
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene oxide immobilized enzymes show high thermal and solvent stability†
Soňa
Hermanová
a,
Marie
Zarevúcká
b,
Daniel
Bouša
c,
Martin
Pumera
d and
Zdeněk
Sofer
*c
aDepartment of Polymers, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: sona.hermanova@vscht.cz
bInstitute of Organic Chemistry and Biochemistry ASCR, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic. E-mail: zarevucka@uochb.cas.cz
cDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic. E-mail: zdenek.sofer@vscht.cz
dDivision of Chemistry & Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: pumera@ntu.edu.sg
First published on 25th February 2015
Abstract
The thermal and solvent tolerance of enzymes is highly important for their industrial use. We show here that the enzyme lipase from Rhizopus oryzae exhibits exceptionally high thermal stability and high solvent tolerance and even increased activity in acetone when immobilized onto a graphene oxide (GO) nanosupport prepared by Staudenmaier and Brodie methods. We studied various forms of immobilization of the enzyme: by physical adsorption, covalent attachment, and additional crosslinking. The activity recovery was shown to be dependent on the support type, enzyme loading and immobilization procedure. Covalently immobilized lipase showed significantly better resistance to heat inactivation (the activity recovery was 65% at 70 °C) in comparison with the soluble counterpart (the activity recovery was 65% at 40 °C). Physically adsorbed lipase achieved over 100% of the initial activity in a series of organic solvents. These findings, showing enhanced thermal stability and solvent tolerance of graphene oxide immobilized enzyme, will have a profound impact on practical industrial scale uses of enzymes for the conversion of lipids into fuels.
Introduction
Effective and highly selective catalysis by enzymes has led to widespread use of enzymes in industrial processes,1 biomedical assays2 (ELISA3) and detection technologies.4 One of the most important enzymes is lipase,5 which is an inexpensive biocatalyst capable of breaking down lipids with very important applications in the conversion of oil into fuel.6 One of the main challenges in the use of enzymes is their stability; they often show a drastic decrease in catalytic activity when exposed to increased temperature or organic solvents. Here we show that when lipase is immobilized on graphene oxide, it is highly resistant to thermal and solvent exposure and it retains its activity under extreme conditions.Graphene and graphene oxide (GO) have been studied as interesting nanosupports for a variety of biologically active agents leading to novel biocatalysts, biosensors, and drug delivery vehicles.7 The morphology and large accessible surface area of GO nanosheets along with the formation of stable aqueous suspensions fulfill the criteria for high enzyme loading on support and thus for the development of catalysts for biotechnological applications.8 Enzymes have been immobilized on GO through covalent bonding due to functional groups on the GO surface or by the use of a cross-linker, and/or non-covalent binding through weak interactions. For example, oxidoreductases, such as horse-radish peroxidase and oxalate oxidase were successfully immobilized on GO surfaces without any pretreatment and the extent of the electrostatic interaction between the enzyme and nanomaterial surface was proved to change according to the degree of the GO's reduction.9,10 The introduction of a glutaraldehyde spacer arm on the GO support enables tethering of enzyme molecules to yield bio-conjugates with improved thermostability, reusability and storage stability.11–13
Herein, we investigated the effect of GOs prepared by two different routes (Brodie and Staudenmaier) as nanomaterial supports for lipase. Lipase from Rhizopus oryzae (ROL) was studied as a model hydrolase for immobilization. Pentane-1,5-dial (glutaraldehyde) modification of the GO support was applied with cross-linking of the enzyme previously adsorbed on GO and through simultaneous addition of the enzyme and glutaraldehyde to GO. We will demonstrate that GO immobilized lipase shows thermal stability retaining up to 65% activity at 70 °C if immobilized on GO, which is approx. 10-fold higher than for native, non-immobilized enzyme.
Experimental
Materials
All of the reagents were of analytical grade. For the GO synthesis, sulfuric acid (98%), nitric acid (98%), potassium chlorate (98%), hydrochloric acid (35%), acetone (99.9%) and isopropanol (99.9%) were purchased from Penta, Czech Republic. Graphite (2–15 μm, 99.9995%) was obtained from Alfa Aesar, Germany. Acetonitrile (p.a.) was purchased from Sigma-Aldrich, Czech Republic. Toluene (p.a.) and n-hexane (p.a.) were supplied by Lach-Ner, Czech Republic. All solvents were dried with molecular sieves before use. Glutaraldehyde (25% v/v in water), p-nitrophenyl laurate (pNPL) and lipase from Rhizopus oryzae (no. 62305, activity 2.96 U mg−1) were supplied by Sigma-Aldrich, Czech Republic and used as received.Two types of graphene oxide: BR GO and ST GO with BET surface areas of 4.3 m2 g−1 and 6.6 m2 g−1, respectively, and differing in surface chemistry and surface charge were studied as immobilization supports. The syntheses for the Brodie graphene oxide and Staudenmaier graphene oxide were performed according to procedures reported previously.14,15 The nanomaterial properties were reported in detail by Pumera et al.16,17
Synthetic procedures
One unit of lipase activity (U) was defined as the amount of enzyme that caused the release of 1 μmol of p-nitrophenol from p-NPL in 1 min under the test conditions.
(i) to the GO dispersion (3 ml) before immobilization
(ii) to non-specifically immobilized enzyme on GO (3 ml)
(iii) to the GO dispersion (3 ml) simultaneously with 1 ml of enzyme solution (0.5 mg ml−1).
Immobilized enzymes were separated by centrifugation, washed three times with phosphate buffer (56 mM, pH 7.0) and stored at 4 °C. The experiments were carried out three times and the standard deviation was up to 9%. The immobilization efficiency was evaluated in terms of enzyme immobilization yield and activity recovery expressed as a percentage as follows:
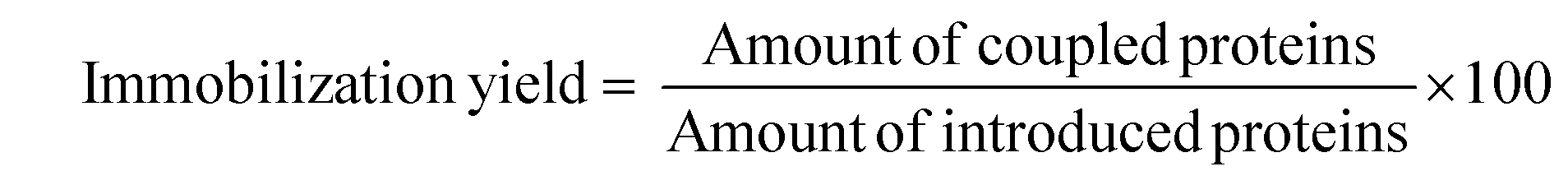
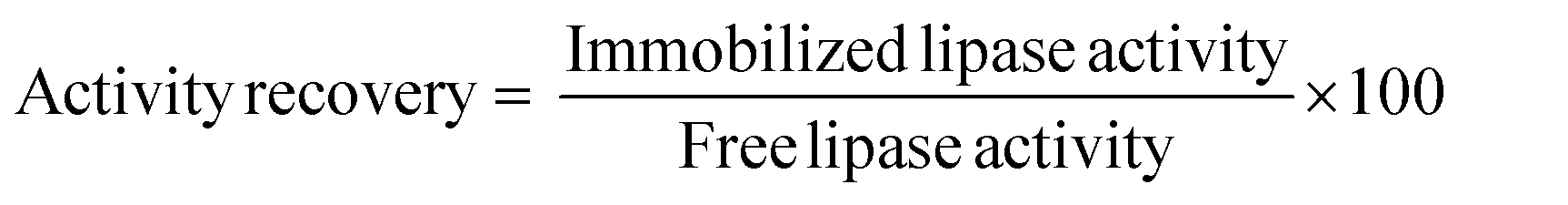
The amount of protein was determined by the method of Bradford using bovine serum albumin as the standard.24
Methods
Combustible elemental analysis (CHNS-O) was performed using a PE 2400 Series II CHNS/O Analyzer (Perkin Elmer, USA). In CHN operating mode (the most robust and interference free mode), the instrument employed a classical combustion principle to convert the sample elements to simple gases (CO2, H2O and N2). The PE 2400 analyzer performed combustion, reduction, homogenization of product gases, separation and detection automatically. A microbalance MX5 (Mettler Toledo) was used for precise weighing of samples (1.5–2.5 mg per single sample analysis). The accuracy of CHN determination was better than 0.30% abs. Internal calibration was performed using N-fenyl urea.FTIR spectra were collected using a FTIR spectrometer Nicolet 6700 (Thermo Scientific, USA) equipped with a diamond crystal GladiATR (PIKE Technologies, USA). The GO samples were placed at the surface of the diamond crystal and were pressed with a press tip flap system. The samples were scanned in the wavenumber range 4000–400 cm−1 and corrected against the background spectrum of air. The spectrum of each sample was obtained by taking the average of 64 scans. Diamond ATR crystal and DTGS detectors were used for the measurements. Binomial 11 points smoothing of the spectra was applied.
An inVia Raman microscope (Renishaw, England) was used for Raman spectroscopy in backscattering geometry with a CCD detector and DPSS laser (532 nm, 5 mW) with 50× magnification objective. Instrument calibration was achieved with a silicon reference which gives a peak position at 520 cm−1 and a resolution of less than 1 cm−1. Samples used for measurements were dispersed in isopropanol (1 mg ml−1) and dried on silicon wafer.
High resolution X-ray photoelectron spectroscopy (XPS) was performed using a ESCAProbeP (Omicron Nanotechnology Ltd, Germany) spectrometer using a monochromatic aluminum X-ray radiation source (1486.7 eV). A freshly cut indium block homogeneously covered with graphene was used for the measurement. An Al X-ray source with a monochromator was applied for the excitation.
The zeta potential of the GO and immobilized ROL was measured using a Zetasizer Nano ZS (Malvern, England) in phosphate buffer (56 mM, pH 7.0). Laser doppler micro-electrophoresis using an interferometric technique was used for the measurement.
Results and discussion
Graphene oxides were prepared by chlorate based methods: Brodie and Staudenmaier. These methods are based on the oxidation of graphite by potassium perchlorate in fuming nitric acid for the Brodie method (termed BR GO)14 and a mixture of fuming nitric acid and sulphuric acid for the Staudenmaier method (termed ST GO).15 Combustion elemental analysis proved that there were different compositions of carbon, oxygen, and hydrogen for both types of support (Tables 1 and S1†). The C/O ratio, calculated according to the C 1s and O 1s peak area, is 5.3 for ST GO and only 1.3 for BR GO. The lower C/O ratio indicates a higher concentration of hydrophilic oxygen functional groups on BR GO in comparison with ST GO.| Sample | At% N | At% C | At% S | At% H | At% O |
|---|---|---|---|---|---|
| BR GO | 0.32 | 62.58 | 0.00 | 15.05 | 22.05 |
| ST GO | 0.25 | 70.47 | 0.00 | 11.34 | 17.94 |
This fact is documented by high resolution XPS spectra of the C 1s peak provided in Table 2. Deconvolution of the C 1s peak was performed in order to resolve different valence states of carbon (see Fig. 1 and 2). The data show that the main peak associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds are located at around 284.5 eV and 285.5 eV, respectively. An asymmetric tail related to oxygen functional groups (like C–O, CO and O–CO) is located in the range 286–290 eV. The high asymmetric tail of the BR-GO sample indicates a high concentration of oxygen functional groups. The presence of a large amount of hydrophobic sp2 hybridized carbon atoms in ST GO is documented by high resolution C 1s peak spectra, where a significantly lower concentration of oxygen functionalities is observed.
C and C–C bonds are located at around 284.5 eV and 285.5 eV, respectively. An asymmetric tail related to oxygen functional groups (like C–O, CO and O–CO) is located in the range 286–290 eV. The high asymmetric tail of the BR-GO sample indicates a high concentration of oxygen functional groups. The presence of a large amount of hydrophobic sp2 hybridized carbon atoms in ST GO is documented by high resolution C 1s peak spectra, where a significantly lower concentration of oxygen functionalities is observed.
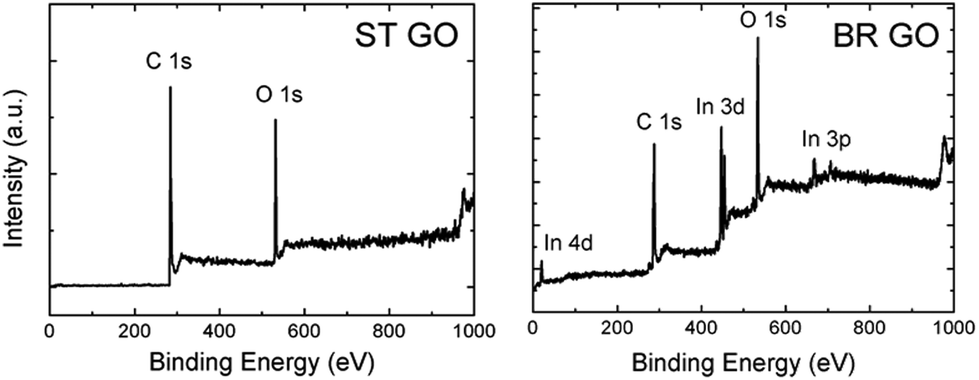 | ||
| Fig. 1 The XPS survey spectra of the BR GO and ST GO samples. | ||
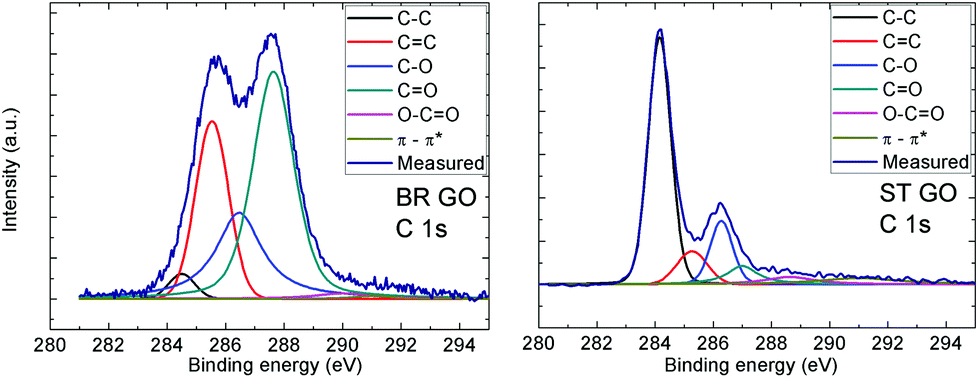 | ||
| Fig. 2 High resolution XPS spectra of the C 1s peak for BR GO and ST GO. Fittings of the individual C 1s spectra shows the possible carbon bonds present in the graphene oxide. | ||
| Functional group | BR GO % | ST GO % |
|---|---|---|
| CC (284.5 eV) |
2.7 | 55.2 |
| C–C (285.4 eV) | 24.5 | 9.6 |
| C–O (286.6 eV) | 24.4 | 14.6 |
| CO (287.6 eV) |
45.8 | 8.1 |
| O–CO (289.8 eV) |
1.9 | 5.2 |
| π–π* interaction (291.0 eV) | 0.7 | 7.4 |
Additionally, Raman and IR spectroscopy analyses of the BR GO and ST GO samples were performed (Fig. 3). A high luminescence background of BR GO indicates a high concentration of oxygen functional groups. According to the FTIR analysis, epoxy groups were also detected on BR GO, which was documented by a peak located at 950 cm−1. The concentration of carboxylic groups (about 0.22 wt% for BR GO and 1.18 wt% for ST GO measured by alkalimetric titration with 0.1 M NaOH) was found to be very low in comparison with other types of GO which are synthesised using permanganate based oxidation methods.
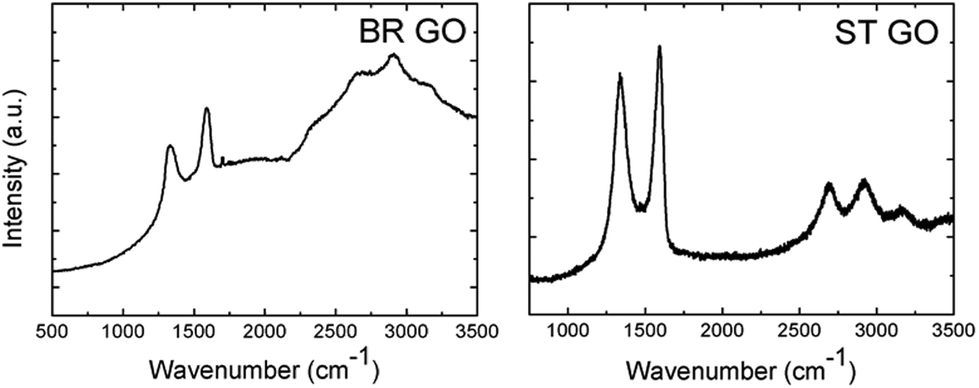 | ||
| Fig. 3 Raman spectra of the graphene oxide samples. The high background of the BR GO sample is related to the strong luminescence of graphene oxide. | ||
For one type of covalent immobilization both types of GO were treated by bifunctional agent-glutaraldehyde to introduce electrophilic functional groups on the surface. The glutaraldehyde modified GOs were characterized by ATR-FTIR spectroscopy and the spectra, with absorption bands ascribed to C–H stretching, are depicted in Fig. 4a.18,19 In an ideal case one aldehyde group of glutaraldehyde is assumed to react with GO's hydroxyl group to form the hemiacetal structure and the pendant one is available to react with primary amine groups of the protein molecules during their covalent attachment to the support.
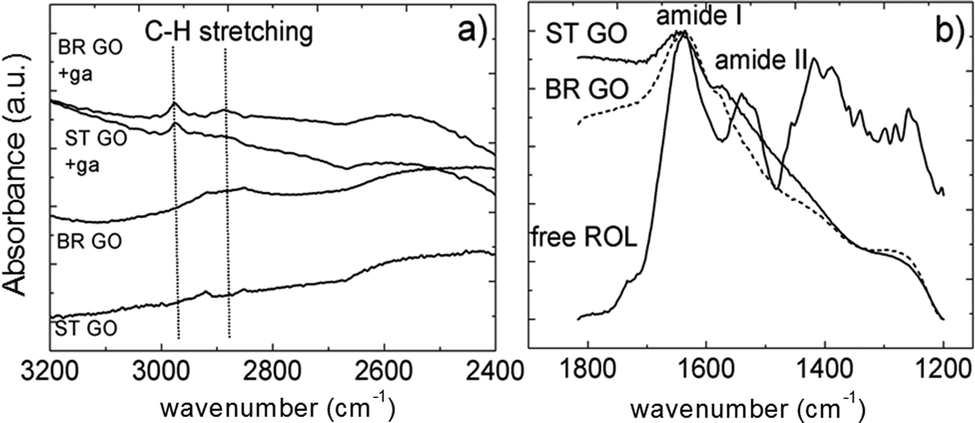 | ||
| Fig. 4 A selected region of the FTIR spectra of glutaraldehyde (ga) modified ST GO and BR GO (a); the amide band region of free ROL and ROL immobilized directly on BR GO and ST GO (b). | ||
In the case of direct enzyme adsorption onto GO, hydrophobic interactions are suggested to dominate as lipases strongly adsorb to hydrophobic surfaces maintaining a functional protein conformation.12 The successful loading of the enzyme protein onto the GO supports was evidenced by FTIR spectroscopy (Fig. 4b).
All immobilization procedures were performed in phosphate buffer solution at pH 7.0 and laboratory temperatures. Under these conditions both GO supports possessed negative charges as documented by zeta potential values (Table 3). Following enzyme immobilization onto the surfaces of the supports the drop in the absolute value of the zeta potential, which was slightly pronounced for BR GO, implies that the carboxylic groups are involved in the interactions between the enzyme molecules and the GO functional surface.9
| Sample | Zeta potential/FWHM of charge distribution in mV | |
|---|---|---|
| BR GO | ST GO | |
| GO | −34.0/12.8 | −27.2/16.1 |
| GO modified by glutaraldehyde | −31.6/12.4 | −26.2/9.7 |
| Direct immobilization | −11.5/13.4 | −11.2/12.9 |
| ROL + GO modified by glutaraldehyde | −13.4/18.0 | −16.0/14.3 |
| ROL + GO followed by crosslinking by glutaraldehyde | −15.1/18.6 | −15.9/13.9 |
| ROL immobilized on GO in the presence of glutaraldehyde | −16.2/14.6 | −15.1/18.6 |
Lipase ROL was directly immobilized onto both BR GO and ST GO supports without any chemical modification at neutral pH in phosphate buffer (56 mM). An incubation time of one hour was proven to be optimal to reach maximum yields and no leaching occurred since both the enzyme concentration and activity remained the same within a week.
The highest immobilization yield was attained at low ROL concentrations (100–500 μg ml−1), where ST GO displayed a higher adsorption capacity than BR GO (Fig. 5a and b). With increasing soluble ROL concentration (7.5–15.0 mg ml−1) a significant drop in the immobilization yield was observed and the values were the same for both studied supports. Activity recovery dramatically decreased with increasing ROL concentration (Table 4), which could be a consequence of protein–protein interactions taking place at high enzyme loading.20 This is also supported by identical immobilization efficiencies achieved independently of the studied support at a soluble ROL concentration range of 7.5–15.0 mg ml−1. The results imply stronger hydrophobic interactions between ROL and ST GO in comparison with BR GO, where non-specific electrostatic interactions are suggested to occur in higher extent, however under non-saturation conditions.
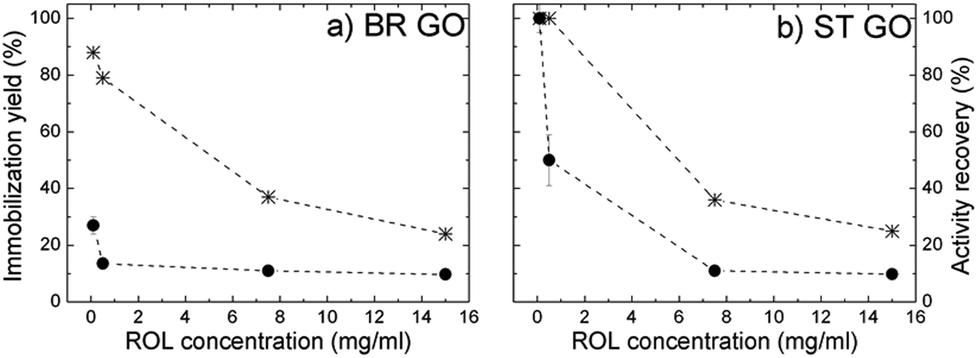 | ||
| Fig. 5 Dependence of the immobilization yield (star) and activity recovery (circle) of ROL directly immobilized on BR GO (a), and ST GO (b) on the soluble ROL concentration used for immobilization. | ||
In all covalent immobilization procedures, where glutaraldehyde chemistry was involved, the quantitative immobilization yield was obtained under conditions optimal for direct immobilization. Complete immobilization was accompanied by a large loss in native enzyme activity, which is a trend frequently reported in the literature.12,21
The thermal stability of ROL was studied within the temperature range 30–70 °C and the results are shown in Fig. 6a and b. The relative activity of free ROL decreased significantly above 40 °C and it retained only 23% of its initial activity at 50 °C. All immobilization procedures resulted in enhanced thermal stability of ROL, which means an increase in the resistance of the immobilized enzyme towards heat-induced conformational changes. The activity of ROL immobilized directly on BR GO decreased more slowly than that of ROL on ST GO in the temperature range from 40 °C to 50 °C, which indicates that interactions stronger than physical ones are contributing.
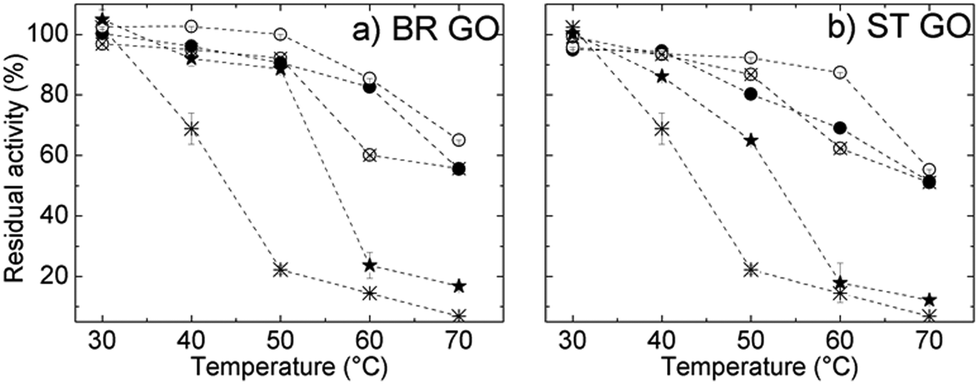 | ||
| Fig. 6 Thermal stability of ROL immobilized on BR GO (a) and ST GO (b): free ROL (star), directly immobilized ROL (solid star), ROL covalently immobilized on glutaraldehyde modified GO (empty circle), cross-linked ROL on GO (solid circle) and the simultaneous addition of glutaraldehyde and ROL to GO (crossed circle). | ||
A significant increase in the thermal stability of all covalently immobilized ROL is ascribed to covalent attachment. The glutaraldehyde treatment enables the formation of covalent bonds – either intermolecular (cross-linking with glutaraldehyde) or covalent attachment to the glutaraldehyde-modified support.
Consequently enzyme un-folding was restricted or rigidification of the enzyme structure by multipoint covalent linkage occurred.22 Free lipase retained only 6% of its initial activity after incubation at 70 °C compared to 65% activity retention of the immobilized lipase under the same conditions. Such enhanced thermal stability improves the catalytic performance of the immobilized enzyme in organic syntheses as at higher temperatures reaction rates are significantly enhanced and the reaction affords high yields.23
The pH optima of free and selected immobilized ROL are depicted in Fig. 7. Free ROL showed a broad pH optimum in the range 7.0–9.0. Directly immobilized ROL on both supports had a pH optimum at 8.0 and displayed improved pH stability in the acidic region. Regarding the pH dependent properties of the GO support itself, in acidic media carboxylic groups at the edges are protonated and the hydrophobic character of the GO sheets increases. Consequently there is a lower proton concentration in the microenvironment of physically adsorbed enzyme than in bulk solution, which correlates well with the measured pH profile. With increasing solution alkalinity carboxylic and also phenolic groups localized at the GO surfaces undergo deprotonation,24 which increases electrostatic repulsion between the sheets. Consequently, the increase in negative charge and its density on the support surface could negatively influence the immobilized enzyme orientation and hence cause a loss in activity (see Fig. S1†).
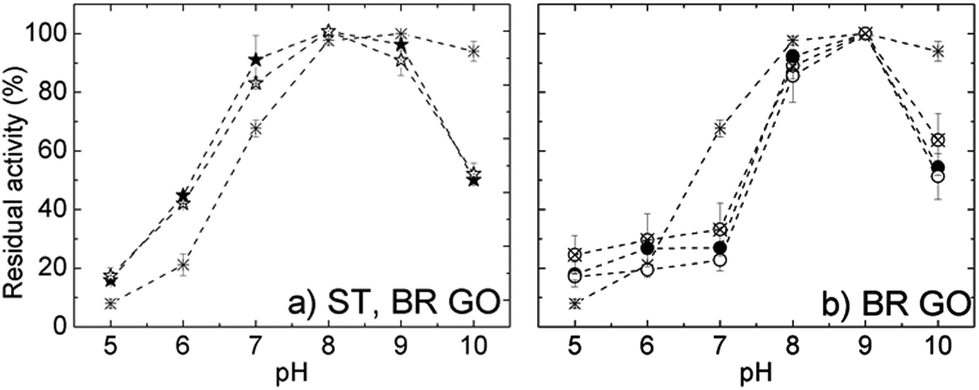 | ||
| Fig. 7 The pH optima of ROL directly immobilized on BR GO and ST GO (a), and ROL covalently immobilized on BR GO (b): free ROL (star), directly immobilized ROL (solid star), ROL covalently immobilized on glutaraldehyde modified GO (empty circle), cross-linked ROL on GO (solid circle) and the simultaneous addition of glutaraldehyde and ROL to GO (crossed circle). | ||
The pH activity profiles of all covalently immobilized ROLs on both GO supports displayed almost identical behavior. The curves possessed a narrower bell shape with a maximum at pH 9.0. Independent of the support or covalent immobilization protocol, the immobilized ROLs showed higher sensitivity towards alkaline (above 9.0) or acidic regions in comparison with soluble enzyme. However, under acidic conditions some activity loss could be connected with enzyme leaching as Schiff bases become unstable and convert back to the aldehyde and amine.
Kinetic parameters of free and immobilized enzyme were determined by measurement of ROL activity for the hydrolysis of the p-NPL substrate with various concentrations (1.75 to 5.00 mM) in phosphate buffer. The interaction between the substrate itself and the GOs was not considered under the study conditions since the hydrolysis reaction did not proceed in the absence of ROL. Almost for all immobilized ROL a decrease in the apparent Km value was observed, indicating higher affinity to enzyme–substrate complex formation (Table 5).
| Sample | K m [mM] | V max [U mg−1] | K m [mM] | V max [U mg−1] |
|---|---|---|---|---|
| BR GO | ST GO | |||
| a Soluble ROL: Km = 0.44 mM, Vmax = 2.60 U mg−1. | ||||
| Direct immobilization | 0.30 | 0.17 | 0.11 | 0.14 |
| GO modified by glutaraldehyde + RO | 0.29 | 0.05 | 0.22 | 0.04 |
| Cross-linked RO + GO | 1.63 | 0.07 | 0.11 | 0.01 |
| RO immobilized on GO in the presence of glutaraldehyde | 0.06 | 0.02 | 0.09 | 0.02 |
The Vmax values indicate the maximum reaction rate, when all of the enzyme sites are saturated with substrate. Physically adsorbed ROL on ST GO and BR GO exhibited 19-fold and 15-fold, respectively, drops in the Vmax value in comparison to soluble enzyme. The decrease was even lower for all immobilized ROL prepared by covalent immobilization as a consequence of multiple bond linkages which, in turn positively increased the thermal stability of the immobilized enzyme. The decrease in Vmax could be ascribed to a lower substrate concentration in the microenvironment of the immobilized lipase, which is caused by diffusion limitations. The same trend for the kinetics parameters of free lipase and lipase immobilized on an activated carbon support has been reported in literature.20,25 A sharp increase in Km observed for cross-linked ROL immobilized on BR GO (Km = 1.63) indicated an undesirable decrease in its affinity for substrate molecules. The results could be ascribed to partial inactivation of active centers.
The effect of several organic solvents on the stability of immobilized ROL, which displayed the highest activity recovery (ROL on ST GO) and storage stability (ROL covalently bound to glutaraldehyde modified ST GO) was determined. The following organic solvents were selected and classified according to their values of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and dipole moment, respectively: acetonitrile (−0.33, 3.2), acetone (−0.23, 2.88), isopropanol (0.28, 1.66), toluene (2.5, 0.36) and n-hexane (3.5, 0). Attempts to establish a correlation with either logP or the dipole moment were not successful. Valivety et al. stated that there is probably no single parameter for solvent polarity according to which the enzyme activity in an organic solvent could be predicted.26
P and dipole moment, respectively: acetonitrile (−0.33, 3.2), acetone (−0.23, 2.88), isopropanol (0.28, 1.66), toluene (2.5, 0.36) and n-hexane (3.5, 0). Attempts to establish a correlation with either logP or the dipole moment were not successful. Valivety et al. stated that there is probably no single parameter for solvent polarity according to which the enzyme activity in an organic solvent could be predicted.26
The hydrolytic activity of ROL on ST GO in non-polar solvents (toluene, n-hexane) after incubation was 109% compared to that of the control without solvent (100%). This behaviour can be attributed to the fact that non-polar organic solvents do not strip off the water layer from the surface of the enzyme. Interestingly, the relative activity retention of ROL covalently immobilized on modified ST GO was low (31% and 18%) after incubation in toluene and n-hexane, respectively. These results show that it is not possible to establish a general rule on the immobilized enzyme behaviour regarding activity and stability in non-polar solvents. Although, the instability of lipases in aprotic polar solvents has been frequently observed and is caused by the stripping of water from the protein surface, along with solvent penetration into the enzyme, leading to protein unfolding and subsequent denaturation.27 The highest activity recovery of both tested immobilized lipases was found after their incubation in acetone (Table 6). Our findings are supported by several reports describing the stability of lipases in aprotic polar solvents. A lipase produced by Pseudomonas sp. had activity ranging from 100 to 110% in acetone, tetrahydrofuran and ethyl acetate.27 Lipase from Mucor javanicus exhibited high stability and increased activity in acetonitrile, ethyl acetate and acetone as well.28 Given that the catalytic performance of lipases is dependent on their tolerance to different solvent systems, the finding that our lipase showed significantly high stability in acetone (activity recovery of 221.4 ± 2.1%) and isopropanol (158.4 ± 7.8%) suggests its potential as a biocatalyst for transesterification reactions and biodiesel production.
| Solvent | Activity recovery in % | |
|---|---|---|
| Direct immobilization ROL on ST GO | ST GO modified by glutaraldehyde + ROL | |
| Acetonitrile | 107.4 ± 2.4 | 4.6 ± 0.5 |
| Acetone | 221.4 ± 2.1 | 58.9 ± 2.4 |
| Control | 100.0 ± 0 | 100.0 ± 0 |
| Isopropanol | 158.4 ± 7.8 | 6.1 ± 0.9 |
| Toluene | 109.7 ± 1 | 31.8 ± 3.4 |
| n-Hexane | 109.9 ± 2.7 | 18.1 ± 1.2 |
Conclusion
We have demonstrated that lipase immobilized on graphene oxide retains its activity at high temperatures (85% of its initial activity at 60 °C and 65% of activity at 70 °C) when compared to native, non-immobilized enzyme (∼65% activity recovery at 40 °C). This has very profound implications to the use of this enzyme for fuel production. The study confirmed successful immobilization of lipase onto graphene oxide supports and its potential applications in synthesis. The tunable presence and distribution of hydrophobic domains with surrounding hydrophilic groups on the graphene oxide surface makes it an advantageous support for straightforward and efficient lipase binding. Lipase adsorption accompanied with minor unspecific interactions contributed to the stabilization of the active protein “open lid” like conformation, which was consequently maintained in organic solvent. From the viewpoint of activity retention, the number of immobilization procedure steps, costs of reagents, etc. physical adsorption has been proven to be the optimal procedure. The pH and thermal stabilities of both types of prepared immobilized lipase were also enhanced (ROL directly immobilized on ST GO, BR GO).Generally, incubation in organic solvents increased the activity of ROL adsorbed on ST GO, which suggests that directly immobilized biocatalysts have great potential in biotechnological processes. In particular, the produced biocatalyst ROL on ST GO shows potential with polar solvents, which have technological advantages such as low toxicity, low boiling points, low costs and the possibility of using polar substrates for novel reactions.
Acknowledgements
Z. S., D. B. and S. H. were supported by Specific University Research grant (MSMT 20/2015) and by Czech Science Foundation (project no. 15-09001S). M. Z. thanks the Academy of Science of the Czech Republic (project No. M200551203) for financial support. M. P. acknowledges Tier 2 grant (MOE2013-T2-1-056) from Ministry of Education, Singapore. Authors acknowledge Dr Stanislava Voběrková for valuable technical assistance during lab work.Notes and references
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC.
- Z. Wang, P. Huang, A. Bhirde, A. Jin, Y. Ma, G. Niu, N. Neamati and X. Chen, Chem. Commun., 2012, 48, 9768 RSC.
- R. Hnasko, A. Lin, J. A. McGarvey and L. H. Stanker, Biochem. Biophys. Res. Commun., 2011, 410, 726 CrossRef CAS PubMed.
- A. Amine, H. Mohammadi, I. Bourais and G. Palleschi, Biosens. Bioelectron., 2006, 21, 1405 CrossRef CAS PubMed.
- D. Sharma, B. Sharma and A. K. Shukla, Biotechnology, 2011, 10, 23–40 CrossRef CAS.
- G. Chen, M. Ying and W. Li, Appl. Biochem. Biotechnol., 2006, 911, 129–132 Search PubMed.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2011, 26, 4637 CrossRef CAS PubMed.
- F. Zhao, H. Li, Y. Jiang, X. Wang and X. Mu, Green Chem., 2014, 16, 2558 RSC.
- J. Zhang, F. Zhang, H. Yang, X. Huang, H. Liu, J. Zhang and S. Guo, Langmuir, 2010, 26, 6083 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, X. Huang, X. Zhou, H. Wu and S. Guo, Small, 2012, 8, 154 CrossRef CAS PubMed.
- R. Su, P. Shi, M. Zhu, F. Hong and D. Li, Bioresour. Technol., 2012, 115, 136 CrossRef CAS PubMed.
- I. V. Pavlidis, T. Vorhaben, T. Tsoufis, P. Rudolf, U. T. Bornscheuer, D. Gournis and H. Stamatis, Bioresour. Technol., 2012, 115, 164–171 CrossRef CAS PubMed.
- D. Kishore, M. Talat, O. N. Srivastava and A. M. Kayastha, PLoS One, 2012, 7, e40708 CAS.
- B. C. Brodie, Philos. Trans. R. Soc. London, 1859, 149, 249 CrossRef.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- C. K. Chua, Z. Sofer and M. Pumera, Chem. – Eur. J., 2012, 18, 13453 CrossRef CAS PubMed.
- H. L. Poh, F. Sanek, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515 RSC.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80 CrossRef CAS PubMed.
- H. Nantao, M. Lei, G. Rungang, W. Yanyan, C. Jing, Y. Zhi, K. Eric Siu-Wai and Z. Yafei, Nano-Micro Lett., 2011, 3, 215 CrossRef.
- K. Ramani, R. Boopathy, C. Vidya, L. J. Kennedy, M. Velan and G. Sekaran, Process Biochem., 2010, 45, 986 CrossRef CAS PubMed.
- J. T. Cang-Rong and G. Pastorin, Nanotechnology, 2009, 20, 255102 CrossRef PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS PubMed.
- L. Fjerbaek, K. V. Christensen and B. Norddahl, Biotechnol. Bioeng., 2009, 102, 1298 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867 CrossRef CAS.
- L. John Kennedy, P. K. Selvi, P. Aruna, K. N. Hema and G. Sekaran, Chemosphere, 2007, 69, 262 CrossRef PubMed.
- R. H. Valivety, G. A. Johnston, C. J. Suckling and P. J. Halling, Biotechnol. Bioeng., 1991, 38, 1137 CrossRef CAS PubMed.
- M. Pogorevc, H. Stecher and K. Faber, Biotechnol. Lett., 2002, 24, 857 CrossRef CAS.
- J. C. Wu, S. S. Lee, M. M. B. Mahmood, Y. Chow, M. M. R. Talukder and W. J. Choi, J. Mol. Catal. B: Enzym., 2007, 45, 108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr00438a |
| This journal is © The Royal Society of Chemistry 2015 |