
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetically triggered release of molecular cargo from iron oxide nanoparticle loaded microcapsules†
Susana
Carregal-Romero‡
ab,
Pablo
Guardia‡
c,
Xiang
Yu
a,
Raimo
Hartmann
a,
Teresa
Pellegrino
*c and
Wolfgang J.
Parak
*ab
aFachbereich Physik, Philipps Universität Marburg, Marburg, Germany. E-mail: teresa.pellegrino@iit.it; wolfgang.parak@physik.uni-marburg.de
bCIC Biomagune, San Sebastian, Spain
cIstituto Italiano di Tecnologia, via Morego 30, Genova, Italy
First published on 10th November 2014
Abstract
Photothermal release of cargo molecules has been extensively studied for bioapplications. For instance, microcapsules decorated with plasmonic nanoparticles have been widely used in in vitro assays. However, some concerns about their suitability for some in vivo applications cannot be easily overcome, in particular the limited penetration depth of light (even infrared). Magnetic nanoparticles are alternative heat-mediators for local heating, which can be triggered by applying an alternating magnetic field (AMF). AMFs are much less absorbed by tissue than light and thus can penetrate deeper overcoming the above mentioned limitations. Here we present iron oxide nanocube-modified microcapsules as a platform for magnetically triggered molecular release. Layer-by-layer assembled polyelectrolyte microcapsules with 4.6 μm diameter, which had 18 nm diameter iron oxide nanocubes integrated in their walls, were synthesized. The microcapsules were further loaded with an organic fluorescent polymer (Cascade Blue-labelled dextran), which was used as a model of molecular cargo. Through an AMF the magnetic nanoparticles were able to heat their surroundings and destroy the microcapsule walls, leading to a final release of the embedded cargo to the surrounding solution. The cargo release was monitored in solution by measuring the increase in both absorbance and fluorescence signal after the exposure to an AMF. Our results demonstrate that magnetothermal release of the encapsulated material is possible using magnetic nanoparticles with a high heating performance.
Introduction
For the in vitro and in vivo delivery of functional molecular cargo (such as drugs or enzymes) it is often beneficial to encapsulate the cargo. Encapsulation can be used to protect the cargo from enzymatic degradation,1 to alter the intracellular distribution of the cargo within cells, as well as the biodistribution within animals,2,3 and to control the release of the cargo.4,5 Polyelectrolyte capsules fabricated by layer-by-layer (LbL) assembly offers a convenient approach for cargo encapsulation.6 Hitherto, this approach has been mainly optimized for in vitro assays, whereby a capsule size on the micrometre scale is beneficial.7–9 However, in vivo applications, for instance vaccination, have also been reported.10,11 Controlled release from such microcapsules has been demonstrated in vitro using a large variety of different stimuli including pH,12 redox potential,13 enzymatic degradation,1 ultrasound,14 microwaves,15 and photothermal local heating.16–20 For externally triggered release, photothermal heating has shown to be of great interest as it can be remotely controlled.Photothermal heating, i.e. the local generation of heat upon optical resonant excitation of plasmonic nanoparticles (NPs),21 has nowadays reached a high level of control and is used in a variety of biological applications.22–24 As an example, photothermal therapy preferentially induces apoptosis in cells by local heating (hyperthermia) of the NPs’ surrounding.24–26 However this is a delicate process since an elevated power dissipation will lead to a high increase of the temperature and thus (above 100 °C) to the formation of bubbles through water evaporation.27 In such a scenario cells would be destroyed by mechanical effects rather than by apoptosis.19,28 It is worth underlining that intracellular effects triggered by light activation of plasmonic NPs are not exclusively/solely based on heat generation, but also on the generation of reactive oxygen species.29 Finally, photothermal heating has also been used for light-triggered destruction of carrier matrixes in which plasmonic NPs are embedded, which can be used for the release of encapsulated molecules.30 Besides polymer capsules and particles, liposomes have also been used as carrier matrixes.31 Such light-triggered release has reached a degree of control that allows for opening of individual capsules inside cells,20 and thus the subsequent in vitro release of different molecular cargo inside cells.5 However, regarding potential in vivo applications, photothermal heating of plasmonic NPs by optical excitation shows a significant drawback due to the strong absorption of light by tissues, slightly reduced in the so-called biological window in the near infrared range.32,33 This issue definitively restricts the use of plasmonic NPs for in vivo applications.
