
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual stimuli polysaccharide nanovesicles for conjugated and physically loaded doxorubicin delivery in breast cancer cells†
P. S.
Pramod
a,
Ruchira
Shah
b and
Manickam
Jayakannan
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER)-Pune, Dr Homi Bhabha Road, Pune – 411008, Maharashtra, India. E-mail: jayakannan@iiserpune.ac.in
bDepartment of Biology, Indian Institute of Science Education and Research (IISER)-Pune, Dr Homi Bhabha Road, Pune – 411008, Maharashtra, India
First published on 9th March 2015
Abstract
The present work reports the development of pH and enzyme dual responsive polysaccharide vesicular nano-scaffolds for the administration of doxorubicin via physical loading and polymer–drug conjugation to breast cancer cells. Dextran was suitably modified with a renewable resource 3-pentadecyl phenol unit through imine and aliphatic ester chemical linkages that acted as pH and esterase enzyme stimuli, respectively. These dual responsive polysaccharide derivatives self-organized into 200 ± 10 nm diameter nano-vesicles in water. The water soluble anticancer drug doxorubicin (DOX·HCl) was encapsulated in the hydrophilic pocket to produce core-loaded polysaccharide vesicles whereas chemical conjugation produced DOX anchored at the hydrophobic layer of the dextran nano-vesicles. In vitro studies revealed that about 70–80% of the drug was retained under circulatory conditions at pH = 7.4 and 37 °C. At a low pH of 6.0 to 5.0 and in the presence of esterase; both imine and ester linkages were cleaved instantaneously to release 100% of the loaded drugs. Cytotoxicity assays on Wild Type Mouse Embryonic Fibroblasts (WTMEFs) confirmed the non-toxicity of the newly developed dextran derivatives at up to 500 μg mL−1 in PBS. MTT assays on fibroblast cells revealed that DOX·HCl loaded nano-vesicles exhibited better killing abilities than DOX conjugated polymer nano-vesicles. Both DOX loaded and DOX conjugated nano-vesicles were found to show significant killing in breast cancer cells (MCF 7). Confocal microscopy images confirmed the uptake of DOX loaded (or conjugated) nano-vesicles by cells compared to free DOX. Thus, the newly developed pH and enzyme dual responsive polysaccharide vesicular assemblies are potential drug vectors for the administration of DOX in both loaded and chemically conjugated forms for the efficient killing of breast cancer cells.
1. Introduction
Polymer scaffolds are emerging as important biomaterials for loading and delivering anticancer drugs to cancer tissues to enhance their treatment efficacies.1 The polymer carriers are capable of accumulating drug molecules selectively at cancer sites through the enhanced permeability and retention (EPR) effect.2,3 The EPR effect enriches the drug content locally at the cancer tissue; however, additional stimuli are required to break the polymer–drug assemblies to administrate the drugs in the intra-cellular compartments.4,5 External stimuli such as temperature,6,7 radiation (for photodynamic therapy),8,9 and magnetic forces10 have been demonstrated for disassembling the scaffolds to release the drugs at the tissue level. Unfortunately, these stimuli were less effective for polymer–drug delivery at the intra-cellular compartments, which are primarily regulated by pH variation in the endosomes/lysosomes (see Fig. 1) and enzymes like esterase and glutathione transferase.11,12 The pH of the endosomes and lysosomes typically varies in the range of 6.0–5.5 which is much lower than that of the cytosol (pH = 7.4).13 Moreover, recent in vivo studies such as optical imaging techniques, MR spectroscopy and PET radiotracers have revealed that tumor tissue and its surrounding environment has a much lower pH compared to normal tissues.14 Pagel's group has developed a paramagnetic CEST agent and reported the pH of breast cancer tissues was 6.0 to 6.5, compared to 7.4 in normal breast.15 Thus, the development of new polymer scaffolds that can undergo cleavage by pH variation at the cancer tissue level (in the range of 6.5 to 5.0) and enzymes (like esterase or glutathione transferase) in intracellular compartments could give potential dual stimuli vectors for efficient drug administration.16 Avidin responsive dendrimer based assemblies,17 trypsin and hepsin cleavable peptide nanoparticles,18 sugar and pH responsive mesoporous silica,19 glucose oxidase, myoglobin and pH responsive cross-linked polymersomes,20 carbohydrate coated graphene nanocarriers21 and glutathione and pH responsive micelles22 are some important example materials for the achieving the above target. Gu and coworkers23 recently reviewed the roles of various enzyme-responsive nanomaterials for controlled drug delivery. These studies emphasized the need for the development of enzyme-responsive drug delivery scaffolds for cancer therapy.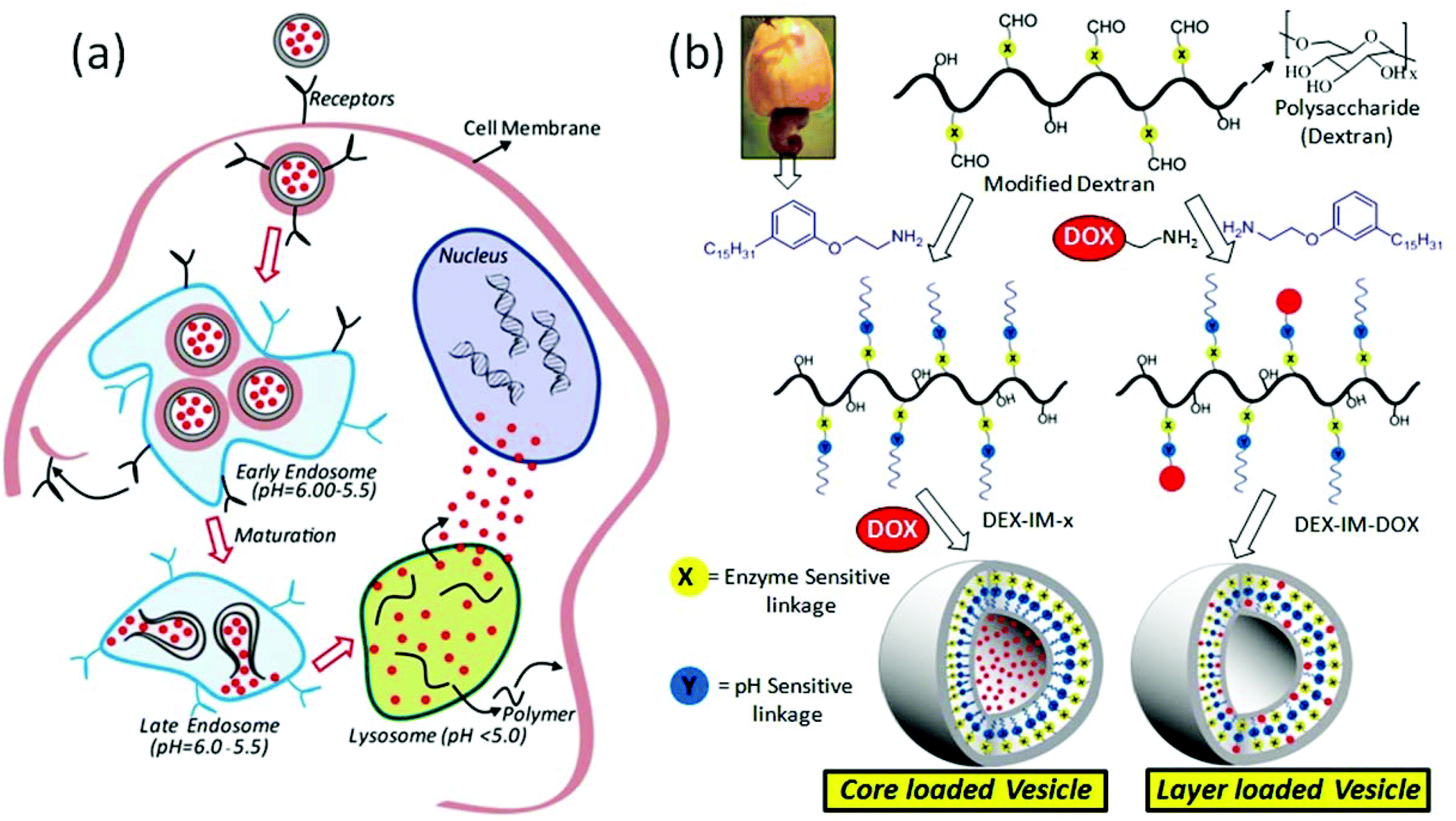 | ||
| Fig. 1 (a) Schematic representation of polymer–drug nanovesicle cellular uptake and their degradation in endocytic compartments. (b) Structural engineering of pH and enzyme dual responsive polysaccharide vesicles. | ||
Polysaccharides are important natural polymers for drug delivery due to their high abundance, structural diversity, biocompatibility and biodegradability by enzymatic pathways.24 Among the polysaccharides, dextran has been extensively utilized as an antithrombolytic agent and MRI contrast component due to its unique advantages of high water solubility, resistance to protein desorption and so on.25,26 In the recent past, dextran was suitably modified using hydrophobic units and the resultant micelles,27 vesicles,28 microgels29 and nano-particles30,31 were employed for delivering drugs. Earlier studies revealed that chemical conjugation of drug molecules in the polymer backbone prevented the premature leakage of drug from the formulation.32–37 The chemical conjugation of drug on the polymer backbone (to produce polymer–drug conjugates) was found to drastically alter the hydrophobicity of the entire matrix. As a result, the drug loaded and conjugated scaffolds were found to be different in size and morphology compared to that of the nascent polymer self-assembly.38,39 This leads to more ambiguity in studying the role of drug release profiles and their action in drug-conjugated versus loaded polymer nano-objects. From our research groups, we reported a unique renewable resource approach for dextran40 (or dextrin)41 modifications and these polysaccharide scaffolds were employed as candidates for dual drug loading for CPT and doxorubicin (DOX) in MCF 7 and DLD 1 cancer cells.42 Efforts were made to address the cellular uptake mechanism, drug ratiometric control and individual cocktail versus dual loaded vesicles and so on and so forth.42 These results suggested that polysaccharide vesicles have preferential uptake for cells that are lacking caveolae, a null (–) type that resembled breast cancer cells (MCF 7) over DLD I type cells (colon cancer). Thus, it would be appropriate to develop new polysaccharide scaffolds to address the following fundamental questions: (i) structural optimization for achieving identical size and shape in both polymer–drug conjugated scaffolds and drug loaded polymer scaffolds (this will facilitate the study of drug release profiles for physically loaded and chemically anchored anticancer drugs in identical nano-assemblies) and (ii) pH and enzyme dual stimuli for drug delivery in the intra-cellular lysosomal compartments to enhance therapeutic efficacy.
The present work is emphasized to develop new dual pH and enzyme responsive polysaccharide vesicles with identical sizes and vesicular morphologies in both drug conjugated and drug loaded nano-assemblies (see Fig. 1). The pH responsiveness was achieved by the imine chemical linkage (stimuli-1, shown in blue) and an enzyme cleavable aliphatic ester bond connected the hydrophobic segment at the dextran backbone (stimuli-2, shown in yellow). A 3-pentadecylphenol hydrophobic unit was suitably modified as a structure director for achieving vesicular geometry in the dextran backbone. The current design provides two vesicular scaffolds: (i) DOX loaded at the core of the nascent polysaccharide vesicles and (ii) DOX conjugated at the hydrophobic layer of the polysaccharide vesicles associated with both pH and enzymatic responsiveness. These DOX containing dextran vesicular scaffolds were tested for cellular uptake and cytotoxicity capabilities in breast cancer cells (MCF7 cells). The overall investigation revealed that the newly developed pH responsive polysaccharide vesicular scaffolds are potential drug vectors for breast cancer treatment.
2. Experimental methods
2.1. Materials
Dextran (Mw = 6000), ethanolamine, di-tert-butyl dicarbonate, 4-hydroxy benzaldehyde, diisopropyl azodicarboxylate, triphenyl phosphine, trifluoroacetic acid, 3-pentadecylphenol, dicyclohexylcarbodiimide, 4-dimethylamino pyridine, pyrene, doxorubicin·HCl (DOX·HCl), Nile red, and rhodamine B were purchased from Aldrich chemicals. Dimethyl sulphoxide was dried over calcium chloride and CaH2 and distilled prior to use. tert-Butyl bromoacetate, K2CO3, KI, and all other reagents and solvents were purchased locally and purified following the standard procedures. Wild type mouse embryonic fibroblasts were maintained in DMEM (phenol red free medium: Gibco) containing 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin–streptomycin at 37° C under a 5% CO2 humidified atmosphere. Cells were trypsinised using 0.05% trypsin (Gibco) and seeded in 96 well or 6 well (as per experiment) flat bottomed plastic plates (Costar) for all assays. Tetrazolium salt, 3-4,5 dimethylthiazol-2,5 diphenyl tetrazolium bromide (MTT), DMSO and paraformaldehyde were obtained from Sigma. Phalloidin conjugated to Alexa 594 was obtained from Molecular Probes (Invitrogen) and fluoromount from Southern Biotech.2.2. General procedures
NMR spectra were recorded using a 400 MHz Jeol NMR spectrometer in CDCl3 or DMSO (d6) containing TMS as the internal standard. FT-IR spectra of all compounds were recorded using a Bruker alphaT Fourier transform infrared spectrometer. The mass of all small molecules was confirmed by using an Applied Biosystems 4800 PLUS MALDI TOF/TOF analyzer. High resolution mass spectra were obtained from a Micro Mass ESI-TOF MS spectrometer. The purity of the modified dextran was determined by gel permeation chromatography (GPC) using a Viscotek VE 1122 pump, Viscotek VE 3580 RI detector, and Viscotek VE 3210 UV/Vis detector in dimethyl formamide. The absorption and emission studies were done by a Perkin-Elmer Lambda 45 UV-Visible spectrophotometer and a SPEX Fluorolog HORIBA JOBIN VYON fluorescence spectrophotometer with a double-grating 0.22 m Spex1680 monochromator and a 450 W Xe lamp as the excitation source at room temperature. The excitation spectra are collected at 375 nm and 420 nm (for the pyrene experiment) and 626 nm (for the Nile red experiment) and the emission spectra are recorded by exciting at the excitation maxima. The dye containing samples were purged with N2 gas for at least 15–20 minutes prior to photophysical experiments. The size determination of the dextran derivatives was carried out by dynamic light scattering (DLS), using a Nano ZS-90 apparatus utilizing a 633 nm red laser (at 90° angle) from Malvern instruments. The reproducibility of the data was checked at least three times using independent polymer solutions. The static light scattering experiment (SLS) was carried out using a 3D-DLS spectrometer, from LS instruments, Switzerland. The instrument consists of a He Ne laser with a wavelength of 632.8 nm attached to a computer using the Lab view interface utilizing toluene as a reference. The measurement was performed in autocorrelation mode from 20° to 130° by steps of 5°. Atomic force microscope (AFM) images were recorded by drop casting the samples on a freshly cleaved mica surface, using a Veeco Nanoscope IV instrument. The experiment was performed in tapping mode using a TAP-190AL-G50 probe from Budget sensors with a nominal spring constant of 48 N m−1 and resonance frequency of 163.5. FE-SEM images were recorded using a Zeiss Ultra Plus scanning electron microscope. For FE-SEM analysis, the samples were prepared by drop casting on silicon wafers and coated with gold. Thermal analysis of dextran derivatives was performed using a TA Q20 differential scanning calorimeter (DSC). The instrument was calibrated using indium standards. TEM images were recorded using a Technai-300 instrument by drop casting the sample on a formvar coated copper grid. The fluorescent micrographs were collected using a Carl Zeiss Axiovert 200 microscope. An LSM710 confocal microscope was used for imaging the cells.2.3. Synthesis of tert-butyl 2-(4-formyl phenoxy) acetate (1)
4-Hydroxy benzaldehyde (6.0 g, 49.1 mmol), anhydrous K2CO3 (13.5 g, 98.3 mmol) and a pinch of KI were taken in dry acetonitrile (60 mL) and stirred at 80 °C under a nitrogen atmosphere for 30 minutes. The contents were cooled and tert-butyl bromoacetate (7.9 mL, 54.0 mmol) was added dropwise. The reaction was continued by stirring at 80 °C for 24 h under a nitrogen atmosphere. Acetonitrile was removed by rotoevaporation and the reaction mixture was poured into water (250 mL). Subsequently the product was extracted with ethylacetate and the organic layer was washed with brine. The contents were dried over anhydrous sodium sulphate and solvent was removed to obtain the product as a low melting solid. It was further purified by passing through a silica gel column using 3% ethyl acetate in hexane as the eluent. Yield = 9.9 g (85%). m.p. = 38 °C. 1H NMR (400 MHz, CDCl3) δ: 9.88 ppm (Ar–CHO), 7.83 ppm (d, 2H, Ar–H), 6.98 ppm (d, 2H, Ar–H), 4.59 ppm (s, 2H,O–CH2), 1.47 ppm (s, 9H, t-butyl). 13C NMR (CDCl3, 100 MHz) δ: 190.84 (Ar–CHO), 167.22 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 162.85, 132.03, 130.66, 114.93 (Ar–C), 83.02 (–C–(CH3)3, 65.61 (Ar–O–CH2–), 28.09(–C–(CH3)3. FT-IR (KBr), cm−1, 3069 (aromatic C–H stretch), 2836, 2750 (C–H stretch of aldehyde), 2978, 2935 (aliphatic C–H stretch), 1743 (CO stretch), 1573, 1506 (ring CC stretch), 1215, 1149 (C(O)–O stretch). MALDI-TOF-TOF, (MW: 216), m/z = 274.3 (M + K+).
O), 162.85, 132.03, 130.66, 114.93 (Ar–C), 83.02 (–C–(CH3)3, 65.61 (Ar–O–CH2–), 28.09(–C–(CH3)3. FT-IR (KBr), cm−1, 3069 (aromatic C–H stretch), 2836, 2750 (C–H stretch of aldehyde), 2978, 2935 (aliphatic C–H stretch), 1743 (CO stretch), 1573, 1506 (ring CC stretch), 1215, 1149 (C(O)–O stretch). MALDI-TOF-TOF, (MW: 216), m/z = 274.3 (M + K+).
2.4. Synthesis of 2-(4-formyl phenoxy) acetic acid (2)
Compound 1 (5.0 g, 21.1 mmol) was dissolved in 25 mL dichloromethane and cooled to 0 °C in an ice bath. Trifluoroacetic acid (16.2 mL, 211.6 mmol) was added dropwise into the above solution and the reaction mixture was brought to room temperature. After 2 hours, dichloromethane was rota evaporated from the mixture and trifluoroacetic acid was removed by co-evaporation with dichloromethane (4× 15 mL). The product was collected as a brownish solid which was further purified by passing through a silica gel column using 2% methanol in chloroform as the eluent to get a white solid. Yield = 3.1 g (81.3%). 1H NMR (400 MHz, d6 DMSO) δ:12.9 ppm (s, 1H, CO–OH), 9.82 ppm (s, 1H, Ar–CHO), 7.82 ppm (d, 2H, Ar–H), 7.06 ppm (d, 2H, Ar–H), 4.78 ppm (s, 2H, O–CH2). 13C NMR (DMSO (d6), 100 MHz) δ: 191.87 (Ar–CHO), 170.17 (–O–CH2–COOH), 163.17, 132.22, 130.51, 115.52, (Ar–C), 65.08(–O–CH2–COOH). FT-IR (KBr), cm−1, 3638 (O–H stretch) 3093 (aromatic C–H stretch), 2843, 2757 (C–H stretch of aldehyde), 2690 (aliphatic C–H stretch), 1750 (aldehyde CO stretch), 1714 (acid CO stretch), 1565, 1505 (ring CC stretch), 1424 (O–H bending), 1210 (C–O stretch of COOH) 1266 (C (O)–O stretch). MALDI TOF-TOF, (MW: 180.1), m/z = 218.2 (M + K+).
2.5. Synthesis of DEX-CHO-x
Dextran (Mw = 6000, 1.0 g, 6.2 mmol of anhydroglucose unit) and compound 2 (1.1 g, 6.2 mmol, for DEX-CHO-10) were dissolved in anhydrous DMSO (50.0 mL) and the solution was purged with dry nitrogen for 15 minutes. Dicyclohexylcarbodiimide (1.5 g, 7.4 mmol) in anhydrous DMSO (3.0 mL) and 4-(dimethylamino) pyridine (0.15 g, 1.2 mmol) in anhydrous DMSO (3.0 mL) and were added into the above reaction mixture. The reaction mixture was stirred for 1 hour under dry nitrogen purging and the reaction was continued at 25 °C for 24 h under a nitrogen atmosphere. The solution was filtered to remove dicyclohexyl urea and the solvent was removed under vacuum distillation (0.1 mm Hg). The thick viscous liquid was precipitated by adding into cold isopropyl alcohol (100 mL). The solid was filtered and washed several times with methanol. It was dissolved again in DMSO and purification by the precipitation technique was done at least twice. The product was filtered out and dried under vacuum at 60 °C to get a yellowish white solid as the product. Yield = 65%. 1H NMR (400 MHz, d6 DMSO) δ: 9.82 ppm (s, 1H, –CHO), 7.82 ppm (m, 2H, Ar–H adjacent to aldehyde group), 7.08 ppm (m, 2H, Ar–H,), 4.47, 4.82, 4.88 ppm (s, hydroxyl of dextran) 4.63 ppm (s, dextran anomeric proton), 3.14–3.69 ppm (dextran glucosidic protons). 13C NMR (DMSO (d6), 100 MHz) δ: 191.98 ppm (aldehyde CO), 168.45 (ester CO), 162.83, 132.26, 130.59, 115.54 (Ar–C), 98.63 (dextran anomeric C), 73.78, 72.29, 70.54, 66.43 (dextran glucosidic carbons), 61.28 ppm (OC–CH2). FT-IR (KBr), cm−1, 3291 (O–H stretch), 2926, 2852 (aliphatic C–H stretch), 1750 (ester CO stretch), 1513 (ring CC stretch), 1422 (O–H bending), 1209 (C (O)–O stretch.
DEX-CHO-x with three different degrees of substitution were synthesized by changing the mole ratios of compound 2 to dextran to 0.5, 1.0 and 3.0 in the feed and their details are provided in the ESI.†
2.6. Synthesis of N-(tert-butoxycarbonyl) ethanolamine (3)
Synthesis was performed using the reported procedures.6 Briefly, ethanolamine (5.0 g, 81.0 moles) was dissolved in 173 mL of sodium carbonate (10%, 17.3 g, 163.7 mmol) solution and tetrahydrofuran (87 mL) was added into it. The reaction mixture was cooled to 0 °C in an ice bath and boc anhydride (19.6 g, 90.0 mmol) dissolved in tetrahydrofuran (86 mL) was added dropwise into the reaction and it was stirred at 25 °C for 12 hours. The solvent was evaporated from the reaction mixture and the remaining aqueous solution was neutralized using 0.1 M HCl solution. The product was extracted with ethyl acetate (100 mL) and the organic layer was washed with brine solution (2× 100 mL) and dried over sodium sulphate. The combined ethyl acetate layer was rota evaporated to get the crude product as a thick colourless liquid. It was further purified by passing through a silica gel column, initially at 5% ethyl acetate in hexane as the eluent to remove all unreacted boc anhydride and followed by isolating the product by passing with ethylacetate. Yield = 10.6 g (80%). 1H NMR (400 MHz, CDCl3) δ: 4.98 ppm (s, 1H, N–H), 3.67 ppm (t, 2H, OH–CH2–CH2–), 3.25 ppm (OH–CH2–CH2–) 1.42 ppm (s, 9H, t-butyl). 13C NMR (CDCl3 (d6), 100 MHz) δ: 156.99 (NH–CO–), 79.78 (C–(CH3)3), 62.73 (OH–CH2–CH2–), 43.24 (OH–CH2–CH2–), 28.44 (C–CH3)3. FT-IR (KBr), cm−1, 3342 (O–H stretch), 2975, 2934 (aliphatic C–H stretch), 1683 (CO stretch), 1393 (O–H bending), 1249 (C–O stretch) 1165 (C–N stretch). MALDI TOF-TOF, (MW: 161.1), m/z = 199.0 (M + K+).
2.7. Synthesis of 2-(3-pentadecylphenoxy) ethanamine (4)
Synthesis was performed using the reported procedures.6 Briefly, 3-pentadecyl phenol (5 g, 16.42 mmol), compound 3 (2.6 g, 16.4 mmol) and triphenyl phosphine (4.3 g, 16.42 mmol) were dissolved in anhydrous tetrahydrofuran (50 mL). The mixture was taken in a 3-neck round bottom flask and cooled in an ice bath purged with dry nitrogen. Diisopropyl azodicarboxylate (3.51 mL, 18.06 mmol) was added dropwise and the reaction was continued under a nitrogen atmosphere for 24 hours at 25 °C. The solvent was removed from the reaction mixture and the remaining thick liquid was dissolved in ethyl acetate (80 mL). The organic layer was washed with brine solution for 3 times (3 × 50 mL) and dried over anhydrous sodium sulphate. It was purified by passing through a silica gel column with 2% ethyl acetate in hexane as the eluent. Yield = 5.8 g (79%). 1H NMR (400 MHz, CDCl3) δ: 7.19 ppm (t, 1H, Ar–H), 6.80 ppm (d, 1H, Ar–H), 6.72 ppm (s,1H, Ar–H), 6.70 ppm (d, 1H, Ar–H), 5.01 ppm (s, 1H, N–H), 4.02 ppm (t, 2H, O–CH2–CH2–NH), 3.53 ppm (t, 2H, O–CH2–CH2–NH), 2.57 ppm (t, 2H, Ar–CH2), 1.60 ppm (m, 2H, Ar–CH2–CH2), 1.46 ppm (s, 9H, t-butyl), 1.28 ppm (m, 27H, aliphatic protons), 0.87 ppm (t, 3H, Ar–(CH2)14–CH3). 13C NMR (CDCl3, 100 MHz) δ: 155.99 (NH–CO–), 58.62, 144.85, 129.30, 121.32, 114.75, 111.40 (Ar–C), 79.56 (C–(CH3)3), 67.08 (OH–CH2–CH2–), 40.26 (OH–CH2–CH2–), 36.10, 32.01, 29.77, 28.48, 22.78, 14.21 (aliphatic carbons). FT-IR (KBr), cm−1, 3395 (N–H stretch), 2918, 2850 (aliphatic C–H stretch), 1691 (CO stretch), 1593, 1514 (ring CC stretch), 1253 (C–O stretch) 1157 (C–N stretch). MALDI TOF-TOF, (MW: 447.6), m/z = 486.3 (M + K+).
2.8. Synthesis of tert-butyl (2-(3-pentadecylphenoxy) ethyl) carbamate (5): (PDP-amine)
Compound 4 (5 g, 11.1 mmol) was dissolved in dichloromethane (25 mL) and cooled to 0 °C in an ice bath. Trifluoroacetic acid (8.54 mL, 111 mmol) was dissolved in dichloromethane (20 mL) and added dropwise into the above solution. The reaction was continued at room temperature for 2 hours. The dichloromethane was removed by rotoevaporation. The remaining trifluoroacetic acid was removed by co-evaporation with dichloromethane (4 × 15 mL) and the product was collected as a yellowish brown low melting solid. Yield = 2.8 g (72%). m.p. = 40 °C. 1H NMR (400 MHz, CDCl3) δ: 7.16 ppm (t, 1H, Ar–H), 6.76 ppm (d, 1H, Ar–H), 6.72 ppm (s, 1H, Ar–H), 6.69 ppm (d, 1H, Ar–H), 3.98 ppm (t, 2H,O–CH2), 3.08 ppm (t, 2H, O–CH2–CH2), 2.55 ppm (t, 2H, Ar–CH2), 1.58 ppm (m, 2H, Ar–CH2–CH2), 1.28 ppm (m, 27H, aliphatic-H), 0.87 ppm (t, 3H, –CH3). 13C NMR (CDCl3, 100 MHz) δ: 157.44, 145.05, 129.31, 122.02, 114.59, 111.43 (Ar–C), 63.21 (Ar–O–CH2), 39.52 (O–CH2–CH2–NH2), 35.91, 31.90, 29.09, 29.34, 22.66, 14.08 (aliphatic carbons in the side chain). FT-IR (KBr), cm−1, 3291 (N–H stretch), 2921, 2852 (aliphatic C–H stretch), 1587 (N–H bending), 1554 (ring CC stretch), 1257 (C–O stretch) 1158 (C–N stretch). MALDI-TOF-TOF, (MW: 347.5), m/z = 385.2 (M + K+).
2.9. Synthesis of Dex-IM-PDP (DEX-IM)
PDP-amine (compound 5) (0.23 g, 0.674 mmol) was dissolved in anhydrous DMSO (10 mL) containing sodium carbonate (0.1 g) and purged with dry nitrogen for 10 min. DEX-CHO-10 (1 g) was dissolved in anhydrous DMSO (80 mL) and added to the above solution. The reaction mixture was purged for another 15 min and then heated at 50 °C for 4 hours under a nitrogen atmosphere. DMSO was removed from the reaction mixture under vacuum and the product was precipitated in cold methanol. The purification was done by dissolving in DMSO and precipitation in methanol for two times. The product was collected by filtration and dried under vacuum at 50 °C to get a brownish yellow solid. Yield = 68%. δ: 8.27 ppm (s, 1H, benzylidene imine proton), 7.63 ppm (d, 2H, Ar–H adjacent to imine linkage), 6.93 ppm (d, 2H, Ar–H), 7.08 ppm (s, 1H, Ar–H of PDP unit), 6.67 ppm (m, 3H, Ar–H of PDP unit), 4.47, 4.82, 4.88 ppm (s, hydroxyl of dextran) 4.63 ppm (s, dextran anomeric proton), 3.14–3.69 ppm (dextran glucosidic protons), 4.14 ppm (t, 2H,O–CH2 of PDP unit), 3.81 ppm (t, 2H, O–CH2–CH2 of PDP unit), 2.45 ppm (t, 2H, Ar–CH2), 1.46 ppm (m, 2H, Ar–CH2–CH2), 1.16 ppm (m, 27H, aliphatic-H), 0.78 ppm (t, 3H, –CH3). FT-IR (KBr), cm−1, 3292 (O–H stretch), 2921, 2852 (aliphatic C–H stretch), 1707 (ester CO stretch), 1644 (CN stretch of imine) 1450 (ring CC stretch), 1342 (O–H bending), 1267 (C (O)–O stretch.
A similar protocol was employed for the synthesis of other derivatives.
2.10. Synthesis of DOX conjugated DEX-IM
Doxorubicin (0.029 g, 0.05 mmol) and PDP-amine (compound 5) (0.042 g, 0.11 mmol) were dissolved in anhydrous DMSO (10 mL). Sodium carbonate (0.1 g) was added to the above solution and the reaction mixture was purged with nitrogen for 10 min. DEX-CHO-10 (0.25 g) was dissolved in anhydrous DMSO (80 mL) and added to the mixture and the nitrogen purging was continued for another 15 min. The reaction mixture was heated to 50 °C for 4 hours under a nitrogen atmosphere. DMSO was removed from the reaction mixture under vacuum and the product was precipitated in cold methanol. Further purification was done by dissolving in DMSO and dialysing (MWCO 3500) against PBS (pH 7.4) for 24 h. The product was collected as a bright red powder after lyophilization. The formation of DOX conjugated DEX-IM was confirmed using gel permeation chromatography, where the UV detector was set to 480 nm. An absorption spectroscopy method was used for the determination of the amount of doxorubicin conjugated on the DEX-IM system.2.11. Preparation of DEX-vesicles and critical vesicular concentration (CVC)
Dextran vesicles were prepared by adopting a nanoprecipitation method. Typically, 20 mg of DEX-IMINE-PDP (DEX-IM) was dissolved in 3 mL of DMSO and 3 mL of PBS (pH 7.4) was added dropwise under moderate stirring at 25 °C. The resulting solution was extensively dialyzed against a buffer solution (pH 7.4) using a regenerated cellulose dialysis membrane (SPECTRA/POR, MWCO 3500) for 48 h.The critical vesicular concentration (CVC) was determined by using hydrophobic pyrene and Nile red as fluorescent probes. The concentrations of pyrene and Nile red were fixed as 0.6 μM and 1.0 μM respectively. In a typical experiment, the required amount of dye in acetone was pipetted into 5 mL glass sample vials and then acetone was allowed to evaporate completely. DEX-IM solutions at different concentrations, varying from 6.2 × 10−7 M to 3.1 × 10−3 M, were added to the vial containing pyrene or Nile red and allowed to equilibrate overnight. The solutions were purged with nitrogen gas before photophysical experiments. In the pyrene-containing experiment, the excitation wavelength was set at 337 nm, the excitation slit at 3 nm, and the emission slit at 3 nm. The ratio of fluorescence intensity in the emission spectra at 375 and 386 nm was calculated and plotted against logarithmic concentrations of polymer. The CVC was estimated at the polymer concentration of the onset of an increase in I375/I386 ratio. In the Nile red experiment, the excitation wavelength was set at 485 nm, the excitation slit at 2 nm, and the emission slit at 3 nm. The emission intensity at a wavelength of 626 nm was plotted versus concentration of polymer added and the deflection point was taken as critical vesicular concentration.
2.12. Encapsulation of hydrophilic rhodamine-B and determination of loading efficiency
Water soluble rhodamine B was encapsulated into the inner core of vesicles by a dialysis method.40 Briefly, 20 mg of DEX-IM-5 and 2 mg of rhodamine B were co-dissolved in 3 mL DMSO. This solution was stirred at 25 °C in a beaker and 3 mL of PBS (pH 7.4) solution was added dropwise into it. The resulting suspension was stirred for 12 h at 25 °C and extensively dialysed by taking in a regenerated cellulose dialysis bag (SPECTRA/POR, MWCO 3500). The dialysis was continued until rhodamine B had completely stopped leaching out from the dialysis bag. DEX-IM-5 vesicles loaded with rhodamine B were collected from the dialysis bag and diluted to 10 mL with PBS. The loaded rhodamine B was allowed to completely release by dissolving 100 μl of the above stock solution in 2.9 mL DMSO solvent. The loading content of rhodamine B was calculated by measuring the absorbance of the above solution at 552 nm and substituting it in Beers law equation where the molar absorption coefficient of rhodamine B was fixed as 115![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The loading content of Rh-B was determined as 2.70 wt%.
000. The loading content of Rh-B was determined as 2.70 wt%.
2.13. Encapsulation of doxorubicin·HCl and drug loading efficiency
Water soluble DNA intercalating doxorubicin HCl (DOX·HCl) was loaded into the inner aqueous cavity of DEX-IM vesicles by a dialysis method.42 Briefly, 100 mg of DEX-IM-5 and 2 mg of DOX·HCl were dissolved in 3 mL of DMSO and stirred moderately at 25 °C under dark conditions. Self assembly of DEX-IM-5 was induced by the slow injection of 3 mL of PBS (pH 7.4) into the above solution. The mixed polymer drug solution was incubated at 25 °C under dark conditions for 12 h. To remove DMSO and unloaded DOX·HCl, the solution was transferred to a dialysis bag (SPECTRA/POR, MWCO 3500) and dialysed against PBS (0.01 M, pH 7.4) for 24 h with 6 times exchange of the dialysis medium with fresh buffer solution. The whole procedure was carried out under dark conditions. The resulting DOX·HCl loaded DEX-IM vesicle solution was filtered through 0.45 μM filters and lyophilized. The DOX·HCl loading content in vesicles was determined by an absorption spectroscopy method. In brief, 3 mg of freeze dried DOX·HCl loaded DEX-IM powder was dissolved in 1.0 mL of DMSO and subsequently 100 μL of this solution was diluted to 3.0 mL with DMSO. The absorbance at 480 nm was measured on a UV-visible spectrophotometer and the amount of DOX·HCl loaded in the vesicles was determined using the molar extinction coefficient of DOX as 11500. Drug loading content (DLC) and drug loading efficiency (DLE) were calculated using the following equations.| DLC (%) = {weight of drug encapsulated in vesicles/weight of drug loaded polymer} × 100% = 2.9 wt% |
| DLE (%) = {weight of drug encapsulated in vesicles/weight of drug in feed} × 100% = 58% |
2.14. In vitro release studies
The drug elution from the vesicles was performed at pH 7.4 (physiological pH), pH 6.5 (tumor tissue pH), pH 6.0, pH 5.5 (endosomal pH) and pH 5.0 (lysosomal pH). A control experiment was also carried out using a solution without DEX-IM polymer by just adding DOX·HCl (60 μg) to 3 mL of PBS (pH 7.4). Briefly, 3.0 mg of DEX-IM vesicles encapsulated with DOX·HCl or DOX conjugated vesicles in 3.0 mL buffer solution of the required pH were placed in a dialysis tube (SPECTRA/POR, MWCO 8000). Control free DOX solution was used for a release experiment without any prior dialysis and showed 100% release in less than 1 h (data not shown). The tube carrying DOX loaded or conjugated DEX-IM vesicles was immersed in 100 mL buffer solution with a similar pH of reconstituted buffer taken in a beaker and the whole solution was incubated at 37 °C. After a suitable time interval (30 min or 60 min) 3.0 mL of dialysis medium was withdrawn and replaced with an equal volume of fresh buffer. The absorbance of each aliquot was determined at 480 nm using UV-Vis absorption spectrophotometry and the amount of DOX released was calculated using Beer's law, where the molar absorption coefficient was kept as 11500. Esterase assisted drug release was performed by adding 10 U esterase enzyme into the dialysis bag prior to the release studies.
2.15. Cell viability assay (MTT assay)
The cytotoxicity of blank DEX-IM, free DOX·HCl, and DOX loaded DEX-IM vesicles was studied in WTMEFs and MCF-7 cells using the tetrazolium salt, 3-4,5 dimethylthiazol-2,5 diphenyl tetrazolium bromide (MTT). The assay was performed using 0.5 μM DOX concentrations. 1 × 103 wild type mouse embryonic fibroblasts or 2 × 103 MCF-7 cells per well were seeded on a fibronectin (2 μg per well) coated 96 well plate (Corning, USA) in 100 μl media and allowed to adhere for 24 h. DMEM with 10% FBS (fetal bovine serum) was used for MCF7 and WTMEFs cells. Prior to drug treatment, the media from the cells was aspirated and 25 μg DEX-IM, 0.5 μM free DOX, or DEX-IM with encapsulated DOX (final concentration 0.5 μM DOX) were prepared in the corresponding media and added accordingly. A blank control, DMEM with FBS in the absence of cells and an untreated control, cells with DMEM containing FBS, were used in each experiment. All control and treated experiment wells were in triplicate. Cells were incubated for 24 h without a change in medium and after 24 h, the drug containing medium was aspirated. A freshly prepared stock of MTT in sterile PBS (5 mg mL−1) was diluted to 50 μg mL−1 in 100 μl DMEM with FBS and added to cells. The cells were then incubated with MTT for 4 h at 37 °C. The medium with MTT was then aspirated from the wells and the purple formazan crystals formed as a result of the reduction of MTT by mitochondrial dehydrogenase enzyme from cells were dissolved in 100 μl of 100% DMSO (added per well). The absorbance from formazan crystals was immediately measured using a microplate reader at 570 nm (Varioskan Flash) and is representative of the number of viable cells per well.Values from the triplicates for each control and treated set were noted and their means used for calculations. If one value from the triplicate had deviated substantially from the other two, it was not considered in the mean calculations. If the triplicate values were all variable the experiment was not used in the final calculations. The mean of the absorbance values for the blank control samples was subtracted from the average of the untreated control and treated samples, respectively. The values thus obtained for the untreated control samples were equated to 100% and relative percentage values for DEX-IM, free DOX·HCl, and DOX physically loaded and chemically conjugated to DEX-IM were calculated accordingly. The percentage values thus obtained for all four cases were subtracted from the untreated control (100%) to determine the percentage loss in cell numbers (relative to control). The results thus obtained from 3 experiments were compiled and the differences between DOX loaded and conjugated vesicles were statistically evaluated using a paired two tailed T-test.
2.16. Cellular uptake of DOX by confocal microscopy
Wild type mouse embryonic fibroblasts (WTMEFs) were seeded at a density of 5 × 104 cells on flame dried cover slips placed in 6 well plates containing DMEM medium with 10% FBS and incubated at 37 °C for 18 hours. The cells were then exposed to the required concentration of free DOX, DOX loaded DEX-IM vesicles and DOX conjugated DEX-IM-5 vesicles (dissolved in PBS) for 4 h in a CO2 incubator at 37 °C. After incubation, the drug-containing medium was aspirated from each well, the cells were washed twice with PBS (1 mL × 2) and fixed with 3.5% paraformaldehyde solution in PBS for 15 min at room temperature. The cells were washed twice with PBS (1 mL) and stained with phalloidin conjugated to Alexa 594 (Invitrogen) diluted 1:500 in 3% BSA solution in PBS. After 1 hour of incubation at room temperature in the dark, the excess dye was washed from the plate and cells were again gently rinsed with PBS. These cover slips were then incubated with DAPI (0.05 μg ml−1) for 2 min each to stain the nucleus. The cover slips were mounted on slides using fluoromount mounting medium (Southern Biotech) and dried overnight at room temperature in the dark. The cells were imaged using an LSM710 confocal microscope using the λ 488 nm (green channel) and λ 568 nm (red channel) lasers.
3. Results and discussion
3.1. Synthesis of pH responsive dextran amphiphiles
The synthesis of modified dextran is shown in Scheme 1. 4-Hydroxybenzaldehyde was reacted with tert-butyl bromoacetate to produce (1). The compound (1) was hydrolyzed and the resultant carboxylic acid derivative (2) was coupled with dextran using DCC/DMAP to get aldehyde substituted dextran (DEX-CHO-x). The renewable resource 3-pentadecyl phenol (PDP) was reacted with Boc protected 2-ethanol amine (3) to make amine functionalized PDP (5). The coupling of the aldehyde group in DEX-CHO-x with PDP-amine (5) produced the pH responsive imine containing dextran derivative DEX-IM-x (see Scheme 1). The free amine group in DOX and PDP amine (5) was simultaneously reacted with DEX-CHO-x to produce drug-conjugated dextran polymer DEX-IM-DOX (see Scheme 1). All intermediates and polymer derivatives were characterized by NMR and FT-IR spectroscopy (see details in the ESI†).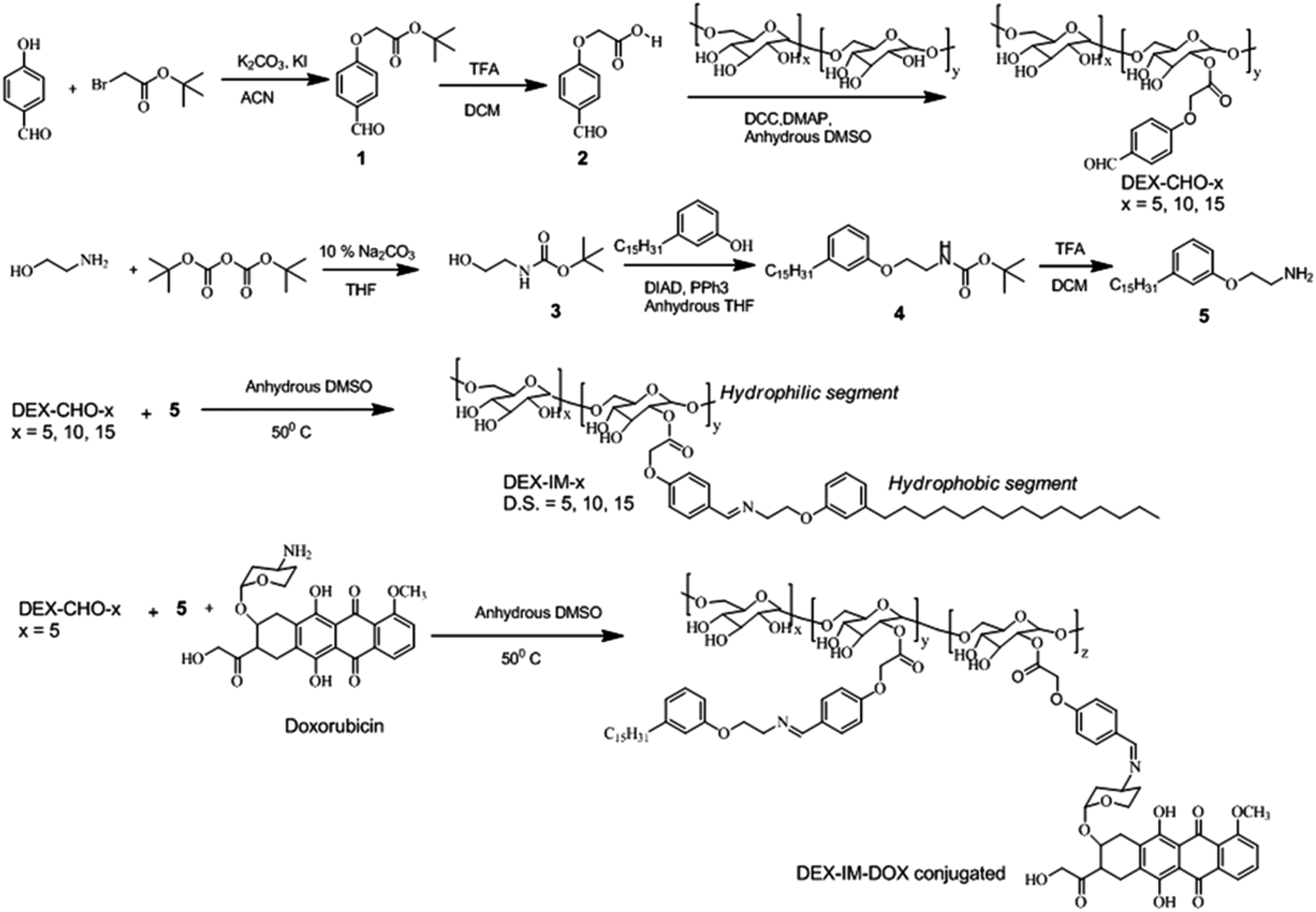 | ||
| Scheme 1 Synthesis of pH and enzyme responsive dextran derivatives. | ||
The 1H-NMR spectrum of the DEX-CHO-x (see Fig. 2a) exhibited peaks at 9.82, and 7.82 to 7.08 ppm for the aldehyde and aryl protons, respectively. The protons of the ester linkage, Ar-OCH2-COO-DEX, appeared at 4.64 ppm, which was merged with the anomeric protons of dextran (see Fig. 2a). All other peaks from 3.00 to 5.00 ppm were assigned to dextran protons. The 13C-NMR spectrum of DEX-CHO showed a peak at 65.11 ppm corresponding to a new ester carbon atom Ar-OCH2-COO-dex (see SF-1†). All the protons and carbons in the structure of the DEX-CHO-x molecule were assigned using a 2D NMR-HSQC technique (see ESI, SF2†). The degree of substitution (DS) in DEX-CHO-x in the dextran back bone was determined by comparing the peak intensities of anomeric protons in dextran at 4.62 ppm with the aryl protons at 7.82 ppm.40,42 The DS in DEX-CHO-x was obtained as x = 5.2, 10.1 and 14.7% (for simplicity x = 5, 10 and 15) for the feed ratio of 0.5, 1.0 and 1.5 equivalents of 4-formyl phenyl acetic acid to the dextran repeating unit. The imine polymer DEX-IM-x (x = 5, 10 and 15) was obtained by reacting DEX-CHO-x with PDP amine (see SF-3 for more 1H-NMR analysis†). The appearance of HCN protons at 8.2 ppm (see proton c in Fig. 2b) and disappearance of the aldehyde proton at 9.82 ppm (see proton c in Fig. 2a) are evidence for the formation of the imine linkage in DEX-IM-x (see Fig. 2b).
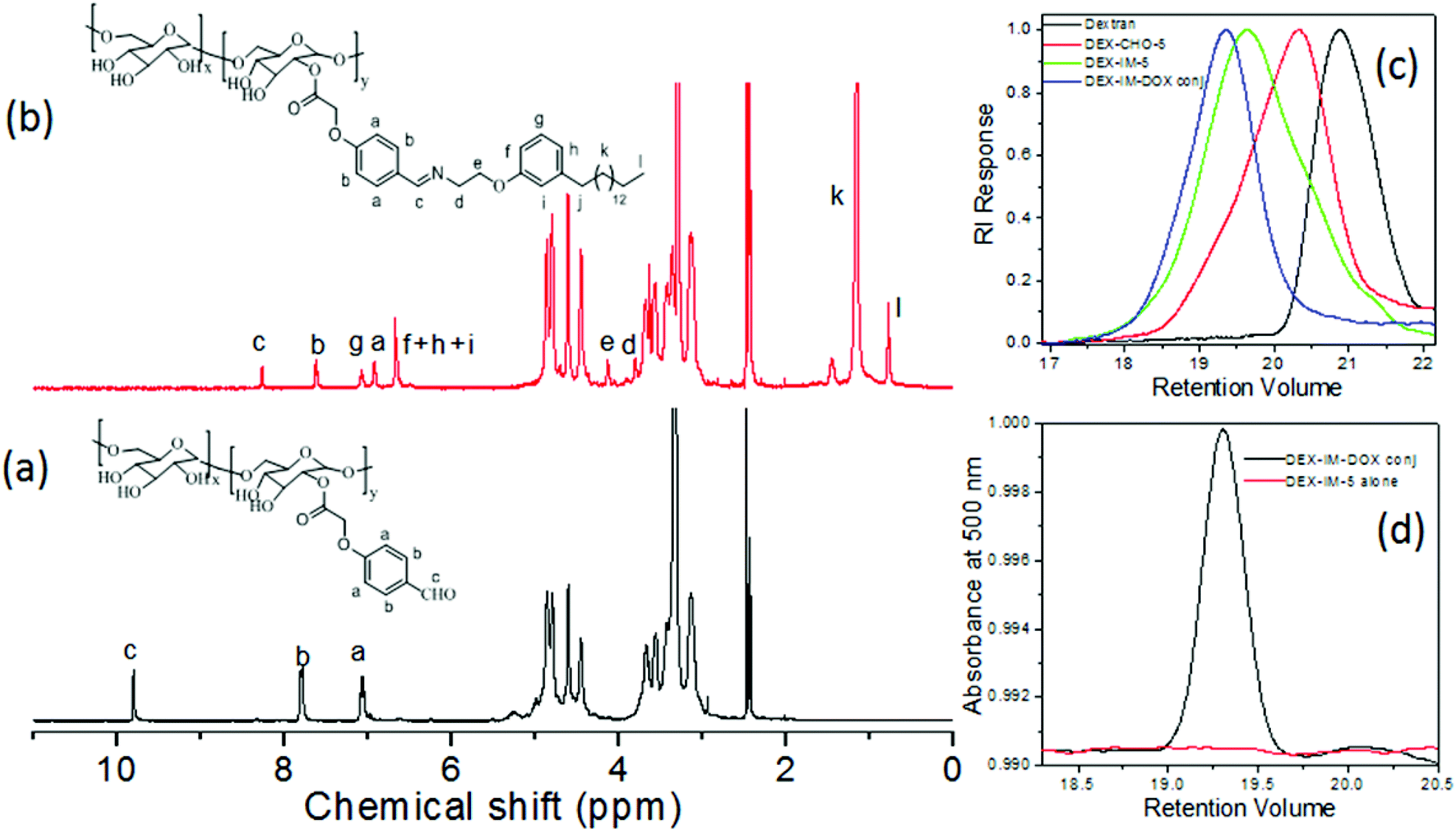 | ||
| Fig. 2 1H NMR spectra of (a) DEX-CHO-x and (b) DEX-IM-x in d6 DMSO. (c) GPC chromatograms of dextran, DEX-CHO-5, DEX-IM-5 and DOX conjugated DEX-IM-x using an RI detector. (d) GPC chromatograms of DEX-IM-5 and DOX conjugated DEX-IM using a UV absorbance detector, detection λ = 480 nm. | ||
1H-NMR spectra of the drug-conjugated polymer DEX-IM-DOX (see SF-4†) showed peaks with respect to DOX; however, the signals are noisy for their actual incorporation. In order to determine the amount of the DOX drug conjugated in the DEX-IM-DOX, the absorbance spectrum of the drug-conjugated polymer was recorded in DMSO (see SF-5†). Based on the equation A = εCl, the amount of DOX conjugated on the dextran backbone was calculated as 29.20 μg mg−1 of polymer.43 The GPC chromatograms of the dextran, DEX-CHO-5 and DEX-IM-5 were recorded using an RI detector and they are shown in Fig. 2c. All the peaks showed mono-modal distribution with respect to the homogeneous grafting on the dextran backbone (see MW in ESI-ST1†). The polymer DEX-PDP-5 (nascent polymer) did not show any signal in the UV-Vis detector. On the other hand, the GPC chromatograms of DEX-IM-DOX showed a peak in the UV-Vis detector response (with λ of detection at 480 nm) with respect to DOX conjugation in the backbone. FT-IR spectra of the polymers are given in the ESI (SF-6†). The appearance of a distinct band at 1597 cm−1 corresponds to the benzoic imine chemical linkage and further validates the formation of the expected chemical structure (see SF-6†).
3.2. Self-assembly of pH responsive scaffolds
The newly synthesized pH responsive dextran derivatives (DEX-IM-5) were self-organized in PBS via a solvent exchange method. The polymer was dissolved in N,N-dimethylsulfoxide (DMSO) and the solution was subjected to dialysis in PBS for 48 h. The resultant DEX-IM-5 solution in PBS was subjected to dynamic light scattering (DLS) analysis. The DLS histograms showed mono-modal distribution (see Fig. 3a for histogram and autocorrelation) with hydrodynamic diameter (Dh) 220 nm ± 10 nm. From this data, the hydrodynamic radius (Dh/2 = Rh) of the polymer assembly was calculated as 110 nm. Static light scattering (SLS) analysis of the polymer assembly provided its radius of gyration (Rg) (Guinier plot, see inset in Fig. 3a), Rg = 110 nm. The ratio of Rg/Rh = 1 revealed that the DEX-IM-5 polymer self-assembled as vesicular structures in solution.40,44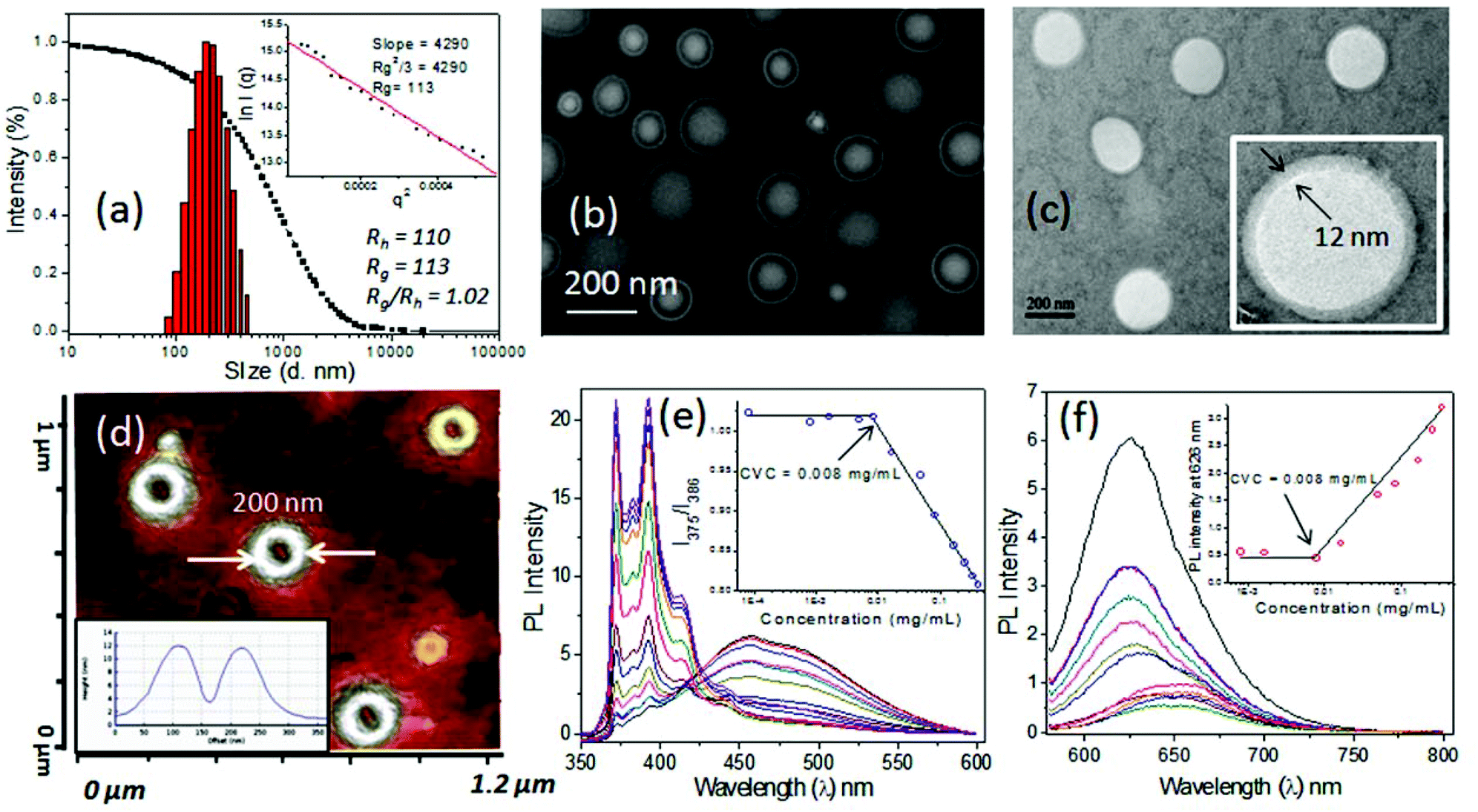 | ||
| Fig. 3 DLS histogram and SLS plot (inset) (a), FE-SEM image (b), TEM image (c) and AFM image (d) of DEX-IM-5. The inset in (c) shows the magnified vesicular structure. Cross sectional analysis of AFM is depicted in the inset in (d). Emission spectra of pyrene (e) and Nile red (f) at different DEX-IM-5 concentrations. I1/I3 of pyrene vs. the concentration of polymer and emission intensity at 626 nm vs. the concentration of polymer are shown as insets in (e) and (f), respectively. | ||
Electron microscopes were employed to visualize the shape and size of the DEX-IM-5 self-assemblies. The FESEM image of DEX-IM-5 polymer shows the formation of spherical objects with a distinct hydrophobic layer and inner cavity with respect to the vesicular assemblies (see Fig. 3b). The average diameter of these vesicles was calculated using ImageJ software as 200 ± 30 nm (see Fig. 3b). HR-TEM images of the DEX-IM-5 aggregates (see Fig. 3c) provided further evidence for the existence of a spherical vesicular morphology with a well defined hydrophobic periphery as seen in Fig. 3c. The average size of the vesicles obtained from the HR-TEM images was 220 ± 25 nm which was in accordance with the FE-SEM analysis. The thickness of the hydrophobic membrane was calculated for individual vesicles as 12 ± 3 nm. Atomic force microscope (AFM) analysis in the tapping mode was employed for the DEX-IM-5 sample on the mica surface. A representative AFM phase image (see Fig. 3d) proved that the vesicles were collapsed on the mica surface and as a result differential responses were provided to the force of the AFM tip from the soft interior and hard peripheral regions of the vesicular structures. AFM images of the vesicles showed a hollow cavity inside, which was further supported by the cross sectional analysis (see Fig. 3d, inset).45,46 The diameter of the vesicles observed in AFM analysis matched with that obtained in the FE-SEM and TEM analysis. Thus the microscope images prove that vesicular assemblies are present in the DEX-IM-5 sample.
The critical vesicular concentrations (CVCs) of DEX-IM-x were determined using pyrene47,48 and Nile red49 as fluorophores. For this purpose, the concentration of pyrene was fixed as 0.6 μM and the ratio of I1/I3 peak intensities was compared to estimate the CVC. In the case of Nile red; the concentration of the fluorophore was fixed as 1.0 μM and the amount of DEX-IM-5 was varied (see inset plot in Fig. 3e and 3f). The encapsulation of Nile red in the hydrophobic cavity enhances the emission intensity (at 626 nm) and this parameter was used for CVC determination (see Fig. 3f). Based on this, the CVC of DEX-IM-5 was calculated as 0.008 mg mL−1 in both the experiments. Similarly, the CVC of DEX-IM-10 was determined to be 0.016 mg mL−1 (see SF-7†) which was almost double that of DEX-IM-5. The DEX-IM-15 was insoluble in water and it restricted the CVC determination. Compared to all the samples, DEX-IM-5 was found to be easily dispersible and produced stable vesicles in water. Thus, DEX-IM-5 was chosen for loading and delivering of anticancer drug molecules in the vesicular scaffold.
3.3. Loading capabilities of dextran vesicles
The loading capability of hydrophilic molecules in the inner cavity of the dextran vesicle was tested using water soluble dye. Water soluble Rhodamine B (RhB) was selectively encapsulated in the hydrophilic corona of the vesicles by a dialysis method (see Fig. 4a). The dye and polymer (DEX-IM-5) were dissolved in DMSO and the solution was dialyzed in PBS using a semi-permeable dialysis membrane. The un-encapsulated Rh-B was removed by continuous replacement of fresh PBS and the dialysis was continued for 3 to 7 days. The dialyzed samples were highly fluorescent and their photographs are shown in Fig. 4a. The DLS histograms of RhB loaded vesicles (see Fig. 4b) revealed that the hydrodynamic diameter of the vesicles was 220 nm (see Fig. 3a). FE-SEM analysis of RhB loaded DEX-IM-5 (see Fig. 4c) showed the existence of a spherical morphology with a distinct hydrophobic layer with respect to the vesicular geometry.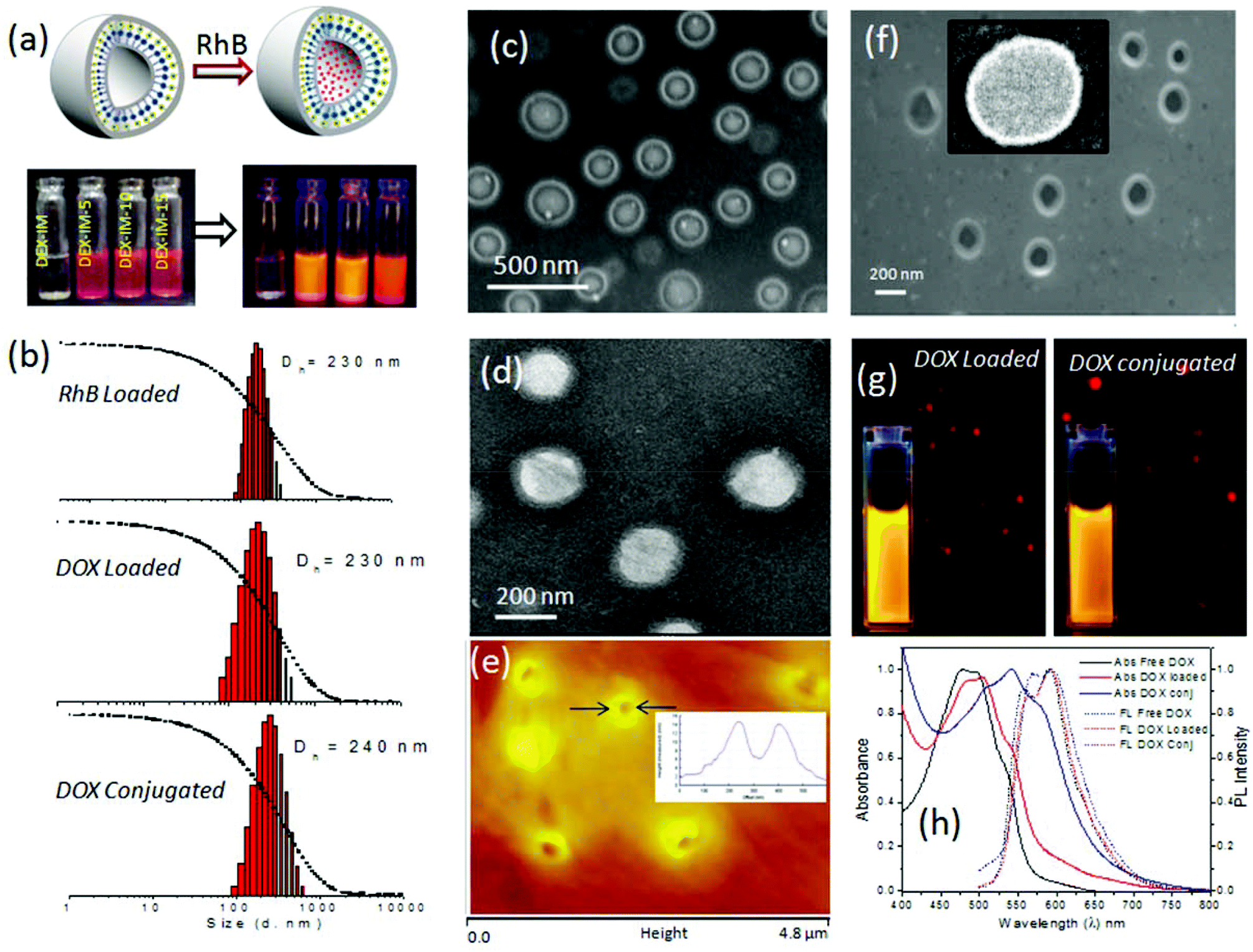 | ||
| Fig. 4 Schematic representation of a rhodamine B (RhB) encapsulated vesicle and photographs of RhB loaded vesicles in vials (a). DLS histogram of RhB loaded, DOX loaded and DOX conjugated DEX-IM-5 vesicles (b). FE-SEM image of RhB loaded DEX-IM-5 vesicles (c). HR-TEM image of DOX loaded DEX-IM-5 vesicles (d). AFM image of DOX loaded DEX-IM-5 vesicles (e). FE SEM image and HR-TEM image (inset) of DOX conjugated vesicles (f). Fluorescence microscope images of DOX loaded and conjugated vesicles (g). Absorbance and fluorescence plots of free DOX, DOX loaded and DOX conjugated DEX-IM vesicles (h). | ||
A similar encapsulation procedure was followed to load the water soluble anticancer drug DOX·HCl (a topoisomerase II inhibitor)50 in the vesicular hydrophilic cavity. The polymer to drug ratio was maintained as 5 wt% and the drug loading content (DLC) in the vesicles was estimated as 2.9 wt% by using absorption spectroscopy. Thus, the drug loading efficiency (DLE) was calculated as 58%. These values are close to those of stearic acid attached dextran amphiphiles, which were reported to load DOX with a DLE of 39–56%.27 The reason for the relatively low DLC and DLE in the dextran vesicular scaffold was attributed to the dialysis method employed over 3 to 7 days to give stable encapsulation of the drugs. This procedure provided very good control over the stability of the loaded drugs (checked for more than 3 months) as well as removal of un-loaded drugs, if any, left out at the periphery of the vesicles. Based on our earlier studies,40,42 we found that stable encapsulation of drugs in the vesicular scaffold is essential for reproducible cellular uptake analysis.
The morphology of DOX loaded vesicles was characterized by both light scattering and electron microscopy methods. In Fig. 4b, the DLS histograms show the size diameter of DOX loaded vesicles as 230 nm, which was similar to the nascent vesicles or Rh-B loaded vesicles (see Fig. 3a and 4b). FE-SEM analysis of the DOX loaded sample also showed the existence of vesicular geometry with an average diameter of 200 ± 5 nm (see SF-8†). In Fig. 4d, the HR-TEM image further confirms the retention of a spherical vesicular structure in the DOX loaded dextran vesicles. The average diameter of vesicles was 210 ± 10 nm (see Fig. 4b). AFM analysis of the DOX loaded sample confirmed the vesicle geometry with an average diameter of 200 ± 5 nm and height of 13 ± 3 nm (see Fig. 4e and cross sectional analysis in the inset). Thus, the above analysis proved that the loading of the DOX·HCl did not alter the morphology of the nascent dextran polymer and the Rh-B and DOX loaded samples were found to retain the vesicular structure. DOX chemically conjugated on the dextran backbone was dissolved in DMSO and allowed to dialyze in PBS (pH 7.4). DLS histograms of this sample showed mono-modal distribution indicating its uniform self-organization (see Fig. 4b). The hydrodynamic diameter of the assembly was obtained as 240 ± 5 nm (see Fig. 4b), which is comparable to the DOX loaded or nascent polymer assemblies (see Fig. 4b and 3a). The FE-SEM image of the DOX conjugated sample showed a spherical vesicular structure and this was further confirmed by the HR-TEM image as well (see Fig. 4f and inset). Since DOX is a fluorescent drug, the DOX loaded and DOX conjugated samples were subjected to photophysical characterization as well as fluorescence microscopy imaging. FL images captured under the red channel proved the existence of intact luminescent spherical structures for both DOX loaded and conjugated systems (see Fig. 4g). The photographs of the vials containing the DOX loaded and DOX conjugated systems showed identical emission following photo-excitation. The absorbance spectra of the free DOX and DOX loaded vesicles displayed almost identical λmax values at 480 to 484 nm (see Fig. 4h). On the other hand, the DOX conjugated dextran showed a 30 nm red-shift with respect to the chemical conjugation. Interestingly, both DOX loaded and DOX conjugated assemblies showed identical emission spectra. This proves that the DOX molecule was very well preserved inside the dextran vesicles. Hence, it may be concluded that the nascent polymer DEX-IM-5, its DOX loaded form and its drug conjugated form all retained identical vesicular morphology with similar sizes. This provides a new opportunity to study the drug release pattern as well the cellular uptake for both DOX loaded and DOX conjugated polysaccharide nano-carriers.
3.4. pH and enzyme responsive in vitro drug release studies
The benzylic imine linkage connecting the dextran backbone with the PDP unit is sensitive to acidic pH conditions. At low pH = 5.0 to 6.5, the imine linkage is cleaved into its components DEX-CHO-5 and PDP amine as shown in Fig. 5a. DEX-IM-5 in d6-DMSO was subjected to 1HNMR analysis after stirring with a drop of trifluoroacetic acid (see Fig. 5b). DEX-IM-5 showed the imine proton HCN at 8.33 ppm which disappeared in the acid treated sample. A new peak at 9.90 ppm appeared in the hydrolyzed sample with respect to the regeneration of the –CHO functional groups in DEX-CHO-5 (see also Fig. 2a). Furthermore, the protons in the PDP-OCH2CH2NCH-Ar-dextran disappeared in the hydrolyzed sample and protons corresponding to PDP-OCH2CH2NH2 (with respect to compound 5) appeared at 4.09 ppm. This control experiment confirmed the cleavage of the imine under acidic conditions. DLS histograms of the DEX-IM-5 were recorded at pH 7.4 and pH 6.0 at different time intervals. At pH 7.4, the DEX-IM-5 retained a mono-modal distribution for a longer duration (checked up to 7 days, see SF-9†). However, under acidic pH conditions (pH 6.0), the uni-modal distribution of DEX-IM-5 was lost and a multi-modal distribution with respect to the existence of non-homogeneous aggregates was obtained. This suggested that DEX-IM-5 was prone to being cleaved under acidic conditions. With increasing time, the distribution of the aggregates became broader and the sizes varied up to 1.0 μm (see Fig. 5c). The pH responsive cleavage of DEX-IM-5 was further investigated at pH = 6.0 by FE-SEM, AFM and HR-TEM analysis (see Fig. 5d, e and f). The FE-SEM image (in Fig. 5d) shows that the vesicles were disintegrated at acidic pH, unlike at pH = 7.4 (see Fig. 3b). A similar observation was made in the AFM image (see Fig. 5e) and TEM image (see Fig. 5f). Thus, the newly designed DEX-IM-5 polymer formed stable vesicular structures at physiological pH = 7.4 (see images in Fig. 3) and was cleaved at acidic pH to the corresponding DEX-CHO and PDP amine units (see images in Fig. 5).
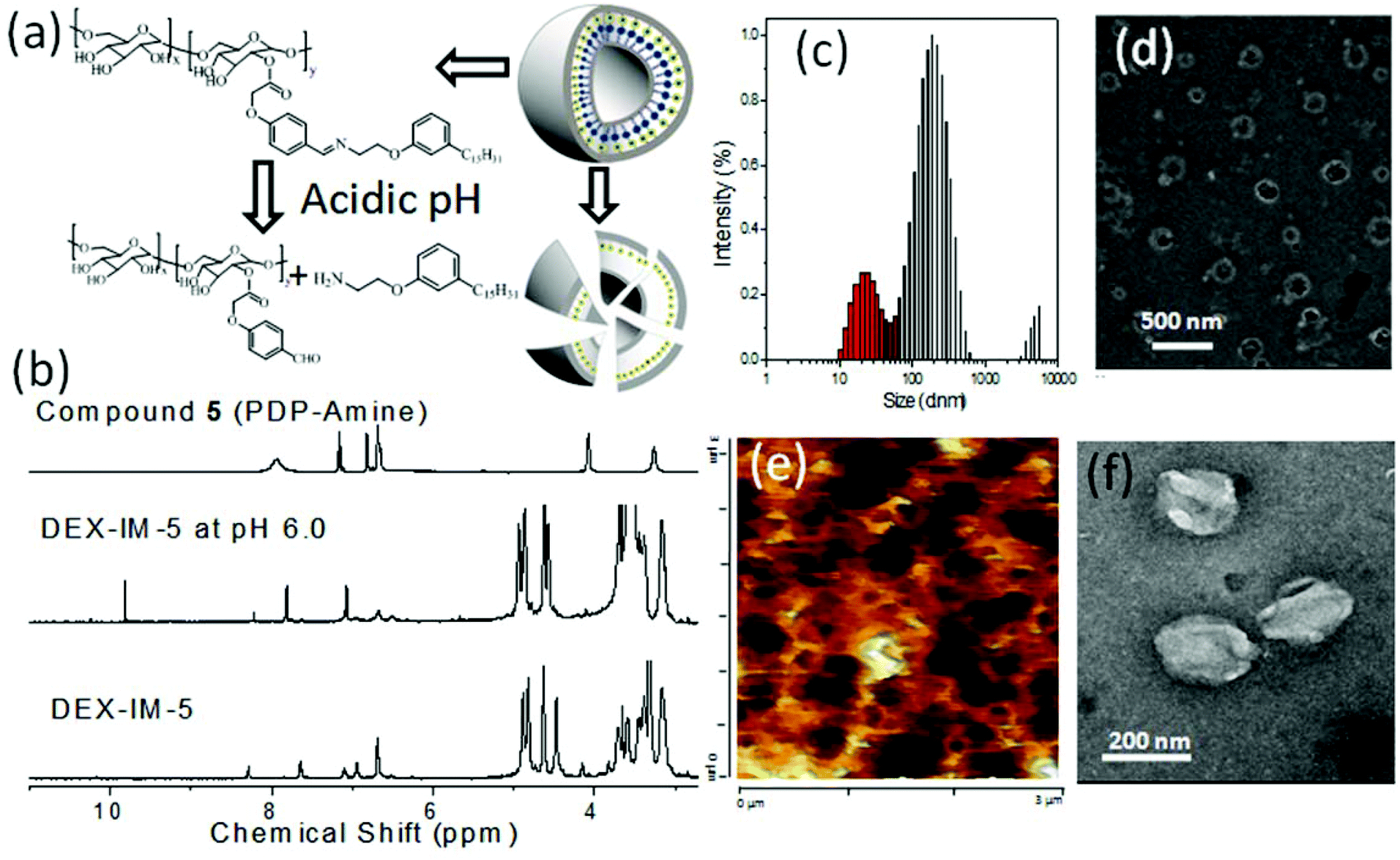 | ||
| Fig. 5 Cleavage of DEX-IM-5 under acidic conditions (a). (b) NMR spectra, (c) DLS histogram, (d) FE-SEM image, (e) AFM image, and (f) TEM image of DEX-IM-5 under acidic pH (6.0) conditions. | ||
The in vitro drug release studies for both DOX loaded dextran vesicles and DOX conjugated dextran vesicles were performed under various pH conditions from pH 7.4 to 5.0 by a dialysis method. In vitro release studies were done in PBS buffer or PB buffer using a dialysis method and the amount of the DOX released was quantified by absorbance spectroscopy up to 48 h. Typical absorbance spectra of DOX released from dextran vesicles are shown in the ESI (see SF-10†) The cumulative release of DOX using pH and esterase as stimuli are shown in Fig. 6a and b, respectively. At pH = 7.4 (normal tissue pH), the vesicles underwent leakage to release about 25 ± 5% of the drugs. A similar trend was observed at pH 6.5 (30 ± 5% drug release) which is similar to cancer tissue pH.
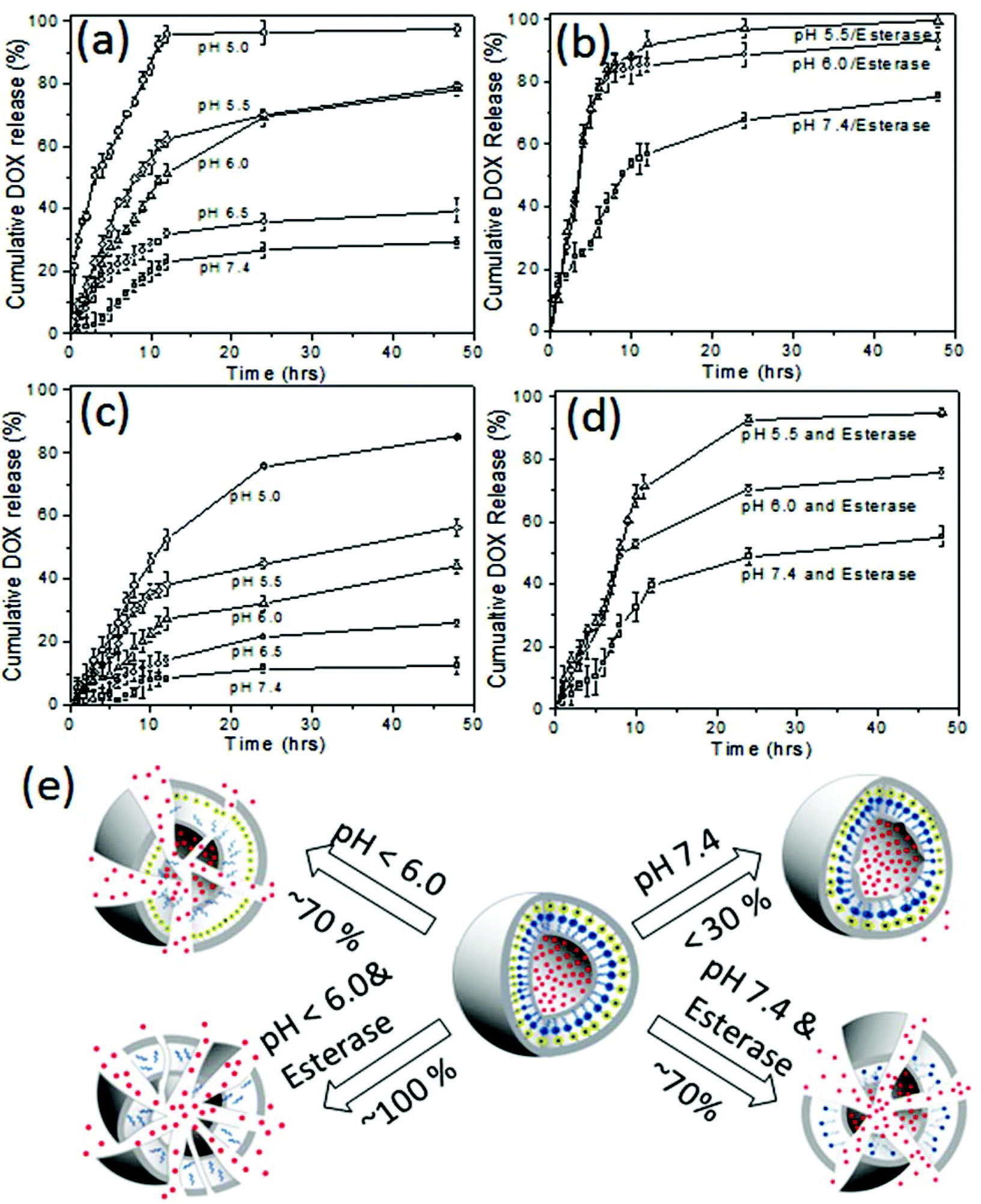 | ||
| Fig. 6 DOX release profiles for DOX loaded DEX-IM-5 vesicles at various pH values (a). DOX release profiles for DOX loaded DEX-IM-5 vesicles in the presence of esterase at various pH values (b). DOX release profiles for DOX conjugated DEX-IM-5 vesicles at various pH values (c). DOX release profiles for DOX conjugated DEX-IM-5 vesicles in the presence of esterase enzyme at various pH values (d). Schematic representation of DOX release under various conditions (e). | ||
Vaupel et al. and others have reported that the pH of mammary carcinoma is 6.7 and in some micro areas it drops to about pH = 5.4.51,52 Thus, a minimum of 20–30% drug leakage from the vesicles was unavoidable under physiological pH conditions. In order to investigate the effect of esterase enzyme; two control experiments were carried out: (i) esterase enzyme administration at pH = 7.4 and (ii) esterase enzyme administration at pH = 6.0 and 5.0. The first experiment created an environment for the dextran vesicles to undergo cleavage by esterase enzymes similar to that of the extracellular environment (if any enzyme is present in the blood plasma). On the other hand, the second experiment provided conditions similar to intracellular conditions in which the enzyme action occurs at a low pH = 6.0 to 5.0 (dual stimuli, abundant in the lysosomes).53 These control experiments indirectly provided more insight into the drug delivery mechanism for the dextran vesicle administration as the model proposed in Fig. 1a.
The drug release profiles for the DOX loaded dextran vesicles are shown in Fig. 6b. The release of DOX was increased from 25% (absence of enzyme, see Fig. 6a) to 70% (in the presence of 10 U enzyme, see Fig. 6b) at pH = 7.4. At pH = 6.0 and pH = 5.5, the esterase enzyme became more active and the vesicles were ruptured completely to release 100% of the loaded drug (see Fig. 6b). To further validate the stability of the DOX loaded dextran vesicles in extracellular conditions, they were subjected to release studies in fetal bovine serum (FBS) and the data are summarized in SF-11† at 37 °C. The DOX loaded vesicles were found to be more stable in FBS and only 18–21% leaching of the drug was noticed at 37 °C. Furthermore, the DOX loaded dextran vesicles were also subjected to DLS analysis in FBS (also FBS + PBS) for 48 h to analyze their stability (see SF-12†). The sizes of the nano-vesicles remained at 220 nm for the period of 48 h without any change. These control experiments revealed that the dextran nano-vesicles are very good at stabilizing DOX in the internal cavity and are selectively ruptured in the presence of esterase enzyme at low pH similar to that of intracellular environment as shown in Fig. 1a.
Similarly, the drug release kinetics of the DOX conjugated dextran vesicles were also studied and the details are shown in Fig. 6c and d. The drug conjugated vesicles showed much less leaching (<10%) under normal circulatory conditions at pH = 7.4 at 37 °C. A similar trend was observed in FBS at 37 °C (see SF-11†). This proved that the imine linker connecting DOX to the dextran back bone was very stable in physiological conditions. With a decrease in pH, the drug releasing ability of the vesicles increased; however, about 30% of the drug was not accessible even at lower pH = 5.0 (see Fig. 6c). The esterase enzyme assisted the much faster release of DOX from 55% to 75% when varying the pH from 7.4 and to pH 6.0. Interestingly, 90–95% of the drug was completely released in the presence of esterase at pH 5.5 (see Fig. 6d). Based on the above data, a schematic presentation of the drug release patterns for DOX loaded dextran vesicles and DOX conjugated dextran vesicles is provided in Fig. 6e. The comparison of drug release trends in both DOX loaded and DOX conjugated dextran vesicles confirmed the intracellular compartment drug release model proposed in Fig. 1a. The rate constant (k) for the drug release was estimated using the first order rate equation:54 ln(At/A0) = −kt, where At and A0 correspond to the drug in the vesicles at time ‘t’ and the initial point, respectively. The rate constants are given in table ST2†. At pH = 7.4 and pH = 6.5 the DOX loaded vesicles showed the rates k = 6.5 to 8.4 μs−1. These values were almost half of those of the DOX conjugated dextran vesicles (k = 2.4 to 4.0 μs−1). At endosomal pH = 6.0 to 5.5, the rate constants were improved three fold (k = 16.5 to 22.6 μs−1) compared to circulatory pH = 7.4. In the presence of esterase, the rate constants that were obtained were much higher with the dual action of pH and enzyme. The above in vitro studies proved that both DOX loaded vesicles and conjugated vesicles preserved the drugs under circulatory pH conditions and released them substantially under intracellular or in vivo acidic tumor conditions.15,51,52
3.5. Cellular uptake and cytotoxicity
The cytotoxicity of the polymer vesicle (DEX-IM-5) was investigated in WTMEFs (fibroblast cells) by the MTT assay method.55 As shown in Fig. 7a, free polymer (DEX-IM-5) showed more than 80% cell viability even at the highest concentration of 500 μg mL−1. This result confirmed the high biocompatibility of the newly designed dextran vesicles for further delivery applications. The cytotoxicity of DOX, DOX loaded DEX-IM-5 vesicles and DOX-conjugated vesicles were tested in both breast cancer MCF 7 cells and normal WTMEFs cell lines. The experiments were carried out under identical conditions and the drug concentration was maintained as 0.289 μg mL−1 (equivalent to 0.5 μM) for free DOX in PBS, DOX loaded and DOX conjugated vesicles in PBS. The cytotoxicity data are summarized for MCF 7 and WTMEFs in Fig. 7b and c, respectively. DOX loaded in vesicles exhibited about 50% killing of MCF-7 cells at 0.5 μM concentration, which was about 15% more than free DOX (see Fig. 7b). The DOX conjugated vesicles showed comparable killing to free DOX. Since the DOX loaded or conjugated nano-vesicles are capable of showing better uptake in the cancer tissues by the EPR effect compared to the free drug; the cytotoxicity data shown by the new scaffolds are very good for drug administration purposes. The comparison of the cytotoxicity among MCF7 cells and normal WTMEFs suggested that the present scaffold design showed significantly better killing in the breast cancer cells. Furthermore, the effect of DOX-conjugated vesicles and DOX loaded vesicles could be enhanced by the surface modification of the nano-carrier using targeting ligands for a better response.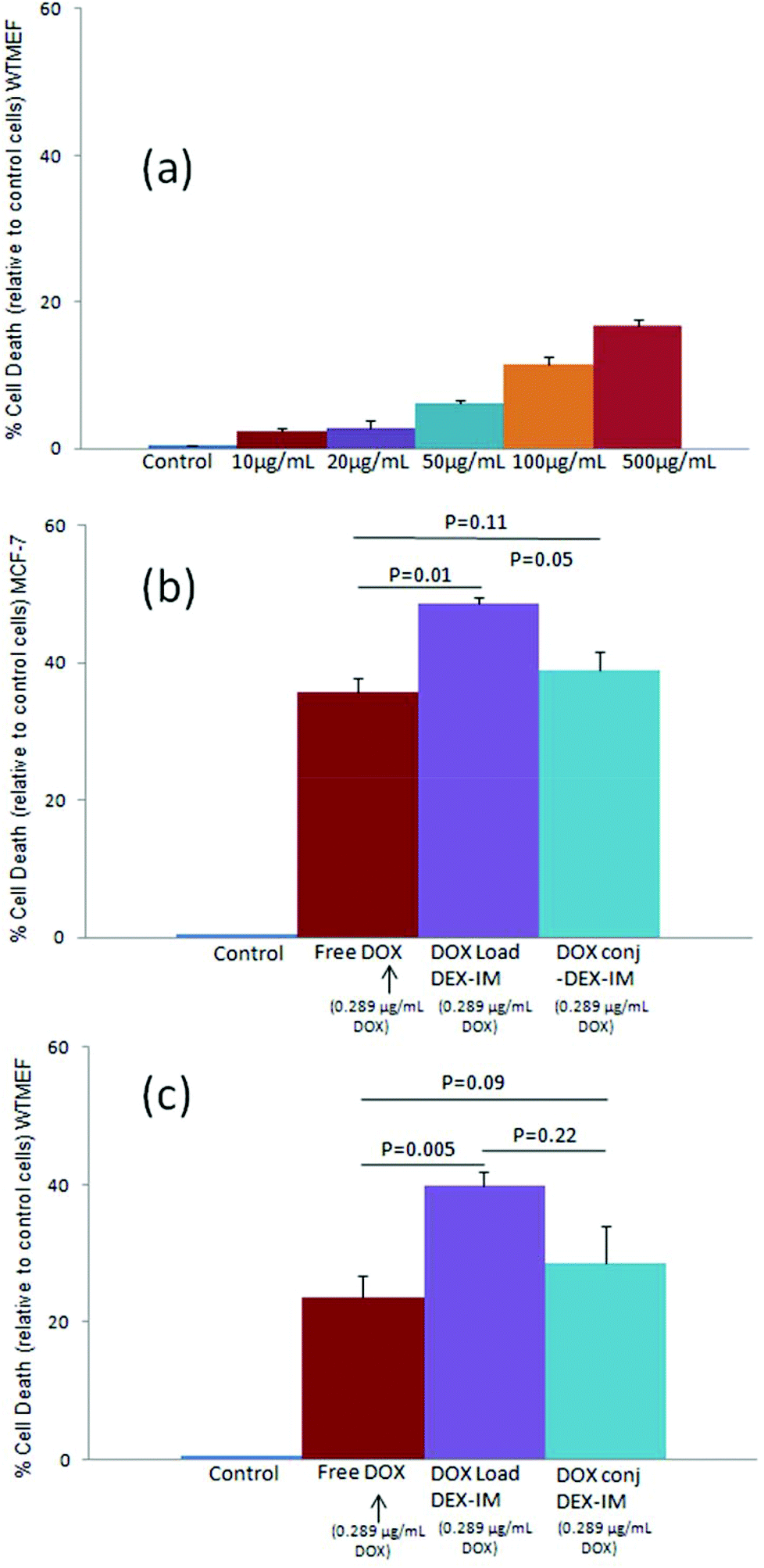 | ||
| Fig. 7 (a) Histogram depicting the cytotoxicity of DEX-IM-5 vesicles in WTMEFs at various concentrations. (b) Cytotoxicity of DOX, DOX loaded DEX-IM-5 and DOX conjugated DEX-IM-5 in MCF-7 cells. (c) Cytotoxicity of DOX, DOX loaded DEX-IM-5 and DOX conjugated DEX-IM-5 in WTMEFs cells. The concentration of the drug was maintained as 0.289 μg mL−1 (0.5 μM) in the free, DOX loaded and DOX-conjugated forms. The graphs represent the mean ± SE of percentage cell death relative to the untreated control in three independent experiments and were statistically analysed (graphs b and c) using a standard t-test. | ||
The cellular uptake studies provide more insight into the DOX delivery efficiency of the vesicles. The red fluorescence of DOX at ∼595 nm assists in studying the uptake of the free drug and drug loaded vesicles into cells.56 Cells were treated with free DOX, DOX loaded DEX-IM-5 vesicles and DOX conjugated DEX-IM-5 vesicles at a 1.0 μM DOX concentration. DOX molecules are known to accumulate in the nucleus of the cell.56 Thus, the nuclei were mapped in these experiments by staining with DAPI. The actin cytoskeletal networks of the cells were also stained with phalloidin conjugated to Alexa 488 dye (green). This staining was used to identify the morphology of the adherent spread cell. The cells were observed through the blue channel (at λ 405 nm) for DAPI – the nucleus, and the green channel (at λ 488 nm) for Phalloidin – the actin cytoskeleton. The collected images were merged together and are shown on left side of each panel in Fig. 8. DOX fluorescence was observed through the red channel (at λ = 568 nm) and these images are shown on the right side in Fig. 8. A closer inspection of the images revealed a very good overlap between the DOX and DAPI staining in the nucleus (compare the left and right side in each row). To demonstrate the reproducibility of the data, two sets of the images were collected and additional images are given in SF-13.† Administration of free drug DOX showed a diffuse staining in the cytosol and some accumulation in the nucleus (see Fig. 8b). DOX loaded in dextran DEX-IM vesicles showed an increased red fluorescence in the nucleus (see Fig. 8c). DOX conjugated vesicles showed higher uptake compared to free DOX (compare Fig. 8b and d); however, the intensity was relatively lower compared to that of DOX loaded vesicles (compare Fig. 8c and d). The fact that the nuclear binding of DOX released from the vesicles is better than that of free DOX seems to suggest the greater availability of DOX at its site of action when in the vesicle than as the free drug. This better binding of DOX showed as higher fluorescence intensity when DOX was administrated in the loaded form.
