
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDendritic polyglycerol sulfate as a novel platform for paclitaxel delivery: pitfalls of ester linkage†
Ana
Sousa-Herves‡
a,
Patrick
Würfel‡
b,
Nicole
Wegner
b,
Jayant
Khandare
ac,
Kai
Licha
b,
Rainer
Haag
a,
Pia
Welker
*b and
Marcelo
Calderón
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustrasse 3, 14195 Berlin, Germany. E-mail: marcelo.calderon@fu-berlin.de
bmivenion GmbH, Robert-Koch-Platz 4, 10115 Berlin, Germany. E-mail: welker@mivenion.com
cMaharastra Institute of Pharmacy, MIT Campus, Kothrud, 411038 Pune, India
First published on 17th December 2014
Abstract
In this study, dendritic polyglycerol sulfate (dPGS) is evaluated as a delivery platform for the anticancer, tubulin-binding drug paclitaxel (PTX). The conjugation of PTX to dPGS is conducted via a labile ester linkage. A non-sulfated dendritic polyglycerol (dPG) is used as a control, and the labeling with an indocarbocyanine dye (ICC) renders multifunctional conjugates that can be monitored by fluorescence microscopy. The conjugates are characterized by 1H NMR, UV-vis measurements, and RP-HPLC. In vitro cytotoxicity of PTX and dendritic conjugates is evaluated using A549 and A431 cell lines, showing a reduced cytotoxic efficacy of the conjugates compared to PTX. The study of uptake kinetics reveals a linear, non saturable uptake in tumor cells for dPGS-PTX-ICC, while dPG-PTX-ICC is hardly taken up. Despite the marginal uptake of dPG-PTX-ICC, it prompts tubulin polymerization to a comparable extent as PTX. These observations suggest a fast ester hydrolysis and premature drug release, as confirmed by HPLC measurements in the presence of plasma enzymes.
Introduction
Paclitaxel (PTX)1 is a commonly used chemotherapeutic of low molecular weight which binds to α- and β-tubulin.2–4 PTX promotes tubulin polymerization and formation of highly stable microtubules, which disrupt the normal tubule dynamics required for cellular division therefore causing cell apoptosis.5 The mode of action of PTX can be visualized by imaging tubulin showing an altered intracellular distribution after incubation with the drug. Despite the wide use of PTX in various carcinomas, such as ovarian and non-small cell lung cancers,6,7 it presents several limitations. PTX is poorly soluble in water and therefore has to be administered with Cremophor EL® and ethanol vehicles, which results in pharmacologic problems and side effects.8–11Macromolecular conjugates, such as those based on polymeric scaffolds, are ideal entities for the delivery of cytotoxic drugs or diagnostic agents. In particular, polymer therapeutics, i.e. polymeric prodrugs, polymer conjugates of proteins, drugs and aptamers, have been widely employed during the last decades to increase the solubility and improve the body distribution and cellular uptake of therapeutic agents.12–17 Moreover, after specific accumulation in the targeted tissue or organ, the cytotoxic drug can be released in a controlled fashion by the effect of internal or external stimuli, leading to an optimization of the therapy. One of the most well-known polymer therapeutics formulations is Opaxio™, currently in phase III for the treatment of ovarian cancer. This polymer therapeutic has a very high drug loading, presents prolonged circulation times in the bloodstream, and can be subsequently accumulated in tumors where PTX is effectively released.18–21 Opaxio™ results from the attachment of PTX to polyglutamic acid through ester linkages, a commonly employed strategy to covalently modify PTX with macromolecules.22–28
Multivalent dendritic polymers constitute a very appealing platform for the preparation of polymer therapeutics. The highly branched, globular architecture of these molecules gives rise to a number of interesting properties when compared to linear polymers of analogous molecular weight. In addition, their surface multifunctionality allows the simultaneous incorporation of drugs, imaging agents, or targeting moieties.15,29,30 Dendritic structures based on highly biocompatible polyglycerol (dPG) are ideal architectures for anti-cancer therapy purposes, because they are water-soluble, can be tuned in size, easily functionalized, and provide multivalently arranged ligands on the surface.31–33 Particularly interesting is dendritic polyglycerol sulfate (dPGS), a highly anionic dendritic polymer which has shown an exceptionally high anti-inflammatory potential due to a high affinity binding to L-selectin.34–36 dPGS has not shown any cytotoxic side effects in vitro and in vivo35,37 and can be easily synthesized on a kilogram scale and in a broad range of sulfation degrees and molecular weights.38,39 In addition, dPGS has demonstrated efficient in vivo anti-inflammatory efficacy (mouse dermatitis model)35 and could be used for in vivo molecular imaging of inflammatory diseases after labeling with a near infrared (NIR) dye.40,41
On the basis of the inflammation-targeting properties of dPGS, we envisioned the preparation of dPGS-PTX conjugates as novel anti-cancer agents with potential self-targeting properties in inflammation-related carcinoma processes. Herein, we present the synthesis of such water-soluble conjugates, their modification with an imaging agent (indocarbocyanine dye, ICC), and the evaluation of their in vitro cellular uptake and activity towards tubulin polymerization (Fig. 1). As a non-targeting control, a non-sulfated, neutral dPG-PTX conjugate was synthesized and compared in vitro with its sulfated counterpart. For the conjugation of PTX to dPG and dPGS we have selected a labile ester linkage, so that PTX release could be triggered by the low pH typically found in the lysosomes and/or by the action of esterases. We have analyzed the PTX release from both conjugates by HPLC at different pHs and in the presence of human plasma. The obtained drug release profiles, together with the in vitro activity observed for PTX, suggest that PTX is released extremely fast in the presence of hydrolytic conditions or esterases. This observation implies that the selection of ester linkages for the covalent attachment of PTX or other cytotoxic drugs to polymeric platforms should be carefully evaluated for each particular system.
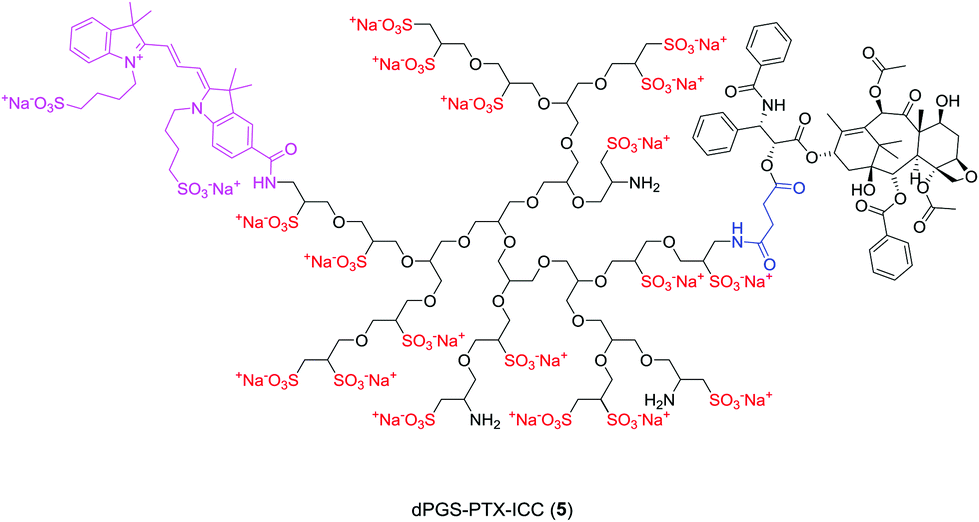 | ||
| Fig. 1 Idealized chemical structure of multifunctional dPGS conjugated to PTX and ICC (dPGS-PTX-ICC (5)). | ||
Experimental
Materials and methods
Chemicals and reagents were obtained from Acros Organics, Sigma-Aldrich, and Merck. They were reagent grade and used as received unless otherwise stated. Milli-Q water was prepared using a Millipore water purification system. Dry reactions were performed in flame-dried glassware under argon atmosphere. Purification by dialysis was performed with membranes of benzoylated cellulose or regenerated cellulose (MWCO 2000; Sigma-Aldrich). Size exclusion chromatography was performed with Sephadex G-25 superfine (GE Healthcare) under ambient pressure and temperature. Reversed phase chromatographic purification was performed using RP-18 Redisep flash columns (ISCO CombiFlash Rf system). Dendritic polyglycerol (dPG, Mn ≈ 6 kDa, PDI < 1.6) was prepared according to literature via an anionic multibranching ring-opening polymerization of glycidol and pentaerythritol as starter.42,43 2S-ICC-NHS and 2S-ICC-NH2 were obtained from mivenion GmbH (Berlin, Germany).441H NMR spectra were recorded on a Jeol ECX 400, Bruker AMX 500, or on a Bruker BioSpin AV 700 spectrometer. Chemical shifts are reported in ppm (δ units) downfield from internal tetramethylsilane (for CDCl3), the HOD solvent peak (for D2O), or residual solvent peak [deuterated dimethyl sulfoxide (DMSO-d6)]. For ESI measurements, a TSQ 7000 (Finnigan Mat) instrument was used. Elemental analysis was performed on a Vario EL III elemental analyzer using sulfanilic acid as standard. Absorption spectra were recorded on a LAMBDA 950 UV/Vis/NIR spectrometer (PerkinElmer, USA). Single human plasma was obtained from a healthy consented unmedicated donor according to German ethical issues. Cell culture experiments were done under sterile conditions. A549 lung-carcinoma cells45 were purchased from DSMZ, Braunschweig. Cells were cultivated with Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% fetal calf serum (FCS) and 1% Penicillin/Streptomycin. Subcultivation was done twice a week using trypsin/ethylenediaminetetraacetic acid (EDTA) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Vulva-carcinoma A431 cells45 were provided by Dr. J. Dernedde (Charité Universitätsmedizin Berlin, Germany) and cultivated as A549 cells but with a subcultivation ratio of 1:12–16.
:10. Vulva-carcinoma A431 cells45 were provided by Dr. J. Dernedde (Charité Universitätsmedizin Berlin, Germany) and cultivated as A549 cells but with a subcultivation ratio of 1:12–16.
Synthesis and characterization
The dendritic conjugates employed in this work are summarized in Table 1.| Compound | Theor. molecular weight | Surface charge | Conjugated PTX (molar ratio)a | Conjugated ICC (molar ratio)b |
|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by UV-vis (λ = 550 nm). | ||||
| dPGS-PTX (3) | ≈14.2 kDa | Negative | ≈1.4 PTX/dPGS | — |
| dPG-PTX (4) | ≈6.2 kDa | Neutral | ≈1.0 PTX/dPG | — |
| dPGS-PTX-ICC (5) | ≈14.3 kDa | Negative | ≈1.4 PTX/dPGS | 0.2 ICC/dPGS |
| dPG-PTX-ICC (6) | ≈6.8 kDa | Neutral | ≈1.0 PTX/dPG | 0.2 ICC/dPG |
| dPGS-ICC (7) | ≈13.3 kDa | Negative | — | 0.4 ICC/dPGS |
dPGS amine (1). Dendritic polyglycerol sulfate (Mn ≈ 13 kDa) containing NH2 groups was prepared following a slightly modified procedure recently reported by our group.46 In brief, dendritic polyglycerol (Mn ≈ 6 kDa) was partially mesylated and the resulting mesyl groups were subsequently substituted for azides by treatment with NaN3. After sulfation with SO3 pyridine complex in N,N-dimethylformamide (DMF), the azide groups were reduced under aqueous conditions with tris(2-carboxyethyl)phosphine (TCEP) to yield dPGS amine (1) with free amino functionalities available for further conjugation. After extensive purification by dialysis, the sulfur content was determined by elemental analysis and corresponded with a degree of sulfation of 80%.
PTX-Suc-NHS ester (2). Paclitaxel (25.0 mg, 0.029 mmol) and succinic anhydride (29.3 mg, 0.29 mmol) were dissolved in dry pyridine (2.1 mL) under argon atmosphere. The mixture was stirred at room temperature (rt) for 24 h protected from light. The reaction was monitored by TLC [silica plates, CHCl2–MeOH (10:1)] until no free PTX was observed. After solvent evaporation, PTX-succinate could be isolated by precipitation with H2O. After extensive washing of the precipitate, the product was redissolved in CHCl2–MeOH and concentrated under vacuum to yield 2′-hemisuccinate derivative of paclitaxel as a white solid (27.4 mg, 99%). 1H NMR (500 MHz, CDCl3) and ESI-MS were consistent with previously reported results.25,26 Subsequently, PTX-hemisuccinate (18 mg, 0.019 mmol) was dissolved in freshly distilled EtOAc (500 μL) under Ar atmosphere. N-hydroxysuccinimide (NHS, 2.46 mg, 0.021 mmol) and N,N′-dicyclohexylcarbodiimide (DCC, 4.33 mg, 0.021 mmol) were added to the solution and the mixture was stirred at rt for 48 h. TLC in CHCl2–MeOH (10:1) was used to monitor the reaction progress. After filtration to remove insoluble ureas, the solvent was evaporated under reduced pressure yielding 2 as a white solid (14.7 mg, 75%). 1H NMR (500 MHz, CDCl3, δ): 8.14 (2H), 7.70 (2H), 7.54 (1H), 7.47–7.23 (10H), 7.10 (1H), 6.23 (1H), 6.17 (1H), 5.93 (1H), 5.63 (1H), 5.47 (1H), 4.96 (1H), 4.38 (1H), 4.24 (1H), 4.13 (1H), 3.74 (1H), 2.91–2.72 (4H), 2.66 (1H), 2.68–2.44 (4H), 2.37 (3H), 2.36 (2H), 2.16 (3H), 1.98 (1H), 1.85 (4H), 1.62 (3H), 1.17 (3H), 1.07 (3H). 13C NMR (125 MHz, CDCl3, δ): 203.95, 171.26, 169.96, 169.14, 168.15, 168.12, 167.84, 167.41, 166.96, 142.78, 133.86, 133.69, 132.79, 132.73, 131.91, 131.79, 130.29, 129.29, 129.09, 128.82, 128.60, 128.56, 128.49, 127.33, 126.83, 84.50, 81.06, 79.01, 76.50, 75.66, 75.08, 74.73, 72.14, 72.03, 58.52, 52.90, 45.59, 43.20, 35.53, 33.46, 31.21, 26.81, 26.22, 25.45, 24.78, 22.68, 22.14, 20.88, 14.81, 9.69. ESI-MS m/z: [M + Na]+ calcd for C55H58N2O19Na, 1073; found, 1073. (Fig. S1, ESI†).
dPGS-PTX (3). PTX-Suc-NHS (2) (7 mg, 0.0067 mmol) and dPGS amine (1) (45.2 mg, 0.0023 mmol) were dissolved in a mixture of DMF–H2O (9:1) (2.21 mL). After 48 h under magnetic stirring at rt, the crude product was purified by dialysis first against acetone–Milli-Q water (1:1) and then against Milli-Q water. After freeze-drying, 37.4 mg (80%) of dPGS-PTX conjugate (3) were obtained. 1H NMR spectra in DMSO-d6–D2O–DCl (85:13.5:1.5, v/v) showed that the conjugate contains approx. 1.4 mol of PTX per mol of dPGS (Fig. S2, ESI†). 1H NMR, [700 MHz, DMSO-d6–D2O–DCl, δ]: 7.90–7.30 (15H), 7.10 (1H), 6.24 (2H), 6.17 (1H), 5.93 (1H), 5.63 (1H), 5.47 (1H), 4.11–3.08 (405H), 2.37 (3H), 2.16 (3H), 1.85 (4H), 1.62 (3H), 1.07 (6H).
dPG-PTX (4). PTX-Suc-NHS (2) (11.8 mg, 0.0113 mmol) and dPG amine37 (22.6 mg, 3.76 mmol) were dissolved in a mixture of DMF–H2O (9:1) (2.21 mL). After 48 h under magnetic stirring at rt, the crude product was purified by dialysis first against acetone–Milli-Q water (1:1) and then against Milli-Q water. After freeze-drying, 23.7 mg (90%) of dPG-PTX conjugate (4) were obtained. 1H NMR spectrum DMSO-d6 showed that the compound has about 1 mol of Paclitaxel per mol of dPG. 1H NMR (700 MHz, DMSO-d6, δ): 8.05–7.34 (15H), 7.01 (1H), 6.50 (2H), 6.30 (1H), 6.03 (1H), 5.43 (1H), 3.90–3.07 (405H), 2.23 (3H), 2.11 (3H), 2.02 (4H), 1.79 (3H), 1.26 (6H).
dPGS-PTX-ICC (5) and dPG-PTX-ICC (6). 3 and 4 (2.5 mg, 0.17 and 0.40 mmol, respectively) were separately dissolved in 0.3 mL 90% DMF aqueous solution. 2S-ICC-NHS dye (0.4 mg, 0.51 mmol for 3; 0.76 mg, 1.2 mmol for 4) was dissolved in 0.1 mL of DMF and added to the solutions in the presence of N,N-diisopropylethylamine (DIPEA, 0.51 mmol and 1.2 mmol, respectively). Reaction mixtures were stirred for 72 h at 41 °C. The reactions were purified by gel-filtration through a Sephadex G-25 column with Milli-Q water. After freeze-drying, 5 and 6 were obtained as pink powders (2.3 and 2.5 mg respectively, 99%). The conjugates were analyzed by UV-vis spectroscopy, which showed that the average molar dye-to-polymer ratio was 0.2 in both cases (extinction coefficient of ICC at 550 nm = 120000 cm−1 M−1).
dPGS-ICC (7) and PTX-ICC (8). dPGS-ICC (7) was prepared as previously reported.38 For the synthesis of PTX-ICC (8), PTX (20 mg, 0.021 mmol), N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 10.34 mg, 0.027 mmol), and DIPEA (9.6 μL, 0.056 mmol) were dissolved in DMF (0.5 mL). After 45 min stirring under argon at rt, 2S-ICC-NH2 dye (22.81 mg, 0.027 mmol) and DIPEA were added (4.8 μL, 0.028 mmol). The mixture was allowed to react for further 48 h and then the solvent was evaporated under vacuum. TLC in CHCl2–MeOH (1:1) was used to monitor the reaction progress. Purification was achieved by RP-18 chromatography (Combi-Flash) using MeOH as eluent. After solvent evaporation 8 (15 mg, 43%) was obtained as a pink solid. MS m/z: [M + Na]+ calcd for C86H100O23S2N5Na2, 1680; found, 1680.
Kinetics of in vitro cellular uptake and elimination
For quantitative cellular uptake kinetics, A431 cells (2.5 × 105 cells per well) were grown in 24-well plates overnight. Subsequently, 1 μM dPGS-PTX-ICC (5), dPG-PTX-ICC (6) and the controls dPGS-ICC (7) and PTX-ICC (8) were diluted in DMEM and incubated for different time intervals (17 measurement points). After incubation, the medium was removed, the cells rinsed with cold phosphate buffered saline (PBS) and harvested with trypsin/EDTA. After further PBS rinsing and centrifugation (250g, 8 °C), the supernatant was discarded and the cells resuspended in a 3% FCS/PBS solution. Measurements were done in duplicate for each measurement point with a fluorescence-activated cell sorting (FACS) Calibur-Cytometer at the Channel FL2-H using CellQuest Pro 4.0.2 (ICC: λex = 515/550, λem = 570 nm). The experiment was replicated once. For elimination kinetics, A431 cells (2 × 105 cells per well) were grown in 24-well plates overnight. After incubation of 1 μM of dPGS-ICC (7) and dPGS-PTX-ICC (5) diluted in DMEM for 24 h, the cell medium was completely removed and the cells washed with warm PBS and incubated with drug free DMEM again. Subsequently, the cells were washed and harvested with trypsin/EDTA at different time intervals (up to 48 h). After rinsing with PBS and centrifugation (350g, 6 °C), the cells were resuspended in 3% FCS/PBS and analyzed by a FACS Calibur-Cytometer using CellQuest Pro 4.0.2. Measurements were done in triplicate for every measurement point and replicated once.In vitro cytotoxicity
For concentration-response curves, A549 and A431 cells were seeded in 96-well plates in 100 μL of medium (0.8–3.5 × 103 cells per well). After attaching overnight, 50 μL were removed and substituted with fresh medium containing different concentrations of PTX or dPGS-PTX (3), dissolved in supplemented medium, and incubated for 24, 48, and 72 h. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used for viability measurements. Briefly, after incubation periods, 10 μL of MTT solution were added and incubated for another 4 h. Subsequently, 50 μL of medium were removed and 150 μL of a 0.04 N HCl/2-propanol solution were added. After dissolving the formazan products, optical density was measured at λ1 = 570 nm and λ2 = 620 nm (reference filter) with a microplate reader (anthos htII). Each concentration was pipetted six times; the experiment was replicated at least thrice. To evaluate the cytotoxicity of dPGS amine (1) and dPG-PTX (4), A431 were seeded in 96-well plates (2.5 × 103 cells per well). After attaching for 4 h, dPGS amine (1), dPGS-PTX (3), PTX and dPG-PTX (4) were diluted in DMEM and incubated for 48 h. MTT assays were subsequently performed. The experiment was repeated at least thrice. Calculation of half maximal inhibitory concentrations (IC50) values was done with GraphPad Prism (version 6.00, GraphPad Software San Diego, USA), using normalization and “log(inhibitor) vs. normalized response – variable slope”.In vitro tubulin polymerization
For tubulin polymerization experiments, A431 cells (2.5 × 105 cells per well) were grown overnight on (3-aminopropyl)triethoxysilane coated coverslips in 24-well plates and subsequently incubated with 100 nM PTX, dPGS amine (1), dPGS-PTX (3), dPG-PTX (4) for 16 h. After incubation, cells were rinsed with PBS, fixated with 4% formaldehyde for 8 min, and stored overnight in 0.1% formaldehyde. After rinsing, the cells were permeabilized with 0.5% Triton X-100/PBS (4 min) and blocked with a sterile filtered Milk/PBS-solution. Primary monoclonal anti-α-tubulin-AB (Sigma-Aldrich. 1:4000 in dry milk) was incubated for 1 h at rt, followed by rinsing and incubation of secondary Cy™3-conjugated AffiniPure donkey anti-mouse antibody (Jackson ImmunoResearch. 1:200 in Milk; 1 h at rt). After nuclear counterstaining with 4′,6-diamidino-2-phenylindole (DAPI; Carl Roth. 1:500 in PBS, 15 min), the slides were mounted in fluorescent mounting medium (Dako).
Analysis of PTX release by HPLC
The study of the release of PTX from the conjugates dPGS-PTX (3) and dPG-PTX (4) in human plasma, pH 7.4, pH 5.0, and pH 2.0 was carried out using a Knauer Smartline-HPLC system with an internal UV absorption detector (λ = 227 nm) and GeminyxSystem software. A Hypersil™ ODS C18 column (Thermo Scientific, 100 mm × 4.6 mm, particle Size: 5 μm) with a direct-connect guard column C18 was employed. Acetonitrile–water (55:45) was used as the mobile phase at a flow rate of 1.0 mL min−1 under isocratic regime. The injection volume was 20 μL and each measurement was performed in triplicate. Stock solutions of PTX in acetonitrile were prepared and assessed by reverse phase HPLC (RP-HPLC) in order to obtain a calibration curve for PTX (0.5–5 μg, R = 0.999) (Retention time: 2.9 min, Fig. S3, ESI†). dPGS-PTX (3) was incubated at a constant PTX concentration of 0.8 mg mL−1 in human plasma (1/3 diluted with PBS), or universal Britton–Robinson buffer47 (BRB) at pH 7.4, 5.0 and 2.0. Samples were maintained at 37 °C under continuous shaking, and aliquots (40 μL) were taken at different time intervals (1, 2, 3.5, 19 and 44 h). The aqueous aliquots were mixed with 320 μL of Et2O–CHCl3 (1:1), vortexed for 2 min, and the phases were separated by centrifugation (10 min, 10000 rpm, rt). 150 μL of the organic phase were taken for each sample, concentrated under vacuum, reconstituted with 90 μL of acetonitrile and analyzed by RP-HPLC. As control experiments, free PTX was incubated at the same concentration, extracted under identical circumstances and then analyzed by RP-HPLC. Release experiments were done thrice, and the release profile was corrected with the mean recovery values obtained for the controls in each solution. PTX extraction efficiency was in the range of 50–75%. In addition, the release profile from dPGS-PTX (3) and dPG-PTX (4) at different pHs was compared. For that purpose, the conjugates (constant PTX concentration) were incubated with BRB of pH 7.4, 5.0, and 2.0. Aliquots (40 μL) at different time intervals were taken, freeze-dried, redissolved in acetonitrile, and analyzed by RP-HPLC. In this case, as no liquid–liquid extraction was needed, control experiments were not necessary.