Besides plasmonic NPs, magnetic NPs are extensively used as heat-mediators for magnetically induced hyperthermia, in which the heat is produced via an alternating magnetic field (AMF).34–37 Technically both modes of excitation are based on the excitation through an electromagnetic field but at different frequencies. While for optical heating the frequency of the electromagnetic field is in the visible and the infrared (IR) range, in the case of magnetic NPs the frequency is in the radiofrequency (RF) range. Similar to plasmonic NPs, the capability of magnetic NPs to act as heat-mediators38,39 has been exploited for magnetically mediated hyperthermia treatment40–42 as well as for controlled drug delivery.43,44 However, magnetic NPs have so far been less exploited to trigger release from carrier matrixes such as polyelectrolyte capsules.4 In this direction permeability changes of the polymeric walls of capsules with embedded magnetic NPs and drug release upon application of an AMF have already been reported.45,46 Besides heat, AMFs can produce mechanical vibrations of magnetic nanoparticles and stretching of the polymers in close proximity. This non-heating effect has been exploited as well to control the enzymatic activity in nanocomposites.47 However, the low heating performance shown by magnetic NPs synthesized by conventional methods so far has set limits to the promising concept of remotely controlled delivery of drugs by magnetothermal heating,48 which requires magnetic NPs with a high heating performance. Recent advances in the synthesis of novel magnetic NPs49 have resulted in iron oxide nanocubes as efficient heat-mediators for magnetically induced hyperthermia.50 Such cubic iron oxide NPs are preferentially prepared in organic solvents, but can be easily transferred to polar solvents,51 which will further allow for their integration into polyelectrolyte capsules. Owing to the high heating capability of these magnetic NPs, they are appealing to be used to trigger the release of molecular cargo from polyelectrolyte capsules and other carrier matrixes via magnetothermal heating. Higher heating capability required the use of less nanoparticles, as the highly uniform size distribution of the nanocubes warrants that most nanoparticles lie within the required (size-dependent) resonance frequency, thus contributing towards heating. The concept of remotely controlled release of encapsulated molecules via magnetothermal heating has recently been demonstrated for liposomes and hybrid capsules containing a fluorescent probe within the lipidic layer of the capsule wall.52,53 While the proof of this concept has already been made, for a more general applicability it still needs to be extended to other types of matrixes as well as improved nanoparticles, which is the objective of this report.
Here we present a proof of concept of magnetically triggered release of a model molecule using polyelectrolyte microcapsules as carrier matrixes. Polyelectrolyte microcapsules bearing iron oxide nanocubes as magnetic NPs in their walls were loaded with Cascade Blue-labelled dextran and the latter released by applying an AMF.
Materials and methods
Magnetic nanoparticle synthesis
Synthesis of water-soluble iron oxide nanocubes with a core size of 18 ± 2 nm were synthesized according to a previously published protocol.51 Iron(III) acetylacetonate (99%), decanoic acid (99%) and dibenzyl ether (99%) were purchased from Acros. Squalane (98%) was purchased from Alfa Aesar. Milli-Q water (18.2 MΩ, filtered with filter pore size 0.22 μM) was from Millipore. All solvents used were of analytical grade and were purchased from Sigma-Aldrich. All chemicals were used as received. Briefly, in a 50 mL three-necked round bottom flask equipped with a water cooled-condenser connected to a standard Schlenk line, 0.353 g (1 mmol) of iron(III) acetylacetonate and 0.78 g (4.5 mmol) of decanoic acid were dissolved in 18 mL of dibenzyl ether (DBE) and 7 mL of squalane. After degassing for 120 minutes at 65 °C, the mixture was heated up to 200 °C (3 °C min−1) and maintained at this temperature for 2.5 h. Finally the temperature was increased (7 °C min−1) up to 310 °C and maintained at this temperature for 1 h. After cooling down to room temperature, 60 mL of acetone were added and the solution was centrifuged at 8500 rpm. The collected black precipitate was dispersed in 2–3 mL of chloroform and then washed repeatedly at least two more times. Finally the collected particles were dispersed in 15 mL of chloroform. For the water transfer, 15 mL of gallol-modified polyethylene glycol (GA-PEG, Mw ≈ 3 kDa)43 solution (0.1 M in chloroform containing 1 mL triethylamine) were added to a solution of the NPs in chloroform and stirred overnight at room temperature. Then, 10 mL of de-ionized water were added resulting in the formation of two phases. After emulsification by means of shaking, the two phases were allowed to separate and the aqueous phase containing the GA-PEG coated magnetic NPs was collected. This step was repeated until all NPs were transferred to water. The excess of GA-PEG was removed by dialysis overnight at room temperature in a de-ionized water bath using a cellulose membrane tubing (molecular weight cut off (MWCO) of 50 kDa). This step was repeated 5 times. Finally, the aqueous solution containing the NPs was concentrated by centrifugation using a centrifuge filter (MWCO of 100 kDa) to obtain an iron concentration of about 15–16 g L−1 (as determined by Induced Coupled Plasma – Atomic Emission Spectroscopy, ICP-AES, Thermo Fisher).Synthesis of capsules
Template microparticles of CaCO3 were obtained by mixing aqueous solutions of CaCl2 (5 mL, 0.33 M) and Na2CO3 (5 mL, 0.33 M) under magnetic stirring at room temperature.20 Polyelectrolyte walls were assembled around the CaCO3 template cores (∼4.5 μm diameter, 10 mL, 165 mg) by LbL coating of alternating layers of poly(allylamine hydrochloride) (PAH, Mw ≈ 56 kDa) as the positive polyelectrolyte and poly(styrene sulfonate) (PSS, Mw ≈ 70 kDa) as the negative polyelectrolyte, using standard protocols reported in the literature.54 For the LbL assembly the CaCO3 particles were mixed with 5 mL of 2 mg mL−1 polymer solutions made in Milli-Q water, with 0.5 M of NaCl. As the 4th layer a stronger positively charged polymer, poly(acrylamide-co-diallyl-dimethylammonium chloride) (P(Am-DDA)), was assembled on the LbL wall. P(Am-DDA) improves the attachment of magnetic NPs.55 In this regard, 1 mL of magnetic NP solution (c(Fe) = 15.65 g L−1) was added to the capsule solution. The concentration of magnetic NPs within the capsule shell can be tailored by decreasing the concentration of this NP solution.55 Magnetic NPs were attached to the capsule wall on the positive layer of P(Am-DDA) via their negative charge (zeta potential of the magnetic NPs ζ = −20.9 ± 0.3 mV) and subsequently LbL coatings of alternating layers of PAH and PSS were added. The final architecture of the LbL wall was (PSS/PAH)(PSS/P(Am-DDA))-NPs (PAH)-(PSS/PAH)2. After dissolution of the CaCO3 core by adding 10 mL of ethylenediamine-tetraacetic acid disodium (EDTA) solution (0.2 M, pH 7) the capsules were filled with Cascade Blue-labelled dextran (10 kDa) via post-loading and further temperature shrinking of the LbL wall at 65 °C for one hour.20 For this purpose 2 mL of an aqueous solution of 1 mg per mL of Cascade Blue-labelled dextran were mixed for 30 minutes with a concentrated solution of capsules (2 mL). The calculated percentage of loading by determining the residual amount of non-encapsulated Blue-labelled dextran via UV-vis spectroscopy was around 60%. The final concentration was 3.3 × 108 capsules mL−1 (in 5 mL) measured with a hemocytometer and 1.6 g L−1 of the Fe content (4.8 × 10−12 g Fe per capsule). The packing fraction, defined as the total volume occupied by the nanocubes divided by the volume within the capsule shell in which the NPs are distributed, is around 100%, as the nanocubes are bigger than the polymer part of the shell. For the calculation we refer to the ESI† and Abbasi et al.55 The capsules were characterized by optical microscopy, transmission electron microscopy (TEM), and dynamic light scattering (DLS) (cf.Fig. 1). The capsule diameter was obtained by analysing optical microscopy images with the software UTHSCSA Image Tool (version 3.0).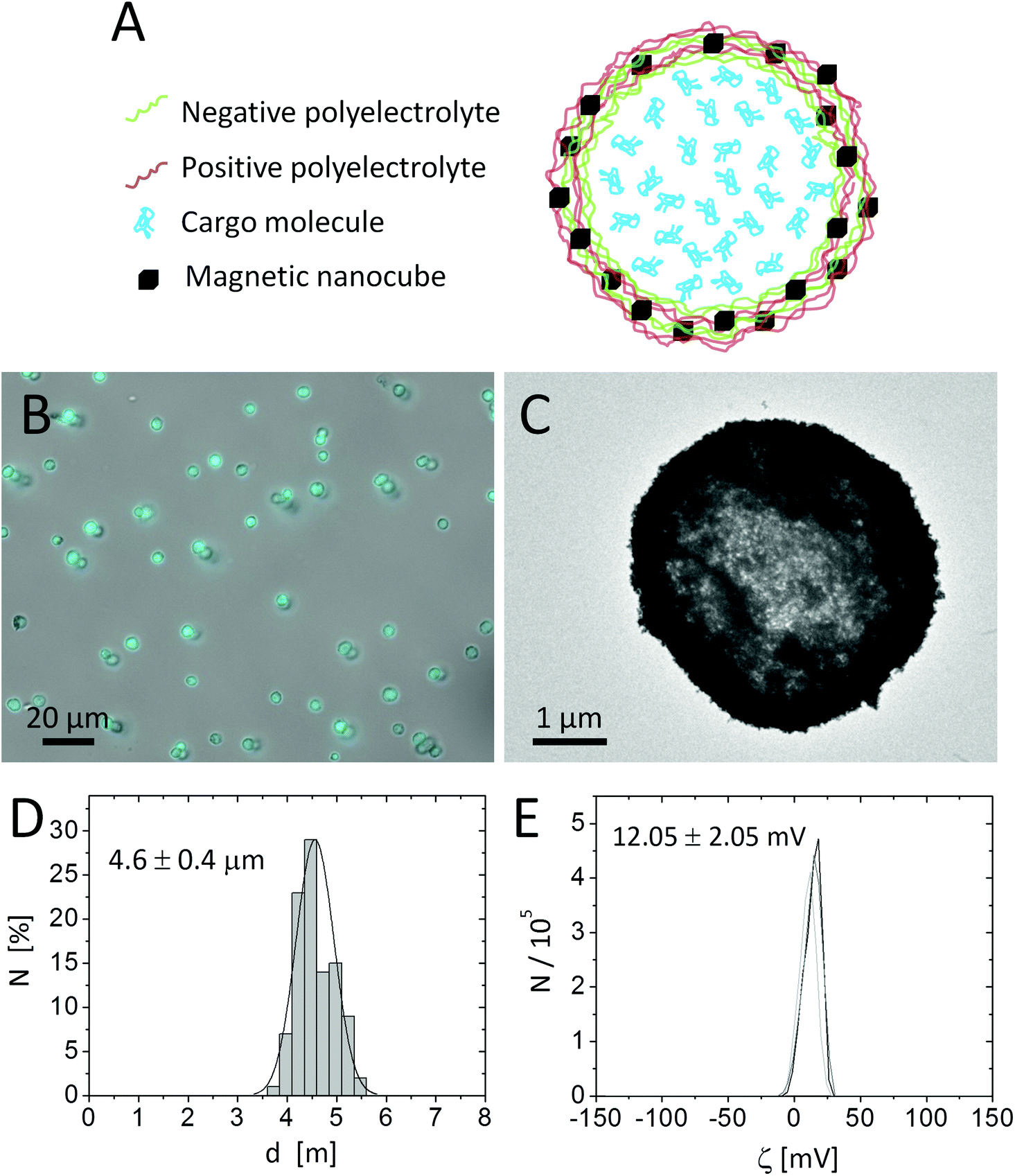 | ||
| Fig. 1 (A) Sketch of one polyelectrolyte capsule comprising Cascade Blue-labelled dextran as fluorescent cargo in the cavity and magnetic NPs in the wall. (B) Optical microscopy image of capsules dispersed in water showing the bright field and blue fluorescence channel (excitation 365/50 nm, beam splitter 395 nm and emission 445/50 nm) corresponding to the emission of Cascade Blue-labelled dextran. The scale bar corresponds to 20 μm. (C) TEM image of dried capsules. The scale bar corresponds to 1 μm. (D) Histogram of the size distribution fitted with a Gaussian curve. (E) ζ-potential measurements of the polyelectrolyte capsules in water. The values are the results of three independent measurements. | ||
Cargo release via magnetothermal heating
For the magnetothermal heating experiments a commercially available set-up was used (DM100 Series, nanoScale Biomagnetics Corp.). Experiments were performed on a concentrated (three times) capsule solution with an iron content of 4.8 g L−1 (9.9 × 108 capsules mL−1; 4.8 × 10−12 g Fe per capsule). For a more detailed explanation about the iron concentration used we refer to the ESI.† Before heating experiments, the capsule solution was purified from Cascade Blue-labelled dextran, which has leaked out of the capsules. For this purpose 350 μL of capsule solution were placed on top of a magnet (0.2 T) for 20 minutes and the capsules were collected (due to the magnetic NPs in their walls) at the bottom, while the supernatant was discarded. Then 350 μL of fresh Milli-Q water was added to the precipitate, followed by careful shaking, in order to redisperse capsules in solution. In order to quantify the possible presence of free Cascade Blue-labelled dextran, after adding fresh Milli-Q water, the solution was again placed on top of a magnet for 20 minutes. After this the capsules were collected at the bottom, the supernatant was withdrawn, and its absorption spectrum was recorded, which did not show significant leaching of Cascade Blue-labelled dextran. The sample was then split into two aliquots: 250 μL of the test-sample to be treated and 100 μL as the control sample. The test-sample was placed under an AMF (300 kHz, 24 kA m−1) for 90 minutes and the temperature of the solution was monitored (cf.Fig. 2). The control sample was kept at room temperature for 90 minutes. Both samples were again placed on a magnet for 20 minutes to collect the capsules. Then supernatants were collected and filtered using microcentrifuge spin cups and columns (MWCO cut-off 100 kDa), in order to remove capsule fragments from the supernatant, which may not have precipitated. Absorption and fluorescence emission spectra of the supernatants were recorded using quartz cuvettes (cf.Fig. 3). In addition TEM images of the precipitate containing the capsules were recorded (cf. the ESI†).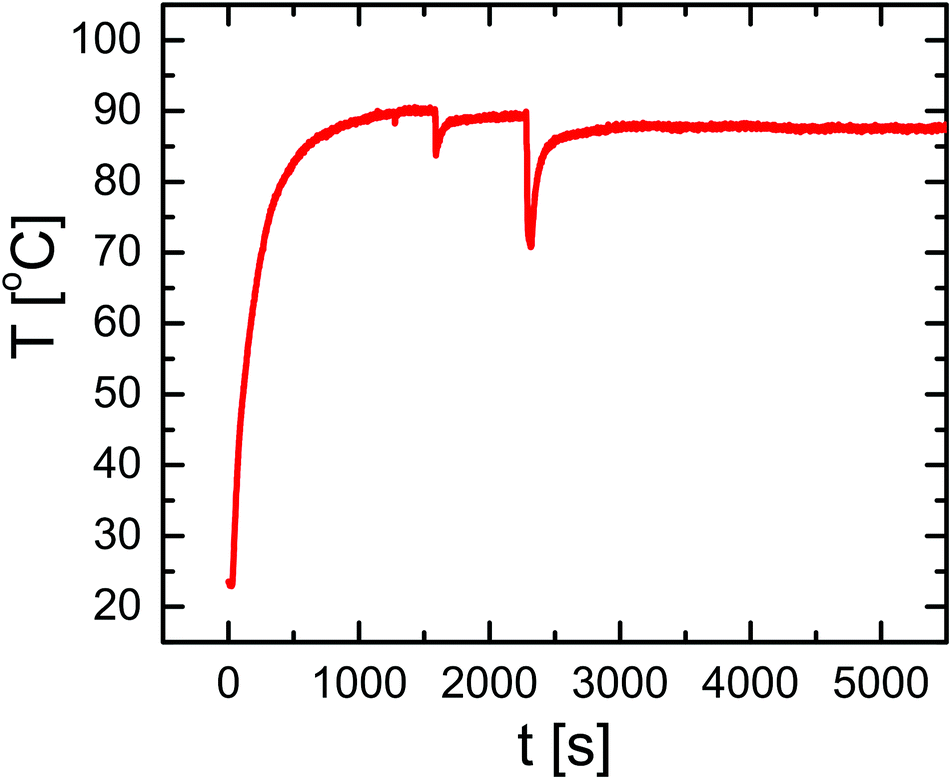 | ||
| Fig. 2 Bulk temperature T(t) as a function of time of a solution of capsules [4.8 g L−1 of Fe] placed under an AMF (300 kHz and 24 kA m−1) for 90 minutes. During the experiment the solution was removed from the cavity and the AMF was applied (dips in the curve at 1500 s and 2250 s) in order to control and shake the solution. | ||
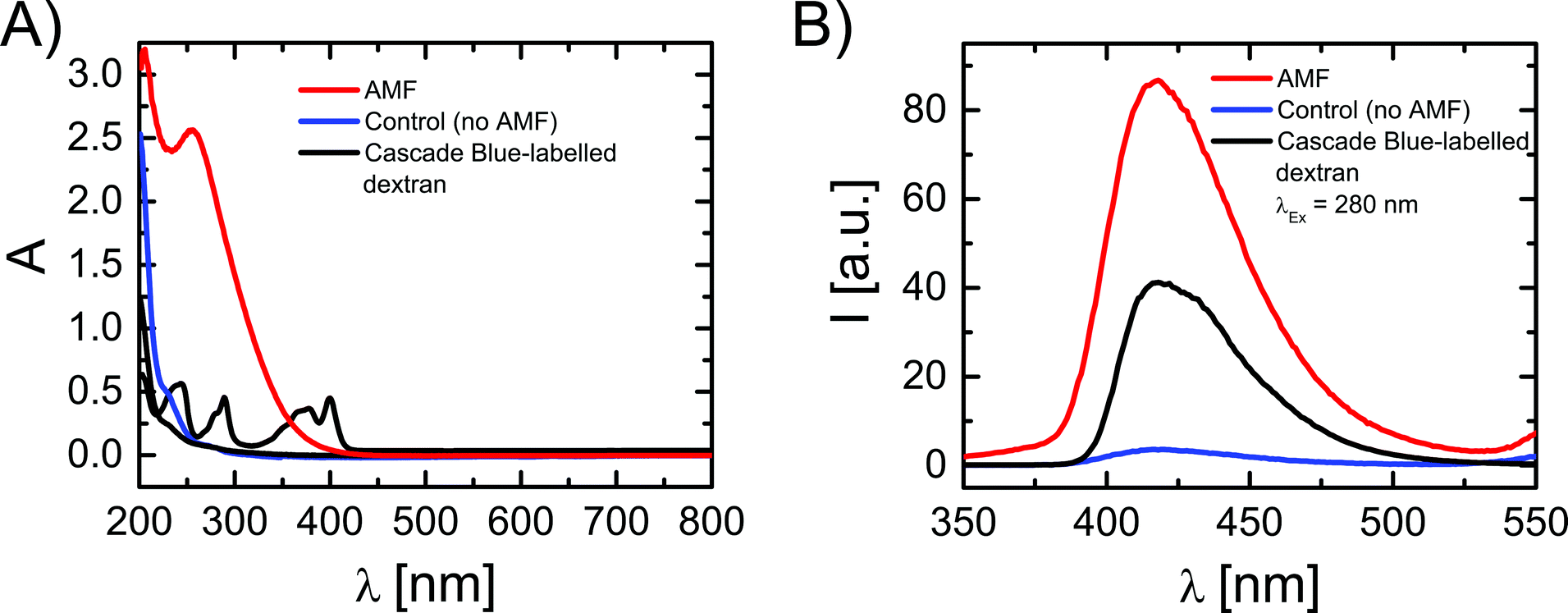 | ||
| Fig. 3 (A) UV-vis absorption spectra, and (B) fluorescence emission spectra (at λexc = 280 nm excitation) of: (i) free Cascade Blue-labelled dextran (black line), (ii) the supernatant of a capsule solution which had been treated for 90 minutes under an AMF (300 kHz and 24 kA m−1) (red line), and (iii) the supernatant of a control sample of capsules which had been kept at room temperature for 90 minutes and thus had not been exposed to an AMF (blue line). | ||
Results and discussion
Capsules of 4.6 ± 0.4 μm diameter with a zeta potential of ζ = +12.05 ± 0.5 mV and decorated with iron oxide nanocubes were successfully synthesized using an LbL approach (Fig. 1A). The presence of iron oxide nanocubes (18 ± 2 nm NP diameter) accounts for the strong interaction of the capsules with a static magnetic field (after less than 5 minutes most of the sample was collected when placed under a magnet of 0.2 T). Despite the incorporation of the magnetic NPs at a high concentration, the geometry of the capsules was rather spherical. The presence of magnetic NPs and the loading of the capsules with Cascade Blue-labelled dextran were confirmed by TEM images and optical microscopy, respectively (cf.Fig. 1 and the ESI†).Because iron oxide nanocubes have shown very efficient heating performances,51 it should be possible to trigger the release of encapsulated Cascade Blue-labelled dextran by an AMF via thermal destruction of the capsule wall in which the nanoparticles are embedded. In order to demonstrate magnetothermal heating, a solution containing capsules with an iron concentration of 4.8 g L−1 was exposed to an AMF for 90 minutes. Upon exposure to the AMF, the used capsule concentration ensured a final temperature of about 90 °C in dynamic equilibrium (cf.Fig. 2). In fact, under the same experimental magnetic field conditions the final temperature reached clearly did depend on the capsule (i.e. iron) concentration. Lower concentrations lead to lower final temperatures (cf. the ESI†). For instance, a capsule solution with a Fe content of 1.5 g L−1 raised the temperature only to 40 °C, whereas with a Fe content of 2.8 g L−1 temperature could be raised to 62 °C. Thus control of the capsules/iron concentration is of paramount importance, as the final temperature plays an important role in the opening of the capsules. It is worth underlining that the final temperature depends not only on the concentration of microcapsules, but also on their heating efficiency, which could be different with respect to “free” iron oxide nanocubes. In this regard, the specific absorption rate (SAR) was calculated from the temperature vs. time curves leading to a value of 427 W gFe−1 (at 300 kHz and 24 kA m−1). This SAR value is below the value for “free” iron oxide nanocubes (824 W gFe−1 at 300 kHz and 24 kA m−1).51 The lower performance can be explained as a result of a low thermal conductivity due to the polymer coating as well as due to magnetic dipole–dipole interactions.56,57 In addition it is worth underlining that the SAR value decreases while decreasing the concentration of microcapsules in solution (cf. the ESI†).
