
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly monodisperse low-magnetization magnetite nanocubes as simultaneous T1–T2 MRI contrast agents†
V. K.
Sharma
*ab,
A.
Alipour
a,
Z.
Soran-Erdem
a,
Z. G.
Aykut
a and
H. V.
Demir
*ab
aUNAM-Institute of Materials Science and Nanotechnology, National Magnetic Resonance Research Center (UMRAM), Department of Electrical and Electronics Engineering, Department of Physics, Department of Molecular Biology and Genetics, Bilkent University, Ankara, 06800, Turkey
bLUMINOUS! Center of Excellence for Semiconductor Lighting and Displays, School of Electrical and Electronic Engineering, School of Mathematical and Physical Sciences, Nanyang Technological University, Singapore 639798, Singapore. E-mail: volkan@bilkent.edu.tr; hvdemir@ntu.edu.sg
First published on 11th May 2015
Abstract
We report the first study of highly monodisperse and crystalline iron oxide nanocubes with sub-nm controlled size distribution (9.7 ± 0.5 nm in size) that achieve simultaneous contrast enhancement in both T1- and T2-weighted magnetic resonance imaging (MRI). Here, we confirmed the magnetite structure of iron oxide nanocubes by X-ray diffraction (XRD), selected area electron diffraction (SAED) pattern, optical absorption and Fourier transformed infrared (FT-IR) spectra. These magnetite nanocubes exhibit superparamagnetic and paramagnetic behavior simultaneously by virtue of their finely controlled shape and size. The magnetic measurements reveal that the magnetic moment values are favorably much lower because of the small size and cubic shape of the nanoparticles, which results in an enhanced spin canting effect. As a proof-of-concept demonstration, we showed their potential as dual contrast agents for both T1- and T2-weighted MRI via phantom studies, in vivo imaging and relaxivity measurements. Therefore, these low-magnetization magnetite nanocubes, while being non-toxic and bio-compatible, hold great promise as excellent dual-mode T1 and T2 contrast agents for MRI.
Introduction
Magnetic nanoparticles have been used as contrast agents for magnetic resonance imaging (MRI),1,2 drug delivery vehicles,3 and in magnetic separation.4 Among them, MRI is one of the most powerful medical diagnostic tools because it can provide images in a noninvasive manner together with real-time monitoring capability featuring excellent anatomical details based on the soft tissue contrast and functional information.5 The sensitivity of MRI can be greatly improved by using contrast agents that enhance the contrast of the region of interest from the background. The MRI contrast agents are generally categorized according to their effects on longitudinal (T1) and transversal (T2) relaxations, and their respective ability is referred to as longitudinal (r1) and transversal (r2) relaxivity. The region where T1 relaxation takes place appears brighter, whereas T2 relaxation results in a darker contrast in the MR images. T1-Based contrast agents are thus also called as positive contrast agents, whereas T2 counterparts are also known as negative contrast agents.Superparamagnetic iron oxide (SPIO) nanoparticles (NPs) with strong magnetic moments are the prevailing T2 contrast agents, especially in the imaging and detection of lesions from normal tissues.6 The significant drawbacks of these T2 contrast nanoparticles are, however, magnetic susceptibility artifacts and negative contrast effects, which may limit their clinical applications. In contrast, T1 imaging, typically using paramagnetic materials as the contrast agents, provides an excellent resolution between tissues due to their high signal intensity. Gadolinium (Gd) and manganese (Mn) based species are the most commonly used T1 contrast agents in clinics.7,8 With unique advantages of their own, combining T1 and T2 imaging capabilities into a single type of contrast agent for MRI attracts considerable interest because this can give accurate diagnostic information. As a result, this creates strong motivation for designing new strategies to obtain synergistically enhanced T1 and T2 dual modal contrast agents (DMCAs) for MRI. There are few reports9–14 on the DMCAs with both T1 and T2 capabilities for MRI. MnxFe1−xO nanocrystals have been reported as potential DMCAs by different groups.9,11 It was found that a specific composition results in simultaneous T1 and T2 contrast enhancement effects, which stems from different magnetic moments of the constituent Mn2+ and Fe2+ ions.15 Gadolinium-labeled magnetite nanoparticles (GMNPs)12 synthesized via conjugation of gadolinium and magnetite nanoparticles have also been reported as potential DMCAs. Zhou et al.10 demonstrated monodisperse gadolinium iron oxide (GdIO) nanoparticles as DMCAs synthesized using a magnetically decoupled core–shell design.16 In this design, GdIO nanoparticles were obtained by embedding the paramagnetic Gd2O3 species into superparamagnetic Fe3O4 nanoparticles. However, although gadolinium (Gd)17 has been the most popular choice among the paramagnetic metals, it has been recently linked to a medical condition known as nephrogenic systemic fibrosis (NSF).7 For obvious reasons, this has led to concerns over the safety of Gd-based T1 contrast agents in MRI applications.
Iron oxide NPs are still considered to be the best materials for MRI applications.18 They are more biocompatible than Gd and Mn based materials because iron species are rich in human blood, which are mostly stored as ferritin in the body. Cytotoxicity investigations also confirmed that the iron oxide NPs are well tolerated by the human body.19–22 However, common iron oxide NPs are not appropriate for T1 MRI contrast agents. Although ferric (Fe3+) ions having 5 unpaired electrons increase the r1 value, the high r2 of iron oxide nanoparticles derived from innate high magnetic moment prevents them from being utilized as T1 contrast agent. This problem can be resolved by decreasing the size of the magnetic nanoparticles. The magnetic moment of magnetic nanoparticles rapidly decreases as their size decreases due to the reduction in the volume magnetic anisotropy and spin disorders on the surface of the nanoparticles. Recently, Kim et al.23 reported 3 nm sized spherical iron oxide nanoparticles as a potential candidate for T1 contrast agents, with high r1 relaxivity of 4.78 mM−1 s−1. On the other hand, Lee et al.22 reported extremely high r2 relaxivity (761 mM−1 s−1) for the ferrimagnetic iron oxide nanocubes of 22 nm size. Very recently, Li et al.24 reported dual modal MRI contrast capabilities of ultrasmall iron oxide nanoparticles. They reported high longitudinal relaxivity r1 = 8.3 mM−1 s−1 but the transverse relaxivity was comparitively lower r2 = 35.1 mM−1 s−1. A careful observation of the result suggests that if we increase the size of the iron oxide NPs, r1 relaxivity will decrease and r2 relaxivity will increase. The MR relaxivity is strongly related to the size and shape of the nanoparticles. Zhen et al.25 observed that iron oxide nanoparticles with cubic geometry possess high relaxivity values (up to 4 times stronger) in comparison with the spherical counterparts. Therefore, size- and shape-controlled synthesis of uniform nanoparticles is critical for the fine control of MR relaxivity. In the previous studies, iron oxide nanoparticles have not been reported as efficient dual modal contrast agents in MRI. The issue is, if we decrease the size too much they compromise the T2 contrast capabilities of these NPs and vice versa. Recently, Zhou et al.26 regulated the balance of T1 and T2 contrast by controlling their structure and surface features, including morphology, exposed facets, and surface coating. Also, iron oxide nanoparticles are commonly known to possess a magnetite (Fe3O4) or maghemite (Fe2O3) crystal structure, which are quite difficult to differentiate only on the basis of XRD measurements. But a careful observation of the previous reports reveals that they also lack detailed characterization to differentiate between a magnetite (Fe3O4) and maghemite (Fe2O3) crystal structure of the iron oxide NPs.
In this article, we report the synthesis of highly monodisperse and crystalline iron oxide nanocubes for simultaneous contrast enhancement in both T1- and T2-weighted MRI. We also performed a detailed characterization to confirm the magnetite structure of the iron oxide nanocubes. These nanocubes were successfully demonstrated as DMCAs in phantom experiments and in vivo MRI. Also, these nanocubes are small in size (9.7 nm) and can be used in most parts of the human body.7,27 These nanocubes are unique in that, being smaller in size, they offer simultaneous T1 and T2 contrast enhancement in MRI while being safer for the body. To the best of our knowledge, this is the first report of dual contrast enhancement in T1- and T2-weighted MR images using magnetite nanocubes.
Experimental section
Materials
Ammonia (28 wt% in water), poly(5)oxyethylene-4-nonylphenyl-ether (Igepal Co 520), tetraethyl orthosilicate (TEOS, 99%), oleic acid (tech 90%), 1-octadecene (tech 90%) and iron(II) chloride hexahydrate (99.99%) were purchased from Sigma-Aldrich. Sodium hydroxide, ethanol, hexane, cyclohexane and other reagents were purchased from Alfa Aesar. All chemicals were used as received without further purification.Synthesis of sodium oleate
Sodium oleate was prepared by adding sodium hydroxide (0.71 g, 17.6 mmol) to oleic acid (5.56 mL, 17.6 mmol) dissolved in ethanol (50 mL). The reaction mixture was stirred overnight at room temperature. Removal of the solvent under vacuum yielded the product as a white soap.Synthesis of iron–oleate complex
In a typical procedure, iron chloride (FeCl2·6H2O ∼ 0.9 g, 5 mmol) and sodium oleate (4.56 g, 15 mmol) were mixed in a round bottom flask with distilled water (60 mL), ethanol (25 mL) and hexane (25 mL) to generate the Fe-oleate complex. The reaction system was allowed to perform at 90 °C for 4 h before cooling to room temperature. When the reaction was complete, the upper organic layer containing the Fe-oleate complex was washed two times with distilled water in a separatory funnel. After washing, hexane was evaporated off, resulting in the Fe-oleate complex in a waxy form.Synthesis of magnetite nanocubes
Iron oleate (0.5 g), oleic acid (0.1 mL) and 1-octadecene (10 mL) were mixed in a three neck bottle flask and degassed under argon for 30 min at 70 °C. The reaction mixture was heated to 320 °C with a constant heating rate of 5.5 °C min−1, and then maintained at that temperature for 30 minutes. When the reaction temperature reached 320 °C, a severe reaction occurred and the initial transparent solution became turbid and brownish black. The resulting solution containing the nanocrystals was then cooled to room temperature, and the synthesized nanocrystals were precipitated using isopropanol and redispersed in hexane for further use.Silica coating on magnetite nanocubes
For the reverse microemulsion synthesis, IgePAL CO-520 (1.3 mL) was dispersed in cyclohexane (10 mL) and stirred for 15 min (500 rpm) to form a stable solution. Subsequently, a dispersion of nanocubes (0.5–1 nmol) in cyclohexane (1 mL) was added, followed by TEOS (80 μL) and ammonia (150 μL). Between the additions, the reaction mixture was stirred for 15 min (500 rpm). Once ammonia was added, the mixture was stirred for 2 days. Finally, the particles were purified by adding 25 mL of ethanol to the reaction mixture and the whole mixture centrifuged for 20 min at 9500 rpm. After the removal of the supernatant, 25 mL of ethanol was added, and the silica particles were sedimented again by centrifugation at 9500 rpm for 20 min. This was repeated once more for 20 min, after which the particles were redispersed in 5 mL double distilled water and stored at 4 °C.Characterization of the magnetite nanocubes
TEM, HR-TEM images and the SAED pattern of nanocubes were obtained using a high resolution transmission electron microscope (TEM – Tecnai G2 F30) operating at 300 kV. UV-Vis absorption spectra were recorded using a UV-Vis spectrophotometer (Varian – Cary 100). FT-IR spectra was recorded by using an FT-IR spectrometer (Bruker-Vertex 70). Magnetic measurements (M–H and M–T curves) were recorded on a Quantum Design MPMS-XL-7 system. MR phantom experiments were performed at room temperature on a 3 T Siemens TrioTim MR scanner. Various concentrations (3 to 60 μM) of magnetite nanoparticles were prepared for MRI phantom study. T1-Weighted and T2-weighted phantom MR images of magnetite nanoparticles were acquired using a spin echo (SE) sequence under the following parameters: TR/TE = 1000/12 ms (T1), TR/TE = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/330 ms (T2), (slice thickness = 3 mm, flip angle = 90°, acquisition matrix = 384 pixels × 384 pixels, FoV = 120 × 120 mm2).
000/330 ms (T2), (slice thickness = 3 mm, flip angle = 90°, acquisition matrix = 384 pixels × 384 pixels, FoV = 120 × 120 mm2).
In vivo MR imaging
Animal experiments were performed using a Sprague Dawley (200–250 g) rat according to a protocol approved by the animal ethics committee of Bilkent University, Turkey. MRI experiments were performed at room temperature on a 3 T Siemens TrioTim MR scanner. Silica coated magnetite nanocubes with the dosage of 1 mg kg−1 were injected into a rat through its tail vein and coronal images of the kidneys were taken before and after the injection of magnetite nanocubes. T1-Weighted and T2-weighted in vivo rat MR images were acquired using a spin echo (SE) sequence under the following parameters: TR/TE = 550/11 ms (T1), TR/TE = 4420/94 ms (T2) (slice thickness = 2 mm, flip angle = 90°, acquisition matrix = 384 pixels × 384 pixels, FoV = 90 × 90 mm2).Cytotoxicity studies
The in vitro cytotoxicity of iron oxides nanocubes was investigated using a L929 mouse cell line. Silica coated iron oxides were added with the concentrations of 0, 25, 100 and 200 μg Fe mL−1 and their toxic responses were evaluated by the Alamar Blue Assay after 24 h. To determine the viability, 2 × 103 L929 cells were seeded into a 96-well plate (n = 3) and silica coated cubic iron oxides were added in different concentrations in ddH2O. For the positive control, the cells were grown without exposure to the nanoparticle solution. In order to understand the fatal effect of less medium on the cells, we added phosphate buffer saline (PBS) with the same amount of ddH2O as the negative control group for each concentration. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C and 5% CO2 for 24 h. Subsequently, the cells were incubated in Alamar Blue solution (10% in DMEM-high glucose colorless medium) at 37 °C for 1 h. After the desired incubation time, the supernatant (200 μL) was transferred into a 96-well plate, and the absorbances at 570 and 595 nm were measured. A calibration curve was constructed using known concentrations of cells (L929) to relate the cell numbers to the dye reduction (%).Results and discussion
Magnetite nanocubes were synthesized using thermal decomposition of the iron–oleate complex using a modified receipe.28 We observed that the shape and size of the iron oxide NPs can be controlled by varying the molar ratio of iron–oletae to oleic acid and the heating rate. In ref. 28, 12 nm sized spherical magnetite NPs were synthesized with the ratio of iron–oleate:oleic acid as 2:1, with a heating rate of 3.3 °C min−1. In our case, cubic shaped magnetite NPs were obtained with increased oleic acid amounts, i.e., equal molar ratio of iron–oleate and oleic acid, with a heating rate of 5.5 °C min−1. The small but critical reduction in the growth rate by the additional oleic acid appears to promote the formation of iron oxide NPs with a nonspherical, faceted shape. Fig. 1a shows the magnetite nanocubes dispersed in hexane and Fig. 1b shows TEM (transmission electron microscopy) images of monodisperse magnetite nanocubes with an average size of 9.7 ± 0.5 nm. The particle size distribution (PSD) of the nanocubes obtained using ImageJ software is shown in Fig. 1d. XRD spectra of as-synthesized iron oxide nanocubes are presented in the ESI (Fig. S1†). From the XRD data, it is found that the reflections are closer to the magnetite structure of the iron oxide NPs (Table S1 in ESI†). To further confirm the structure of these nanocubes, we have also recorded, SAED, FT-IR and absorption spectra. The magnetite structures of the iron oxide nanocubes were confirmed by selected area electron diffraction (SAED) pattern,29 Fourier transformed infrared spectra (FT-IR)28,30 and optical absorption measurements.30 We carried out SAED (Fig. 1e) of these nanocubes and found that the rings can be assigned to the spinel structure of magnetite (JCPDS#19-0629). The 220 (d = 2.9683 Å) and 400 (d = 2.0956 Å) phases in the SAED pattern are exclusive to the structure of magnetite.29 The highly crystalline nature of these nanocubes is demonstrated by the high-resolution (HR) TEM images as shown in Fig. 1c. HR-TEM also confirms that the spacing between the planes (d ∼ 0.295 nm) is close to the magnetite structure of iron oxide nanocubes.
 | ||
| Fig. 1 (a) Magnetite nanocubes dispersed in hexane. (b) TEM, (c) HR-TEM, (d) PSD and (e) SAED pattern of the as-synthesized magnetite nanocubes. | ||
To further confirm the crystal structure of the as-synthesized iron oxide nanocubes, we performed absorption and FT-IR measurements. The nanocubes are easily dispersed in hexane to form transparent colloids, with a characteristic vivid color corresponding to the color of the bulk material. The absorption data are generally consistent with the characteristic color of the sample and are, therefore, considered as a reliable way of differentiating magnetite and maghemite structures of iron oxide. For Fe3O4 nanocubes, the absorption spectrum exhibits a full absorption band in the visible region 400–700 nm, which corresponds to the black color of the dispersion.30 For α-Fe2O3, the strongest absorption peak appears at 400–450 nm and corresponds to the red color. In our case, the absorption spectrum exhibits a full absorption band in the visible area (Fig. 2a) along with the black color of the dispersion (Fig. 1a). Therefore, from the absorption data, it is clear that in our case the nanocubes possess a magnetite structure.
 | ||
| Fig. 2 (a) Absorption spectra of the as-synthesized magnetite nanocubes. (b) FT-IR spectra of the as-synthesized magnetite nanocubes and the iron–oleate complex. | ||
FT-IR spectra of the iron–oleate complex and iron oxide nanocubes are presented in Fig. 2b. FT-IR was used to identify the functional groups present in the nanocubes. The wide band at 3130–3630 cm−1 is assigned to O–H vibrations. The sharp bands at 2923 and 2853 cm−1 are assigned to the asymmetric methyl stretching and the asymmetric and symmetric methylene stretching modes, respectively. The sharpness of the bands is attributed to the well-ordered, long hydrocarbon chain of oleic acid. The characteristic bands at 1560 and 1443 cm−1 can be attributed to the asymmetric and symmetric COO− stretches, respectively, indicating that the oleic acid chain is attached in a bidentate fashion, with both oxygens symmetrically coordinated to the surface.31 Based on the FT-IR spectra, oleic acid is thought to coat the surface of the nanocubes. TEM results, in conjunction with FT-IR data, suggest that, in our case we have a core–shell structure, with an iron oxide core and an oleate shell (∼1.6 nm). This is also confirmed by the uniform spacing between the nanocubes (see Fig. 1b). FT-IR is also used as a tool to distinguish magnetite and maghemite structures from each other through their distinct lattice absorption peaks.14 The lattice absorption peaks of the iron oxide nanocubes centered at ∼595 cm−1 (Fig. 2b) indicate that the nanocubes are most probably magnetite.32,33 Therefore, on the basis of the SAED pattern, optical absorption data and FT-IR measurements, we confirm that these iron oxide nanocubes possess a magnetite structure.
We also studied the magnetic properties of these nanocubes using a Quantum Design MPMS-XL-7 system. The magnetization dependence on the magnetic field (M–H curve) of the magnetite nanocubes was measured at body temperature (310 K). We performed the measurements at body temperature because we wanted to use these materials in humans as MRI contrast agents. M–H curves as shown in Fig. 3a indicate that the saturated magnetization (Ms) of the as-synthesized magnetite nanocubes (∼18 emu g−1) is much lower than that of magnetite NPs with a similar size (∼65 emu g−1) measured at room temperature.34 Moreover, the continuous growth of magnetization along with the applied magnetic field for magnetite nanocubes is probably due to the enhanced spin canting effect on the surface layer of these nanocubes because of the size and shape,35 which may be responsible for the partially paramagnetic properties of these nanocubes. The characteristic M–H curves of these nanocubes are similar to those of the high-spin paramagnetic rare-earth materials and superparamagnetic nanoparticles,36 suggesting that these nanocubes exhibit both superparamagnetic and paramagnetic behaviors. The presence of mixed magnetic phases is further confirmed by fitting the M–H curves with the following relation:
 | (1) |
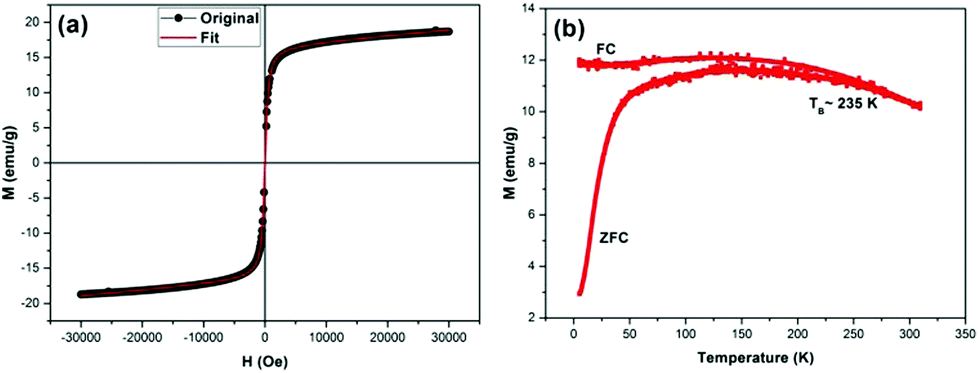 | ||
| Fig. 3 Magnetic properties; (a) M–H and (b) M–T curve of the as-synthesized magnetite nanocubes. | ||
We also studied the magnetization (M) dependence (zero field cooled – ZFC and field cooled – FC curves) on temperature (T) of the as-synthesized magnetite nanocubes. The ZFC and FC curves, which coincide initially, but start to separate and follow different trends as the temperature is decreased from 310 to 5 K. In the FC mode at the field level of H = 100 Oe, the magnetization increases slightly and then levels off (Fig. 3b), whereas the ZFC magnetization reaches a maximum followed by a steady decrease to a value approaching zero in the low temperature region. The shape of the FC curves is the result of the presence of dipole–dipole interactions between the oleate-capped magnetite nanocubes.37 Moreover, the variation of the magnetization in the ZFC and FC modes indicates a dominant superparamagnetic behavior for the magnetite nanocubes. The value of the blocking temperature for the nanocubes is estimated to be 235 K, obtained from the Stoner–Wohlfarth relationship:
 | (2) |
Magnetite nanocubes were made water soluble for MRI applications by silica coating using a recipe reported elsewhere.38 The encapsulated nanoparticles showed excellent colloidal stability in water. The hydrodynamic diameter of the silica coated nanocubes in deionized (DI) water, measured by dynamic light scattering (DLS), was 27.8 nm (Fig. S2 of ESI†). DLS measurements reveal that the nanocubes are monodisperse with no aggregation. The hydrodynamic diameter value is less than 30 nm. Therefore, these nanocubes come in the category of ultra-small iron oxide nanocubes (USIONs). Hydrodynamic diameter is an important parameter for the use of contrast agents in the human body. Our nanocube hydrodynamic size lies between 43 nm22 (maximum r2 relaxivity ∼ 761 mM−1 s−1 reported) and 15 nm23 sized nanoparticles (r1 relaxivity ∼ 4.78 mM−1 s−1). Our coated nanocube size (∼27.8 nm) is close to the median of these two values. Therefore, we believe that because of the size and shape (enhanced spin-canting effect) of our nanocubes, they have the ability to enhance the contrast in both T1-and T2-weighted MRI.
The utility of the water-soluble magnetite nanocubes as DMCAs for MR phantom studies was investigated in solution. Nanocubes were studied by using a 3 T Siemens MR scanner to observe the contrast enhancement in both T1- and T2-weighted MR images. Fig. 4a shows the T1-weighted MR images of silica capped magnetite nanocubes at different concentrations. We can clearly observe the increase in the image contrast (bright) with the increase in nanocubes concentration.
 | ||
| Fig. 4 (a) T1-weighted and (c) T2-weighted MR phantom images of the as-synthesized magnetite nanocubes. (b) T1 and (d) T2 relaxivity plots of the as-synthesized magnetite nanocubes obtained at 3 Tesla @ 25 °C. | ||
To examine the feasibility of using magnetite nanocubes as simultaneous T1 and T2 MRI contrast agents, the relaxation time was measured. The relaxation time T1,2 was measured at 3 T @ 25 °C using a spin echo sequence. The longitudinal (r1) and transverse (r2) relaxivities were determined from the following relation:
 | (3) |
Fig. 4c shows the T2-weighted MR images of silica capped magnetite nanocubes at different concentrations. Here, we can clearly observe the decrease in the image contrast (dark) with the increasing nanocubes concentration.The transversal relaxivity (r2) value of nanocubes obtained from the slope of (1/T2–1/T0) versus nanocubes concentration (Fig. 4d) is 89.68 mM−1 s−1. Magnetite nanocubes exhibit low T2 relaxivity as compared to the larger sized particles because the low magnetic moment induces weak magnetic inhomogeneity around the particles.22
Thus, with increased concentrations of magnetite nanocubes, we observed reduced signals in T2-weighted MR images and increased signal in T1-weighted MR images, indicating that magnetite nanocubes can act as both negative and positive contrast agents simultaneously. Iron oxide NPs are well known for their excellent T2 contrast enhancement effect with no obvious T1 contrast effect. By decreasing the size of the magnetic NPs, they are also reported as potential T1 contrast agents.23,40
By fine-tuning the shape of the iron oxide nanoparticles into cubes and making their size ultra-small, here we aim at achieving simultaneous enhancement in both positive and negative MR contrast images. In our case, we conclude that our nanocubes shape and dimension combinedly result in the simultaneous contrast enhancement in both T1- and T2-weighted MRI, which we do not observe otherwise individually.
The in vitro cytotoxicity of magnetite nanocubes was investigated using the L929 mouse cell line with the concentrations of 0, 25, 100 and 200 μg Fe mL−1 in ddH2O. No appreciable toxicity was observed even at very high concentrations of 100 μg Fe mL−1 (Fig. S3 in the ESI†), which is consistent with the recent report by Wortmann et al.41 On the other hand, further addition of cubic iron oxide decreased the viability of the L929 cell. The result of cell assays confirmed that the silica coated iron oxide nanocubes are not significantly cytotoxic, up to high concentrations of 100 μg Fe ml−1.
We further studied the in vivo MR imaging of the rat kidneys using these nanocubes. For in vivo MR imaging, T1 and T2 dual-mode abdominal images before and after the injection were obtained by using a 3 T MR scanner at room temperature. Silica coated magnetite nanocubes with a dosage of 1 mg kg−1 were injected into the rat through its tail vein and the coronal images of the kidneys were taken before injection, immediately after injection, and after 30 and 60 min of injection (Fig. 5). Since the kidney is an important member of the urinary system and one of its functions is a filtration of waste products from the body, we focused on the kidneys in the MR imaging. With the post injection time, the blood vessels going into kidneys gradually turned brighter and darker in T1 and T2 coronal planes, respectively. Color images of the kidney are shown in the insets of Fig. 5 for clarity. These results demonstrate that although our silica coated cubic nanoparticles have a hydrodynamic diameter (HD) of 27.8 nm, they can be observed in the kidneys where the renal cut-off is 5–6 nm. This may be due to the coating material, “silica”. There are several reports42,43 on the renal clearance of silica coated nanoparticles, which revealed intact and larger particles in the urine; however, the exact excretion process remained unclear. In order to understand the clearance mechanism involving silica, Lu et al.44 investigated the biodistribution of silica nanoparticles with diameters of ca. 100–130 nm. They observed a rapid excretion of almost all of the nanoparticles from the body through urine and feces. Similar results were also observed by He et al.45 revealing that the silica nanoparticles of ca. 45 nm accumulated mainly in the liver, kidney, and urinary bladder a few hours after intravenous injection and consequently silica nanoparticles are safely removed through the renal route. All of these previous studies clearly show that very large nanoparticles can be efficiently removed from the body via renal excretion. In the light of these studies, we can attribute the excretion of our nanoparticles to the silica coating which may help particles to escape RES recognition by possibly limiting the opsonization of nanoparticles46 and guiding them to renal clearance. Furthermore, intravenous injection might also take a role in the rapid renal excretion of our nanoparticles as reported by He et al.45 In addition, similar to ref. 44, it is also possible that our nanoparticles degrade quickly in the bloodstream and the smaller particles may then prefer renal clearance. However, a more detailed analysis on the clearance mechanism of the silica coated iron oxide nanoparticles larger than 6 nm should be investigated as a subject of another study for a better and deeper understanding.
 | ||
| Fig. 5 (a) T1- and (b) T2-weighted in vivo MR images obtained before and after the nanocubes injection into the rat, at 3 Tesla @ 25 °C. In the inset, the kidney images in color are shown for the clear enhancement in the contrast. | ||
In summary, our experiments have demonstrated that these nanocubes are suitable as a contrast agent for MRI owing to their strong MR contrast enhancement in both T1- and T2-weighted imaging. Because of their dual-mode contrast feature and high biocompatibility, they allow access to comprehensive information with higher accuracy in medical diagnosis.
Conclusions
In this work, we have synthesized highly crystalline, monodisperse and low-magnetization magnetite nanocubes that achieve simultaneously enhanced contrast in T1- and T2-weighted MR images. The dual-mode MR contrast enhancement capabilities of these nanocubes are a direct result of the simultaneous presence of superparamagnetic and paramagnetic phases as confirmed by the magnetic measurements. Also, these nanocubes are small in size (∼9.7 nm) and almost harmless for use in the human body. These nanocubes while being non-toxic and bio-compatible, hold great promise as DMCAs for better diagnosis of patients using MRI.Acknowledgements
The authors would like to thank the EU-FP7 Nanophotonics4Energy NoE, TUBITAK EEEAG 109E002, 109E004, 110E010, 110E217, NRF-RF-2009-09, NRF-CRP-6-2010-02, and A*STAR of Singapore for the financial support. H.V.D. acknowledges support from ESF-EURYI and TUBA-GEBIP. We would like to acknowledge Biomaten (METU), in particular Prof. Vasıf Hasırcı, Dr Arda Buyuksungur and Tugba Dursun for cytotoxicity experiments. Also, we would like to acknowledge Dr U. O. S. Seker (UNAM) for his valuable discussions.Notes and references
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC.
- M. Srinivas, E. H. J. G. Aarntzen, J. W. M. Bulte, W. J. Oyen, a. Heerschap, I. J. M. de Vries and C. G. Figdor, Adv. Drug Delivery Rev., 2010, 62, 1080–1093 CrossRef CAS PubMed.
- Y. Piao, J. Kim, H. Bin Na, D. Kim, J. S. Baek, M. K. Ko, J. H. Lee, M. Shokouhimehr and T. Hyeon, Nat. Mater., 2008, 7, 242–247 CrossRef CAS PubMed.
- E. I. Galanzha, E. V. Shashkov, T. Kelly, J.-W. Kim, L. Yang and V. P. Zharov, Nat. Nanotechnol., 2009, 4, 855–860 CrossRef CAS PubMed.
- H. Bin Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef PubMed.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- D. Pan, A. H. Schmieder, S. a. Wickline and G. M. Lanza, Tetrahedron, 2011, 67, 8431–8444 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- D. Choi, A. Han, J. P. Park, J. K. Kim, J. H. Lee, T. H. Kim and S.-W. Kim, Small, 2009, 5, 571–573 CrossRef CAS PubMed.
- Z. Zhou, D. Huang, J. Bao, Q. Chen, G. Liu, Z. Chen, X. Chen and J. Gao, Adv. Mater., 2012, 24, 6223–6228 CrossRef CAS PubMed.
- W. Chen, F. Lu, C.-C. V. Chen, K.-C. Mo, Y. Hung, Z.-X. Guo, C.-H. Lin, M.-H. Lin, Y.-H. Lin, C. Chang and C.-Y. Mou, NMR Biomed., 2013, 26, 1176–1185 CrossRef CAS PubMed.
- K. H. Bae, Y. B. Kim, Y. Lee, J. Hwang, H. Park and T. G. Park, Bioconjugate Chem., 2010, 21, 505–512 CrossRef CAS PubMed.
- S.-J. Cho, B. R. Jarrett, A. Y. Louie and S. M. Kauzlarich, Nanotechnology, 2006, 17, 640–644 CrossRef CAS.
- F. Hu, Q. Jia, Y. Li and M. Gao, Nanotechnology, 2011, 22, 245604 CrossRef PubMed.
- G. Huang, H. Li, J. Chen, Z. Zhao, L. Yang, X. Chi, Z. Chen, X. Wang and J. Gao, Nanoscale, 2014, 6, 10404–10412 RSC.
- J.-S. Choi, J.-H. Lee, T.-H. Shin, H.-T. Song, E. Y. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015–11017 CrossRef CAS PubMed.
- M. Cho, R. Sethi, J. S. Ananta narayanan, S. S. Lee, D. N. Benoit, N. Taheri, P. Decuzzi and V. L. Colvin, Nanoscale, 2014, 6, 13637–13645 RSC.