Results and discussion
Synthesis of the conjugates
dPGS was selected as the dendritic platform for conjugation to PTX due to its non-toxicity, excellent inflammation-targeting properties, and easy availability with functional groups.38 dPGS amine (1) (Scheme 1) was prepared from dPG following a sequential synthetic strategy recently reported by our group.46 This approach involves partial mesylation of dPG OH groups, substitution of the mesyl groups with azides by treatment with NaN3, sulfation of the remaining OH groups in the presence of SO3 pyridine complex and, finally, reduction of the N3 groups with TCEP under aqueous conditions. This methodology allowed the synthesis of a large amount of dPGS amine (1) in high purity and yield. Elemental analysis of dPGS amine (1) showed a 80% degree of sulfation, while 1H NMR measurements revealed a 15% of free amino functionalities available for further conjugation.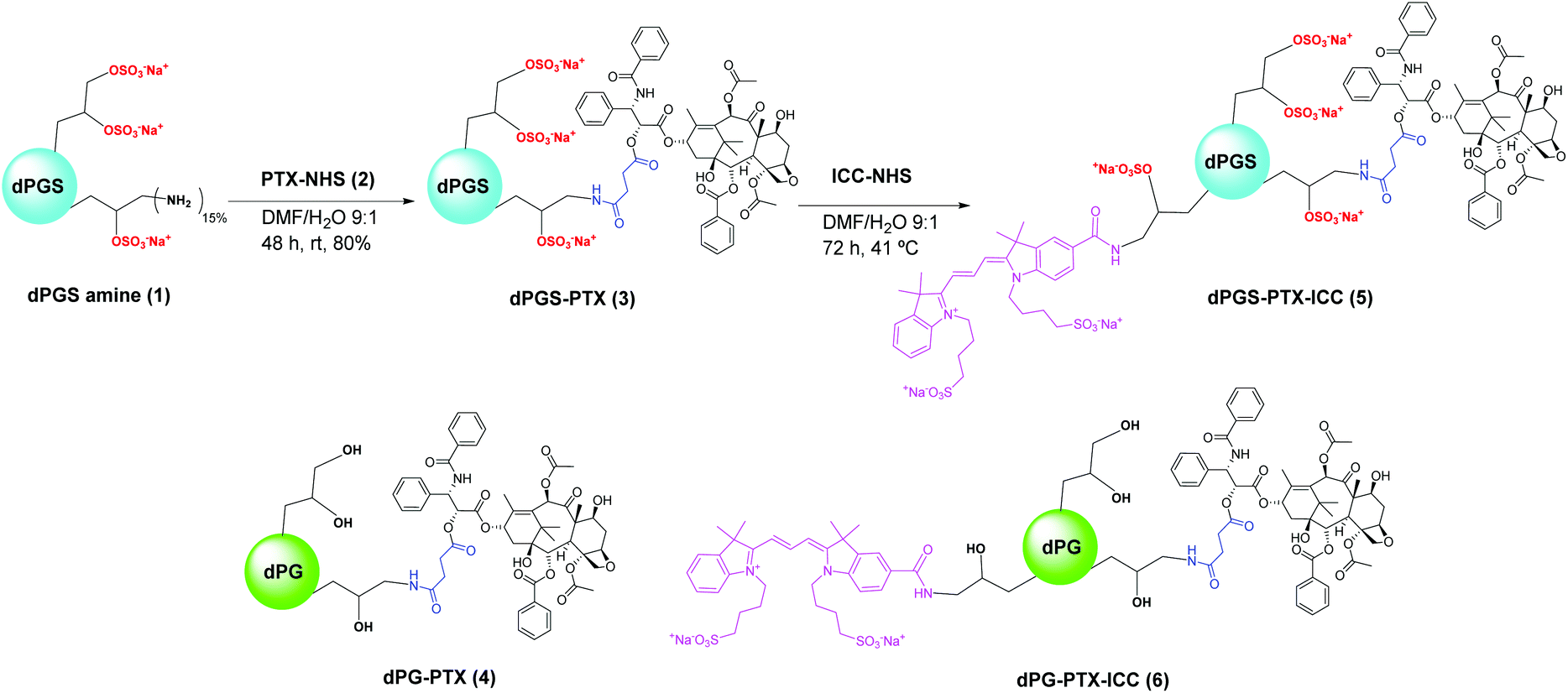 | ||
| Scheme 1 Synthesis of dPGS-PTX-ICC (5) and schematic representation of non-sulfated controls 4 and 6. | ||
To evaluate the ability of dPGS to efficiently internalize and release PTX into cancer cells, we selected a pH-labile ester linkage for the conjugation of the drug to dendritic platform. This strategy should enable a controlled release through ester hydrolysis in tumor tissues (pH ≈ 6) or after internalization in the acidic cellular compartments endosomes/lysosomes (pH ≈ 5.5–5). Ester linkages can also be degraded by the action of hydrolytic enzymes. To this aim, PTX was reacted with succinic anhydride and the resulting product was linked to NHS in the presence of the coupling agent DCC to yield activated PTX-Suc-NHS (2) (Scheme 1). The product was characterized by ESI-MS, 13C and 1H NMR, which showed the expected peaks at 2.5, 2.6, and 2.7 ppm corresponding to the succinimide-NHS protons. Likewise, the perfect match between experimental and calculated m/z values confirmed the identity and the purity of the compound (Fig. S1, ESI†). The coupling of compounds 2 to and 1 had to be accomplished in a mixture of DMF–H2O (9:1), because dPGS amine is only soluble in water or aqueous mixtures and PTX is completely insoluble in water. The synthesized conjugate was purified by dialysis in acetone and water to remove unreacted PTX-Suc-NHS ester. As expected, the conjugate dPGS-PTX (3) was water soluble, which overcame the need for a surfactant, such as Cremophor EL®, currently used to solubilize PTX in a therapeutic formulation. The amount of PTX present in the conjugate could not be determined by UV absorption at 227 nm, as typically done for PTX-polymer conjugates, because the dendritic scaffold presented absorption in that region as well. Furthermore, 1H NMR quantification could not be performed for different aqueous mixtures (D2O had to be present in order to solve dPGS-PTX) because the HOD peak overlapped with the corresponding protons in the dPGS amine scaffold. This matter was finally solved by shifting the HOD peak by adding DCl (1.5%) to a mixture of DMSO-d6 and D2O immediately before recording the 1H NMR spectrum. This way, the amount of PTX present in the conjugate could be determined by integration of the dPGS amine and PTX signals in the 1H NMR spectrum (Fig. S2, ESI†). The molar ratio PTX/dPGS was found to be approx. 1.4, which represented a PTX average loading of 8.45% (weight PTX/weight conjugate). The purity of the conjugate could also be determined by RP-HPLC, which showed a well-defined, single peak and no signs of free drug (Fig. S3 (b), ESI†).
In order to analyze the effect of the peripheral sulfate groups of the conjugate on its cellular uptake and PTX cytotoxic activity, hydroxyl-terminated dPG was conjugated to the drug following the same synthetic approach and was employed as a non-sulfated control (4, Scheme 1). The lack of sulfate groups in the control conjugate 4 extremely facilitated the characterization of the final compound, because the conjugate was completely soluble in common polar organic solvents such as DMSO (Fig. S4, ESI†).
Finally, the peripheral amine groups displayed on these structures allowed an easy simultaneous incorporation of PTX and ICC dyes, which afforded multifunctional platforms for drug delivery. Labeling of 3 and 4 was accomplished by a straightforward coupling of the remaining amine groups with ICC-NHS ester. The incorporation of 0.2 ICC molecules on average, as determined by UV absorbance, resulted in multifunctional dendritic-drug structures 5 and 6 that could be monitored by fluorescence microscopy. Importantly, previous studies from our group have demonstrated that ICC dye conjugation does not have any significant influence on the target affinity of dPGS towards L-selectin.41
Cellular uptake
The quantitative cellular uptake of the conjugates dPGS-PTX-ICC (5) and dPG-PTX-ICC (6) was studied using flow cytometry in A431 cells. In addition, in order to perform a comparison of the uptake kinetics of the drug and the dendritic platform, PTX and dPGS amine (1) were labeled with the same ICC dye [PTX-ICC (8) and dPGS-ICC (7), respectively]. Since dPGS-PTX-ICC (5) was envisioned as a potential drug delivery platform for in vivo situations, the uptake analysis was extended to 48 h. As shown in Fig. 2, PTX-ICC (8) and the anionic conjugates dPGS-ICC (7) and dPGS-PTX-ICC (5) showed a strong internalization at 12 h, while neutral dPG-PTX-ICC (6) had a comparable response to the negative control (medium).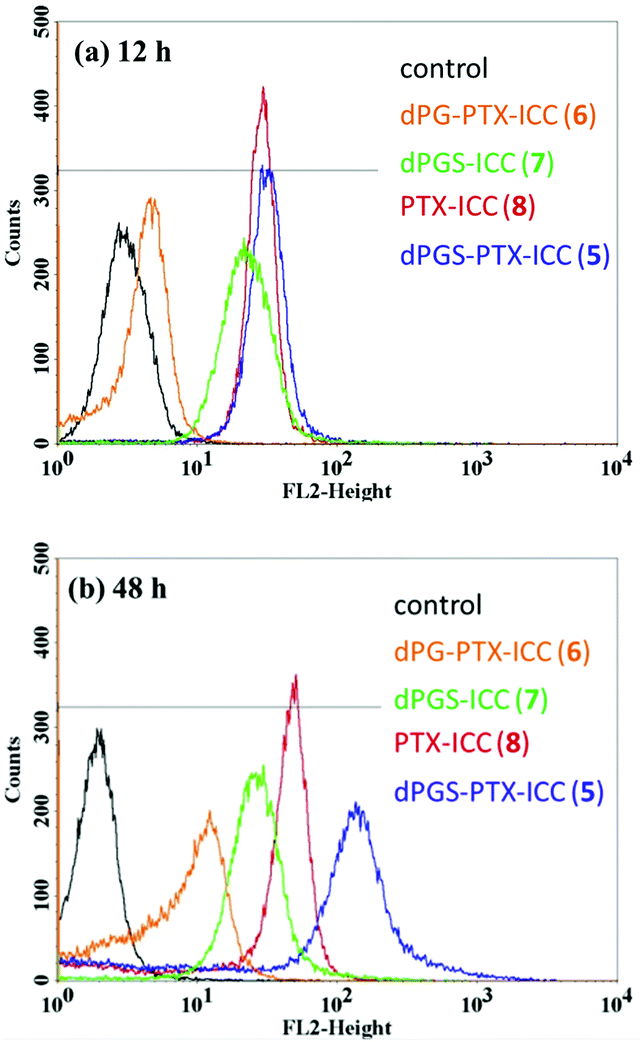 | ||
| Fig. 2 Representative distribution of A431 cells in FL2-H histograms. (a) Incubation time = 12 h, (b) incubation time = 48 h. | ||
A strong difference was also observed for the internalization kinetics of the various compounds. The analysis of the PTX-ICC (8) internalization revealed a logarithmic uptake kinetic with a saturation plateau, while dPGS-PTX-ICC (5) was taken up linearly without any saturation plateau over 48 h (Fig. 3). This indicates a non-saturated transport of the conjugate. Notably, the observed logarithmic uptake for PTX was much slower than what has been described in the literature.48 The different behaviors might be attributed to the conjugation of PTX to the ICC moiety. Interestingly, as seen in Fig. 3, the lack of PTX in dPGS-ICC (7) resulted in a completely different uptake profile. This can be explained by the strong decrease in the hydrophobicity of this conjugate along with the absence of the active PTX fraction (ongoing proliferation). Remarkably, in concordance with previous results,37 only a minor uptake of dPG-PTX-ICC (6) was observed after several hours, confirming that cellular uptake is strongly charge and size dependent.49 Our group has previously shown that neutral dPG was not uptaken by different cell lines when the molecular weights were below 20 kDa, while dPGS with similar molecular weights was rapidly internalized in those cells.37,41,49 Besides flow cytometry, fluorescence microscopy was also employed to monitor the internalization of dPGS-PTX-ICC (5) in A431 cells. According to microscopic analysis, dPGS-PTX-ICC (5) was predominantly localized in the surrounding of cell nuclei and displayed increasing fluorescence during the incubation period, which indicated cellular accumulation of the conjugate (Fig. S5, ESI†).
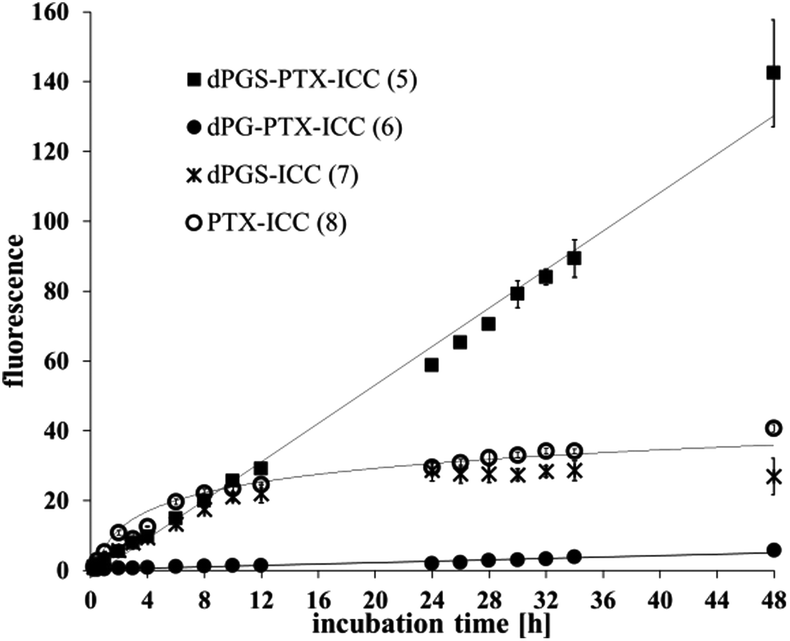 | ||
| Fig. 3 Internalization kinetic profiles of PTX-ICC (8), dPGS-ICC (7), dPGS-PTX-ICC (5) and dPG-PTX-ICC (6) in A431 cells (concentration = 1 μM). | ||
Elimination kinetics revealed a prolonged fluorescence of dPGS-PTX-ICC (5) in A431 cells compared to drug-free dPGS-ICC (7), indicating a slow release and intracellular accumulation of dPGS-PTX-ICC (5) (Fig. S6, ESI†). This accumulation profile seems to be ideal for the delivery of cytotoxic agents whose effects are cell cycle- or time-dependent. We hypothesize that this observation might be due to the presence of the active agent PTX, which reduces the proliferation and therefore the distribution of fluorescence to posterior generations.
In vitro cytotoxicity in A549 and A431 cells
In vitro toxicity was assessed in A549 and A431 cell lines. A549 cell line was selected because PTX is a chemotherapeutic agent commonly used for the treatment of lung carcinoma. A549 cells represent a human adenocarcinoma of the lung with typical features of pneumocytes type II. On the other hand, A431 cells are derived from a squamous cell carcinoma and highly express EGF-receptors. As shown in Fig. 4, free PTX was more toxic than dPGS-PTX (3) in both cell lines. dPGS-PTX (3) presented higher IC50 values (related to the PTX equivalents) than free PTX for each time point measured (Fig. 4 and Table S1 in ESI†). This is a typical observation when a free drug, which enters cells by diffusion, is compared to a multifunctionalized polymer-drug conjugate that is taken up by endocytosis.50,51 Specifically, at 48 h, IC50 values of dPGS-PTX (3) were 7.6 nM for A431 cells, which is about 2.3-fold higher than that of free PTX. In the case of dPG-PTX (4), IC50 values were 1.5-fold higher than that of dPGS-PTX (3). The obtained IC50 values of PTX in A549 cells fit with those described in the literature,52–56 while the published IC50 values of A431 differed between the sources57–59 and those presented here. The concentration-response curves obtained for both PTX and the conjugate were nevertheless conclusive (Fig. 4). In addition, experiments performed with dPGS amine (1) showed that there is no reduced viability of A431 cells after 48 h of incubation (IC50 value not evaluable; Fig. S7, ESI†). This is consistent with previous reports37 and confirms that the cytotoxic effect is related to the PTX fraction of the conjugate.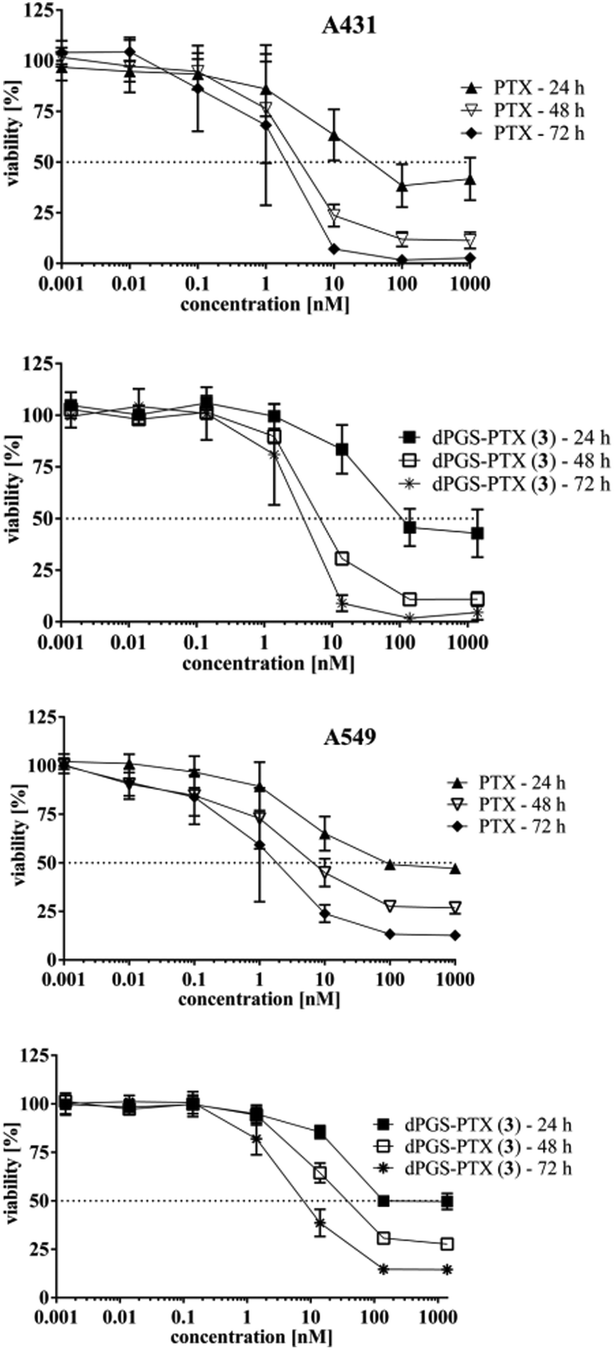 | ||
| Fig. 4 Concentration-response curve of PTX and dPGS-PTX (3) incubated on A549 and A431 cells at different time intervals. dPGS-PTX (3) conjugates present higher IC50-values compared to free PTX at every measurement point. | ||
In vitro activity of PTX and dPGS-PTX conjugates in A431 cells
The in vitro activity of the conjugates towards tubulin polymerization was studied by confocal microscopy. Fig. 5 shows representative confocal microscopy images of tubulin aggregation and reduction of free cytosolic tubulin staining induced by PTX and by dPGS-PTX (3). Treatment with the nude dendritic platform dPGS amine (1) did not cause tubulin aggregation. However, when the activity of control conjugate dPG-PTX (4) was analyzed, it was seen that the non-sulfated conjugate induced aggregation of tubulin to a similar extent as the activity observed for PTX and dPGS-PTX (3). Cell viability measurements were in concordance with these observations. This type of activity was not expected for the neutral conjugate, because dPG-PTX (4) was marginally internalized by A431 cells, as clearly demonstrated in the above sections. One plausible explanation for these findings could be the hydrolysis of the ester linkage, which would result in a premature PTX release from the conjugates before they were internalized in the cells. This hypothesis would explain the similar activity of PTX found in both conjugates, since the free PTX would enter the cells regardless of the nanocarrier. To further demonstrate this data interpretation, we analyzed the release of the drug from both conjugates at different pHs and in the presence of esterases and other hydrolytic enzymes (human plasma).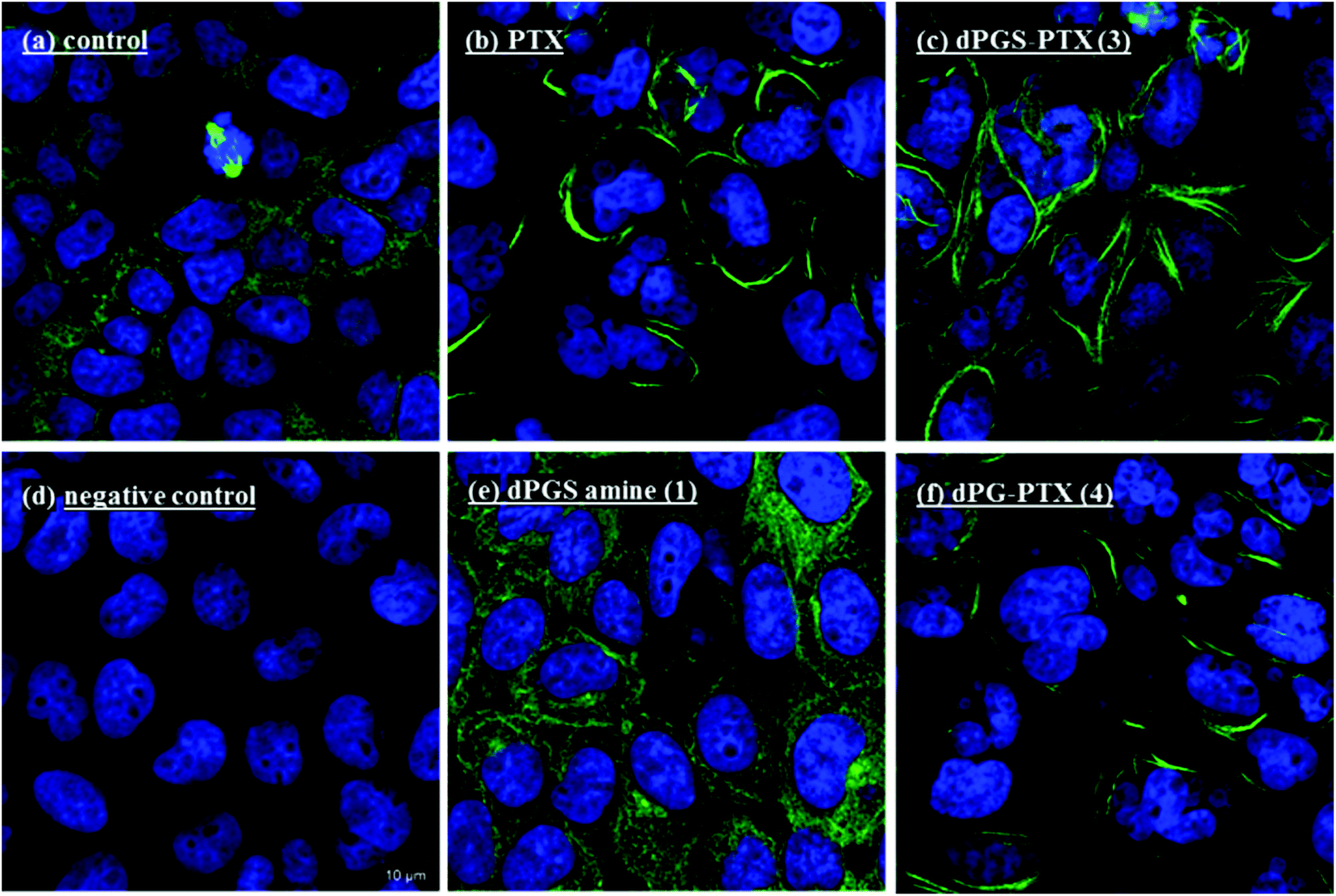 | ||
| Fig. 5 Representative confocal microscopy images of A431 cells incubated with (a) media, (b) PTX, (c) dPGS-PTX (3), (e) dPGS amine (1), and (f) dPG-PTX (4). (d) Represents a negative control. PTX, dPGS-PTX (3) and dPG-PTX (4) promote histologically visible tubulin alteration. dPGS amine (1) does not influence tubulin. C = 100 nM, incubation time = 16 h. Immunocytochemical staining of α-tubulin (green) and nuclear staining with DAPI (blue). Mitosis is seen in (a). | ||
Study of the release of PTX from dPGS-PTX (3) and dPG-PTX (4) by HPLC
In order to better understand the results obtained in the analysis of the activity towards tubulin polymerization induced by both sulfated and non-sulfated conjugates, we decided to study the release profile of PTX from the conjugate dPGS-PTX (3) at different pHs and in human plasma. We aimed to evaluate the stability of the ester linkage in the presence of esterases and other endogenous hydrolytic enzymes present in plasma. For that purpose, a liquid-liquid extraction protocol was chosen to isolate free PTX from dPGS and plasma proteins. Control experiments with known concentrations of the free drug were performed in order to validate the method and establish the extraction efficiency. PTX was stable during the time of analysis, and no degradation products were observed. The obtained release profile at 37 °C over a 44 h period is shown in Fig. 6. It can be clearly observed that the PTX release in human plasma is extremely fast, with values of 75% of PTX release within 3.5 h. These observations are in concordance with our premature drug release hypothesis and would explain the results obtained for tubulin polymerization. Indeed, the hydrolysis rate of the ester bond was much higher in buffer at pH 7.4 (85% at 24 h) than in pH 5 (15% at 24 h). This behavior has been reported for other polymer-PTX conjugates26,28 and is expected for succinimidyl esters hydrolysis.60 Nevertheless, in the case of neutral dPG-PTX (4) we did not observe such an effect, and PTX release was higher at acid pH (only 15% of release in buffer at pH 7.4 at 24 h). PTX release via succinimidyl ester hydrolysis from other dendritic scaffolds has been previously studied, with absolutely different results. Thus, in the case of hydroxyl-terminated poly(amido amine) PAMAM conjugated to PTX, only 20% of free drug was observed after 24 h of incubation in the presence of esterases,24 for hyaluronic acid-PTX conjugates, approx. 40% of the drug was released at 24 h,27 while a 100% release in the same time interval was reported for dendritic poly(ethylene glycol) bearing PTX and alendronate.28 In addition, the supramolecular organization of the conjugates in solution, i.e., the formation of micelles or supramolecular aggregates, is known to significantly affect the drug release kinetics.28 These diverse PTX release profiles suggest that the employment of ester linkages for the covalent attachment of PTX or other cytotoxic drugs to polymeric platforms should be carefully evaluated for each particular system.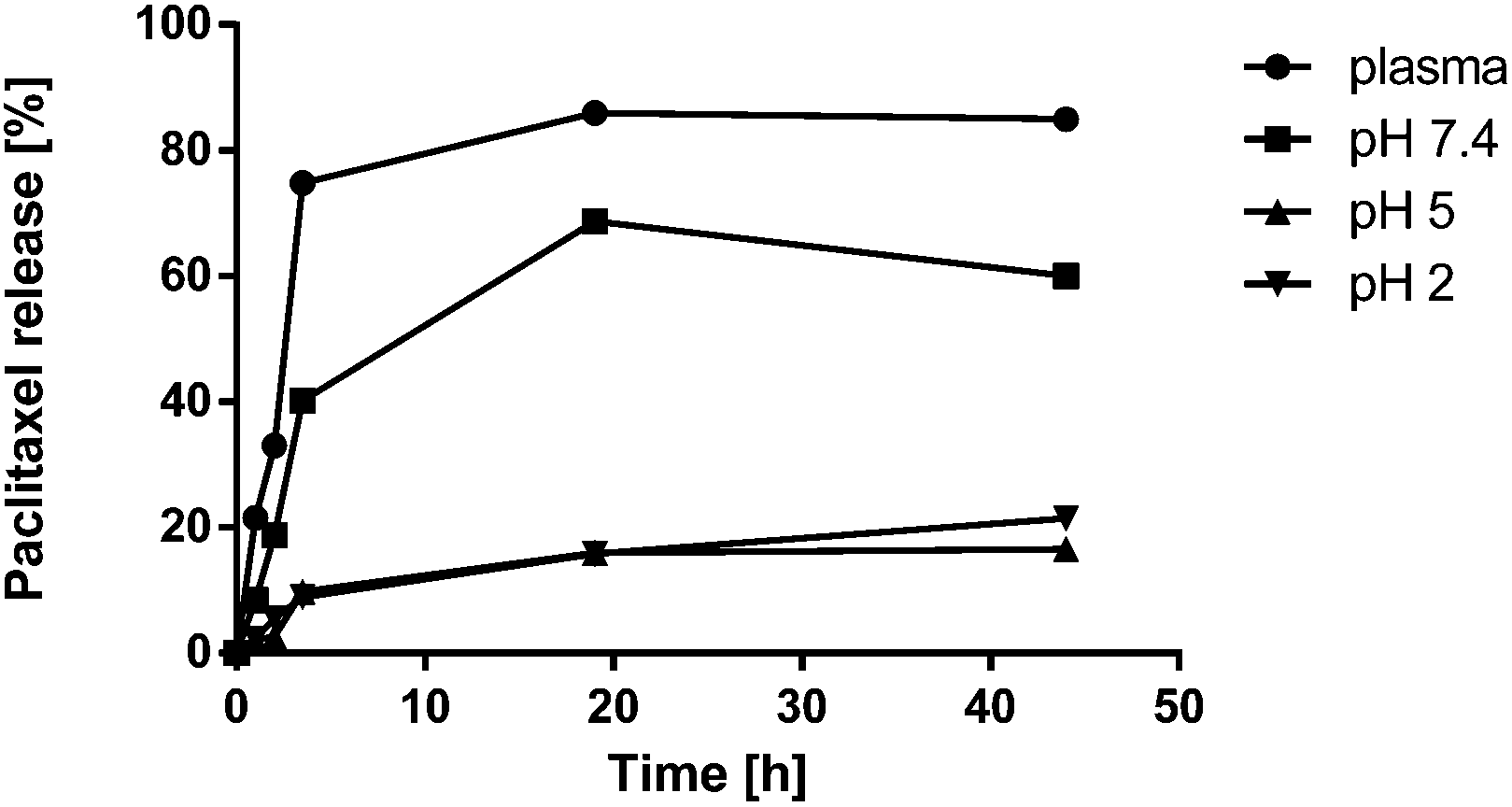 | ||
| Fig. 6 Cumulative release (in %) of PTX from dPGS-PTX (3) at 37 °C in human plasma and at different pH buffers. | ||
Conclusions
In this paper we describe the synthesis and characterization of multifunctional dendritic conjugates for the delivery of PTX. dPGS was chosen as the dendritic platform based on its excellent inflammation targeting properties and was conjugated to PTX through a succinyl ester linkage. Hydroxyl-terminated dPG-PTX conjugates were prepared as non-targeting controls, and both conjugates were further functionalized with an ICC dye. Cellular uptake experiments showed that anionic dPGS-PTX-ICC was rapidly internalized into A431 cells, while the uptake of dPG-PTX-ICC was only marginal. However, both conjugates generated the same activity towards tubulin polymerization. Their similar toxic profile could be explained by a premature drug release by the action of hydrolytic enzymes, as demonstrated by HPLC. Our results suggest that information on the kinetics of the release is critical for designing polymer-drug conjugates and must be carefully evaluated for each particular case. We are currently focusing on a different linker to improve the therapeutic properties of this conjugate. Nevertheless, the multifunctional conjugates described herein may find application in processes where a fast drug release is required.Acknowledgements
We thank the collaborative research center SFB765 sponsored by the Deutsche Forschungsgemeinschaft (DFG) and the BMBF-project MODIAMD for financial support. We acknowledge the focus area Nanoscale of the Freie Universität Berlin (http://www.nanoscale.fu-berlin.de). A. S.-H. is grateful for a Marie Curie Intra-European Fellowship (Project 302717). J. K. acknowledges the Alexander von Humboldt program. We are grateful to Dr Pamela Winchester for proofreading the manuscript.Notes and references
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- S. Rao, N. E. Krauss, J. M. Heerding, C. S. Swindell, I. Ringel, G. A. Orr and S. B. Horwitz, J. Biol. Chem., 1994, 269, 3132–3134 CAS.
- S. Rao, G. A. Orr, A. G. Chaudhary, D. G. Kingston and S. B. Horwitz, J. Biol. Chem., 1995, 270, 20235–20238 CrossRef CAS PubMed.
- C. Loeb, C. Combeau, L. Ehret-Sabatier, A. Breton-Gilet, D. Faucher, B. Rousseau, A. Commercon and M. Goeldner, Biochemistry, 1997, 36, 3820–3825 CrossRef CAS PubMed.
- P. B. Schiff and S. B. Horwitz, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 1561–1565 CrossRef CAS.
- M. M. Abu-Khalaf and L. N. Harris, in DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology, ed. V. T. DeVita, S. Hellman and S. A. Rosenberg, Wolters Kluwer, Lippincott Williams & Wilkins, Philadelphia, PA USA, 9th edn, 2011, ch. 38, pp. 413–421 Search PubMed.
- M. Joshi, X. Liu and C. P. Belani, Anti-Cancer Drugs, 2014, 25, 571–583 CrossRef CAS PubMed.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235, 179–192 CrossRef CAS PubMed.
- P. H. Wiernik, E. L. Schwartz, J. J. Strauman, J. P. Dutcher, R. B. Lipton and E. Paietta, Cancer Res., 1987, 47, 2486–2493 CAS.
- U. De Giorgi, G. Rosti, M. Monti, G. L. Frassineti and M. Marangolo, Ann. Oncol., 2003, 14, 1588–1589 CrossRef CAS PubMed.
- T. L. O'Connor and E. Kossoff, Pharmacotherapy, 2009, 29, 993–996 CrossRef PubMed.
- R. Duncan and M. J. Vicent, Adv. Drug Delivery Rev., 2013, 65, 60–70 CrossRef CAS PubMed.
- M. J. Vicent, H. Ringsdorf and R. Duncan, Adv. Drug Delivery Rev., 2009, 61, 1117–1120 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- V. Chandolu and C. R. Dass, Curr. Drug Discovery Technol., 2013, 10, 170–176 CrossRef CAS.
- J. W. Singer, J. Controlled Release, 2005, 109, 120–126 CrossRef CAS PubMed.
- S. D. Chipman, F. B. Oldham, G. Pezzoni and J. W. Singer, Int. J. Nanomed., 2006, 1, 375–383 CrossRef CAS PubMed.
- C. Li and S. Wallace, Adv. Drug Delivery Rev., 2008, 60, 886–898 CrossRef CAS PubMed.
- V. L. Galic, J. D. Wright, S. N. Lewin and T. J. Herzog, Expert Opin. Invest. Drugs, 2011, 20, 813–821 CrossRef CAS PubMed.
- J. Lim, A. Chouai, S.-T. Lo, W. Liu, X. Sun and E. E. Simanek, Bioconjugate Chem., 2009, 20, 2154–2161 CrossRef CAS PubMed.
- B. Thierry, P. Kujawa, C. Tkaczyk, F. M. Winnik, L. Bilodeau and M. Tabrizian, J. Am. Chem. Soc., 2005, 127, 1626–1627 CrossRef CAS PubMed.
- J. J. Khandare, S. Jayant, A. Singh, P. Chandna, Y. Wang, N. Vorsa and T. Minko, Bioconjugate Chem., 2006, 17, 1464–1472 CrossRef CAS PubMed.
- X. Bi, X. Shi, I. J. Majoros, R. Shukla and J. R. Baker, J. Comput. Theor. Nanosci., 2007, 4, 1179–1187 CAS.
- G. Cavallaro, M. Licciardi, P. Caliceti, S. Salmaso and G. Giammona, Eur. J. Pharm. Biopharm., 2004, 58, 151–159 CrossRef CAS PubMed.
- Y. Luo, M. R. Ziebell and G. D. Prestwich, Biomacromolecules, 2000, 1, 208–218 CrossRef CAS PubMed.
- C. Clementi, K. Miller, A. Mero, R. Satchi-Fainaro and G. Pasut, Mol. Pharm., 2011, 8, 1063–1072 CrossRef CAS PubMed.
- A. Sousa-Herves, D. Groger, M. Calderon, E. Fernandez-Megia and R. Haag, in Dendrimers in Biomedical Applications, The Royal Society of Chemistry, 2013, pp. 56–72 Search PubMed.
- R. K. Tekade, P. V. Kumar and N. K. Jain, Chem. Rev., 2009, 109, 49–87 CrossRef CAS PubMed.
- A. F. Hussain, H. R. Krüger, F. Kampmeier, T. Weissbach, K. Licha, F. Kratz, R. Haag, M. Calderón and S. Barth, Biomacromolecules, 2013, 14, 2510–2520 CrossRef CAS PubMed.
- M. Calderón, P. Welker, K. Licha, I. Fichtner, R. Graeser, R. Haag and F. Kratz, J. Controlled Release, 2011, 151, 295–301 CrossRef PubMed.
- M. Calderón, S. Reichert, P. Welker, K. Licha, F. Kratz and R. Haag, J. Biomed. Nanotechnol., 2011, 10, 92–99 CrossRef.
- M. Weinhart, D. Gröger, S. Enders, J. Dernedde and R. Haag, Biomacromolecules, 2011, 2502–2511 CrossRef CAS PubMed.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS PubMed.
- H. Türk, R. Haag and S. Alban, Bioconjugate Chem., 2004, 15, 162–167 CrossRef PubMed.
- J. Khandare, A. Mohr, M. Calderón, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- D. Gröger, F. Paulus, K. Licha, P. Welker, M. Weinhart, C. Holzhausen, L. Mundhenk, A. D. Gruber, U. Abram and R. Haag, Bioconjugate Chem., 2013, 24, 1507–1514 CrossRef PubMed.
- M. Weinhart, D. Gröger, S. Enders, S. B. Riese, J. Dernedde, R. K. Kainthan, D. E. Brooks and R. Haag, Macromol. Biosci., 2011, 11, 1088–1098 CrossRef CAS PubMed.
- S. Biffi, S. Dal Monego, C. Dullin, C. Garrovo, B. Bosnjak, K. Licha, P. Welker, M. M. Epstein and F. Alves, PLoS One, 2013, 8, e57150 CAS.
- K. Licha, P. Welker, M. Weinhart, N. Wegner, S. Kern, S. Reichert, I. Gemeinhardt, C. Weissbach, B. Ebert, R. Haag and M. Schirner, Bioconjugate Chem., 2011, 22, 2453–2460 CrossRef CAS PubMed.
- A. Sunder, R. Mülhaupt, R. Haag and H. Frey, Adv. Mater., 2000, 12, 235–239 CrossRef CAS.
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- K. Licha, C. Hessenius, A. Becker, P. Henklein, M. Bauer, S. Wisniewski, B. Wiedenmann and W. Semmler, Bioconjugate Chem., 2001, 12, 44–50 CrossRef CAS PubMed.
- D. J. Giard, S. A. Aaronson, G. J. Todaro, P. Arnstein, J. H. Kersey, H. Dosik and W. P. Parks, J. Natl. Cancer Inst., 1973, 51, 1417–1423 CAS.
- D. Gröger, M. Kerschnitzki, M. Weinhart, S. Reimann, T. Schneider, B. Kohl, W. Wagermaier, G. Schulze-Tanzil, P. Fratzl and R. Haag, Adv. Healthcare Mater., 2014, 3, 375–385 CrossRef PubMed.
- H. T. S. Britton and R. A. Robinson, J. Chem. Soc., 1931, 1456–1462 RSC.
- H.-J. Kuh, S. H. Jang, M. G. Wientjes and J. L.-S. Au, J. Pharmacol. Exp. Ther., 2000, 293, 761–770 CAS.
- S. Reichert, P. Welker, M. Calderón, J. Khandare, D. Mangoldt, K. Licha, R. K. Kainthan, D. E. Brooks and R. Haag, Small, 2011, 7, 820–829 CrossRef CAS PubMed.
- F. Kratz, U. Beyer, T. Roth, N. Tarasova, P. Collery, F. Lechenault, A. Cazabat, P. Schumacher, C. Unger and U. Falken, J. Pharm. Sci., 1998, 87, 338–346 CrossRef CAS PubMed.
- P. C. A. Rodrigues, U. Beyer, P. Schumacher, T. Roth, H. H. Fiebig, C. Unger, L. Messori, P. Orioli, D. H. Paper, R. Mülhaupt and F. Kratz, Bioorg. Med. Chem., 1999, 7, 2517–2524 CrossRef CAS PubMed.
- T. Yamori, S. Sato, H. Chikazawa and T. Kadota, Cancer Sci., 1997, 88, 1205–1210 CrossRef CAS PubMed.
- S. Padar, C. Van Breemen, D. W. Thomas, J. A. Uchizono, J. C. Livesey and R. Rahimian, Br. J. Pharmacol., 2004, 142, 305–316 CrossRef CAS PubMed.
- H. Wang, H. Li, M. Zuo, Y. Zhang, H. Liu, W. Fang and X. Chen, Cancer Lett., 2008, 268, 89–97 CrossRef CAS PubMed.
- I. Ojima, X. Geng, X. Wu, C. Qu, C. P. Borella, H. Xie, S. D. Wilhelm, B. A. Leece, L. M. Bartle, V. S. Goldmacher and R. V. J. Chari, J. Med. Chem., 2002, 45, 5620–5623 CrossRef CAS PubMed.
- I. Ojima, J. C. Slater, E. Michaud, S. D. Kuduk, P.-Y. Bounaud, P. Vrignaud, M.-C. Bissery, J. M. Veith, P. Pera and R. J. Bernacki, J. Med. Chem., 1996, 39, 3889–3896 CrossRef CAS PubMed.
- R. B. Lichtner, A. Rotgeri, T. Bunte, B. Buchmann, J. Hoffmann, W. Schwede, W. Skuballa and U. Klar, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11743–11748 CrossRef CAS PubMed.
- R. Miyata, M. Ueda, H. Jinno, T. Konno, K. Ishihara, N. Ando and Y. Kitagawa, Int. J. Cancer, 2009, 124, 2460–2467 CrossRef CAS PubMed.
- N. Sain, B. Krishnan, M. G. Ormerod, A. De Rienzo, W. M. Liu, S. B. Kaye, P. Workman and A. L. Jackman, Mol. Cancer Ther., 2006, 5, 1197–1208 CrossRef CAS PubMed.
- M. C. Carter and M. E. Meyerhoff, J. Immunol. Methods, 1985, 81, 245–257 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the conjugates, HPLC chromatograms, internalization images of dPGS-PTX-ICC (5), elimination kinetics of dPGS-PTX-ICC (5) and dPGS-ICC (7), comparison of IC50 values of PTX and dPGS-PTX (3) in A431 and A549 cell lines and cell viability of dPGS amine (1). See DOI: 10.1039/c4nr04428b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |