
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell uptake, intracellular distribution, fate and reactive oxygen species generation of polymer brush engineered CeO2−x NPs†
Yuan
Qiu
a,
Elena
Rojas
a,
Richard A.
Murray
a,
Joseba
Irigoyen
a,
Danijela
Gregurec
a,
Pablo
Castro-Hartmann
b,
Jana
Fledderman
c,
Irina
Estrela-Lopis
c,
Edwin
Donath
c and
Sergio E.
Moya
*a
aSoft Matter Nanotechnology Laboratory, CIC biomaGune, Paseo Miramón 182 C, 20009, San Sebastián, Spain. E-mail: smoya@cicbiomagune.es; Tel: +34 943 00 53 11
bUniversitat Autónoma de Barcelona, 08193 Cerdanyola del Vallés, Spain
cInstitute of Medical Physics and Biophysics, Leipzig University, Härtelstrasse 16-18, D-04107, Leipzig, Germany
First published on 12th March 2015
Abstract
Cerium Oxide nanoparticles (CeO2−x NPs) are modified with polymer brushes of negatively charged poly (3-sulfopropylmethacrylate) (PSPM) and positively charged poly (2-(methacryloyloxy)ethyl-trimethylammonium chloride) (PMETAC) by Atom Transfer Radical Polymerisation (ATRP). CeO2−x NPs are fluorescently labelled by covalently attaching Alexa Fluor® 488/Fluorescein isothiocyanate to the NP surface prior to polymerisation. Cell uptake, intracellular distribution and the impact on the generation of intracellular Reactive Oxygen Species (ROS) with respect to CeO2−x NPs are studied by means of Raman Confocal Microscopy (CRM), Transmission Electron Microscopy (TEM) and Inductively Coupled Plasma Mass Spectroscopy (ICP-MS). PSPM and PMETAC coated CeO2−x NPs show slower and less uptake compared to uncoated Brush modified NPs display a higher degree of co-localisation with cell endosomes and lysosomes after 24 h of incubation. They also show higher co-localisation with lipid bodies when compared to unmodified CeO2−x NPs. The brush coating does not prevent CeO2−x NPs from displaying antioxidant properties.
Introduction
Spherical brushes are polymer brushes tethered to a colloidal particle. Polyelectrolyte chains in the brush are attached at one end of the polymer chain to the particle while the other end remains unattached. In this molecular arrangement, the polymer chains retain a significant degree of freedom, similar to being in solution. An important application of spherical polyelectrolyte brushes is their use in the dispersion of particles in aqueous media due to the multiple charges provided by the polymer chains.1–4 Indeed, brushes can be very useful for providing charge and stabilising nanoparticles (NPs) in aqueous media while coating the NPs with a thin polymer shell. The thickness and density of this shell can be controlled by the polymerisation conditions and reaction time.NPs modified with polyelectrolyte brushes are an interesting system for studying the role of charge in the interaction of NPs with cells and biological media for the above mentioned reasons. By means of a brush, a densely charged coating on the nanoparticle is ensured, which is difficult to achieve by other means.
Cerium oxide nanoparticles (CeO2−x NPs) are amongst the most widely used metal oxide NPs with a large number of applications in solar cells, as catalysts, polishing agents, and fuel additives.5–13 In addition, CeO2−x NPs are drawing increasing attention in the biomedical field due to their antioxidant properties. These are based on the redox properties of the CeO2−x owing to the existence of two oxidation states of cerium: Ce3+ and Ce4+, on the surface of the cerium oxide lattice. Moreover, at the nanoscale, cerium oxide can contain intrinsic oxygen defects. These oxygen defects are actually ‘hot spots’ for catalytic reactions. The concentration of oxygen defects and the concentration of Ce3+ ions increases as particle size decreases.14,15 For this reason, CeO2−x NPs have improved antioxidant properties with respect to the corresponding bulk material. CeO2−x NPs have also shown different protective effects on cells both in vitro and in vivo, against radiation exposure, oxidative stress, and as anti-inflammatories.16–19
Despite the diverse applications of CeO2−x NPs, their potential impact on human health is not fully understood. In vitro studies indicate that CeO2−x NPs are toxic to different cell lines. For example, CeO2−x NPs were able to reduce cell viability and induce apoptosis and autophagy of human monocytes via mitochondrial damage and over expression of apoptosis inducing factors.20 CeO2−x NPs of various sizes caused certain cytotoxicity when exposed to human lung epithelial cells (BEAS-2B) and eventually led to cell death, an increase in Reactive Oxygen Species (ROS), GSH decrease, and the activation of oxidative stress-related genes such as heme oxygenase-1, catalase, glutathione S-transferase, and thioredoxin reductase takes place.21 When human lung cancer cells were exposed to CeO2−x NPs, cell viability decreased significantly as a function of NP dose and exposure time.22 However, there are also many other studies that show that CeO2−x NPs are non-toxic and have a protective effect regarding cell apoptosis. For instance, CeO2−x NPs were internalised in endosomal compartments in the macrophage cell line RAW 264.7 and BEAS-2B cells without inflammation or cytotoxicity. Prior incubation with CeO2−x NPs could protect these cells against the cytotoxic effects of exogenous stress stimulus.23 In another case, CeO2−x NPs with diameters in the range of 7 to 94 nm were found to be non toxic to the human monocytic cell line U937 and could scavenge intracellular ROS.24 The impact of CeO2−x NPs on cell viability and apoptosis depends on their effect on the intracellular oxidation process. When the NPs behave as an antioxidant, they have a pro-survival effect and may protect cells from oxidative stress, whereas when the CeO2−x NPs exhibit pro-oxidant effect behaviour, ROS and apoptosis are induced.25 Cell type, NP surface characteristics, NP size and intracellular pH may all play a role in the intracellular behaviour of Cerium oxide NPs. However, how these factors influence the anti- or pro-oxidant behaviour and cellular uptake of CeO2−x NPs has not yet been determined.26,27
In this work commercially available CeO2−x NPs were surface modified by coating them with polyelectrolyte brushes via in situ Atom Transfer Radical Polymerisation (ATRP). ATRP is one of the most effective and versatile techniques to introduce macromolecules onto the surface of inorganic NPs.28–31 The charge of the uncoated NPs estimated from ζ-potential measurements was positive but very close to 0 and the NPs can be considered as being almost neutral. Because the polymer chains in a spherical brush are attached on one end to the NP and each chain has several charges the coating of the NPs with a brush ensures a high charge density on the NPs, which is difficult to achieve by other functionalisation procedures.
CeO2−x NPs with polyelectrolyte brushes allow us to address the issue of surface charge on the uptake and fate of the CeO2−x NPs. The use of the ‘grafted from’ polyelectrolyte brushes to modify CeO2−x NPs has another important advantage over other functionalisation strategies. For uptake studies it is often necessary to attach a dye to the surface of the NPs. Fluorescent dyes are often charged but at the same time they usually display conjugated aromatic rings that give them a certain hydrophobic character. By attaching a dye to the surface of the NP, the charge and hydrophobicity of the NP surface may be altered, which in turn will affect the uptake and intracellular fate of the NPs. Toxicity, due to the presence of NPs, may also be altered by the presence of the dye as the dye itself may have an inherent toxicity. We will show that brushes can be used to shield dyes at the surface of NPs. Fluorescent dyes will be directly attached to amine terminated silanes. Following brush synthesis, the dyes will be entrapped in the brushes. In other words, the NPs will be labelled but the label will not be placed on the outer surface of the NPs, thus not affecting the surface charge or degree of surface hydrophobicity. Flow cytometry (FACS) and Confocal Laser Scanning Microscopy (CLSM) will be applied to study uptake kinetics, uptake routes and intracellular localisation. In parallel, the uptake of non labelled brush coated CeO2−x NPs will be followed by Inductively Coupled Plasma Mass Spectrometry (ICPMS), Transmission Electron Microscopy (TEM) and Confocal Raman Microscopy (CRM). The results from the labelled and label free NPs will be compared to assess the impact of labelling on the intracellular fate of NPs.
In addition, brushes are thin organic layers that can behave as a barrier around the CeO2−x NPs. This barrier can affect the redox equilibrium between Ce3+ and Ce4+, which depends on electron transfer processes between the NPs and cell molecules. The use of brushes to functionalise the surface of the NPs allows us to study the effect of an organic coating in this equilibrium.
Experimental section
Materials
CeO2−x NPs were purchased from PLASMACHEM A/G, Berlin, Germany.ATRP polymerisation: dichloromethane (99%), 2-bromo-2-methylpropionic acid (98%), 3-aminopropyltris(triethoxy)silane (APTES), 2,2′-bipyridyl (99%) (bipy), copper chloride (99.99%), 4-dimethylaminopyridine (DMAP), dicyclohexylcarbodiimide (99%) (DCC), [2-]methacryloyloxy)ethyl] trimethylammonium chloride 80% wt in H2O (METAC), 3-sulfopropylmethacrylate potassium salt (99%) (SPM), dimethylformamide (99%) (DMF), acetone and sodium chloride were purchased from Sigma-Aldrich. Ethanol (99.95%) was purchased from Scharlau (Spain).
Labelling: Fluorescein isothiocyanate isomer I (90%) (FITC) was purchased from Sigma-Aldrich, and Alexa Fluor 488 carboxylic acid succinimidyl ester was purchased from molecular probes.by life technologies.
Two different cell culture lines were used in these studies: A549 (human lung cancer) and HEK 293 (embryonic kidney) both from ATTC. A549 cells were cultured in RPMI 1640 medium from Lonza, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and HEK293 were cultured in Dulbecco's Modified Eagle Medium (DMEM) from Sigma Aldrich (same supplementation). For all experiments cells were seeded after reaching a confluence of 80% 24 hours before the treatment.
Uptake inhibitors: amiloride, cytochalasin D, filipin, genistein and chlorpromazine were purchased from Sigma Aldrich. tert-Butyl hydroperoxide (TBHP) was purchased from Sigma Aldrich and 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA) and Cellmask™ deep red from Invitrogen-Life Technologies.
Grafting from polymerisation of polymer brushes onto the surface of cerium oxide NPs
The surface of cerium oxide NPs was modified via Atomic Transfer Radical Polymerisation (ATRP) as shown in Scheme 1.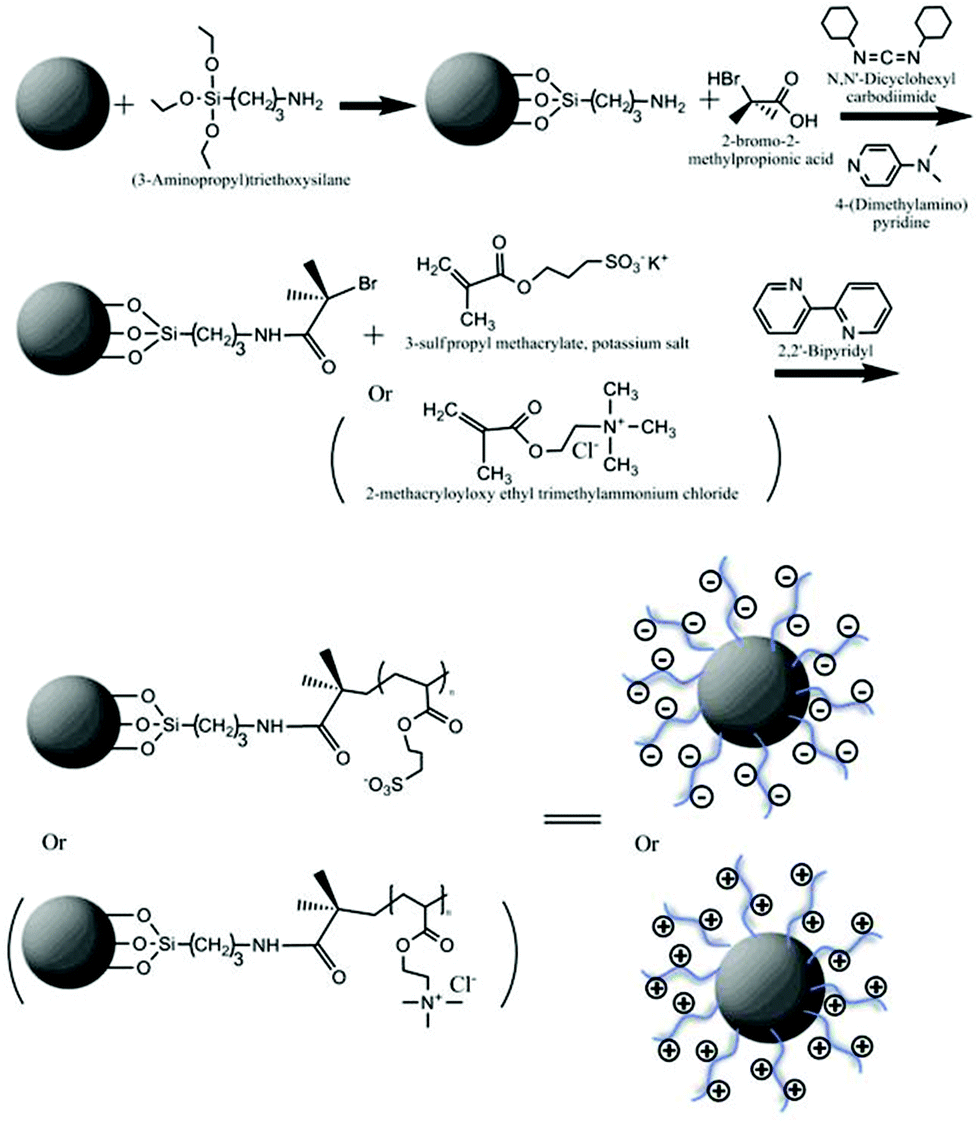 | ||
| Scheme 1 (a) Scheme of ‘grafting from’ polymer brush synthesis at the surface of cerium oxide NPs. | ||
Polyelectrolyte brushes of poly (3-sulfopropylmethacrylate) (PSPM) and poly (2-(methacryloyloxy)ethyl-trimethylammonium chloride) (PMETAC) were synthesised in situ through a ‘grafting from’ method to the surface of cerium oxide NPs. Firstly, 100 mg of NPs were well dispersed in dimethylformamide, (DMF) and mixed with 20 μL (3-aminopropyl) triethoxysilane. This reaction was carried out for 18 h in a N2 gas environment. NPs were then washed thoroughly by centrifugation to remove chemical residues from the reaction. After attachment of the silane to the NP surface, the radical polymerisation initiator 2-bromo-2 methylpropionic acid was bound to the amine group of the silane via a condensation reaction.1 NPs were resuspended in CH2Cl2 and placed in a round bottom flask. 2-Bromo-2-methylpropionic acid, was added to the suspension along with dimethyl aminopyridine (DMAP). The mixture was cooled to 0° C with vigorous stirring. N,N′-Dicyclohexylcarbodiimide (DCC), another catalyst, was then added to the flask. The reaction continued for another 18 h in a N2 atmosphere. After carefully washing; twice in CH2Cl2, twice in acetone, and twice in DMF, NPs coated with initiators were dispersed in N,N-dimethylformamide DMF–water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) solution with monomer, 2,2-bipyridine and CuCl (molar ratio, 10:2:1). The polymerisation reaction was carried out for 5 h in a N2 atmosphere.54
:2, v/v) solution with monomer, 2,2-bipyridine and CuCl (molar ratio, 10:2:1). The polymerisation reaction was carried out for 5 h in a N2 atmosphere.54
For the fluorescent labelling of NPs, 100 mg of silanised NP were first mixed with 257 μg of Alexa 488 in DMF and allowed to react for 4 h. After five washes via centrifugation, polymer brushes were synthesised on the surface of the NPs (as described before).