- G. Huang, X. Zhu, H. Li, L. Wang, X. Chi, J. Chen, X. Wang, Z. Chen and J. Gao, Nanoscale, 2015, 7, 2667–2675 RSC.
- E. a. Neuwelt, P. Várallyay, a. G. Bagó, L. L. Muldoon, G. Nesbit and R. Nixon, Neuropathol. Appl. Neurobiol., 2004, 30, 456–471 CrossRef CAS PubMed.
- A. H. Kaim, T. Wischer, T. O'Reilly, G. Jundt, J. Fröhlich, G. K. von Schulthess and P. R. Allegrini, Radiology, 2002, 225, 808–814 CrossRef PubMed.
- K. H. Bae, M. Park, M. J. Do, N. Lee, J. H. Ryu, G. W. Kim, C. Kim, T. G. Park and T. Hyeon, ACS Nano, 2012, 6, 5266–5273 CrossRef CAS PubMed.
- N. Lee, Y. Choi, Y. Lee, M. Park, W. K. Moon, S. H. Choi and T. Hyeon, Nano Lett., 2012, 12, 3127–3131 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. Il Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. Bin Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- Z. Li, P. W. Yi, Q. Sun, H. Lei, H. Li Zhao, Z. H. Zhu, S. C. Smith, M. B. Lan and G. Q. (Max) Lu, Adv. Funct. Mater., 2012, 22, 2387–2393 CrossRef CAS PubMed.
- G. Zhen, B. W. Muir, B. a. Moffat, P. Harbour, K. S. Murray, B. Moubaraki, K. Suzuki, I. Madsen, N. Agron-Olshina, L. Waddington, P. Mulvaney and P. G. Hartley, J. Phys. Chem. C, 2011, 115, 327–334 CAS.
- Z. Zhou, Z. Zhao, H. Zhang, Z. Wang, X. Chen, R. Wang, Z. Chen and J. Gao, ACS Nano, 2014, 8, 7976–7985 CrossRef CAS PubMed.
- J. Huang, L. Bu, J. Xie, K. Chen, Z. Cheng, X. Li and X. Chen, ACS Nano, 2010, 4, 7151–7160 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- a. Shavel, B. Rodríguez-González, M. Spasova, M. Farle and L. M. Liz-Marzán, Adv. Funct. Mater., 2007, 17, 3870–3876 CrossRef CAS PubMed.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS PubMed.
- A. a. Khaleel, Chemistry, 2004, 10, 925–932 CrossRef CAS PubMed.
- Y.-W. Jun, Y.-M. Huh, J.-S. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Yoon, K.-S. Kim, J.-S. Shin, J.-S. Suh and J. Cheon, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- F. Hu, Z. Li, C. Tu and M. Gao, J. Colloid Interface Sci., 2007, 311, 469–474 CrossRef CAS PubMed.
- D. Caruntu, G. Caruntu and C. J. O'Connor, J. Phys. D: Appl. Phys., 2007, 40, 5801–5809 CrossRef CAS.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, L. Casas, B. Martínez and F. Sandiumenge, Chem. Mater., 1999, 11, 3058–3064 CrossRef CAS.
- G. F. Goya, T. S. Berquó, F. C. Fonseca and M. P. Morales, J. Appl. Phys., 2003, 94, 3520 CrossRef CAS PubMed.
- L. Suber, P. Imperatori, G. Ausanio, F. Fabbri and H. Hofmeister, J. Phys. Chem. B, 2005, 109, 7103–7109 CrossRef CAS PubMed.
- E. M. Hutter, F. Pietra, R. J. A. van Dijk - Moes, D. Mitoraj, J. D. Meeldijk, C. de Mello Donegá and D. Vanmaekelbergh, Chem. Mater., 2014, 26, 1905–1911 CrossRef CAS.
- E. A. Weitz, C. Lewandowski, E. D. Smolensky, M. Marjańska and V. C. Pierre, ACS Nano, 2013, 7, 5842–5849 CrossRef CAS PubMed.
- L. Zeng, W. Ren, J. Zheng, P. Cui and A. Wu, Phys. Chem. Chem. Phys., 2012, 14, 2631–2636 RSC.
- L. Wortmann, S. Ilyas, D. Niznansky, M. Valldor, K. Arroub, N. Berger, K. Rahme, J. Holmes and S. Mathur, ACS Appl. Mater. Interfaces, 2014, 6, 16631–16642 CAS.
- X. Huang, L. Li, T. Liu, N. Hao, H. Liu, D. Chen and F. Tang, ACS Nano, 2011, 5, 5390–5399 CrossRef CAS PubMed.
- V. Mamaeva, C. Sahlgren and M. Lindén, Adv. Drug Delivery Rev., 2013, 65, 689–702 CrossRef CAS PubMed.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS PubMed.
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr00752f |
| This journal is © The Royal Society of Chemistry 2015 |