
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA hydrogel delivery vehicle for light-triggered and synergistic cancer therapy†
Jaejung
Song
a,
Kyuhyun
Im
b,
Sekyu
Hwang
c,
Jaehyun
Hur
d,
Jutaek
Nam
c,
G-One
Ahn
e,
Sungwoo
Hwang
b,
Sungjee
Kim
*ac and
Nokyoung
Park
*f
aSchool of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science & Technology (POSTECH), San 31, Hyojadong, Pohang 790-784, Korea. E-mail: sungjee@postech.ac.kr; Tel: +82-54-279-2108
bSamsung Advanced Institute of Technology, Samsung Electronics, Yongin, Gyeonggi-do 446-712, Korea
cDepartment of Chemistry, POSTECH, Korea
dDepartment of Chemical and Biological Engineering, Gachon University, Seongnam, Gyeonggi-do 461-701, Korea
eDivision of Integrative Biosciences and Biotechnology, POSTECH, Korea
fDepartment of Chemistry, Myongji University, Yongin, Gyeonggi-do 449-728, Korea. E-mail: pospnk@mju.ac.kr; Tel: +82-31-330-6188
First published on 11th May 2015
Abstract
A DNA hydrogel is reported as a delivery vehicle for gold nanorods and doxorubicin. The two photothermal and chemo cancer agents were co-loaded using electrostatic and DNA binding interactions, respectively. Light-triggered and highly synergistic combination cancer therapy was demonstrated in cellular and animal models.
Gold nanostructures of which the surface plasmon resonance (SPR)1 is tuned at near-infrared (NIR) wavelengths such as nanorods,2 nanoshells,3 and nanocages,4 have demonstrated potential for many deep-tissue penetrating biomedical applications that include photothermal therapy, Raman imaging and radio-sensitization. Gold nanorods (AuNRs) have been widely used for photothermal effects because of their relative simplicity in synthesis, their strong local heating upon NIR irradiation that cannot be paralleled by conventional organic dye-based agents, and resistance against photobleaching.5 Typically, many photothermal agents including gold nanoparticles (NPs) can carry a number of small molecules by their flexible surface functionalities for efficient drug delivery.6 However, most of the AuNR surfaces are covered by surfactants such as cetyltrimethylammonium bromide (CTAB) which is essential for NR morphology control but lacks conjugation capability. As the result, it is difficult to attain a AuNR hybrid system that integrates conventional anticancer agents in the proximity of AuNR and permits multimodal thermo-chemo combination cancer therapy.7 To achieve the maximal synergistic effect by combination therapy, the proximity between the two agents is crucial.8 In cases where chemo-agents were combined with NPs by physical mixtures7a,b or by loosely bound conjugates through hydrophobic interactions,7c the weak binding between the two agents often results in undesired separation upon therapeutic administrations which can significantly hamper the spatially concerted action for synergistic effects. Herein, we report a novel AuNR-based thermo-chemo combination therapy platform with a DNA hydrogel (Dgel) (Scheme 1). Dgels are three-dimensional structures composed of internally cross-linked DNA monomers with many nanoscale pores (Scheme 1a).9 Because the DNA backbone is highly negatively charged, AuNRs which are positively surface-charged can be stably incorporated into the Dgel pores by electrostatic attractions. Dgels are also exploited as templates for small anticancer drug loading. Dgels can be used as delivery vehicles for DNA-binding anticancer drugs with strong binding affinity preventing biodegradation or uncontrolled release of the payloads.9a Recently, we have reported a Dgel-AuNP assembly system for synergistic cancer therapy.10 Dgels can be used as a biocompatible scaffold with unique melting characteristics by thermal denaturation, which can be used for triggered drug release and/or programmed disassembly after therapy (Scheme 1b). The Dgel platform with NIR-responsive AuNRs and DNA-binding anticancer drugs allow stable NR and drug loadings and ‘on-demand’ type activation of the therapeutic combination action by external light triggering.
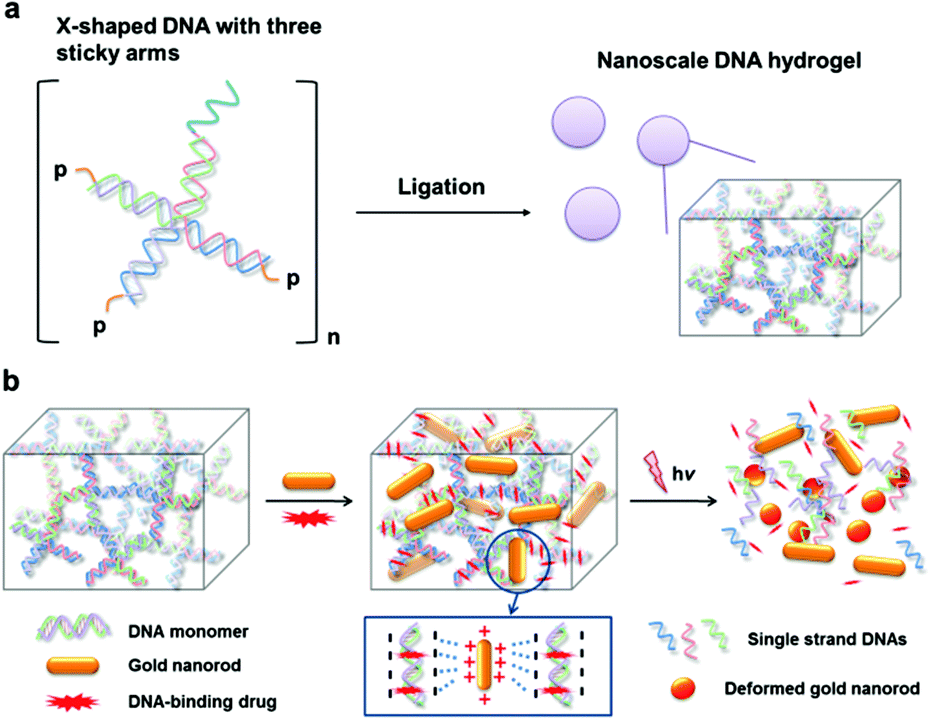 | ||
| Scheme 1 (a) Schematic drawing of Dgel nanoscaffold synthesis. The X-DNAs with one disabled arm (left) are mixed and ligated to produce nanoscale Dgel (right). (b) Schematic illustration of the DNA gel, gold nanorod incorporation, and cancer drug loading. External light triggering induces the drug release by DNA melting, which permits synergistic thermo-chemo combination cancer therapy. | ||
Positively charged AuNRs (length 35.6 (±2.6) nm, width 20.9 (±9.9) nm) were prepared by following a seedless growth method described in the literature (see Fig. 1a for the TEM image).11 Excess free CTAB in the AuNR solution was rigorously removed by centrifugation and decanting. X-shaped DNA (X-DNA) was designed to have three sticky ends and one disabled end to control the size of Dgel upon ligation (Scheme 1a, Table 1). The self-confined geometry by the disabled arm provided control over the crosslinking and thus yielded Dgels around one hundred nanometers in size. In the case of X-DNA with all four sticky ends, the enzymatic crosslinking resulted in bulk Dgels of over tens of microns in size. Freeze dried Dgel was swelled under AuNR aqueous solution. Then, the mixture was centrifuged to remove unassembled free Dgels and AuNRs. AuNR incorporated Dgel (AuNR-Dgel) was prepared by mixing Dgels with AuNRs in deionized water. When a Dgel sample prepared by the ligation of 10 nmol X-DNA was co-incubated with 2.5 pmol AuNR, near complete incorporation was confirmed spectrophotometrically by the absence of free AuNR in the supernatant after centrifugation. The Dgel retained colloidal stability after AuNR loading. TEM measurement showed tens to hundreds of AuNRs densely incorporated into a Dgel (Fig. 1b). The average hydrodynamic (HD) sizes determined by dynamic light scattering were 32 (±14) nm for the AuNR, 87 (±6) nm for the Dgel, and 148 (±67) nm for the AuNR-Dgel (Fig. S1†). The HD size increase for the Dgel upon AuNR loading suggests partial inclusion of the AuNRs in the Dgel, which is expected when considering the significant size of the AuNRs relative to the Dgel. This was supported by the partial charge compensation of the Dgel upon AuNR incorporation. The Dgel initially showed a Zeta potential of −46 mV which reduced (in absolute value) to −27 mV upon the incorporation of the AuNRs of which the Zeta potential was 34 mV (Fig. S2†). Fig. 1c shows the absorption profiles of the AuNRs, Dgel, and AuNR-Dgel. The AuNRs initially show two distinct plasmon peaks at 515 and 650 nm which correspond to the transverse and longitudinal modes, respectively. Upon incorporation into the Dgel, the AuNRs showed broadened and red-shifted plasmon peaks. The transverse peak showed ∼20 nm red-shift, whereas the longitudinal peak did not show much shift although it manifested a noticeable tailing towards longer wavelengths. It has been reported by others that a side-by-side assembly of eight AuNRs (which showed a similar aspect ratio to what we have used in this paper) resulted in a 20 nm transverse peak shift.12 The larger red-shift of the transverse peak over the longitudinal peak is presumably due to dominant side-by-side plasmon couplings between the AuNRs inside the Dgel. The DNA peak at 260 nm remained unchanged after the AuNR incorporation, suggesting that the AuNR loading does not severely perturb the DNA stacking backbones in the Dgel.
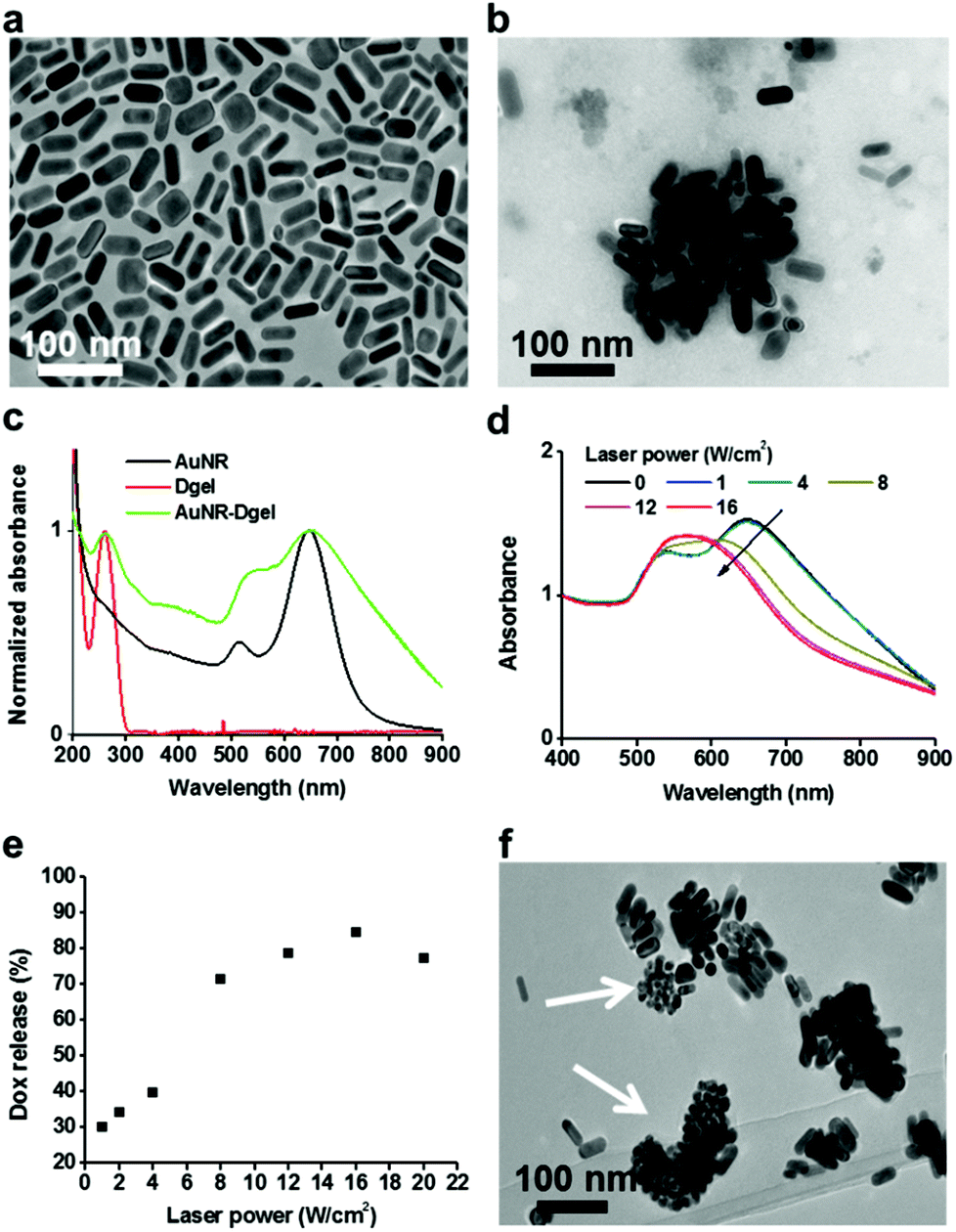 | ||
| Fig. 1 TEM images of the AuNRs (a) and AuNR-Dgel (b). (c) Absorption spectra of AuNRs, Dgel, and AuNR-Dgel. Laser power dependent absorption spectra (d) and Dox release percentages (e) of the Dox-AuNR-Dgel upon irradiation for 5 min. (f) TEM image of Dox-AuNR-Dgel after laser irradiation at 16 W cm−2 for 5 min. | ||
| Strand | End segment | Main body segment |
|---|---|---|
| a 5′-p represents the phospholylated 5′ end of oligonucleotide. | ||
| X01 | 5′-p-ACGTa | CGA CCG ATG AAT AGC GGT CAG ATC CGT ACC TAC TCG-3′ |
| X02 | 5′-p-ACGT | CGA GTA GGT ACG GAT CTG CGT ATT GCG AAC GAC TCG-3′ |
| X03 | 5′-p-ACGT | CGA GTC GTT CGC AAT ACG GCT GTA CGT ATG GTC TCG-3′ |
| X04 | 5′-(GT)20 | CGA GAC CAT ACG TAC AGC ACC GCT ATT CAT CGG TCG-3′ |
The high-density DNA strands of the Dgel was used to co-load a DNA binding drug. To obtain doxorubicin (Dox) and AuNR co-loaded Dgel (Dox-AuNR-Dgel), Dox was co-incubated for 1 h with AuNR-Dgel in deionized water and unloaded free Dox was removed by centrifugation. The amount of loaded Dox (obtained by subtracting the amount of unloaded Dox from the co-incubated Dox) and the number of AuNRs in the AuNR-Dgel sample were obtained by a spectrophotometric method using extinction coefficients reported elsewhere.13 On average, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Dox molecules were loaded per AuNR (Fig. S3†). Based on an earlier hypothesis that one million X-DNAs construct a Dgel, each Dox-AuNR-Dgel was co-loaded with 250 AuNRs and 2.8 million Dox molecules which corresponds to 2.8 Dox molecules being incorporated into each X-DNA building block.
000 Dox molecules were loaded per AuNR (Fig. S3†). Based on an earlier hypothesis that one million X-DNAs construct a Dgel, each Dox-AuNR-Dgel was co-loaded with 250 AuNRs and 2.8 million Dox molecules which corresponds to 2.8 Dox molecules being incorporated into each X-DNA building block.
Dox-AuNR-Dgel is designed for the light-triggered release of Dox by plasmonic heat shock. To test if Dox could be released upon photothermally-triggered DNA melting, a 660 nm laser which matches the major plasmonic peak of the AuNR was irradiated to the Dox-AuNR-Dgel samples at different laser power densities for 5 min each. As the laser power increased, the absorption blue-shifted progressively. The longitudinal peak of the AuNR shifted abruptly above 8 W cm−2, which indicates that the AuNRs generated sufficient energy to melt the Dgel (Fig. 1d).14 This also accords with the drastically increased Dox release at or above 8 W cm−2 laser power (Fig. 1e). The amount of Dox released by the light-triggering was investigated by the supernatant absorption measurements after centrifugation. A linear increase by the laser power up to 16 W cm−2 followed by a plateau region was observed (Fig. 1e and Fig. S4†). Efficient Dox release was demonstrated with a percentage release as high as 85%. The TEM measurements show fragmentation of the Dgel template and shape transformations of the AuNRs into more spherical particles (Fig. 1f). Such AuNR shape deformation is known to occur at a local temperature of above 200 °C,14 which explains well the efficient DNA melting and subsequent Dox release. Nearby, the complete release of Doxs from Dgel is expected as the Dgel turns into single strands above the melting temperature from the Dox intercalated B-form of the DNA double stranded helix.15
The successful demonstration of light-triggered release of heat shock and Dox allowed us to investigate cellular level combination cancer therapy. B16 F10 cells were incubated with Dox-AuNR-Dgel which had been prepared by the ligation of 0.24 nmol X-DNA and subsequent co-loading of 0.06 pmol AuNR and 0.03 nmol Dox. Control samples of AuNR-Dgel and Dox only were also prepared using identical concentrations. Cellular accumulations of AuNRs were monitored over time using dark-field microscopy. The Dox-AuNR-Dgel and AuNR-Dgel showed similar accumulations over time as indicated by the increased orange-color scatterings visualizing the cell morphologies (Fig. 2a). Cell viabilities were also measured over time by MTT assay. Under no laser light, no significant cytotoxicity was observed up to 12 h in all cases (Fig. 2b). At 24 h, slight cytotoxicity was observed for the Dox only control. Light-triggered release of Dox was confirmed by the appearance of Dox red fluorescence as recovered from the quenched state in the Dox-AuNR-Dgel (Fig. 2c). Quantitative measurements for the fluorescence can be found in Fig. S5.† Since internalization of the Dox-AuNR-Dgel and Dox release were confirmed, we further performed thermo-chemo combination cancer therapy tests. The co-incubation conditions used for the dark-field microscopy and MTT assays were adopted, and a 660 nm laser was used for illumination. The circles in Fig. 2d represent the position of the 660 nm laser spots. The spot has a ∼500 μm radius, as measured using photosensitive paper. Each sample was illuminated for 5 min using different laser power densities of 12, 16, 20, 24, and 28 W cm−2. Trypan blue was used to reveal the mortality of cells by blue staining. In the case of the Dox-AuNR-Dgel, cell mortality appeared at a laser power of 16 W cm−2 or higher. The circular areas of damaged cells increased linearly with the irradiation power density. AuNR-Dgel showed a significantly larger threshold, showing mortality at 20 W cm−2 or higher. On the other hand, no noticeable cell mortality was found for the Dox and non-treated controls. It is noted that the cells outside of the laser spot also showed no mortality. As the Dox release concurrently burst with the heat shock, the thermo and chemo effects boosted each other. This synergistic effect resulted in the reduction of the laser threshold for the Dox-AuNR-Dgel over the AuNR-Dgel. Conversely, this combined therapy would require a smaller amount of Dox than the uncombined case. The synergistic effect can potentially mitigate the side effect of patients by reducing the dosage (or laser power).
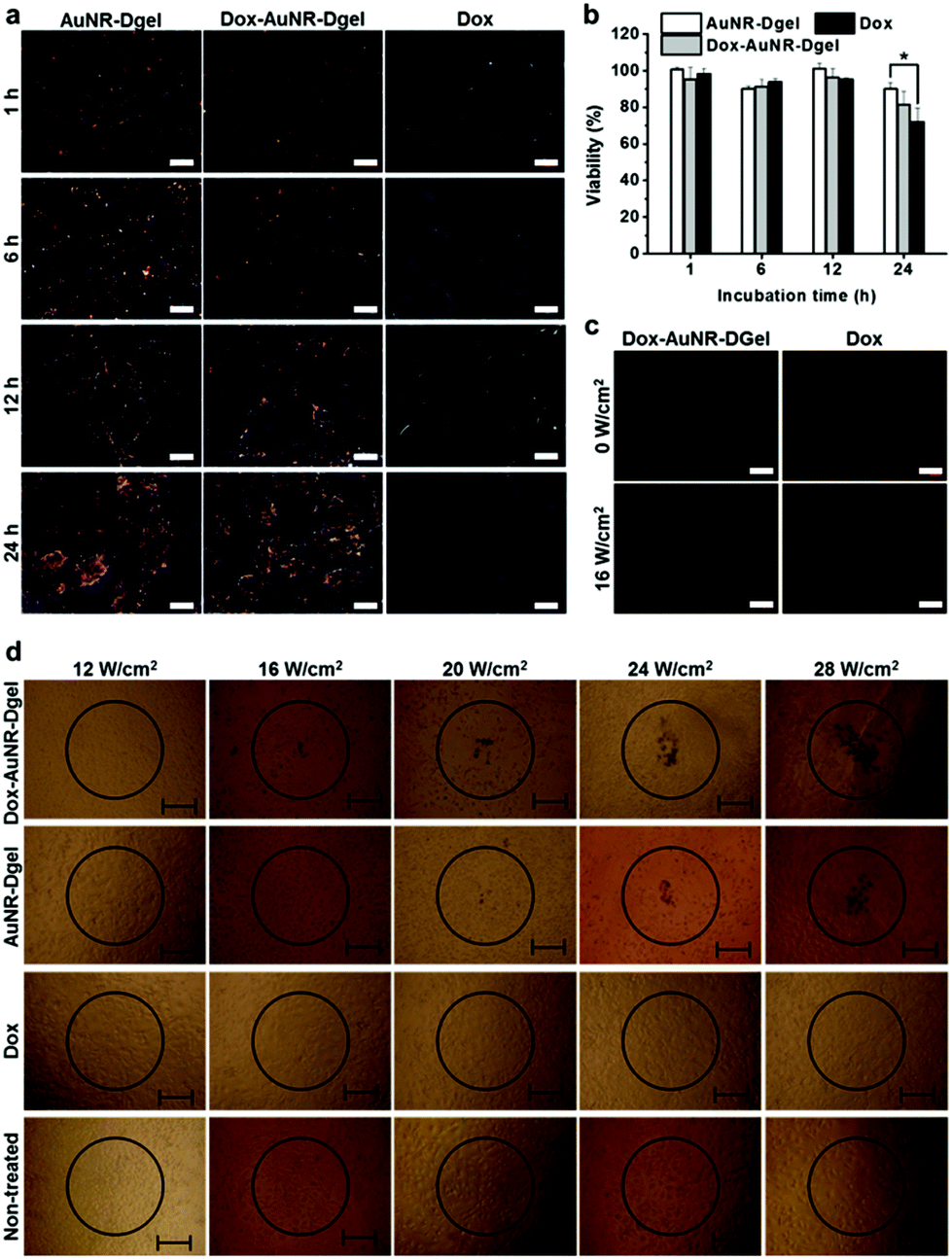 | ||
| Fig. 2 (a) Dark-field microscopy images of B16 F10 cells co-incubated with AuNR-Dgel, Dox-AuNR-Dgel, or Dox for 1, 6, 12, or 24 h (scale bar: 20 μm). (b) Viabilities of the B16 F10 cells co-incubated with each sample for 1, 6, 12 or 24 h (*P = 0.019). (c) Fluorescence microscopy images of B16 F10 cells co-incubated with Dox-AuNR-Dgel or with Dox and irradiated with no laser (0 W cm−2) or with laser power of 16 W cm−2 (scale bar: 40 μm). (d) Photothermal destruction of the cells co-incubated with Dox-AuNR-Dgel, AuNR-Dgel, Dox, or no sample (non-treated) for 12 h followed by laser irradiation for 5 min at different power densities (scale bar: 100 μm). | ||
To quantify the synergistic effect, viabilities were studied for cells co-incubated with Dox-AuNR-Dgel and separately with the AuNR-Dgel, Dox only, and non-treated controls for 12 h by varying the laser irradiation time from 0 (no exposure), 1, 3, 5, to 10 min at 16 W cm−2 (Fig. 3a). Dox-AuNR-Dgel showed the most radical change in the viability by the irradiation time, dropping to 34.2% at 10 min, which was followed by AuNR-Dgel showing 60.5% viability under the same irradiation conditions. This almost two-fold viability reduction of the Dox-AuNR-Dgel over AuNR-Dgel indicates the highly synergistic effect of the co-loaded Dox. It is noted that the Dox alone only showed ∼4% decrease in the viability because of the low dosage level. Synergistic effects were obtained by dividing the predicted additive viability by the measured viability (Fig. 3b).8 The additive viability was obtained from the expression VAdditive = VChemo × VThermo, where VAdditive is the predicted viability as a result of the purely additive interaction between the chemo and thermo therapies, and VChemo and VThermo are the viabilities after the chemo and thermo therapies alone, respectively.7a,16 The synergistic factor was ∼1 for all the samples prior to laser irradiation, which indicates the absence of any synergistic effect. The largest synergistic increase of 70.0% was observed for the Dox-AuNR-Dgel sample after 10 min laser irradiation. The synergistic factor is comparable with maximal values observed for previous combination treatments using gold nanostructures with Dox.7c,17 The highly synergistic effect of the Dox-AuNR-Dgel can be explained by (1) the high local concentration of Dox released from the Dgel scaffold by thermal denaturation and (2) the photothermally enhanced induction of the cytotoxic effect of Dox, i.e., thermal chemopotentiation.18
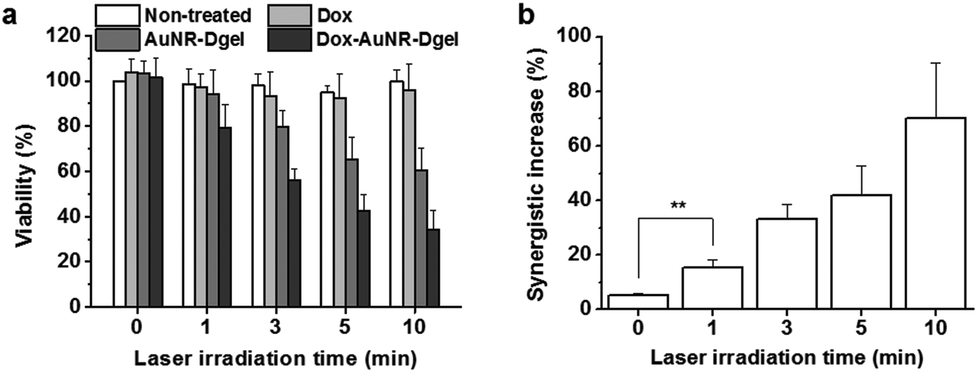 | ||
| Fig. 3 (a) Viabilities of cells co-incubated with each or no sample and exposed to laser irradiation. (b) Synergistic effects for the Dox-AuNR-Dgel (**P = 0.0039). | ||
The efficacy of the combination therapy was further demonstrated in vivo using a mouse model bearing B16 F10 melanoma xenografts. PBS buffer, Dox, AuNR-Dgel, or Dox-AuNR-Dgel were intratumorally injected, and the tumor growth was monitored with/without laser irradiation. To monitor the local heating effect, a thermocouple needle was inserted into the tumor region 10 min post injection, and the local temperature was measured for 5 min during irradiation under 660 nm laser (1 W cm−2) and for another 3 min after the laser was turned off (Fig. 4a). Both the AuNR-Dgel and Dox-AuNR-Dgel showed a rapid temperature increase upon laser irradiation, which was significantly larger than that of the PBS or Dox control. This temperature increase should be sufficient for tumor ablation19 and thermal enhancement of Dox cytotoxicity.18,20 When the laser was turned off, the tumor quickly cooled down, implying that the heat generation was localized. After the intratumoral administration, the mice were laser-irradiated only once for 5 min on day one. The tumor suppression efficacy was evaluated by recording the relative tumour volumes every two days for two weeks (Fig. 4b). Dox-AuNR-Dgel treated mice showed the most effective tumor suppression, which was followed by AuNR-Dgel and Dox. The Dox-AuNR-Dgel case showed that the relative tumor volume was 3.9 times smaller than the PBS control. It was also 1.8 times and 2.9 times smaller than those of the AuNR-Dgel and Dox, respectively, which clearly indicates the synergistic effect. The experiment was repeated by omitting the laser irradiation, where no significant tumor suppression was observed (Fig. 4c). The tumors were dissected after the experiments, and showed well-correlated size patterns with the temporal volume change (Fig. 4d). The dissected tumors were measured and compared by weight, which matched well with the volume data on the last day (Fig. S6†). These results demonstrate light-controlled activation of thermo-chemo combination therapy, which shows synergistically enhanced efficiency over single treatment.
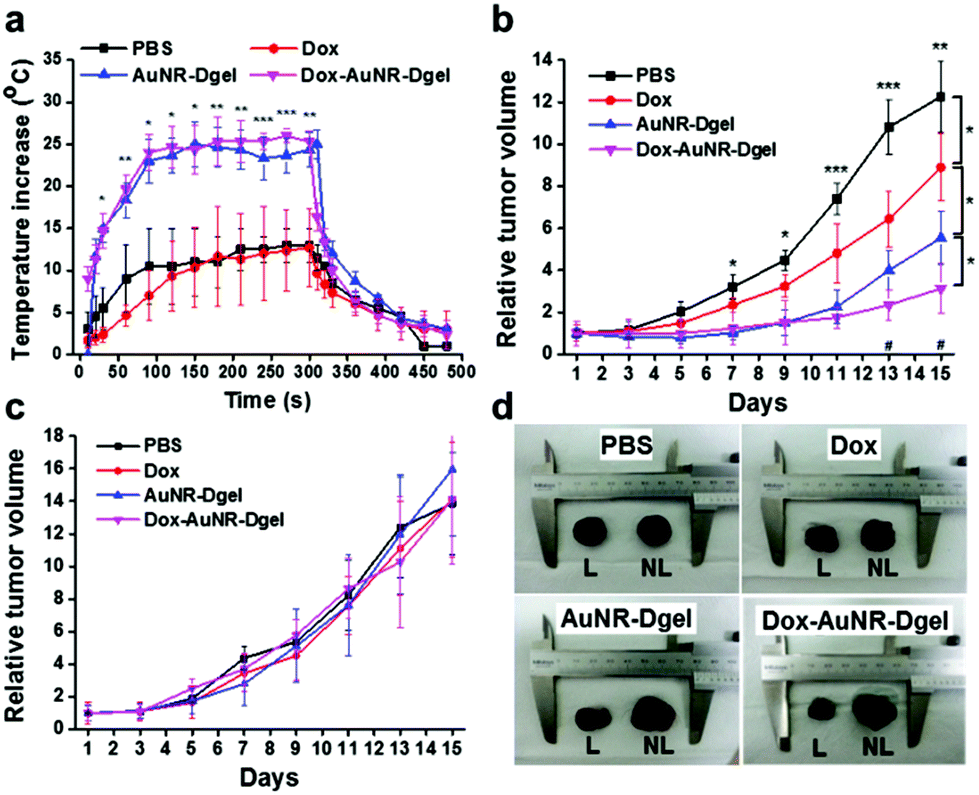 | ||
| Fig. 4 (a) Local temperature of the tumor core during and after laser irradiation at 10 min after injection of PBS, Dox, AuNR-Dgel, or Dox-AuNR-Dgel (*P < 0.05 and **P < 0.005 for comparison with Dox and PBS). Relative tumor volume change over 15 days with (b) or without (c) laser irradiation (*P < 0.05, **P < 0.005 and ***P < 0.001 are noted between PBS and Dox-AuNR-Dgel, #P < 0.05 is noted between AuNR-Dgel and Dox-AuNR-Dgel). (d) Images of dissected tumors after the experiment with laser irradiation (L) or without (NL) laser irradiation. | ||
In summary, to overcome the limited conjugation capability of AuNRs, we have employed Dgel as a scaffold to assemble AuNRs with Doxs in nanoscale proximity. This platform can be potentially applied for CT contrast imaging, radiosensitization, and so forth.
Acknowledgements
This research was supported by Korea Health Technology R&D Project through KHIDI (HI12C1642), the National R&D Program for Cancer Control (1320220), Ministry for Health and Welfare, and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MEST) (No. 2011-0019635), Republic of Korea.Notes and references
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS PubMed.
- (a) W. I. Choi, J. Y. Kim, C. Kang, C. C. Byeon, Y. H. Kim and G. Tee, ACS Nano, 2011, 5, 1995 CrossRef CAS PubMed; (b) G. von Maltzahn, A. Centrone, J.-H. Park, R. Ramanathan, M. J. Sailor, T. A. Hatton and S. N. Bhatia, Adv. Mater., 2009, 21, 3175 CrossRef CAS PubMed; (c) X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115 CrossRef CAS PubMed.
- (a) C. Loo, A. Lowery, N. Halas, J. West and R. Drezek, Nano Lett., 2005, 5, 709 CrossRef CAS PubMed; (b) L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549 CrossRef CAS PubMed.
- (a) S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587 CrossRef CAS PubMed; (b) L. Au, D. Zheng, F. Zhou, Z.-Y. Li, X. Li and Y. Xia, ACS Nano, 2008, 2, 1645 CrossRef CAS PubMed.
- C. Sonnichsen, B. M. Reinhard, J. Liphardt and A. P. Alivisatos, Nat. Biotechnol., 2005, 23, 741 CrossRef PubMed.
- (a) E. S. Shibua, M. Hamadaa, N. Murasea and V. Biju, J. Photochem. Photobiol., C, 2013, 15, 53 CrossRef PubMed; (b) J. Nam, N. Won, J. Bang, H. Jin, J. Park, S. Jung, S. Jung, Y. Park and S. Kim, Adv. Drug Delivery Rev., 2013, 65, 622 CrossRef CAS PubMed.
- (a) T. S. Hauck, T. L. Jennings, T. Yatsenko, J. C. Kumaradas and W. C. W. Chan, Adv. Mater., 2008, 20, 3832 CrossRef CAS PubMed; (b) J.-H. Park, G. von Maltzahn, M. J. Xu, V. Fogal, V. R. Kotamraju, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 981 CrossRef CAS PubMed; (c) H. Park, J. Yang, J. Lee, S. Haam, I.-H. Choi and K.-H. Yoo, ACS Nano, 2009, 3, 2919 CrossRef CAS PubMed.
- J. Nam, W.-G. La, S. Hwang, Y. S. Ha, N. Park, N. Won, S. Jung, S. H. Bhang, Y.-J. Ma and Y.-M. Cho, ACS Nano, 2013, 7, 3388 CrossRef CAS PubMed.
- (a) S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797 CrossRef CAS PubMed; (b) J. B. Lee, S. Peng, D. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. Chen, R. Long and M. Wu, Nat. Nanotechnol., 2012, 7, 816 CrossRef CAS PubMed.
- J. Song, S. Hwang, K. Im, J. Hur, J. Nam, S. Hwang, G. O. Ahn, S. Kim and N. Park, J. Mater. Chem. B, 2015, 3, 1537 RSC.
- P. Zijlstra, C. Bullen, J. W. M. Chon and M. Gu, J. Phys. Chem. B, 2006, 110, 19315 CrossRef CAS PubMed.
- (a) W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4 Search PubMed; (b) L. Wang, Y. Zhu, L. Xu, W. Chen, H. Kuang, L. Liu, A. Agarwal, C. Xu and N. A. Kotov, Angew. Chem., Int. Ed., 2010, 49, 5472 CrossRef CAS PubMed.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990 CrossRef CAS PubMed.
- H. Petrova, J. Perez Juste, I. Pastoriza-Santos, G. V. Hartland, L. M. Liz-Marzan and P. Mulvaney, Phys. Chem. Chem. Phys., 2006, 8, 814 RSC.
- (a) C. A. Frederick, L. D. Williams, G. Ughetto, G. A. Van der Marel, J. H. Van Boom, A. Rich and A. H. Wang, Biochemistry, 1990, 29, 2538 CrossRef CAS; (b) O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157 CrossRef CAS PubMed; (c) J. B. Lee, A. S. Shai, M. J. Campolongo, N. Park and D. Luo, ChemPhysChem, 2010, 11, 2081 CrossRef CAS PubMed.
- G. M. Hahn, J. Braun and I. Har-Kedar, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 937 CrossRef CAS.
- J. You, G. Zhang and C. Li, ACS Nano, 2010, 4, 1033 CrossRef CAS PubMed.
- (a) P. Jacquet, A. Averbach, O. A. Stuart, D. Chang and P. H. Sugarbaker, Cancer Chemother. Pharmacol., 1997, 41, 147 CrossRef; (b) P. Wust, B. Hildebrandt, G. Sreenivasa, B. Rau, J. Gellermann, H. Riess, R. Felix and P. M. Schlag, Lancet Oncol., 2002, 3, 487 CrossRef CAS.
- (a) S. M. Lee, H. Park and K. H. Yoo, Adv. Mater., 2010, 22, 4049 CrossRef CAS PubMed; (b) E. B. Dickerson, E. C. Dreaden, X. Huang, I. H. El-Sayed, H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57 CrossRef CAS PubMed.
- J. M. C. Bull, Cancer Res., 1984, 44, 4853 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c5nr00858a |
| This journal is © The Royal Society of Chemistry 2015 |