In order to evaluate the influence that the AMF had on the capsules, and hence the temperature increase, TEM images of the aliquots of the solution with AMF exposed capsules and the control sample were compared (cf. the ESI†). In the AMF treated capsules some damage in the form of partially broken walls could be observed, together with the presence of some free magnetic NPs which had been released from the capsule walls. Both effects could not be found in the control capsules. However, even if some pieces of evidence of the effect of AMF exposure were observed, these results are clearly purely qualitative and the observable difference between both samples is at best relatively small. We thus performed an additional study based on recording the absorption and fluorescence spectra of the supernatant, in order to probe release of Cascade Blue-labelled dextran from the capsules upon AMF exposure (cf.Fig. 3). Note that before experiments eventually leached dye had been removed and in an additional control no significant further leaching was observed on the time scale of experiments. For the control sample, after 90 minutes at room temperature (no AMF) a rather small enhancement in the absorption and fluorescence spectra was observed, which is likely ascribed to a slow leakage of Cascade Blue-labelled dextran from the capsules (cf.Fig. 3). On the other hand, in the case of AMF exposure remarkable changes in the absorption and fluorescence signals of the supernatant were observed. The fluorescence peak was centred at 420 nm, corresponding to the spectral feature of the fluorescence signal of free Cascade Blue-labelled dextran. In absorption however, for the AMF exposed sample, there was a strong and broad signal with a peak at 270 nm, and the characteristic absorption peaks of the free Cascade Blue-labelled dextran were partially hidden under this signal. This signal was clearly not present in the spectra of the control sample and not in the initial supernatant. The absorption peak at 270 nm might be attributed to the presence of small polymeric fragments derived from the broken capsule walls, which show a strong absorption below 300 nm (cf. the ESI†). Indeed, this hypothesis is supported by the TEM images of the AMF treated capsules in which a surface damage was observed. We thus conclude that, besides the release of Cascade Blue-labelled dextran, as observed by the fluorescence spectra, the heat generated under an AMF by the magnetic NPs in the capsule walls might also partially damage the polymeric wall in a way that polymer fragments, which absorb in the UV region, are released. In this context, Lu et al. studied the change of permeability of polyelectrolyte capsules containing magnetic nanoparticles using frequencies from 100 to 1000 Hz, while keeping the strength of the magnetic field constant at 95.5 kA m−1.45 They observed that low frequency AMFs (150 Hz, 95.5 kA m−1, 30 minutes) increased the permeability of the polyelectrolyte walls and the temperature up to 50 °C. However, frequencies beyond 300 Hz did not increase the permeability even after 1 hour of irradiation. The change of permeability was associated with NP agitation at low frequency AMFs and temporal disturbance of the polyelectrolyte wall. The temperature effect on the permeability was dismissed after performing control experiments with capsules heated at 50 °C. In contrast, in our experiments the capsules released cargo molecules after irradiation with an AMF of 300 Hz and the disruption of the wall must be ascribed to the higher heat production (90 °C) due to the presence of iron nanocubes and the rupture of the polymeric wall observed in TEM images.
Finally, the fluorescence intensities of the capsules before and after treatment of AMF irradiation were recorded with confocal microscopy. In a population of over 400 capsules an evident decrease of the mean fluorescence intensity was observed for the AMF treated capsules as compared to the control sample (cf. the ESI†). However, the AMF treatment did not release all the cargo molecules within the polyelectrolyte capsules, as observed in photothermal controlled release in vitro.20 This is due to the structure of the polyelectrolyte capsules. Polyelectrolyte capsules made on CaCO3 templates can be described as a gel-like matrix, as (due to the porosity of CaCO3) polyelectrolytes permeate within the template during the LbL assembly, and after the core dissolution a polyelectrolyte matrix is formed within the inner cavity of the capsules.58 Some cargo molecules can stay attached to the charged polyelectrolytes of the matrix, even after disruption of the wall. The decrease of fluorescence upon AMF exposure measured with confocal microscopy is an additional indication of the release of Cascade Blue-labelled dextran to the environment.
Conclusions
The proof-of-concept experiment shown in this study suggests that release of the molecular cargo from polyelectrolyte capsules by NP-mediated heating is not only possible by light-irradiation of plasmonic NPs, but also by AMF-irradiation of magnetic NPs. Owing to the high heating performance of iron oxide nanocubes, damage in the polymer wall was prompted through an AMF, which allowed for the release of the encapsulated molecular cargo. For successful remotely controlled release of cargo by magnetothermal heating the content of magnetic NPs has to be carefully tuned. In our case iron concentrations above 2.7 g L−1 (4.8 g L−1) allowed for a suitable temperature increase for opening the wall of the capsules. In comparison with optical excitation, AMF excitation is barely absorbed by tissues, which should allow for convenient externally triggered release. This issue opens up new and exciting possibilities of cargo release for in vivo applications, though the geometry of the capsules would need to be optimized towards such applications. In this regard, the future direction of this research should deal with the requirement of in vivo applications which preferentially will involve smaller capsules that can be fabricated using the LbL technique.59,60Acknowledgements
The authors are grateful to Dr. Pablo del Pino and Prof. Heinz Jänsch for helpful discussions, and to Simone Nitti and Giammarino Pugliese for helping with sample preparation. This work was supported by HSFP (project RGP0052/2012 to WJP), by the European project Magnifyco (contract NMP4-SL-2009-228622 to TP) and by the Italian AIRC project (contract no. 14527 to TP). X.Y. acknowledges support by the Chinese government (CSC, no. 2010691036).References
- P. Rivera_Gil, S. D. Koker, B. G. De_Geest and W. J. Parak, Nano Lett., 2009, 9, 4398–4402 CrossRef CAS PubMed.
- S. De Koker, B. G. De Geest, C. Cuvelier, L. Ferdinande, W. Deckers, W. E. Hennink, S. De Smedt and N. Mertens, Adv. Funct. Mater., 2007, 17, 3754–3763 CrossRef CAS.
- H. Shen, H. Shi, M. Xie, K. Ma, B. Li, S. Shen, X. Wang and Y. Jin, J. Mater. Chem. B, 2013, 1, 3906–3917 RSC.
- A. Johnston, G. Such and F. Caruso, Angew. Chem., Int. Ed., 2010, 49, 2664 CrossRef CAS PubMed.
- M. Ochs, S. Carregal-Romero, J. Rejman, K. Braeckmans, S. C. De Smedt and W. J. Parak, Angew. Chem., Int. Ed., 2013, 52, 695–699 CrossRef CAS PubMed.
- G. B. Sukhorukov, A. L. Rogach, B. Zebli, T. Liedl, A. G. Skirtach, K. Köhler, A. A. Antipov, N. Gaponik, A. S. Susha, M. Winterhalter and W. J. Parak, Small, 2005, 1, 194–200 CrossRef CAS PubMed.
- F. Caruso, Chem. – Eur. J., 2000, 6, 413–419 CrossRef CAS.
- W. Tong, X. Song and C. Gao, Chem. Soc. Rev., 2012, 41, 6103–6124 RSC.
- Y. Yan, M. Bjornmalm and F. Caruso, Chem. Mater., 2014, 26, 452–460 CrossRef CAS.
- A. Sexton, P. G. Whitney, S.-F. Chong, A. N. Zelikin, A. P. R. Johnston, R. De Rose, A. G. Brooks, F. Caruso and S. J. Kent, ACS Nano, 2009, 3, 3391 CrossRef CAS PubMed.
- S. De Koker, T. Naessens, B. G. De Geest, P. Bogaert, J. Demeester, S. De Smedt and J. Grooten, J. Immunol., 2010, 184, 203–211 CrossRef CAS PubMed.
- G. B. Sukhorukov, A. A. Antipov, A. Voigt, E. Donath and H. Mohwald, Macromol. Rapid Commun., 2001, 22, 44–46 CrossRef CAS.
- K. Liang, G. K. Such, Z. Zhu, S. J. Dodds, A. P. R. Johnston, J. Cui, H. Ejima and F. Caruso, ACS Nano, 2012, 6, 10186–10194 CrossRef CAS PubMed.
- A. M. Pavlov, V. Saez, A. Cobley, J. Graves, G. B. Sukhorukov and T. J. Mason, Soft Matter, 2011, 7, 4341–4347 RSC.
- L. L. del Mercato, E. Gonzalez, A. Z. Abbasi, W. J. Parak and V. Puntes, J. Mater. Chem., 2011, 21, 11468–11471 RSC.
- B. Radt, T. A. Smith and F. Caruso, Adv. Mater., 2004, 16, 2184–2189 CrossRef CAS.
- A. S. Angelatos, B. Radt and F. Caruso, J. Phys. Chem. B, 2005, 109, 3071–3076 CrossRef CAS PubMed.
- A. G. Skirtach, A. M. Javier, O. Kreft, K. Köhler, A. P. Alberola, H. Möhwald, W. J. Parak and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2006, 45, 4612–4617 CrossRef CAS PubMed.
- A. Muñoz Javier, P. del Pino, M. F. Bedard, A. G. Skirtach, D. Ho, G. B. Sukhorukov, C. Plank and W. J. Parak, Langmuir, 2008, 24, 12517–12520 CrossRef PubMed.
- S. Carregal-Romero, M. Ochs, P. Rivera_Gil, C. Ganas, A. M. Pavlov, G. B. Sukhorukov and W. J. Parak, J. Controlled Release, 2012, 159, 120–127 CrossRef CAS PubMed.
- G. Baffou and R. Quidant, Laser Photonics Rev., 2013, 7, 171–187 CrossRef CAS.
- X. Huang, P. Jain, I. El-Sayed and M. El-Sayed, Lasers Med. Sci., 2007, 23, 217–228 CrossRef PubMed.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef CAS PubMed.
- D. P. O'Neal, L. R. Hirsch, N. J. Halas, J. D. Payne and J. L. West, Cancer Lett., 2004, 209, 171–176 CrossRef PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- D. Hühn, A. Govorov, P. Rivera_Gil and W. J. Parak, Adv. Funct. Mater., 2012, 22, 294–303 CrossRef.
- S. Peeters, M. Kitz, S. Preisser, A. Wetterwald, B. Rothen-Rutishauser, G. N. Thalmann, C. Brandenberger, A. Bailey and M. Frenz, Biomed. Opt. Express, 2012, 3, 435–446 CrossRef CAS PubMed.
- Z. Krpetic, P. Nativo, V. See, I. A. Prior, M. Brust and M. Volk, Nano Lett., 2010, 10, 4549–4554 CrossRef CAS PubMed.
- A. Topete, M. Alatorre-Meda, E. M. Villar-Alvarez, S. Carregal-Romero, S. Barbosa, W. J. Parak, P. Taboada and V. Mosquera, Adv. Healthcare Mater., 2014, 3, 1309–1325 CrossRef CAS PubMed.
- S. J. Leung and M. Romanowski, ACS Nano, 2012, 6, 9383–9391 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- A. Vogel and V. Venugopalan, Chem. Rev., 2003, 103, 577–644 CrossRef CAS PubMed.
- A. Jordan, R. Scholz, P. Wust, H. Fahling and R. Felix, J. Magn. Magn. Mater., 1999, 201, 413–419 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- H. Huang, S. Delikanli, H. Zeng, D. M. Ferkey and A. Pralle, Nat. Nanotechnol., 2010, 5, 602–606 CrossRef CAS PubMed.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306–4334 RSC.
- R. E. Rosensweig, J. Magn. Magn. Mater., 2002, 252, 370–374 CrossRef CAS.
- R. Hiergeist, W. Andrä, N. Buske, R. Hergt, I. Hilger, U. Richter and W. Kaiser, J. Magn. Magn. Mater., 1999, 201, 420–422 CrossRef CAS.
- R. Hergt, S. Dutz, R. Mueller and M. Zeisberger, J. Phys.: Condens. Matter, 2006, 18, S2919–S2934 CrossRef CAS.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- J. P. Fortin, C. Wilhelm, J. Servais, C. Menager, J. C. Bacri and F. Gazeau, J. Am. Chem. Soc., 2007, 129, 2628–2635 CrossRef CAS PubMed.
- A. Riedinger, P. Guardia, A. Curcio, M. A. Garcia, R. Cingolani, L. Manna and T. Pellegrino, Nano Lett., 2013, 13, 2399–2406 CrossRef CAS PubMed.
- T. T. T. N'Guyen, H. T. T. Duong, J. Basuki, V. Montembault, S. Pascual, C. Guibert, J. Fresnais, C. Boyer, M. R. Whittaker, T. P. Davis and L. Fontaine, Angew. Chem., Int. Ed., 2013, 52, 14152–14156 CrossRef PubMed.
- Z. H. Lu, M. D. Prouty, Z. H. Guo, V. O. Golub, C. Kumar and Y. M. Lvov, Langmuir, 2005, 21, 2042–2050 CrossRef CAS PubMed.
- S.-H. Hu, C.-H. Tsai, C.-F. Liao, D.-M. Liu and S.-Y. Chen, Langmuir, 2008, 24, 11811–11818 CrossRef CAS PubMed.
- N. L. Klyachko, M. Sokolsky-Papkov, N. Pothayee, M. V. Efremova, D. A. Gulin, N. Pothayee, A. A. Kuznetsov, A. G. Majouga, J. S. Riffle, Y. I. Golovin and A. V. Kabanov, Angew. Chem., Int. Ed., 2012, 51, 12016–12019 CrossRef CAS PubMed.
- R. Hergt and S. Dutz, J. Magn. Magn. Mater., 2007, 311, 187–192 CrossRef CAS PubMed.
- J. H. Lee, J. T. Jang, J. S. Choi, S. H. Moon, S. H. Noh, J. W. Kim, J. G. Kim, I. S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS PubMed.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- P. Guardia, A. Riedinger, S. Nitti, G. Pugliese, S. Marras, A. Genovese, L. Manna and T. Pellegrino, J. Mater. Chem. B, 2014, 2, 4426–4434 RSC.
- K. Katagiri, Y. Imai, K. Koumoto, T. Kaiden, K. Kono and S. Aoshima, Small, 2011, 7, 1683–1689 CrossRef CAS PubMed.
- K. Katagiri, M. Nakamura and K. Koumoto, ACS Appl. Mater. Interfaces, 2010, 2, 768–773 CAS.
- L. L. del Mercato, A. Z. Abbasi and W. J. Parak, Small, 2011, 7, 351–363 CrossRef CAS PubMed.
- A. Z. Abbasi, L. Gutierrez, L. L. del Mercato, F. Herranz, O. Chubykalo-Fesenko, S. Veintemillas-Verdaguer, W. J. Parak, M. P. Morales, J. M. Gonzalez, A. Hernando and P. de la Presa, J. Phys. Chem. C, 2011, 115, 6257–6264 CAS.
- C. Haase and U. Nowak, Phys. Rev. B: Condens. Matter, 2012, 85, 045435 CrossRef.
- C. Martinez-Boubeta, K. Simeonidis, D. Serantes, I. Conde-Leborán, I. Kazakis, G. Stefanou, L. Peña, R. Galceran, L. Balcells, C. Monty, D. Baldomir, M. Mitrakas and M. Angelakeris, Adv. Funct. Mater., 2012, 22, 3737–3744 CrossRef CAS.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS.
- J. Schwiertz, W. Meyer-Zaika, L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, M. Vallet-Regi and M. Epple, J. Mater. Chem., 2008, 18, 3831–3834 RSC.
- S. Krol, S. del Guerra, M. Grupillo, A. Diaspro, A. Gliozzi and P. Marchetti, Nano Lett., 2006, 6, 1933–1939 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr04055d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |