
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGoverning the morphology of Pt–Au heteronanocrystals with improved electrocatalytic performance†
Stefanos
Mourdikoudis
ab,
Mariana
Chirea
 a,
Daniele
Zanaga
c,
Thomas
Altantzis
c,
Manasis
Mitrakas
b,
Sara
Bals
c,
Luis M.
Liz-Marzán
ade,
Jorge
Pérez-Juste
*a and
Isabel
Pastoriza-Santos
*a
a,
Daniele
Zanaga
c,
Thomas
Altantzis
c,
Manasis
Mitrakas
b,
Sara
Bals
c,
Luis M.
Liz-Marzán
ade,
Jorge
Pérez-Juste
*a and
Isabel
Pastoriza-Santos
*a
aDepartamento de Química Física, CINBIO, Universidade de Vigo, 36310 Vigo, Spain. E-mail: pastoriza@uvigo.es; juste@uvigo.es
bAnalytical Chemistry Laboratory, Department of Chemical Engineering, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece
cEMAT, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium
dBionanoplasmonics Laboratory, CIC biomaGUNE, Paseo de Miramon 182, 20009 Donostia-San Sebastian, Spain
eIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 12th March 2015
Abstract
Platinum–gold heteronanostructures comprising either dimer (Pt–Au) or core–satellite (Pt@Au) configurations were synthesized by means of a seeded growth procedure using platinum nanodendrites as seeds. Careful control of the reduction kinetics of the gold precursor can be used to direct the nucleation and growth of gold nanoparticles on either one or multiple surface sites simultaneously, leading to the formation of either dimers or core–satellite nanoparticles, respectively, in high yields. Characterization by electron tomography and high resolution electron microscopy provided a better understanding of the actual three-dimensional particle morphology, as well as the Au–Pt interface, revealing quasi-epitaxial growth of Au on Pt. The prepared Pt–Au bimetallic nanostructures are highly efficient catalysts for ethanol oxidation in alkaline solution, showing accurate selectivity, high sensitivity, and improved efficiency by generating higher current densities than their monometallic counterparts.
Introduction
Bimetallic nanomaterials are of great interest, as they display multiple functionalities and superior catalytic activity, selectivity and stability over monometallic ones.1 They perform not only a simple combination or improvement of the properties attributed to their single metal counterparts, but may also give rise to new properties due to synergic effects, thus broadening the potential application fields.2 Reported architectures for bimetallic nanostructures include crown-jewel structures, as well as hollow shapes, heterostructured dimers, alloys, core–shell, core–satellite, hybrid and porous configurations.1,3,4 A number of recent review articles have summarized the wide range of existing synthetic protocols, associated properties and uses of bimetallic nanostructures.5–11Generally, heterostructured nanoparticles are obtained by seeded growth methods, i.e. the growth of a second metal on preformed metal nanoparticles. The final configuration of a bimetallic structure is governed mainly through kinetic control in the reaction pot, which can be carried out by slowing down the reduction rate of a salt precursor. A weaker reductant, a more stable precursor, lower temperature, decreased reagent concentration or a combination of these parameters can be used for this purpose.12 In particular, the synthesis of dimer and core–satellite structures relies on promoting heterogeneous nucleation.
Focusing on Pt–Au heterostructures, several studies have reported the synthesis of dimers based on the nucleation and growth of Au on Pt seeds.13–15 The lattice spacing of both metals is generally matched at the interface to lower the energy required for overgrowth of the second component.16 Other configurations, showing homogeneous distribution of both metals throughout the whole nanostructure volume, such as Pt–Au nanotubes17 as well as Pt–Au nanocages18 have been very recently reported. However, heterostructured dimers and multimers of Pt–Au in particular possess a larger amount of catalytic sites that can be employed for sensing, electron transport or catalysis, thanks to the coupling effect resulting from interfaces between two or more distinct types of metallic crystals directly exposed to the solution.19
We describe in this paper the tailored, high yield synthesis of Pt–Au nanodimers and Pt@Au core–satellite nanostructures in aqueous medium. Pre-synthesized Pt nanodendrites were used as seeds and two different protocols were used for Au deposition, with distinct nucleation and growth kinetic modes. Characterization by advanced electron microscopy techniques (electron tomography, high resolution transmission electron microscopy and scanning transmission electron microscopy) enabled a better understanding of the actual particle morphology and elucidation of their growth patterns. The interface between Au and Pt was investigated to provide evidence of epitaxial growth. This interface seems to have an important influence on the electrocatalytic response of the Pt–Au bimetallic nanoparticles. Both Pt–Au dimers and Pt@Au core–satellite particles present an excellent electrocatalytic performance in the ethanol oxidation reaction (EOR), which is improved as compared to the corresponding behavior of either Pt dendrites or Au particles on their own. Such differences reveal a very interesting and unusual electrochemistry taking place at the surface of Pt–Au dimers and Pt@Au core–satellite nanoparticles.
Results and discussion
Control over the morphology of Pt–Au heterostructures was achieved by the selective nucleation of gold nanoparticles on Pt nanodendrites. As illustrated in Scheme 1 two strategies were followed to obtain Pt–Au dimers or Pt@Au core–satellites. Both strategies were based on the heteronucleation (single-site or multiple-site) and subsequent growth of Au on preformed platinum nanodendrites.20 The main difference was the nature of the capping agent (CTAB or PVP), which was found to play a fundamental role in the kinetics of the process. | ||
| Scheme 1 Electron tomography rendering images showing the morphology of Pt nanodendrites and the result of single-site and multiple-site Au growth. | ||
Platinum nanostructures of ca. 20 nm in diameter, with dendritic morphology and two different porosity degrees (see Fig. S1, ESI†) were synthesized by thermolytic reduction of Pt(acac)2 in DMF, in the presence of PEI.20 As previously reported, by changing the concentration of metal precursor Pt dendrites with different ratios of branch volume to total volume (30% and 22%) were obtained (see ref. 20). The particles are also known to be polycrystalline, with interconnected arms branching in various orientations.20 The presence of PEI on the particle surface provided a highly positive ζ-potential (+44 mV) and prolonged colloidal stability in water.
Pt–Au nanodimers
The synthesis of Pt–Au dimers was performed by addition of PEI-stabilized Pt nanodendrites to an aqueous growth solution containing HAuCl4, CTAB and ascorbic acid, which led to immediate nucleation and growth of Au on the Pt seeds. The precise control of the experimental conditions (see the Experimental part) allowed selective control over Au nucleation and ultimately formation of Pt–Au nanodimers in high yield. Fig. 1a shows a representative TEM image of Pt–Au dimers prepared in the presence of 50 μL of Pt medium-compact dendrites (Fig. S1a, ESI†) with an average Au particle size of ∼20 nm. The yield for dimer formation is around 75% (Fig. S2, ESI†). It should be pointed out that Pt–Au dimers in high yield could also be obtained using Pt dendrites with different degrees of porosity (data not shown). In addition, the Au particle size could be tuned within a limited range by simply changing the amount of Pt dendrite seeds. For example, Au particle size varied from 20 to 10 nm by increasing the amount of added Pt from 50 to 125 μL. Beyond these limits either unattached Au nanoparticles with a mixture of dimers, trimers and higher order nanostructures (for lower Pt volumes), or single Pt nanodendrites (for higher Pt volumes), together with a majority of Pt–Au dimers, were observed. Interestingly, the dimers showed high colloidal stability, due to the high positive ζ-potential (+50 mV) provided by CTAB and PEI. Regarding the optical properties of such Pt–Au nanodimers, the extinction spectra show one band located around 530–540 nm corresponding to the localized surface plasmon resonance (LSPR) of Au nanoparticles (Fig. S3, ESI†). | ||
| Fig. 1 (a) Low-magnification TEM image of Pt–Au nanodimers, (b) STEM image of a Pt–Au nanodimer, (c) HRTEM image of a Pt–Au nanodimer and (d) 3D volume representation of a reconstructed Pt–Au nanodimer. | ||
Finally, X-ray photoelectron spectroscopy (XPS) was performed to obtain information about the surface chemistry of the dimers. Fig. S4a (ESI†) shows a high resolution Au 4f spectrum where the Au 4f7/2 peak with a binding energy of 83.6 eV was assigned to Au0.21 In the high resolution Pt 4f spectrum, deconvolution analysis revealed the presence of a small peak at 71.0 eV attributed to Pt2+, which coexisted with Pt0 (72.7 eV). (Fig. S4b, ESI†). Besides, the comparison of the peak intensities in the deconvoluted spectra showed atomic percentages of ∼79% and 21% for Pt0 and Pt2+, respectively.
To obtain a deeper understanding concerning the formation mechanism of the Pt–Au dimers, complete morphological characterization is required. Three-dimensional characterization was carried out by using electron tomography. Visualization of a typical 3D-reconstruction is presented in Fig. 1d and the corresponding movie is available as ESI.† The 3D representation of the reconstructed volume shows a dimer particle composed of a spherical part and a porous part. STEM-EDX analysis of the Pt–Au dimers enabled us to determine the elemental distribution in the nanostructure, confirming that the spherical particles are made of gold and the dendritic ones contain platinum (see Fig. S5, ESI†). Furthermore, high resolution HAADF-STEM images (Fig. 1b) and HRTEM images (Fig. 1c and S6, ESI†) show the presence of twinning in the formed Au particles. High resolution HAADF-STEM images of the Pt–Au interface in different nanodimers were acquired (Fig. S7a, ESI†) and visualization of the atomic columns shown in Fig. S8 (ESI†) proved epitaxial growth. EDX mapping (Fig. S7b, ESI†) of the same interface was also acquired, denoised through the application of a Gaussian filter and overlaid onto the high resolution HAADF-STEM image to generate the combined map shown in Fig. 2. It was then possible to qualitatively distinguish the Pt dendrite (green), the Au particle (red) and their overlap along the observation direction (yellow). The Fourier transform of the two regions confirms the different lattice spacing along a {110} zone axis (Fig. S8 and Table S1 in the ESI†). The lattice mismatch can be visualized by observing the dots in the Fourier transforms, which get closer to the center while moving from Pt areas to Au areas (Fig. S9 in the ESI†). The shortening of the distances in the reciprocal space reflects the expansion of the crystal structure as expected.
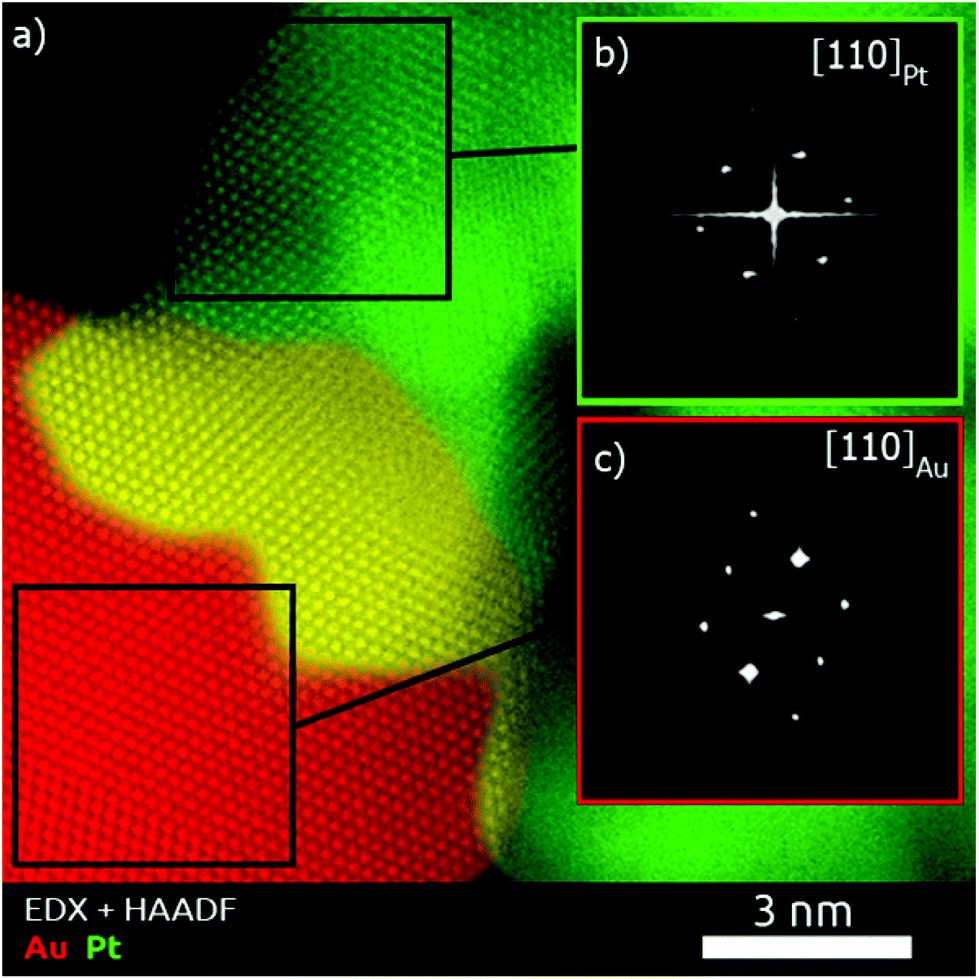 | ||
| Fig. 2 (a) EDX map overlaid to the corresponding high resolution HAADF-STEM image of the Au–Pt interface in a dimer. (b, c) FFT patterns from the highlighted regions, showing the [110] zone axis of the fcc crystal structure. | ||
Pt@Au core–satellite nanostructures
Pt@Au core–satellite nanostructures were obtained when AuCl4− was reduced by ascorbic acid in the presence of KBr and PVP. Typically, a gold salt solution was added to an aqueous solution containing ascorbic acid, KBr, PVP and Pt nanodendrites, giving rise to a fast violet coloration. Fig. 3 shows representative TEM images of the obtained nanostructures consisting of gold petals around Pt nanodendrite cores. The size of the Au nanoparticles could be marginally tuned by modifying the amount of Pt seeds (see Fig. S10 and Table S2, ESI†). We however noticed that small amounts of Pt seeds (high Au to Pt molar ratios) led to the formation of an almost continuous Au shell around the Pt cores through coalescence of the growing Au nanoparticles (Fig. S10a, ESI†). In contrast, a very low Pt amount provided isolated gold particles and/or aggregated Pt@Au core–satellite structures (data not shown). Finally, Pt@Au core–satellite nanostructures could also be synthesized in high yield from Pt nanodendrites with different degrees of porosity.20 | ||
| Fig. 3 (a) TEM image of typical Pt@Au core–satellite particles, (b) HRTEM image of a single Pt@Au core–satellite NP, (c) High resolution HAADF-STEM image of a single Au ‘petal’ located around the Pt dendrite core. Twinning is observed. (d) EDX map of a Pt@Au core–satellite particle. | ||
The Pt@Au core–satellite nanostructures display a negative ζ-potential (ca. −35 mV). Although initially Pt nanodendrites were stabilized by PEI and therefore showed a positive charge, once the Au nanoparticles were deposited on the Pt surface the PVP coating on the metal surface reverted the overall surface charge to negative values.23 Regarding the optical properties, Fig. S11 (ESI†) shows the extinction spectrum of the aqueous dispersion of a representative Pt@Au core–satellite sample with an average size of ∼57 nm (see Table S2, ESI†). A relatively broad LSPR band centered at ∼580 nm can be observed. Certainly, the position and the width of this band differ from that corresponding to Pt–Au dimers (see Fig. S3, ESI†). Such differences have been ascribed to a strong plasmon coupling between the Au particles in the core–satellite system.24 In addition, XPS analysis for such core–satellite structures showed Au 4f7/2 and Pt 4f7/2 peaks, assigned to Au0 and Pt0 (Fig. S12, ESI†).
STEM-EDX analysis was also performed and indeed confirmed the Pt@Au core–shell structure. Fig. 3d and S13 (ESI†) evidence Pt dendrites homogeneously coated by gold particles. Both HAADF-STEM and HRTEM images (Fig. 3c, b and S14,† respectively) illustrate the presence of defects (twinning) in the Au particles surrounding the Pt seeds. The complexity and strong overlap of the structures prevented us from acquiring high resolution HAADF-STEM images of sufficient quality at the interface between Pt and Au. Nevertheless, based on the above described observations for the Pt–Au dimers, we hypothesize that the growth of Au onto Pt in the case of the Pt@Au core–satellite particles is also epitaxial.
Confirmation of the peculiar morphology of the Pt@Au core–satellite structures required 3D characterization. Representative electron tomography reconstructions of various nanostructures are displayed in Fig. 4a and b showing a Pt@Au particle viewed from different angles, which was synthesized from Pt nanodendrites with a different degree of porosity as compared to those in Fig. 4c and d. Movies illustrating the morphology of these structures can be downloaded from the ESI† material. We therefore conclude that, regardless of the porosity of the Pt dendrite, this synthetic methodology leads to the formation of Pt@Au core–satellite nanostructures.
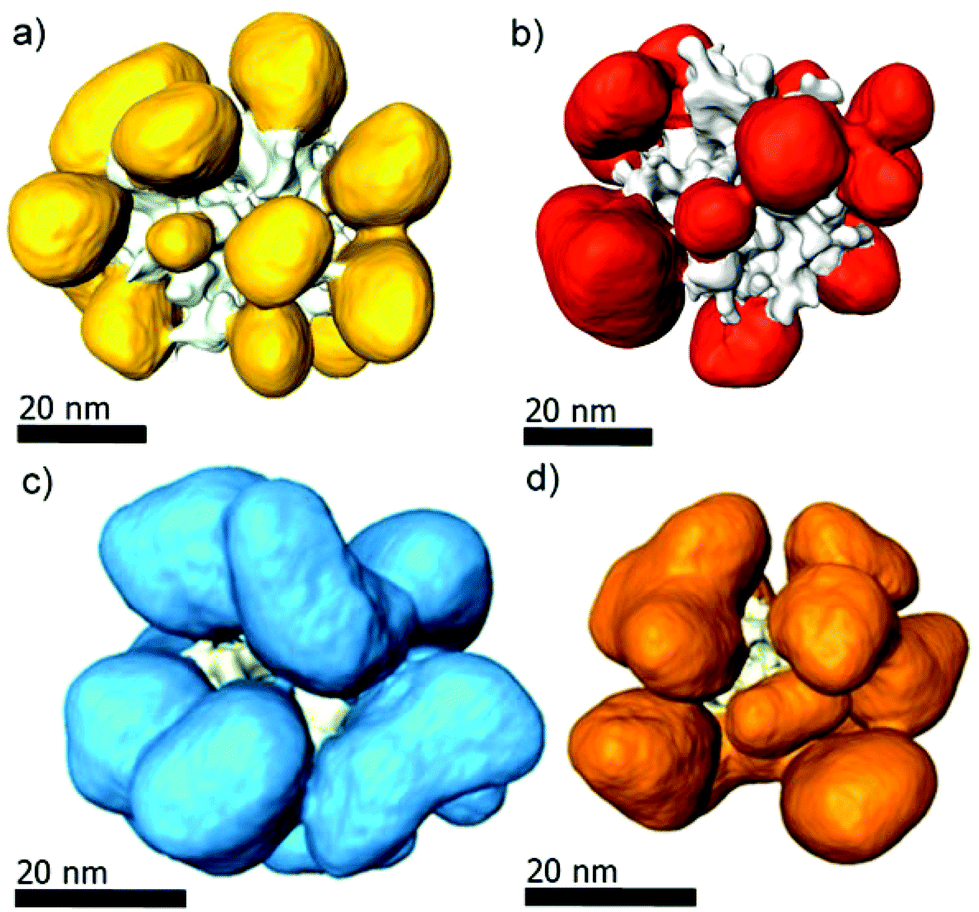 | ||
| Fig. 4 3D-representations along different views of Pt@Au core–satellite particles grown from ‘medium-compact’ Pt dendrite seeds (a, b), and from ‘porous’ Pt dendrites (c, d). | ||
Growth mechanism
Our approach clearly demonstrates that HAuCl4 can be selectively and catalytically reduced on Pt dendrites to generate either Pt–Au dimers or Pt@Au core–satellites, depending on the specific conditions. In an attempt to figure out the mechanism governing the site-selective heterogeneous growth of Au on Pt, we must consider several parameters: lattice matching and surface energies,25 as well as the balance between Au nucleation and growth and surface charge of the Pt nanodendrites.It is well established that lattice parameters for Pt are smaller than those for Au. However, the small lattice mismatch (3.9%) still allows epitaxial growth of Au onto Pt. Another interesting point to be considered is whether gold deposition should occur on weakly bound atoms at sharp convex tip ends or at surface defects present in concave areas. On the one hand, the relatively sharp angles at the tips inherently impose crystallographic edges with the existence of more reactive atoms/sites. On the other hand, the concave curvature between the different arms/tips of the dendrite must impose the existence of surface defects, such as atomic steps, kinks, vacancies and/or dangling bonds, commonly associated with higher chemical reactivity due to their unsaturated coordination. In this particular case, we postulate that the presence of a relatively high concentration of the polyelectrolyte PEI during the synthesis of Pt dendrites could block the porous cavities of the dendrites, thus hindering the diffusion of gold atoms towards them. Therefore, nucleation at the tips of Pt dendrites should be favored.
It is well known that the addition of capping agents, such as surfactants or polymers, can change the kinetics of reduction. In this work the main difference between the strategies followed to obtain dimer or core–satellite structures was the type of capping agent used, CTAB or PVP. In any case, we have to take into account that the overall process takes place in less than five minutes, so we are dealing with a relatively fast process. In what follows we analyze both strategies to address the role of these capping agents in the determination of the final nanostructure.
In the presence of a quaternary ammonium surfactant, such as CTAB, AuCl4− ions will be complexed with CTA+ ions, forming an ion-pair that will be readily solubilized within CTAB micelles. This complexation leads to an increase of the Au3+ to Au0 reduction potential,26 and as a consequence the addition of ascorbic acid (in the absence of any metal seed) will lead to reduction from AuCl4-CTA to AuCl2-CTA, thereby hindering the nucleation process. Notwithstanding, the subsequent addition of metal seeds leads to catalytic reduction to Au0 on their surface.27 Such an approximation has been applied, for instance, to synthesize gold nanospheres and nanorods with size control or core–shell (metal@Au) nanoparticles.28,29 We demonstrate here that Pt dendrites can also catalyze this reaction, but more importantly, under controlled conditions this process gives rise to the formation of Pt–Au dimers in high yield. The entire process could be explained in terms of the relatively slow reaction rate under our specific experimental conditions (CTAB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio ∼60, low ascorbic acid concentrations and room temperature), resulting in the growth process being favored over nucleation. We should also consider that there is a thermodynamic preference for the additional growth of Au on Au nuclei rather than on exposed Pt (forming new Au nuclei) because of the lattice mismatch. Even under conditions favorable to nucleation just some trimers or tetramers might form but not core–satellites (see Fig. S2, ESI†).
:Au ratio ∼60, low ascorbic acid concentrations and room temperature), resulting in the growth process being favored over nucleation. We should also consider that there is a thermodynamic preference for the additional growth of Au on Au nuclei rather than on exposed Pt (forming new Au nuclei) because of the lattice mismatch. Even under conditions favorable to nucleation just some trimers or tetramers might form but not core–satellites (see Fig. S2, ESI†).
The use of PVP instead of CTAB was found to induce multiple-site growth. While CTAB prevents Au nucleation by forming a complex with AuCl4− ions, PVP might even induce the nucleation, apart from preventing the final heterostructures from agglomeration. The formation of Pt@Au core–satellites could be explained in terms of an increase of the reduction rate, which favors the nucleation of Au at multiple sites of the Pt nanodendrite surface. Since the experimental conditions were such that they favored Au reduction, the gold salt should be added after the Pt nanodendrites to avoid nucleation of free Au nanoparticles. In fact, mixing AuCl4− ions with ascorbic acid before Pt dendrite addition led to the formation of free Au nanoparticles (see Fig. S15b and S15c ESI†).
Finally, another parameter to take into consideration is the surface charges of the Pt nanodendrites and Au precursor ionic charge in both systems. In the presence of CTAB, Pt nanodendrites and Au precursor are both positively charged, the former due to the PEI coating, and the latter due to the complexation of AuCl4− with CTA+ ions and solubilization of the complex within CTAB micelles. In the presence of PVP however, Pt nanodendrites and Au precursor present opposite charges. The electrostatic adsorption of PVP on positively charged Pt nanodendrites does not revert the surface charge (as confirmed by zeta potential measurements), therefore the particles still present a positive charge (ca. +15 mV) while the Au precursor (AuCl4−) is anionic. Since Pt nanodendrites and Au precursor present opposite charges, electrostatic interactions between Au precursor and Pt nanodendrite surface occur and therefore contribute to the multiple Au nucleation onto the Pt surface.
Electrocatalytic application for EOR
In the present work we studied the electrocatalytic activity of Pt–Au dimers (Fig. 1) and Pt@Au core–satellites (Fig. 3) for ethanol oxidation in alkaline solution and it was compared with the performance of pure Pt nanodendrites and pure Au nanospheres. In the case of Pt@Au core–satellite particles the Au particle size can be tuned within a small size range (see Table S2, ESI†), which allowed us to study the effect of Au size in the electrochemical activity of the Pt@Au core–satellite samples for EOR. Glassy carbon (GC) electrodes were modified with Pt–Au dimers and Pt@Au core–satellite nanoparticles with overall sizes of 47 nm (GC–Pt@Au1, Au size ∼13 nm), 57 nm (GC–Pt@Au2, Au size ∼17.5 nm) and 61 nm (GC–Pt@Au3, Au size ∼20 nm).Cyclic voltammetry measurements were performed at GC electrodes modified with Pt–Au dimers (GC–Pt–Au, Fig. 5a) and Pt@Au core–satellite nanoparticles (GC–Pt@Au, Fig. 5b) in alkaline solution of 1 M ethanol and 1 M NaOH. The cyclic voltammograms show two main oxidation steps, as previously reported for Pt nanodendrites:20 acetaldehyde formation depicted by an anodic peak current density at positive potentials (0.15 V and 0.17 V for GC–Pt–Au and GC–Pt@Au1, respectively) and CO2 production depicted by an anodic peak current density at negative potentials (−0.19 V). The cyclic voltammograms from Fig. 5 were normalized to the mass content of catalyst and showed an excellent mass activity for EOR. The electrochemically active area per gram of catalyst for all samples varied as follows: 206.3 m2 g−1 (GC–Pt–Au), and 53.0 m2 g−1 (GC–Pt@Au1), 69.75 m2 g−1 (GC–Pt@Au2), and 50.8 m2 g−1 (GC–Pt@Au3). Analysis of mass activity revealed the production of 10 A mg−1 at Pt–Au catalyst and on average 3 to 4 A mg−1 at Pt@Au catalyst (Fig. 5). The influence of Au nanoparticles grown on Pt nanodendrites is reflected in the shift of the acetaldehyde and CO2 peak potentials toward more positive values and enhancement in the current densities with respect to the CV features observed for the GC–Pt nanodendrite modified electrode (see Table S3, ESI† and ref. 20). These shifts reveal an improvement of electrocatalytic activity17,18 toward EOR. For consistency, current densities were expressed in mA cm−2 rather than mA mg−1 in order to compare the catalytic activity of Pt–Au dimers and Pt@Au core–satellite particles with the results previously reported for Pt nanodendrites (in ref. 20). Accordingly, the GC–Pt–Au modified electrode generated a maximum current density of 520 mA cm−2 after 105 CVs at 50 mV s−1 scan rate, whereas pure Pt nanodendrites generated 197 mA cm−2 after 320 CVs at 50 mV s−1 (see Fig. S17 and Table S3, ESI†). The catalytic activity for a similar time frame with Pt nanodendrites (300 CVs) showed a decreased yield of a ca. 100 mA cm−2 at the GC–Pt–Au modified electrode for EOR (Fig. S17, ESI†), the remaining current density still being higher (double) than that obtained at GC–Pt nanodendrites. In the case of GC–Pt@Au modified electrodes, maximum current densities of 340.6 mA cm−2 (GC–Pt@Au1), 448.24 mA cm−2 (GC–Pt@Au2) and 252.5 mA cm−2 (GC–Pt@Au3) after 158 CVs, 125 CVs and 90 CVs, respectively (all at 50 mV s−1 scan rate), were observed (Fig. S18, ESI†), whereas the pure Pt nanodendrites (medium compact ones) presented a much lower current density (61 mA cm−2).20 In other words, the electrocatalytic properties of the Pt nanodendrites were improved by growing either one or multiple gold spheres on their surface.
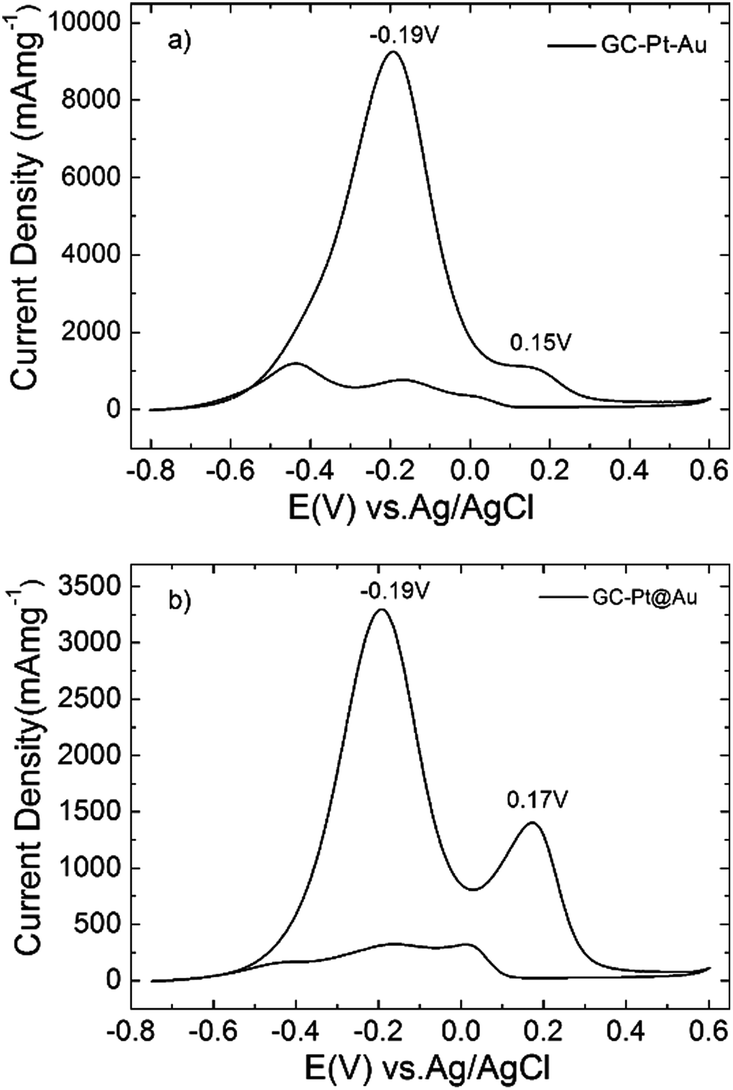 | ||
| Fig. 5 Cyclic voltammograms recorded at GC–Pt–Au (a) and GC–Pt@Au (b) in 1 M ethanol and 1 M NaOH aqueous solution. The scan rate was 50 mV s−1. | ||
All this seems to indicate that Pt–Au dimers are better catalysts than Pt@Au-core–satellite particles because of their higher current densities for EOR generated for a lower number of scans. XPS analysis (Fig. S4 and S12, ESI†) revealed that Pt–Au dimers contained Pt0 as well as Pt+2 whereas only Pt0 was found for Pt@Au core–satellites. The presence of Pt+2 could contribute to the improvement of the catalytic performance. Moreover, the Au-rich structure of Pt@Au core–satellites has a stronger contribution to the production of acetaldehyde (higher peak current densities at +0.17 V than at dimers), which in turn is reflected in a higher poisoning rate of the Pt core. This could explain the lower current densities observed at the GC–Pt@Au modified electrode during EOR than at the GC–Pt–Au electrodes (Fig. 5 and Fig. S17 and S18, ESI†). Chronoamperometry tests (Fig. S19, ESI†) demonstrate that the Pt–Au dimers are indeed more resistant to poisoning with small organic molecules than Pt@Au core–satellite particles. The chronoamperogram (CA) measured at the GC–Pt–Au modified electrode for 3000 s and an applied potential of −0.19 V (black curve in Fig. S19, ESI†) showed a pronounced decay of current densities in the first 250 s with a loss of activity of 30% and a slow decay for the remaining 2750 s. The CA measured under the same conditions at GC–Pt@Au1, GC–Pt@Au2 and GC–Pt@Au3 catalysts (Fig. S19, ESI†) revealed a 35.7% activity loss in 250 s, and 33% and 46.0% activity loss respectively after 150 s with a slow decay of current density for the remaining reaction time. This is consistent with the CVs measured at the modified electrodes, indicating that Pt–Au dimers are more resistant against poisoning.
In addition, by comparing EOR at our bimetallic structures with EOR at an Au nanoparticle-modified electrode (Fig. S20, ESI†) one can easily understand the contribution of gold spheres. CVs with one oxidation peak at +0.18 V in the EOR were recorded at GC–Au nanoparticle-modified electrodes reaching a maximum current density of 129 mA cm−2 after 60 cycles. This single oxidation peak at +0.18 V may be attributed to the formation of acetaldehyde as the final product (a sign of incomplete ethanol oxidation), which supports the above described electrocatalytic response of our Pt–Au hybrids, i.e. high production of acetaldehyde by both Au spheres and Pt nanodendrites20 being efficiently converted to carbonate by the Pt nanodendrites. In other words, there is a synergic effect of both metals in the EOR.
One remarkable feature should be mentioned when comparing EOR at different sized Pt@Au core–satellite nanoparticles: the middle sized particles (57 nm) display the best electrocatalytic performance (Fig. S18, ESI†). Although it is known that the electron transfer for monometallic nanoparticles occurs more efficiently for smaller sizes,30–34 the core–satellite structure with 17.5 nm Au spheres revealed the best electron transfer abilities which are most probably related to the efficiency of electron transfer at the interface between Pt dendrite and epitaxially grown Au spheres. Moreover, the decrease of the electrocatalytic activity of GC–Pt@Au3 could be explained in terms of Au nanoparticles’ coalescence, as indicated by the TEM characterization, as well as the broad and red-shifted LSPR band (see Fig. S11b, ESI†). These data demonstrate that an optimal size of gold chemically bonded to Pt is necessary to achieve the highest conversion of ethanol to carbonate.
Regarding their catalytic efficiency, Pt–Au dimers and Pt@Au core–satellite nanoparticles generate current densities of hundreds of mA cm−2 for EOR, demonstrating a better catalytic activity (under similar experimental conditions; 1 M ethanol and 50 mV s−1 scan rate) than Pt black (5 mA cm−2),35 bimetallic Pd–Au (<14 mA cm−2),35,36 dendritic Au–Pd (40 mA cm−2),37 concave Pt@Au nanocubes (18 mA cm−2)38 or Pd–Au core–shell nanoparticles (10 mA cm−2).39
Moreover, all GC modified electrodes maintained high ratios of forward to backward anodic peak current density of jf1/jb1 = 7.76, jf2/jb2 = 5.51 (GC–Pt–Au, Fig. S17, ESI†), jf1/jb1 = 5.23, jf2/jb2 = 4.75 (GC–Pt@Au1, Fig. S18a, ESI†), jf1/jb1 = 5.2, jf2/jb2 = 4.75 (GC–Pt@Au2, Fig. S18b, ESI†), jf1/jb1 = 8.3, jf2/jb2 = 3.45 (GC–Pt@Au3, Fig. S18c, ESI†), revealing excellent resistance to poisoning with by-products resulting from the EOR. Therefore, it is demonstrated that Pt–Au bimetallic nanostructures are highly efficient catalysts for ethanol oxidation in alkaline solution showing accurate selectivity, high sensitivity, and improved efficiency by generating high current densities for a lower number of cycles.
Conclusions
We have shown that careful control of the experimental conditions allows tuning the seeded growth of Pt–Au heterostructures to obtain either dimer or core–satellite configurations, using platinum dendrites as seeds. The process was explained in terms of reduction kinetics and surface charges together with a thermodynamic preference for additional Au to grow on Au than on exposed Pt because of the lattice strain (3.9%) caused by the mismatch in lattice parameters. The actual configurations of the heterostructures were elucidated by electron tomography and epitaxial growth of Au on Pt was demonstrated through high resolution TEM and STEM measurements. The prepared Pt–Au bimetallic nanostructures are highly efficient catalysts for ethanol oxidation in alkaline solution demonstrating accurate selectivity, high sensitivity, and better efficiency by producing higher current densities than their monometallic counterparts.Experimental
Materials
Gold(III) chloride (HAuCl4), L-ascorbic acid (AA, 99+%, ACS reagent), N,N-dimethylformamide (DMF, >98%) branched polyethyleneimine (PEI, Mw 25000), absolute ethanol, and polyvinylpyrrolidone (PVP, Mw 55000) were purchased from Sigma-Aldrich. Potassium bromide (KBr, 99+%, IR grade) was bought from Acros Organics, while CTAB (>96%) was provided by Fluka and Pt(acac)2 (98%) from Strem Chemicals Inc. Milli-Q grade water was used in all preparations. All the chemicals were used without further purification. Glassware and stirring magnets were rinsed with aqua regia and dried well prior to use.
Synthesis of Pt nanodendrites
The platinum nanodendrites used as seeds were synthesized as previously reported by our group.20 Briefly, a 0.6 M solution of PEI (concentration refers to the monomer unit, ethyleneimine) in 30 mL of DMF was stirred, and then a certain amount of Pt(acac)2 was added. The concentration of Pt(acac)2 was varied (15 mM and 30 mM) in order to obtain dendrites of different porosities. This yellow solution was heated to 150 °C and left overnight. After precipitation and washing, the dendrites were dispersed in 17 mL of water forming a stable black colloidal dispersion. TEM revealed that the particles were monodispersed Pt nanodendrites of ∼21 nm in size.Synthesis of Pt–Au nanodimers
In a typical synthesis, 35.2 μL of 0.143 M HAuCl4 in water is added to 20 mL of an aqueous solution of 15 mM CTAB under mild stirring, followed by addition of 100 μL of 0.1 M ascorbic acid in water. The yellow-orange reaction mixture turns transparent, reflecting the reduction of Au3+ to Au+. Subsequently 5 mL of the colorless solution is isolated and a selected amount (between 50 and 125 μL) of an aqueous dispersion of Pt nanodendrites is added under stirring. A reddish color gradually develops within a few minutes, indicating the nucleation and growth of gold nanoparticles. After 5 min of reaction at room temperature, the stirring is stopped and the particles are precipitated by centrifugation (5000 rpm, 30 min) and redispersed in 5 mL of water.Synthesis of Pt@Au core–satellite structures
In a typical synthesis, to 4.14 mL of water, the following reagents are sequentially added under stirring: a designated amount of an aqueous solution of Pt dendrites, 262 μL of ascorbic acid (0.1 M in water), 30 μL of 6.72 mM KBr (in water) and 180 μL of 0.1 M PVP (in water – the concentration of PVP was calculated in terms of its repeating unit). The mixture is stirred at room temperature for 20 min and then 10 μL of an aqueous solution of 0.143 M HAuCl4 is quickly added. The reaction is then maintained under stirring for 2 min as a violet-purple color appears. Finally the product is collected by centrifugation (8500 rpm, 15 min) and washed with water for another two times. The particles are finally dispersed into 4.7 mL of water.Characterization
High resolution transmission electron microscopy (HRTEM) images were acquired using a JEOL 3000F electron microscope operated at 300 kV. High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, electron tomography series and EDX maps were acquired using a FEI Tecnai G2 electron microscope operated at 200 kV and an aberration corrected cubed FEI Titan 60–300 electron microscope, operated at 200 kV. The EDX acquisition was carried out using a ChemiSTEM system and analyzed using the Bruker ESPRIT software. The acquisition of the tomography series was performed using a Fischione model 2020 single tilt tomography holder, within a tilt range from −76° to +76° and an angular increment of 2°. The alignment of the series was performed using FEI Inspect 3D and IMOD.22 For the reconstructions, the simultaneous iterative reconstruction technique (SIRT) algorithm as implemented in FEI Inspect 3D was used. Conventional TEM images were acquired with a JEOL JEM 1010 microscope operated at 100 kV. ζ-potentials were measured via electrophoretic mobility by taking the average of five measurements at the stationary level, using a Zetasizer Nano S (Malvern Instruments) equipped with a He–Ne laser, operating at 633 nm and with a detection angle of 173° (4 mW). Optical characterization was performed using an Agilent 8453 UV-Visible spectrophotometer. XPS experiments were performed in a SPECS Sage HR 100 spectrometer with a non-monochromatic X-ray source (Magnesium Kα line of 1253.6 eV energy and 250 W, calibrated using the 3d5/2 line of Ag with a full width at half maximum (FWHM) of 1.1 eV. The selected resolution for the spectra was 15 eV of pass energy and 0.15 eV per step. All measurements were made in an ultra-high vacuum (UHV) chamber at a pressure below 8 × 10−8 mbar. In the fittings, Gaussian–Lorentzian functions were used (after a Shirley background correction) where the FWHM of all the peaks was constrained while the peak positions and areas were set free. ICP (inductively coupled plasma) elemental analysis measurements were performed at the CACTI institute in Vigo.Electrochemical measurements
All electrochemical measurements were performed on a PGSTAT 302N potentiostat (EcoChemie B.V., The Netherlands) at room temperature. A standard three-electrode system was used with a glassy carbon (GC) electrode (0.0314 cm2 area) as the working electrode, a platinum wire as the counter electrode and Ag/AgCl (3 M KCl) as the reference electrode. GC electrodes were cleaned by polishing on Micro-cloth pads with alumina powder dispersed in Milli-Q water, followed by sonication in 50/50 (v/v%) water–ethanol for 3 min, and electrochemically cleaned by cyclic voltammetry in 0.1 M H2SO4 aqueous solution. Freshly cleaned GC electrodes were dried under a stream of N2 and subsequently modified with Pt–Au nanomaterials by drop casting.20 For this purpose 10 μL (1.0 mg mL−1) of Pt–Au dimer or Pt@Au core–satellite nanostructures dispersed in Milli-Q water were drop-casted on each cleaned GC electrode, dried in air and fixed with Nafion film.The electrochemical reaction studied by cyclic voltammetry was ethanol oxidation in alkaline aqueous solution using 1 M ethanol and 1 M NaOH. The objective was to evaluate the electrocatalytic properties of the Pt–Au nanostructures as compared to their Pt dendrites20 and Au nanosphere components. Consecutive cyclic voltammograms (CV) were recorded for a potential window of −0.75 V to 0.6 V vs. Ag/AgCl (3 M KCl), at a scan rate of 50 mV s−1, a step potential of 4 mV and up to 300 scans. Chronoamperometry tests on different catalysts were performed at −0.19 V for 3000 s. The electrochemically active surface areas (EASA) of the modified electrodes were determined by cyclic voltammetry measurements performed in 0.1 M HClO4 aqueous solution (Fig. S16 in ESI†). A charge density of 210 μC cm−2 for clean polycrystalline Pt and 390 μC cm−2 for clean polycrystalline Au was used to estimate the EASA of the Pt–Au and Pt@Au nanostructure modified GC electrodes. For the Pt–Au dimer modified GC electrode the EASA value was 0.00018 cm2, whereas for the Pt@Au core–satellite modified GC electrode (GC–Pt@Au) it was 0.0025 cm2 for GC–Pt@Au1, 0.0055 cm2 for GC–Pt@Au2 and 0.0068 cm2 for GC–Pt@Au3.
For comparison, PVP-stabilized AuNPs were drop casted on GC electrodes (1 mg mL−1), dried and fixed with Nafion film (EASA was 0.0045 cm2). The average diameters of Au NPs were 15 nm as determined from TEM images. In this case, a maximum of 60 CVs for EOR were recorded at the GC–AuNP modified electrode in 1 M ethanol and 1 M NaOH from −0.5 V to 0.5 V, at a scan rate of 50 mV s−1 and step potential 4 mV. Deaeration of electrolyte solutions was ensured by purging N2 gas in the solution (15 min before starting each experiment) and over the solution during measurements.20
Acknowledgements
This work was supported by funding from the European Research Council under the Seventh Framework Program (FP7) (ERC grant N335078 – COLOURATOMS), also the European Union under the FP7 (Integrated Infrastructure Initiative no. 262348 European Soft Matter Infrastructure, ESMI), the Spanish MINECO (MAT2013-45168-R and CTQ 2010-1639), the European Social Fund (ESF) and the Greek State (PE4(1546)). S.M. acknowledges the General Secretariat for Research and Technology of Greece for his Postdoctoral Grant (Action “Supporting Postdoctoral Researchers” of the Operational Program “Education and Lifelong Learning”). The authors thank Luis Yate for performing the XPS measurements.Notes and references
- X. Liu, D. Wang and Y. Li, Nano Today, 2012, 7, 448 CrossRef CAS PubMed.
- S. Duan and R. Wang, Prog. Nat. Sci., 2013, 23, 113 CrossRef PubMed.
- Nanoplatform-Based Molecular Imaging, ed. X. Chen, Wiley, 2011 Search PubMed.
- D. Rodríguez-Fernández and L. M. Liz-Marzán, Part. Part. Syst. Charact., 2013, 30, 46 CrossRef PubMed.
- D. Wang and Y. Li, Adv. Mater., 2013, 23, 1044 CrossRef PubMed.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240 CrossRef CAS PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- B. Lim and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 76 CrossRef CAS PubMed.
- C. N. R. Rao, H. S. S. Ramakrishna Matte, R. Voggu and A. Govindaraj, Dalton Trans., 2012, 41, 5089 RSC.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS PubMed.
- P. R. Sajanlal, T. S. Sreeprasad, A. K. Samal and T. Pradeep, Nano Rev., 2011, 2, 5883 Search PubMed.
- G. He, J. Zeng, M. Jin, H. Zhang, N. Lu, J. Wang, M. J. Kim and Y. Xia, ChemCatChem, 2012, 4, 1668 CrossRef CAS PubMed.
- C. Wang, W. Tian, Y. Ding, Y.-Q. Ma, Z.-L. Wang, N. M. Markovic, V. R. Stamenkovic, H. Daimon and S. Sun, J. Am. Chem. Soc., 2010, 132, 6524 CrossRef CAS PubMed.
- S. I. Lim, M. Varon, I. Ojea-Jiménez, J. Arbiol and V. Puntes, J. Mater. Chem., 2011, 21, 11518 RSC.
- C. A. García-Negrete, T. C. Rojas, B. R. Knappett, D. A. Jefferson, A. E. H. Wheatley and A. Fernández, Nanoscale, 2014, 6, 11090 RSC.
- K. W. Kwon and M. Shim, J. Am. Chem. Soc., 2005, 127, 10269 CrossRef CAS PubMed.
- X. Guo, W. Ye, R. Zhu, W. Wang, F. Xie, H. Sun, Q. Zhao, Y. Ding and J. Yang, Nanoscale, 2014, 6, 11732 RSC.
- M. Xue and Y. Tan, Nanoscale, 2014, 6, 12500 RSC.
- Z. Zheng, H. Li, Y. Shen and R. Cao, Chem. – Eur. J., 2011, 17, 8440 CrossRef CAS PubMed.
- S. Mourdikoudis, M. Chirea, T. Altantzis, I. Pastoriza-Santos, J. Pérez-Juste, F. Silva, S. Bals and L. M. Liz-Marzán, Nanoscale, 2013, 5, 4776 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, 1995 Search PubMed.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71 CrossRef CAS PubMed.
- I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Chem. Mater., 2006, 18, 2465 CrossRef CAS.
- S. Vial, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Langmuir, 2007, 23, 4606 CrossRef CAS PubMed.
- M. R. Buck, J. F. Bondi and R. E. Schaak, Nat. Chem., 2012, 4, 37 CrossRef CAS PubMed.
- J. Rodríguez-Fernández, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, J. Phys. Chem. B, 2005, 109, 14257 CrossRef PubMed.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542 CrossRef CAS PubMed.
- J. Rodríguez-Fernández, J. Pérez-Juste, F. J. García de Abajo and L. M. Liz-Marzán, Langmuir, 2006, 22, 7007 CrossRef PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957 CrossRef CAS.
- M. Chirea, V. García-Morales, J. A. Manzanares, C. M. Pereira, R. Gulaboski and F. Silva, J. Phys. Chem. B, 2005, 109, 21808 CrossRef CAS PubMed.
- M. Chirea, C. M. Pereira and F. Silva, J. Phys. Chem. C, 2007, 111, 9255 CAS.
- M. Chirea, A. Cruz, C. M. Pereira and F. Silva, J. Phys. Chem. C, 2009, 113, 13077 CAS.
- M. Chirea, J. Borges, C. M. Pereira and F. Silva, J. Phys. Chem. C, 2010, 114, 9478 CAS.
- D. V. Talapin, J.-S. Lee, M. K. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- L. Y. Chen, N. Chen, Y. Hou, Z. C. Wang, S. H. Lv, T. Fujita, J. H. Jiang, A. Hirata and M. W. Chen, ACS Catal., 2013, 3, 1220 CrossRef CAS.
- F. Ksar, L. Ramos, B. Keita, L. Nadjo, P. Beaunier and H. Remita, Chem. Mater., 2009, 21, 3677 CrossRef CAS.
- Y. W. Lee, M. Kim, Y. Kim, S. W. Kang, J.-H. Lee and S. W. Han, J. Phys. Chem. C, 2010, 114, 7689 CAS.
- H. Li, H. Wu, Y. Zhai, X. Xu and Y. Jin, ACS Catal., 2013, 3, 2045 CrossRef CAS.
- C. N. Brodsky, A. P. Young, K. Chon Ng, C.-H. Kuo and C.-K. Tsung, ACS Nano, 2014, 8, 9368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tabulated data, TEM, HAADF-STEM, HRTEM images, UV-visible measurements, XPS spectra, ICP elemental analysis, EDX-mapping, cyclic voltammetry and chronoamperometry measurements and tomography videos. See DOI: 10.1039/c4nr07481e |
| This journal is © The Royal Society of Chemistry 2015 |