In order to leave sufficient amine available for attachment of the initiator, a calculation of the amount of amine on the surface of one CeO2−x NP was performed on the basis of the size of the CeO2−x NPs and the size of a unit cell of cerium oxide crystal as describe by S. Lee et al.17 The average size of the CeO2−x NPs is 15 nm (based on the result from over 200 CeO2−x NPs from TEM images, see ESI†).
Confocal laser scanning microscopy (CLSM)
Cellular uptake and intracellular distribution of the CeO2−x NPs were studied using an LSM 510-META CLSM (Zeiss, Germany). The cell membrane was stained with Cellmask™ deep red. In order to reveal the intracellular localisation of CeO2−x NPs, acidic cell compartments (e.g. endosomes and lysosomes) were stained with LysoTracker® Red.Flow cytometry (FACS)
Flow cytometry data were analysed using WinMDI 2.9 software. First, a threshold of fluorescence was generated using untreated HEK293 cells as a control sample. All events corresponding to the control sample were located at intensities below this threshold. Each experiment was set to count 10000 events per run and each run was recorded using identical parameters. The number of cells bearing fluorescently labelled CeO2−x NPs is obtained from the area corresponding to the events located at higher intensities than the threshold. The cellular uptake ratio was calculated as the (no. of events over the threshold/total no. of events) × 100%. The cell uptake ratio may be understood as the number of cells that have uptaken nanoparticles. The amount of nanoparticles per cell is given by the fluorescent intensity as this is proportional to the number of particles present in the cell. The fluorescence intensity was set as the maximum of the fluorescence intensity versus cell number distribution. The relative fluorescence intensity is calculated with respect to the fluorescence intensity of the NPs measured by FACS.
Cell uptake mechanisms
Several different cell uptake pathway inhibitors were used in order to determine the uptake mechanism of NPs. Cells were cultured at low temperature (4 °C) for one hour without any inhibitor to determine if the uptake is energy-dependent.55For all the uptake pathway experiments, cells were pretreated with different endocytosis pathway inhibitors for 1 h and then incubated with Alexa 488 labelled CeO2−x NPs, and CeO2−x NPs with PSPM and PMETAC for another 4 h and finally thoroughly rinsed with PBS. The average cell fluorescence intensity was measured by FACS. Relative fluorescence intensity was determined by defining the fluorescence intensity of control cells (cells incubated with NPs without inhibitors treatment) as 100.
Inductively coupled plasma mass spectroscopy (ICP-MS)
Cellular uptake was quantitatively studied via ICPMS. For IPCMS experiments, HEK 293 cells were exposed to NPs without fluorescence labelling and collected following the same procedures as for flow cytometry. Collected cells were then dissolved in nitric acid up to a final concentration of 0.1 mg ml−1. Then, the elemental concentrations of cerium in cells were measured by Perkin Elmer Analyst 800 ICP-MS.Confocal Raman microscopy (CRM)
Raman spectra were recorded along a square or circle defined to cover the whole cell area. Between two subsequent measurements there was a distance increment of 0.5 μm. The distributions of different components were analysed focusing on signals of NPs, nucleic acids, proteins and lipids.Raman spectroscopy was employed to investigate the uptake of unlabelled CeO2−x NPs. Raman spectroscopic data of CeO2−x NPs have a distinct peak in Raman spectra at approximately 465 cm−1. This band is assigned to a symmetric breathing mode of the oxygen atoms around the cerium ions.56 After polymer coating, this strong peak is still observable without a significant shift of the spectral position of the band. The broad peak present at 2900 cm−1 is assigned to a combination band mainly involving the C–H stretching mode of CH2 from the polymer backbone. And the peaks in the 1200 cm−1 to 1600 cm−1 region are assigned to the C–H deformation mode.57
Cells were seeded onto a CaF2 crystal substrate with a diameter of 20 mm. After 24 h, CeO2−x NPs were added to the cell medium to a final concentration of 20 μg ml−1 and incubated for 24 h. Cells were washed with PBS several times, fixed with 3.7% formaldehyde and characterised by CRM. Measurements were performed with a Renishaw inVia Raman Microscope with 532 nm excitation and a grating of 1800 mm−1. Spectra were taken using a 40× water immersion objective. The size of the focal spot was approximately 1 μm. Raman spectra were recorded over the range 800–3200 cm−1 with a resolution of approximately 7 cm−1. The system was calibrated to the spectral line of crystalline silicon at 520.7 cm−1.
The tendency of co-localisation of two components (e.g. CeO2−x NPs and lipid) can be quantified by the Pearson product-moment correlation coefficient, ρX,Y. The Pearson coefficient is a correlation coefficient used to measure the linear dependence between two variables X and Y, yielding a value between −1 and +1. The absolute value 1 indicates a 100% linear dependence between X and Y while 0 shows a total independence.58 The correlation between distributions of CeO2−x NPs and cell lipid rich compartments were calculated as follows:
 | (1) |
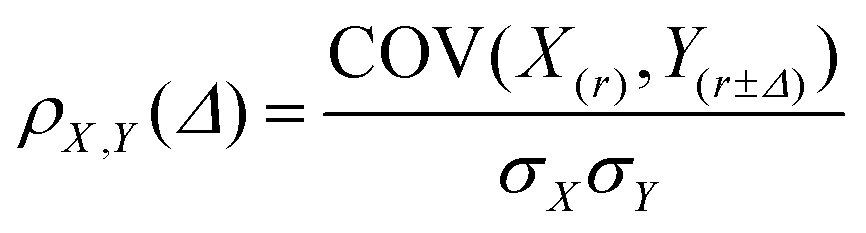 | (2) |
The maximum value of ρX,Y(Δ) can be used as an evaluation of the correlation between CeO2−x, NPs and lipids.
Transmission electron microscopy (TEM)
For TEM experiments, 100000 HEK 293 cells were seeded in a 35 mm diameter petridish and grown for 24 h. Different types of CeO2−x NPs were then added at concentrations of 10 μM ml−1. After 1 h and 24 h co-incubation, cells were washed with PBS and collected by trypsinisation.
For TEM imaging, cells were further fixed with 3.7% formaldehyde, washed with PBS and stained with 1% osmium tetroxide. Afterwards, samples were dehydrated in a group of acetone solutions with gradient concentrations (from 50 to 100%) and followed by EPO resin embedment. The resin blocks were cut into ultrafine slices of 70 nm by an ultramicrotome. Each slide was placed on a copper grid and contrasted by the Reynolds technique for appropriate visualisation.
Reactive oxygen species (ROS)
HEK293 cells were first incubated with CeO2−x NPs at different concentrations. In order to avoid CeO2−x NPs toxicity, only very low concentrations were used: 5 μg ml−1 and 10 μg ml−1. After 24 hours, cells were washed with PBS 3 times to remove the NPs that were not internalised and incubated with 0.5 mM tert-butyl hydroperoxide (TBHP), which can induce ROS inside cells for a further 2 hours. After washing out the redundant TBHP, all samples were then incubated with 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA) for 30 minutes. Carboxy-H2DCFDA is commonly used as an indicator for ROS in cells. This non fluorescent molecule can be converted to a green-fluorescent form when the acetate groups are removed by intracellular esterases and oxidation occurs within the cell, which is an indication of ROS. The green fluorescence intensity was measured by FACS in the FITC channel. The level of the reactive oxygen species inside cells was calculated as following: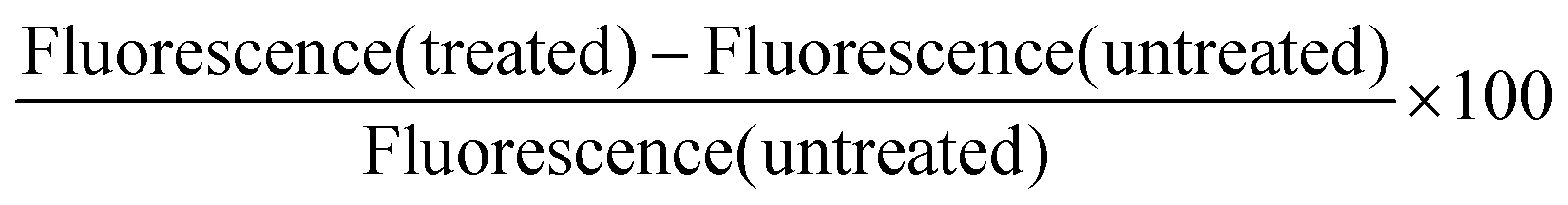
Here, untreated cells refer to control cells exposed to TBHP for induction of ROS but without NPs. The oxidative stress observed in cells without NPs and not exposed to TBHP will be taken as a zero value. Oxidative stress after exposing the cells to TBHP with and without NPs will be related to the ROS values displayed by the cells without TBHP.
Results and discussion
Characterisation of modified CeO2−x nanoparticles
The surface modification of CeO2−x NPs with brushes was demonstrated by TEM and ζ-potential measurements. Fig. S1 (ESI†), shows TEM images of brush coated NPs. CeO2−x NPs display a crystalline structure with a single particle size of approximately 15 nm. By staining with uranyl acetate, the polymer brush layer around the NPs was revealed as a faint shell surrounding the NPs, which is absent in the uncoated NPs. PSPM brushes had an average thickness of approximately 6 nm (Fig. S1b†) while PMETAC brushes displayed a thickness of approximately 2 nm in the dry state (Fig. S1c†). The hydrodynamic size and ζ-potentials of CeO2−x NPs with different polymer coatings are shown in Table S1 (ESI†). Unmodified CeO2−x NPs (CeO2−x NPs) show a large hydrodynamic size stemming from the formation of agglomerates in aqueous solution due to low surface charge and limited colloidal stability, as previously reported, whereas the PSPM coated NPs (CeO2@PSPM NPs) and PMETAC coated NPs (CeO2@PMETAC NPs) presented much smaller hydrodynamic sizes in aqueous solution, which indicates that the polymer coating effectively reduces agglomeration.32 CeO2−x NPs show a slightly positive ζ-potential, close to zero, while the CeO2@PSPM NPs show a highly negative potential around −30 mV which can be attributed to the sulfonate groups in PSPM. CeO2@PMETAC NPs show a high positive charge (∼+25 mV) coming from the quaternary group in PEMTAC. The polymer brush coating provided a high charge density, with larger charges, as revealed by ζ-potential, improving the colloidal stability of NPs in aqueous solution. When the NPs are dispersed in cell media, irrespective of charge, ζ-potential reports negative values with potentials close to −10 mV due to the formation of a protein corona.Unlike most of the rare earth metals that exist in trivalent states, cerium can also occur in a tetravalent state and may shift between these two states in a redox equilibrium. XPS analysis of uncoated NPs results in the assignment of the Ce 3d 5/2 region to two oxidation states, Ce(III) and Ce(IV). Spectra of Ce 3d 5/2 and O 1s regions are shown in Fig. S2.†33
In the case of CeO2@PSPM NPs (see Fig. S3 ESI†) the XPS spectra show the existence of a sulphonate group in the 2p region of sulphur, at 168.1 eV, which indicates the presence of PSPM brushes on top of the NPs.34,35 The nitrogen signal recorded in the N 1s region at 400.1 eV arises from the NH2 group. This is a result of the surface functionalisation of CeO2 with the silane linker terminated with primary amines.
As shown in Fig. S4 (ESI†), in the case of CeO2@PMETAC, the photoelectron peak in the N 1s region can be resolved into two species; NH2 at 400 eV and quaternary nitrogen at 402.8 eV respectively.35 The first peak emanates from the amine group at the end of the silane linker, as seen for CeO2@PSPM NPs. The quaternary ammonium structure arises from the PMETAC polymer, and here serves as proof of the existence of the brushes.
The fluorescence emission spectra of the labelled NPs were compared with the fluorescence emission spectra of Alexa 488 in water. The peak emission wavelength of the dye attached to the NPs shifted only a few nanometres (Fig. S5†), meaning that the attachment of the dye to the NPs did not alter its spectroscopic properties.
In Fig. S6† we can observe the Raman spectra of the unmodified CeO2−x NPs and the CeO2−x NPs with brushes. The Raman spectra reveals a strong band at 465 cm−1, which is practically unaffected by the brush coating irrespectively of the charge.
Cellular uptake and intracellular distribution
Uptake of fluorescently labelled CeO2−x NPs was characterised by CLSM and FACS. As has been previously explained the labelling of PSPM and PMETAC coated CeO2−x NPs is performed by attaching Alexa 488 to the surface of the NPs through silanisation prior to the synthesis of the brushes. In this way the dye remains shielded in the brushes and does not affect the surface characteristics of the NPs. To apply CLSM and FACS to uncoated NPs it was necessary to modify the NP with silanes but with a low grafting density. 0.2 μL of (3-aminopropyl)triethoxysilane (ATPS) to 100 mg CeO2−x NPs, 50 times less than that used for the synthesis of polymer brushes. Under these conditions silanisation will consume approximately 2% of the oxygen on the surface of CeO2−x NPs. By doing this we aim to keep the surface of the NPs as close as possible to the unmodified NPs while also introducing a fluorescent label. The ζ-potential of Alexa 488 labelled CeO2−x NPs without polymer brush is 3 mV, which is very similar to the value of unmodified CeO2−x NPs. Alexa 488 labelled CeO2−x NPs without a polymer brush coating will be denoted as CeO2 NPs.Confocal laser scanning microscopy (CLSM)
CLSM images in XYZ planes reveal that CeO2−x NPs are internalised by HEK293 after 24 h. In Fig. 1, CeO2−x NPs, showing green fluorescence, are located within the cell membrane labelled in red. The lower left image in Fig. 1a–c corresponds to the middle plane of a cell. The upper image was produced by ZX-stacking and the lower right image corresponds to ZY stacking. 3D stacking of images proved that after 24 h of incubation with HEK293 cells, all three NP variations are completely internalised. In addition, some CeO2@PMETAC NPs could be found adhering to the cell membrane rather than being internalised. | ||
| Fig. 1 CLSM image of HEK293 cells after incubation with (a) CeO2 NPs (b) CeO2@PSPM NPs (c) CeO2@PMETAC NPs for 24 h. Cell membrane was stained by Cellmask™ deep red (red), and CeO2−x NPs were labelled by Alexa 488 (green). | ||
In order to investigate the intracellular distribution of the NPs, acidic cell compartments were labelled by LysoTracker® Red. Experiments were carried out with HEK 293 cells incubated for 24 h with fluorescently labelled NPs. Acidic organelles include endosomes and lysosomes, which have a complex maturation process. The formation and maturation of acidic organelles begins with the endocytosis of extracellular material and continues with the transfer of internalised cargo from early endosomes to late endosomes, and then to the lysosomes. During the maturation process, acidic organelles can fuse and mix their contents, which then results in higher amounts of endocytosed material and progressively larger dimensions of the organelles. Another unique characteristic of acidic organelles is that they experience a progressive decrease in pH during the maturation process, leading to much lower pH values inside mature acidic organelles than that in the cytoplasm.36 As shown in Fig. 2, the potential co-localisation between NPs (green) and lysosomes (red) will yield a yellow/orange overlap in the merged images. CLSM showed that NPs with and without polymer brush coatings were normally co-localised with Lysotracker. This indicates that most NPs were internalised by HEK 293 cells through common endosome and lysosome involved endocytosis and located within lysosomes 24 h after uptake. However, CeO2 NPs without brushes show a lower degree of co-localisation compared to CeO2@PSPM and CeO2@PMETAC NPs·CeO2 NPs could be present in the cytosol or in non acid lysosomes. However, we cannot disregard the possibility of a slower trafficking of the unmodified NPs as they are significantly more aggregated than the modified ones. This result can be attributed to the nearly neutral surface charge of CeO2 NPs without brushes and to the high surface charge of polymer coated CeO2−x NPs reported.37
 | ||
| Fig. 2 CLSM image of HEk293 cells after incubated with (a) to (c) CeO2−x NPs, (d) to (f) CeO2@PSPM NPs, (g) to (i) CeO2@PMETAC NPs for 24 h. Cell acidic compartments were stained with LysoTracker® Red (red) and NPs were labelled with Alexa 488 (green). | ||
Flow cytometry (FACS)
FACS data shown in Fig. 3(a and b) clearly reveal that unmodified CeO2 NPs have the fastest uptake. In the first 24 h, CeO2@PMETAC NPs show the slowest uptake among all three CeO2−x NPs, both in uptake ratio and fluorescence intensity. However, after 48 h CeO2@PMETAC NPs show a higher value of the number of cells with internalised NPs and a higher number of NPs per cell (fluorescence intensity) than that of CeO2@PSPM NPs, and still have a tendency to increase unlike the other two NPs, which have already reached a plateau after 12 h. This is understandable given that the uptake of CeO2@PSPM NPs is very slow and the number of NPs per cell is much lower than that of unmodified CeO2 NPs since the negative surface charge can hinder the uptake of NPs. However, positively charged NPs have been reported to adhere to the negatively charged cell membrane implying improved uptake but this is not observed for the uptake of CeO2@PMETAC NPs. One possible reason for this could be that although the positive charge helps CeO2@PMETAC NPs to adhere to the cell surface, the uptake kinetics are rather slow and a significant uptake can only happen after 24 h. This process may be relatively slow with vigorous uptake taking place after 24 h. After 48 h, both uptake ratio and the fluorescence intensity of CeO2@PMETAC NPs are higher than that of CeO2@PSPM NPs.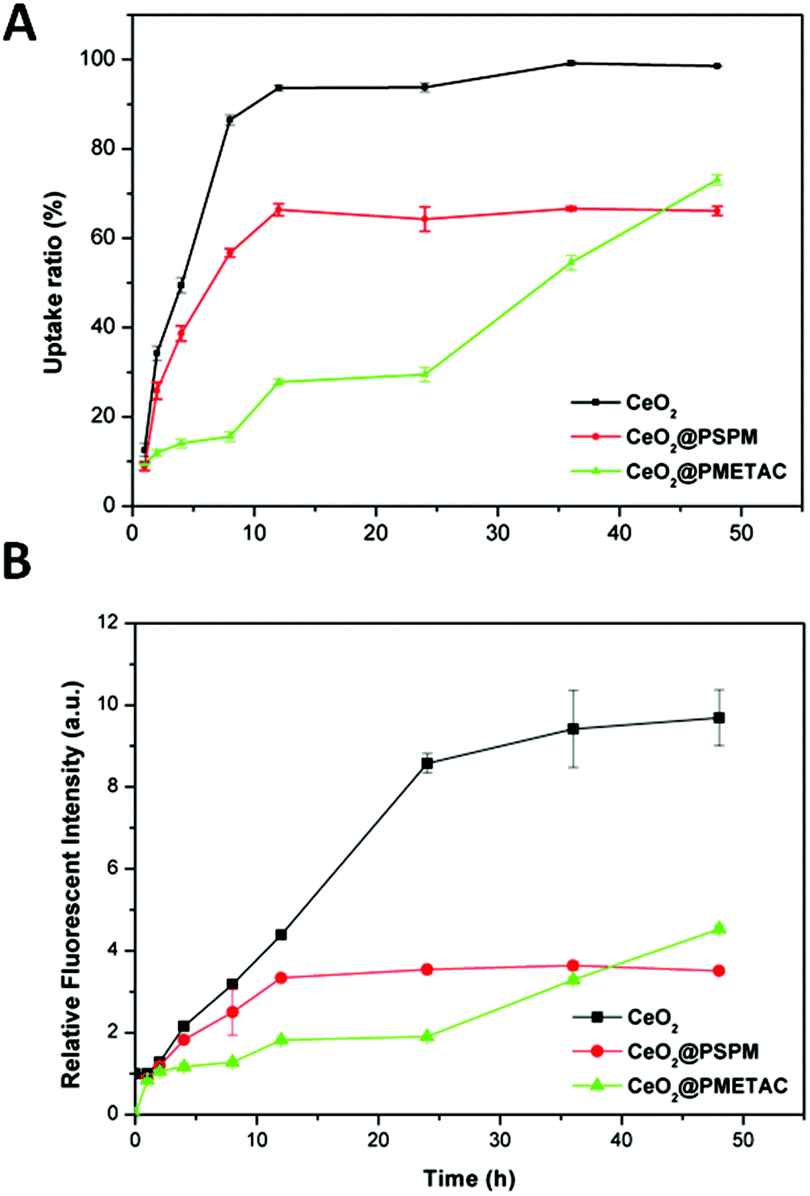 | ||
| Fig. 3 (a) Cellular uptake ratio and (b) relative fluorescence intensity of cerium oxide with different coatings as a function of the incubation time with HEK293 cells. | ||
Extracellular substances enter the cell via a process termed ‘endocytosis’. In this process, macromolecules and particles are carried into cells in vesicles derived by the invagination and pinching-off of pieces of the plasma membrane. Endocytosis includes several different mechanisms, which can be mainly divided into ‘phagocytosis’ (cell eating) and ‘pinocytosis’ (cell drinking).38
The amiloride-HCl drug inhibits uptake via macropinocytosis by blocking the Na+/H+ exchanger.39,40 Chlorpromazine (CPM) is a clathrin-mediated pathway inhibitor, which inhibits the formation of clathrin-coated pits.41–44 Filipin is a cholesterol binding agent that can inhibit caveolae-mediated uptake by the flattening of caveolae and the disruption of caveolin protein coating.45,46 Cytochalasin D is a actin filament depolymerising, which can block cell uptake via membrane ruffling involving actin filament (macropinocytosis).47,48
Inhibition experiments were conducted with the A549 cell line by means of flow cytometry.49 Results are shown in Fig. 4. The uptake of all three CeO2−x NPs is significantly reduced in presence of sodium azide, below 40%. From this it can be concluded that the uptake of the CeO2−x NPs with and without brushes, is energy-dependent. Treatment with amiloride affects the uptake of CeO2 coated with PSPM, which is reduced below 60%, hinting that their uptake mechanism takes place through macropinocytosis. There is no significant inhibition of CeO2−x coated with PMETAC, which remains over 80%. In all cases the error bar is high. Uptake of CeO2−x NPs with PSPM is also inhibited by amantadine HCl supporting the idea that the uptake occurs via macropinocytosis. All other inhibitors used do not cause a significant reduction in the uptake.
 | ||
| Fig. 4 Relative uptake of cerium oxide NPs by A549 cells with pretreatment with endocytosis pathway inhibitors: sodium azide (100 mM), amantadine-HCl (1 mM) amiloride (2.5 mM), cytochalasin D (10 μg ml−1), filipin (5 μg ml−1). | ||
Inductively coupled plasma mass spectroscopy (ICP-MS)
ICP-MS showed similar results to FACS for the non labelled NPs (see Fig. S7 in ESI†), suggesting that labelling does not have an influence on the uptake as expected due to placing the dye inside the brush. At the beginning of the incubation time (1 h), the three NPs showed very similar uptake. After 48 h, the CeO2−x NPs showed the highest uptake compared to the other two NPs. Nevertheless, the differences in the amount of NP uptake between CeO2@PSPM and CeO2@PMETAC NPs after 48 h as measured by ICP-MS are much larger than the differences between these two types of NPs in fluorescence intensity from FACS (Fig. 3a and b). One possible reason is that the fluorescence intensity can reveal the quantity of NPs but it is not necessary linearly associated with it.Confocal Raman microscopy (CRM)
The strong and distinct Raman peak of cerium oxide, at approximately 465 cm−1, allows us to distinguish the presence of the CeO2−x NPs in the cells without fluorescently labelling them This is particularly important since it allow us to compare the uptake of unmodified CeO2 NPs with polymer brushes coated NPs and gain additional information on NP localisation.Fluorescence techniques required the labelling of the CeO2−x NPs without brushes using a procedure that minimises changes to the surface characteristics of the NPs but nevertheless the surface of the CeO2−x NPs is to a certain extent modified. As has been shown in the ESI,† coating CeO2−x NPs with polymer brushes does not affect the Raman spectra of the NPs, allowing us to have a direct comparison between the impact of the polymer modification of the NPs and changes in their uptake in relation to the unmodified NPs. Raman also allows us to verify if the intracellular localisation of the labelled NPs is different from the unlabelled NPs and consequently if the labelling has had an impact on the intracellular localisation and uptake. The molecules present in the cells: proteins, lipids and nucleic acids have distinctive Raman bands, which are indicative of their molecular characteristics. Since the proportion of lipids, proteins and nucleic acids varies in the different cell compartments the relative intensities of their bands can be used to distinguish regions within the cell from the Raman spectra measured intracellularly, without labelling the cell. When nanomaterials, with a measurable Raman spectra, are uptaken by cells the Raman spectra recorded within the cells result in a combination of the Raman bands of the NPs and that of the cellular components. If the bands of the nanomaterials do not overlap with those of the cellular components it will be possible to identify the cellular environment of the NPs.50Fig. 5 shows a Raman spectrum from cells after incubation with CeO2−x NPs, unmodified and with polymer brushes (red line), the Raman spectrum of NPs (green line) and a Raman spectrum from untreated cells (blue line). The spectra from cells were taken from the central plane of the cell in both cases. For cells exposed to NPs, the peak of cerium oxide at 465 cm−1 can be found together with the signal from cell components such as protein and lipids, which can also be visualised in the unexposed cells.
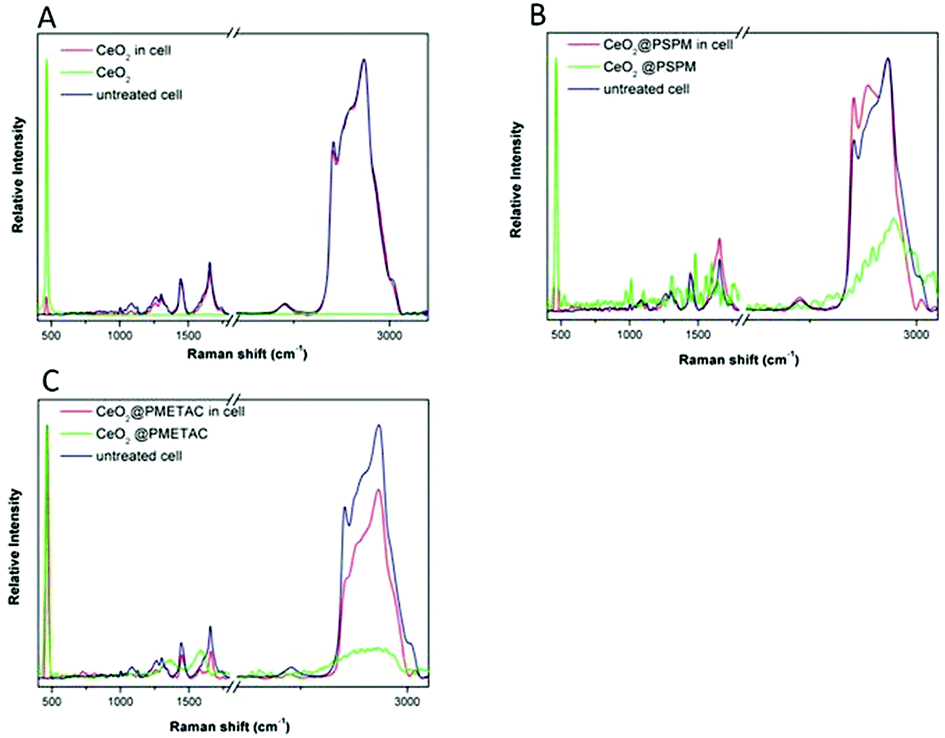 | ||
| Fig. 5 Raman spectra taken in HEK293 cells after 24 h incubation with (a) CeO2 NPs, (b) CeO2@PSPM NPs, (c) CeO2@PMETAC NPs. | ||
Cells with internalised NPs were mapped by applying CRM recording Raman spectra point by point. We performed Raman mapping at the middle plane of each individual cell. XY Raman mapping was performed to obtain information about the distribution and intracellular localisation of CeO2−x NPs.
The maximum value of the Pearson product-moment correlation coefficient, ρx,y(Δ), as described in the Experimental section can be used as an evaluation of the correlation between CeO2−x NPs and lipids. The correlation of Raman signals of CeO2−x NPs and lipids along the line of X axis and the line of Y axis were calculated for the unmodified and brush coated CeO2−x NPs. The maximum intensity of CeO2 NPs and lipids do not coincide along the X and Y axes meaning a limited co-localisation (Fig. 5a). On the contrary, cells with CeO2@PSPM and CeO2@PMETAC NPs show a higher degree of co-localisation of both maximum and minimum intensity of NPs and lipids (Fig. 5b and c).This indicates that CeO2 NPs do not have a tendency to co-localise with the lipid rich regions inside cells while modified CeO2−x NPs show a preference to co-localise with lipid rich intracellular regions. Moreover, the calculated maximum values of ρx,y(Δ) for CeO2 NPs are 0.42 and 0.45 along the X and Y axes respectively, while that of CeO2@PSPM NPs are 0.79 and 0.60 along the X and Y axes respectively (Fig. 6) and that of CeO2@PMETAC NPs are 0.79 along the X axis and 0.59 along the Y axis (Fig. 6). A ρx,y(Δ) value closer to 1 means a higher linear dependence of the two components while closer to 0 means a lower linear dependence. Therefore, it can be concluded that CeO2−x NPs have a lower linear dependence with lipids inside the cell than CeO2−x NPs modified with polymer brushes.
 | ||
| Fig. 6 Raman mapping data inside a HEK293 cell incubated with CeO2 NPs for 24 h. (a, d, and f) show typical cells in which mapping was conducted; (b, e, and h) show the intensity distribution of CeO2 NPs corresponding to the CeO2 band (red line) and lipids (black line) inside HEK293 cells along the X axis. (c, f, and i) show the intensity distribution of CeO2 NPs corresponding to the CeO2 band (red line) and lipids (black line) inside HEK293 along the Y axis. The X and Y lines were chosen arbitrarily in the centre of the cell. | ||
Lipid rich intracellular regions in the cell can be associated with the presence of lipid bodies. Lipid bodies are cytoplasmic organelles found in most cell types, formed by neutral lipids, such as triacylglycerides, diacylglycerol and cholesterol esters, and a surrounding phospholipid monolayer. The high density of lipids in the lipid bodies results in a strong Raman band corresponding to CH2, which cannot be detected from the cell membrane or the membranes of the lysosomes.
CRM proves co-localisation of the brush coated NPs while CLSM shows co-localisation with the lysosomes. Therefore, it can then be concluded that the lysosomes and lipid bodies are also in close vicinity in the cytoplasm. A similar result was observed by Khatchadourian et al. for CdTe NPs in pheochromocytoma cells.51 In the presence of these NPs the amount of lipid bodies seems to increase in parallel with an increase in oxidative stress in the cell. The authors use a dual labelling of lysosomes and the body lipids for the visualisation of both organelles. Here we use two independent techniques for the visualisation of lipid bodies and lysosomes. The localisation of the brush coated NPs in the lysosomes could also be proven with TEM.
Transmission electron microscopy (TEM)
In the TEM images in Fig. 7, CeO2−x NPs can be recognised inside the cell by their shape and very high contrast. The image clearly shows that complete cellular internalisation of all three types of CeO2−x NPs occurs after 1 h incubation. CeO2−x NPs inside cells are mostly present in clusters from several hundreds of nanometres to 2 μm. Closer inspection of the images reveals that the majority of NPs are enclosed in intracellular vacuoles. These vacuoles are the lysosomal cell compartments. This indicates that CeO2−x NPs were internalised typically through endocytosis, which involves the formation of vesicles from the plasma membrane. The TEM results are in good agreement with the CLSM results, where most of internalised NPs were inside lysosomal compartments. Vacuoles larger than 1 μm can be associated with macropinosomes, which are the large, irregular primary endocytic vesicles formed during macropinocytosis.52 The intracellular fate of macropinosomes varies depending on the cell type but in most cases ends up fusing with lysosomes and shrinking.53 It is worth noting that, besides the NP clusters, there are also some single NPs or very small clusters of NPs present in the cytoplasm which are probably single NPs that penetrated the cell membrane through passive diffusion. | ||
| Fig. 7 TEM images of HEK293 cells treated with CeO2 NPs for (a) 1 h, (d) 24 h; with CeO2@PSPM NPs for (b) 1 h, (e) 24 h; with CeO2@PMETAC NPs for (c) 1 h, (f) 24 h. The insets are the closer view of the corresponding image. | ||
As ICP-MS confirms the FACS data, the patterns of intracellular localisation observed by CLSM are in agreement with the TEM data for the unlabelled NPs.
Reactive oxygen species assay
Cerium oxide NPs are well known for their superoxide scavenging capacity by the reversible binding of oxygen and reversible equilibrium between Ce3+ and Ce4+. Therefore, the oxidative stress level of the cells treated with cerium oxide NPs with PSPM and PMETAC brushes was studied.FACS measurements showed that cells not exposed to the NPs but exposed to tert-butyl hydrogen peroxide (TBHP) showed a relatively high level of reactive oxygen species whereas cells treated with NPs exhibited negative oxidative stress level (Fig. 8). Negative ROS means, in this case, that the ROS values are lower than those cells not exposed to TBHP. This indicates that all the cerium oxide NPs possess the capacity to scavenge ROS and protect cells from oxidative stress generated by TBHP. Compared to unmodified CeO2−x NPs, cells exposed to polymer brush coated NPs exhibited higher ROS levels but still much lower than untreated cells. This means that the polymer brush on the surface of the NPs hinders the oxygen binding and the redox changes between Ce3+ and Ce4+ to some extent, but the antioxidant capacity of the NPs is still retained when compared to cells without treatment. For future experiments it is planned to synthesize thicker and denser brushes increasing either the polymerization time or the density of the initiators. The denser the brush around the NPs the more the brush should impact on the transport of the oxygen species and the less the antioxidant effect of the CeO2−x NPs should be noticeable.
 | ||
| Fig. 8 Level of the reactive oxygen species of HEK293 cells treated with NPs. The 0 corresponds to the cells without TBHP and nanoparticles. Untreated refers to the cells exposed only to TBHP and not to the nanoparticles. | ||
Conclusions
The use of brushes for NP modification increases the surface charge of the NPs providing greater aqueous stability and reducing NP aggregation. The brush functionalisation also allows attachment of a fluorescent dye close to the surface of the CeO2−x that remains shielded in the polymer coating without affecting the surface characteristics of the NPs, which could affect the intracellular localisation and uptake of the NPs. The results from CLSM and FACS were corroborated by TEM and ICPMS with unlabelled NPs proving that the labelling does not indeed impact on uptake and intracellular localisation.The surface coating of CeO2−x NPs with polyelectrolyte brushes has a clear impact on the uptake and localisation of the NPs intracellularly. CeO2@PMETAC NPs result in a higher degree of uptake compared to CeO2@PSPM NPs. However, in both cases uptake is significantly less than for unmodified NPs. The uptake rate seems to proceed more slowly for the CeO2@PMETAC NPs but uptake continues progressively, even after the uptake of CeO2@PSPM NPs and CeO2 NPs has reached plateau. Interestingly, coating the NPs with brushes and the charge of the brushes affects the uptake routes. CeO2−x NPs and CeO2@PSPM are mainly internalised by cells preferentially through macropinocytosis but not CeO2@PMETAC NPs.
Moreover, polymer brush coated CeO2−x NPs showed higher co-localisation with acidic cell compartments in the cell, meaning that these CeO2−x NPs were mostly internalised through endosomal and lysosomal involved endocytosis. In addition, the brush coated NPs showed co-localisation with lipid bodies, which seem to be associated with the lysosomes in the presence of these NPs. For uncoated NPs no co-localisation with lipid bodies was found and the co-localisation with lysosomes are limited.
Finally, the brush coating does not prevent the NPs from having antioxidant properties; though these are slightly decreased in relation with the unmodified CeO2−x NPs.
Acknowledgements
This work was supported by the European Commission in the framework of FP7 NMP-2008 Project HINAMOX Proposal no: CP-FP28825-2, FP7 People-Pirses-Brasinoeu Grant agreement 319816 and the FP7 capacities project QNANO Grant reference: 262163. The authors also acknowledge the project MAT2013-48169-R from the Spanish Ministry of Economy (MINECO) and the Department of Industry of the Basque Country (grant ETORTEK 2011).Notes and references
- G. Romero, I. Estrela-Lopis, P. Castro-Hartmann, E. Rojas, I. Llarena, D. Sanz, E. Donath and S. E. Moya, Soft Matter, 2011, 7, 6883–6890 RSC.
- M. Ballauff, Prog. Polym. Sci., 2007, 32, 1135–1151 CrossRef CAS PubMed.
- X. Guo, A. Weiss and M. Ballauff, Macromolecules, 1999, 32, 6043–6046 CrossRef CAS.
- J. Irigoyen, V. B. Arekalyan, Z. Navoyan, J. Iturri, S. E. Moya and E. Donath, Soft Matter, 2013, 9, 11609–11617 RSC.
- S. Yang and L. Gao, J. Am. Chem. Soc., 2006, 128, 9330–9331 CrossRef CAS PubMed.
- A. Srinivas, P. J. Rao, G. Selvam, P. B. Murthy and P. N. Reddy, Toxicol. Lett., 2011, 205, 105–115 CrossRef CAS PubMed.
- C. Wang and S. Lin, Appl. Catal., A, 2004, 268, 227–233 CrossRef CAS PubMed.
- Q. Zhuang, Y. Qin and L. Chang, Appl. Catal., 1991, 70, 1–8 CrossRef CAS.
- A. M. Silva, R. R. Marques and R. M. Quinta-Ferreira, Appl. Catal., B, 2004, 47, 269–279 CrossRef CAS PubMed.
- V. D. Kosynkin and A. a. Arzgatkina, J. Alloys Compd., 2000, 303–304, 421–425 CrossRef CAS.
- P. Janoš and M. Petrák, J. Mater. Sci., 1991, 26, 4062–4066 CrossRef.
- F. R. Cassee, E. C. van Balen, C. Singh, D. Green, H. Muijser, J. Weinstein and K. Dreher, Crit. Rev. Toxicol., 2011, 41, 213–229 CrossRef PubMed.
- B. Park, K. Donaldson, R. Duffin, L. Tran, F. Kelly, I. Mudway, J.-P. Morin, R. Guest, P. Jenkinson, Z. Samaras, M. Giannouli, H. Kouridis and P. Martin, Inhalation Toxicol., 2008, 20, 547–566 CrossRef CAS PubMed.
- S. Deshpande, S. Patil, S. V. N. T. Kuchibhatla and S. Seal, Appl. Phys. Lett., 2005, 87 Search PubMed.
- C. Korsvik, S. Patil, S. Seal and W. T. Self, Chem. Commun., 2007, 1056–1058 RSC.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Nano Lett., 2005, 5, 2573–2577 CrossRef CAS PubMed.
- S. S. Lee, W. Song, M. Cho, H. L. Puppala, P. Nguyen, H. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS PubMed.
- A. Karakoti, S. Singh, J. M. Dowding, S. Seal and W. T. Self, Chem. Soc. Rev., 2010, 39, 4422–4432 RSC.
- S. M. Hirst, A. S. Karakoti, R. D. Tyler, N. Sriranganathan, S. Seal and C. M. Reilly, Small, 2009, 5, 2848–2856 CrossRef CAS PubMed.
- S. Hussain, F. Al-Nsour, A. B. Rice, J. Marshburn, B. Yingling, Z. Ji, J. I. Zink, N. J. Walker and S. Garantziotis, ACS Nano, 2012, 6, 5820–5829 CrossRef CAS PubMed.
- E.-J. Park, J. Choi, Y.-K. Park and K. Park, Toxicology, 2008, 245, 90–100 CrossRef CAS PubMed.
- W. Lin, Y. Huang, X.-D. Zhou and Y. Ma, Int. J. Toxicol., 2006, 25, 451–457 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Ma, B. Gilbert, Ќ. H. Shi, J. I. Yeh, J. I. Zink, A. E. Nel, L. Mädler and H. Shi, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- M. S. Lord, M. Jung, W. Y. Teoh, C. Gunawan, J. A. Vassie, R. Amal and J. M. Whitelock, Biomaterials, 2012, 33, 7915–7924 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- S. Singh, A. Kumar, A. Karakoti, S. Seal and W. T. Self, Mol. BioSyst., 2010, 6, 1813–1820 RSC.
- M. Wason and J. Zhao, Am. J. Transl. Res., 2013, 5, 126–131 CAS.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- L. K. Limbach, Y. Li, R. N. Grass, T. J. Brunner, M. a. Hintermann, M. Muller, D. Gunther and W. J. Stark, Environ. Sci. Technol., 2005, 39, 9370–9376 CrossRef CAS.
- C. K. Kim, T. Kim, I.-Y. Choi, M. Soh, D. Kim, Y.-J. Kim, H. Jang, H.-S. Yang, J. Y. Kim, H.-K. Park, S. P. S. Park, T. Yu, B.-W. Yoon, S.-H. Lee and T. Hyeon, Angew. Chem., Int. Ed., 2012, 51, 11039–11043 CrossRef CAS PubMed.
- S. Das, J. M. Dowding, K. E. Klump, J. F. McGinnis, W. Self and S. Seal, Nanomedicine, 2013, 8, 1483–1508 CrossRef CAS PubMed.
- J. Jiang, G. Oberdörster and P. Biswas, J. Nanopart. Res., 2008, 11, 77–89 CrossRef.
- M. Engelhard, S. Azad and S. Thevuthasan, Surf. Sci. Spectra, 2005, 11, 73–81 Search PubMed.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. J. Rumble, NIST Standard Reference Database 20 version 3.4, 2004 Search PubMed.
- J. J. Pireaux, J. Electron Spectrosc. Relat. Phenom., 1993, 62, 371–372 CrossRef.
- Y. Chen, G. Xiong and E. a. Arriaga, Electrophoresis, 2007, 28, 2406–2415 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis, J. M. Perez and O. Florida, ACS Nano, 2010, 4, 5321–5331 CrossRef CAS PubMed.
- S. D. Conner and S. L. Schmid, Nature, 2003, 422, 37–44 CrossRef CAS PubMed.
- N. S. Dangoria, W. C. Breau, H. A. Anderson, D. M. Cishek and L. C. Norkin, J. Gen. Virol., 1996, 77(Pt 9), 2173–2182 CrossRef CAS.
- D. Werling and J. Hope, J. Leukocyte Biol., 1999, 66, 50–58 CAS.
- K. Saha, S. T. Kim, B. Yan, O. R. Miranda, F. S. Alfonso, D. Shlosman and V. M. Rotello, Small, 2013, 9, 300–305 CrossRef CAS PubMed.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468–474 CrossRef CAS PubMed.
- K. Tahara, T. Sakai, H. Yamamoto, H. Takeuchi, N. Hirashima and Y. Kawashima, Int. J. Pharm., 2009, 382, 198–204 CrossRef CAS PubMed.
- A. W. Cohen, R. Hnasko, W. Schubert and M. P. Lisanti, Physiol. Rev., 2004, 84, 1341–1379 CrossRef CAS PubMed.
- M. R. Jackman, W. Shurety, J. a. Ellis and J. P. Luzio, J. Cell Sci., 1994, 107(Pt 9), 2547–2556 CAS.
- Y.-L. Chiu, Y.-C. Ho, Y.-M. Chen, S.-F. Peng, C.-J. Ke, K.-J. Chen, F.-L. Mi and H.-W. Sung, J. Controlled Release, 2010, 146, 152–159 CrossRef CAS PubMed.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailänder, Macromol. Biosci., 2008, 8, 1135–1143 CrossRef CAS PubMed.
- G. Romero, I. Estrela-Lopis, J. Zhou, E. Rojas, A. Franco, C. S. Espinel, A. G. Fernández, C. Gao, E. Donath and S. E. Moya, Biomacromolecules, 2010, 11, 2993–2999 CrossRef CAS PubMed.
- A. Khatchadourian and D. Maysinger, Mol. Pharm., 2009, 6, 1125–1137 CrossRef CAS PubMed.
- B. J. Nichols and J. Lippincott-Schwartz, Trends Cell Biol., 2001, 11, 406–412 CrossRef CAS.
- H. Meng, S. Yang, Z. Li, T. Xia, J. Chen, Z. Ji, H. Zhang, X. Wang, S. Lin, C. Huang, Z. H. Zhou, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4434–4447 CrossRef CAS PubMed.
- I. Llarena, J. J. I. Ramos, E. Donath and S. E. Moya, Macromol. Rapid Commun., 2010, 31, 526–531 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- E. K. Goharshadi, S. Samiee and P. Nancarrow, J. Colloid Interface Sci., 2011, 356, 473–480 CrossRef CAS PubMed.
- H. Willis, V. J. Zichy and P. Hendra, Polymer, 1969, 10, 737–746 CrossRef CAS.
- I. Estrela-Lopis, G. Romero, E. Rojas, S. E. Moya and E. Donath, J. Phys.: Conf. Ser., 2011, 304 CrossRef CAS , 012017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr00884k |
| This journal is © The Royal Society of Chemistry 2015 |