Trikalitis, P. J. Chupas, G. S. Armatas and M. G. Kanatzidis, Science, 2007, 317, 490–493 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2007, 129, 1840–1841 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, Acc. Chem. Res., 2007, 40, 801–809 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2006, 128, 7964–7971 CrossRef CAS PubMed.
- A. Eychmüller, Angew. Chem., Int. Ed., 2005, 117, 4917–4919 CrossRef PubMed.
- N. Gaponik, A. Wolf, R. Marx, V. Lesnyak, K. Schilling and A. Eychmüller, Adv. Mater., 2008, 20, 4257–4262 CrossRef CAS PubMed.
- J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 397–400 CAS.
- T. Hendel, V. Lesnyak, L. Kühn, A.-K. Herrmann, N. C. Bigall, L. Borchardt, S. Kaskel, N. Gaponik and A. Eychmüller, Adv. Funct. Mater., 2013, 23, 1903–1911 CrossRef CAS PubMed.
- V. Lesnyak, S. V. Voitekhovich, P. N. Gaponik, N. Gaponik and A. Eychmüller, ACS Nano, 2010, 4, 4090–4096 CrossRef CAS PubMed.
- S.-Y. Chang, L. Liu and S. A. Asher, J. Am. Chem. Soc., 1994, 116, 6739–6744 CrossRef CAS.
- M. Darbandi, R. Thomann and T. Nann, Chem. Mater., 2005, 17, 5720–5725 CrossRef CAS.
- R. Koole, M. M. van Schooneveld, J. Hilhorst, C. de Mello Donegá, D. C. Hart, A. van Blaaderen, D. Vanmaekelbergh and A. Meijerink, Chem. Mater., 2008, 20, 2503–2512 CrossRef CAS.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693–6700 CrossRef CAS.
- Y. Kobayashi, T. Nozawa, T. Nakagawa, K. Gonda, M. Takeda, N. Ohuchi and A. Kasuya, J. Sol–Gel Sci. Technol., 2010, 55, 79–85 CrossRef CAS PubMed.
- M. A. Correa-Duarte, M. Giersig and L. M. Liz-Marzán, Chem. Phys. Lett., 1998, 286, 497–501 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- H. Yamauchi, T. Ishikawa and S. Kondo, Colloids Surf., 1989, 37, 71–80 CrossRef.
- K. Osseo-Asare and F. J. Arriagada, Colloids Surf., 1990, 50, 321–339 CrossRef CAS.
- C.-L. Chang and H. S. Fogler, Langmuir, 1997, 13, 3295–3307 CrossRef CAS.
- Q. Wang, N. Iancu and D.-K. Seo, Chem. Mater., 2005, 17, 4762–4764 CrossRef CAS.
- S. Jun, J. Lee and E. Jang, ACS Nano, 2013, 7, 1472–1477 CrossRef CAS PubMed.
- C. Li and N. Murase, Langmuir, 2004, 20, 1–4 CrossRef CAS.
- P. Yang, C. L. Li and N. Murase, Langmuir, 2005, 21, 8913–8917 CrossRef CAS PubMed.
- L. Sorensen, G. F. Strouse and A. E. Stiegman, Adv. Mater., 2006, 18, 1965–1967 CrossRef CAS PubMed.
- Y. Chan, P. T. Snee, J.-M. Caruge, B. K. Yen, G. P. Nair, D. G. Nocera and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 3146–3147 CrossRef CAS PubMed.
- Z. Popović, W. Liu, V. P. Chauhan, J. Lee, C. Wong, A. B. Greytak, N. Insin, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, Angew. Chem., Int. Ed., 2010, 49, 8649–8652 CrossRef PubMed.
- Y. Ma, Y. Li, S. Ma and X. Zhong, J. Mater. Chem. B, 2014, 2, 5043–5051 RSC.
- P. Yang, M. Ando and N. Murase, Langmuir, 2011, 27, 9535–9540 CrossRef CAS PubMed.
- P. N. Gaponik, S. V. Voitekhovich and O. A. Ivashkevich, Russ. Chem. Rev., 2006, 75, 507–539 CrossRef CAS PubMed.
- G. Aromí, L. A. Barrios, P. Gamez and O. Roubeau, Coord. Chem. Rev., 2011, 255, 485–546 CrossRef PubMed.
- H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC.
- L. Ma, Y.-C. Qiu, G. Peng, J.-B. Cai and H. Deng, Eur. J. Inorg. Chem., 2011, 3446–3453, DOI:10.1002/ejic.201100213.
- J. H. Lee, H. Lee, S. Seo, J. Jaworski, M. L. Seo, S. Kang, J. Y. Lee and J. H. Jung, New J. Chem., 2011, 35, 1054–1059 RSC.
- H. H.-Y. Wei, C. M. Evans, B. D. Swartz, A. J. Neukirch, J. Young, O. V. Prezhdo and T. D. Krauss, Nano Lett., 2012, 12, 4465–4471 CrossRef CAS PubMed.
- A. Wolf, V. Lesnyak, N. Gaponik and A. Eychmüller, J. Phys. Chem. Lett., 2012, 3, 2188–2193 CrossRef CAS.
- Y. Yang, L. Jing, X. Yu, D. Yan and M. Gao, Chem. Mater., 2007, 19, 4123–4128 CrossRef CAS.
- P. Yang, M. Ando and N. Murase, J. Colloid Interface Sci., 2007, 316, 420–427 CrossRef CAS PubMed.
- A. van Blaaderen and A. P. M. Kentgens, J. Non-Cryst. Solids, 1992, 149, 161–178 CrossRef CAS.
- A. van Blaaderen and A. Vrij, J. Colloid Interface Sci., 1993, 156, 1–18 CrossRef CAS.
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, Langmuir, 1998, 14, 3740–3748 CrossRef CAS.
- S. Li, Q. Wan, Z. Qin, Y. Fu and Y. Gu, Langmuir, 2014, 31, 824–832 CrossRef PubMed.
- P. Yang and N. Murase, ChemPhysChem, 2010, 11, 815–821 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr01880c |
| This journal is © The Royal Society of Chemistry 2015 |