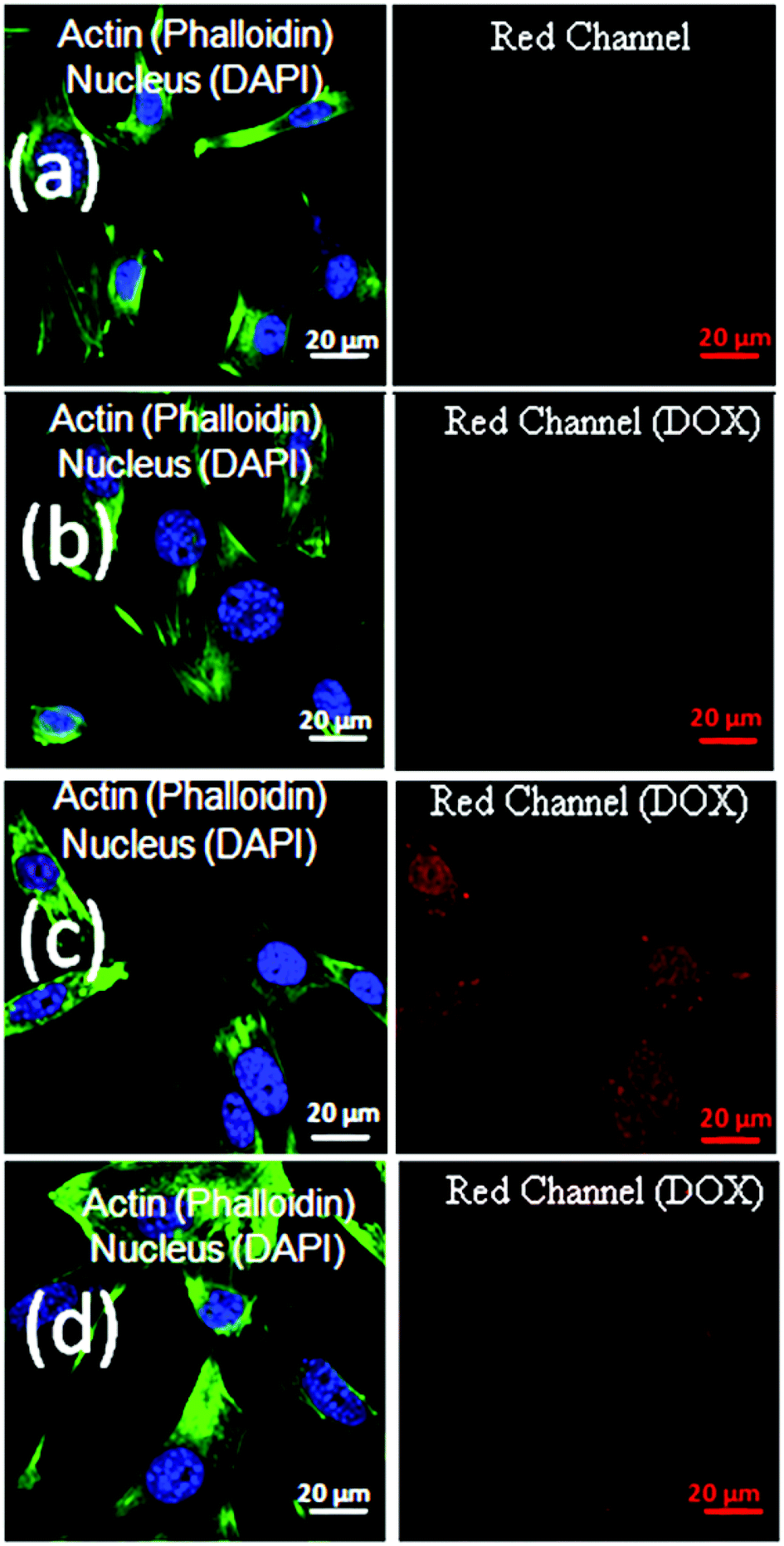 | ||
| Fig. 8 Confocal microscopic images of (a) control, (b) free DOX, (c) DOX loaded DEX-IM vesicles and (d) DOX conjugated DEX-IM vesicles treated WTMEFS cells. The nucleus was counter stained with DAPI (blue), the actin cytoskeletal network in cells is stained with phalloidin (green). The cells were observed through the red channel to locate DOX fluorescence (red). The scale bar in each panel represents 20 μm. | ||
The difference in the uptake between the DOX loaded and DOX conjugated vesicles is attributed to the difference in their release profiles (see Fig. 6). The DOX loaded vesicles showed much faster release compared to the DOX conjugated vesicles; as a result, the physically loaded DOX showed enhanced intensity in cellular imaging. Based on the cellular imaging studies alone, it is rather difficult to prove the pathways under which the nano-vesicles entered and ruptured in the intracellular compartments. More experimental proof is required in terms of the organelle specific accumulation of nano-vesicles such as in endosomes, lysosomes, etc. To address this problem, current research is focused on developing pH responsive dye probes which could be either physically loaded or chemically conjugated in the dextran vesicles. Nevertheless, the detailed in vitro drug release studies, cytotoxicity data and cellular imaging proved that the current dextran vesicles are potential vectors for releasing loaded cargoes in intracellular compartments in breast cancer cells. Furthermore, the combined effect of esterase and pH on the cytotoxicity data would be more visible only in in vivo studies in cancer tissues.
4. Conclusion
In conclusion, new polysaccharide vesicles were designed and developed based on dextran and a renewable resource hydrophobic unit. Both pH and enzyme dual responsiveness was in-built in the structural engineering to deliver drugs exclusively in a cancer tissue environment. The pH and enzyme responsiveness were achieved through acid labile imine linkages and the lysosomal esterase enzyme cleavable aliphatic ester linkage. The dextran derivative was self-organized into vesicular scaffolds in which the water soluble anticancer drug DOX·HCl was successfully encapsulated for delivery into the cancer cells. The imine chemistry was further exploited to anchor DOX in the dextran backbone which produced DOX conjugated dextran vesicles. Remarkably, the vesicular geometry in the dextran scaffold was retained even after the loading and anchoring of DOX molecules. The vesicular assemblies were characterized by light scattering, electron microscopy, atomic force microscopy and photophysical studies. In vitro studies confirmed the stability of dextran vesicular assemblies under physiological pH conditions in PBS and FBS. NMR and microscopic analysis confirmed the cleavage of the acid labile benzylic imine linkage at acidic pH (≤6.0). In vitro studies further revealed that the esterase enzyme (abundant in lysosomal compartments of cells) assisted the vesicular rupture so that 100% of the loaded or conjugated drugs were released in intracellular environments. The MTT assay showed that the dextran vesicle was non toxic to cells up to 500 μg mL−1. The DOX loaded (and also conjugated) vesicles exhibited better cell killing efficiency than free drug in breast cancer cells (MCF7) compared to normal cells. The approach demonstrated here provides a new design principle to suitably modify dextran as a pH and enzyme responsive nano-carrier; however, this approach is not restricted to dextran alone, and in principle, it may be applicable to a wide variety of other polysaccharides. Furthermore, in the present studies only one water soluble anticancer drug, DOX·HCl, was explored; in principle, the approach may be expanded to load other water soluble drugs as well as water insoluble drugs in dextran vesicles. The present approach provides new insights into the design of pH and enzyme responsive polysaccharide vesicles for physically loaded and chemically conjugated DOX and its delivery in breast cancer cells.Authors contribution
All authors contributed equally. The authors declare no competing financial interests.Acknowledgements
The authors thank a research grant from Department of Science and Technology (DST), New Delhi, INDIA, under the nano-mission initiative project SR/NM/NS-42/2009 and SB/S1/OC-37/2013 under the SERB scheme. The authors thank Dr Nagaraj Balasubramanian, Department of Biology, IISER-Pune for discussions and providing cell culture facilities and cellular uptake studies. Pramod thanks IISER Pune for a research fellowship. The authors thank National Chemical laboratory, Pune, India for HR-TEM, and SLS facilities.Notes and references
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- S. Kashyap and M. Jayakannan, J. Mater. Chem. B, 2014, 2, 4142–4152 RSC.
- K. L. Hamner, C. M. Alexander, K. Coopersmith, D. Reishofer, C. Provenza and M. M. Maye, ACS Nano, 2013, 7, 7011–7020 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- S. Y. Park, H. J. Baik, Y. T. Oh, K. T. Oh, Y. S. Youn and E. S. Lee, Angew. Chem., Int. Ed., 2011, 50, 1644–1647 CrossRef CAS PubMed.
- J.-H. Lee, K.-J. Chen, S.-H. Noh, M. A. Garcia, H. Wang, W.-Y. Lin, H. Jeong, B. J. Kong, D. B. Stout, J. Cheon and H.-R. Tseng, Angew. Chem., Int. Ed., 2013, 52, 4384–4388 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharm., 2010, 7, 1913–1920 CrossRef CAS PubMed.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- X. Zhang, Y. Lin and R. J. Gillies, J. Nucl. Med., 2010, 51, 1167–1170 CrossRef CAS PubMed.
- G. Liu, Y. Li, V. R. Sheth and M. D. Pagel, Mol. Imaging, 2012, 11, 47–57 CAS.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- M. M. Molla, P. Rangadurai, G. M. Pavan and S. Thayumanavan, Nanoscale, 2015, 7, 3817–3837 RSC.
- A. V. Fuchs, N. Kotman, J. Andrieu, V. Mailander, C. K. Weiss and K. Landfester, Nanoscale, 2013, 5, 4829–4839 RSC.
- M. D. Yilmaz, M. Xue, M. W. Ambrogio, O. Buyukcakir, Y. Wu, M. Frasconi, X. Chen, M. S. Nasser, J. F. Stoddart and J. I. Zink, Nanoscale, 2015, 7, 1067–1072 RSC.
- D. Grafe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- A. R. Maity, A. Chakraborty, A. Mondal and N. R. Jana, Nanoscale, 2014, 6, 2752–2758 RSC.
- J. Dai, S. Lin, D. Cheng, S. Zou and X. Shuai, Angew. Chem., Int. Ed., 2011, 50, 9404–9408 CrossRef CAS PubMed.
- Q. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- S. Mizrahy and D. Peer, Chem. Soc. Rev., 2012, 41, 2623–2640 RSC.
- J. Behravan, B. S. Fazly Bazzaz and Z. Salimi, Biotechnol. Appl. Biochem., 2003, 38, 267–269 CrossRef CAS PubMed.
- R. Mehvar, J. Controlled Release, 2000, 69, 1–25 CrossRef CAS.
- Y.-Z. Du, Q. Weng, H. Yuan and F.-Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS PubMed.
- C. Schatz, S. Louguet, J.-F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS PubMed.
- S. V. Ghugare, E. Chiessi, B. Cerroni, M. T. F. Telling, V. G. Sakai and G. Paradossi, Soft Matter, 2012, 8, 2494–2502 RSC.
- Y.-L. Li, L. Zhu, Z. Liu, R. Cheng, F. Meng, J.-H. Cui, S.-J. Ji and Z. Zhong, Angew. Chem., Int. Ed., 2009, 48, 9914–9918 CrossRef CAS PubMed.
- A. Sinha, A. Chakraborty and N. R. Jana, ACS Appl. Mater. Interfaces, 2014, 6, 22183–22191 CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
- C. C. Lee, A. T. Cramer, F. C. Szoka and J. M. J. Fréchet, Bioconjugate Chem., 2006, 17, 1364–1368 CrossRef CAS PubMed.
- B. Chen, D. G. van der Poll, K. Jerger, W. C. Floyd, J. M. J. Fréchet and F. C. Szoka, Bioconjugate Chem., 2011, 22, 617–624 CrossRef CAS PubMed.
- J. Cui, Y. Yan, Y. Wang and F. Caruso, Adv. Funct. Mater., 2012, 22, 4718–4723 CrossRef CAS.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS PubMed.
- A. S. Mikhail and C. Allen, Biomacromolecules, 2010, 11, 1273–1280 CrossRef CAS PubMed.
- P.-F. Gou, W.-P. Zhu and Z.-Q. Shen, Biomacromolecules, 2010, 11, 934–943 CrossRef CAS PubMed.
- P. S. Pramod, K. Takamura, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Biomacromolecules, 2012, 13, 3627–3640 CrossRef CAS PubMed.
- U. Sridhar, P. S. Pramod and M. Jayakannan, RSC Adv., 2013, 3, 21237–21241 RSC.
- P. S. Pramod, R. Shah, S. Chaphekar, N. Balasubramanian and M. Jayakannan, Nanoscale, 2014, 6, 11841–11855 RSC.
- Q.-D. Hu, H. Fan, Y. Ping, W.-Q. Liang, G.-P. Tang and J. Li, Chem. Commun., 2011, 47, 5572–5574 RSC.
- C. Houga, J. Giermanska, S. Lecommandoux, R. Borsali, D. Taton, Y. Gnanou and J.-F. Le Meins, Biomacromolecules, 2008, 10, 32–40 CrossRef PubMed.
- H. Iatrou, H. Frielinghaus, S. Hanski, N. Ferderigos, J. Ruokolainen, O. Ikkala, D. Richter, J. Mays and N. Hadjichristidis, Biomacromolecules, 2007, 8, 2173–2181 CrossRef CAS PubMed.
- M. Yang, W. Wang, F. Yuan, X. Zhang, J. Li, F. Liang, B. He, B. Minch and G. Wegner, J. Am. Chem. Soc., 2005, 127, 15107–15111 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- A. Zhang, Z. Zhang, F. Shi, J. Ding, C. Xiao, X. Zhuang, C. He, L. Chen and X. Chen, Soft Matter, 2013, 9, 2224–2233 RSC.
- D. Agudelo, P. Bourassa, G. Bérubé and H.-A. Tajmir-Riahi, Int. J. Biol. Macromol., 2014, 66, 144–150 CrossRef CAS PubMed.
- P. W. Vaupel, S. Frinak and H. I. Bicher, Cancer Res., 1981, 41, 2008–2013 CAS.
- J. L. Wike-Hooley, J. Haveman and H. S. Reinhold, Radiother. Oncol., 1984, 2, 343–366 CrossRef CAS.
- G. Xu, W. Zhang, M. K. Ma and H. L. McLeod, Clin. Cancer Res., 2002, 8, 2605–2611 CAS.
- J. Xu, Q. Fu, J. M. Ren, G. Bryant and G. G. Qiao, Chem. Commun., 2013, 49, 33–35 RSC.
- Y. Urasaki, G. S. Laco, P. Pourquier, Y. Takebayashi, G. Kohlhagen, C. Gioffre, H. Zhang, D. Chatterjee, P. Pantazis and Y. Pommier, Cancer Res., 2001, 61, 1964–1969 CAS.
- P. Mohan and N. Rapoport, Mol. Pharm., 2010, 7, 1959–1973 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR of DEX-CHO, 2D NMR spectra of DEX-CHO, 1H NMR of DEX-IM, 1H NMR of DEX-IM-DOX conjugated, absorbance spectra of DEX-IM-DOX conjugated, DLS, FE-SEM and TEM image of DEX-CHO-5, emission spectra of pyrene and Nile red with DEX-IM-10, FE-SEM image of DEX-IM-DOX loaded, FE-SEM image of acid treated DEX-IM-5, absorbance spectra of DOX released, in vitro DOX release from drug loaded and conjugated vesicles in the presence of serum (FBS), DLS data depicting stability of DEX-IM vesicles in serum (FBS), 1HNMR, 13C NMR and HR-MS spectra of all intermediates are provided. See DOI: 10.1039/c5nr00799b |
| This journal is © The Royal Society of Chemistry 2015 |