
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional graphene-based composites for energy applications
Shun
Mao†
a,
Ganhua
Lu†
 b and
Junhong
Chen
*a
b and
Junhong
Chen
*a
aDepartment of Mechanical Engineering, University of Wisconsin-Milwaukee, 3200 North Cramer Street, Milwaukee, Wisconsin 53211, USA. E-mail: jhchen@uwm.edu; Fax: +1-414-2296958; Tel: +1-414-2292615
bDepartment of Mechanical Engineering, University of Alaska Anchorage, 3100 Providence Drive, Anchorage, Alaska 99508, USA
First published on 26th December 2014
Abstract
Three-dimensional (3D) graphene-based composites have drawn increasing attention for energy applications due to their unique structures and properties. By combining the merits of 3D graphene (3DG), e.g., a porous and interconnected network, a high electrical conductivity, a large accessible surface area, and excellent mechanical strength and thermal stability, with the high chemical/electrochemical activities of active materials, 3DG-based composites show great promise as high-performance electrode materials in various energy devices. This article reviews recent progress in 3DG-based composites and their applications in energy storage/conversion devices, i.e., supercapacitors, lithium-ion batteries, dye-sensitized solar cells, and fuel cells.
 Shun Mao | Shun Mao received his Ph.D. in Mechanical Engineering from the University of Wisconsin-Milwaukee in 2010 for the study of hybrid nanomaterials for biosensing applications. He is currently a research associate at the University of Wisconsin-Milwaukee. His research focuses on hybrid 2D/0D nanomaterials for environmental and energy applications. |
 Ganhua Lu | Ganhua Lu is an Assistant Professor of Mechanical Engineering at the University of Alaska Anchorage. His research is focused on the synthesis of hybrid nanostructures consisting of nanocarbons (i.e., graphene and CNTs) decorated with functional nanocrystals and their use for environmental and energy devices. |
 Junhong Chen | Junhong Chen is a Professor of Mechanical Engineering, a Professor of Materials Science and Engineering at the University of Wisconsin-Milwaukee, and a Fellow of the American Society for Mechanical Engineers (ASME). He is also the Director of the Industry-University Cooperative Research Center (I/UCRC) on Water Equipment and Policy, supported by the US National Science Foundation (NSF) and water-based industrial partners. His research interests lie in nanocrystal synthesis and assembly; nanocarbons (i.e., graphene and CNTs) and hybrid nanomaterials; nanostructure-based gas sensors, water sensors, and biosensors; and nanocarbon-based hybrid nanomaterials for sustainable energy and environment (http://www.uwm.edu/nsee/). |
1. Introduction
Graphene is a crystalline allotrope of carbon with a two-dimensional (2D) structure, in which carbon atoms are densely packed in a regular sp2-bonded atomic-scale hexagonal pattern.1,2 Due to its unique structure (atomic thick carbon layer) and properties, e.g., a remarkably high electron mobility at room temperature, a high specific surface area, a high thermal conductivity, excellent mechanic strength, etc.,3,4 graphene has attracted tremendous attention for a wide range of applications, e.g., energy storage/conversion devices.5–7Although many promising energy applications have been demonstrated with 2D graphene-based nanostructures, a huge challenge and need still remains for the efficient use of graphene's large specific surface area and extraordinary electrical, chemical, and mechanical properties. For instance, the main reason behind the interest and relevance of graphene in energy devices is its large specific surface area and high electrical conductivity. However, the specific surface area of graphene depends on its solid surface and not the porosity. Since graphene sheets tend to form irreversible agglomerates or restack due to the strong π–π stacking and van der Waals interactions between the graphene sheets, graphene normally used as an electrode material has a dramatically decreased surface area, which limits its practical application in energy devices. To increase the accessible surface area of graphene, studies have been carried out to use “spacers”, e.g., carbon nanotubes,8,9 nanoparticles (NPs),10,11 and polymers,12,13 between graphene sheets to separate them. These types of graphene composites have solved the restacking issues of graphene sheets and led to enhanced performance of graphene in energy applications; however, their ability to load a sufficient amount of active materials is still unsatisfactory to deploy these materials in practical devices.
Recently, macroscopic three-dimensional (3D) graphene structures have been successfully synthesized by various methods, e.g., self-assembly, template-assisted synthesis, aerosolization, direct deposition, etc. This type of 3D graphene (3DG) has a very unique structure different from the graphenes normally used, i.e., a 3D porous/hollow structure. Due to their unique structures, 3DGs have outstanding properties, e.g., a large accessible surface area, excellent mechanical strength, and good flexibility. A lot of effort has been devoted to controlling the morphologies and properties of 3DGs in order to fully utilize their potential in energy applications. For example, a graphene hydrogel, made from the self-assembly of graphene oxide (GO) sheets, has a 3D porous structure and excellent mechanical stability and flexibility.14 It shows great promise for application in supercapacitors since it not only facilitates the access of the electrolyte to its entire surface, but also provides electrically conductive channels for active materials anchored on it.
Although several reviews have summarized the up-to-date achievements of 3DGs and their applications in some energy devices, most of them focused on the 3DG synthesis methods15–19 or their applications in supercapacitors.20,21 By creating 3DG/active material composites, the unique properties of 3DGs and active materials and the synergic effects between them can be utilized, thereby greatly expanding the potential of 3DG-based materials in energy applications. A timely summary focusing on 3DG-based composites and their use in a wide range of energy applications, e.g., supercapacitors, lithium-ion batteries, dye-sensitized solar cells, and fuel cells, is thus deemed necessary for a better understanding and further improvement of 3DG-based composites in the abovementioned energy systems/devices. In this review, we highlight recent progress in 3DG-based composites with an emphasis on their applications in energy storage/conversion devices. The synthesis methods of 3DG composites as well as typical 3DG composite-based electrode materials in energy devices are introduced and discussed.
2. Preparation methods and structures
Due to their unique structures and appealing properties, tremendous effort has been devoted to the fabrication of 3DG-based composites with controlled morphologies and structures. In this section, the fabrication methods as well as the resulting 3DG composite structures are discussed.2.1 Self-assembly methods
The chemical preparation of graphene from GO is promising for the mass production of graphene-based materials since GO can be synthesized in large quantities and easily reduced by different methods.22–26 Self-assembly of GO sheets is one of the commonly used methods to obtain 3DGs. As a typical example, 3D graphene structures can be produced through the gelation process of a GO suspension. In a stable GO suspension, individual GO sheets are well dispersed due to the van der Waals attractions from the basal planes of GO sheets and the electrostatic repulsions from the functional groups on GO sheets. Once the force balance is broken, the gelation of the GO suspension starts, and GO sheets interconnect with each other to form a hydrogel, which has a 3D porous architecture. A previous study shows that the formation of the graphene hydrogel was driven by either the linkers or π–π stacking interactions of graphene sheets.14 For the π–π stacking interactions, the recovery of a π-conjugated system from GO sheets upon the gelation process was confirmed by the decreased interlayer spacing of graphene hydrogel (3.76 Å) compared with GO (6.94 Å), which resulted from a reduction process.14 Moreover, the hydrophilic oxygenated functional groups on GO or reduced GO (RGO) sheets can encapsulate water in the process of self-assembly. This factor together with the π-stacking of graphene sheets resulted in the successful construction of the hydrogel. There are many ways to trigger the gelation of a GO suspension, for example, adding cross-linkers,27–33 changing the pH value of the GO dispersion,34–37 or using chemical reactions.38,39 Interestingly, without adding any chemicals, self-assembly of GO sheets into 3DGs has also been achieved by physical treatments of the GO suspension, e.g., hydrothermal treatment,14,40,41 direct freeze-drying,42–45 ultrasonication,46 controlled filtration/centrifugation/baking,47–50 temperature/light induction,36 electrochemical deposition,51,52etc. Fig. 1a–c show the typical structures of self-assembled graphene hydrogel (SGH) prepared by hydrothermal reduction of a GO suspension.14 The SEM image clearly shows that the hydrogel is composed of a 3D porous graphene network, which is mechanically stronger than conventional self-assembled hydrogels due to the graphene skeleton. Similar to the thermal reduction of GO in a vacuum or an inert gas environment, the high pressure and temperature in a hydrothermal process could remove the oxygenated functional groups in GO by breaking the bonds between the carbon and oxygen groups. The hydrothermal process-derived graphene hydrogel usually has a higher electrical conductivity than the GO precursor.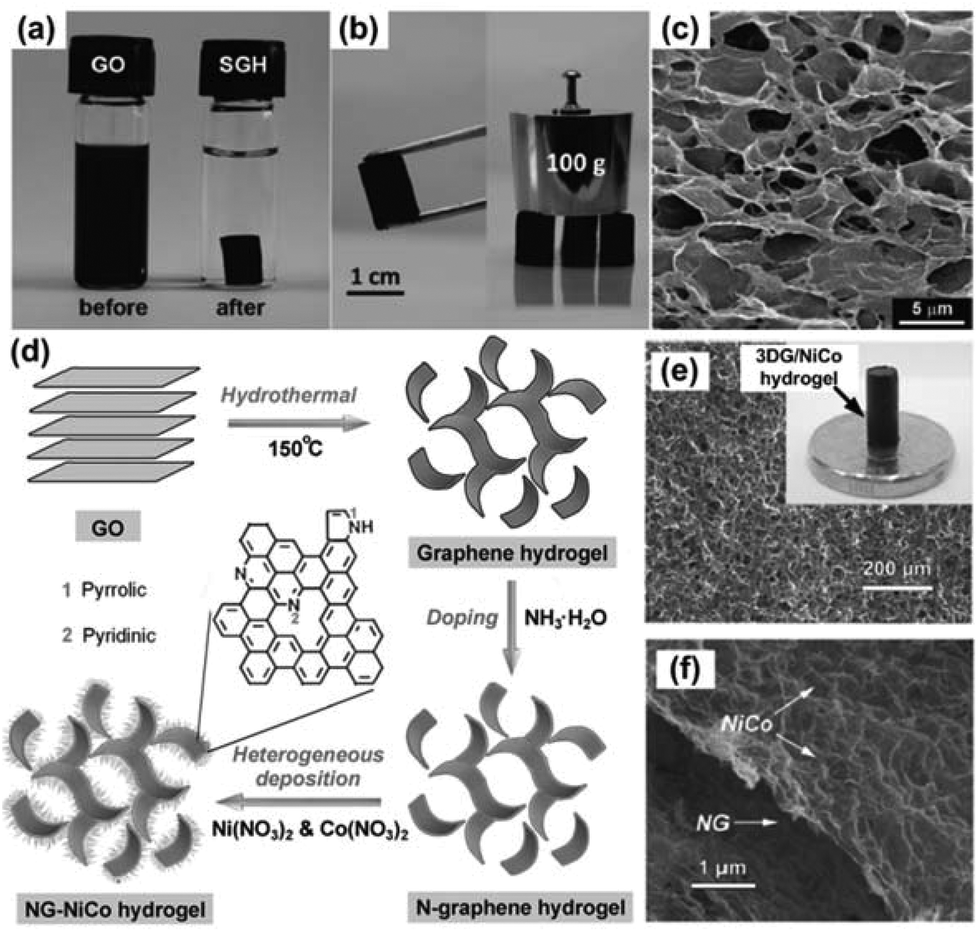 | ||
| Fig. 1 (a) Photographs of a 2 mg mL−1 homogeneous GO aqueous dispersion before and after hydrothermal reduction at 180 °C for 12 h. (b) Photographs of a strong SGH allowing easy handling and supporting weight. (c) SEM image of the SGH interior microstructures. Reproduced with permission from ref. 14. (d) A schematic showing the fabrication of the NG–NiCo composite. (e–f) SEM images of the NG–NiCo composite; the inset in (e) shows a digital image of the composite hydrogel. Reproduced with permission from ref. 60. | ||
To create 3DG-based composites, precursor ions are usually added during or after the GO gelation process for in situ decoration of NPs/nanomaterials on 3DGs. This method was successfully demonstrated in decorating various nanomaterials on 3DGs, including metallic particles,53–57 alloys,58 inorganic compounds,59–66 and organic polymers.39,67–69 In one study, NiCo double hydroxide decorated N-doped graphene hydrogels, denoted NG-NiCo, were produced by hydrothermal treatment of a GO suspension and subsequent heterogeneous deposition of NiCo double hydroxides from Ni2+ and Co2+ precursors.60 The black NG–NiCo composite hydrogel is a self-supported macroscopic cylinder, which is highly hydrophilic, containing 95.3 wt% of water and only 4.7 wt% of the NG–NiCo composites (Fig. 1d and e). As shown in Fig. 1e and f, the composite hydrogel exhibits a well-developed 3D interconnected porous network of NG with NiCo double hydroxides decorated uniformly on the surface. The major feature of a graphene hydrogel is the large porous 3D graphene network, which has a pore size up to several micrometers. The porous structure leads to a large accessible surface area, which is suitable for active material decoration. Moreover, because of the strong intersheet linking, the graphene hydrogel also has excellent mechanical strength and thermal stability,14 which brings tremendous opportunities for its application in energy devices.
2.2 Template methods
Compared with self-assembly methods that produce 3DG composites with a large amount of pores in the graphene network, template-assisted methods have also been used to produce 3DG composites with more controlled and uniform morphologies and structures. This strategy has been well demonstrated by the direct growth of graphene on 3D templates using chemical vapour deposition (CVD) methods. For example, by using porous Ni foam as both the template and the catalyst, a graphene network could be successfully synthesized on the Ni surface. After the etching of the Ni template (usually in HCl), 3DG networks were obtained.70–74 3DGs produced by this method have a higher specific surface area (as high as 850 m2 g−1)71 than those produced through chemical methods (normally 100–700 m2 g−1).22,75,76 Using CVD or annealing methods, 3DG structures have also been produced with other templates, e.g., Ni NPs,77,78 Ni film/amorphous carbon,79,80 and anodic aluminium oxide.81 In addition to the chemical growth of graphene on the template surface, direct mixing of GO with various templates, e.g., spongy,82,83 Ni foam,84–86 polystyrene spheres,87–90 silica spheres,91,92 and cellulose,93 is also an efficient route for creating porous and hollow graphene structures. Compared with the conventional CVD process, which produces a limited amount of graphene, large amounts of 3DG materials can be produced using the template-assisted direct mixing methods. This greatly benefits the applications of 3DGs that require a large quantity of materials.Similar to the self-assembly methods, the template-synthesized 3DG composites are usually produced by chemical or physical deposition of NP/nanomaterials onto the 3DG surface.87,89,90,94–96Fig. 2a shows one of the procedures to produce a 3D RGO film with a polystyrene sphere template. The experiment was carried out using a humid air flow to blow a mixture of GO and polystyrene spheres dispersed in an organic solvent. Because of the endothermic evaporation of the volatile organic solvent, the suspension condensed and closely packed into a 3D film composed of polystyrene sphere-grafted GO. Then upon subsequent pyrolysis, a mechanically flexible and robust macroporous 3D RGO film was produced (as shown in Fig. 2b). This study shows that, through the in situ reduction of metal salts (HAuCl4), gold particles could be synthesized and deposited on the surface of the 3D RGO film (Fig. 2c) and used for different applications. The template-assisted method is attractive for fabricating highly ordered, mechanically flexible macroporous graphene assemblies with tuneable open porous morphologies. The pore size and the number of graphene layers could be effectively controlled by the size of the templates and the GO suspension concentration or growth conditions. Further structural modification, e.g., doping, could be used to tune the 3DG properties for various applications.
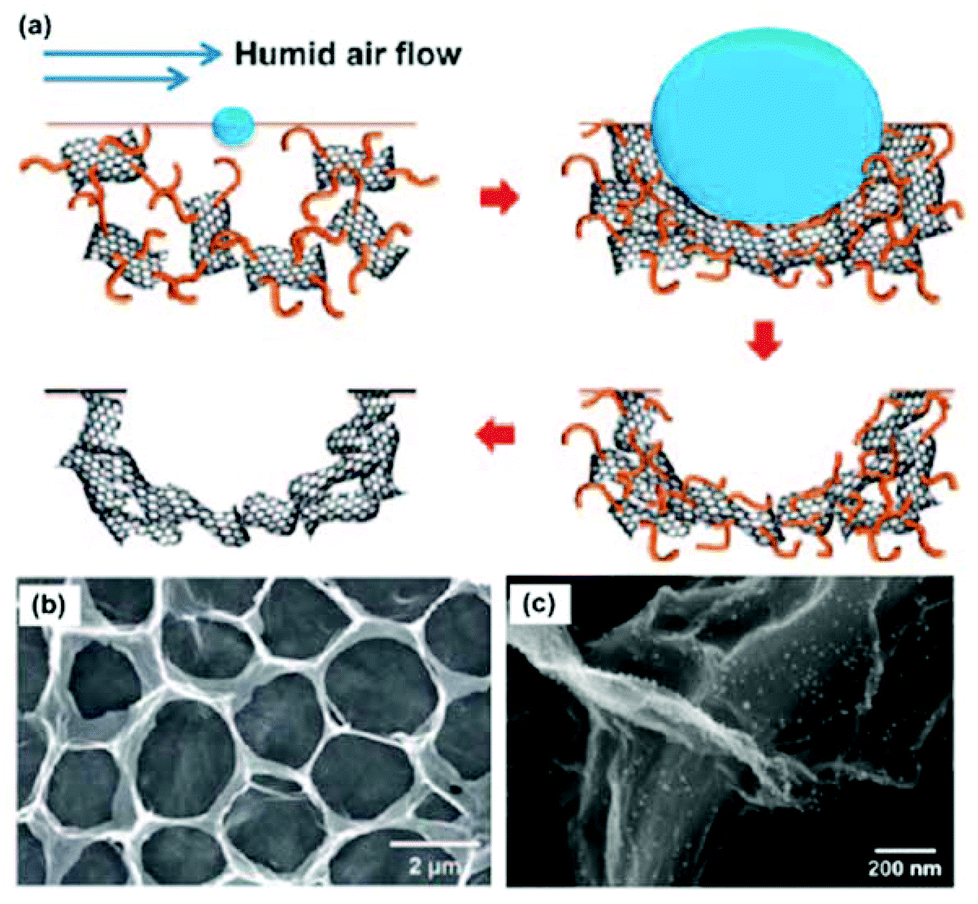 | ||
| Fig. 2 (a) Procedure for the self-assembly of RGO into macroporous carbon films with polystyrene sphere templates. (b) Plane-view SEM image of an RGO film. (c) SEM image of the N-doped porous RGO assembly decorated with gold NPs by ionic interaction. Reproduced with permission from ref. 89. | ||
2.3 Aerosolization methods
Recently, it was reported that aqueous GO sheets can be converted into crumpled graphene (CG) balls by the capillary force associated with rapid solvent loss and physical deformation through an aerosolization process, in which GO was transformed from a 2D sheet to a 3D hollow structure.97–100 Huang's and Zangmeister's groups have applied this method for synthesizing CGs and studied their structures and properties.97,98,101,102 Their studies show that when a GO aqueous solution was sprayed/aerosolized, due to the rapid evaporation of the solvent of GO droplets, GO sheets were compressed and aggregated to form a 3D graphene structure similar to a crumpled ball. Although the crumpling process could happen at room temperature, the GO could be thermally reduced to RGO simultaneously if the crumpling was carried out at an elevated temperature.101 Importantly, their studies showed that CGs had a high compressive strength, which gave them an interesting aggregation-resistant property, and the CGs could be redispersed in a solvent even after compression.97 The as-produced CG is of great interest for fundamental scientific research and practical applications due to their unique structures and properties.To expand its potential applications, CG has been combined with various functional nanomaterials, and such CG-based composites have provided tremendous opportunities for energy applications. For instance, various CG-based composites were produced by aerosolizing the mixtures of GO and pre-synthesized NPs (Pt, Ag, Au, Si, carbon black, Fe3O4, TiO2, etc.).103–109 In addition to aerosolizing mixed GO and pre-made NPs, an approach using precursor ions mixed with a GO suspension for local crystallization and growth of NPs on CG was reported.110–112 In a typical synthesis procedure, precursor ions (e.g., Mn2+, Sn4+, Ag+, and [PtCl6]2+) were dissolved in a GO suspension, which was then aerosolized and dried through a tube furnace using an ultrasonic nebulizer (Fig. 3a).112 CG-based composites with different nanocrystals (Mn3O4, SnO2, Ag, and Pt) have been fabricated during this process. SEM images of the CG/Mn3O4 composite shows that NPs were uniformly deposited on the surface of CG balls, indicating the local growth of NPs during the GO aerosolization and crumpling process (Fig. 3b and c). Based on the TEM image (Fig. 3d), the CG has a 3D hollow structure with NPs decorated on both the inner and outer surfaces. Of practical significance, through the aerosolization method, the CG-based composites can be continuously produced and collected onto various types of substrates, enabling the direct fabrication of binder-free electrodes for various energy storage and conversion devices.
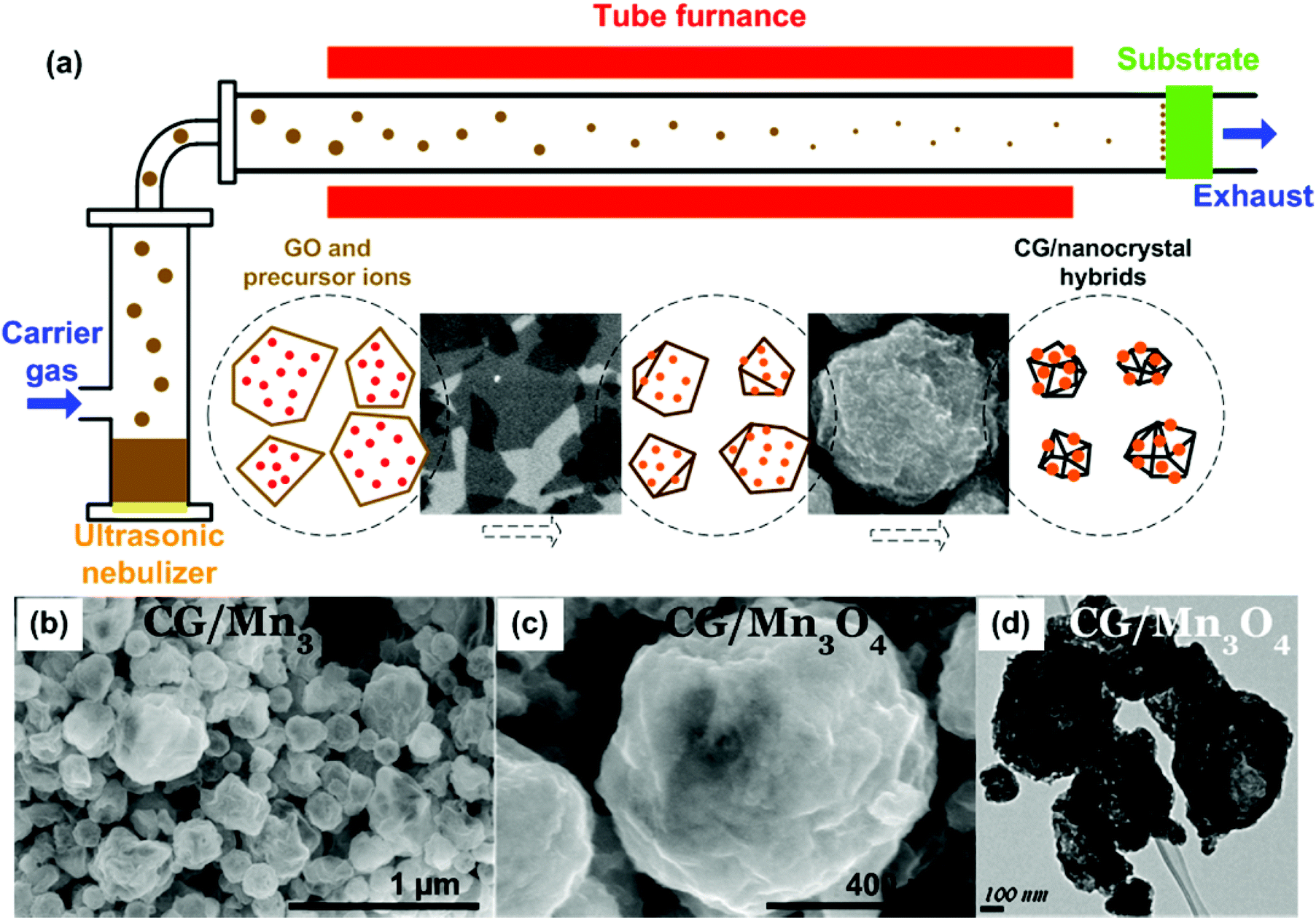 | ||
| Fig. 3 (a) Preparation of CG/nanocrystal hybrids by rapid compression of GO sheets in evaporating aerosol droplets and simultaneous chemical reactions for the growth of nanocrystals on the CG surface. Schematic illustration of the experimental setup and the aerosolization/high-temperature-induced GO crumpling and nanocrystal growth process. (b–d) SEM and TEM images of CG/Mn3O4 composites. Reproduced with permission from ref. 112. | ||
2.4 Direct deposition methods
Graphene nanosheets arranged perpendicularly to the substrate surface, i.e., vertically-oriented graphene (VG) sheets, are another type of 3DGs. VGs are normally produced by a plasma-enhanced chemical vapor deposition (PECVD) method.113–121 With the assistance of plasma, the growth of VG can be carried out in a low-temperature, highly-efficient, and catalyst-free manner, with controllable structure and properties by tuning the growth conditions. Compared with RGO produced from wet chemical methods, this type of 3DGs possess a number of unique features, e.g., vertical orientation on the substrate, non-agglomerated 3D morphology, open inter-sheet channels, and exposed ultra-thin and ultra-long edges. By incorporating other nanomaterials into them, VG-based composites were widely studied and presented great potential in energy and environmental applications.122–128 For example, Seo et al. reported a supercapacitor device with MnO2/VG composites as the electrode material.126 In their study, VGs grown on a nickel foil were used as conductive templates for the deposition of MnO2. The vertical orientation and open space between the graphene sheets facilitate ion diffusion, and the covalent bonding between MnO2 and VG leads to a highly stable structure.In addition to the PECVD growth of VG, a 3DG structure could also be prepared through electrochemical reduction of GO in an aqueous dispersion, in which the RGO sheets are deposited directly onto the surface of the electrode, which shows a similar structure to the graphene hydrogel.129 3DG composites could be fabricated by subsequent electrochemical deposition of the active materials on the 3DG surface.
3. Energy applications
Compared with 2D graphene/RGO from the chemical processes, 3DG networks normally possess a number of unique features, such as a non-agglomerated 3D inter-networked morphology, a large pore volume, a high accessible surface area, a controlled inter-sheet connectivity, as well as excellent stability and mechanical strength. By incorporating 3DGs with other nanostructures, e.g., NPs, nanorods, nanotubes, nanosheets, etc., 3DG-based composites have shown great potential in energy applications. For instance, the 3D hollow structure, the large accessible surface area, and the high in-plane electrical conductivity of graphene, and the high activity of the incorporated nanomaterials lead to many advances in nanomaterial-based supercapacitors, lithium-ion batteries, dye-sensitized solar cells, and fuel cells. A detailed review of 3DG composites’ applications in energy storage/conversion devices is presented in this section.3.1 Supercapacitor applications
Supercapacitors are advanced electrochemical devices suited for energy storage that undergo frequent charge/discharge cycles at high power and short duration and have a much higher capacitance than regular capacitors.130–132 Based on the charge storage mechanisms, supercapacitors are categorized into electric double-layer capacitors (EDLCs) and pseudocapacitors. EDLCs operate with the electric double-layer mechanism, where electrode materials are charged for the rapid separation and surface adsorption of ions with opposite charges. Pseudocapacitors incorporate pseudo-capacitance derived from reversible Faradaic-type charge transfer in electrode materials. Although supercapacitors are commercially available, there is still a need for further improvement in supercapacitor performance, e.g., in mass/volume-specific capacitance, rate capability, and power and energy densities. Nanocarbon-based materials, such as activated carbons, carbon nanotubes, and graphene, have been widely studied as supercapacitor electrode materials, due to their high electrical conductivity, high surface area, and low cost.76,132–134 To further enhance the capacitor performance of graphene, 3DG-based materials have been employed recently.20,135–137 The porous structure of 3DGs not only improves the accessibility of the electrolyte to the electrode surface, but also provides electrically conductive channels for the active material decoration, which enhances the performance of both EDLCs and pseudocapacitors.As pure carbon electrodes, most 3DGs, e.g. graphene hydrogels,14,138 template-based 3DGs,84,139,140 hollow CGs,135,141 and VGs,126,142 were used in EDLCs. Some of the demonstrated EDLCs with 3DGs showed a greatly enhanced performance compared with other carbon-based electrodes. In one report from Shi's group,84 a graphene hydrogel/Ni foam (G-Gel/NF) composite electrode was prepared by direct deposition and in situ reduction of GO on the framework of Ni foam with subsequent free-drying of the hydrogel. The graphene gel had a 3D porous structure with a pore size in the range of sub-micrometer to several micrometers. To test the performance of G-Gel/NF EDLC, symmetric cells with a two-electrode configuration were constructed. Their study showed that the G-Gel/NF EDLC can be operated at 100 ms level, which is among the most rapid graphene-based EDLCs. The capacitance of G-Gel/NF is 45.6 mF cm−2 at a discharge current of 0.67 mA cm−2, which is about one order of magnitude higher than those of the high-rate EDLCs based on other carbon materials such as onion-like carbon and laser-scribed graphene films. The excellent performance of G-Gel/NF EDLCs is attributed to the high conductivity and electrochemical stability of the graphene gel, the 3D porous microstructure of the G-Gel/NF electrode, and the short distances of charge transfer from the graphene hydrogel to the current collector (Ni foam).
Because of the redox reactions, pseudocapacitors usually have higher capacitance but lower power density and poorer cycling stability than those of EDLCs.20,131,143 By combining the merits of 3DGs and pseudo-species, e.g., transition metal oxides, repeated redox reactions can be realized. In 3DG-based composite electrodes, the roles of 3DG mainly include: increasing the specific loading of pseudo-species for higher energy density, enhancing the charge transport between pseudo-species and electrode for higher power and rate capabilities, and improving the adhesion of pseudo-species for enhanced cycling stability. Commonly used pseudo-species include Mn,63–65,87,112,144–147 Ni,70,148–151 Co,152 and V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 153,154-based transition metal oxides or compounds with various morphologies.
153,154-based transition metal oxides or compounds with various morphologies.
In one study, a MnO2-coated free-standing, flexible, lightweight, and highly conductive 3DG network was fabricated for supercapacitor electrodes.145 The 3DG network had a remarkably high specific surface area (392 m2 g−1), allowing an extremely high MnO2 mass loading of 9.8 mg cm−2 (∼92.9% of the entire electrode mass). To fabricate the 3DG network, a Ni foam was used as the template for CVD growth of the graphene film. After removal of the Ni foam, a 3DG with a foam-like network was obtained, with a weight of 0.70–0.75 mg cm−2 and a thickness lower than 200 μm. Their study showed that the free-standing 3DG network obtained from compressed Ni foam showed greater flexibility and superior mechanical strength in comparison with those from non-compressed Ni foam. Moreover, the 3DG network exhibited a high electrical conductivity of 55 S cm−1 and possessed a hollow internal structure similar to the Ni foam template. The nanostructured MnO2 materials were deposited on the 3DGs through an electrochemical deposition process. The mass loading of MnO2 could be well controlled by adjusting the deposition current and deposition time. Fig. 4b shows the SEM images of a typical 3DG/MnO2 composite with a mass loading of 0.85 mg cm−2. The MnO2 nanomaterials were uniformly coated on the entire surface of the 3DG networks. The MnO2 had a hierarchical nanosphere structure, consisting of MnO2 nanosheets, as shown in the TEM images (Fig. 4c).
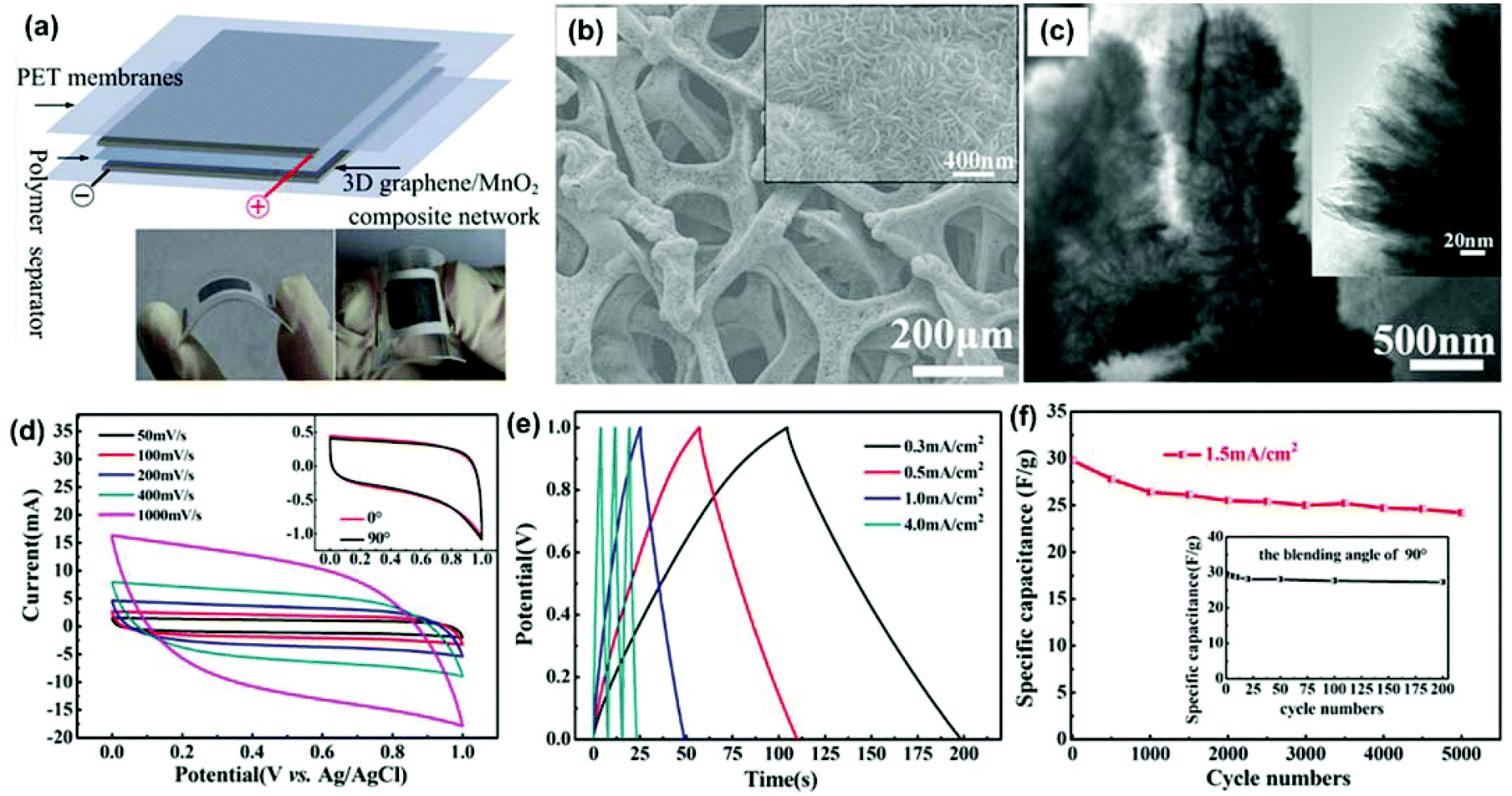 | ||
| Fig. 4 (a) Schematic of flexible supercapacitors consisting of two symmetrical graphene/MnO2 composite electrodes, a polymer separator, and two PET membranes. The two digital photographs show the flexible supercapacitors when bended. (b) SEM image of a typical sample with mass loadings of 0.85 mg cm−2. The inset is the corresponding high-magnification SEM image of the sample. (c) TEM image of the nanoporous structure of MnO2 with a deposition time of 120 min. The inset is the high-magnification TEM image. (d) CVs of the flexible supercapacitors at scan rates of 50, 100, 200, 400, and 1000 mV s−1. The inset shows the CVs of the flexible supercapacitors with bending angles of 0° and 90° at a fixed scan rate of 10 mV s−1. (e) Galvanostatic charge/discharge curves of the supercapacitor at different current densities. (f) Cycling performance of the flexible supercapacitors for charging and discharging at a current density of 1.5 mA cm−2. The inset shows cycling performance of the flexible supercapacitors for bending cycles with a bending angle of 90°. Reproduced with permission from ref. 145. | ||
To explore the advantages of the 3DG support, a supercapacitor was assembled from two pieces of 3DG/MnO2 composites, as shown in Fig. 4a. The rectangular shapes and symmetry of the cyclic voltammetry (CV) scans indicate its ideal pseudocapacitive nature, even at a high scan rate of 1000 mV s−1 (Fig. 4d). In order to demonstrate the flexibility of the device, they compared the CV scans of the device with a bending angle of 90°. As shown in the inset of Fig. 4d, there was no significant difference between the CV scans before and after bending, suggesting the high flexibility and stability of the 3DG/MnO2 composite electrodes in the symmetrical supercapacitor.
A galvanostatic charge/discharge test is usually performed to study the capacitor performance under different current densities. The galvanostatic charge/discharge results of the 3DG/MnO2 capacitor over a voltage window of 0–1 V are shown in Fig. 4e. The linear voltage versus time profiles, the symmetrical charge/discharge characteristics, and a quick I–V response represent good capacitive characteristics for the 3DG/MnO2 supercapacitor. The cycling performance of a supercapacitor is also critical for its practical application. Fig. 4f shows the cycling stability of the supercapacitor over the 0–1 V range at a current density of 1.5 mA cm−2. The specific capacitance of the 3DG/MnO2 supercapacitor decreased slightly from 29.8 F g−1 to 27.8 F g−1 after 500 cycles. After 5000 cycles, it still retained a high specific capacitance of 24.2 F g−1. The decrease of the capacitance is probably due to the mechanical expansion of MnO2 during the redox reactions or the dissolution of some MnO2 during the charge/discharge cycling. The cycling performance of the flexible supercapacitors for bending cycles (bending angle of 90°) was also tested, as shown in Fig. 4f (inset). The specific capacitance retained 92% of its initial value after 200 bending actions, indicating its excellent mechanical and flexible properties. With a MnO2 mass loading of 0.1 mg cm−2 and at a scan rate of 2 mV s−1, the 3DG/MnO2 supercapacitor had a specific capacitance of 465 F g−1, higher than that of MnO2-carbon nanotube-textile supercapacitors.155
In addition to single-component pseudo-species, hybrid pseudo-species can also be integrated with 3DGs for supercapacitor applications. For instance, hybrid Ni3S2 and Ni(OH)2 nanostructures were grown on a 3DG network (3DGN) and used as the electrodes in supercapacitors (Fig. 5a).156 The 3DGN was produced by CVD growth of graphene on a Ni foam. The as-produced 3DGN was sealed in an autoclave containing precursors for Ni3S2 and Ni(OH)2. By controlling the hydrothermal reaction duration, three types of composites were produced, i.e., Ni3S2/3DGN (6 h), Ni3S2@Ni(OH)2/3DGN (12 h), and Ni(OH)2/3DGN (24 h). In the composite, the Ni3S2 has a nanorod structure and the Ni(OH)2, which has a nanosheet morphology, covers the nanorod to form a core–shell heterostructure (Fig. 5b).
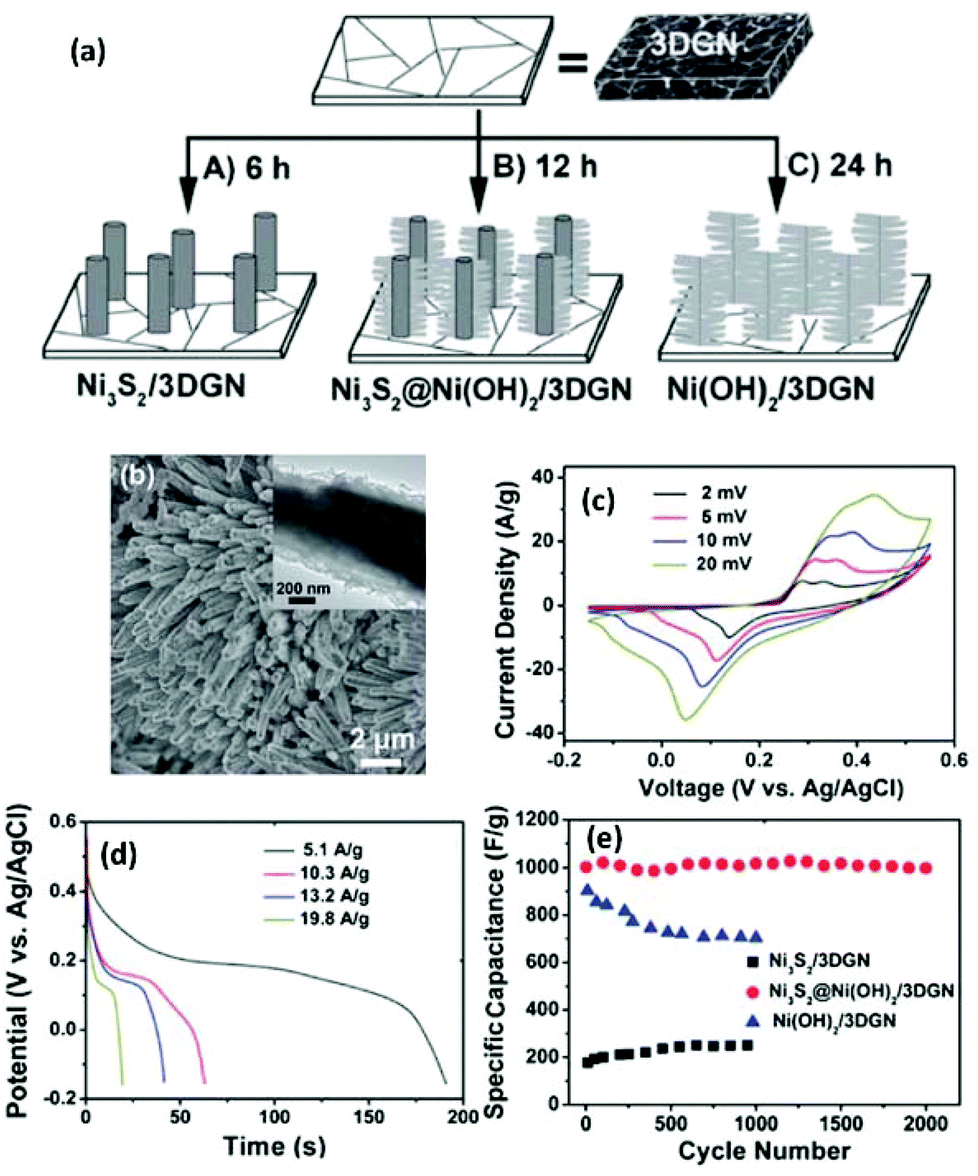 | ||
| Fig. 5 (a) Schematic illustration of the hydrothermal growth of different Ni3S2@Ni(OH)2 structures on 3DGN. (b) SEM image of Ni3S2@Ni(OH)2/3DGN (reaction time: 12 h); inset: a magnified image of the hybrid structure. (c) CVs of the Ni3S2@Ni(OH)2/3DGN at different scan rates of 2, 5, 10 and 20 mV s−1. (d) Discharge curves for the Ni3S2@Ni(OH)2/3DGN at various current densities. (e) Cycling stability of the Ni3S2/3DGN, Ni3S2@Ni(OH)2/3DGN, and Ni(OH)2/3DGN at a current density of 5.9 A g−1. Reproduced with permission from ref. 151. | ||
CV curves of the Ni3S2@Ni(OH)2/3DGN electrode at different scan rates were tested (Fig. 5c). With increasing scan rate, the current response increased accordingly, and the shapes of CV curves were retained, indicating a good rate capability. Fig. 5d shows the constant current galvanostatic charge/discharge curves of the Ni3S2@Ni(OH)2/3DGN electrode at different current densities. A high specific capacitance (1037.5 F g−1) was obtained at a discharge current density of 5.1 A g−1. Even at a relatively high current density of 19.8 A g−1, a specific capacitance of 398 F g−1 was obtained. These specific capacitances are higher than those of supercapacitors based on mesoporous carbons, e.g., lignin-derived mesoporous carbon (102.3 and 91.7 F g−1 for CO2- and KOH-activated carbons, respectively).157Fig. 5e shows the cycling performances of the three electrodes at a current density of 5.9 A g−1. The Ni3S2@Ni(OH)2/3DGN electrode had the highest specific capacitance and better durability than those of the other two electrodes. Specifically, the Ni3S2@Ni(OH)2/3DGN electrode maintained more than 99% of its initial capacitance after 2000 cycles of operation. They also studied the morphology of the Ni3S2@Ni(OH)2/3DGN electrode after 2000 cycles and found no obvious change in its structure, indicating good electrochemical stability of the materials during the charge and discharge process.
Using 3DG as the support for pseudo-species has been demonstrated to be an efficient method to prepare high performance supercapacitor electrodes. The outstanding performance of the 3DG-based composites is mainly attributed to the following reasons: (1) the porous structure and high surface area of 3DG increases the loading of pseudo-species and leads to easy access for the electrolyte to the electrode surface; (2) the highly conductive 3DG network serves as an excellent channel for electron transfer; and (3) the good contact between the 3DG and active pseudo-species ensures low contact resistance.
3.2 Lithium-ion battery applications
Lithium-ion batteries (LIBs) have attracted considerable interest due to their high energy density with high cell voltage (>3.2 V) and low maintenance (low self-discharge and no periodic discharge needed). They have found applications ranging from renewable energy, portable electronic devices, to electric or hybrid vehicles, and even to grid storage.158 But the widespread use of LIBs as transportation and stationary power systems remains challenging due to the high cost and inadequate performance. There has been tremendous effort at exploring new, inexpensive lithium intercalation materials with superior performance for both anodes and cathodes.159,160 In this section, we discuss the potential of 3DG-based composites as LIB anode materials.The ideal candidate for LIB anode materials should have characteristics such as high lithium storage capability and robust structure for repeated charge/discharge cycles. Sn, SnO2, and Si have been demonstrated as potential anode materials for LIBs due to their high specific capacities (981 mA h g−1 for Sn, 1491 mA h g−1 for SnO2, and 4200 mA h g−1 for Si) compared with those of carbon-based materials (372 mA h g−1 for graphite).161–165 However, these anodes usually suffer from severe capacity degradation (aging) due to the aggregation of Sn/Si-based particles and the huge volume change (over 300%) during Li+ insertion/extraction cycles, which causes the pulverization of the anodes and electrical detachment of the active materials from the current collector. Reducing the material size to the nanoscale and dispersing the material onto porous structures have proved very effective in solving the problems of similar systems.166 Meanwhile, due to its single-atom thickness, large surface area, and ballistic conductivity, graphene has emerged as an attractive conducting support for NPs, which can not only facilitate the transport of electrons and electrolyte in the electrode, but also lower the stress of the collective electrode during charge/discharge.167,168 Progress has been made in using hybrids of NPs supported on planar RGO for LIBs.169,170 However, those reported NP/RGO composite materials are still far from practically serving as anodes due to low Coulombic efficiency in the first cycle and unsatisfactory cycling performance. The 2D graphene or RGO sheets easily restack due to the van der Waals forces, significantly reducing the accessible surface area and limiting electron and ion transport. In addition, the mass loading per unit area of nanostructured anode materials has typically been low, limited by either the fabrication process or the structural design itself.
Constructing 2D graphene sheets into 3D architectures can effectively avoid the restacking of graphene and provide a resulting 3DG with a large specific surface area, porous structure, and fast electron transport.71,171 Many novel composites combining 3DG with various high-capacity anode materials (e.g., Sn, SnO2, Si, and Fe3O4) have been demonstrated for LIB applications. In this section, we provide a brief overview of 3DG/Sn, SnO2, Si, or Fe3O4 composites as LIB anode materials. It is worth mentioning that many other low-cost, high-capacity materials have also emerged as functional components that can be integrated with 3D graphene to serve as LIB anode materials. These materials include Fe2O3,172,173 MnO,174 MnO2,175,176 TiO2,177,178 NiO,179 MoO2,180 V2O5,181 Co3O4,182 CoO,183 MoS2,184,185 Ni3S2,186 Ge,183 S,187 and so on.
Qin et al. reported a scalable in situ CVD approach for one-step fabrication of a 3DG-Sn NP composite as a LIB anode material.188 In this composite (designated as 3D Sn@G-PGNW), Sn NPs (5–30 nm) encapsulated by conformal graphene shells (∼1 nm) were uniformly and tightly anchored on the surfaces of thin walls (∼3 nm) of the 3D porous graphene network (Fig. 6a). The graphene shells effectively prevented Sn NPs from direct exposure to the electrolyte, preserved their structural and interfacial stabilization, suppressed their aggregation, and buffered their volume expansion. The 3D porous graphene network provided high electrical conductivity, large surface area, and high mechanical flexibility to the core–shell Sn@G structure, resulting in enhanced electrical conductivity and structural integrity of the overall electrode. As shown in Fig. 6b and c, the 3DG/Sn composite anode exhibited high rate performance (1022 mA h g−1 at 0.2 C, 865 mA h g−1 at 0.5 C, 780 mA h g−1 at 1 C, 652 mA h g−1 at 2 C, 459 mA h g−1 at 5 C, and 270 mA h g−1 at 10 C, 1 C = 1 A g−1) and long cycling stability even at high rates (a capacity of 682 mA h g−1 was achieved at 2 A g−1 and maintained approximately 96.3% after 1000 cycles). The high specific capacity and good capacity retention achieved by the 3DG/Sn composite anode at a high rate (2 A g−1) exceed those of many other carbon/Sn nanostructures, e.g., nano-Sn/carbon composites189 and porous core–shell Sn@carbon composites.190
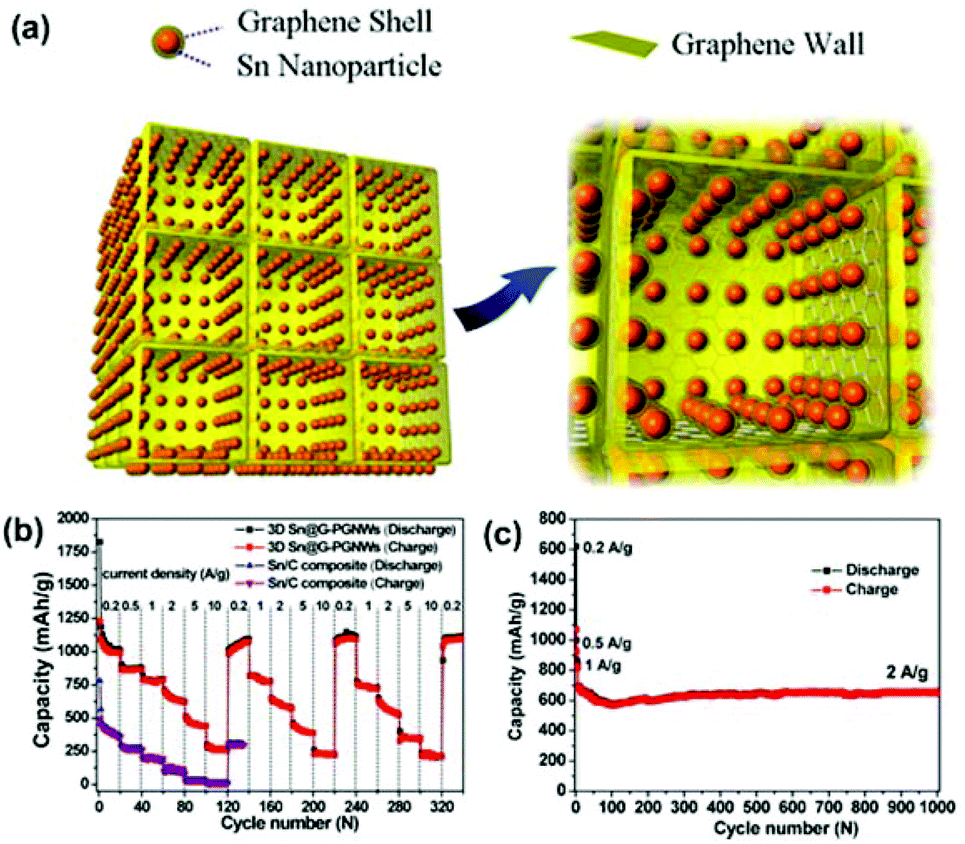 | ||
| Fig. 6 (a) Schematic of the 3DG-Sn NP composite. (b) Rate cycle performance of the electrodes of 3D Sn@G-PGNWs and Sn/C composites at charge/discharge rates from 0.2 to 10 C (1C = 1 A g−1) for 340 cycles. (c) Cycle performance of the 3D Sn@G-PGNW electrode at current densities of 0.2, 0.5, and 1 A g−1 for the initial six cycles and then 2 A g−1 for the subsequent 1000 cycles. Reproduced with permission from ref. 188. | ||
Crumpled GO/graphene has attracted increasing attention due to its 3D open structure and excellent stability in an aqueous solution. We reported a general and one-step approach for producing CG–nanocrystal composites by direct aerosolization of a GO suspension mixed with precursor ions.112 Nanocrystals spontaneously grow from precursor ions and assemble on both the external and internal surfaces of CG balls during the solvent evaporation and GO crumpling process (Fig. 3). The 3D CG–nanocrystal composites could be directly deposited onto various current-collecting substrates, facilitating their potential for energy applications such as in LIBs. As a proof of concept, we demonstrated the use of 3D CG–SnO2 as the anode material in LIBs. CG–SnO2 nanohybrids were directly deposited on a copper current collector (copper foil) without any binder or conductive carbon (Fig. 7a). The performance of the resulting battery was attractive and outperformed conventional flat graphene-based hybrid devices. By comparing the initial two charge–discharge curves (Fig. 7b and c), the CG–SnO2 nanohybrids displayed a much smaller change, signifying a much smaller capacity loss in the initial two cycles than flat GO–SnO2 nanohybrids. Fig. 7d compares the cycling performance and the rate capability of CG–SnO2 nanohybrids and flat graphene–SnO2. The CG–SnO2 nanohybrids exhibit a remarkable improvement in various performance parameters, including specific capacity, rate capability, and stability. Our study provided a new and facile route for fabricating high-performance CG–nanocrystal composite electrodes for LIB systems.
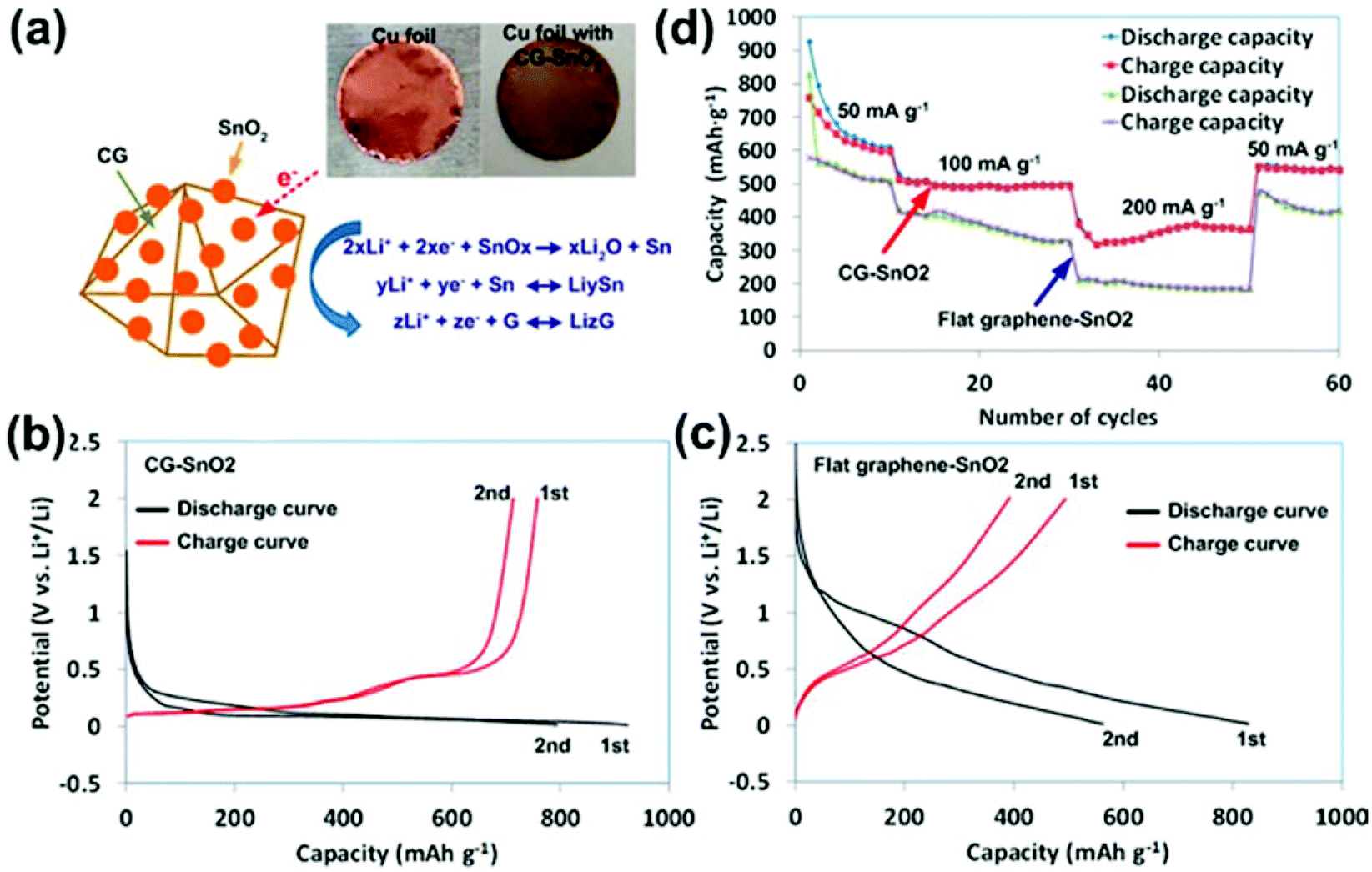 | ||
| Fig. 7 (a) Schematic of the LIB anode test for CG–SnO2 hybrid on a Cu foil. The initial two charge (red) and discharge (black) curves at a current density of 50 mA g−1 for (b) CG–SnO2 and (c) flat graphene–SnO2. (d) Capacity retention of CG–SnO2 and flat graphene–SnO2 at various current densities. Reproduced with permission from ref. 112. | ||
Several other groups also synthesized 3DG/SnO2 composites using hydrothermal processes191,192 or a template method95 and applied these composites to LIBs. For example, Huang et al. reported a two-step assembly method for producing 3D graphene–SnO2 composites with SnO2 NPs uniformly immobilized on the graphene surface.191 SnO2 NPs were first formed on GO sheets by in situ synthesis, and then 3D SnO2/graphene frameworks (SnO2/GFs) were formed through the hydrothermal self-assembly of the 2D SnO2/GO nanosheets. The resulting 3D SnO2/GFs had a large surface area, numerous macropores, and a low mass density. The 3D SnO2/GFs structure could effectively accommodate the volume change of SnO2 NPs during the cycling process and provide multidimensional channels for electron transport and electrolyte access, enabling the rapid diffusion of lithium ions from the electrolyte to the electrode. SnO2/GFs exhibited a high capacity of 830 mA h g−1 for up to 70 charge–discharge cycles at 100 mA g−1. At a current density of 500 mA g−1, SnO2/GFs still maintained a reversible capacity as high as 621 mA h g−1 with good cycling stability. Wang et al. reported a solvothermal-induced self-assembly process using SnO2 colloid sol as the precursor to form a 3D aerogel composed of self-assembled nitrogen-doped graphene networks homogeneously coated with SnO2 NPs (3–7 nm).192 Nitrogen-doping could further tune the electronic properties of graphene and enhance its lithium storage properties. The resulting 3D nitrogen-doped graphene-SnO2 showed good rate capability and prolonged cyclic stability (905 mA h g−1 after 1000 cycles at 2000 mA g−1). Liu et al. demonstrated a 3D macroporous graphene framework (GFs)-based SnO2 composite (3D SnO2/GFs) by a self-assembly process, in which polystyrene (PS) nanospheres were used as sacrificial templates.95 The 3D SnO2/GF electrode displayed a capacity of 1244 mA h g−1 for 50 cycles at 100 mA g−1 and a rate capability of 753 mA h g−1 for up to 200 cycles at 1000 mA g−1. The good cycling performance and rate capability of 3D SnO2/GFs were attributed to the synergistic effects of graphene, SnO2, and the 3D porous structure.
Various approaches have been proposed to produce 3DG/Si composites as high-performance anode materials for LIBs.108,176,193–196 Zhou et al. designed an Si/carbon/graphene composite (abbreviated as Si–C/G). In this structure, carbon NPs were used to improve the electronic conductivity of the composite, while graphene sheets combined with carbon NPs resulted in a 3D conducting network to buffer the large volume changes of Si during battery cycling.197 The Si–C/G composite exhibited a reversible capacity of 1521 mA h g−1 at 0.2 C after 200 cycles and a good high-rate capability as well. Xin et al. reported a 3D porous composite of Si/graphene using a spray-drying method. The Si/graphene microspheres showed a capacity as high as 1000 mA h g−1 for 30 cycles.196 Chen et al. synthesized 3D carbon-coated mesoporous Si nanospheres@graphene foam (C@Si@GF) by a thermal bubble ejection assisted CVD and magnesiothermic reduction process.193 C@Si@GF nanocomposites showed good electrochemical performance including a specific capacity as high as 1200 mA h g−1 at the current density of 1 A g−1, excellent high rate capabilities, and an outstanding cyclability (Fig. 8a and b). The morphology of 3D C@Si@GF electrodes was well maintained after 200 cycles. Wu et al. synthesized a 3D interconnected network of graphene-wrapped porous silicon spheres (Si@G network) via layer-by-layer assembly and a subsequent in situ magnesiothermic-reduction process.195 When tested as a LIB anode material, the Si@G composite showed enhanced lithium-storage capabilities in terms of specific capacity, cycling stability, and rate capability in comparison with bare Si spheres (Fig. 8c and d). When current density was varied from 0.05 to 0.1, 0.2, 0.5 C, and finally back to 0.05 C, the discharge capacity of the Si@G composite changed from 1467.5 to 1210.6, 970.7, and 697.8 mA h g−1 and finally reversibly recovered to 1096.8 mA h g−1. Huang's group produced submicrometer-sized capsules of Si NPs wrapped by CG shells using an aerosolization method.108 Si NPs could be protected from excessive deposition of the insulating solid electrolyte interphase as the folds and wrinkles in the CG coating could buffer the volume expansion of Si upon lithiation. The CG/Si composite capsules showed better performance than the bare Si particles as LIB anodes in terms of capacity, cycling stability, and Coulombic efficiency (Fig. 8e and f).
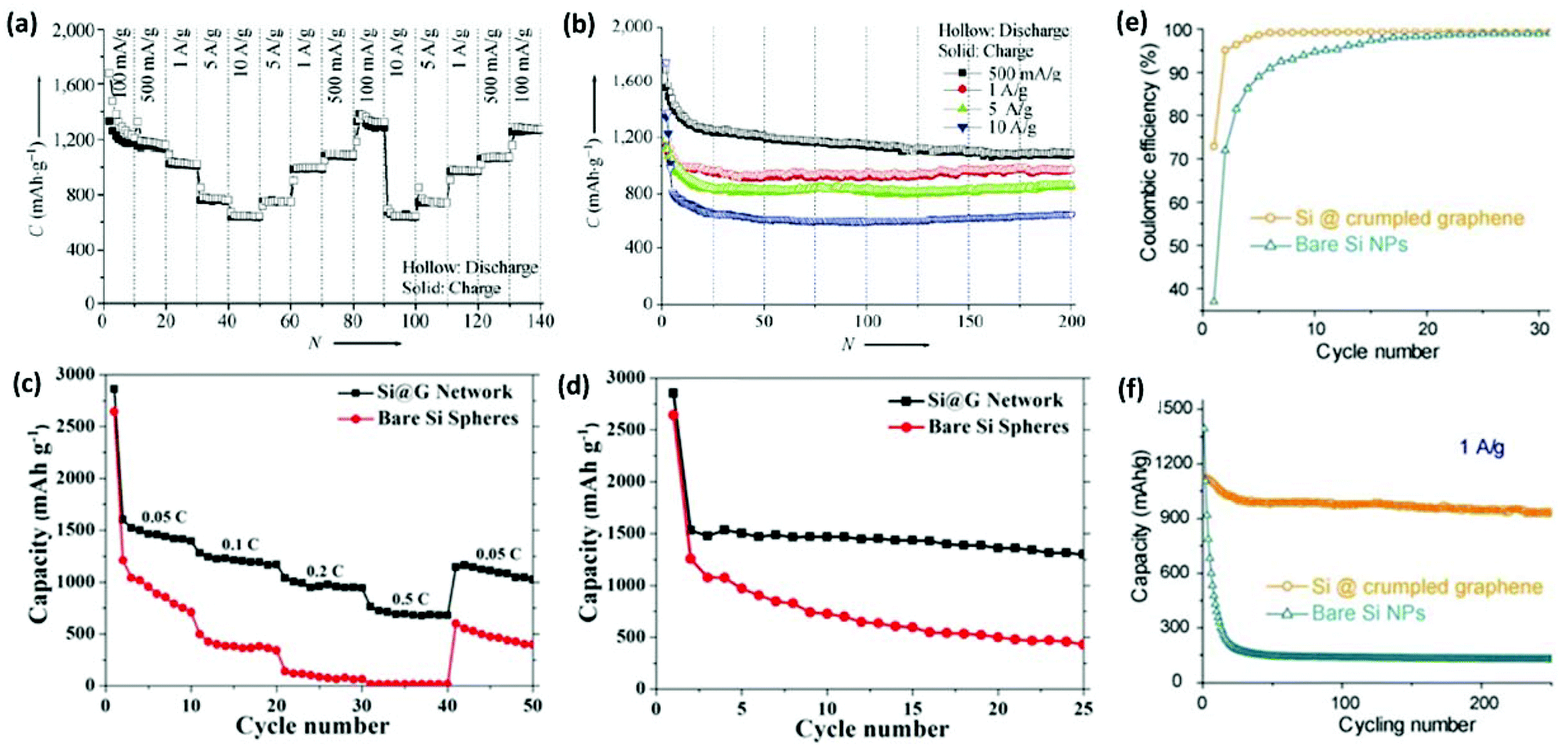 | ||
| Fig. 8 (a) Step-wise rate performance of C@Si@GF composites at different current densities. (b) Cycling performances at high current densities. (c) Cycling performance and (d) rate capability of the Si@G network and bare Si spheres. (e) Coulombic efficiency and (f) charge/discharge cycling test of the composite capsules in comparison with the unwrapped Si NPs at a constant current density of 1 A g−1. Reproduced with permissions from ref. 108, 193, and 195. | ||
Fe3O4 is a promising LIB anode material owing to its high theoretical capacity, earth abundance, low cost, and nontoxicity. 3DG/Fe3O4 composites have been synthesized using various methods and investigated as LIB anode materials.59,110,198–201 For instance, Luo et al. reported a bottom-up approach assisted by atomic layer deposition to graft bicontinuous mesoporous nanostructure Fe3O4 onto 3DG foams and directly used the composite as the LIB anode.199 A capacity as high as 785 mA h g−1 was obtained at a 1 C rate and could be maintained without decay up to 500 cycles. Wei et al. reported a method to fabricate 3DG foams cross-linked with Fe3O4 nanospheres (NSs) encapsulated with graphene shells (Fe3O4@GS/GF).200 The structure of Fe3O4@GS/GF resulted in double protection against the volume change of Fe3O4 NSs during charge/discharge: the graphene shells could suppress the aggregation of Fe3O4 NSs and buffer the volume expansion, while the 3D graphene networks could strengthen the core–shell Fe3O4@GS structure and thus enhance the electrical conductivity of the overall electrode. Fe3O4@GS/GF had a high reversible capacity of 1059 mA h g−1 over 150 cycles and good rate capability, exhibiting potential as an anode material for LIBs.200
3.3 Dye-sensitized solar cell applications
Dye-sensitized solar cells (DSSCs) have been considered as a technical and economical alternative to Si-based p–n junction photovoltaic devices.202,203 The key components of a DSSC generally include a dye-sensitized photoanode, an electrolyte solution with a redox mediator, and a counter electrode (or cathode). Typically, the photoanode is made of a fluorine-doped SnO2 (FTO) transparent electrode with a sintered TiO2 (anatase) NP scaffold that is sensitized with organometallic dye molecules; an iodide/triiodide redox couple dissolved in acetonitrile serves as the electrolyte and a platinized FTO electrode as the counter electrode. The DSSC works in the following sequence: (1) generation of electron–hole pairs through the light-absorbing dye; (2) quick separation of electrons and holes with electrons injected into TiO2 and holes into the electrolyte through redox species; and (3) transport of the charge carriers (i.e., electrons and holes) in the respective phases to opposing electrodes.Integration of new materials and processing techniques has been actively pursued to improve the performance and lower the cost of DSSCs. As a “star” material with exceptional electrical, optical, and mechanical properties, graphene has been vigorously explored for each part of a DSSC (i.e., the photoanode, the electrolyte, and the counter electrode).204 Graphene materials (e.g., CVD graphene and RGO) have been studied as transparent conductors,205 in the semiconducting layer,206 and even as the sensitizer itself for the photoanode of a DSSC.207,208 The combination of the excellent electrical conductivity and the porous structure makes 3DG a promising anode/cathode material for building DSSCs with enhanced photovoltaic properties and superior performance.
3DG has been incorporated into the semiconducting layer (e.g., TiO2 scaffold) in the photoanode to improve photocurrent density by enhancing electron transport, dye adsorption, and light scattering. Tang et al. prepared 3DG by CVD with Ni foam rods as the template and then mixed 3DG with P25 (a traditional photoanode material made of TiO2 NPs) to form the photoanode of DSSCs.209 The continuous structure between graphene sheets in the 3DG led to a good transport channel for the photogenerated electrons from TiO2 NPs to conductive glass. The power conversion efficiency (η) of the as-prepared DSSCs was observed to increase, which was attributed to the excellent electron transport properties and large surface area of the 3DG. The amount of 3DG in the photoanode ranged from 0.2 to 2 wt%. The DSSC with a photoanode of 1 wt% 3DG showed the best photovoltaic performance (η as high as 6.87%): the short circuit current density (Jsc) and power conversion efficiency increased by up to 29.4% and 32.7%, respectively, in comparison with the DSSC based on pure P25 photoanodes. Additionally, DSSCs with the 3DG based counter electrode and photoanode were also studied (Fig. 9); however, the performance of the resulting DSSCs was inferior to that of DSSCs with a Pt counter electrode. It is believed that enhancing the contact between the 3DG and conductive glass could improve the 3DG counter electrode and thus the whole DSSC.
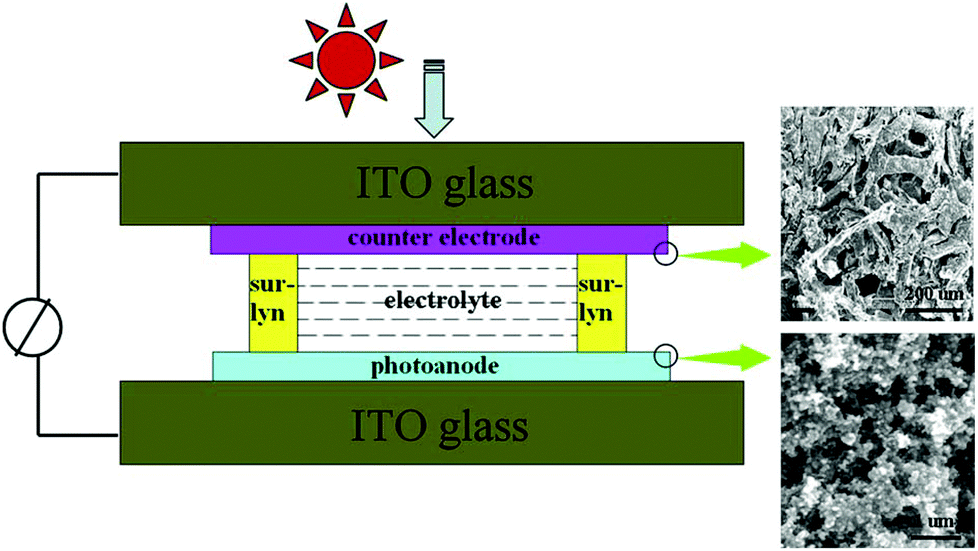 | ||
| Fig. 9 Left: schematic of the DSSC with 3DGN-based photoanode and counter electrode; right: SEM images of the electrodes. Reproduced with permission from ref. 209. | ||
3DG hydrogel and aerogel are robust hosts for anchoring various functional NPs. Han et al. modified a hydrothermal method that was previously reported for synthesizing graphene hydrogel/aerogel and prepared a ternary aerogel composed of an interconnected 3DG aerogel embedded with CdS and TiO2 NPs (Fig. 10a).210 In addition to the advantages of each component in the ternary aerogel, synergistic interactions among the three components could also be expected. Indeed, the ternary aerogel showed some promising properties, such as enhanced light absorption and photocurrent (Fig. 10b), efficient charge separation, and good durability, showing its potential in DSSCs and quantum dot-sensitized solar cells (QDSSCs).
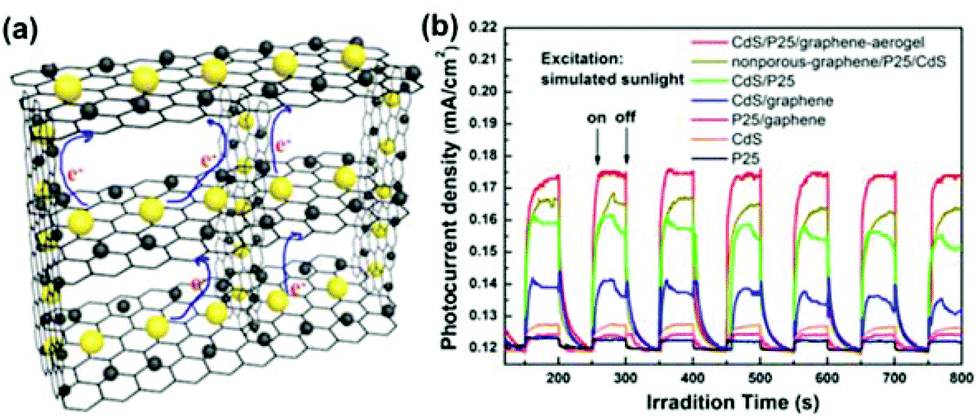 | ||
| Fig. 10 (a) Schematic of the 3D CdS/P25/graphene aerogel network. (b) Comparison of the photocurrent responses from CdS/P25/graphene aerogel, nonporous graphene/P25/CdS composite, CdS/P25, CdS/graphene aerogel, P25/graphene aerogel, CdS NPs, and P25 NPs in a sulfide–sulfite electrolyte with the applied potential of 0 V. Reproduced with permission from ref. 210. | ||
Yin et al. developed a scalable, solution-processable technique to form free-standing 3D ordered porous structures made of functionalized graphene sheets via an “on water spreading” method (Fig. 11a–c).211 The free-standing graphene honeycomb film with a large-area, uniform, periodic structure could be transferred onto various substrates of interest. Functional NPs such as TiO2 NPs were easily introduced into the 3DG honeycomb films by premixing. The formed composite honeycomb film electrode showed a fast, stable, and reversible photocurrent response with each switch-on and switch-off of simulated sunlight irradiation. The graphene-based honeycomb scaffold enhanced the light-harvesting efficiency and improved the photoelectric conversion behaviour; the photocurrent of the composite film was about two times as high as that of the pure TiO2 film electrode (Fig. 11d). The composite porous films could combine the good electrochemical performance of graphene, a large electrode/electrolyte contact area, and good stability during the photo-conversion process, holding promise for further applications in solar energy conversion.
 | ||
| Fig. 11 (a) Photograph and (b) SEM image of the free-standing honeycomb film using the GO/dimethyldioctadecylammonium (DODA) complex. (c) Photograph of the self-supporting honeycomb film transferring onto Parafilm. (d) Current versus time under chopped irradiation of RGO/DODA/P25 (red), RGO/DODA (green), and P25 (black), at a bias voltage of 0 V (vs. Ag/AgCl). Reproduced with permission from ref. 211. | ||
The function of the counter electrode in a DSSC is usually twofold: to regenerate the redox couples by a catalyst and to collect electrons from the external circuit as a current collector. Counter electrode materials thus should possess excellent catalytic activity and electrical conductivity. Conventionally, a counter electrode consists of Pt (as the catalyst) deposited on FTO conductive glass (as the electron collector). Although Pt has been proved to be an excellent catalyst due to its high catalytic activity and stability, it is not suitable for large-scale applications because of its high cost and limited reserves. Considerable effort has been devoted to finding high-performance, low-cost counter electrode materials to replace Pt and Pt/FTO.
Inspired by the unique properties of graphene (e.g., high surface area, high conductivity, and flexibility in tuning defects and composites), various graphene materials, including 3DG and 3DG-based composites, have been recently used in the DSSC counter electrode.121,212–215 Wang et al. synthesized a 3D honeycomb-like structured graphene (HSG) by a simple reaction between Li2O and CO and used the HSG/FTO as a counter electrode for DSSCs.215 HSG showed good catalytic performance for DSSC with an energy conversion efficiency as high as 7.8%, which is comparable to that of a Pt/FTO electrode. CVD-grown p-doped 3DG nano-networks also offered promising performance as a counter electrode material in DSSCs, which had a maximum photoconversion efficiency of 8.46%.212
Graphene-based composites have also been investigated as counter electrode materials for DSSCs. Metal sulfides (MS, M = Ni, Co, Cu, Mo, W, etc.) are a group of efficient counter-electrode catalysts for DSSCs.216 A composite structure with SnS2 NPs uniformly dispersed on RGO (SnS2/RGO) was synthesized and used as a counter electrode in a DSSC system.217 The SnS2/RGO composite showed promising electrocatalytic properties in reducing the triiodide, due to the synergetic effect between SnS2 NPs and graphene sheets and the improved conductivity. The DSSC with the SnS2/RGO counter electrode achieved a power conversion efficiency of 7.12%, higher than those cells using SnS2 nanoparticles (5.58%) or RGO alone (3.73%) as counter electrodes, and comparable to that of the DSSC with a Pt counter electrode (6.79%).217
Bi et al. demonstrated the composite film of metal sulfide (MS, M = Ni, Co) NPs/3DG as a novel transparent conductive counter electrode with high electrocatalytic activity for DSSCs. Graphene sheets were grown on dielectric SiO2 substrates by CVD and then deposited with MS NPs by dip coating of MS precursors and further annealing at 400 °C for 30 min in Ar. The DSSCs with NiS/3DG and CoS/3DG counter electrodes showed good electrocatalytic activity and photovoltaic conversion efficiencies of 5.04% and 5.25%, respectively, which are higher than or comparable to the DSSC with a Pt/FTO counter electrode (5.00%). The good DSSC efficiencies were ascribed to the good electrocatalytic activity of the MS and the high electrical properties of 3DG films. However, it is debatable whether the graphene synthesized with the method detailed in this work is indeed 3D. Future work is needed to explore the integration of 3DG-based composites as counter electrode materials in DSSCs. Nevertheless, this work suggests that electrodes based on MS NPs/graphene (particularly 3D) composites could be a promising alternative to Pt and Pt/FTO counter electrodes for low-cost and high performance DSSCs.
3.4 Fuel cell applications
Fuel cells are devices that convert chemical energy from fuels, e.g., hydrogen, methanol, ethanol, etc., into electricity through chemical reactions with oxidizing agents, e.g., oxygen.218–220 There are many types of fuel cells, but they all consist of an anode, a cathode, and an electrolyte that allows charges to move between the two sides of the fuel cell. In a typical fuel cell system, chemical reactions occur at the interfaces of anode/cathode electrode and electrolyte. At the anode, a catalyst is normally employed to assist the oxidation of the fuels, while at the cathode, a catalyst is used to assist the reduction of oxidizing agents. The catalytic behaviour of the catalyst strongly influences fuel cell performance and applications.Precious metals are the most commonly used catalysts in fuel cells for fuel oxidation reactions and oxygen reduction reaction (ORR). For instance, Pt and Ru particles are widely used for methanol/ethanol oxidation reactions.221–223 Pt and its alloys, e.g., Pt–Ni, Pt–Co, and Pt–Cu, are the most popular ORR catalysts.224–226 Although precious metal catalysts have the best catalytic activities in fuel cell reactions, they suffer from high cost and poor stability. Alternative catalysts based on non-precious metals, e.g., Co, Fe, and V-based catalysts,227–232 and metal-free materials, e.g., carbon nanotubes, graphene, and C3N4,233–238 have been actively studied.
Although defected or doped graphene has good catalytic activities,239,240 graphene is normally used as the catalyst support due to its high conductivity and large specific surface area.241–244 Compared with conventional graphene, the unique features of 3DGs make them even more appealing catalyst supports for such reactions. The non-agglomerated morphology and porous structure of 3DGs facilitate the loading and dispersion of catalysts. Moreover, the interconnected graphene networks promote fast electron transport between the reaction sites and the electrode. 3DG-based composites are widely used as catalysts in fuel cell reactions, e.g., methanol oxidation reaction (MOR),245–247 ethanol oxidation reaction (EOR),248,249 biofuel oxidation reactions,250,251 and ORR.96,243,252,253
Specifically, for the EOR in direct-ethanol fuel cells, Pt/PdCu nanocubes supported by a 3DG framework (3DGF) exhibited better electrocatalytic performance in activity and stability than Pt-based catalysts. The composites, prepared by a dual solvothermal process, have a well-defined and interconnected 3D porous network of graphene sheets and uniformly decorated Pt/PdCu nanocubes with an average size of 30 nm (Fig. 12a and b). The catalytic activity of ternary Pt/PdCu nanocubes on 3DGF was investigated by the electro-oxidation reaction of ethanol in a basic medium. Fig. 12c shows the CVs of ethanol oxidation in a 1.0 M KOH and 1.0 M C2H5OH solution at a scan rate of 50 mV s−1 with Pt/3DGF, PdCu/3DGF, Pt/PdCu/3DGF, and 20% Pt/C electrodes, respectively. The onset potential for the ethanol oxidation with the Pt/PdCu/3DGF electrode is at −0.6 V, which is more negative than the other three catalysts. Moreover, the Pt/PdCu/3DGF electrode has the highest peak current density, which is a few times higher than those of the other electrodes. Based on the electrochemical results, the Pt/PdCu/3DGF has been proved to have the highest catalytic activity towards ethanol oxidation among the tested catalysts. The function of the 3DG support in the composite catalyst is that it provides high accessibility of active species to the fuel to ensure an effective mass transfer of reactants and/or products.
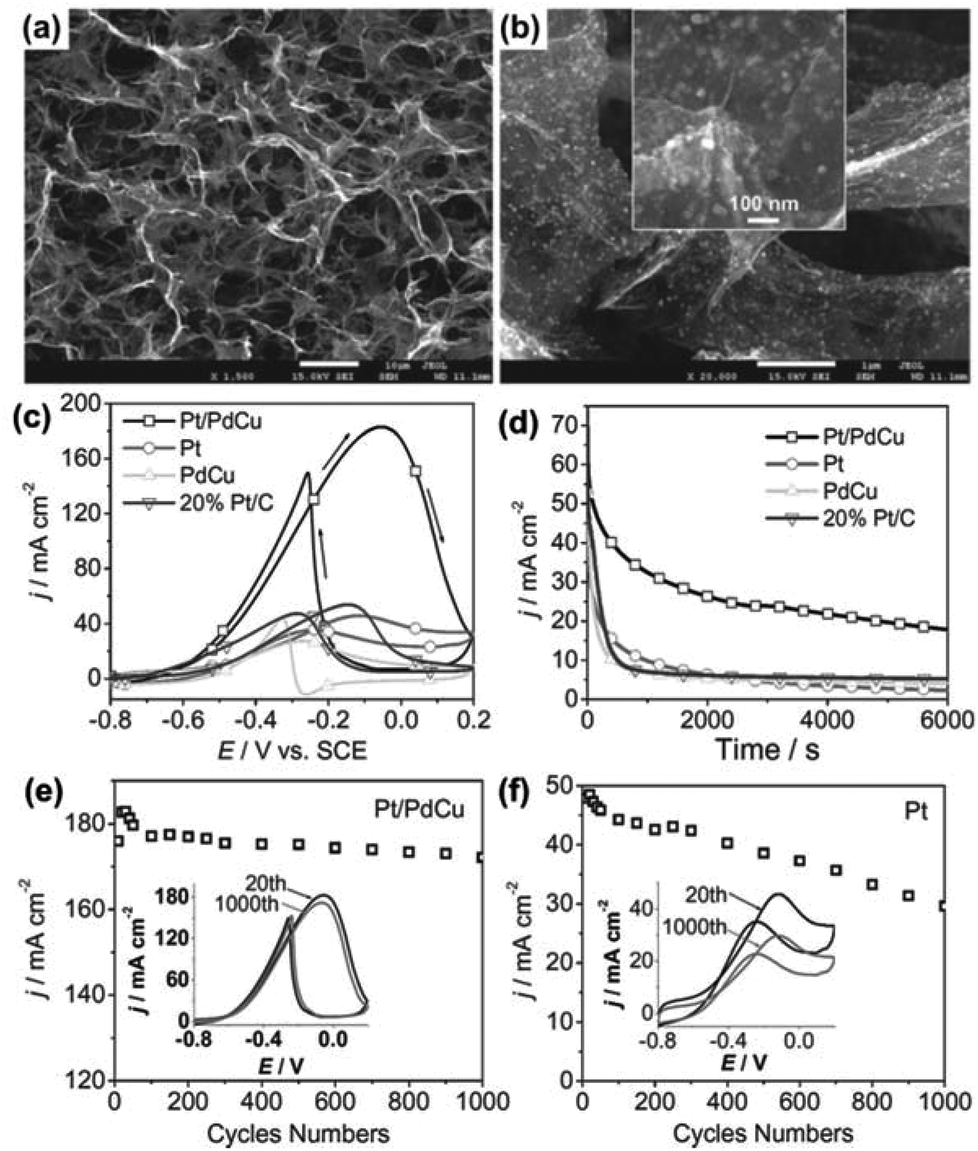 | ||
| Fig. 12 (a, b) SEM images of the Pt/PdCu on 3DGF. (c) CVs of the Pt/PdCu, Pt, PdCu on 3DGF, and commercial 20% Pt/C catalysts. (d) The chronoamperometric curves of the Pt/PdCu, Pt, PdCu on 3DGF, and 20% Pt/C electrodes at a given potential of −0.3 V. (e, f) Forward peak current density (j) as a function of potential scanning cycles of Pt/PdCu and Pt on 3DGF between −0.8 and 0.2 V, respectively. The insets in (e) and (f) correspond to the 20th and 1000th CV curves of Pt/PdCu and Pt on 3DGF, respectively. All tests were carried out in 1 M CH3CH2OH and 1 M KOH aqueous solution. Reproduced with permission from ref. 248. | ||
The electrochemical stability of the catalysts for ethanol oxidation was investigated by chronoamperometric tests at a given potential (–0.3 V). The reaction current for the ethanol oxidation reaction shows a rapid decay in the first few hundred seconds for all catalysts (Fig. 12d), which is due to the formation of the intermediate species during the ethanol oxidation reaction in alkaline media. Nevertheless, the current decay in the Pt/PdCu/3DGF electrode is much slower than those of the other electrodes, indicating a better durability of the Pt/PdCu/3DGF catalyst for long-term operation. The high stability of the Pt/PdCu/3DGF electrode was also demonstrated in Fig. 12e–f in comparison with the Pt/3DGF electrode. The Pt/PdCu/3DGF electrode shows a very small degradation (∼6%) in the reaction current and almost no shift in onset potential compared with the Pt/3DGF electrode (35% degradation in the reaction current) after 1000 cycles of operation.
At the cathode of fuel cells, the reduction of oxygen is the dominant reaction, and great effort has been made to develop highly active and stable catalysts for ORR. 3D N-doped graphene hydrogel-supported Fe3O4 NPs (Fe3O4/3DG) is one of the typical composites used for the ORR catalyst.253 By combining the good ORR catalytic activities of Fe3O4 NPs and N-doped graphene, the good conductivity and large surface area of 3DG, and the synergistic effects between the two components, high performance ORR catalysts were designed and synthesized. The SEM and TEM images in Fig. 13a and b show that the composite has a 3D porous structure with Fe3O4 NPs decorated on the graphene network surface. The Fe3O4 NP has a well-defined crystalline texture that is encapsulated by graphene sheets.
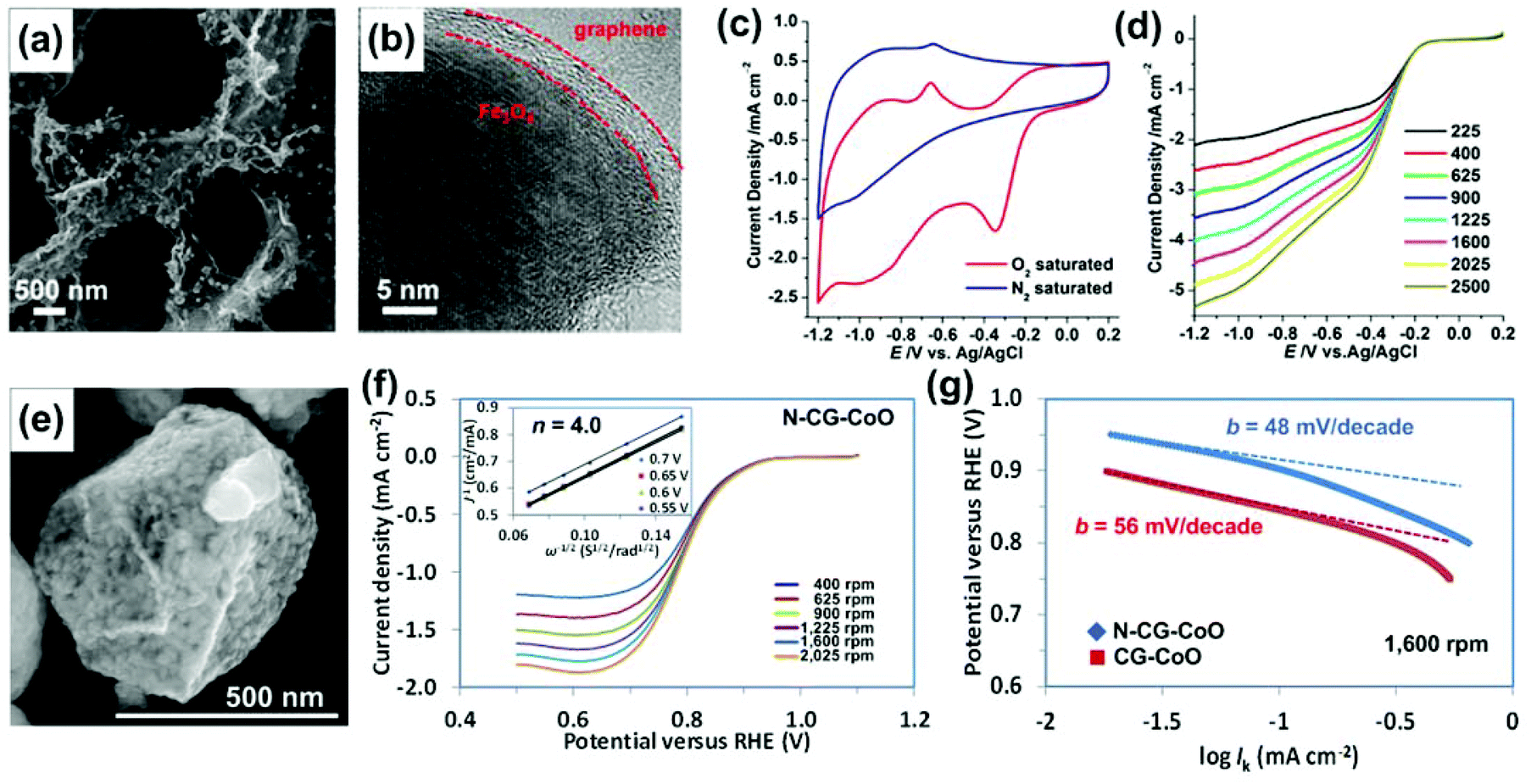 | ||
| Fig. 13 (a, b) SEM and TEM images of Fe3O4/3DG. (c) CVs of Fe3O4/3DG in N2- and O2-saturated 0.1 M aqueous KOH electrolyte solution at a scan rate of 100 mV s−1. (d) LSVs of Fe3O4/3DG in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 at different RDE rotation rates. Reproduced with permission from ref. 253. (e) SEM image of CG–CoO composites. (f) LSVs of N-CG–CoO composites in an O2-saturated 1 M KOH with a sweep rate of 5 mV s−1 at different RDE rotation rates. The inset in (f) shows the corresponding Koutecky–Levich plots (J−1versus ω−0.5) at different potentials. (g) Tafel plots of the CG–CoO and N-CG–CoO composites derived by the mass-transport correction of corresponding RDE data. Reproduced with permission from ref. 243. | ||
The ORR activity of the catalyst was first evaluated by examining the electrocatalytic properties of Fe3O4/3DG in a N2- or O2-saturated 0.1 M aqueous KOH electrolyte solution using CV (Fig. 13c). In the N2-saturated electrolyte, there was no oxygen reduction peak found in the CV curve. In contrast, a significant oxygen reduction peak with an onset potential at −0.19 V was found with the O2-saturated electrolyte, indicating that the oxygen reduction happens on the electrode surface. To study the reaction kinetics of the catalyst, linear sweep voltammograms (LSVs) were recorded in an O2-saturated electrolyte using a rotating disk electrode (RDE). As shown in Fig. 13d, by applying the Koutecky–Levich equation, the LSVs of Fe3O4/3DG revealed a direct four-electron transfer pathway from −0.19 to −1.2 V for producing OH− ions. The direct four-electron transfer indicates a high catalytic activity of the catalysts in ORR since catalysts usually have a two-electron transfer pathway to produce hydroperoxide anions (HO2−) as intermediates in alkaline media.
Nitrogen-doped 3D crumpled graphene and CoO (N-CG–CoO) composites (Fig. 13e) were also reported as ORR catalysts.243 These types of composites were produced by the previously introduced aerosolization method. The unique 3D and hollow graphene structure offers a large surface area and stable anchoring sites for CoO nanocrystals, which enables the full use of the catalyst surface area by minimizing the agglomeration/restacking of the graphene sheets. This catalyst also showed a direct four-electron oxygen reduction process (Fig. 13f). Based on the RDE results, the Tafel slope (Fig. 13g) of the N-CG–CoO was calculated as 48 mV per decade, which is among the smallest Tafel slopes reported for ORR catalysts so far.
Due to its high conductivity, 3D porous structure, and high specific surface area, 3DG has shown great promise as a catalyst support for fuel cell applications and may replace the commonly used catalyst supports such as carbon black and activated carbon when large-scale production of 3DG is realized.
4. Conclusions and outlook
In recent years, many research studies have focused on the design and fabrication of 3DG-based hybrid structures to exploit their potential in energy devices. This article reviews recent progress in the synthesis methods for 3DG-based composites, discusses representative applications of the composites in energy devices, and compares their performance with conventional graphene-based counterparts. Although 3DGs could be fabricated into various morphologies for different applications, the key features of these materials are their 3D porous structure and high electrical conductivity. These unique features have enabled various promising energy applications of 3DGs and 3DG-based composites.A number of future directions can be explored and are expected to further enhance the 3DG-based composites’ unique properties. The adaptability of 3DG-based composites can be improved so that they can accommodate the diverse demands in various energy applications, where the different aspect(s) of the 3DG composites may be emphasized. For example, 3DGs with a high electrical conductivity are desired in some devices (e.g., supercapacitors and LIBs, where 3DG serves as the support); however, since most of the 3DGs in these applications are currently made from GO, the conductivity of the 3DG needs to be further enhanced with easier and more efficient reduction routes. Even so, due to its inherently defective nature, RGO has an electrical conductivity that is much lower than intrinsic graphene. Therefore, in addition to pursuing more effective reduction methods, some “healing” processes, such as annealing254 and plasma255 treatments, may be options for repairing RGO-derived 3DGs by reducing the defects. CVD is widely used to produce single-layered graphene sheets with quality close to intrinsic graphene. However, at present, the component graphene sheets in 3DGs made from CVD are mostly few-layered. One direction is thus to explore the CVD process to obtain 3DG composed of mostly single-layered graphene sheets. On the other hand, defective graphene with high catalytic reactivity is favourable in some energy applications (e.g., in fuel cells), where graphene materials with better-controlled defects (e.g., graphene nanomesh) are needed as building blocks for 3DGs. Better control of the 3DG structures (e.g., porosity, surface area, conductivity, and thickness of component graphene sheets) by tuning the synthesis conditions and choosing appropriate precursors can also result in the easy incorporation of active materials into the 3DG network, potentially improving the overall performance of the 3DG composites. Large-scale and low-cost fabrication of the 3DG-based composites is another critical issue for their practical applications since most of the energy devices need a significant amount of materials for operation.
The performance of the 3DG-based composites in energy devices is essentially determined by their electronic, electrochemical, and structural properties. The control of the composition of the 3DG-based composites (i.e., the mass ratio of 3DG to active materials in a composite) is important for realizing the optimum structures for specific applications. More experimental and theoretical studies are needed to reveal information on the composite system, particularly, the interactions between the two components, i.e., 3DG and loaded active materials (e.g., NPs). In addition, a more profound understanding of the functional performance of 3DG and active materials is essential for the optimum material design and device performance optimization. Examples of such mechanisms include charge/ion storage and transfer at the interface of electrolyte and electrode materials for supercapacitors and lithium-ion batteries, and electron transport during the interactions of electrode materials with the electrolyte for DSSCs and fuel cells.
Due to its high specific surface area, tunable surface behaviour, and high mechanical strength, 3DG has also been used in water remediation, such as the cleanup of oil and removal of heavy metal ions and soluble organic contaminants.256 It is foreseeable that many more energy and environmental applications from 3DG and 3DG-based composites will emerge in the near future.
Acknowledgements
Chen gratefully acknowledges the financial support from the US National Science Foundation (EECS-1001039 and IIP-1128158) and Research Growth Initiative Program of UWM. Lu thanks UAA for an Innovate Award and a Faculty Development Grant for financial support.Notes and references
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 CAS.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480–3498 CrossRef CAS PubMed.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- M.-Q. Zhao, X.-F. Liu, Q. Zhang, G.-L. Tian, J.-Q. Huang, W. Zhu and F. Wei, ACS Nano, 2012, 6, 10759–10769 CAS.
- J. Chang, X. Huang, G. Zhou, S. Cui, P. B. Hallac, J. Jiang, P. T. Hurley and J. Chen, Adv. Mater., 2014, 26, 758–764 CrossRef CAS PubMed.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS PubMed.
- J. L. Vickery, A. J. Patil and S. Mann, Adv. Mater., 2009, 21, 2180–2184 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- L. Jiang and Z. Fan, Nanoscale, 2014, 6, 1922–1945 RSC.
- C. Li and G. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- L. Liu, Z. Niu, L. Zhang and X. Chen, Small, 2014, 10, 2200–2214 CrossRef CAS PubMed.
- S. Nardecchia, D. Carriazo, M. Luisa Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794–830 RSC.
- S. Yin, Z. Niu and X. Chen, Small, 2012, 8, 2458–2463 CrossRef CAS PubMed.
- X. Cao, Z. Yin and H. Zhang, Energy Environ. Sci., 2014, 7, 1850–1865 CAS.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- S. Mao, K. Yu, S. Cui, Z. Bo, G. Lu and J. Chen, Nanoscale, 2011, 3, 2849–2853 RSC.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- S. Mao, H. Pu and J. Chen, RSC Adv., 2012, 2, 2643–2662 RSC.
- H. Bai, C. Li, X. Wang and G. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CAS.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- X. Jiang, Y. Ma, J. Li, Q. Fan and W. Huang, J. Phys. Chem. C, 2010, 114, 22462–22465 CAS.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang, S. Ozden, P. K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, A. K. Roy, M. R. Anantharaman and P. M. Ajayan, ACS Nano, 2013, 7, 7034–7040 CrossRef CAS PubMed.
- L. Van Hoang, T. Huynh Ngoc, H. Le Thuy, H. Nguyen Thi Minh, E.-S. Oh, J. Chung, E. J. Kim, W. M. Choi, B.-S. Kong and S. H. Hur, J. Mater. Chem. A, 2013, 1, 208–211 Search PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- J. Cao, Y. Wang, P. Xiao, Y. Chen, Y. Zhou, J.-H. Ouyang and D. Jia, Carbon, 2013, 56, 389–391 CrossRef CAS PubMed.
- A. Sahu, W. I. Choi and G. Tae, Chem. Commun., 2012, 48, 5820–5822 RSC.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- M. A. Worsley, T. Y. Olson, J. R. I. Lee, T. M. Willey, M. H. Nielsen, S. K. Roberts, P. J. Pauzauskie, J. Biener, J. H. Satcher Jr. and T. F. Baumann, J. Phys. Chem. Lett., 2011, 2, 921–925 CrossRef CAS.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher Jr. and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067–14069 CrossRef CAS PubMed.
- Z. Dong, C. Jiang, H. Cheng, Y. Zhao, G. Shi, L. Jiang and L. Qu, Adv. Mater., 2012, 24, 1856–1861 CrossRef CAS PubMed.
- Z. Han, Z. Tang, P. Li, G. Yang, Q. Zheng and J. Yang, Nanoscale, 2013, 5, 5462–5467 RSC.
- L. Qiu, J. Z. Liu, S. L. Y. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- Y. Su, Y. Zhang, X. Zhuang, S. Li, D. Wu, F. Zhang and X. Feng, Carbon, 2013, 62, 296–301 CrossRef CAS PubMed.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554–2560 CrossRef CAS PubMed.
- X. Xie, Y. Zhou, H. Bi, K. Yin, S. Wan and L. Sun, Sci. Rep., 2013, 3, 2117 Search PubMed.
- O. C. Compton, Z. An, K. W. Putz, B. J. Hong, B. G. Hauser, L. C. Brinson and S. T. Nguyen, Carbon, 2012, 50, 3399–3406 CrossRef CAS PubMed.
- H. S. Ahn, J. M. Kim, C. Park, J.-W. Jang, J. S. Lee, H. Kim, M. Kaviany and M. H. Kim, Sci. Rep., 2013, 3, 1960 Search PubMed.
- F. Liu and T. S. Seo, Adv. Funct. Mater., 2010, 20, 1930–1936 CrossRef CAS.
- Z. Niu, J. Chen, H. H. Hng, J. Ma and X. Chen, Adv. Mater., 2012, 24, 4144–4150 CrossRef CAS PubMed.
- X. Yang, L. Qiu, C. Cheng, Y. Wu, Z.-F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325–7328 CrossRef CAS PubMed.
- Y. Meng, Y. Zhao, C. Hu, H. Cheng, Y. Hu, Z. Zhang, G. Shi and L. Qu, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- K. Sheng, Y. Sun, C. Li, W. Yuan and G. Shi, Sci. Rep., 2012, 2, 247 Search PubMed.
- C. Du, Z. Yao, Y. Chen, H. Bai and L. Li, RSC Adv., 2014, 4, 9133–9138 RSC.
- L. Estevez, A. Kelarakis, Q. Gong, E. H. Da'as and E. P. Giannelis, J. Am. Chem. Soc., 2011, 133, 6122–6125 CrossRef CAS PubMed.
- Z. Fan, B. Liu, J. Wang, S. Zhang, Q. Lin, P. Gong, L. Ma and S. Yang, Adv. Funct. Mater., 2014, 24, 3933–3943 CrossRef CAS.
- J. Li, C.-y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603–4607 CrossRef CAS PubMed.
- M. Yuan, A. Liu, M. Zhao, W. Dong, T. Zhao, J. Wang and W. Tang, Sens. Actuators, B, 2014, 190, 707–714 CrossRef CAS PubMed.
- Y. Chang, J. Li, B. Wang, H. Luo, H. He, Q. Song and L. Zhi, J. Mater. Chem. A, 2013, 1, 14658–14665 CAS.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 13567–13570 CrossRef CAS PubMed.
- Y. Chen, Q. Zhang, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 13101–13110 CAS.
- Y. Hou, B. Zhang, Z. Wen, S. Cui, X. Guo, Z. He and J. Chen, J. Mater. Chem. A, 2014, 2, 13795–13800 CAS.
- L. Li, Z. Hu, Y. Yang, P. Liang, A. Lu, H. Xu, Y. Hu and H. Wu, Chin. J. Chem., 2013, 31, 1290–1298 CrossRef CAS.
- S. Wu, W. Chen and L. Yan, J. Mater. Chem. A, 2014, 2, 2765–2772 CAS.
- J. Yuan, J. Zhu, H. Bi, Z. Zhang, S. Chen, S. Liang and X. Wang, RSC Adv., 2013, 3, 4400–4407 RSC.
- M. Zhang, Y. Wang and M. Jia, Electrochim. Acta, 2014, 129, 425–432 CrossRef CAS PubMed.
- P. Huang, W. Chen and L. Yan, Nanoscale, 2013, 5, 6034–6039 RSC.
- X. Qi, C. Tan, J. Wei and H. Zhang, Nanoscale, 2013, 5, 1440–1451 RSC.
- S.-Z. Zu and B.-H. Han, J. Phys. Chem. C, 2009, 113, 13651–13657 CAS.
- X. Cao, Y. Shi, W. Shi, G. Lu, X. Huang, Q. Yan, Q. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- W. Li, S. Gao, L. Wu, S. Qiu, Y. Guo, X. Geng, M. Chen, S. Liao, C. Zhu, Y. Gong, M. Long, J. Xu, X. Wei, M. Sun and L. Liu, Sci. Rep., 2013, 3, 2125 Search PubMed.
- P. M. Wilson, G. N. Mbah, T. G. Smith, D. Schmidt, R. Y. Lai, T. Hofmann and A. Sinitskii, J. Mater. Chem. C, 2014, 2, 1879–1886 RSC.
- L. Wu, W. Li, P. Li, S. Liao, S. Qiu, M. Chen, Y. Guo, Q. Li, C. Zhu and L. Liu, Small, 2014, 10, 1421–1429 CrossRef CAS PubMed.
- Z. Bo, X. Shuai, S. Mao, H. Yang, J. Qian, J. Chen, J. Yan and K. Cen, Sci. Rep., 2014, 4, 4684 Search PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- Z. Chen, W. Ren, B. Liu, L. Gao, S. Pei, Z.-S. Wu, J. Zhao and H.-M. Cheng, Carbon, 2010, 48, 3543–3550 CrossRef CAS PubMed.
- S.-M. Yoon, W. M. Choi, H. Baik, H.-J. Shin, I. Song, M.-S. Kwon, J. J. Bae, H. Kim, Y. H. Lee and J.-Y. Choi, ACS Nano, 2012, 6, 6803–6811 CrossRef CAS PubMed.
- X. Xiao, T. E. Beechem, M. T. Brumbach, T. N. Lambert, D. J. Davis, J. R. Michael, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, ACS Nano, 2012, 6, 3573–3579 CrossRef CAS PubMed.
- X. Xiao, J. R. Michael, T. Beechem, A. McDonald, M. Rodriguez, M. T. Brtunbach, T. N. Lambert, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, J. Mater. Chem., 2012, 22, 23749–23754 RSC.
- M. Zhou, T. Lin, F. Huang, Y. Zhong, Z. Wang, Y. Tang, H. Bi, D. Wan and J. Lin, Adv. Funct. Mater., 2013, 23, 2263–2269 CrossRef CAS.
- N. Duc Dung, N.-H. Tai, S.-B. Lee and W.-S. Kuo, Energy Environ. Sci., 2012, 5, 7908–7912 Search PubMed.
- H.-B. Yao, J. Ge, C.-F. Wang, X. Wang, W. Hu, Z.-J. Zheng, Y. Ni and S.-H. Yu, Adv. Mater., 2013, 25, 6692–6698 CrossRef CAS PubMed.
- J. Chen, K. Sheng, P. Luo, C. Li and G. Shi, Adv. Mater., 2012, 24, 4569–4573 CrossRef CAS PubMed.
- H. Wang, G. Wang, Y. Ling, F. Qian, Y. Song, X. Lu, S. Chen, Y. Tong and Y. Li, Nanoscale, 2013, 5, 10283–10290 RSC.
- S. Ye, J. Feng and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 7122–7129 CAS.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- D. Fan, Y. Liu, J. He, Y. Zhou and Y. Yang, J. Mater. Chem., 2012, 22, 1396–1402 RSC.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff and S. O. Kim, Angew. Chem., Int. Ed., 2010, 49, 10084–10088 CrossRef CAS PubMed.
- H. Wang, D. Zhang, T. Yan, X. Wen, J. Zhang, L. Shi and Q. Zhong, J. Mater. Chem. A, 2013, 1, 11778–11789 CAS.
- X. Huang, K. Qian, J. Yang, J. Zhang, L. Li, C. Yu and D. Zhao, Adv. Mater., 2012, 24, 4419–4423 CrossRef CAS PubMed.
- G.-h. Moon, Y. Shin, D. Choi, B. W. Arey, G. J. Exarhos, C. Wang, W. Choi and J. Liu, Nanoscale, 2013, 5, 6291–6296 RSC.
- W. Ouyang, J. Sun, J. Memon, C. Wang, J. Geng and Y. Huang, Carbon, 2013, 62, 501–509 CrossRef CAS PubMed.
- X. Cao, Z. Zeng, W. Shi, P. Yep, Q. Yan and H. Zhang, Small, 2013, 9, 1703–1707 CrossRef CAS PubMed.
- X. Liu, J. Cheng, W. Li, X. Zhong, Z. Yang, L. Gu and Y. Yu, Nanoscale, 2014, 6, 7817–7822 RSC.
- S. Sattayasamitsathit, Y. Gu, K. Kaufmann, W. Jia, X. Xiao, M. Rodriguez, S. Minteer, J. Cha, D. B. Burckel, C. Wang, R. Polsky and J. Wang, J. Mater. Chem. A, 2013, 1, 1639–1645 CAS.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943–8949 CrossRef CAS PubMed.
- X. Ma, M. R. Zachariah and C. D. Zangmeister, Nano Lett., 2012, 12, 486–489 CrossRef CAS PubMed.
- R. Ruoff, Nature, 2012, 483, S42–S42 CrossRef PubMed.
- W.-N. Wang, Y. Jiang and P. Biswas, J. Phys. Chem. Lett., 2012, 3, 3228–3233 CrossRef CAS.
- X. Ma, M. R. Zachariah and C. D. Zangmeister, J. Phys. Chem. C, 2013, 117, 3185–3191 CAS.
- C. D. Zangmeister, X. Ma and M. R. Zachariah, Chem. Mater., 2012, 24, 2554–2557 CrossRef CAS.
- Y. Chen, F. Guo, A. Jachak, S.-P. Kim, D. Datta, J. Liu, I. Kulaots, C. Vaslet, H. D. Jang, J. Huang, A. Kane, V. B. Shenoy and R. H. Hurt, Nano Lett., 2012, 12, 1996–2002 CrossRef CAS PubMed.
- Y. Chen, F. Guo, Y. Qiu, H. Hu, I. Kulaots, E. Walsh and R. H. Hurt, ACS Nano, 2013, 7, 3744–3753 CrossRef CAS PubMed.
- H. D. Jang, S. K. Kim, H. Chang, J.-W. Choi, J. Luo and J. Huang, Aerosol Sci. Technol., 2013, 47, 93–98 CrossRef CAS.
- H. D. Jang, S. K. Kim, H. Chang, K.-M. Roh, J.-W. Choi and J. Huang, Biosens. Bioelectron., 2012, 38, 184–188 CrossRef CAS PubMed.
- Y. Jiang, W.-N. Wang, P. Biswas and J. D. Fortner, ACS Appl. Mater. Interfaces, 2014, 6, 11766–11774 CAS.
- J. Luo, X. Zhao, J. Wu, H. D. Jang, H. H. Kung and J. Huang, J. Phys. Chem. Lett., 2012, 3, 1824–1829 CrossRef CAS.
- K. Sohn, Y. J. Na, H. Chang, K.-M. Roh, H. D. Jang and J. Huang, Chem. Commun., 2012, 48, 5968–5970 RSC.
- S. H. Choi and Y. C. Kang, Carbon, 2014, 79, 58–66 CrossRef CAS PubMed.
- S. Ci, S. Mao, T. Huang, Z. Wen, D. A. Steeber and J. Chen, Electroanalysis, 2014, 26, 1326–1334 CrossRef CAS.
- S. Mao, Z. Wen, H. Kim, G. Lu, P. Hurley and J. Chen, ACS Nano, 2012, 6, 7505–7513 CrossRef CAS PubMed.
- Z. Bo, Y. Yang, J. Chen, K. Yu, J. Yan and K. Cen, Nanoscale, 2013, 5, 5180–5204 RSC.
- Z. Bo, K. Yu, G. Lu, P. Wang, S. Mao and J. Chen, Carbon, 2011, 49, 1849–1858 CrossRef CAS PubMed.
- Z. Bo, W. Zhu, W. Ma, Z. Wen, X. Shuai, J. Chen, J. Yan, Z. Wang, K. Cen and X. Feng, Adv. Mater., 2013, 25, 5799–5806 CrossRef CAS PubMed.
- K. Davami, M. Shaygan, N. Kheirabi, J. Zhao, D. A. Kovalenko, M. H. Rümmeli, J. Opitz, G. Cuniberti, J.-S. Lee and M. Meyyappan, Carbon, 2014, 72, 372–380 CrossRef CAS PubMed.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113–224 CrossRef CAS.
- D. H. Seo, S. Kumar and K. Ostrikov, Carbon, 2011, 49, 4331–4339 CrossRef CAS PubMed.
- C. Yang, H. Bi, D. Wan, F. Huang, X. Xie and M. Jiang, J. Mater. Chem. A, 2013, 1, 770–775 CAS.
- K. Yu, P. Wang, G. Lu, K.-H. Chen, Z. Bo and J. Chen, J. Phys. Chem. Lett., 2011, 2, 537–542 CrossRef CAS.
- K. Yu, Z. Wen, H. Pu, G. Lu, Z. Bo, H. Kim, Y. Qian, E. Andrew, S. Mao and J. Chen, J. Mater. Chem. A, 2013, 1, 188–193 CAS.
- H.-C. Chang, H.-Y. Chang, W.-J. Su, K.-Y. Lee and W.-C. Shih, Appl. Surf. Sci., 2012, 258, 8599–8602 CrossRef CAS PubMed.
- S. Jin, N. Li, H. Cui and C. Wang, Nano Energy, 2013, 2, 1128–1136 CrossRef CAS PubMed.
- S. Mao, Z. Wen, S. Ci, X. Guo, K. Ostrikov and J. Chen, Small, 2014 DOI:10.1002/smll.201401598.
- S. Mao, K. Yu, J. Chang, D. A. Steeber, L. E. Ocola and J. Chen, Sci. Rep., 2013, 3, 1696 Search PubMed.
- D. H. Seo, Z. J. Han, S. Kumar and K. Ostrikov, Adv. Energy Mater., 2013, 3, 1316–1323 CrossRef CAS.
- D. H. Seo, A. E. Rider, S. Kumar, L. K. Randeniya and K. Ostrikov, Carbon, 2013, 60, 221–228 CrossRef CAS PubMed.
- G. Xiong, K. P. S. S. Hembram, R. G. Reifenberger and T. S. Fisher, J. Power Sources, 2013, 227, 254–259 CrossRef CAS PubMed.
- K. Chen, L. Chen, Y. Chen, H. Bai and L. Li, J. Mater. Chem., 2012, 22, 20968–20976 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed.
- S. Mao, Z. Wen, Z. Bo, J. Chang, X. Huang and J. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 9881–9889 Search PubMed.
- L.-h. Song, S. N. Lim, K.-K. Kang and S. B. Park, J. Mater. Chem. A, 2013, 1, 6719–6722 CAS.
- S. Ye and J. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 9671–9679 CAS.
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Muellen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed.
- Z. Xu, Z. Li, C. M. B. Holt, X. Tan, H. Wang, B. S. Amirkhiz, T. Stephenson and D. Mitlin, J. Phys. Chem. Lett., 2012, 3, 2928–2933 CrossRef CAS.
- J. Zhi, W. Zhao, X. Liu, A. Chen, Z. Liu and F. Huang, Adv. Funct. Mater., 2014, 24, 2013–2019 CrossRef CAS.
- J. Luo, H. D. Jang and J. Huang, ACS Nano, 2013, 7, 1464–1471 CrossRef CAS PubMed.
- J. R. Miller, R. A. Outlaw and B. C. Holloway, Science, 2010, 329, 1637–1639 CrossRef CAS PubMed.
- Z.-S. Wu, G. Zhou, L.-C. Yin, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS PubMed.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801–2810 CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef CAS PubMed.
- Y. Shao, H. Wang, Q. Zhang and Y. Li, J. Mater. Chem. C, 2013, 1, 1245–1251 RSC.
- T. Zhai, F. Wang, M. Yu, S. Xie, C. Liang, C. Li, F. Xiao, R. Tang, Q. Wu, X. Lu and Y. Tong, Nanoscale, 2013, 5, 6790–6796 RSC.
- U. M. Patil, J. S. Sohn, S. B. Kulkarni, S. C. Lee, H. G. Park, K. V. Gurav, J. H. Kim and S. C. Jun, ACS Appl. Mater. Interfaces, 2014, 6, 2448–2456 Search PubMed.
- Y. Wei, S. Chen, D. Su, B. Sun, J. Zhu and G. Wang, J. Mater. Chem. A, 2014, 2, 8103–8109 CAS.
- C. Wu, S. Deng, H. Wang, Y. Sun, J. Liu and H. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 1106–1112 CAS.
- W. Zhou, X. Cao, Z. Zeng, W. Shi, Y. Zhu, Q. Yan, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2216–2221 CAS.
- J. Yuan, J. Zhu, H. Bi, X. Meng, S. Liang, L. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2013, 15, 12940–12945 RSC.
- H. Wang, H. Yi, X. Chen and X. Wang, J. Mater. Chem. A, 2014, 2, 1165–1173 CAS.
- H. Zhang, A. Xie, C. Wang, H. Wang, Y. Shen and X. Tian, ChemPhysChem, 2014, 15, 366–373 CrossRef CAS PubMed.
- L. Hu, W. Chen, X. Xie, N. Liu, Y. Yang, H. Wu, Y. Yao, M. Pasta, H. N. Alshareef and Y. Cui, ACS Nano, 2011, 5, 8904–8913 CrossRef CAS PubMed.
- Z. Zhang, F. Xiao, Y. Guo, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2227–2233 CAS.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer III, S. Dai, M. P. Paranthaman and A. K. Naskar, Langmuir, 2014, 30, 900–910 CrossRef CAS PubMed.
- J. Kumagai, IEEE Spectrum, 2012, 49, 45–46 CrossRef.
- P. G. Bruce, Solid State Ionics, 2008, 179, 752–760 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- B. A. Boukamp, G. C. Lesh and R. A. Huggins, J. Electrochem. Soc., 1981, 128, 725–729 CrossRef CAS PubMed.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- I. A. Courtney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045–2052 CrossRef CAS PubMed.
- Y. Huang, D. Wu, J. Wang, S. Han, L. Lv, F. Zhang and X. Feng, Small, 2014, 10, 2226–2232 CrossRef CAS PubMed.
- H. X. Zhang, C. Feng, Y. C. Zhai, K. L. Jiang, Q. Q. Li and S. S. Fan, Adv. Mater., 2009, 21, 2299–2304 CrossRef CAS.
- M. S. Park, S. A. Needham, G. X. Wang, Y. M. Kang, J. S. Park, S. X. Dou and H. K. Liu, Chem. Mater., 2007, 19, 2406–2410 CrossRef CAS.
- T. Bhardwaj, A. Antic, B. Pavan, V. Barone and B. D. Fahlman, J. Am. Chem. Soc., 2010, 132, 12556–12558 CrossRef CAS PubMed.
- D. H. Wang, R. Kou, D. Choi, Z. G. Yang, Z. M. Nie, J. Li, L. V. Saraf, D. H. Hu, J. G. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS PubMed.
- Q. H. Yang, F. Y. Su, C. H. You, Y. B. He, W. Lv, W. Cui, F. M. Jin, B. H. Li and F. Y. Kang, J. Mater. Chem., 2010, 20, 9644–9650 RSC.
- G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2014, 26, 849–864 CrossRef CAS PubMed.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Angew. Chem., Int. Ed., 2014, 126, 1428–1433 CrossRef.
- L. Xiao, D. Wu, S. Han, Y. Huang, S. Li, M. He, F. Zhang and X. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 3764–3769 CAS.
- S. M. Lee, S. H. Choi, J.-K. Lee and Y. C. Kang, Electrochim. Acta, 2014, 132, 441–447 CrossRef CAS PubMed.
- Y. Y. Li, Q. W. Zhang, J. L. Zhu, X. L. Wei and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3163–3168 CAS.
- X. Zhu, P. Zhang, S. Xu, X. Yan and Q. Xue, ACS Appl. Mater. Interfaces, 2014, 6, 11665–11674 CAS.
- X. Jiang, X. L. Yang, Y. H. Zhu, H. L. Jiang, Y. F. Yao, P. Zhao and C. Z. Li, J. Mater. Chem. A, 2014, 2, 11124–11133 CAS.
- L. Shen, X. Zhang, H. Li, C. Yuan and G. Cao, J. Phys. Chem. Lett., 2011, 2, 3096–3101 CrossRef CAS.
- S. H. Choi, Y. N. Ko, J. K. Lee and Y. C. Kang, Sci. Rep., 2014, 4, 5786 CrossRef PubMed.
- S. H. Choi and Y. C. Kang, ChemSusChem, 2014, 7, 523–528 CrossRef CAS PubMed.
- S. H. Choi and Y. C. Kang, Chem. – Eur. J., 2014, 20, 6294–6299 CrossRef CAS PubMed.
- B. G. Choi, S. J. Chang, Y. B. Lee, J. S. Bae, H. J. Kim and Y. S. Huh, Nanoscale, 2012, 4, 5924–5930 RSC.
- J. J. Ma, J. L. Wang, Y. S. He, X. Z. Liao, J. Chen, J. Z. Wang, T. Yuan and Z. F. Ma, J. Mater. Chem. A, 2014, 2, 9200–9207 CAS.
- X. H. Cao, Y. M. Shi, W. H. Shi, X. H. Rui, Q. Y. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433–3438 CrossRef CAS PubMed.
- J. Wang, J. Liu, D. Chao, J. Yan, J. Lin and Z. X. Shen, Adv. Mater., 2014, 26, 7162–7169 CrossRef CAS PubMed.
- Y. Zhao, J. Feng, X. Liu, F. Wang, L. Wang, C. Shi, L. Huang, X. Feng, X. Chen, L. Xu, M. Yan, Q. Zhang, X. Bai, H. Wu and L. Mai, Nat. Commun., 2014, 5, 4565 CAS.
- C. Wang, K. Su, W. Wan, H. Guo, H. Zhou, J. Chen, X. Zhang and Y. Huang, J. Mater. Chem. A, 2014, 2, 5018–5023 CAS.
- J. Qin, C. N. He, N. Q. Zhao, Z. Y. Wang, C. S. Shi, E. Z. Liu and J. J. Li, ACS Nano, 2014, 8, 1728–1738 CrossRef CAS PubMed.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed.
- X. Li, A. Dhanabalan, L. Gu and C. Wang, Adv. Energy Mater., 2012, 2, 238–244 CrossRef CAS.
- Y. Huang, D. Wu, S. Han, S. Li, L. Xiao, F. Zhang and X. Feng, ChemSusChem, 2013, 6, 1510–1515 CrossRef CAS PubMed.
- R. Wang, C. Xu, J. Sun, L. Gao and H. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 3427–3436 CAS.
- S. Chen, P. Bao, X. Huang, B. Sun and G. Wang, Nano Res., 2014, 7, 85–94 CrossRef CAS.
- N. Li, S. Jin, Q. Liao, H. Cui and C. X. Wang, Nano Energy, 2014, 5, 105–115 CrossRef CAS PubMed.
- P. Wu, H. Wang, Y. Tang, Y. Zhou and T. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 3546–3552 CAS.
- X. Xin, X. F. Zhou, F. Wang, X. Y. Yao, X. X. Xu, Y. M. Zhu and Z. P. Liu, J. Mater. Chem., 2012, 22, 7724–7730 RSC.
- X. S. Zhou, Y. X. Yin, A. M. Cao, L. J. Wan and Y. G. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 2824–2828 CAS.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- J. S. Luo, J. L. Liu, Z. Y. Zeng, C. F. Ng, L. J. Ma, H. Zhang, J. Y. Lin, Z. X. Shen and H. J. Fan, Nano Lett., 2013, 13, 6136–6143 CrossRef CAS PubMed.
- W. Wei, S. Yang, H. Zhou, I. Lieberwirth, X. Feng and K. Müllen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- J. Zhu, D. Yang, X. Rui, D. Sim, H. Yu, H. E. Hoster, P. M. Ajayan and Q. Yan, Small, 2013, 9, 3390–3397 CrossRef CAS PubMed.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 CrossRef CAS PubMed.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- N. L. Yang, J. Zhai, D. Wang, Y. S. Chen and L. Jiang, ACS Nano, 2010, 4, 887–894 CrossRef CAS PubMed.
- K. J. Williams, C. A. Nelson, X. Yan, L.-S. Li and X. Zhu, ACS Nano, 2013, 7, 1388–1394 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2012, 6, 9777–9789 CrossRef CAS PubMed.
- B. Tang, G. Hu, H. Gao and Z. Shi, J. Power Sources, 2013, 234, 60–68 CrossRef CAS PubMed.
- W. Han, L. Ren, L. Gong, X. Qi, Y. Liu, L. Yang, X. Wei and J. Zhong, ACS Sustainable Chem. Eng., 2013, 2, 741–748 CrossRef.
- S. Yin, Y. Goldovsky, M. Herzberg, L. Liu, H. Sun, Y. Zhang, F. Meng, X. Cao, D. D. Sun, H. Chen, A. Kushmaro and X. Chen, Adv. Funct. Mater., 2013, 23, 2972–2978 CrossRef CAS.
- H. J. Ahn, I. H. Kim, J. C. Yoon, S. I. Kim and J. H. Jang, Chem. Commun., 2014, 50, 2412–2415 RSC.
- W. Hong, Y. Xu, G. Lu, C. Li and G. Shi, Electrochem. Commun., 2008, 10, 1555–1558 CrossRef CAS PubMed.
- L. Kavan, J. H. Yum, M. K. Nazeeruddin and M. Gratzel, ACS Nano, 2011, 5, 9171–9178 CrossRef CAS PubMed.
- H. Wang, K. Sun, F. Tao, D. J. Stacchiola and Y. H. Hu, Angew. Chem., Int. Ed., 2013, 52, 9210–9214 CrossRef CAS PubMed.
- H. K. Mulmudi, S. K. Batabyal, M. Rao, R. R. Prabhakar, N. Mathews, Y. M. Lam and S. G. Mhaisalkar, Phys. Chem. Chem. Phys., 2011, 13, 19307–19309 RSC.
- B. Yang, X. Zuo, P. Chen, L. Zhou, X. Yang, H. Zhang, G. Li, M. Wu, Y.-q. Ma, S. Jin and X. S. Chen, ACS Appl. Mater. Interfaces, 2014 DOI:10.1021/am5040522.
- B. E. Logan, B. Hamelers, R. A. Rozendal, U. Schrorder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- D. Wang, Y. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- B. Huang, S. R. Wang, R. Z. Liu and T. L. Wen, J. Power Sources, 2007, 167, 288–294 CrossRef CAS PubMed.
- T. Huang, S. Mao, G. Zhou, Z. Wen, X. Huang, S. Ci and J. Chen, Nanoscale, 2014, 6, 9608–9613 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- N. Soin, S. S. Roy, T. H. Lim and J. A. D. McLaughlin, Mater. Chem. Phys., 2011, 129, 1051–1057 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Mueller, R. Schloegl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165–2173 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. a. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- X. Chen, Z. Cai, X. Chen and M. Oyama, J. Mater. Chem. A, 2014, 2, 315–320 CAS.
- A. K. Das, R. K. Layek, N. H. Kim, D. Jung and J. H. Lee, Nanoscale, 2014, 6, 10657–10665 RSC.
- S. Mao, Z. Wen, T. Huang, Y. Hou and J. Chen, Energy Environ. Sci., 2014, 7, 609–616 CAS.
- J.-J. Shao, Z.-J. Li, C. Zhang, L.-F. Zhang and Q.-H. Yang, J. Mater. Chem. A, 2014, 2, 1940–1946 CAS.
- H. D. Jang, S. K. Kim, H. Chang, J.-W. Choi, J. Luo and J. Huang, Aerosol Sci. Technol., 2013, 47, 93–98 CrossRef CAS.
- T. Maiyalagan, X. Dong, P. Chen and X. Wang, J. Mater. Chem., 2012, 22, 5286–5290 RSC.
- X. Wang, C. Li and G. Shi, Phys. Chem. Chem. Phys., 2014, 16, 10142–10148 RSC.
- C. Hu, H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. Dai and L. Qu, Adv. Mater., 2012, 24, 5493–5498 CrossRef CAS PubMed.
- C. Hu, X. Zhai, Y. Zhao, K. Bian, J. Zhang, L. Qu, H. Zhang and H. Luo, Nanoscale, 2014, 6, 2768–2775 RSC.
- K. P. Prasad, Y. Chen and P. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 3387–3393 CAS.
- Y. Zhang, M. Chu, L. Yang, Y. Tan, W. Deng, M. Ma, X. Su and Q. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 12808–12814 CAS.
- J. Tian, R. Ning, Q. Liu, A. M. Asiri, A. O. Al-Youbi and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 1011–1017 CAS.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Muellen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- V. Lopez, R. S. Sundaram, C. Gomez-Navarro, D. Olea, M. Burghard, J. Gomez-Herrero, F. Zamora and K. Kern, Adv. Mater., 2009, 21, 4683–4686 CrossRef CAS.
- M. Cheng, R. Yang, L. Zhang, Z. Shi, W. Yang, D. Wang, G. Xie, D. Shi and G. Zhang, Carbon, 2012, 50, 2581–2587 CrossRef CAS PubMed.
- Z. Niu, L. Liu, L. Zhang and X. Chen, Small, 2014, 10, 3434–3441 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |