
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterizing nanoparticles in complex biological media and physiological fluids with depolarized dynamic light scattering
S.
Balog†
*a,
L.
Rodriguez-Lorenzo†
a,
C. A.
Monnier
a,
M.
Obiols-Rabasa
b,
B.
Rothen-Rutishauser
a,
P.
Schurtenberger
b and
A.
Petri-Fink
*ac
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: sandor.balog@unifr.ch; alke.fink@unifr.ch
bPhysical Chemistry, Department of Chemistry, Lund University, 221 00 Lund, Sweden
cChemistry Department, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland
First published on 19th January 2015
Abstract
Light scattering is one of the few techniques available to adequately characterize suspended nanoparticles (NPs) in real time and in situ. However, when it comes to NPs in multicomponent and optically complex aqueous matrices – such as biological media and physiological fluids – light scattering suffers from lack of selectivity, as distinguishing the relevant optical signals from the irrelevant ones is very challenging. We meet this challenge by building on depolarized scattering: Unwanted signals from the matrix are completely suppressed. This approach yields information with an unprecedented signal-to-noise ratio in favour of the NPs and NP-biomolecule corona complexes, which in turn opens the frontier to scattering-based studies addressing the behaviour of NPs in complex physiological/biological fluids.
Introduction
The appeal of nanomedicine lies in its potential of addressing some of the most important and current challenges in diagnosis and treatment by exploiting the unique properties of nanoparticles (NPs).1 This vast field includes designing NPs capable of targeting certain cells to deliver drugs, genetic material, NPs or nanofibers used for tissue engineering or nanoscale devices or sensors.2 Significant progress has been made in the materials field in recent years, and NPs have been designed in virtually all shapes and sizes3–7 to take advantage of their physico-chemical properties to a maximum degree. However, before NPs can actually interact with living cells/organisms, their surfaces are inevitably confronted to biological fluids – such as cell culture medium, blood or lung fluid – whose components (i.e., bio- and small molecules, such as proteins, antibodies, salts/ions, vitamins, lipids)8 will inevitably interact with the particle surfaces.9 This has several critical consequences; for example, a tightly bound immobile protein layer is known to form on the particle surface (i.e., the so-called hard corona) and possibly a weakly associated mobile layer (i.e., the soft corona).10 NP aggregation, which is a common phenomenon in this complex environment, might also be induced and consequently has to be taken into account.11 All these various factors underline the complexity of these systems and importance of precisely understanding NP behavior in biologically relevant surroundings at a basic level, which is indispensable in developing any kind of nanomaterial for subsequent medical application.Currently, it is widely accepted that the cellular fate, as well as the subsequent absorption, distribution, metabolism, and clearance is dictated by the NP behavior in the biological environment.12,13 In this context, the interaction of NPs with living matter has been heavily addressed over the past years.14–16 Although NP behavior in biological fluids is fundamental for either exploiting their potentially beneficial properties or mitigating the risks they may pose to human health and the environment,17,18 our general understanding is still lagging behind current NP development.19 Given the promises, concerns and commercial aspects of NPs, developing reliable and fast experimental protocols dedicated to the characterization of suspended NPs in biological media is pressing.19
Among the dozen of techniques available, priority is usually given to a handful only, i.e., to methods that are non-invasive and capable of performing beyond static and single-point measurements – including in situ and real time data acquisition – allowing the investigation of kinetic phenomena20,21 and the observation of transient states.8,22 In this regard, dynamic light scattering (DLS) is particularly popular and widely adopted for its straightforward use and its highly quantitative nature. However, the subtleties of scattering theory,23 the founding pillars of the technique, may be easily obscured by the experimental simplicity, especially when dealing with multicomponent and optically complex systems.24 An inherent – yet often neglected – characteristic feature of scattering experiments is that the measured primary signal, e.g., the scattering intensity trace, may also contain contributions from the biological environment. Unfortunately, these contributions to the scattering intensity are generally too significant compared to the NPs and NP-biomolecule complexes,11 and thus should not be omitted. Otherwise, the primary data may easily lead to wrong interpretation.25,26 However, selectively separating the relevant signals from the irrelevant ones is virtually impossible, and thus jeopardizes the effective usage of DLS for investigating nanoparticles in application-relevant surroundings.
In this article, we present an approach to circumvent this obstacle and show that regardless of the presence of a complex matrix, unwanted scattering signals from dissolved free biomolecules, e.g., proteins that are not associated to the NPs, can be completely suppressed, resulting in an extraordinary signal-to-noise ratio. Our approach relies on the optical anisotropy found in many NPs and can be easily extended to systems without apparent colloidal stability. In addition, this method does not rely on the presence of fluorescent tags,27 which are known to substantially alter the NP surface properties.28 To demonstrate the power of our approach, we designed a representative model system consisting of Au NPs in commonly used buffers and cell culture media. As-synthesized citrate Au NPs were investigated and compared to three types of poly(ethylene glycol) (PEG) coated Au NPs to examine the influence of surface functionality. Thiolated PEG carrying either an amine (PEG-NH3+), a carboxylic acid (PEG-COO−), or a methoxy- (PEG-CH3) end group, respectively, was used. Each particle type was then incubated in four different and increasingly complex biological media: (1) Phosphate buffer (PBS, 10 mM sodium phosphate monobasic/disodium phosphate hydrogen, pH 7), (2) Bovine Serum Albumin (BSA, a high molecular weight protein and major component of serum), in PBS (5 mg mL−1), (3) Dulbecco's Modified Eagle Medium (DMEM, a commonly used cell culture medium containing amino acids, salts, glucose, and vitamins), and (4) Fetal Bovine Serum (FBS) supplemented DMEM (10%).
Theory of depolarized light scattering
Owing to their polycrystalline nature, round metallic NPs have an inhomogeneous internal structure and are not perfectly spherical.29–32 These imperfections are strong enough to result in a small but highly relevant optical anisotropy.33,34 When excited by electromagnetic waves, Au NPs support coherent oscillations of the surface conduction electrons. This phenomenon, i.e., the confined oscillations of the charge density, is referred to as localized surface plasmon resonance (LSPR).35 It has been shown that upon scattering, LSPR coupled with such optical anisotropy results in a depolarized speckle pattern, whose temporal fluctuations yield precise information on the Au NP size.34 The depolarized field auto-correlation function decreases exponentially in time g1,vh(t) = e−Γt,23 and for uniform spherical particles, the decay constant Γ is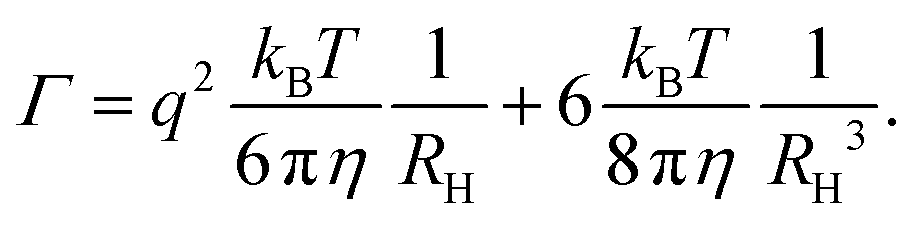 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin (θ/2) the momentum transfer, θ the scattering angle, λ the wavelength of the scattered waves and n the refractive index of the solution. The first and second term represents translational and rotational diffusion, respectively. The second term is independent of the angle of observation and dominates at low angles. Owing to the RH−3 dependence, rotational diffusion is far more sensitive to changes in particle size than translational diffusion.34 For polydisperse samples, the field correlation function is expressed as the Laplace transform of the probability density function describing the dispersion in the relaxation rate. Accordingly, the correlation function is written as
sin (θ/2) the momentum transfer, θ the scattering angle, λ the wavelength of the scattered waves and n the refractive index of the solution. The first and second term represents translational and rotational diffusion, respectively. The second term is independent of the angle of observation and dominates at low angles. Owing to the RH−3 dependence, rotational diffusion is far more sensitive to changes in particle size than translational diffusion.34 For polydisperse samples, the field correlation function is expressed as the Laplace transform of the probability density function describing the dispersion in the relaxation rate. Accordingly, the correlation function is written as | (2) |
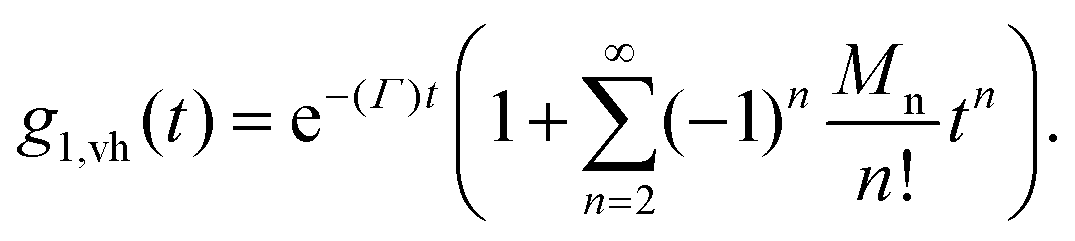 | (3) |
 | (4) |
 | (5) |
At early correlation times
| lng1,vh (t) ≅ −〈Γ〉t. | (6) |
The average size, therefore, can be estimated with eqn (1) and (6).
Results and discussion
UV-Vis spectroscopy
Au NPs possess unique optical properties, resulting in a very particular UV-Vis spectrum which corresponds to a typical deep ruby color.37 LSPR is sensitive to the local dielectric environment, and thus has been used to obtain information about protein adsorption and possible NP aggregation.35,38 Therefore, certain conclusions can be drawn solely from the UV-Vis spectrum of the NP suspension. This approach is justified for as-synthesized citrate Au NPs, and changes in the LSPR can be easily identified in the UV-Vis extinction spectrum (Fig. 1).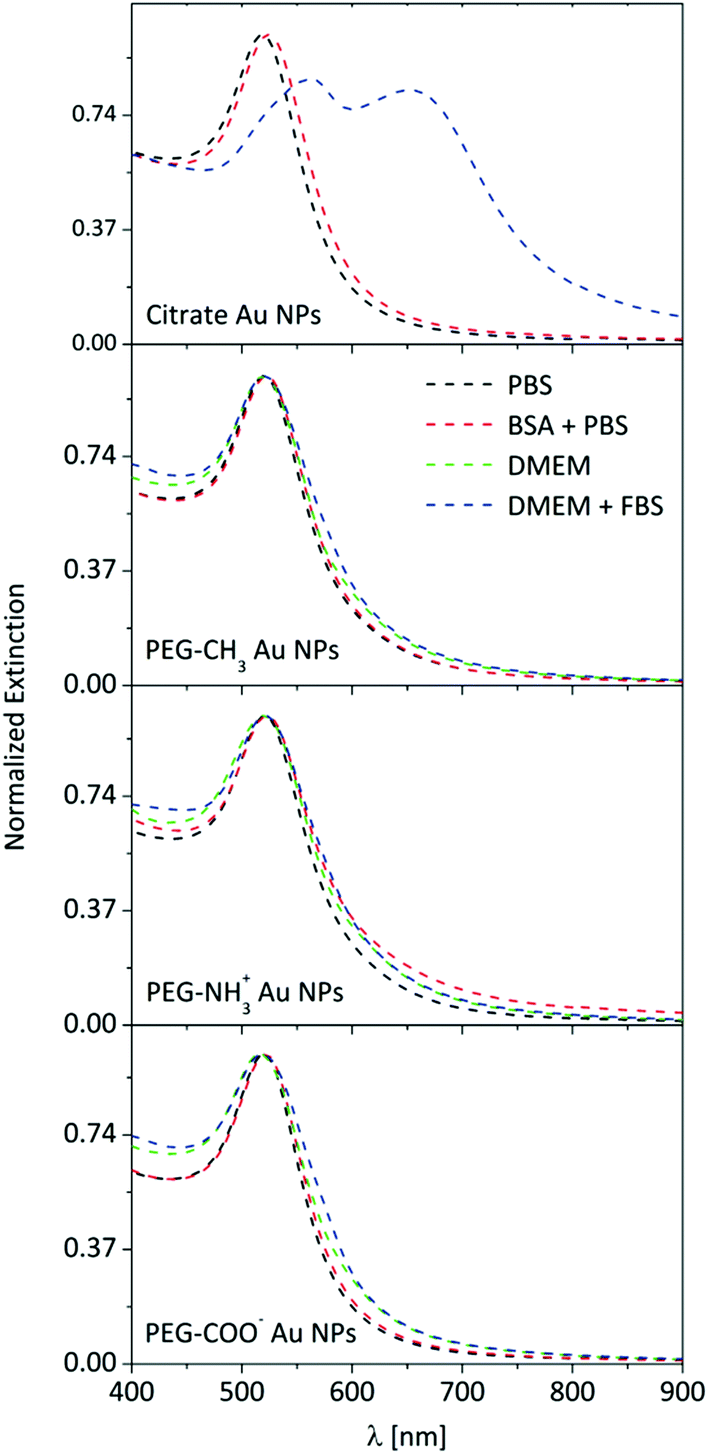 | ||
| Fig. 1 UV-Vis extinction spectra of Au NPs in PBS, in a BSA-PBS mixture, in DMEM, and in FBS supplemented DMEM. | ||
The extinction spectra of the citrate Au NPs are indicative of interactions. While the LSPR in PBS is centered at 519 nm, a redshift is observed (524 nm) after incubation with 5 mg mL−1 BSA, which is attributed to the change in the refractive index of the particle surface, upon the adsorption of BSA.39 The suspension of citrate Au NPs collapsed in DMEM. Precipitation and rapid sedimentation in the order of minutes were visible even by naked eye. Consequently, no UV-Vis spectrum was obtained due to this strong aggregation and rapid particle sedimentation. However, incubating the citrate Au NPs in DMEM with 10% FBS brought peculiar features into the extinction spectrum: the amplitude of the LSPR at 520 nm decreased, its center shifted, and a new resonance band developed in the near-infrared region. It has previously been shown that these features can be attributed to plasmonic coupling between closely packed NPs.40 Interestingly, the presence of FBS prevented the collapse of spinning out of control and re-stabilized the already-formed aggregates, most likely via a mechanism similar to surface-charge stabilization. Several groups reported that particle aggregation can be prevented by the addition of albumin or serum, owing to protein adsorption onto the particle surface.41–44 Therefore, it is strongly indicated that the adsorption/conjugation of proteins on the NP surface is rapid, starting as soon as the NPs are dispersed in cellular medium. In contrast to the citrate Au NPs, none of the PEGylated Au NPs exhibited any significant changes in the UV-Vis spectrum (i.e., either peak shift or peak broadening or rise in the baseline of the spectrum, which was independent of the media). The absence of significant changes in the LSPR curve can be explained by the presence of the polymer shell, which acts as a dielectric spacer and is an effective insulator, and thus hinders additional coupling of LSPR oscillations between associated particles as well as decreases the sensitivity to the refractive index changes.45 Therefore, UV-Vis becomes practically insensitive, and monitoring the behavior (e.g., protein adsorption or particle aggregation) of such surface-functionalized NPs in biological media is not conclusive.
Dynamic light scattering
DLS may overcome the limitations of UV-Vis spectroscopy, but only if the scattering from the complex bio-matrix populated by proteins, vitamins, lipids and salts/ions is negligible compared to the NPs. However, the generally investigated NP concentrations are usually moderate, and thus, standard (polarized) DLS must deal with the presence of this complex background.25,26 Indeed, as shown in Fig. 2, for some of the media, the magnitude of coherent scattering from biomolecules can be of the same order as of the Au NPs. If this scattering is not separated from that of the particles, the analysis will be biased.25,26 Depolarized scattering from the biological matrix is virtually invisible compared to the depolarized scattering of the NPs; while several thousands of photons are detected from the Au NPs in time units and for unit power of the incident laser, the photon count rate corresponding to depolarized scattering from the biological matrix does not exceed the dark count rate of photon detectors. Accordingly, in depolarized configuration, scattering information originating exclusively from the NPs on an essentially zero-background can selectively be detected, as depicted in Fig. 3.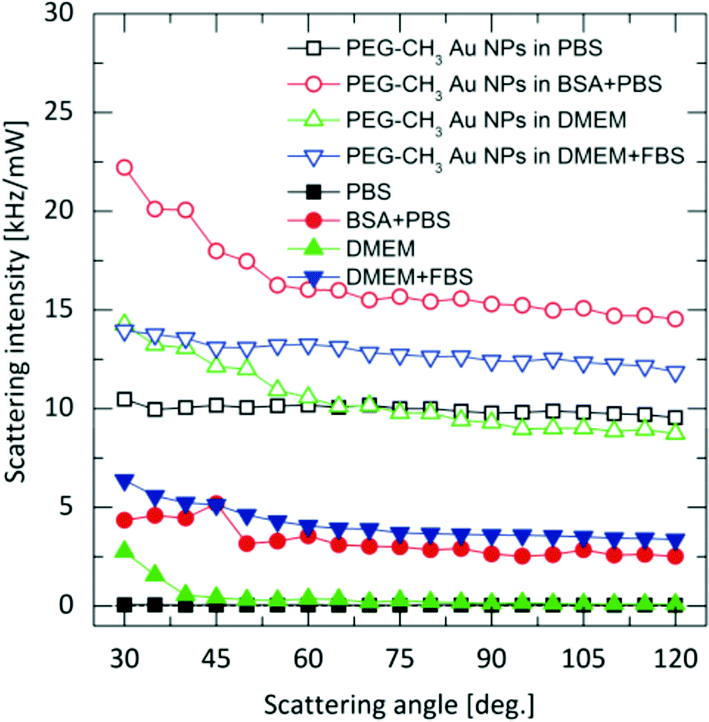 | ||
| Fig. 2 Polarized scattering from the Au NPs and from the biological media (Intensity is defined as the number of detected photons per unit time and unit power of the incident laser). | ||
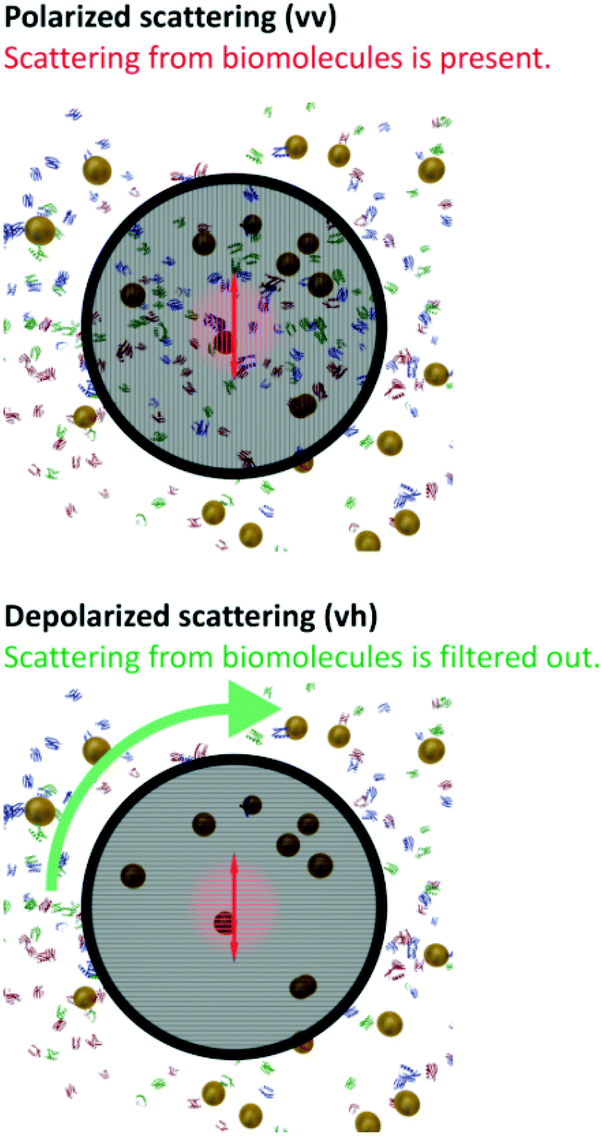 | ||
| Fig. 3 Depiction of NPs and the bio-matrix background as seen in standard polarized (top) and depolarized (down) dynamic light scattering experiments, respectively. While in the first experiment scattering from the complex bio-matrix is clearly present, in the latter, depolarized scattering from the biological matrix is not visible, and thus, entirely negligible compared to the depolarized scattering of the nanoparticles. Owing to this, scattering exclusively from the NPs on an essentially zero-background is detected. | ||
By relying on depolarized dynamic light scattering (DDLS), we find that the size of the methoxy or carboxylate terminated PEGylated Au NPs remains unchanged under all conditions (Fig. 4), suggesting that these particles do not respond to the present biomolecules.
 | ||
| Fig. 4 Hydrodynamic radii of the functionalized Au NPs incubated in the biological media for 24 hours. Hydrodynamic radii were estimated viaeqn (1) and (6). | ||
This is also confirmed by the Zeta-potential measurements that do not display any significant change in any of the media (Table 1), and agree with the absence of significant changes in the UV-Vis spectra.
| Particle system | PBS | BSA + PBS | DMEM | DMEM + FBS |
|---|---|---|---|---|
| Citrate Au NP | −32.0 (1.7) | −15.0 (0.4) | −21.0 (0.4) | −18.0 (0.3) |
| PEG-CH3 Au NP | −4.0 (1.4) | −6.0 (0.4) | −2.0 (1.7) | −9.0 (0.7) |
| PEG-NH3+ Au NP | 5.0 (0.7) | −3.0 (0.3) | 4.0 (0.4) | −5.0 (0.4) |
| PEG-COO– Au NP | −10.0 (0.6) | −6.0 (0.3) | −8.0 (2.0) | −10.0 (0.8) |
DDLS analysis of the PEG-NH3+ Au NPs, however, showed features that were not quantifiable by UV-Vis. The hydrodynamic radius increased by nearly 14 nm in BSA + PBS, indicating strong interaction with BSA. Zeta-potential measurements revealed a negatively charged surface despite the presence of protonated amine groups –NH3+. This is explained by the adsorption of BSA onto the particles, since BSA exhibits a negative charge at pH 7.3,48 In DMEM and in DMEM + FBS, no considerable size change was observed for the PEG-NH3+ Au NPs. These NPs exhibited a positive surface charge in pure DMEM (Table 1) and a negative Zeta-potential was measured in DMEM + FBS, which strongly indicates a certain interaction with the FBS, influenced by the presence of thiolated molecules. Both Larson et al. and Maus et al. demonstrated that, at physiological concentrations, cysteine can displace methoxy-PEG-thiol molecules on the Au NP surface, which in turn leads to protein adsorption.49,50 This possible loss of PEG density on the gold surface explains the similar hydrodynamic radius for PEG-NH3+ as well as for PEG-CH3 Au NPs. In agreement with DDLS, TEM micrographs confirm a stable suspension of PEG-NH3+ Au NPs (Fig. 6B, D, F and H). It was shown above that the behaviour of the citrate Au NPs is modified in BSA + PBS, in DMEM, and in DMEM + FBS. In BSA + PBS, the average apparent hydrodynamic radius increases by approximately 5 nm. The order of this increase agrees well with the hydrodynamic radius of the BSA protein and suggests the formation of a protein monolayer adsorbed onto the surface of the Au NPs.39,51 It is plausible that this observation was the result of two dominating but competing processes: One destabilizes the suspension, while the other balances the electrostatic screening of mobile charges52 and preserves the aggregated or non-aggregated NPs, e.g., by the onset of a protein shell which acts as a protective layer against further aggregation.41,53 By performing time-resolved DDLS, we are able to monitor changes in the hydrodynamic size from the point of incubation. Fig. 5 displays the results of consecutively executed three-second-long measurements.
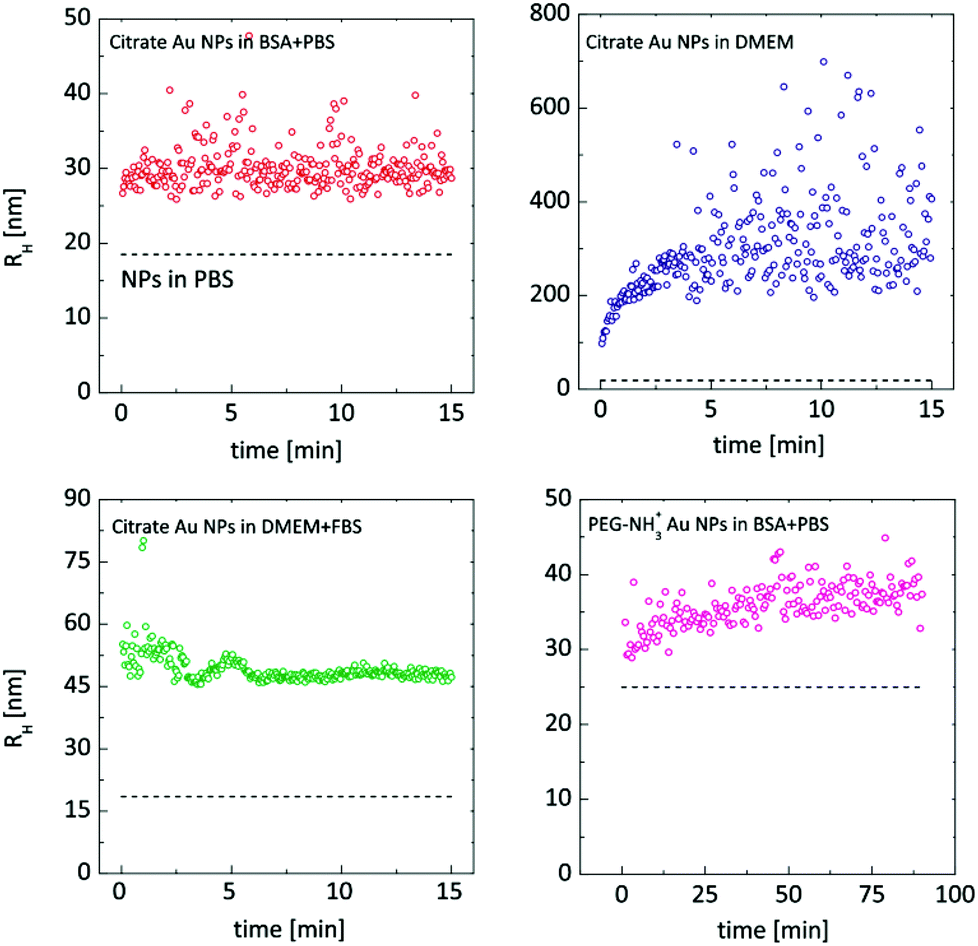 | ||
| Fig. 5 Time-resolved DDLS study started promptly after incubating the Au NPs in the biological media. The dashed lines correspond to the Au NPs in PBS buffer. Hydrodynamic radii are estimated viaeqn (1) and (6). | ||
 | ||
| Fig. 6 TEM micrographs of citrate and PEG-NH3+ Au NPs in PBS (A, B), incubated in BSA solution (C, D), in DMEM (E, F), and in FBS supplemented DMEM (G, H). Scale bar = 500 nm. | ||
When citrate Au NPs were incubated in BSA + PBS, a very rapid response was apparently happening. Even such short measurements were not sufficient to capture the initial phase of the protein monolayer formation. In DMEM, the course was clearly captured: a rapid initial phase – the first 30 seconds – resulted in an increase of almost 200 nm, after which the process slowed down. The most interesting case was that of the Au NPs incubated in DMEM + FBS: clear oscillations were present until eight minutes passed, indicating a complex, possibly competitive, process. Compared to the citrate Au NPs, PEG-NH3+, Au NPs in BSA + PBS exhibit a slower process that levels only after an hour.
TEM micrographs (Fig. 6C) further support the formation of a protein monolayer adsorbed onto the surface of the Au NPs, as non-aggregated NPs were visualized, which indicates a stable colloidal suspension. However, this stability was completely jeopardized in DMEM, resulting in rapid sedimentation within minutes. As predicted, large aggregates were visible by TEM (Fig. 6E). Nonetheless, the addition of 10% FBS to DMEM changed the case considerably. A moderate degree of aggregation with about a threefold increase in size was observed in DMEM + FBS (Fig. 4) and also confirmed by TEM (Fig. 6G).
Conclusions
While the characterization of NPs in optically complex biological/physiological matrices is challenging, we have shown that the limitations can be overcome by using depolarized light scattering. Quantitative information with an unprecedented signal-to-noise ratio can be recorded in real time, no matter how optically complex the surrounding environment is. This non-invasive approach offers new opportunities to the wide range of studies addressing the behaviour of NPs in complex physiological/biological fluids; it is straightforward and robust, and comes with considerable advantages over UV-Vis and fluorescence correlation spectroscopy.Experimental
Citrate-stabilized gold nanoparticles (Au NPs) ([Au] = 0.5 mM) were synthesized as reported by Turkevich et al.54 Aqueous solutions (3.4 × 10−3 mM) of thiolated amine- (PEG-NH3+), carboxylic- (PEG-COO−), methoxy- (PEG-CH3) poly(ethylene glycol) (PEG) were sonicated for 15 min and subsequently mixed with 100 mL of the Au NP suspension. This concentration ratio provided approximately 10 PEG chains per nm2 of particle surface. These mixtures were left to react at 25 °C for 24 h. To remove any excess polymer, the PEGylated NPs were centrifuged twice at 104 g for 1 h and then re-dispersed in 10 mL water. Each particle type, at a concentration of 50 μg mL−1, was incubated in four different, increasingly complex, biological media at 25 °C for 24 h: (1) Phosphate buffer (PBS, 10 mM sodium phosphate monobasic/disodium phosphate hydrogen, pH 7), (2) Bovine Serum Albumin (BSA), which is a high molecular weight protein and major component of serum, in PBS (5 mg mL−1), (3) Dulbecco's Modified Eagle Medium (DMEM), a commonly used cell-culture medium containing amino acids, salts, glucose, and vitamins, and (4) Fetal Bovine Serum (FBS) supplemented DMEM (10%). UV-Vis extinction spectra were recorded at 25 °C on a Jasco V-670 spectrophotometer, using quartz cuvettes of 10 mm path length. For performing electron microscopy, suspensions were spin-coated and dried on carbon-film square-mesh copper grids (Electron Microscopy Sciences, CF-300-Cu) and micrographs of the Au NPs were recorded with a Morgagni transmission electron microscope (FEI) operating at 80 kV. The effective surface charge was characterized at 25 °C, using phase amplitude light scattering (Brookhaven, ZetaPALS) and NP tracking analysis (NanoSight, NS500, Z-NTA Software version 2.3, Salisbury, UK). Suspensions of 50 μg Au per mL were prepared in the different biological media. For ZetaPALS, forty-five cycles of electrophoretic mobility measurements were replicated tenfold and the mean and the standard deviation were estimated. For NP tracking analysis, the electrophoretic velocities were recorded by a digital video microscope system (10 videos of 90 s). Light scattering measurements were performed at constant temperature (21 °C) using a commercial goniometer instrument (3D LS Spectrometer, LS Instruments AG, Switzerland). The primary beam was formed by a linearly polarized and collimated laser beam (HeNe, 632.8 nm, 21 mW) and the scattered light was collected by single-mode optical fibers equipped with integrated collimation optics. Collected light was coupled into two high-sensitivity APD detectors (Perkin Elmer, Single Photon Counting Module) and their outputs were fed into a two-channel multiple-tau correlator (Correlator.com). Signal-to-noise ratio was improved by cross-correlating these two channels. With respect to the primary beam, depolarized scattering was observed via cross-polarizers. The incoming laser beam passed through a Glan–Thompson polarizer with an extinction ratio of 10−6, and another Glan–Thompson polarizer, with an extinction ratio of 10−8, was mounted in front of the collection optics. The instrumental depolarization was controlled by measuring a suspension of FBS, (Invitrogen, Switzerland) diluted in PBS (10% Vol./Vol.).Acknowledgements
S. Balog, L. Rodriguez-Lorenzo, C. A. Monnier, B. Rothen-Rutishauser and A. Petri-Fink are grateful for the financial support of the Swiss National Science Foundation (PP00P2_123373), the Adolphe Merkle Foundation, and the University of Fribourg. L. Rodriguez-Lorenzo acknowledges the financial support from the L'Oreal Switzerland and UNESCO's fellowship program “For Women in Science 2013”. This research was also supported by the Swiss National Science Foundation through the National Centre of Competence in Research Bio-Inspired Materials. Access to TEM was kindly provided by the Microscopy Imaging Centre of the University of Bern. M. Obiols-Rabasa and P. Schurtenberger acknowledge financial support from the Swedish Research Council VR through the Linnaeus Center of Excellence on Organizing Molecular Matter. The authors also acknowledge financial support from the European Commission under the Seventh Framework Program by means of the grant agreement for the Integrated Infrastructure Initiative N. 262348 European Soft Matter Infrastructure (ESMI).Notes and references
- R. L. Juliano, Sci. Public Policy, 2012, 39, 99–104 CrossRef.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed.
- S. P. Boulos, T. A. Davis, J. A. Yang, S. E. Lohse, A. M. Alkilany, L. A. Holland and C. J. Murphy, Langmuir, 2013, 29, 14984–14996 CrossRef CAS PubMed.
- M. L. Personick and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- L. Zhang, W. F. Dong and H. B. Sun, Nanoscale, 2013, 5, 7664–7684 RSC.
- Y. Chen, H. R. Chen and J. L. Shi, Adv. Mater., 2013, 25, 3144–3176 CrossRef CAS PubMed.
- J. Wang, J. D. Byrne, M. E. Napier and J. M. DeSimone, Small, 2011, 7, 1919–1931 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- I. Lynch, D. Langevin and K. A. Dawson, Nanoscience: Colloidal and Interfacial Aspects, Lessons for Bionanointeractions from Colloidal Science, CRC Press, 2010 Search PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef CAS PubMed.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- J. Ferin, G. Oberdörster and D. P. Penney, Am. J. Respir. Cell Mol. Biol., 1992, 6, 535–542 CrossRef CAS PubMed.
- G. Oberdörster, V. Stone and K. Donaldson, Nanotoxicology, 2007, 1, 2–25 CrossRef.
- K. Donaldson, V. Stone, C. L. Tran, W. Kreyling and P. J. A. Borm, Occup. Environ. Med., 2004, 61, 727–728 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- C. Gunawan, M. Lim, C. P. Marquis and R. Amal, J. Mater. Chem. B, 2014, 2, 2060–2083 RSC.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- G. Pyrgiotakis, C. O. Blattmann and P. Demokritou, ACS Sustainable Chem. Eng., 2014, 2, 1681–1690 CrossRef CAS PubMed.
- P. d. Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Materials Horizons, 2014, 1, 301–313 RSC.
- R. Pecora, Dynamic Light Scattering: Applications of Photon Correlation Spectroscopy, Plenum Press, New York, 1985 Search PubMed.
- A. K. Pal, I. Aalaei, S. Gadde, P. Gaines, D. Schmidt, P. Demokritou and D. Bello, ACS Nano, 2014, 8, 900–9015 CrossRef PubMed.
- C. Graf, Q. Gao, I. Schütz, C. N. Noufele, W. Ruan, U. Posselt, E. Korotianskiy, D. Nordmeyer, F. Rancan, S. Hadam, A. Vogt, J. Lademann, V. Haucke and E. Rühl, Langmuir, 2012, 28, 7598–7613 CrossRef CAS PubMed.
- N. Hondow, R. Brydson, P. Wang, M. Holton, M. R. Brown, P. Rees, H. Summers and A. Brown, J. Nanopart. Res., 2012, 14, 1–15 CrossRef.
- L. Treuel, K. A. Eslahian, D. Docter, T. Lang, R. Zellner, K. Nienhaus, G. U. Nienhaus, R. H. Stauber and M. Maskos, Phys. Chem. Chem. Phys., 2014, 16, 15053–15067 RSC.
- L. Rodriguez-Lorenzo, K. Fytianos, F. Blank, C. von Garnier, B. Rothen-Rutishauser and A. Petri-Fink, Small, 2014, 10, 1341–1350 CrossRef CAS PubMed.
- V. Petkov, Y. Peng, G. Williams, B. Huang, D. Tomalia and Y. Ren, Phys. Rev. B: Condens. Matter, 2005, 72, 195402 CrossRef.
- C. Zhou, J. Yu, Y. Qin and J. Zheng, Nanoscale, 2012, 4, 4228–4233 RSC.
- M. C. Scott, C.-C. Chen, M. Mecklenburg, C. Zhu, R. Xu, P. Ercius, U. Dahmen, B. C. Regan and J. Miao, Nature, 2012, 483, 444–447 CrossRef CAS PubMed.
- A. S. Barnard, N. P. Young, A. I. Kirkland, M. A. van Huis and H. Xu, ACS Nano, 2009, 3, 1431–1436 CrossRef CAS PubMed.
- R. Watanabe-Tamaki, A. Ishikawa and T. Tanaka, Appl. Phys. Lett., 2013, 102, 043110 CrossRef PubMed.
- S. Balog, L. Rodriguez-Lorenzo, C. A. Monnier, B. Michen, M. Obiols-Rabasa, L. Casal-Dujat, B. Rothen-Rutishauser, A. Petri-Fink and P. Schurtenberger, J. Phys. Chem. C, 2014, 118, 17968–17974 CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- D. E. Koppel, J. Chem. Phys., 1972, 57, 4814–4820 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- V. Myroshnychenko, J. Rodriguez-Fernandez, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzan and F. J. G. de Abajo, Chem. Soc. Rev., 2008, 37, 1792–1805 RSC.
- S. Dominguez-Medina, S. McDonough, P. Swanglap, C. F. Landes and S. Link, Langmuir, 2012, 28, 9131–9139 CrossRef CAS PubMed.
- D. Aili, P. Gryko, B. Sepulveda, J. A. G. Dick, N. Kirby, R. Heenan, L. Baltzer, B. Liedberg, M. P. Ryan and M. M. Stevens, Nano Lett., 2011, 11, 5564–5573 CrossRef CAS PubMed.
- P. Bihari, M. Vippola, S. Schultes, M. Praetner, A. Khandoga, C. Reichel, C. Coester, T. Tuomi, M. Rehberg and F. Krombach, Part. Fibre Toxicol., 2008, 5, 14 CrossRef PubMed.
- Z. E. Allouni, M. R. Cimpan, P. J. Høl, T. Skodvin and N. R. Gjerdet, Colloids Surf., B, 2009, 68, 83–87 CrossRef CAS PubMed.
- B. Chanteau, J. Fresnais and J. F. Berret, Langmuir, 2009, 25, 9064–9070 CrossRef CAS PubMed.
- S. Kittler, C. Greulich, J. S. Gebauer, J. Diendorf, L. Treuel, L. Ruiz, J. M. Gonzalez-Calbet, M. Vallet-Regi, R. Zellner, M. Koller and M. Epple, J. Mater. Chem., 2010, 20, 512–518 RSC.
- A. Vanderkooy, Y. Chen, F. Gonzaga and M. A. Brook, ACS Appl. Mater. Interfaces, 2011, 3, 3942–3947 CAS.
- D. C. Henry, Proc. R. Soc. London, Ser. A, 1931, 133, 106–129 CrossRef CAS.
- M. V. Smoluchowski, J. Phys. Théor. Appl., 1907, 6, 659–660 CrossRef.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS PubMed.
- T. A. Larson, P. R. Joshi and K. Sokolov, ACS Nano, 2012, 6, 9182–9190 CrossRef CAS PubMed.
- L. Maus, O. Dick, H. Bading, J. P. Spatz and R. Fiammengo, ACS Nano, 2010, 4, 6617–6628 CrossRef CAS PubMed.
- R. Huang, R. P. Carney, K. Ikuma, F. Stellacci and B. L. T. Lau, ACS Nano, 2014, 8, 5402–5412 CrossRef CAS PubMed.
- V. Hirsch, J. Salaklang, B. Rothen-Rutishauser and A. Petri-Fink, IEEE Trans. Magn., 2013, 49, 402–407 CrossRef.
- Z. Ji, X. Jin, S. George, T. Xia, H. Meng, X. Wang, E. Suarez, H. Zhang, E. M. V. Hoek, H. Godwin, A. E. Nel and J. I. Zink, Environ. Sci. Technol., 2010, 44, 7309–7314 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |