
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional polymer-capped mesoporous silica nanoparticles for pH-responsive targeted drug delivery†‡
Stefan
Niedermayer§
a,
Veronika
Weiss§
a,
Annika
Herrmann§
b,
Alexandra
Schmidt
a,
Stefan
Datz
a,
Katharina
Müller
b,
Ernst
Wagner
b,
Thomas
Bein
*a and
Christoph
Bräuchle
*a
aDepartment of Chemistry, Nanosystems Initiative Munich (NIM) and Center for Nano Science (CeNS), University of Munich (LMU), Butenandtstr. 11 (E), 81377 Munich, Germany. E-mail: bein@lmu.de; Christoph.Braeuchle@cup.uni-muenchen.de
bDepartment of Pharmacy, Pharmaceutical Biotechnology, Nanosystems Initiative Munich (NIM) and Center for Nano Science (CeNS), University of Munich (LMU), Butenandtstr. 5-13, 81377 Munich, Germany
First published on 13th April 2015
Abstract
A highly stable modular platform, based on the sequential covalent attachment of different functionalities to the surface of core–shell mesoporous silica nanoparticles (MSNs) for targeted drug delivery is presented. A reversible pH-responsive cap system based on covalently attached poly(2-vinylpyridine) (PVP) was developed as drug release mechanism. Our platform offers (i) tuneable interactions and release kinetics with the cargo drug in the mesopores based on chemically orthogonal core–shell design, (ii) an extremely robust and reversible closure and release mechanism based on endosomal acidification of the covalently attached PVP polymer block, (iii) high colloidal stability due to a covalently coupled PEG shell, and (iv) the ability to covalently attach a wide variety of dyes, targeting ligands and other functionalities at the outer periphery of the PEG shell. The functionality of the system was demonstrated in several cell studies, showing pH-triggered release in the endosome, light-triggered endosomal escape with an on-board photosensitizer, and efficient folic acid–based cell targeting.
1. Introduction
Polymers can be attractive for biological applications due to their biocompatibility, tuneable properties and production from sustainable sources.1,2 Therefore, polymers are widely used as responsive coatings of surfaces for selective ion-permeability,3 surface patterning4,5 and drug delivery with polyplexes,1,6,7 hybrid dendrimers,8,9 and other drug-polymer conjugates.10,11 Regarding drug-polymer conjugates, which offer relatively small particle sizes and good biocompatibility, possible issues may arise from low loading capacities and potentially poor stability, depending on the strength of the drug-polymer interactions.12,13 The drug-polymer interactions need to be optimized for different kinds of drugs to ensure stable drug-polymer conjugates. Aimed at creating a more general delivery platform, much effort has been extended towards the development of inorganic nanoparticles such as mesoporous silica nanoparticles (MSNs) for the delivery of bioactive compounds to cells and tumors.14–18 MSNs have a high loading capacity and can be functionalized internally and on the external surface.19 To prevent premature release from these mesoporous systems, different strategies including covalent attachment of the cargo inside the mesopore or complete capping of the whole particle have been developed.20–26 The release mechanisms include changes in reduction potential,27,28 pH,20–22,29–31 temperature,32–34 and irradiation with light.16,35–37Combining the high loading capacity and stability of mesoporous nanoparticles with the biocompatibility and tuneable properties of polymers offers the possibility to create stimuli-responsive and reversible delivery systems. Drug delivery vehicles based on nanoparticles in a size range of around 100 nm are likely to be taken up by cells via endocytosis.38 During this process, the endosome is being acidified by the action of proton pumps.39,40 Therefore, pH-responsive polymer coatings around mesoporous silica nanoparticles offer the possibility to use the changes in pH during endocytosis as trigger for controlled release. For instance, attachment of poly(N-isopropylacrylamide) (PNiPAM)34,41 poly(acrylic acid),42 chitosan,43,44 polyethylenimine (PEI),45 gelatin46 or poly(2-(diethylamino)ethyl methacrylate)47 amongst others48 showed the ability of pH-responsive functional polymer coatings around mesoporous silica nanoparticles for release applications. Liu et al. reported on different release kinetics from mesoporous silica by the use of poly(4-vinylpyridine) as pH-responsive shell created by multipoint anchoring.22 Poly(2-vinylpyridine) (PVP) has also been used for the pH-sensitive micropatterning of surfaces, due to the pronounced transition between hydrophilicity and hydrophobicity upon protonation and deprotonation.4
Here, we describe a multifunctional drug delivery vehicle that combines the high loading capacity of a mesoporous silica core with the environmental sensitivity of a pH-responsive polymer (poly(2-vinylpyridine). The highly promising feature of our system is the covalent attachment of the polymer which makes the system more stable and the opening mechanism presumably reversible. Additionally, the bi-functional polymer can be functionalized on its ends with various groups, e.g. polymers like poly(ethylene glycol) (PEG) to increase the enhanced permeability and retention (EPR) effect, targeting ligands or additional endosomal opening groups like photosensitizers. At pH values around 5.5 or higher, the polymer is deprotonated producing a hydrophobic state, which causes a collapse of the polymer onto the silica surface, thus preventing release of the cargo. After endocytic uptake of the particles by a cell, the endosomes are acidified and the change in pH can automatically lead to a switch of the particle into its open state. Thus cargo loaded into the mesopores is released into the endosomal compartment. For membrane-impermeable cargo, an additional release mechanism to open the endosome is necessary; therefore we also introduced a photosensitizer (AlPcS2a) in our system. To maintain colloidal dispersibility for these particles, even in the hydrophobic closed state, PEG blocks were attached to the ends of the PVP cap system. The use of α-, ω-bis-functionalized polymers offers the possibility for anchoring any functionality of interest (photosensitizer, targeting ligands) to the MSNs. With this multifunctional system we perform cellular uptake and targeting experiments and show the triggered release induced by pH-changes and photoactivation in living cells.
2. Experimental section
2.1. Chemicals
All reagents were purchased from commercial suppliers: tetraethyl orthosilicate (TEOS, Sigma-Aldrich, >98%), cetyltrimethylammonium chloride (CTAC, Fluka, 25 wt% in H2O), triethanolamine (TEA, Sigma-Aldrich, 98%), (3-aminopropyl)triethoxysilane (APTES, Fluka, 95%), (3-mercaptopropyl)-triethoxysilane (MPTES, Gelest, 95%), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, Sigma-Aldrich, 97%), ammonium nitrate (Sigma-Aldrich, 99%), conc. hydrochloric acid (Sigma-Aldrich, >95%, 37 wt%), α-amino-ω-carboxy terminated poly(2-vinylpyridine) (NH2-PVP-COOH, Polymer source, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, PDI = 1.08), α,ω-polyethyleneglycolbisamine (NH2-PEG-NH2, Sigma-Aldrich, Mn = 2000), Boc anhydride (Aldrich, 95%), folic acid (FA, Sigma, >97%), aluminium (lll) phthalocyanine chloride tetrasulfonic acid (AlPcS2a, Frontier Scientific), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS, Sigma-Aldrich, 98%), 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich, ≥98%), calcein (Sigma-Aldrich), fluorescein sodium salt (Sigma-Aldrich, BioReagent), dimethyl sulfoxide (DMSO, Sigma-Aldrich, >99.5%), ethanol (EtOH, Sigma-Aldrich, >99.5%), dichloromethane (anhydrous, Sigma-Aldrich, >99.5%), tetrahydrofuran (anhydrous, Sigma-Aldrich, ≥99.9%), trifluoroacetic acid (TFA, Acros Organics, 99%), oxalic acid dihydrate (Sigma-Aldrich, >99%), magnesium sulfate (MgSO4, anhydrous, Sigma-Aldrich, ≥99.5%), triethylamine (Sigma-Aldrich, ≥99%), saline-sodium citrate buffer concentrate (SSC-buffer (20×), Sigma-Aldrich), citric acid buffer solution (citric acid/HCl/NaCl buffer, pH2, Sigma-Aldrich), doxorubicin hydrochloride (Dox, Sigma-Aldrich, D1515, 98–102%).
000, PDI = 1.08), α,ω-polyethyleneglycolbisamine (NH2-PEG-NH2, Sigma-Aldrich, Mn = 2000), Boc anhydride (Aldrich, 95%), folic acid (FA, Sigma, >97%), aluminium (lll) phthalocyanine chloride tetrasulfonic acid (AlPcS2a, Frontier Scientific), N-hydroxysulfosuccinimide sodium salt (sulfo-NHS, Sigma-Aldrich, 98%), 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich, ≥98%), calcein (Sigma-Aldrich), fluorescein sodium salt (Sigma-Aldrich, BioReagent), dimethyl sulfoxide (DMSO, Sigma-Aldrich, >99.5%), ethanol (EtOH, Sigma-Aldrich, >99.5%), dichloromethane (anhydrous, Sigma-Aldrich, >99.5%), tetrahydrofuran (anhydrous, Sigma-Aldrich, ≥99.9%), trifluoroacetic acid (TFA, Acros Organics, 99%), oxalic acid dihydrate (Sigma-Aldrich, >99%), magnesium sulfate (MgSO4, anhydrous, Sigma-Aldrich, ≥99.5%), triethylamine (Sigma-Aldrich, ≥99%), saline-sodium citrate buffer concentrate (SSC-buffer (20×), Sigma-Aldrich), citric acid buffer solution (citric acid/HCl/NaCl buffer, pH2, Sigma-Aldrich), doxorubicin hydrochloride (Dox, Sigma-Aldrich, D1515, 98–102%).
All chemicals were used as received without further purification. Doubly distilled water from a Millipore system (Milli-Q Academic A10) was used for all synthesis and purification steps.
Wheat germ agglutinin (WGA) Alexa Fluor 488 conjugate (life technologies), Atto 633 maleimide (ATTO-TEC).
2.2. Materials synthesis
000 rpm (43146 rcf), redispersed in ethanol and heated under reflux at 90 °C in a solution of 8 mL concentrated HCl and 32 mL ethanol for 45 minutes. After centrifugation, the particles were re-dispersed in ethanol, resulting in a colloidal suspension.
000 rpm, 43146 rcf, 20 min) and washed 3 times by repeated centrifugation and redispersion in 30 mL THF.
000 rpm, 43146 rcf, 20 min) and redispersion in a mixture of 0.01 M HCl and EtOH.
000 rpm, 43146 rcf, 20 min) and redispersion in a mixture of 0.01 M HCl and EtOH.
000 rpm, 16873 rcf) with a mixture of EtOH–water 1:1. The washing step was repeated three times with 1 mL each.
:1) were partially reacted with folic acid (0.44 μg, 1 nmol). Subsequently EDC (0.5 μL, 2.8 μmol) and a catalytic amount of Sulfo-NHS (∼1 mg) were added. After 1 h, the sample was washed 3 time by subsequent centrifugation (4 min, 14000 rpm, 16873 rcf) and re-dispersion with a mixture of EtOH–water 1:1.
:1 20 μL of AlPcS2a (1 mg mL−1 in DMSO) was added. The resulting mixture was stirred at room temperature in the dark for 24 hours. The sample was washed extensively with DMSO–water 1:1 by repeated centrifugation (4 min, 14000 rpm, 16873 rcf) and re-dispersion until no photosensitizer could be detected in the supernatant.
2.3. Release experiments
000 g mol−1) and placed on top of the fluorescence cuvette, which was also filled with SSC buffer. The “release” of fluorescein out of the closed particles was monitored for 2 h (λex = 490 nm, λem = 512 nm). Subsequently, the cap was removed and the particles were centrifuged and redispersed in 200 μL citrate–phosphate buffer (pH 5), before being put into the cap of the release setup. The fluorescence cuvette was also filled with citrate–phosphate buffer (pH 5). After reassembling the release setup, the release of fluorescein was continuously measured for 10 h. For comparison of the obtained curves, calibration curves of fluorescein in SSC buffer (pH 7) and in citrate–phosphate buffer (pH 5) were recorded.
2.4. Characterization
All samples were investigated with a FEI Titan 80–300 operating at 300 kV with a high-angle annular dark field detector. A droplet of the diluted MSN solution in absolute ethanol was dried on a carbon-coated copper grid. Nitrogen sorption measurements were performed on a Quantachrome Instruments NOVA 4000e. All samples (15 mg each) were heated to 60 °C for 12 h in vacuum (10 mTorr) to outgas the samples before nitrogen sorption was measured at 77 K. Pore size and pore volume were calculated by a NLDFT equilibrium model of N2 on silica, based on the desorption branch of the isotherms. In order to remove the contribution of the interparticle textural porosity, pore volumes were calculated only up to a pore size of 8 nm. A BET model was applied in the range of 0.05–0.20 p/p0 to evaluate the specific surface area of the samples. Centrifugation was performed using a Sorvall Evolution RC equipped with a SS-34 rotor or an Eppendorf centrifuge 5418 for small volumes. Raman spectra were recorded on a Jobin Yvon Horiba HR800 UV Raman microscope using a He–Ne laser emitting at λ = 633 nm. Dynamic light scattering (DLS) measurements were performed on a Malvern Zetasizer-Nano instrument equipped with a 4 mW He–Ne laser (633 nm) and an avalanche photodiode. The hydrodynamic radius of the particles was determined by dynamic light scattering in ethanolic suspension. For this purpose, 100 μL of an ethanolic suspension of MSN (ca. 10 mg mL−1) was diluted with 3 mL of ethanol prior to the measurement. Zeta potential measurements of the samples were performed on a Malvern Zetasizer-Nano instrument equipped with a 4 mW He–Ne laser (633 nm) and an avalanche photodiode. Zeta potential measurements were performed using the add-on Zetasizer titration system (MPT-2) based on diluted NaOH and HCl as titrants. For this purpose, 1 mg of the particles was diluted in 10 mL bi-distilled water. IR measurements were performed on a Bruker Equinox 55 FTIR/FTNIR Spectrometer in absorbance mode (spectra were background substracted). UV-VIS spectra were recorded with a NanoDrop ND 1000 spectrometer. Usually, 2 μL of sample were used and all presented spectra are background corrected for water absorption.2.5. Cell culture and in vitro experiments
:1) (life technologies) with Glutamax I medium supplemented with 10% FBS. Tubulin-GFP expressing HuH7 cells were grown in Dulbecco's modified Eagle's medium–F12 (1:1) supplemented with 10% FBS. All cells were cultured at 37 °C in a 5% CO2 humidified atmosphere. KB cells were seeded on ibidiTreat μ-Slides (IBIDI). HeLa and tubulin green fluorescent protein (GFP) expressing HuH7 cells were seeded on collagen A – coated LabTek chambered cover glass (Nunc). For live cell imaging the cells were seeded 48 or 24 h before measuring, at a cell density of 1 × 104 or 2 × 104 cells cm−2, respectively.
3. Results and discussion
3.1. Synthesis and particle characterization
Colloidal core–shell mesoporous silica nanoparticles (MSNs) with amino-moieties on the shell were synthesized via a delayed co-condensation approach according to a published synthesis procedure with CTAC as structure directing agent and TEOS as silica source.19 The outer surface of the template-free MSNs was subsequently modified with bi-functional poly(2-vinylpyridine) (PVP) via an EDC-assisted amidation reaction (cf. Experimental section, Fig. 1 and ESI Fig. S1‡). The bi-functionality of the polymer allows the subsequent attachment of PEG, fluorescent dyes and targeting ligands. | ||
| Fig. 1 Concept of the pH-responsive delivery system. The pores can be reversibly opened and closed through changes in the water solubility of the polymer. | ||
According to transmission electron microscopy (TEM), the shell functionalized mesoporous silica nanoparticles show an average particle diameter of 90 nm with a wormlike pore structure (Fig. S13a‡). The stepwise functionalization with poly(2-vinylpyridine) and PEG was monitored with several methods (dynamic light scattering (DLS), zeta potential, and nitrogen adsorption, Fig. 2A–C, Fig. S4‡) and is discussed in the following (further spectroscopic details, IR and Raman spectra, in ESI Fig. S2 and S3‡).
 | ||
| Fig. 2 Characterization of the functionalized MSN: (A) dynamic light scattering data of MSN-NH2 (black), MSN-PVP-NH2 (red) and MSN-PVP-PEG-NH2 (blue), (B) titration experiments showing the zeta potential of MSN-PVP (black), and dynamic light scattering of MSN-PVP (red) and MSN-PVP-PEG (blue), respectively, (C) nitrogen physisorption measurements of MSN-NH2 (black), MSN-PVP in the open state of the polymer (red) and MSN-PVP-closed (blue). | ||
DLS measurements revealed the need for PEG as stabilizing agent. The hydrodynamic diameter of amino shell-functionalized MSN in aqueous media reveals an average particle size of 160 nm (Fig. 2A, black curve), whereas the polymer-modified sample MSN-PVP-NH2 (red curve) shows strong aggregation in aqueous media due to their hydrophobicity at pH 7, indicated by an apparent size increase to 550 nm. However, subsequent PEGylation of PVP recovers the colloidal nature of the system, which is indicated by a decrease of the average apparent particle size (blue curve). Additionally, zeta potential titration experiments of the samples MSN-NH2, MSN-PVP-NH2, MSN-PVP-COOH and MSN-PVP-PEG-NH2 were carried out to prove the functionalization of the synthesized inorganic–organic hybrid material (Fig. 2B and Fig. S4‡). The high zeta potential (+50 mV) of MSN-PVP at low pH reflects the polymer cap system in its fully protonated and thus open state. At pH = 5.5, close to the pKa value of protonated pyridine, deprotonation of the polymer cap system occurs, leading to a significant drop in zeta potential with increasing pH. Together with the zeta potential, DLS measurements were performed simultaneously at the same pH values. For MSN-PVP the particles showed good colloidal dispersability as long as the polymer remained in its protonated state (Fig. 2B). Upon deprotonation, the polymer collapses at the particle surface, forming a hydrophobic shell around the particles, which is revealed in the increase in size due to agglomeration. In contrast, attachment of PEG blocks to the PVP-functionalized particles led to the suppression of aggregation, as no changes in size were observed over the whole pH-range for MSN-PVP-PEG. In order to show the ability of poly(2-vinylpyridine) to act as a pH responsive cap system, nitrogen sorption measurements were performed (Fig. 2D). To observe the open form (MSN-PVPopen), the particles where dried from a 0.01 M HCl dispersion at 60 °C; this was expected to keep the protonated PVP functionalized MSN in an open state. The closed state could be measured after drying from a dispersion in Millipore water (pH 7). In the open state, the mesopores are highly accessible, whereas the polymer completely blocks the pores in the closed state, even for very small molecules like nitrogen. The BET surface area of 51 m2 g−1 in the closed state is in very good agreement with an estimated external BET surface area of 45 m2 g−1 of spheres with similar density and diameter. The BET surface area of 51 m2 g−1 for MSN-PVPclosed corresponds to a reduction in surface area of 95% compared with MSNs without polymer cap system (MSN-NH2), indicating that the surface of the pore walls after blocking is not even accessible for nitrogen. The key features of the different MSNs are summarized in Table 1.
| Sample | BET surface area [m2 g−1] | NLDFT pore size [nm] | NLDFT pore volume [cm3 g−1] |
|---|---|---|---|
| MSN-NH2 | 1097 | 3.78 | 0.79 |
| MSN-PVPopen | 617 | 3.42 | 0.36 |
| MSN-PVPclosed | 51 | — | — |
The decrease of the specific surface area in the sample MSN-PVPopen can be explained by the addition of non-porous PVP and the blocking of some pores by frozen polymer on the surface layer of the MSNs. This effect is comparably large due to the length of the PVP.
3.2. Targeting
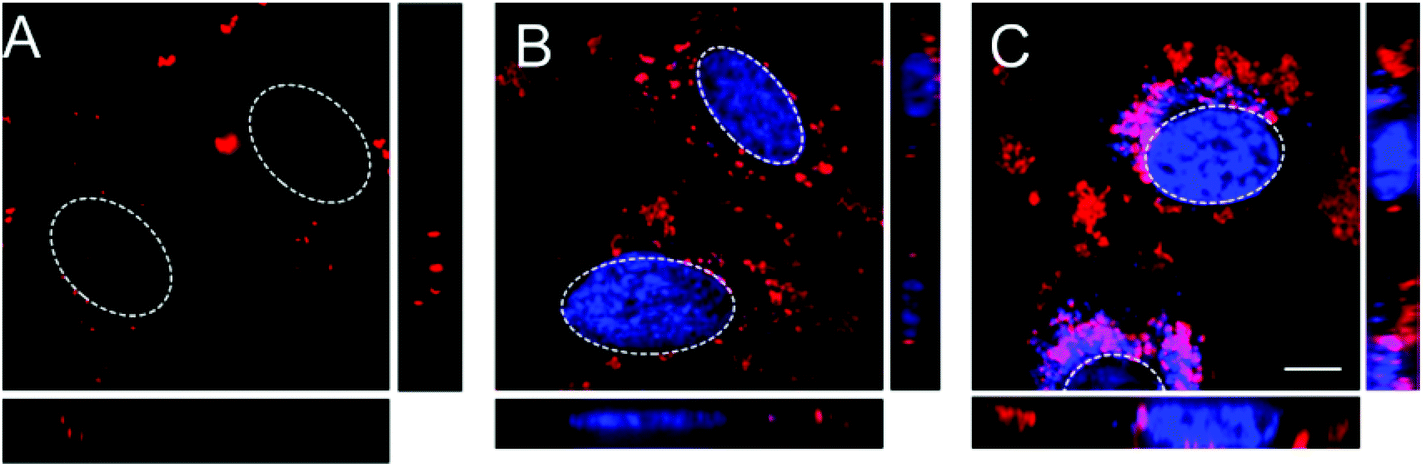
To design an effective drug delivery system, a targeting system is highly desirable. Generally one can distinguish between passive and active targeting. Modifications such as attaching PEG at the surface of the delivery system provide shielding against undesired interactions, and therefore favor the enhanced permeability and retention effect (passive targeting). Active targeting, on the other hand, takes advantage of the overexpression of certain receptors on diseased cells by attachment of the corresponding targeting ligand to the delivery vehicle.49 Accordingly, active targeting was shown in vitro using the example of folic acid (FA) receptor targeting on KB-cells.The attachment of active targeting ligands is important for creating a functional drug delivery system.49 Folic acid was chosen as targeting ligand because it is an ideal model system for targeting with a small molecule and is also believed to be a promising candidate for clinical applications.50 Furthermore, the covalent binding of folic acid to the particles can be achieved with an EDC-mediated coupling and could be easily substituted by other targeting ligands without changing the coupling chemistry. To demonstrate the functionality of the particles, they were incubated with KB-cells, which are known to overexpress FA-receptors.50 In a competition experiment, with either free folic acid to block the FA-receptors or with uninhibited FA-receptors, the uptake of MSN-PVP-PEG-NH2-FA particles and hence the targeting abilities were monitored. For the visualization of nanoparticle-uptake, z-stacks of the cells were recorded with a spinning disk confocal fluorescence microscope, which allowed us to determine MSNs inside or outside the cells after staining the cell membrane.
In Fig. 3, typical confocal cuts through the cells together with the orthogonal projection are shown. In the case of pre-incubation with free folic acid, only a few individual particles are present inside the cells (Fig. 3A), and also in the full stacks the quasi-absence of nanoparticles is noted, independent of the chosen focal plane. In contrast, incubation of MSN-PVP-PEG-NH2-FA on KB-cells without pre-incubation of FA resulted in significant uptake of particles (Fig. 3B). The particles appear in the representative z-slice as well as in the entire height of the cell as shown in the orthogonal views.
 | ||
| Fig. 3 Unspecific and receptor-mediated uptake of MSN-PVP-PEG-NH2-FA particles by KB cells. (A) Incubation of MSN-PVP-PEG-NH2-FA particles with FA pre-incubated KB cells for 2 h at 37 °C. (B) Incubation of MSN-PVP-PEG-NH2-FA particles with KB cells, no free FA in cell culture medium, for 2 h at 37 °C. The KB cell membrane is shown in green (dye WGA 488) and the particles in red (dye Atto 633). Orthogonal views are given to illustrate the uptake more clearly. The scale bars represent 10 μm. | ||
In the case of blocked receptors, only unspecific uptake is expected to occur, therefore the results demonstrate the specific folate-receptor mediated endocytosis within two hours with only a minor rate of unspecific uptake.
3.3. Release experiments
To verify the release properties of the particle-system, several experiments were performed. First, the pH dependency was tested in vial, in a cuvette system, at pH 7 and pH 5 (Fig. 4). In a second approach, the multifunctional MSNs were tested in vitro. For these experiments, membrane-permeable (DAPI, colchicine, doxorubicin) and impermeable (calcein) dyes/drugs were adsorbed in the mesopores of the MSNs and the cargo-loaded nanoparticles were incubated with cells to prove the functionality of the system on a cellular level. Membrane-impermeable cargos cannot escape from the endosomal compartments, therefore a photosensitizer (AlPcS2a) was covalently attached to the outer surface of the sample MSN-PVP-PEG.16 The pH-responsive opening of the polymer cap system was demonstrated by time-based fluorescence release experiments with our custom-made release setup.51,52 For this purpose, a distinct amount of fluorescein-loaded MSN-PVP-NH2 (Fig. 4, blue curve, or MSN-PVP-PEG-NH2 (red), respectively) was examined in the closed state of the polymer for 2 h before the sample was acidified, allowing a subsequent opening of the pores and the diffusion of cargo molecules. | ||
| Fig. 4 Time based fluorescence release experiment of MSN-PVP-NH2 (blue) and MSN-PVP-PEG-NH2 (red) with fluorescein as cargo. Only after change (from pH 7) to acidic pH (PH 5) a significant cargo release can be detected. | ||
All intensity values are normalized to the highest obtained fluorescent counts of the respective sample. Therefore, the highest amount released is set to 100% relative release. The pH-dependent release curves show the striking capping effect of the poly(2-vinylpyridine) cap system. At pH 7, the polymer is in its hydrophobic state and thus successfully blocks the pores of the MSN until the release is triggered by decreasing the pH. In acidic media, the polymer changes into its water soluble hydrophilic state through protonation, which allows the cargo to diffuse out of the pores. No significant changes in release kinetics are observed by further attachment of PEG compared with sample MSN-PVP (Fig. 4) The total amount of cargo released from the MSN could be calculated to 21 μg dye per mg MSN. Long-term release experiments showed that even after 16 h, the premature release is less than 4% (see ESI Fig. S6‡).
The controlled release of cargo inside the desired tissue is one of the greatest challenges in drug delivery. Many systems have been developed and characterized, including fusion, redox-sensitive systems and photoactivation.40 Some of these systems exhibit already a good temporal and spatial control of the cargo-release. But once they are activated, the release of the cargo is continuous, even if the particles leave the tissue of interest. One advantage of our newly developed system is the reversibility of the pore opening. If the pH of the environment turns neutral again, the PVP capping system should be able to close the pores again and the delivery system can safely circulate inside the blood system.
To safely deliver membrane-permeable cargos like DAPI,53 doxorubicin or colchicine54 inside cells with our newly developed system, the implementation of additional endosomal escape mechanisms is not necessary. It is anticipated that the particles will be taken up in endosomal compartments, and in the following the endosomes get acidified. This drop in pH from around 7 to approximately 5 results in the solvation of poly(2-vinyl pyridine) and thus in release of the cargo from the pores into the endosome, and consequently into the cytosol if it diffuses through the endosomal membrane. On the other hand, if the particles get exocytosed the pH will increase and poly(2-vinyl pyridine) will turn insoluble again. In this case the cargo will stay inside the particles and the latter can circulate again and possibly find a new target cell. DAPI and colchicine were used as model systems to examine the delivery of membrane permeable cargo. DAPI preferentially stains dsDNA and thereby produces a fluorescence enhancement of around 20-fold.55
Fig. 5 shows the staining of the nucleus after several time intervals. Shortly after incubation only a weak staining of the nucleus can be seen, after 15 h the nucleus is clearly visible and after 46 h even the region of the endoplasmic reticulum gets stained. Because of the slowly increasing and very strong DAPI-fluorescence signal after several hours of incubation, it could be excluded that the staining is due to free DAPI in the particle solution. In the case of free DAPI in the solution, the staining would be expected to be detectable shortly after addition, as the typical incubation time for nucleus staining with free DAPI in solution is only between 1–5 minutes.
 | ||
| Fig. 5 Fluorescence microscopy of HeLa cells incubated with DAPI-loaded MSN-PVP-PEG. MSN-PVP-PEG loaded with DAPI (blue) and labeled with Atto 633 (red) after (A) 15 min (B) 15 h and (C) 46 h incubation with the cells. The nucleus region is indicated with dashed white lines. The scale bar represents 10 μm. | ||
The second cargo colchicine hinders the polymerization of microtubuli as it binds to free tubulin.54 It is blocking the formation of the mitotic spindle but not the mitosis itself. For this study stable GFP-transfected liver carcinoma cells (HuH7) were used, such that GFP is attached to the tubulin network. In Fig. S12a‡ a picture of untreated HuH7 cells with GFP tagged on the tubulin network is depicted; one can clearly see the fibrous tubulin network. As shown in Fig. S12b,‡ MSN-PVP-PEG particles loaded with colchicine were incubated for 18 h, and as clearly seen, the fibrous structure has vanished completely, because the formation of the tubulin network is hindered.
The delivery behavior of cell-membrane impermeable cargos was investigated in a second step. Due to the acidic pH inside late endosomes or lysosomes,56 cargo encapsulated inside the PVP-capped MSN should be able to diffuse out of the mesopores after reaching these compartments. The inability of calcein to cross endosomal membranes was demonstrated with calcein-loaded MSN-PVP-PEG particles by incubating them for several hours on HeLa cells (cf. ESI Fig. S9‡). As supposed, the calcein fluorescence stayed co-localized with the dye-labeled particles, which demonstrates that the decrease in pH is not able to induce endosomal escape. Therefore the system had to be further enhanced for the induction of endosomal escape.
Thus the photosensitizer AlPcS2a (PS) was covalently linked to the PEG-chains, in addition to the pH-sensitive polymer on the particle surface. Endosomal escape was expected to occur after acidification of the endosomes and activation of the photosensitizer with red laser light. After activation with red laser light (639 nm, 1.2 W mm−2) singlet oxygen is produced, which can destroy the endosomal membrane. We emphasize that the lifetime of the singlet oxygen is very short (around 2 × 10−7 s57), thus its diffusion range is limited and therefore the cargo and also the cell membrane should stay intact, due to the spatial separation of cargo, cell membrane and PS. Moreover, the red light used for activation of the photosensitizer is in a useful region for medical applications and is already used in photodynamic therapy.58 Previous studies from our groups have already established “surgical” endosomal release using covalently attached photosensitizers. It was demonstrated that the activation of singlet oxygen only influences endosomes containing MSN with covalently bound photosensitizer, while the movement along microtubules for non-particle containing endosomes was unaffected.59
For the photoactivated release experiment, MSN-PVP-PEG-AlPcS2a/FA particles were incubated on HeLa cells for 18 h. As seen in Fig. 6, calcein and PS are co-localized (yellow spots, Fig. 6C). Directly after 1 min photoactivation with 1.2 W mm−2 of 639 nm light, calcein-spreading over the whole cell can be observed, and the calcein-fluorescence gets brighter over time due to de-quenching of the dye upon escape from the MSN host (Fig. 6E, I, M). In comparison to the emission from calcein, the fluorescence of the photosensitizer stays localized in the same dot-like pattern as prior to the photoactivation, only the intensity is reduced because of photo-bleaching effects after the activation (Fig. 6F, J and N). The intensity profile along the white line, indicated in the merged pictures, depicts the fluorescence increase due to calcein more clearly (Fig. 6D, H, L and P). The cargo release can also be visualized with a constant PS-activation and simultaneous observation of calcein fluorescence (over a period of 8 min; Fig. S10‡). Strikingly, a significant part of the cargo appears to be released inside the cell already within two minutes.
 | ||
| Fig. 6 Fluorescence microscopy of MSN-PVP-PEG-AlPcS2a-FA nanoparticles loaded with calcein after 18 h incubation on a HeLa cell. (A–C) Calcein (green) and AlPcS2a (red) are co-localized (yellow) prior to photoactivation. (D) Intensity profile along the white line in the merged image. The red line indicates photoactivation with 1.2 W mm−2 of red light (639 nm). (E–H) After 1 min photoactivation, (I–L) 5 min after photoactivation, (M–P) 10 min after photoactivation. The scale bar represents 10 μm. | ||
The above results were compared to those of incubation experiments of cells with a solution of free calcein and PS. As seen in Fig. S11,‡ the amount of internalized calcein is negligible. We also observe that the free PS is spreading over the entire cell. If the PS is not covalently bound to an MSN, the molecules can diffuse inside the whole cell and can cause much more damage to the cell membrane and other organelles due to their greater proximity.
The release of a pharmacologically relevant cargo was also investigated. For this purpose we performed MTT assays of bladder carcinoma T24 cells with MSN-PVP-PEG-NH2, uncoated MSN, MSN-PVP-PEG-NH2 loaded with doxorubicin and free doxorubicin for 24 and 48 hours. The concentration of particles was calculated on the basis of loaded doxorubicin, such that at the end similar amounts of doxorubicin were delivered. As compared with free doxorubicin, the cell killing kinetics by the doxorubicin loaded MSN particle was delayed (Fig. 7). After 24 hours the cell viability was 54.5 ± 2.8% for free doxorubicin, but 70.9 ± 4.7% for the doxorubicin loaded particles (MSN-NH2: 99.3 ± 7.9%; MSN-PVP-PEG-NH2: 77.6 ± 5.6%). However, after 48 hours the cell viability for doxorubicin-loaded particles had decreased to 39.1 ± 1.6% (Dox: 31.0 ± 2.4%; MSN-NH2: 97.0 ± 10.8%; MSN-PVP-PEG-NH2: 79.7 ± 7.6%). Apparently our MSN-based system needs time to open up the pores in the acidic endolysosomal compartments and slowly releases the drug over time. Variations in the dox-loading and release kinetics of MSN-PVP-PEG-NH2 can lead to a slight differences in the cell viability compared to free dox after 48 h. Nevertheless a comparable reduction in cell viability can be observed for free Dox and the Dox loaded particles.
 | ||
| Fig. 7 MTT assay of different MSNs. Unfunctionalized (MSN-NH2), MSN-PVP-PEG-NH2 loaded with doxorubicin and unloaded as well as free doxorubicin (Dox) were incubated on T24 cells for 24 and 48 hours. The particle concentration was calculated regarding doxorubicin concentration per well. The viability was calculated as percentage of wells treated with HBG. The statistical significance was determined by ANOVA followed by Bonferroni posttests (***, p < 0.01; ns, p > 5, no statistical significance). | ||
3.4. Stability tests of the nanoparticles
Previous studies on the effect of functionalization of MSN on their bio-degradation demonstrated the fast degradation of pure, unprotected MSN in simulated body fluids.11,60 The attachment of poly(ethylene glycol) significantly increased the stability up to several days, whereas the un- or organo-silane functionalized MSN lost their porosity in the first hours of incubation. During the partial hydrolysis and dissolution of the silica material, hydroxyapatite layers precipitated on the surface of the silica by adsorption of Ca2+ ions in the porous system and via hydrogen bonding of PO43− ions.11,60 The functionalization with PEG already enhanced the stability of the MSNs, but the PEG-layer does not close the pores of the MSNs. In contrast, in the case of PVP-functionalization the particles are completely protected from the environment as long as they stay in neutral or basic milieu. The stability of the MSN was tested (i) in bio-fluids and (ii) for storage purposes in order to estimate shelf life. Biostability assays were performed to test whether the polymer cap system will prevent the mesoporous silica nanoparticles from degradation. This was done using Dulbecco's Modified Eagle's Medium (DMEM) at 37 °C at two different pH values (5 and 7). As a widely used basal medium for growing many mammalian cells, DMEM contains various amino acids, vitamins and inorganic salts, amongst others and is used here to simulate a biological environment. Samples were removed after specific times, washed and dried. The nanoparticles were then characterized with transmission electron microscopy and nitrogen sorption. TEM images show significant degradation of the nanoparticle structure for unfunctionalized MSN after 24 h immersion at pH 7 (see ESI Fig. S12b‡), which is in good agreement with results of similar experiments reported earlier.11 In contrast, nanoparticles functionalized with PVP offer very good stability in DMEM at pH 7. At this pH, the polymer cap system forms a tight, hydrophobic, and therefore impermeable envelope for water, proteins and salts around the MSN. Additionally, the polymer shell improves the stability of the MSN in DMEM at pH 5, where the cap system is in its open state. In comparison with uncoated nanoparticles, MSN-PVP particles show less degradation, which was also confirmed by nitrogen sorption experiments (Fig. S14 and S15‡). In both cases, in the open and the closed state of the cap system, no changes in the pore size distribution were observed, indicating that the pores are not affected by the medium.In order to test the stability of the MSN under storage conditions, the functionality of 4-week-old particles (stored at 4 °C) could be verified with in vitro release experiments (Fig. S16‡). After storage in water (and therefore in a closed state of the PVP) the particles retain the same calcein release behavior in cells as already demonstrated in Fig. 5.
Drying and re-dispersion of MSN-PVP-PEG-NH2 in water exhibits no significant agglomeration of the particles or any other visible change of their solution behaviour (DLS measurement in Fig. S17‡). This remarkable behavior is exceptional for mesoporous silica nanoparticles; the common response to drying is serious agglomeration that cannot even be broken with ultrasound. Based on the redispersibility of the nanoparticles after drying, a long shelf life of the nanoparticles is expected.
4. Outlook
In this work, we have developed a multifunctional drug delivery vehicle based on polymer-functionalized mesoporous silica nanoparticles. This system combines attractive features such as the high loading capacity of MSNs with the tuneable functionality of a polymer. The use of bis-functionalized polymer blocks allowed us to covalently attach step-by-step many functionalities of interest. We demonstrate that covalently bound poly(2-vinylpyridine) in combination with a PEG-block acts as a pH-responsive cap system for MSN. With this system, we show the successful delivery of different cargos into cells. Membrane permeable cargos were delivered without any external trigger, because such cargos could penetrate the endosomal membrane. Covalent attachment of a red-light-photosensitizer offered the possibility to open the endosome through irradiation with laser light, and to deliver membrane-impermeable cargos. Effective targeting of folic acid receptor expressing cells was enabled by the covalent attachment of folic acid. Stability tests revealed the effectiveness of poly(2-vinylpyridine) not only as cap system, but also as a highly effective protecting agent for MSN against aggressive body fluids. Strikingly, the polymer-coated nanoparticles can even be dried and then re-dispersed in water without causing agglomeration, which bodes well for a long shelf life of the dried particles. Preliminary biodistribution experiments of MSN-PVP-PEG nanoparticles in vivo revealed no particle agglomeration and showed preferred liver accumulation. The modified and unmodified MSN were well tolerated in mice without causing visible side effects, which was also confirmed by blood parameter and histopathological examination (cf. ESI‡). With this new system we have developed a multifunctional toolbox based on MSNs that is able to open and close its cap system depending on the environment. One major advantage of our system derives from the covalent attachment of all components, which allows the system to operate for several cycles. We anticipate that the possible integration of almost any functionality of interest as well as the efficient synthesis make this system promising for wide-ranging biological and medical applications, especially in cancer therapy.Acknowledgements
The authors acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) in the context of SFB 749 and the Excellence Clusters Nanosystems Initiative Munich (NIM) and Center for Integrated Protein Science Munich (CIPSM). S. N. thanks the “Verein der Chemischen Industrie” (VCI) for his Kekulé grant. S. D. thanks the “Römer Stiftung” for financial support. We thank Dr Bastian Rühle for 3D graphics design.References
- E. Brewer, J. Coleman and A. Lowman, J. Nanomater., 2011, 1–10 CrossRef PubMed.
- J. Y. Shu, B. Panganiban and T. Xu, Annu. Rev. Phys. Chem., 2013, 64, 631–657 CrossRef CAS PubMed.
- G. W. de Groot, M. G. Santonicola, K. Sugihara, T. Zambelli, E. Reimhult, J. Vörös and G. J. Vancso, ACS Appl. Mater. Interfaces, 2013, 5, 1400–1407 CAS.
- A. Synytska, M. Stamm, S. Diez and L. Ionov, Langmuir, 2007, 23, 5205–5209 CrossRef CAS PubMed.
- J.-i. Hotta, H. Uji-i and J. Hofkens, Opt. Express, 2006, 14, 6273–6278 CrossRef CAS.
- M. Meyer, C. Dohmen, A. Philipp, D. Kiener, G. Maiwald, C. Scheu, M. Ogris and E. Wagner, Mol. Pharm., 2009, 6, 752–762 CrossRef CAS PubMed.
- D. Edinger and E. Wagner, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 33–46 CrossRef CAS PubMed.
- A. Vetter, K. S. Virdi, S. Espenlaub, W. Rödl, E. Wagner, P. S. Holm, C. Scheu, F. Kreppel, C. Spitzweg and M. Ogris, Mol. Pharm., 2013, 10, 606–618 CrossRef CAS PubMed.
- C. Minard-Basquin, T. Weil, A. Hohner, J. O. Rädler and K. Müllen, J. Am. Chem. Soc., 2003, 125, 5832–5838 CrossRef CAS PubMed.
- S. Arpicco, B. Stella, O. Schiavon, P. Milla, D. Zonari and L. Cattel, Int. J. Pharm., 2013, 454, 653–659 CrossRef CAS PubMed.
- V. Cauda, C. Argyo and T. Bein, J. Mater. Chem., 2010, 20, 8693–8699 RSC.
- Z. Zhou, X. Ma, E. Jin, J. Tang, M. Sui, Y. Shen, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Biomaterials, 2013, 34, 5722–5735 CrossRef CAS PubMed.
- S. Kim, J.-H. Kim, O. Jeon, I. C. Kwon and K. Park, Eur. J. Pharm. Biopharm., 2009, 71, 420–430 CrossRef CAS PubMed.
- K. K. Coti, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS PubMed.
- S. A. Mackowiak, A. Schmidt, V. Weiss, C. Argyo, C. von Schirnding, T. Bein and C. Bräuchle, Nano Lett., 2013, 13, 2576–2583 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- F. Zhang, E. Lees, F. Amin, P. Rivera_Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- A. Schlossbauer, C. Dohmen, D. Schaffert, E. Wagner and T. Bein, Angew. Chem., Int. Ed., 2011, 50, 6828–6830 CrossRef CAS PubMed.
- A. Schlossbauer, C. Dohmen, D. Schaffert, E. Wagner and T. Bein, Angew. Chem., Int. Ed., 2011, 123, 6960–6962 CrossRef PubMed.
- R. Liu, P. Liao, J. Liu and P. Feng, Langmuir, 2011, 27, 3095–3099 CrossRef CAS PubMed.
- D. Tarn, M. Xue and J. I. Zink, Inorg. Chem., 2013, 52, 2044–2049 CrossRef CAS PubMed.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- V. Cauda, H. Engelke, A. Sauer, D. Arcizet, C. Bräuchle, J. Rädler and T. Bein, Nano Lett., 2010, 10, 2484–2492 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- I. I. Slowing, J. L. Vivero-Escoto, C.-W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- J. M. Rosenholm, E. Peuhu, L. T. Bate-Eya, J. E. Eriksson, C. Sahlgren and M. Lindén, Small, 2010, 6, 1234–1241 CrossRef CAS PubMed.
- S.-H. Cheng, W.-N. Liao, L.-M. Chen and C.-H. Lee, J. Mater. Chem., 2011, 21, 7130–7137 RSC.
- R. Liu, Y. Zhang, X. Zhao, A. Agarwal, L. J. Mueller and P. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS PubMed.
- C. Wang, Z. Li, D. Cao, Y.-L. Zhao, J. W. Gaines, O. A. Bozdemir, M. W. Ambrogio, M. Frasconi, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Angew. Chem., Int. Ed., 2012, 51, 5460–5465 CrossRef CAS PubMed.
- J. Croissant and J. I. Zink, J. Am. Chem. Soc., 2012, 134, 7628–7631 CrossRef CAS PubMed.
- Y.-Z. You, K. K. Kalebaila, S. L. Brock and D. Oupický, Chem. Mater., 2008, 20, 3354–3359 CrossRef CAS.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. C. Wang, G. Zou and X.-J. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- C. A. Strassert, M. Otter, R. Q. Albuquerque, A. Höne, Y. Vida, B. Maier and L. De Cola, Angew. Chem., Int. Ed., 2009, 48, 7928–7931 CrossRef CAS PubMed.
- C. A. Strassert, M. Otter, R. Q. Albuquerque, A. Höne, Y. Vida, B. Maier and L. De Cola, Angew. Chem., Int. Ed., 2009, 121, 8070–8073 CrossRef PubMed.
- O. Harush-Frenkel, N. Debotton, S. Benita and Y. Altschuler, Biochem. Biophys. Res. Commun., 2007, 353, 26–32 CrossRef CAS PubMed.
- M. Lakadamyali, M. J. Rust, H. P. Babcock and X. Zhuang, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9280–9285 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- Y. Chen, W. Yang, B. Chang, H. Hu, X. Fang and X. Sha, Eur. J. Pharm. Biopharm., 2013, 85, 406–412 CrossRef CAS PubMed.
- C.-Y. Hong, X. Li and C.-Y. Pan, J. Mater. Chem., 2009, 19, 5155–5160 RSC.
- A. Popat, J. Liu, G. Q. Lu and S. Z. Qiao, J. Mater. Chem., 2012, 22, 11173–11178 RSC.
- F. Chen and Y. Zhu, Microporous Mesoporous Mater., 2012, 150, 83–89 CrossRef CAS PubMed.
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS PubMed.
- Z. Zou, D. He, X. He, K. Wang, X. Yang, Z. Qing and Q. Zhou, Langmuir, 2013, 29, 12804–12810 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CAS.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2013, 26, 435–451 CrossRef.
- N. T. Huynh, E. Roger, N. Lautram, J.-P. Benoît and C. Passirani, Nanomedicine, 2010, 5, 1415–1433 CrossRef CAS PubMed.
- M. A. Salazar and M. Ratnam, Cancer Metastasis Rev., 2007, 26, 141–152 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 121, 3138–3141 CrossRef PubMed.
- D. Zink, N. Sadoni and E. Stelzer, Methods, 2003, 29, 42–50 CrossRef CAS.
- M. E. Lalande, V. Ling and R. G. Miller, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 363–367 CrossRef CAS.
- J. Kapuscinski, J. Histochem. Cytochem., 1990, 38, 1323–1329 CrossRef CAS PubMed.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- K. I. Salokhiddinov, I. M. Byteva and G. P. Gurinovich, J. Appl. Spectrosc., 1981, 34, 561–564 CrossRef.
- N. Brasseur, R. Ouellet, C. La Madeleine and J. E. van Lier, Br. J. Cancer, 1999, 80, 1533–1541 CrossRef CAS PubMed.
- A. Schloßbauer, A. M. Sauer, V. Cauda, A. Schmidt, H. Engelke, U. Rothbauer, K. Zolghadr, H. Leonhardt, C. Bräuchle and T. Bein, Adv. Healthcare Mater., 2012, 1, 316–320 CrossRef PubMed.
- V. Cauda, A. Schlossbauer and T. Bein, Microporous Mesoporous Mater., 2010, 132, 60–71 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Dr Klaus Römer on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Materials and methods used for the preparation of the nanoparticles, transmission electron microscopy, dynamic light scattering, nitrogen sorption isotherms, IR and Raman spectroscopy, in situ release experiments, UV-VIS spectroscopy, fluorescence microscopy and in vivo experiments, Fig. S1–S18 including supplementing text. See DOI: 10.1039/c4nr07245f |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |