
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic effects in friction and adhesion through cooperative rupture and formation of supramolecular bonds†
Johanna
Blass
ab,
Marcel
Albrecht
c,
Bianca L.
Bozna
a,
Gerhard
Wenz
c and
Roland
Bennewitz
*ab
aINM – Leibniz Institute of New Materials, Campus D2 2, Saarland University, 66123 Saarbrücken, Germany. E-mail: Roland.Bennewitz@inm-gmbh.de
bPhysics Department, Campus D2 2, Saarland University, 66123 Saarbrücken, Germany
cDepartment of Chemistry, Campus C4 2, Saarland University, 66123 Saarbrücken, Germany
First published on 24th March 2015
Abstract
We introduce a molecular toolkit for studying the dynamics in friction and adhesion from the single molecule level to effects of multivalency. As experimental model system we use supramolecular bonds established by the inclusion of ditopic adamantane connector molecules into two surface-bound cyclodextrin molecules, attached to a tip of an atomic force microscope (AFM) and to a flat silicon surface. The rupture force of a single bond does not depend on the pulling rate, indicating that the fast complexation kinetics of adamantane and cyclodextrin are probed in thermal equilibrium. In contrast, the pull-off force for a group of supramolecular bonds depends on the unloading rate revealing a non-equilibrium situation, an effect discussed as the combined action of multivalency and cantilever inertia effects. Friction forces exhibit a stick-slip characteristic which is explained by the cooperative rupture of groups of host–guest bonds and their rebinding. No dependence of friction on the sliding velocity has been observed in the accessible range of velocities due to fast rebinding and the negligible delay of cantilever response in AFM lateral force measurements.
1. Introduction
Friction is the dissipative force acting in sliding contacts. Contributions to friction arise from the plastic or viscoelastic deformation of the contacting bodies and from surface adhesion. This distinction has been the basis of successful scientific investigation into friction phenomena, in particular since Bowden and Tabor's studies on plasticity and adhesion in metallic contacts by an ensemble of molecular bonds,1 which has been introduced in a model based on force-dependent off rates and aging-related on rates by Filippov et al.2 We follow a molecular toolkit concept, where adhesion between the contacting surfaces is caused by supramolecular host–guest interactions in aqueous environment. The novelty of our system lies in its versatility achieved by the symmetric functionalization of the opposing surfaces by assemblies of host molecules. Various ditopic connector molecules with two guest end groups form a specific supramolecular bond between the CDs on the two surfaces leading to adhesion and friction. Our study addresses the adhesion and friction dynamics of a group of supramolecular complexes attached to a microscopic asperity, realized by the tip of an atomic force microscope (AFM) (see Fig. 1). The curved shape of the AFM tip leads to a non-equal load sharing between the bonds which is a key aspect in the discussion of the results. This situation is more complex than the equal load sharing described in previous studies and reflects the situation at the interface between rough surfaces. | ||
| Fig. 1 Schematic representation of a β-CD hosts monolayer attached to an AFM tip and silicon wafer. Hydrophobic interactions lead to inclusion of the connector molecules into the hosts and to the tip-sample interaction. | ||
Supramolecular interactions, in particular the inclusion of hydrophobic groups into the cavities of cyclodextrin molecules, have been employed for the development of a variety of molecular materials, ranging from supramolecular structures3 and molecular print boards4 to self-assembling macroscopic blocks of gels.5 At the single complex level, atomic force microscopy has revealed the force which inclusion complexes can sustain, typically of the order of several tens of piconewton.6,7 Due to the high binding and unbinding rates of inclusion complexes, single molecule force experiments typically probe the system in thermal equilibrium leading to no observable dependence on the force loading rate.7 Macroscopic measurements of adhesion recorded in thermal equilibrium result in values for the work of adhesion which are proportional to the density of bonds and to the free energy found for single molecule detachment experiments.8 The situation can become different when the force sustained by multiple inclusion complexes. The kinetics change dramatically and lead to significant rate-dependent adhesion when the several bonds contribute equally to the interaction.9 Such effects of multivalency play an important role in supramolecular chemistry10 and beyond in biological adhesion and recognition.11
The results of this study quantify the friction caused by multiple inclusion complexes. They reveal the conditions under which the complexes, which share the external force non-equally due to attachment to a curved surface, exhibit multivalency effects in adhesion. The bond failure in our system is characterized by a cooperative behavior as described by Evans and Williams12 and Filippov et al.2 Such physical cooperative behavior is encountered even in the absence of a chemical cooperativity in the sense of effects of binding in adjacent receptors, and may then also called non-cooperative multivalency.9
We have functionalized the surfaces of AFM tips and silicon wafer substrates by dense monolayers of β-cyclodextrin (β-CD) molecules. Adhesion and friction between tip and surface, probed by normal and lateral force vs. distance curves, arise from complexation of the two adamantane end groups of the ditopic connector molecules into the cavities of opposing β-CD molecules. The design of a ditopic system allows the quantification of non-specific interactions between the opposing surfaces in direct control experiments. Moreover, different connector molecules can bind to the same surface functionalization which allows to control friction and adhesion using switchable connector molecules13 or to adapt to surface roughness by varying the lengths of the connectors. In this study, we use a connector molecule with adamantyl end groups because of its high binding constant and the well-known molecular kinetics for the monotopic guests.9,14 For sake of simplicity, we will refer to the specific interaction between one connector molecule and two opposing β-CDs as bond throughout the manuscript and ask the reader to keep in mind the non-covalent, supramolecular nature of the bond.
2. Experimental section
2.1. Instrumentation
Isothermal titration calorimetry (ITC) was performed on a Nano ITC2G from TA Instruments at 25 °C. 1H and 13C NMR spectra were recorded on a Bruker Magnet System 400 MHz Ultra shield plus (1H: 400.00 MHz) using CDCI3 at 25 °C. The solvent signals were used as internal standards. The program MestReNova 6.0.2 was used for data processing and deconvolution of the spectra. The following abbreviations were used for multiplicities: s for singlet, t for triplet. Overlapping signals and multiplets were labeled with m, broad singlets with bs.2.2. Synthesis of the ditopic connector (3)
1,11-Diiodo-3,6,9-trioxoundecane (5 mmol, 2.070 g, synthesized according to ref. 15) and 1-aminoadamantane (11 mmol, 1.663 g) were dissolved in dry methanol (20 mL). Sodium carbonate (20 mmol, 2.1 g) was added and the mixture was stirred at reflux temperature for 46 h. After filtration of the solid the solvent was evaporated and the residue was purified by column chromatography (SiO2, dichloromethane–methanol 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The product was obtained as a white solid (2.25 g, 63%).
:1). The product was obtained as a white solid (2.25 g, 63%).
1 HNMR (CDCl 3 , 400 MHz): δ 8.35 (bs, 4 H, NH2-H), 4.00 (t, J = 4.6 Hz, 4 H), 3.68 (m, 8 H), 3.16 (t, J = 4.6 Hz, 4 H), 2.16 (m, 6 H), 2.07 (m, 12 H), 1.67 (m, 12 H) ppm.
13 CNMR (CDCl 3 , 100 MHz): δ 70.3, 69.7, 65.5, 58.5, 39.7, 38.2, 35.3, 28.9 ppm.
IR(ATR): 2907, 2851, 1611, 1453, 1362, 1307, 1074 cm−1.
2.3. Preparation of monolayer
As substrates commercial available Si (111) wafers were used (Si-Mat, Kaufering, Germany) which were first cleaned using piranha solution (3:2 mixture of sulforic acid and hydrogen peroxide) to remove organic contamination, rinsed with ultrapure water and dried with N2. Subsequently the samples were kept for two hours in the silane solution (0.1 vol% of 3-isothiocyanatopropyl-triethoxysilane, diluted in tetrahydrofuran (THF). The silane 3-isothiocyanatopropyl-triethoxysilane was synthesized according to ref. 16. After a washing procedure with THF and water the wafer remained in 1 mM solution of mono(6-deoxy-6-amino)-β-CD diluted in water over night at room temperature. All samples were freshly prepared for the AFM measurements and stored in water. The silicon AFM tips (Nanosensors(TM) PPP-Cont AFM Probes, NanoandMore, Wetzlar, Germany) were functionalized in the same way.
2.4. AFM measurements
All AFM measurements were performed with a Nanowizard 3 setup (JPK Instruments, Berlin, Germany) in water or connector solution at room temperature. A connector molecule concentration of 10 μM was chosen in order to work in the saturation regime of the Langmuir curve (see ESI† for more details). Silicon cantilevers with nominal normal spring constant of 0.2 N m−1 and typical lateral spring constants of 50 N m−1 have been calibrated using the thermal noise analysis. Lateral force measurements were calibrated using the wedge method in air17 (sample TGG01 from Micromash, Sofia, Bulgaria) and applying the correction factor for experiments in liquid introduced by Tocha et al.18 To avoid surface damage and unspecific contributions to the friction force the maximum normal force was kept below 0.5 nN. The shown adhesion values were obtained on at least three different surface positions, more than 100 force–distance curves each. Friction experiments were performed with low feedback gains by scanning perpendicular to the axis of the cantilever.2.5. Data analysis
For the force spectroscopy analysis the implemented Data Processing software (JPK instruments, Berlin, Germany) was used. Each force curve was examined either for the maximum pull-off force or the last rupture, depending on the experiment. The results were plotted in a histogram and the most probable rupture force was obtained by a Gaussian curve fitted to the data.3. Results
The synthesis of the ditopic connector 3 was performed in a straightforward approach (Fig. 2) starting from commercially available materials. In the first step tetraethylene glycol was directly converted into the corresponding diiodo compound using PPh3, imidazole, and iodine.15 Subsequent substitution reaction with 1-aminoadamantane resulted in the formation of the ditopic connector 3 as its diammonium iodide salt as indicated by a characteristic signal of the ammonium protons in CDCl3 at 8.35 ppm. Based on its ionic structure product 3 is well soluble in water which is essential for our molecular toolkit approach. The ditopic connector 3 forms an 1:2 inclusion complex with β-CD. The binding constant K = 11300 M−1 equivalent to a binding free energy ΔG = −23.1 kJ mol−1 and a binding enthalpy of ΔH = −39.4 kJ mol−1 was determined by ITC in aqueous solution (see ESI† for details). These values compare well with values for the corresponding monotopic 1-adamantylammonium ΔG = −22.2 kJ mol−1 and ΔH = −20.1 kJ mol−1 (ref. 19) indicating independent complexation of both adamantane groups.
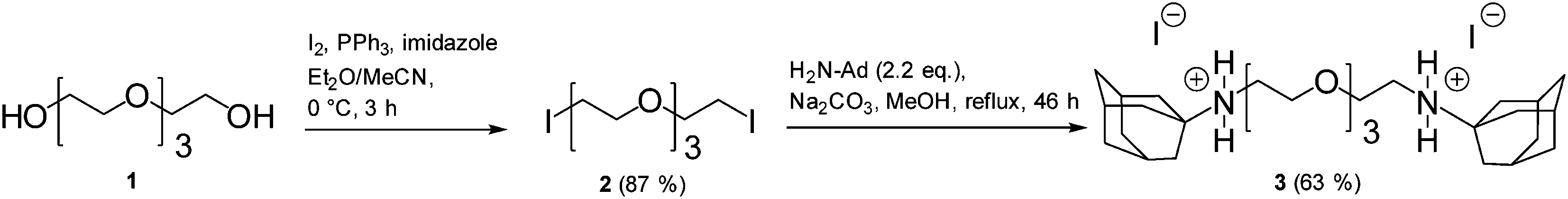 | ||
| Fig. 2 Synthesis of the ditopic connector 3. | ||
The molecular interactions contributing to friction and adhesion were studied by means of AFM. A β-CD monolayer was attached to the AFM tip and substrate in a two step process. In the first step, a silane adlayer with an isothiocyanate group was self assembled on the silicon oxide surface.20 Subsequently, the β-CD molecules which carry an amine functionality21 were attached by forming a thiourea bond with the isocyanate groups at the surface.13 The specific interaction between the β-CDs on the AFM tip and surface is mediated by ditopic connector molecules via the hydrophobic effect. The self-passivation of the surface by inclusion of both end groups into CDs at one surface is prevented by using a short tetraethylene glycol chain to connect the end groups of the connector molecule.
The effect of the connector molecules on adhesion was revealed in force distance curves as shown in Fig. 3. The pull-off force between a β-CD covered AFM tip and surface increases significantly in the presence of connector molecules in comparison to the control experiments without connector molecules. During retraction of the AFM tip, a step wise rupture is observed in Fig. 3(b) which reflects the subsequent rupture of single and multiple β-CD – adamantane complexes. For a statistical evaluation, the rupture forces of only the last rupture event (labeled Frupture in Fig. 3(b)) of 300 force curves were summarized in the histogram in Fig. 3(c). By analyzing only the last event, we ensure that the cantilever is fully relaxed after the rupture. The periodic distribution in Fig. 3(c) reveals the rupture force for one or more bonds. The first maximum quantifies the rupture force of a single bond with 82 ± 10 pN, the second a parallel rupture of two bonds etc. The experiment has been repeated for various unloading rates. No rate dependency is observed for the most probable single rupture force in the experimentally accessible range from 1.5 to 2500 nN s−1 (see Fig. 3(d)).
 | ||
| Fig. 3 (a) Force curve recorded during retraction of a β-CD functionalized AFM-tip from the surface in solution of the ditopic connector 3 and in water (control); (b) zoom into the separation curve in adamantane connector solution, the last rupture event is marked which is included into histogram in (c); (c) histogram derived from the last rupture forces at an unloading rate of 150 nN s−1, the solid line is the sum of Gaussian functions fitted to the peaks; (d) the single rupture force at different unloading rates, the error bars indicate the standard deviation of the Gaussian curve fitted to the first peaks, respectively. | ||
In contrast, the maximum pull-off force and, interestingly, also the overall shape of the force curves are highly loading rate dependent when connector molecules are present (see Fig. 4(a)). Without connector molecules, no rate dependency is observed. At low unloading rates, the force curve exhibits a plateau at adhesion forces of below 0.5 nN. With increasing unloading rate, a transition in shape occurs from the force plateau to a triangular shape with a high maximum pull-off force. After rupture at the maximum pull-off force, the curve turns into a plateau with forces similar to the plateau force observed for slow pulling. Note that all triangularly shaped adhesion curves last for several milliseconds, much longer than the response time of the cantilever of about 120 μs. Fig. 4(b) shows the unloading rate dependence of the maximum pull-off force.
 | ||
| Fig. 4 (a) Representative force vs. distance curves at different unloading rates in nN s−1; (b) maximum adhesion force depending on the unloading rate in connector solution and control experiment, each data point represents the average of 300 force curves recorded at three independent surface positions. The error bars indicate the standard deviation of the three positions; (c) and (d) Details of the force curve in (a) recorded with 1.5 nN s−1, the dotted lines indicate the effective contact stiffness. | ||
A detail of the force curve recorded at a low unloading rate of 1.5 nN s−1 in Fig. 4(c) shows that the plateau consists of several force drops with a similar spacing of about 0.15 nm. Between the force drops, the force curve develops a constant slope which reveals the effective stiffness of the contact which is close to the cantilever stiffness of 0.2 N m−1. In Fig. 4(d), a detail of the force curve at higher separation, the distance between force drops is about 1 nm, the peak force only around 80 pN, and the effective stiffness only kneff = 0.02 N m−1.
The friction force is the force acting opposite to the direction of sliding when scanning across a surface. During sliding, adhesive bonds between the β-CDs on the AFM tip and surface cause a torsion of the cantilever. Scanning in forward and backward direction we can derive the friction force from the average torsion. The friction force caused by unspecific interactions between AFM tip and surface can be revealed from the control experiment without guest molecules in Fig. 5. In order to minimize these contributions the normal force was kept below 0.5 nN so that friction was dominated by the specific host–guest interaction. When connector molecules are present, the friction force increased by up to a factor of five in comparison to the control experiment as shown in Fig. 5(a). Contrary to adhesion, friction is not rate dependent. The sliding velocity has no significant impact on the measured friction force in the experimentally accessible range from 3 nm s−1 to 1000 nm s−1. Representative friction loops recorded with a tip velocity of 100 nm s−1 are shown for adamantane solution and the control experiment in Fig. 5(b). The characteristic sawtooth shape reveals an irregular stick-slip motion of the tip apex which is observed for all sliding speeds (see Fig. 5(c)). In the control experiment no stick-slip motion is observed. The dotted line indicates the effective stiffness in lateral direction kleff = 0.13 ± 0.01 N m−1 which is two orders of magnitude smaller than the torsional spring constant. A detail of the force curve recorded with a scan velocity of 10 nm s−1 in Fig. 5(d) shows that the large triangular force drops consists of smaller drops with a distance of 0.5 nm to 1 nm.
 | ||
| Fig. 5 (a) Average friction force vs. scan velocity of a β-CD functionalized AFM-tip and surface in solution of the ditopic connector 3 and in water (control), the error bars represent the standard deviation of 50 friction loops; (b) representative friction loops recorded with 100 nm s−1, the dashed line indicates the lateral contact stiffness; (c) forward scan of a friction loop recorded with different velocities, the unit of the velocity values in the legend is nm s−1, force curves are offset for better readability, the average does not change with velocity; (d) detail of a forward scan of (c) recorded with a velocity of 10 nm s−1. | ||
4. Discussion
In the following we will discuss the dynamics of adhesion, normal and lateral, which are governed not only by rupture and rebinding of individual bonds, but also by the cooperative dynamics of multiple bonds at a given tip-sample distance.The force required to rupture the bond between two β-CD guests established by the ditopic adamantane connector is revealed by the force drop measured when the last bond is ruptured in a pull-off experiment. The value for the most probable rupture force is 82 ± 10 pN, comparable but significantly lower than the values of 102 ± 15 pN7 and 97 ± 19 pN9 reported for single adamantane hosts bound to β-CD guests. The most probable force to rupture one of the two complexes formed at the end of a ditopic connector molecule is expected to be lower than the most probable force to rupture a single complex. The probability distribution p(F) of rupture forces of one host–guest bond in Fig. 3(d) of ref. 7 can be approximated by a Gaussian normal distribution with a mean value of μ = 100 pN and a standard deviation of σ = 15 pN. The probability to rupture one of two such bonds in series at a force F is then given as
 | (1) |
For a standard deviation of σ = 15 pN in the single bond rupture force, the most probable force for rupturing one of two bonds in series is 93 pN, for σ = 10 pN the value is 95 pN and for σ = 20 pN it is 90 pN. A small reduction of the most probable rupture force for two bonds in series by only a fraction of the width of the rupture force distribution is in agreement with a model by Williams and Evans12 and recent experimental results.22 In our experiment, the measured rupture force of 82 pN for the connection by the ditopic adamantane molecule is lower than expected from the probability argument. The lower value may be explained by interactions between the two β-CD hosts, whose distance is estimated to be less than 2 nm when the ditopic guest molecule is complexed. Furthermore, the ditopic connector used in this work carries one positive charge close to each adamantane end group, which is known to reduce the affinity for complexation in comparison to the uncharged adamantane derivatives.19 The lower affinity is reflected in the lower binding free energy of −5.5 kcal mol−1 resulting from ITC measurements in comparison to that for single uncharged adamantane guest molecules of −6.5 kcal mol−1.7 The binding free energy and the rupture force for our connector molecule actually lie in between the respective values reported by Auletta et al. for ferrocene and (1,1-dimethylethyl)benzene guests, confirming that the lower rupture force of 82 pN is rather due to a lower affinity than to the effects of two bonds in series.
In the following we will discuss the dynamics of adhesion and start with the rupture of a single bond. The loading rate was varied over more than three orders of magnitude (see Fig. 3(d)) and in agreement with previous studies on adamantane-β-CD interactions no loading rate dependence was observed for single bonds, indicating that the complexes are probed in thermodynamic equilibrium.7,9,23 We determined the binding constant of ditopic guests with β-CD-hosts in solution by isothermal titration calorimetry as K = 1.1 × 104 M−1 which is slightly lower than value reported by Mulder et al.14 for a similar system. The small difference is in line with the lower rupture force in our ditopic system. By assuming that kon is diffusion limited and thus in the order of 108 M−1 s−1,24 one concludes that koff is 8000 s−1. In surface bound cyclodextrin systems, koff is expected to be the same, but the binding constants are even higher because the high concentration of guest molecules at the surface increases kon.14 We can draw the conclusion that we have very fast binding kinetics in the scale of the koff = 8000 s−1 and are certainly in equilibrium for our experimental pulling rates.
We turn now to the system with multiple bonds and discuss the strong loading rate dependence of the pull-off force and the overall shape of the force distance curves. We will start with the force curve recorded at slow pulling speeds and focus on the elasticity of the supramolecular assembly. The force curve exhibits a force plateau over which the bonds between tip and surface are successively ruptured. The length of the force plateau is unexpected large. The expected upper bound for the range of interaction between tip and surface is given by the thickness of the two β-CD monolayers, which are deformed upon contact formation plus the length of the ditopic connector molecule. Elastic deformation of silicon AFM tip and sample are of the order of tens of pm for the forces applied here and can be neglected. The thickness of the β-CD monolayer is estimated from the size of molecular constituents to be 3 nm, this thickness has been confirmed in AFM height measurements of areas where the monolayer was removed by scanning at higher load. The length of the ditopic connector is estimated to be 2 nm. Thus, the force plateau is longer than expected by up to 12 nm. We conclude that our supramolecular assembly offers more elastic flexibility than expected.
The shape of the force curves in Fig. 4(a) are strikingly similar to those recorded by Pussak et al.25 for the interaction between an soft elastic probe and a flat surface mediated by specific mannose–ConA interactions. In our system, such elastic flexibility can only be provided by the silane network layer binding the β-CD molecules to the oxidized silicon surface. This layer is known to be strongly cross-linked, but may provide flexibility by ring formation and a limited number of covalent anchor points to the oxidized surface.26,27 Thus, the stiffness of the bonds is not only determined by the stiffness of the linker and CD molecules but additionally by the flexibility of the silane network.
The force plateau exhibit force drops in irregular distances, around 0.1–0.2 nm in the earlier part of the curve (Fig. 4(c)) and 1 nm in the later part (Fig. 4(d)). The height differences agree well with a bond density close to the maximum density of 0.33 nm−2 of β-CD molecules on a curved surface from which we can reveal the number of bonds of around sixty, a number highly dependent on the radius of the AFM tip. The slope of the force curve between the rupture events reflects the effective contact stiffness which is equal to the spring constant of the cantilever in the early part of the force curve because it is softer than the molecular springs in parallel. Due to the curvature of the AFM tip, the bonds are not equally distributed along the pulling distance but the height difference between the CD molecules increases closer to the tip apex. Thus, less bonds contribute to the force in the later part of the curve resulting in a significant lower contact stiffness and a larger distance between the rupture events. However, the low effective stiffness of only 0.02 N m−1 can only be explained by an elastic deformation of the silane network. The development of the effective contact stiffness is reflected by the noise of the force curve. The noise originates from thermal vibrations of the force probe which is damped in the early part of the force curve where the tip is bound by several parallel springs. In the later part of the force curve, the contact stiffness is low due to the lesser number of bonds stretched in parallel resulting in a weaker damping of the cantilever.
With increasing loading rate, the detachment process transforms from a zipper-like peeling of bonds, which resembles the macroscopic equilibrium situation described by Raman et al.,8 to a more parallel rupture of bonds with an increasing pull-off force indicating that the system moves out of equilibrium.
We will address three different aspects which can lead to a rate dependency of the curve shape and pull-off force and start with the dynamics of the cantilever. The force probe has a limited reaction time τ = 1/4f caused by inertia and viscous damping which is in the order of 120 μs. In contrast, typical force curves in the high unloading rate regime last at about 3 ms so that we can exclude inertia causing the rate dependency of the pull-off force. However, inertia does cause a delay in the cantilever movement whose possible role in the detachment kinetics is discussed below.
The second aspect leading to rate dependence of the pull-off force refers to the elasticity of the overall contact. For viscoelastic contacts, the pull-off force is determined by a crack opening process. With increasing pulling speed, the viscoelastic losses at the edge of the contact increase, leading to an increase of the pull-off force.25 For our system, scratch tests revealed a thickness of the organic layer composed of CD and silane of about 3 nm. The relaxation of the even thinner silane layer is presumably too fast on the time scale of the force curve measurement to explain an increase in energy dissipation by a factor of 10 during a force measurement by viscous effects. Nevertheless, the elasticity of the attachment allows a parallel stretching of several bonds which then leads to a cooperative rupture behavior.
In literature, multiple bond failure is described for either a parallel arrangement of bonds with equal load sharing or a zipper-like subsequent failure of independent bonds.12,28,29 Due to its curvature, the situation is more complex for an asperity like the AFM tip. In the beginning of the force curve, the height difference between the bonds is less than the maximum stretching length of one bond. For this case, the overlap of bonds results in cooperative dynamics which can increase the life time of the ensemble of bonds by several orders of magnitudes.12 This effect is referred to as multivalency, for example by Gomez-Casado et al. in the discussion of a rather weak increase of pull-off force with increasing loading rate for two and three parallel adamantane – β-CD bonds.9 Because of the longer life time, and thus the lower off rate of the ensemble of bonds compared to a single bond, the bonds are probed out of equilibrium and a rate dependency of the pull-off force can be observed while the rupture force for a single bond is constant.9 In our specific supramolecular system, the on rate is orders of magnitudes higher than the off rate so that the multivalency effect needs to be extremely effective to explain the significant loading rate dependence of the pull-off force. We suggest that, inertia of the cantilever enhances the multivalency effect. After one bond is ruptured, inertia causes a delay in the cantilever movement which is significant on the time scale of the on rate so that a rebinding of already broken bonds is favored. The higher the unloading rate, the higher the contribution of inertia to cooperative dynamic effects. Thus, the finite response time of the cantilever can lead to a higher pull-off force by a combined effect of multivalency and cantilever inertia. After the force drop at the pull-off force, the gap between subsequent bonds increases and a shape transition occurs from a parallel to a zipper-like peeling of bonds which reveals itself as a force plateau at a constant adhesion force.
In the following section we will discuss the dynamics resulting in the friction force with respect to the difference between friction and adhesion. The irregular saw-tooth modulation of the lateral force, as observed in the friction loop, indicates that the repeated breaking of groups of irregularly distributed bonds determines the measured friction force. A stick-slip friction behavior caused by the cooperative rupture of subsystems of molecular bonds and subsequent rebinding was already predicted by the model introduced by Filippov et al.2 From the effective stiffness in lateral direction we can derive the force pulling rate which ranges from 0.4 nN s−1 to 130 nN s−1, about one order of magnitude smaller than in our adhesion experiment. For intermediate pulling rates, adhesion and friction exhibit very similar rupture characteristics: multiple complexes are ruptured at the same time resulting in the triangular shape of the force curve in adhesion and in the characteristic stick-slip shape in friction, with comparable peak forces of about 1 nN. The slip events consist of a series of smaller force drops indicating that several bonds contribute to the peak force. Similar to the adhesion measurements, we can assume that the lateral distance between the host molecules is smaller than the maximum stretching length of bonds in lateral direction Flrup/kleff = 1 nN/0.13 N m−1 ≈ 7.5 nm due to the flexibility of the silane network and the overall length of the β-CD-adamantane-β-CD complex.
However, a different development of the forces in lateral and normal direction is observed when varying the scanning speed and unloading rate, respectively. The friction force as well as the shape of the friction loop remain the same when varying the scan velocity over three orders of magnitude. To discuss the difference between adhesion and friction, we will address the same three aspects discussed above for the adhesion measurements. The first aspect is the response time τl = 1/4fl = 5 μs of the cantilever. Because of the high spring constant, the response time of the cantilever in lateral direction is two orders of magnitude smaller than in normal direction so that any direct contribution of inertia to the measured shape of the friction loop can be neglected. The second aspect is the viscoelasticity of the contact. As already discussed for the adhesion measurement, the relaxation of the molecular layer is presumably too fast compared to the time scale of the measurement to lead to a significant energy dissipation. Nevertheless, its elastic flexibility facilitates the simultaneous stretching of several bonds leading to the cooperative rupture behavior of the molecular ensemble. To discuss the contribution of multivalency in our friction measurements, we first focus on the kinetics of an individual bond and compare the reaction kinetics with the time scale of the experiment by considering the contact time for a single bond. With the fastest sliding velocity of 1000 nm s−1 and the stretching length of the bond of 7.5 nm, we arrive at a minimum contact time in the order of milliseconds, one order of magnitude higher than the survival time of a single bond τ = 1/koff ≈ 0.125 ms. Because of the fast reaction kinetics the individual bonds are ruptured in thermodynamic equilibrium as observed for the single pull-off force. This interpretation of the data is supported by the observation of a saturation regime for friction as function of the connector molecule concentration (see ESI† for the respective Langmuir curve). The monotonous increase of friction with concentration and its saturation indicate that the connector molecules do not passivate the surface and that we probe the specific adamantane β-CD interaction. For a non-equilibrium situation with slow reaction kinetics, a high concentration of connector molecules would block the CDs on the surface resulting in a decrease of friction which is not observed for our system.
In contrast to adhesion experiments, the cantilever inertia does not contribute to multivalency effects in friction due to the fast response in lateral direction. Furthermore, broken bonds are permanently rebound across the contact in sliding friction experiments. As result, the fast reaction kinetics lead to an equilibrium situation in friction for the ensemble of bonds and the friction force remains constant for the range of sliding speeds investigated.
5. Conclusion
In summary, we quantified the dynamic effects in friction and adhesion caused by cooperative rupture of supramolecular bonds. For our system based on the inclusion of ditopic adamantane connectors in cyclodextrin hosts and probed by AFM, we discovered a remarkable difference in the dynamics of friction and adhesion. In force spectroscopy, the pull-off force for multiple bonds increased dramatically with loading rate although the rupture force for an individual complex remained constant indicating that a single bond is probed in thermodynamic equilibrium. We assign the loading rate dependence to a cooperative rupture of multiple bonds, where the multivalency effect is enhanced by the inertia of the cantilever, leading to a non-equilibrium situation. The friction force was found to be constant over three orders of magnitude in sliding velocity. In friction, equilibrium is maintained by the permanent fast rebinding of broken bonds and the fast response time of the force sensor in lateral direction. These results confirm the importance of microscopic time scales for macroscopic friction and adhesion results, as predicted in ref. 2. The molecular design of our approach provides the opportunity for studies of the microscopic dynamics underlying friction and adhesion because the wide range of possible connector molecules offers to tune the kinetics of the system. Connector molecules with different binding constants or different complexation kinetics open a pathway to systematically explore the influence of complexation strength and kinetics on friction.Acknowledgements
This work was financially supported by the Volkswagen Foundation. We thank Anand Jagota for valuable discussions. Co-authors at the INM thank Eduard Arzt for continuing support of this study.References
- F. P. Bowden and D. Tabor, The friction and lubrication of solids, Clarendon Press, Oxford, 2008 Search PubMed.
- A. Filippov, J. Klafter and M. Urbakh, Phys. Rev. Lett., 2004, 92, 135503 CrossRef CAS.
- G. Wenz, Angew. Chem., Int. Ed. Engl., 1994, 33, 803–822 CrossRef PubMed.
- T. Auletta, B. Dordi, A. Mulder, A. Sartori, S. Onclin, C. M. Bruinink, M. Pter, C. A. Nijhuis, H. Beijleveld, H. Schönherr, G. J. Vancso, A. Casnati, R. Ungaro, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2004, 43, 369–373 CrossRef CAS PubMed.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2010, 3, 34–37 CrossRef PubMed.
- H. Schönherr, M. W.-J. Beulen, J. Bügler, J. Huskens, F. C. van Veggel, D. N. Reinhoudt and G. J. Vancso, J. Am. Chem. Soc., 2000, 122, 4963–4967 CrossRef.
- T. Auletta, M. R. de Jong, A. Mulder, F. C. J. M. van Veggel, J. Huskens, D. N. Reinhoudt, S. Zou, S. Zaopotoczny, H. Schönherr, G. J. Vancso and L. Kuipers, J. Am. Chem. Soc., 2004, 126, 1577–1584 CrossRef CAS PubMed.
- S. Raman, T. Utzig, T. Baimpos, B. R. Shrestha and M. Valtiner, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- A. Gomez-Casado, H. H. Dam, M. D. Yilmaz, D. Florea, P. Jonkheijm and J. Huskens, J. Am. Chem. Soc., 2011, 133, 10849–10857 CrossRef CAS PubMed.
- J. D. Badjic, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10419–10419 CrossRef CAS PubMed.
- E. Evans and P. Williams, in Physics of bio-molecules and cells. Physique des biomolécules et des cellules, Springer, 2002, pp. 145–204 Search PubMed.
- J. Blass, B. L. Bozna, M. Albrecht, J. A. Krings, B. J. Ravoo, G. Wenz and R. Bennewitz, Chem. Commun., 2015, 51, 1830–1833 RSC.
- A. Mulder, T. Auletta, A. Sartori, S. D. Ciotto, A. Casnati, R. Ungaro, J. Huskens and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 6627–6636 CrossRef CAS PubMed.
- X.-L. Qui, G. Li, G. Wu, J. Zhu, L. Zhou, P.-L. Chen, A. R. Chamberlin and W.-H. Lee, J. Med. Chem., 2009, 52, 1757–1767 CrossRef PubMed.
- T. Yamamoto, A. Terada, T. Muramatsu and K. Ikeda, Org. Prep. Proced. Int., 1994, 26, 555–557 CrossRef CAS.
- H. Wang and M. L. Gee, Ultramicroscopy, 2014, 136, 193–200 CrossRef CAS PubMed.
- E. Tocha, J. Song, H. Schönherr and G. J. Vancso, Langmuir, 2007, 23, 7078–7082 CrossRef CAS PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- J. Kinkel, B. Anspach, K. Unger, R. Wieser and G. Brunner, J. Chromatogr. A, 1984, 297, 167–177 CrossRef CAS.
- G. Wenz, C. Strassnig, C. Thiele, A. Engelke, B. Morgenstern and K. Hegetschweiler, Chem. – Eur. J., 2008, 14, 7202–7211 CrossRef CAS PubMed.
- A. Embrechts, H. Schönherr and G. J. Vancso, J. Phys. Chem. B, 2011, 116, 565–570 CrossRef PubMed.
- S. Zapotoczny, T. Auletta, M. R. de Jong, H. Schönherr, J. Huskens, F. C. van Veggel, D. N. Reinhoudt and G. J. Vancso, Langmuir, 2002, 18, 6988–6994 CrossRef CAS.
- M. Novo, D. Granadero, J. Bordello and W. Al-Soufi, J. Inclusion Phenom. Macrocyclic Chem., 2011, 70, 259–268 CrossRef CAS.
- D. Pussak, D. Ponader, S. Mosca, T. Pompe, L. Hartmann and S. Schmidt, Langmuir, 2014, 6142–6150 CrossRef CAS PubMed.
- P. Silberzan, L. Leger, D. Ausserre and J. Benattar, Langmuir, 1991, 7, 1647–1651 CrossRef CAS.
- V. V. Naik, M. Crobu, N. V. Venkataraman and N. D. Spencer, J. Phys. Chem. Lett., 2013, 4, 2745–2751 CrossRef CAS.
- P. M. Williams, Anal. Chim. Acta, 2003, 479, 107–115 CrossRef CAS.
- R. W. Friddle, A. Noy and J. J. De Yoreo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13573–13578 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C5NR00329F |
| This journal is © The Royal Society of Chemistry 2015 |