
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquid-assisted exfoliation and dispersion: stripping graphene and its two-dimensional layered inorganic counterparts of their inhibitions
Sudhir
Ravula
,
Sheila N.
Baker
,
Ganesh
Kamath
and
Gary A.
Baker
*
Department of Chemistry, University of Missouri–Columbia, Columbia, MO 65211-7600, USA. E-mail: bakergar@missouri.edu; Tel: +1 573-882-1811
First published on 22nd January 2015
Abstract
Research on graphene—monolayers of carbon atoms arranged in a honeycomb lattice—is proceeding at a relentless pace as scientists of both experimental and theoretical bents seek to explore and exploit its superlative attributes, including giant intrinsic charge mobility, record-setting thermal conductivity, and high fracture strength and Young's modulus. Of course, fully exploiting the remarkable properties of graphene requires reliable, large-scale production methods which are non-oxidative and introduce minimal defects, criteria not fully satisfied by current approaches. A major advance in this direction is ionic liquid-assisted exfoliation and dispersion of graphite, leading to the isolation of few- and single-layered graphene sheets with yields two orders of magnitude higher than the earlier liquid-assisted exfoliation approaches using surface energy-matched solvents such as N-methyl-2-pyrrolidone (NMP). In this Minireview, we discuss the emerging use of ionic liquids for the practical exfoliation, dispersion, and modification of graphene nanosheets. These developments lay the foundation for strategies seeking to overcome the many challenges faced by current liquid-phase exfoliation approaches. Early computational and experimental results clearly indicate that these same approaches can readily be extended to inorganic graphene analogues (e.g., BN, MoX2 (X = S, Se, Te), WS2, TaSe2, NbSe2, NiTe2, and Bi2Te3) as well.
 Sudhir Ravula | Sudhir Ravula received his B.Sc. in 2006 and M.Sc. (Physical Organic Chemistry) in 2008 from Osmania University, in Hyderabad, India. Until 2011, he worked as a research scientist at Advinus Therapeutics Ltd. (Pune, India). He is presently a graduate student in the department of chemistry at the University of Missouri under the supervision of Prof. Gary A. Baker. His research interests focus primarily on the bottom-up synthesis and top-down exfoliation of two-dimensional nanosheets using various solvents, including ionic liquids. To date, he has published six papers in high-impact journals. |
 Sheila N. Baker | Sheila Baker obtained her Ph.D. from University at Buffalo, The State University of New York, in 2002 under the supervision of Prof. Frank V. Bright. She is currently an Assistant Professor in the Department of Chemistry at the University of Missouri. Her primary research interests include novel solvents and nanostructures for sensory, energy, and sustainable chemistry applications. |
 Ganesh Kamath | Ganesh Kamath received his M.S. (2003) and Ph.D. (2006) degrees in Chemical Engineering from Wayne State University. He completed postdoctoral appointments at Oak Ridge National Laboratory and the University of Missouri before moving to InterX Inc. During his career, he has performed a range of molecular modeling studies and developed Monte Carlo, MD simulations, and free energy analysis programs for determining phase diagrams and the thermodynamics properties of varied materials. |
 Gary A. Baker | Gary A. Baker received his Ph.D. in Analytical Chemistry from the University at Buffalo, The State University of New York, under the direction of Prof. Frank V. Bright. After postdoctoral training at Los Alamos National Laboratory, he was a research staff member at Oak Ridge National Laboratory. In 2011, he moved to the University of Missouri where he is currently an Assistant Professor. His research interests have been mainly concerned with nano-enabled sustainable energy, ionic liquids and related deep eutectic solvents, and green nanoscience. To date, he has published over 200 original research papers on these topics and holds six patents. |
“Graphene, graphene, with nature unforeseen, discovered by Novoselov and Andre Geim.
Wood from highland forests, coal from valley seams, diamonds from the deepest mines of Finsch and Koffiefontein,
Of all the carbon allotropes, the strangest on the scene, an atomic sheet of chicken wire is N and G's graphene.
Graphene graphene, with nothing in between, a single-layer lattice looking something like benzene.
Take a carbon nanotube and slit it like a bean, roll it flat as a Berber mat with a very small machine or take a piece of Sellotape and peel a layer clean, as Novoselov and Geim did (they're Russian not Slovene).
Graphene graphene, much thinner than a gene. If carbon is squaddie, then you're a crack marine.
Graphene can switch electrons, can modulate light beams. A sheet can filter biofuel, make cheaper solar screens, but, best of all, by blocking gas while filtering, like baleen, it can cut the distillation cost of vodka or Jim Bean.
Graphene graphene, as thin as bubble sheen, the same array of atoms as Buckmaster Fullerene.”
–In praise of Graphene by Charles Michael, Tacchi-Morris Institute.
1. Introduction
Considered to be the Holy Grail for physicists and material scientists, the elusive material-graphene is touted to be the future silicon of the microelectronics industry. This strictly two-dimensional material (the thinnest known in the universe), consisting of a one atom thick perfect honeycomb network of sp2 carbon atoms, is the simplest allotrope of carbon, see Fig. 1, already has a number of superlatives associated with it, despite a relatively brief history. For an excellent account on the rise of graphene as the superstar allotrope of carbon and the genealogy of graphene, the readers are redirected to the treatises by Boehm1 and Ruoff2 respectively. Indeed, graphene is the strongest material (fracture strength, 125 GPa)3 and has a high Young's modulus (∼1100 GPa)3 ever measured. It can sustain current densities six orders of magnitude higher than that of copper, is impermeable to gases, has a huge specific surface area (calculated value, 2630 m2 g−1),4 shows record thermal conductivity (∼5000 W m−1 K−1),5 and exhibits an anomalous quantum Hall effect.6 Amazingly, its charge carriers exhibit giant intrinsic mobility, have zero effective mass, and can travel for microns without scattering at ambient temperature, making this material a boon to next-generation electronics and devices.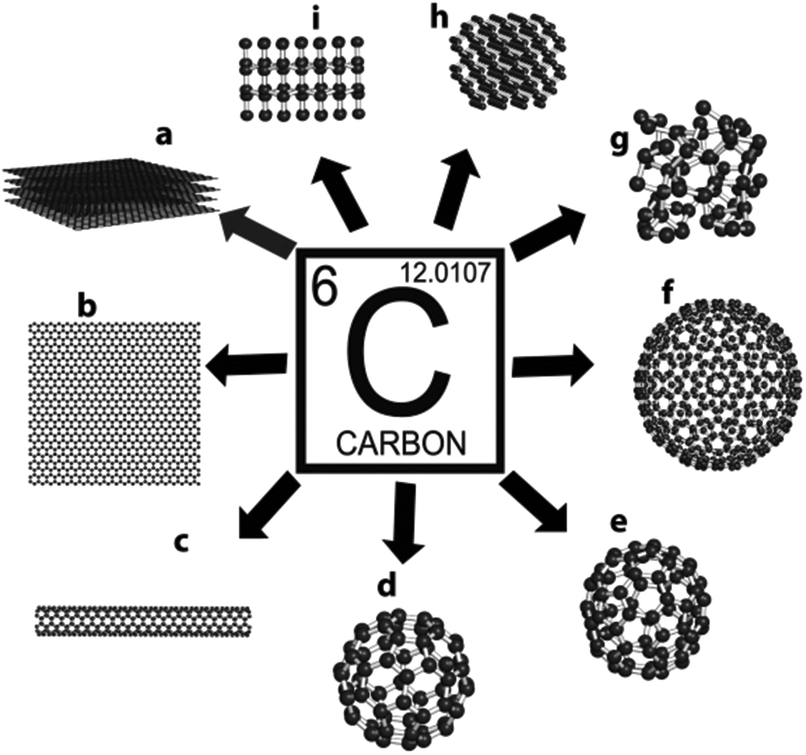 | ||
| Fig. 1 Different allotropes of carbon: (a) graphite, (b) graphene, (c) carbon nanotube, (d) C60, (e) C70, (f) C540, (g) amorphous carbon, (h) lonsdaleite, and (i) diamond. | ||
Subsequent to the Scotch tape method (micromechanical cleavage),7 a plethora of methods have emerged for production of graphene nanosheets (GNs) ranging from epitaxy methods to chemical vapour deposition,8 and separation of GNs from a variety of graphitic starting materials (e.g. highly ordered pyrolytic graphite (HOPG), expanded graphite, graphene oxide (GO)) using liquid-phase exfoliation.9 Among the different methods for production of GNs, liquid-phase exfoliation9–11 presents a clear advantage owing to the simplicity of the process, relatively lower variable cost, and immense potential for large scale up. However, this method is marred by certain drawbacks; primarily the production of poor quality of GNs due to the presence of residual solvents on the surface and the nature of the volatile organics used, which tend to be environmentally toxic.
With this in mind, ionic liquids (ILs)—molten salts comprising of polyatomic organic or inorganic ions that have melting temperatures below 100 °C—have a number of attractive features indicating that they would rise to this challenge.12 Consider these facts: (1) ILs have a versatile chemistry that can be molecularly tuned in many ways, as opposed to the conventional molecular solvents; see Fig. 2, for an illustration of ILs as a molecular toolkit; (2) a growing number of ILs exist which have essentially negligible vapour pressure and are thermally stable at high temperatures, radiation-resistant, non-flammable, and chemically inert; (3) imidazolium-based ILs have successfully served as dispersion media for single-walled carbon nanotubes13 and IL-based bucky gels, a gelatinous paste of single-walled carbon nanotubes and ILs,14 (4) ILs have surface tensions closely matching the surface energy of graphite; (5) ILs can allow homogeneous GN dispersions via stabilization against re-aggregation by Coulomb repulsion of the surface charges introduced by the adsorbed (intrinsically charged) IL. Given these attributes, ILs, find an avant-garde application as excellent solvent media for the exfoliation and stabilization of GNs.
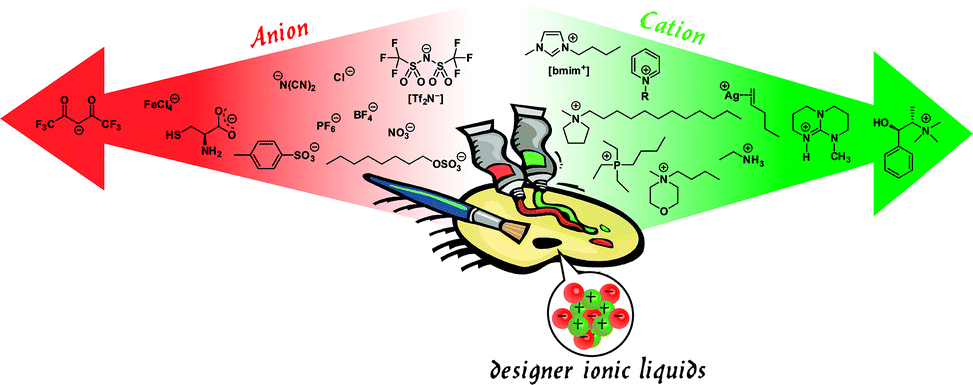 | ||
| Fig. 2 Ionic liquid chemistry represents a ‘designer palette’ due to the fact that their constituent ions can be readily tuned in mix-and-match fashion and may even carry special functionality (the chiral and metal-containing ions shown are classic examples of this), allowing the resulting ionic liquid to satisfy specific performance requirements. | ||
This review examines the ability of ILs for practical exfoliation, dispersion, and modification of GNs as well as GO, reduced GO (RGO), and inorganic graphene analogues (IGAs). The pros and cons of using ILs will be discussed as well as demonstrated and potential applications of IL-functionalized materials keeping in mind a further goal of industrial scale up. Computer simulations, complementing and expanding upon experimental findings will be examined as both a screening tool and a method to provide molecular insights into the mechanism of ILs for exfoliation and dispersion.
2. Overview of IL-assisted exfoliation methods
Since the micromechanical cleavage of graphene from graphite,7 considerable efforts have been put toward dispersing and stabilizing GNs from a variety of starting graphitic materials including reduced GO (RGO) sheets, HOPG, and thermally-expanded graphitic sheets utilizing different stabilizers such as non-ionic-biocompatible block copolymers,15 ionic surfactants,16,17 and DNA.18 For IL-based stabilizers, many methods including mechanical, chemical, microwave, hydrothermal, electrochemical, and sonochemical techniques have been employed to successfully exfoliate and disperse GNs into the ILs and their mixtures. Further, IL-stabilized GNs provide a suitable platform to fabricate GNs decorated with polymer and metal nanoparticles toward enhanced material applications. This section chronicles and critiques the methods used until date for exfoliation of GNs using ILs. It also includes the use of ILs for dispersion and reduction of GO into RGO, a graphene material with defects left-over from oxidation and incomplete reduction.2.1. Sonication and grinding methods
For efficient exfoliation and successful dispersion of GNs, and similar counterparts, a minimization of the Gibbs free energy (ΔG) for the process is necessitated. Given the Second Law of Thermodynamics, a small enthalpy of mixing would result in the entropic contributions playing a dominant contributing factor towards a negative ΔG. Such entropic contributions can arise due to increase in temperature (normally solvothermal process which includes the heating of the graphite, resulting in expandable graphene layers) or providing external energy to the surface of the graphite sheets in the solvent media using mechanical methods such as grinding or ultrasound where energy is imparted to the solvent molecules which collide with the graphene sheets transferring energy. This energy aids in the relative displacement of the graphene sheets through a shear force, overcoming the π–π stabilizing interactions between the graphene sheets. Further, successful exfoliation also relies on the proper choice of solvent (or surfactant, or other compound) through surface interactions such as surface matching providing sufficient solvent–graphene interaction to balance the energy cost for graphite layer expansion and prevent re-aggregation of the graphene sheets.The pioneering work of Wang et al.19 demonstrated the direct exfoliation of graphite flakes assisted by a conventional IL, 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide [C4mim][Tf2N], acting as a solvent and stabilizer producing GNs suspensions with concentrations as high as 0.95 mg mL−1, see Fig. 3. In their process, natural graphite flakes (20 mg) were dispersed in 10 mL of [C4mim][Tf2N], which was then subjected to tip ultra-sonication for 60 min (using 5–10 min intervals). The obtained GNs dispersion was centrifuged at 10,000 rpm for 20 min and the supernatant analysed using scanning transmission electron microscopy (STEM) and Raman spectroscopy; which indicated dispersion of GNs with <5 layers and low levels of defects. Further, X-ray photoelectron spectroscopy (XPS) studies showed the GNs were stabilized by strong non-covalent interactions of the IL with the GNs thus indicating that IL-functionalized GNs (IL-GNs) were produced. The IL functionalization prevented the re-aggregation of the sheets allowing for the stable GNs dispersion observed. Following this work, Mariani and co-workers20 exfoliated and dispersed graphene in a similar fashion using the IL, 1-hexyl-3-methylimidazolium hexafluorophosphate [C6mim][PF6]. By using a bath sonication time for over 24 h, followed by centrifugation, they obtained GNs suspensions with concentrations as high as 5.33 mg mL−1.
 | ||
| Fig. 3 Images illustrating a dispersion of graphite flakes in [C4mim][Tf2N] before (A) and after (B) ultrasonication. Panel (C) shows the Tyndall effect for a dilute graphene suspension using a green laser pointer. | ||
During sonication, cavitation, the formation of small bubbles from dissolved gases or vapors due to pressure changes and shear stresses imparted by the sonication waves, often, if not always, occurs. These bubbles (liquid-free zones) have a high internal Laplace pressure which, upon explosion on the graphene surface can induce cyclic stress resulting in fatigue and defects in the GNs. A way to overcome the cavitation effects normally seen with sonication processes, is to instead use methods that relies on shearing forces to exfoliate graphite such as mechanical grinding21 (mortar and pestle, ball milling, etc.) or the use of a vortex fluidic device.22 Shang et al.21 utilized mechanical grinding in conjunction with small quantities of IL to exfoliate graphene from pristine graphitic flakes. In this method, graphite flakes (<20 μm2 in size) were mixed and ground with 1-butyl-3-methylimidazolium hexafluorophosphate [C4mim][PF6] for 4 h using a mortar and pestle to form a gel. Then dimethylformamide (DMF) and acetone were added with subsequent centrifugation to remove any excess IL followed by sedimentation and drying. Grinding times and the initial ratios of graphite![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IL were vital for the type of end product. When grinding times were less than 30 min (using ratios of graphite flakes (mg) to IL (mL) of 1:10–1:4), graphene sheets of 0.006–0.0125 μm2 having 2–5 layers were dominant. However, increasing the grinding time and decreasing the amount of IL resulted in the formation of graphene nanodots with diameters, controllable by grinding times, of 9–29 nm and heights of 1–16 nm.
:IL were vital for the type of end product. When grinding times were less than 30 min (using ratios of graphite flakes (mg) to IL (mL) of 1:10–1:4), graphene sheets of 0.006–0.0125 μm2 having 2–5 layers were dominant. However, increasing the grinding time and decreasing the amount of IL resulted in the formation of graphene nanodots with diameters, controllable by grinding times, of 9–29 nm and heights of 1–16 nm.
Based on the prior knowledge of using ILs for the exfoliation and functionalizing of carbon nanotubes,23 Tour and co-workers24 used a mortar and pestle to grind thermally converted to graphene (TCG), <10-layered-graphene stacks with oxygen content <3% produced by rapidly heating GO by heating above 1000 °C in an inert atmosphere, in the presence of various diazonium salts with either the IL 1-octyl-3-methylimidazolium tetrafluoroborate [C8mim][BF4] or hexylpyridinium tetrafluoroborate [C6pyr][BF4] along with the base K2CO3 followed by washing steps to remove any excess salt, IL, or base. The resultant GNs were shown to be functionalized by the diazonium salt used. They experimented with 14 different diazonium salts and measured the solubility of the salt-functionalized GNs in DMF. Although, a wide gamut of functionalized nanosheets is achieved by this process, the final dispersion of GNs in DMF was only 0.02 mg mL−1.
2.2. Electrochemical methods
Electrochemical methods have also been used to exfoliate graphite precursors to form GNs. Electrochemical methods have potential advantages such as, a simple one-step process, easy operation and control during the course of the process, mass production of large sheets ranging from few nanometres to micrometres of length under ambient conditions and without introducing a large number of defects or oxygenation on the surface of the GNs.25 The quality of the end product allows for use in widespread applications such as imaging, sensing, dye sensitized solar cells (DSSCs), electro catalyst, and fuel cells.In 2008, Liu et al.26 first reported the electrochemical synthesis of GNs to form IL-functionalized GNs. In their work, a mixture of imidazolium-based ILs (1-octyl-3-methylimidazolium hexafluorophosphate [C8mim][PF6], 1-octyl-3-methylimidazolium tetrafluoroborate [C8mim][BF4], 1-octyl-3-methylimidazolium chloride [C8mim][Cl], and [C4mim][PF6]) and water served as the electrolyte. Upon application of a 15 V potential between two graphite rods, a black precipitate of IL-functionalized GNs was obtained at the bottom of the reactor. The IL-functionalized GNs flakes were up to ∼700 nm length and 500 nm width as observed through TEM and FESEM images with an average thickness 1.1 nm confirmed by AFM studies. These IL-functionalized GNs could be homogeneously distributed into polar aprotic solvents such as DMF. Further, the [C8mim][PF6]-functionalized GNs were combined with polystyrene to form composites with enhanced (3–15 times greater) electrical conductivities as compared to their single-walled carbon nanotube-polystyrene counterparts. The mechanism of IL-functionalized GNs formation was speculated to occur by the IL cation reducing on the cathode forming a free radical at the C2 position of the IL cation which then combined with the a π-bond electron of a GNs. However, this conjecture was brought into question by subsequent work.
Loh and co-workers25 further investigated the mechanism for electrochemical GNs exfoliation and functionalization by ILs using a similar experimental construct as Liu et al.26 They found the amount of water content within the ILs was key for controlling the resultant shape and type of carbon nanostructure formed which varied from nanoparticles to nanoribbons and nanosheets. A water content greater than 10 wt% resulted in the formation of carbon nanoparticles, while nanoribbons or nanosheets were predominant at lower water contents. Further, IL-functionalization of the GNs was only observed at water contents less than 10% and higher water contents were associated with higher oxidation of the resulting structures. They also observed exfoliation of the graphene flakes occurring only at the anode, which seemed counter to the process reported by Liu et al.26 Instead, Loh and co-workers25 proposed a three stage process involving, in order, electrochemical oxidation, intercalation/expansion, and cleavage of the graphite anode (Fig. 4). In Stage 1, water is oxidized to form hydroxyl and oxygen radicals which cause corrosion at the anodic graphite edge sites, grain boundaries, or defect sites causing the dissolution of 8–10 nm of fluorescent carbon nanocrystals with hexagonal shape with concurrent yellowing/browning of the electrolyte. This edge oxidation facilitates the opening of space between graphite sheets allowing for intercalation (Stage 2) by the IL anion and water. In this stage, approximately 10 nm × 60 nm fluorescent nanoribbons with flat rectangular edges are formed with some GNs also observed. Further expansion finally leads to Stage 3 where graphite flakes, 200 nm × 500 nm GNs, peel off from the anode forming black slurry with the electrolyte.
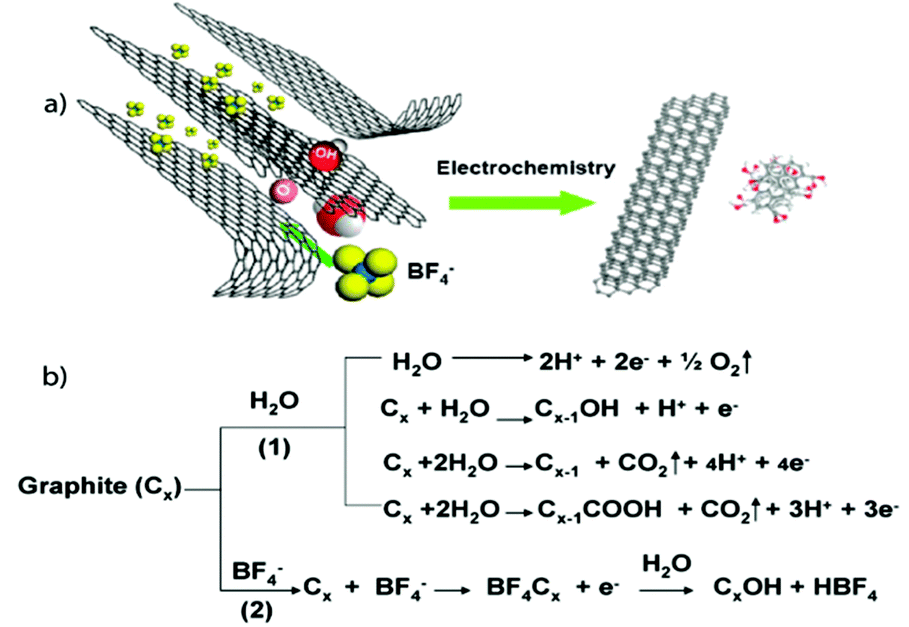 | ||
| Fig. 4 (a) Illustration of the exfoliation process showing the attack of the graphite edge planes by hydroxyl and oxygen radicals, which facilitate the intercalation of [BF4]− anion. The dissolution of hydroxylated carbon nanoparticles gives rise to the fluorescent carbon nanoparticles. Oxidative cleavage of the expanded graphite produces graphene nanoribbons. (b) The interplay of anodic oxidation of water as well as intercalation of [BF4]−. Reproduced with permission from ref. 25. | ||
Exfoliation experiments carried out using neat [bmim][BF4] IL, with no water or dissolved oxygen present, resulted in a highly viscous electrolyte from which a bucky gel, composed of IL-functionalized GNs and carbon nanoparticles, could be isolated after centrifugation with the supernatant containing soluble exfoliates comprising fluorescent carbon structures of nanoparticles and nanoribbons.25 This “one-pot” synthesis shows enormous promise for industrial scalability and mass production, including applications toward biological labelling and imaging applications. Similarly, Singh and co-workers27 used a three-electrode, as opposed to the conventional two-electrode system for exfoliation, consisting a graphite pencil and of two platinum electrodes immersed in triethylsulfonium bis(trifluoromethylsulfonyl)imide [S222][Tf2N] IL. Applying a potential of 16 V was applied resulted in the formation of black precipitate, typically a bucky gel as seen by Loh and co-workers in the presence of pure IL.25 GO could also be produced by this set-up by ramping the applied potential from 0 to 8 V.
To further elucidate the role of IL ions and the mass production of the GNs synthesized through the electrochemical process, Najafabadi et al.28 used a novel IL–acetonitrile (IL–ACN) binary mixture electrolyte with low IL content (∼1:50 IL:ACN vol. ratio) to produce high yields of GNs in addition to smaller quantities of carbonaceous particles and rolled sheets. Four different ILs (1-ethyl-3-methylimidazolium tetrafluoroborate [C2mim][BF4], 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide [C2mim][Tf2N], butyltrimethylammonium bis(trifluoromethanesulfonyl)amide [N1114][Tf2N], 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)amide [C4mPyrr][Tf2N]) were chosen to study the roles played by both the IL cation and anion during the process. The highest yield, as determined by electrode volume changes, of exfoliated IL-functionalized GNs were obtained for [C4mPyrr][Tf2N] with a yield of 86% and the lowest for [C2mim][BF4] with a yield of only 29% after 4 h of electrolysis (at 7 V). The higher yield was due to the extensive reactivity of graphite anode with the oxygenated [Tf2N] anion and the low yield likely due to strong absorption of [emim]+ on the cathode surface, where the C2 proton can initiate H2 evolution and also produce decomposition products that adsorb to the anode hindering IL anion intercalation.29,30 Moreover, the electrolyte color change was shown to be attributable to the occurrence of reaction products from the IL not necessarily attributable to different stages of exfoliation as reported by Loh and co-workers.25 The method of using non-aqueous electrolytes with low volumes of IL improved electrochemical stability, resulted in higher exfoliation rates, reduces costs associated with larger quantities of ILs and opens up avenues for IL binary and ternary mixtures. Zeng and co-workers31 used electrochemical AFM to monitor the morphological changes in HOPG when subjected to intercalation by two types of ILs. The hydrophilic and acidic IL 1-butyl-3-methyl hydrogen sulfate [C4mim][HSO4] with its [HSO4]− anion provided a charge induction resulting in a greater morphological change, but still reversible expansion of the graphitic layers as compared to the [Tf2N]− anion of [C4mim][Tf2N]. An increase in the potential beyond 0.8 V resulted in surface degradation of the HOPG resulting in the formation of carbon nanoparticles.
Electrochemical exfoliation procedures, given their ability to produce controlled uniform size, shape, and enhanced specific energy capacity, are gaining momentum for GNs targeted for battery applications. Wei and co-workers32 used the electrochemical exfoliation of graphite in mixtures of [C4mim][BF4], deionized water, and lithium salts to produce GNs with conductivities as high as 750 mA h g−1 in comparison to graphite typically used in batteries (200 mA h g−1).
2.3. IL as surfactant/stabilizer
Han and co-workers34 used formed GO dispersed in water by Hummers method then poly(1-vinyl-3-butylimidazolium chloride) a polymer ionic liquid (PIL) was added followed by the chemical reduction of GO with hydrazine. The PIL acted as a stabilizer by interacting with the GO through non-covalent π–π interactions. The resulting PIL-functionalized RGO aqueous dispersion was mixed with hydrophobic [C4mim][PF6], and the PIL-functionalized RGO was extracted into the IL phase due to the excellent compatibility of the PIL and IL and the IL containing PIL-functionalized RGO was isolated. An increase in the conductivity (∼0.25 κ S−1 m−1) of the IL system was obtained at low concentrations (0.8 mg mL−1) of PIL-functionalized RGO complex in the IL. This was attributed due the π–π interactions between the IL and the RGO complex.
Zhang et al.35 reported the formation of a stable dispersion of RGO in three hydrophilic ILs [C4mim][BF4], 1-allyl-3-methylimidazolium chloride [Amim][Cl] and N-butylpyridinium ([C4py][BF4] at high concentrations. Here, the IL was added to an aqueous solution of GO, obtained via Hummer's method. Following removal of the water, the GO was reduced using 85% hydrazine monohydrate at 80 °C for 12 h to form RGO suspensions (easily up to 7.0 mg mL−1) within ILs which were stable over several months. The stabilization effect from the IL was attributed to an IL molecular ordering layer on the surface of the RGO likely through cation–π or π–π interactions. Rheological studies showed that at 0.21 mg mL−1, RGO in [C4mim][BF4], showed the viscous modulus (G′′) was always larger than the elastic modulus (G′) as function of oscillation frequency and as the RGO concentration increased to 4.2 mg mL−1, G′ predominated over G′′, indicating the formation of a gel structure. Ma et al. also reported homogenous dispersions and good compatibility of [Amim][Cl]-functionalized RGO prepared by reducing GO in the presence of the IL in DMF. This material could further be incorporated into polyurethane matrix to form IL-G/PU nanocomposites which exhibited excellent mechanical and thermal properties.36
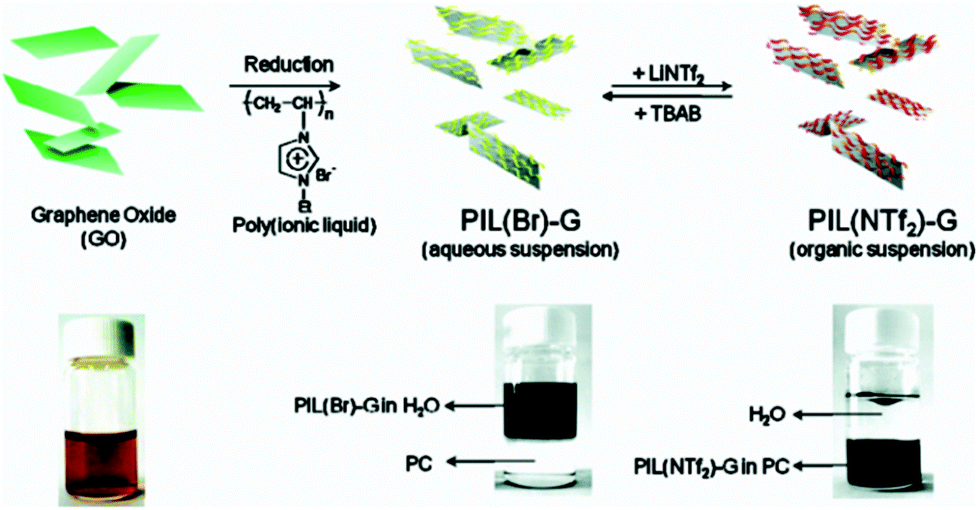 | ||
| Fig. 5 Schematic illustration of the synthetic process for the PIL-modified graphene sheets (PIL-G). Chemical reduction of the graphene oxide (GO) dispersion with hydrazine in the presence of water-soluble PIL(Br) produced a stable aqueous suspension of the PIL(Br)-G. Anion exchange of PIL leads to the phase transfer of the PIL-modified graphene sheets between aqueous and organic solvent media (propylene carbonate). Fig. 5 was reproduced with permission from ref. 38. Copyright: American Chemical Society, 2010. | ||
Yan et al.40 also reported a simple and effective method of phase transfer of dispersed aqueous GO in the presence of the IL 1,3-didodecylimidazolium bromide [C12C12im][Br] from the aqueous phase into a non-polar organic solvent such as ortho-dichlorobenzene. The two dodecyl groups [C12C12im][Br] were crucial for allowing the phase transfer process due the higher solubility in the organic solvent they imparted as compared to other shorter alkyl chain imidazolium bromide ILs such as [C4mim][Br] and [C12mim][Br]. The transfer of GO from the aqueous phase to the organic phase also depended on experimental parameters such as pH and the ratio of GO to the IL with the optimal results shown at pH = 9 and weight ratio of 1:8 GO:DDIB. After phase transfer, hydrazine could be used to produce IL-functionalized RGO. Heat treatment from 300 °C to 1000 °C was performed which resulted in a better restoration of the sp2 nature of the RGO at higher temperature with resultant RGO conductivities up to 823 S cm−1. The reasoning provided was that the thermal annealing and surface stabilization provided by the long IL alkyl chains aided in the healing of the defects. Such novel strategies could enable the development of suitable post processing techniques.
Towards this, Safavi and co-workers,42 used the ILC 1,1′-didoceyl-4,4′-bipyridinium bis(trifluoromethanesulfonyl)amide to exfoliate GNs. This ILC has a smectic liquid crystal phase at room temperature and is stable up to 356 °C. Graphite-ILC composites, of varying weight ratios, were heated in a tube furnace under the flow of Ar to prevent oxidation of graphite at different temperatures and exposure times. As the temperature was increased up to 356 °C, the viscosity of the ILC decreased allowing for ILC intercalation to occur but no exfoliation followed. Further increasing in temperature from 356 to 700 °C resulted in the decomposition of the ILC resultant evolution of the gases. This gas evolution, assisted in the exfoliation of graphite and subsequent exfoliation of GNs. Having the ratio of the graphite–ILC to 30/70 (w/w) resulted in maximization of the exfoliation process. The temperature-dependent process of ILC intercalation and decomposition followed by gas release and ultimately graphite exfoliation is shown in Fig. 6. Although this dry GNs synthesis method has potential for industrial scale up, this method results in the formation of defects in the GNs owing to the gas evolution.
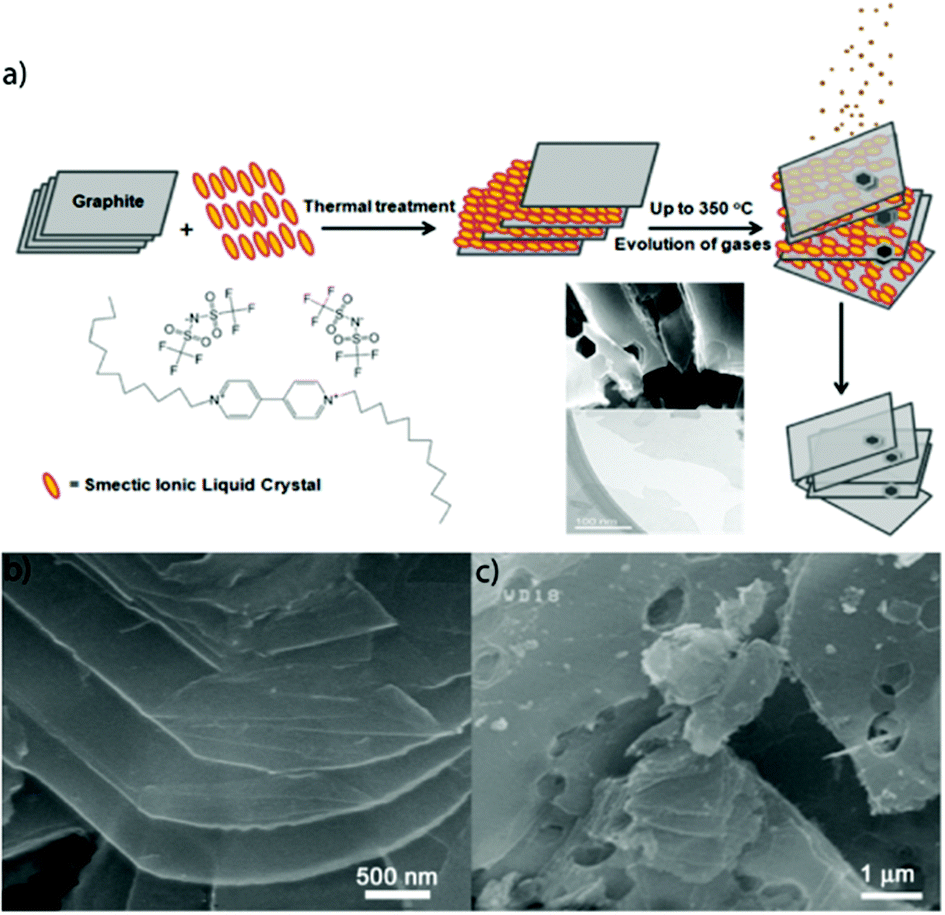 | ||
| Fig. 6 (a) Schematic presentation of the synthesis process of GNs. Intercalation of ILCs between graphite layers, their decomposition and evolution of gases assist in exfoliation of graphite and separation of layers (b) and (c) FESEM images of GNSs. Reproduced from ref. 42 with permission from The Royal Society of Chemistry. | ||
Hydrothermal methods are simple and easy ways for GO exfoliation or expansion of graphite into single or few layers of graphene sheets at high temperatures in confined volumes. Using this method, GO can be simultaneously reduced and decorate with inorganic nanoparticles to form highly crystalline nanocomposites. Using one-pot hydrothermal synthesis involving [C4mim][PF6], Ye and co-workers44 formed Ag-RGO nanocomposites that showed excellent anti-bacterial properties. Subsequently, the same group45 reported a hydrothermal route to TiO2-RGO composites in ILs that exhibited excellent photocatalysis of hydrogen evolution at a rate of 20 μmol h−1.
Hao and co-workers46 used a microwave-assisted ionothermal treatment to reduce GO within [C4mim][BF4]. A 3 mg mL−1 colloidal dispersion of GO in [C4mim][BF4] was treated by microwave heating at 600 W up to 1 h while stirring. After cooling, absolute alcohol was added resulting in a black precipitate which isolated and dried at 70 °C for 24 h to obtain thermally RGO nanosheets (T-RGOs) as illustrated in Fig. 7. Typically, to thermally reduce and exfoliate GO in the absence of intercalating agents, a critical temperature of 550 °C must be exceeded often to temperatures over 1000 °C to ensure the complete exfoliation and reduction of GOs. However, when using [C4mim][BF4] as in this study, the IL serves to pre-exfoliate and GO reduction can occur at the much lower temperature of 200 °C under atmospheric pressure within 40 min. The T-RGOs produced had a relatively high carbon to oxygen ratio and four orders of magnitude increase in electrical conductivity. These properties coupled the lower temperatures required, makes this an immensely promising method for economical and green industrial scale up. Similarly, Suh and co-workers47 used microwave irradiation to reduce GO in the presence of the IL [C2mim][Tf2N], forming RGO within 15 s. An electrode made from this RGO immersed in an [C2mim][Tf2N]/acetonitrile electrolyte (50/50, w/w) was stable up to 3.5 V, and exhibited a specific capacitance (Csp) of 135 F g−1 at a current density of 2 A g−1. RGO with surface modification by ILs and poly(ionic liquid) and its use as effective capacitors has also been reported by other groups.23,48,49
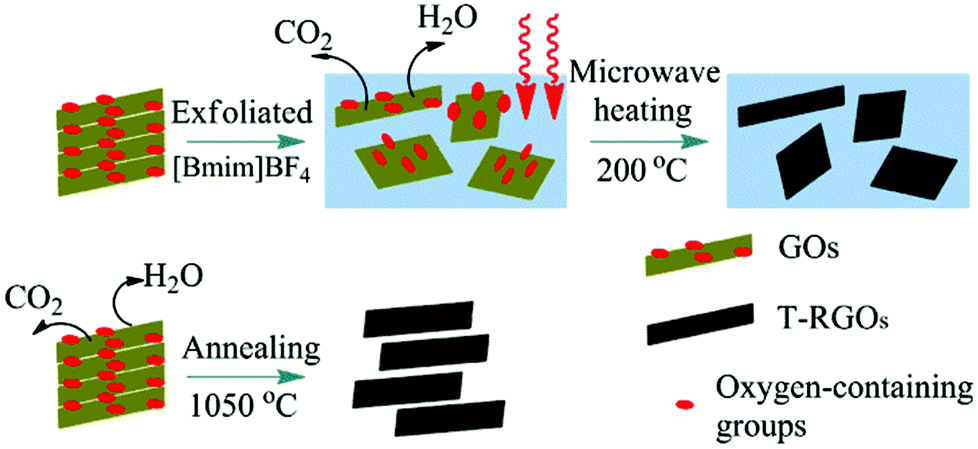
Yang et al.50 introduced a viable method for the poly-dispersion of chemically converted graphene sheets, with the use of an amine-terminated IL, 1-(3-aminopropyl)-3-methylimidazolium bromide ([NH2C3mim][Br]) to functionalize GO, through a nucleophilic ring-opening reaction. The grafted IL on the GO sheets increased their stabilization and dispersion (in multiple solvents including DMSO, DMF, and water) through electrostatic repulsion and increasing the distance between the GO sheets from 9.6 to 14.9 Å. This improved stability is advantageous toward electrochemical applications. For instance, P. Bhunia et al.,51 reported a non-volatile memory device based on the IL, [NH2C3mim][Br], covalently bonded to partially reduced GO in polar organic solvents for stable write–read–erase–read devices with multiple cycles and retention times of both the ON and OFF states with stabilities over 1000 s. The improved memory was attributed to the presence of IL attachment to the RGO overcoming electrical short circuiting and leakage of ions.
Y. Fu et al.52 reported a novel approach to improve the wetting properties of exfoliated and functionalized RGO produced by pH-triggered electrolysis. In this process, graphite was pre-treated with [C4mim][Cl] by grinding to form a gel that was then washed and dried. The IL-treated graphite was then oxidized using a modified Hummer's method to form GO. Then the simultaneous 3-aminopropyltriethoxysilane (APTES) functionalization of the GO was carried out by adding the GO to trihexyltetradecylphosphonium decanoate dissolved in ethanol followed by sonication, APTES addition, and adjustment of the pH to 5. After 30 min of sonication, the mixture was refluxed for 24 h at 80 °C to produce the final APTES-modified GO product. The author proposed the IL aided in the accessibility of functional groups on the surface of graphite sheets to allow for improved interaction with other modifiers such as APTES.
IL-graphene complexes can also be strategically used in the fabrication of graphene/polymer nanocomposites in a different method. Han and co-workers53 reported manufacturing graphene-polyaniline composite (GPC) nanosheets using a PIL. A homogenous suspension of graphene in DMF–H2O was mixed with poly(1-vinyl-3-butylimidazolium chloride) followed by DMF removal by dialysis resulting in PIL-stabilized graphene sheets (PIL-G) in aqueous solution. The absence of PIL in the process leads to re-aggregation of the graphene sheets. Anion exchange from Cl− to S2O82− of the PIL-G could also be accomplished by addition of ammonium peroxydisulfate forming PIL-G-S2O8. Upon addition of aniline monomer in HCl, the S2O82− initiated aniline polymerization, resulting in the replacement of the PIL with PANI on the surface of graphene sheets. This facile polymer exchange route has potential applications in the areas of microelectronics, energy storage, and anticorrosion.
Similarly, Lonkar and co-workers54 used aqueous GO suspended in the amphiphilic IL, 1-(11-hydroxyundecyl)-3-methylimidazolium bis(trifluoromethanesulfonyl)amide ([C11(OH)mim][Tf2N]) which, upon reduction produced an IL-functionalized RGO complex that was highly dispersible in a range of different organic solvents. The hydroxyl functionalities (provided by the IL) at the surface of the IL-functionalized RGO were used to initiate in situ ring-opening polymerization of 3-caprolactone, illustrating a “grafting-from” method as to successfully achieve finely dispersed RGO sheets within the a poly(3-caprolactone) matrix. Such supramolecular approaches could pave the way for the production of graphene-embedded polymer composites with superior mechanical and thermal stabilities.
In addition, Mai and co-workers55 developed IL-functionalized RGO by chemically reducing RGO using NaBH4 and NaOH in the presence of the IL 1,6-bis[3-(vinyl benzyl)imidazolium-1-yl] hexane chloride ([bVB-bim-C6] [Cl]). This IL functionalized and stabilized the resulting RGO which could be well dispersed in water DMSO, and DMF. The presence of vinyl groups of the [bVB-bim-C6] [Cl] further served as cross linking reactive sites for the polymerization of methyl methacrylate (MMA) resulting in RGO-embedded PMMA polymer composites. The IL-functionalized RGO sheets were found to be uniformly dispersed within the PMMA matrix which contributed to large increases in the storage modulus (by 58.3%) and glass transition temperature (by 19.2 °C) at only 2.08 vol% RGO loading. While PMMA is a natural insulator, these composites with RGO loading levels beyond 1 vol% exhibited high electrical conductivity near 2.55 S m−1 with a low percolation threshold (0.25 vol.%). For further details on the recent advances on graphene- (or RGO-) filled polymer composites, the readers are directed to the review by Kuilla et al.56
Composites of GNs and biological polymers have also been studied. Peng et al.,57 reported a facile route for synthesizing biocompatible RGO from GO in the presence of [C4mim][Cl] IL media while using cellulose (CL) as both a reducing and stabilizing agent. CL is a polysaccharide, difficult to dissolve in water or organic solvents because of its intense hydrogen bonding network, but CL has been shown to readily dissolve in some ILs including [C4mim][Cl].12,58–60 Cellulose dissolved in [C4mim][Cl] was added to GO dispersed in the same IL followed by heating to 80 °C for a period of 12 h. The resultant composite was purified by washing with a NaOH–thiourea aqueous solution and water. An aqueous solution of the IL-RGO-CL was filtered through a nylon membrane and after drying, flexible IL-RGO-CL papers could be peeled off from the membrane. A biocompatibility study based on cell viability of HeLa cells on IL-RGO-CL and IL-RGO control films showed the IL-RGO-CL composite film had remarkably improved the cell biocompatibility. This significantly improved biocompatibility, in addition mechanical flexibility and conductivity properties of the IL-RGO-CL films shows potential for applications such as biomedical scaffolds, tissue engineering, and medical devices.
Ma and co-workers61 developed IL-functionalized GNs coated with gold nanoparticles (AuNPs). They first produced IL-functionalized GNs using an electrochemical method similar to Liu et al.26 by immersing two graphite rods parallel to each other (6.0 cm apart) in an aqueous solution of the IL 1-aminopropyl-3-methylimidazolium chloride ([NH2C3mim][Cl]) and applying a potential of 15 V for 6 h. The IL-GNs were then added to aqueous solutions of citrate-stabilized Au NPs to make IL-GNs and AuNP composites (IL-GN-Au). Through imaging and spectroscopic studies, the showed the IL-GN-Au composited displayed homogeneous and dense coverage of the AuNPs on the IL-GN surface. An electrochemical immuno-sensor for carcinoembryonic antigen (CEA) was fashioned using the IL-GN-Au composite. The IL-GS-Au was placed on surface of the glassy carbon electrode which was further appended with carcinoembryonic antibody (anti-CEA). Using this electrode, CEA could be detected at an ultralow detection limit of 0.1 fg mL−1 with a wide linear range (1 fg mL−1 to 100 ng mL−1). Similarly, different IL-GN containing nanoparticles have been developed for sensors to detect inorganic and organic molecules at low concentrations. For example, N. Zhou et al.62 recently reported the detection of Hg2+ at 0.03 nM concentration based on IL-functionalized GO decorated with AuNPs. Tunckol et al.63 recently reviewed carbon nanomaterial-IL hybrids which highlights different IL-graphene based sensors including those with containing other nanoparticles.
Electrochemical deposition routes to nanoparticle-modified IL-GNs have also been used. Park and co-workers64 reported the electrochemical deposition of a hierarchical flower-like structure of MnO2 on IL-RGO. The GO was reduced in [C4mim][BF4] IL media to form the IL-RGO composite. This composite was used as a working electrode in an electrolytic bath with platinum (counter) and Ag/AgCl (reference) electrodes with an electrolyte of 1 M MnSO4. Upon subjecting the bath to an electrical potential of 0.8 V, MnO2 in a hierarchical flower-like structure was deposited on the IL-RGO composite. The morphological properties of the IL-RGO-MnO2 composite such as pore size (4.28 nm), surface area (422.06 m2 g−1) and pore volume (0.45 cm3 g−1) were enhanced in comparison to the original IL-RGO composite. Finally, these IL-RGO-MnO2 composites also showed high specific capacitance (266 F g−1) and capability rate (61% retention at 30 A g−1) as compared to MnO2 and IL-RGO films alone. This opens avenues for rational design and manufacture of hierarchical nanostructures combining the benefits of graphene nanomaterials and metal oxides with immense potential in the energy sector.
Using [C8mim][PF6] and an electrochemical graphite exfoliation method, Y. Liu et al.65 prepared IL-GNs on which copper (Cu) nanoparticles were deposited by a chemical reduction methods. Following Cu nanoparticle deposition onto the IL-GNs, H2PtCl6 was added and reacted with Cu atoms to form a monolayer of Pt shell (Cu@Pt). These IL-GNs-Cu@Pt composites were tested for electrocatalytic activity toward methanol oxidation using cyclic voltammetry, chronoamperometry and CO-stripping voltammetry methods. The electrocatalytic activity and stability of IL-GNs-Cu@Pt for methanol oxidation was determined to be better than for commercial carbon blacks. This was attributed to the increase of the graphene conductivity by the IL and the facile removal of intermediate poisoning species with the aid of Cu. This work shows IL-GNs-Cu@Pt can serve as alternative support for Pt immobilization in direct methanol fuel cell applications. Another IL-RGO material with potential for fuels cell was prepared, based on the Yang et al. method,50 by Niu and co-workers.66 In this work, a one-step synthesis of a novel hollow flower like AuPd (hAuPd) with rough surface was decorated on IL-RGO sheets, prepared using an amine-terminated IL, to form IL-RGO-hAuPd through electrostatic interactions. These IL-RGO-hAuPd composites demonstrated better electrocatalytic activity in comparison to neat hAuPd NPs for direct oxidation of formic acid with potential applications in fuel cells.
Recently, Xiao and co-workers67 used the IL [C4mim][cholate] exfoliate pristine graphite to graphene by sonication. The resulting IL-GNs were used in conjunction with small (<2 nm diameter) noble metal (metal (M) = (Pt, Pd, Ru, or Rh)) nanoparticles to form IL-GN-M composites that were uniformly decorated with the metal nanoparticle with no free-standing metal aggregates or agglomerates. These IL-GN-M composites showed superior catalytic activity for arene hydrogenation with the IL-GN-Ru (Ru content of 5%) the most active for the hydrogenation of benzene to hexane with a turnover frequency (TOF) as high as 6000 h−1 G-Ru at 110 °C and an H2 pressure of 8 MPa. Even at room temperatures, the TOF for IL-GN-Ru was 270 h−1 which is higher than the typical TOF of 100–200 h−1 for Ru on Al2O3 or carbon powder.68 The high performance of IL-GN-M composites was attributed to the uniform distribution of the metal nanoparticles on the GNs.
3. Graphene's inorganic cousins
Layered transition metal dichalcogenides (TMDs) and transition metal oxides (TMOs) and other 2-D layered materials such as BN, Bi2Te3, and Bi2Se3 possess graphite-like layered structures in which the layers are held together by weak van der Waals forces and are considered as inorganic graphene analogues (IGAs). A recent review by Rao and co-workers highlights different IGAs as well as bottom-up chemical approaches to produce single- or few-layer TMDs and TMOs.69 As these layered materials are similar to graphene, exfoliation approaches like those used to make graphene from graphite, should also be applicable to these analogues, yet few such exfoliation studies exist. The Coleman group pioneered the recent investigations of liquid-phase exfoliation on a myriad of IGAs.70 Using sonication techniques, they investigated many organic solvent, but no ILs, to determine the key solvent characteristics for inducing efficient exfoliation of either BN, WS2 or MoS2 as well as MoSe2, MoTe2, TaSe2, NbSe2, NiTe2, and Bi2Te3. They found successful solvents were those that minimized the energy of exfoliation; thus, those that possessed dispersive, polar, and H-bonding components with surface tensions close to γ-40 mJ m−2. N-Methyl-pyrrolidone (NMP) and isopropanol (IPA) were the more promising solvents leading to concentrations as high as 0.3 mg mL−1 for MoS2 (in NMP), 0.15 mg mL−1 for WS2 (in NMP), and 0.06 mg mL−1 for BN (in IPA) which were stable for hundreds of hours. Many of the exfoliated materials could be further processes into thin films, composites with polyurethane, graphene, or CNTs, or sprayed as flakes onto a substrate. Zhang and co-workers further expanded on this work use by using an ethanol (EtOH)–water mixed-solvent strategy for exfoliating IGAs.71 The used sonication methods to exfoliate MoS2, WS2, or BN in mixtures of different volume fractions of EtOH in water. The highest dispersion concentrations ranged from 0.018 mg mL−1 for MoS2 (45 vol% EtOH), to 0.032 mg mL−1 for WS2 (35 vol% EtOH) and 0.075 mg mL−1 for BN (55 vol% EtOH). While these values seem low, they are still up to 68 times higher than could be achieved in either neat EtOH or water lending credence to a mixed solvent approach. Further improvement for exfoliation was also found by using ball-milling as a pre-treatment method followed by sonication in aqueous solutions containing sodium dodecyl sulphate (SDS) surfactant.72Given the ability of solvents such as NMP, ionic surfactants, and mixed-solvent systems for dispersing IGAs, it stands to reason ILs hold great potential for exfoliation of IGAs as well. Recently our group,73 using ABF-MD simulations, demonstrated ILs are a most likely viable media for assisting and exfoliating h-BN nanosheets; with negative free energies of exfoliation comparable to the negative free energy of exfoliation of h-BN in NMP.
Recently, Sutto and co-workers,74 studied a series of 1,2-dimethyl-3-N-R-imidazolium BF4 ILs (R = propyl, butyl, decyl, or hexadecyl alkane chains) for their ability to intercalate within TiS2 layers. TiS2 layers normally have distances between two repeat sheets of 5.77 Å and are often characterized by weak sulphur–sulphur interactions. Three types of methods for intercalation were carried out: electrochemically-induced intercalation, solution reactions in IL–acetonitrile mixtures, and the direct thermal reaction of the IL with liquid with TiS2 (Fig. 8). The most promising methods were the direct thermal route using either decyl- or hexadecyl-substituted ILs. The decyl-substituted ILs oriented as a single layer within the TiS2 van der Waals gap, while the hexadecyl-substituted IL were arranged as a bi-layer between the sheets, giving rise to a c-axis expansion of over 49 Å.
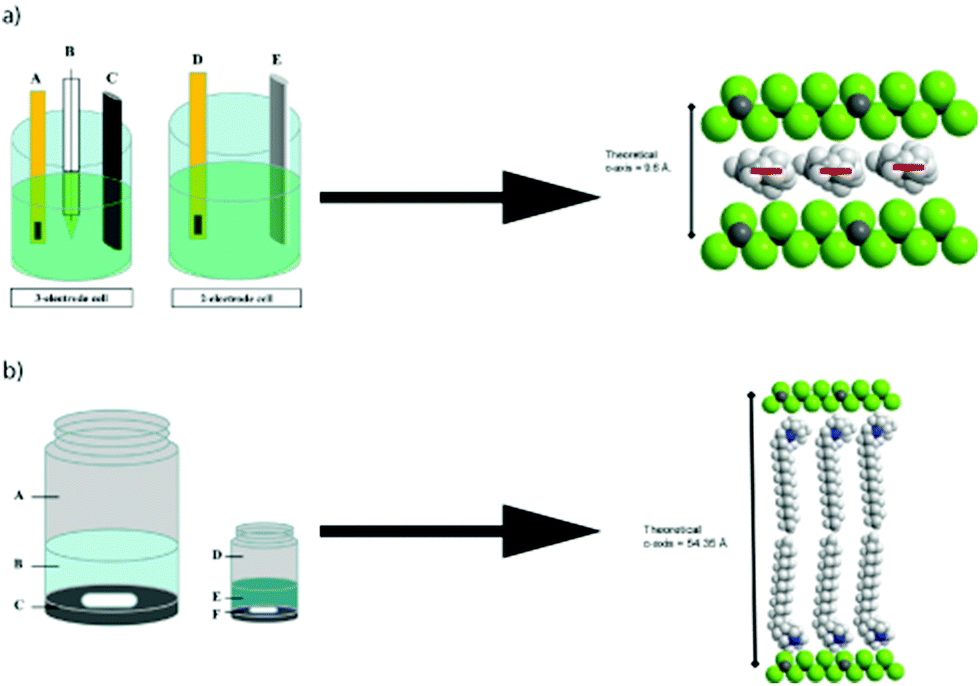 | ||
| Fig. 8 (a) Left, The cell on the left is the three electrode cell used for collecting cyclic voltammetry data for TiS2 in the ionic liquids. Here, (A) is the working electrode, consisting of gold foil with TiS2 pressed into it, (B) is the Vycor-tipped Ag/Ag+ reference electrode, and (C) is the graphite rod counter electrode. The cell on the right is the two electrode cell used for intercalating the ionic liquid into TiS2, where (D) is again the gold foil with TiS2 pressed into it and (E) is a Li metal rod anode for the charge/discharge studies. (a) Right, structural schematic for TiS2 intercalated by MMPIBF4 if the plane of the imidazolium ring is parallel to the a–b plane of TiS2. The red bar indicates the orientation of the plane of the imidazolium ring. Atom colors: green = sulfur; dark gray = titanium; white = hydrogen; light gray = carbon; blue = nitrogen. (b) Left, the schematic on the left is the solution preparation of the ionic liquid intercalation of TiS2, where A is the sample vial without the cap, B is the 60 mL solution of ionic liquid in acetonitrile, and C is the layer of TiS2 and the stir bar. On the right is the direct thermal reaction schematic where D is the sample vial, E is 7 mL of the pure ionic liquid and F is the TiS2 layer with a stir bar. (b) Right, structural schematic for TiS2 intercalated by MMPIBF4 if the plane of the imidazolium ring is at an angle of 45° relative to the a–b plane of TiS2. Fig. 8 reproduced with permission from ref. 74 Copyright: Elsevier Ltd. 2012. | ||
4. Computational insights
From a microscopic point of view, researchers postulate π–π interactions or cation–π interactions are the surface interactions responsible for the GNs stabilization various solvents and ILs. Computer simulations offer an avenue towards understanding the forces responsible for exfoliation and stabilizing of GNs in different solvents while also being a priori screening tool for ranking the exfoliation efficiency of various solvents for GNs and IGAs. While simulation studies have been carried out on solvent interactions with graphite surfaces since the 1980′s, these solvents have been limited to water75 and various organic solvents.76,77 In the past decade, looking into interactions of graphite and various other surfaces with ILs has started to grow.78,79 While these simulations present a static picture of the interactions of different surfaces with ILs, (primarily since the sheets or surfaces were considered to be static in nature) the energetics and dynamics of the interactions during the exfoliation and dispersion of 2-D materials in ILs is lacking.Towards this, our group recently evaluated the free energies for graphene exfoliation from bilayer graphene using IL based on various cations paired with the Tf2N anion from adaptive bias force-molecular dynamics (ABF-MD) simulations.80 As a part of the ABF algorithm, an external biasing force, estimated locally from the sampled conformations of the system and updated continuously, is applied at each step to facilitate the system in overcoming significant energy barriers, if present along the reaction coordinate. This force is applied to one layer of the graphene sheet in the tangential z-direction with respect to the other layer, as illustrated in Fig. 9. This approach is selected to emulate the shearing force provided by solvent media in the physical exfoliation of graphene monolayers from graphite. The first derivative of the free energy is related to the partial derivative of the Hamiltonian of the system with the reaction coordinate and is related to the constraint force acting along the reaction coordinate. These free energy predictions comport with experiment; a test case of water exfoliating graphene sheet from the bilayer constituted a positive free energy suggesting the well-known fact that water is a poor solvent for graphene exfoliation, while predicting a negative free energy of exfoliation for IL-assisted graphene exfoliation, see Fig. 9. Alkyl–π interactions with the graphene surface dominate for imidazolium- and pyrrolidinium-based ILs while π–π interactions played a major role for the pyridinium-based ILs. This was derived from the relative magnitudes of the radial distribution functions which provide number density of molecules within a given radius from a reference point and the ordering factor (S) which provides a quantitative feature for the relative orientation of the cations and anions with respect to the graphene surface. The alkyl–π interactions being dominant (relatively lower π–π interactions seen) for the case of the imidazolium ring interacting with the graphene surface. This, while being relatively incongruous with the simulations of Bell and co-workers,78 (wherein their results showed a dominance of the cation ring being parallel to the graphite surface thereby implying π–π interactions), seem to comply with sum-frequency vibrational-spectroscopy experiments on imidazolium-based ILs on silica81 and graphene surfaces.82 In these studies, the cations preferentially orient at an angle with the alkyl groups perpendicular to the surface. Nevertheless, computational methods are proving to be an excellent supplement to experiments and analysis at the atomic-scale level. Using normal-mode analysis and solvation-shell structure around the nanosheets could further enhance our understanding of the dynamics of exfoliation and stabilization processes in addition to better understanding gained from the kinetic theory of colloid aggregation,83 DVLO, and Flory Higgins theory adapted for the dispersion of nanotubes84 and nanosheets. Last, but not the least, results from molecular simulations help unravel the shape of the curve for differential capacitance of the graphite–IL interface versus the electrode potential can provide insights into the surface roughness of the electrode, further suggesting that controlling the topography of the electrode could improve the energy densities in electric double-layer capacitors.85
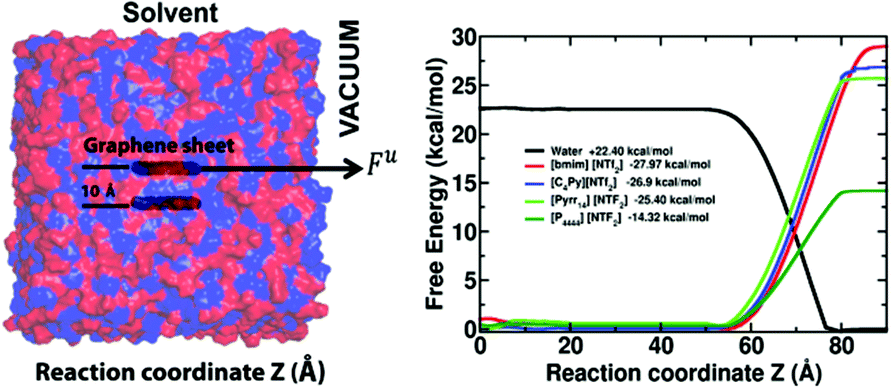 | ||
| Fig. 9 Free energies for graphene exfoliation from bilayer graphene using ionic liquids based on various cations paired with the bis(trifluoromethylsulfonyl)imide anion were determined from adaptive bias force-molecular dynamics (ABF-MD) simulation and fall in excellent qualitative agreement with experiment. This method has notable potential as an a priori screening tool for performance based rank order prediction of novel ionic liquids for the dispersion and exfoliation of various nanocarbons and inorganic graphene analogues. Reproduced from ref. 80 by permission of PCCP Owner Societies. | ||
5. Comparison of methods and ILs
Table 1 shows the concentrations of dispersed GNs and RGO in different ILs using the variety of techniques discussed above. For comparison, the concentration of dispersed GNs in organic solvents vary from 0.16 μg mL−1 in pentane to 8.5 μg mL−1 in cyclopentanone (A table comprising of 50 solvents and ranking them based on their Hansen and Hildebrand parameters and graphene dispersions can be found in the works of Coleman and co-workers86). While the best yield in neat organic solvents is typically below 10 μg mL−1 and involves sonication for as long as 450 h, in the case of ILs the yield of graphene can be above 5 mg mL−1.20 Only when mixtures of conventional organic solvents are used can GNs dispersions reach concentrations in the range of IL media. For example, chlorosulfonic acid and hydrogen peroxide mixtures can be used to produce a 3 mg mL−1 dispersion of GNs.87 While a battery list of organic solvents have been used (>50), the number of ILs tried are far less, not to forget that with the multitude of ions, a vast number of permutations and combinations could be carried out and “task specific” ILs can be developed based on the fundamental understanding of interactions of ILs with graphene to even further improve the yield of dispersed GNs.| Starting material | Method | Dispersible solvents | Initial conc. (mg mL−1) | Final conc. (mg mL−1) | Thickness (nm) | Lateral size | Reference |
|---|---|---|---|---|---|---|---|
| GO + poly (1-vinyl-3-imidazolium chloride) | Chemical (oxidation/reduction) | [C4mim][PF6] | 0.8 + 100 mg (PIL) | 0.8 | 3.0 | n/a | 34 |
| GO + poly (1-vinyl-3-imidazolium bromide) + Li[Tf2N] | Chemical (oxidation/reduction) | Water, PC, ACN, DMF, THF, NMP | 1.5 + 400 mg (PIL) + 1.5 equiv. ([Li][Tf2N]) | n/a | 1.9 | n/a | 38 |
| GO + PTPSS | Chemical (oxidation/reduction) | Water–ethanol | 1.3 + 1 gm. (PTPSS) | n/a | n/a | n/a | 39 |
| GO + [C4mim][BF4]+ [Amim][Cl] + [C4Py][BF4] | Chemical (oxidation/reduction) | [C4mim][BF4], [Amim][Cl], [C4Py][BF4] | 6 | 4.2–7 | 0.85 | Few hundred nm to μm | 35 |
| 2.1 | |||||||
| 0.5 | |||||||
| GO + [N4448][MS] | Chemical, thermal annealing | Water, PC | 1:2 (wt/wt) |
n/a | n/a | n/a | 43 |
| GO + water + [C4mim][BF4] | Microwave-assisted ionothermal treatment | [C4mim][BF4], NMP, DMF | 1 | 3 | 0.72 | Tenth to several mm | 46 |
| 0.5 | |||||||
| 0.5 | |||||||
| GO + [C4mim][BF4] + water + hydrazine | Sonication (1 h) + chemical method (95 °C for 12 h) | [C4mim][BF4] | 5 mg mL−1 GO, 5 wt% IL, 35 wt% hydrazine | n/a | 1.3 | n/a | 64 |
| GO + NaOH + NaBH4 + [bVB-bim-C6][Cl] + DMSO | Chemical method (18 h at 35 °C) | H2O, DMSO, DMF | 300 mg GO, 200 mL NaOH, 1.2 g NaBH4, 10 mg mL−1 IL in DMSO | n/a | 1.28 | Several hundred nanometers to micrometers | 55 |
| GO + [C4mim][Cl] + water + cellulose + ammonia + NaOH + thiourea | Chemical method (80 °C for 12 h) + sonication (20 min) | [C4mim][Cl] | 30 g IL, 0.8 mg mL−1 GO, 5 wt%, 20 mL cellulose | n/a | 1.9 | n/a | 57 |
| GO + [C11(OH)mim][Tf2N] + water + hydrazine | Chemical method (80 °C overnight) | EtOH, CHCl3, THF, toluene | 1 mg mL−1 GO, 20 mg IL, 10 μL hydrazine | 1 mg mL−1 | n/a | n/a | 54 |
| GO/TCG + [C4mim][BF4] + diazonium salt + K2CO3 + [C6Py][BF4] | Thermal and mechanical | DMF | 12 | 0.01 | 0.7–1.3 | 0.45 μm pore size | 24 |
| Graphite + [C4mim][Tf2N] | Micromechanical (sonication) | [C4mim][Tf2N] | 2 | 0.95 | n/a | μm | 19 |
| Graphite + [C6mim][PF6] | Sonication | [C6mim][PF6] | n/a | 5.33 | 2 | 3–4 μm | 20 |
| Graphite + [C4mim][PF6] | Grinding | DMF | 100–500 | 20 wt% | n/a | Nanosheets/nanodots | 21 |
| Graphite + ILC | Thermal treatment and exfoliation based on gas evolution | No solvent | 30/70 wt% | n/a | ∼1–1.8 | Few μm | 42 |
| Graphite rods + [C4mim][BF4]/[Cl] + water | Electrochemical oxidation, intercalation, exfoliation | 10% IL/90% water, 90% IL/10% water | 1 cm × 1 cm × 1 mm rods | n/a | n/a | (a) 8–10 nm carbon nanocrystals | 25 |
| 10% IL/90% water | (b) 10 nm × 60 nm nanoribbons | ||||||
| 90% IL/10% water | (c) 200 nm × 500 nm nanosheets | ||||||
| Graphite rods + [C4mim][BF4] | Electrochemical oxidation, intercalation, exfoliation | [C4mim][BF4] | 1 cm × 1 cm × 1 mm rods + IL | n/a | n/a | Bucky gel | 25 |
| Graphite rods + [C8mim][PF6]/[BF4]/[Cl] + water + [C4mim][PF6] + water | Electrochemical exfoliation | DMF | 10 mL IL + 10 mL water + graphite rods | n/a | 1.1 | 700 nm × 500 nm | 26 |
| Graphite + [C4mim][cholate] | Bath Sonication | [C4mim] [cholate] | 5 mg mL−1 | n/a | n/a | n/a | 67 |
| Pencil + [S222][Tf2N] | Electrochemical method | [Triethyl sulfonium bis(trifluoromethyl sulfonyl)imide] | 10 mL (IL) | n/a | 0.8–1.3 | A few nm | 27 |
| Iso-molded graphite rod | Electrochemical method | ([C2mim][BF4]/[C2mim][Tf2N],/[N1114][Tf2N]/[C4mpyrr][Tf2N] + acetonitrile (ACN) | ∼1:50 (IL–ACN, volume ratio) |
29–86% | n/a | ∼500 nm | 28 |
One such key parameter towards this is the understanding of the interaction of ILs with graphene for successful exfoliation/dispersion is negative free energy for the process, particularly decreasing the enthalpy of mixing, which implies a matching of the surface energy of the 2-D material to the surface tension of the IL. As shown in Fig. 10, a plot of the dispersibility against surface tension, the ideal surface tension occurs near 40 mJ m−2 for greatest dispersibility in both ILs and organic solvents implying that the surface tension of a solvent is an excellent a priori property for determining the successful exfoliation and dispersion of GNs. As illustrated in Fig. 10, high exfoliation/dispersion concentrations of GNs near 4–5.5 mg mL−1 can be reached using the ILs [C6mim][PF6] and [C4mim][PF6] with surface tensions of 43.4 and 43.6 mJ m−2, respectively.20 The concentration GNs dispersions in the other ILs decreases as the IL surface tensions differs further away from 40 mJ m−2, see Fig. 10. Based on the above yields, it may be concluded that the exfoliation of graphene was either stabilized by the hydrophilic or hydrophobic nature of organic solvents, surface tension, and π–π/cation–π interactions.
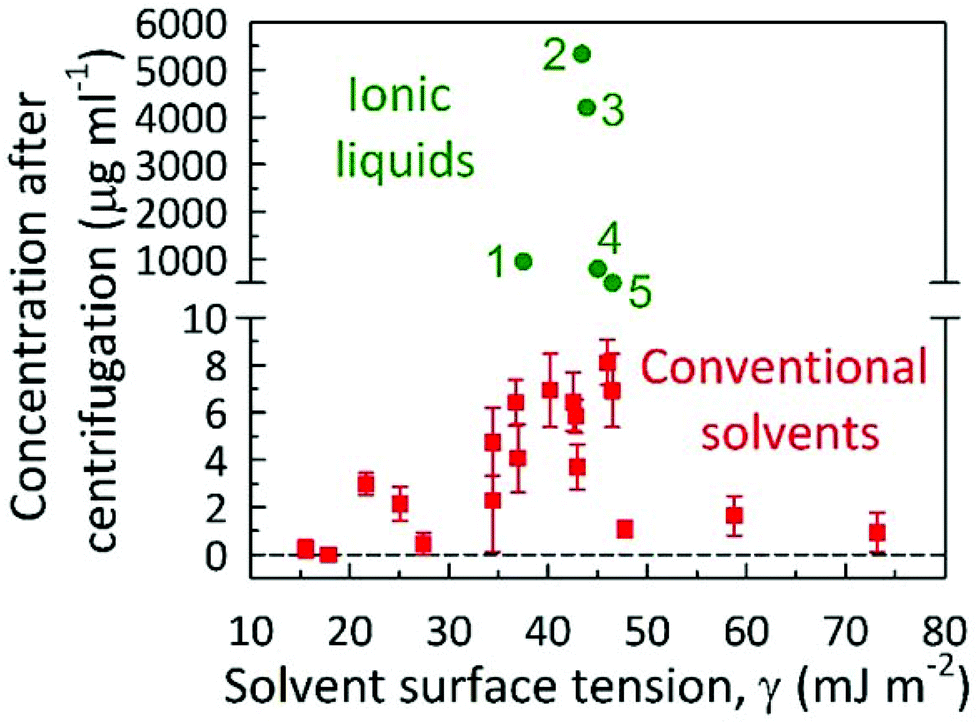 | ||
| Fig. 10 Graphene concentration measured after centrifugation for different solvents (organics, water and ILs) against the surface tension of the solvent. Red squares (values of surface tension of organic solvents and the corresponding yields of graphene dispersed in those solvents taken from ref. 9) and green circles (values of surface tension of ILs and the corresponding yields of graphene in the presence of ILs (1) = [bmim][NTf2],19 (2) = [hmim][PF6],20 (3) = [bmim][BF4],35 (4) = [bmim][PF6],34 and (5) = [C4Py][BF4]35). | ||
For the case of hydrophilic GO, which has a comparable dispersion of 1 mg mL−1 in organic solvents and 7 mg mL−1 in water,88 it can be well stabilized by water and hydrophilic organic solvents. However, when reduced to RGO, it tends to have many structural defects which causes a disruption of the π-electron and hampering the electronic properties as compared to graphene. ILs can help overcome these problems through the presence of hydrophilic and hydrophobic groups which stabilizes both GO and RGO through ionic, π–π, and cation–π interactions and further heals many structural defects by the presence of the IL long-chain alkyl groups.
Other than the choice of IL, a variety of protocols ranging from different sonication methods (bath sonication, tip or probe sonication), periods of sonication followed by centrifugation have been considered. However, while semi-empirical methods have been developed by Coleman to correlate the size of the flakes to the different experimental parameters while using data from various organic solvents, a consensus and optimization of the various parameters controlling the dispersion amount and the size of the flakes is desired. The final concentration of the graphene is dependent on various operational parameters such as volume of dispersion, initial ratio of the feed, sonication or grinding time and centrifugation rate. Nuvoli et al. performed liquid phase exfoliation of graphite using [C6mim][PF6],20 suggests the concentration of the GNs dispersion is correlated to the square root of the sonication time. As Hersam puts it in his perspective,89 “The polydispersity of graphene dispersions can lead to unpredictable or non-ideal behaviour once they are incorporated into devices since the properties of graphene vary as a function of its structural parameters”. At the present, a significant fraction of researchers are just investigating different methods and various solvent media to polydisperse graphene with less oversight towards deriving the parameters and conditions that would enable uniform size and defect-free GNs dispersion and with an eye towards large scale-up.
Further, a major concern of using ILs as a solvent or aid for graphene exfoliation and dispersion, lies in the removal of the IL from the exfoliated GNs, RGO, or IGAs, especially the fluorinated ILs which possess higher thermal stability ranges. Other than heating, one avenue could be centrifugation at high rpms to remove excess IL followed by repeated washing of organic solvents. However, depending on the application, such post-processing may not be needed, for example, if the materials are to be used as electrodes and/or with IL electrolytes or in hybrid nano-assemblies.
6. Conclusions and future prospects
In this Minireview, we have overviewed the production of graphene sheets in the presence of various ILs as assisting solvent media including brief applications of these IL-GNs composites towards avenues such as sensing, separation, fuel cells, photovoltaics, catalysis, environmental remediation, and other energy applications. Further, this IL-based exfoliation approach will enable the use of low-cost solution processing techniques such as drop-casting, electrostatic adsorption, spraying, filtration, and dip- or spin-coating of dispersions to fabricate graphene-based devices, broadening their applications. The advantages of using ILs are highlighted, with a multitude of permutations and combinations possible in the anions and cations, demonstrating immense potential for production capabilities of exfoliated GNs, RGO, and IGAs. In stark contrast to other dispersants for carbon nanomaterials, ILs tend to be less obtrusive to the properties inherent to graphene. The use of ILs as solvents for liquid-phase exfoliation and stabilization of the colloids, while still being in its infancy, most often outperforms many traditional solvents. This warrants further investigation including, in particular, advantages of using mixtures of ILs, phase thermodynamics for optimized exfoliation, optimization of technique parameters, and post-processing procedures.Keeping in mind the goal of producing GNs “by the pound”, a better fundamental understanding of the interactions between graphene and IL components is essential, including a systematic effort looking at novel functional cations/anions with an eye toward enhanced intercalation and dispersion. A vital component is the involvement of molecular dynamics and potential of mean force simulations, with in silico feedback informing the experimental results. In addition, novel experimental methods such as sum frequency generation (SFG) spectroscopy are increasingly being used to probe into the interface of electrodes and electrolytes and especially extrapolation of such methods for graphene and its analog electrodes with ILs, emphasising the need for refined theories such as Gouy–Chapman model instead of the popular Debye–Huckel theory.90,91 Given the simplicity and facile control during single-pot nanocarbon synthesis, coupled with its potential for expansion to an industrial scale, this approach will find much activity in the coming years. The urgency for additional and more specialized experimental tools and simulation approaches to probe the interface, however, clearly remains. Significantly, these fundamental approaches and insights can be transferred to the exfoliation of inorganic 2-D analogs, including BN, MoS2, WS2, and Bi2Te3. Certainly, efforts along this pathway have begun in a number of laboratories already.
Acknowledgements
Financial support from an ACS PRF grant (51865-DNI10) to GAB is gratefully acknowledged.Notes and references
- H.-P. Boehm, Angew. Chem., Int. Ed., 2010, 49, 9332–9335 CrossRef CAS PubMed.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- Y. Zhang, Y.-W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- X. Li, C. W. Magnuson, A. Venugopal, J. An, J. W. Suk, B. Han, M. Borysiak, W. Cai, A. Velamakanni, Y. Zhu, L. Fu, E. M. Vogel, E. Voelkl, L. Colombo and R. S. Ruoff, Nano Lett., 2010, 10, 4328–4334 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- X. Cui, C. Zhang, R. Hao and Y. Hou, Nanoscale, 2011, 3, 2118–2126 RSC.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- S. Tang, G. A. Baker and H. Zhao, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS PubMed.
- J. Lee and T. Aida, Chem. Commun., 2011, 47, 6757–6762 RSC.
- J.-W. T. Seo, A. A. Green, A. L. Antaris and M. C. Hersam, J. Phys. Chem. Lett., 2011, 2, 1004–1008 CrossRef CAS.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- S. Vadukumpully, J. Paul and S. Valiyaveettil, Carbon, 2009, 47, 3288–3294 CrossRef CAS PubMed.
- A. J. Patil, J. L. Vickery, T. B. Scott and S. Mann, Adv. Mater., 2009, 21, 3159–3164 CrossRef CAS.
- X. Wang, P. F. Fulvio, G. A. Baker, G. M. Veith, R. R. Unocic, S. M. Mahurin, M. Chi and S. Dai, Chem. Commun., 2010, 46, 4487–4489 RSC.
- D. Nuvoli, L. Valentini, V. Alzari, S. Scognamillo, S. B. Bon, M. Piccinini, J. Illescas and A. Mariani, J. Mater. Chem., 2011, 21, 3428–3431 RSC.
- N. G. Shang, P. Papakonstantinou, S. Sharma, G. Lubarsky, M. Li, D. W. McNeill, A. J. Quinn, W. Zhou and R. Blackley, Chem. Commun., 2012, 48, 1877–1879 RSC.
- X. Chen, J. F. Dobson and C. L. Raston, Chem. Commun., 2012, 48, 3703–3705 RSC.
- P. Tamailarasan and S. Ramaprabhu, J. Phys. Chem. C, 2012, 116, 14179–14187 CAS.
- Z. Jin, J. R. Lomeda, B. K. Price, W. Lu, Y. Zhu and J. M. Tour, Chem. Mater., 2009, 21, 3045–3047 CrossRef CAS.
- J. Lu, J.-x. Yang, J. Wang, A. Lim, S. Wang and K. P. Loh, ACS Nano, 2009, 3, 2367–2375 CrossRef CAS PubMed.
- N. Liu, F. Luo, H. Wu, Y. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18, 1518–1525 CrossRef CAS.
- V. V. Singh, G. Gupta, A. Batra, A. K. Nigam, M. Boopathi, P. K. Gutch, B. K. Tripathi, A. Srivastava, M. Samuel, G. S. Agarwal, B. Singh and R. Vijayaraghavan, Adv. Funct. Mater., 2012, 22, 2352–2362 CrossRef CAS.
- A. T. Najafabadi and E. Gyenge, Carbon, 2014, 71, 58–69 CrossRef CAS PubMed.
- L. S. Ott, M. L. Cline, M. Deetlefs, K. R. Seddon and R. G. Finke, J. Am. Chem. Soc., 2005, 127, 5758–5759 CrossRef CAS PubMed.
- M. Feroci, I. Chiarotto, M. Orsini, G. Sotgiu and A. Inesi, Adv. Synth. Catal., 2008, 350, 1355–1359 CrossRef CAS.
- P. R. Singh and X. Zeng, J. Phys. Chem. C, 2011, 115, 17429–17439 CAS.
- D. Wei, L. Grande, V. Chundi, R. White, C. Bower, P. Andrew and T. Ryhanen, Chem. Commun., 2012, 48, 1239–1241 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- X. Zhou, T. Wu, K. Ding, B. Hu, M. Hou and B. Han, Chem. Commun., 2010, 46, 386–388 RSC.
- B. Zhang, W. Ning, J. Zhang, X. Qiao, J. Zhang, J. He and C.-Y. Liu, J. Mater. Chem., 2010, 20, 5401–5403 RSC.
- W.-S. Ma, L. Wu, F. Yang and S.-F. Wang, J. Mater. Sci., 2014, 49, 562–571 CrossRef CAS.
- Y. Liang, D. Wu, X. Feng and K. Mullen, Adv. Mater., 2009, 21, 1679–1683 CrossRef CAS.
- T. Kim, H. Lee, J. Kim and K. S. Suh, ACS Nano, 2010, 4, 1612–1618 CrossRef CAS PubMed.
- Y. Men, X.-H. Li, M. Antonietti and J. Yuan, Polym. Chem., 2012, 3, 871–873 RSC.
- W. Yan, Y. Huang, Y. Xu, L. Huang and Y. Chen, J. Nanosci. Nanotechnol., 2012, 12, 2270–2277 CrossRef CAS PubMed.
- K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed.
- A. Safavi, M. Tohidi, F. A. Mahyari and H. Shahbaazi, J. Mater. Chem., 2012, 22, 3825–3831 RSC.
- M. Acik, D. R. Dreyer, C. W. Bielawski and Y. J. Chabal, J. Phys. Chem. C, 2012, 116, 7867–7873 CAS.
- J. Shen, M. Shi, B. Yan, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21, 7795–7801 RSC.
- J. Shen, M. Shi, B. Yan, H. Ma and M. Ye, Nano Res., 2011, 4, 795–806 CrossRef CAS PubMed.
- B. Wang, X. Wang, W. Lou and J. Hao, New J. Chem., 2012, 36, 1684–1690 RSC.
- T. Kim, H. C. Kang, T. T. Tung, J. D. Lee, H. Kim, W. S. Yang, H. G. Yoon and K. S. Suh, RSC Adv., 2012, 2, 8808–8812 RSC.
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2010, 5, 436–442 CrossRef PubMed.
- J. Kim and S. Kim, Appl. Surf. Sci., 2014, 295, 31–37 CrossRef PubMed.
- H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC.
- P. Bhunia, E. Hwang, M. Min, J. Lee, S. Seo, S. Some and H. Lee, Chem. Commun., 2012, 48, 913–915 RSC.
- Y. Fu, J. Zhang, H. Liu, W. C. Hiscox and Y. Gu, J. Mater. Chem. A, 2013, 1, 2663–2674 CAS.
- X. Zhou, T. Wu, B. Hu, G. Yang and B. Han, Chem. Commun., 2010, 46, 3663–3665 RSC.
- S. P. Lonkar, A. Bobenrieth, J. D. Winter, P. Gerbaux, J.-M. Raquez and P. Dubois, J. Mater. Chem., 2012, 22, 18124–18126 RSC.
- Y.-K. Yang, C.-E. He, R.-G. Peng, A. Baji, X.-S. Du, Y.-L. Huang, X.-L. Xie and Y.-W. Mai, J. Mater. Chem., 2012, 22, 5666–5675 RSC.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS PubMed.
- H. Peng, L. Meng, L. Niu and Q. Lu, J. Phys. Chem. C, 2012, 116, 16294–16299 CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- H. Zhao, G. A. Baker, Z. Song, O. Olubajo, T. Crittle and D. Peters, Green Chem., 2008, 10, 696–705 RSC.
- H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, J. Biotechnol., 2009, 139, 47–54 CrossRef CAS PubMed.
- N. Liu, X. Chen and Z. Ma, Biosens. Bioelectron., 2013, 48, 33–38 CrossRef CAS PubMed.
- N. Zhou, J. Li, H. Chen, C. Liao and L. Chen, Analyst, 2013, 138, 1091–1097 RSC.
- M. Tunckol, J. Durand and P. Serp, Carbon, 2012, 50, 4303–4334 CrossRef CAS PubMed.
- B. G. Choi, Y. S. Huh, W. H. Hong, H. J. Kim and H. S. Park, Nanoscale, 2012, 4, 5394–5400 RSC.
- Y. Liu, Y. Huang, Y. Xie, Z. Yang, H. Huang and Q. Zhou, Chem. Eng. J., 2012, 197, 80–87 CrossRef CAS PubMed.
- J. Chai, F. Li, Y. Hu, Q. Zhang, D. Han and L. Niu, J. Mater. Chem., 2011, 21, 17922–17929 RSC.
- W. Xiao, Z. Sun, S. Chen, H. Zhang, Y. Zhao, C. Huang and Z. Liu, RSC Adv., 2012, 2, 8189–8193 RSC.
- S. Miao, Z. Liu, B. Han, J. Huang, Z. Sun, J. Zhang and T. Jiang, Angew. Chem., Int. Ed., 2006, 45, 266–269 CrossRef CAS PubMed.
- C. N. R. Rao and A. Nag, Eur. J. Inorg. Chem., 2010, 2010, 4244–4250 CrossRef.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- K.-G. Zhou, N.-N. Mao, H.-X. Wang, Y. Peng and H.-L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed.
- Y. Yao, Z. Lin, Z. Li, X. Song, K.-S. Moon and C.-p. Wong, J. Mater. Chem., 2012, 22, 13494–13499 RSC.
- G. Kamath and G. A. Baker, RSC Adv., 2013, 3, 8197–8202 RSC.
- T. E. Sutto and T. T. Duncan, Electrochim. Acta, 2012, 77, 204–211 CrossRef CAS PubMed.
- C.-Y. Lee, J. A. McCammon and P. J. Rossky, J. Chem. Phys., 1984, 80, 4448–4455 CrossRef CAS PubMed.
- C. J. Shih, S. Lin, M. S. Strano and D. Blankschtein, J. Am. Chem. Soc., 2010, 132, 14638–14648 CrossRef CAS PubMed.
- C. Fu and X. Yang, Carbon, 2013, 55, 350–360 CrossRef CAS PubMed.
- C. Pinilla, M. G. Del Pópolo, R. M. Lynden-Bell and J. Kohanoff, J. Phys. Chem. B, 2005, 109, 17922–17927 CrossRef CAS PubMed.
- M. V. Federov and R. M. Lynden–Bell, Phys. Chem. Chem. Phys., 2012, 14, 2552–2556 RSC.
- G. Kamath and G. A. Baker, Phys. Chem. Chem. Phys., 2012, 14, 7929–7933 RSC.
- B. D. Fitchett and J. C. Conboy, J. Phys. Chem. B, 2004, 108, 20255–20262 CrossRef CAS.
- S. Baldelli, J. Bao, W. Wu and S.-s. Pei, Chem. Phys. Lett., 2011, 516, 171–173 CrossRef CAS PubMed.
- C.-J. Shih, S. Lin, M. S. Strano and D. Blankschtein, J. Am. Chem. Soc., 2010, 132, 14638–14648 CrossRef CAS PubMed.
- S. D. Bergin, V. Nicolosi, P. V. Streich, S. Giordani, Z. Sun, A. H. Windle, P. Ryan, N. P. P. Niraj, Z.-T. T. Wang, L. Carpenter, W. J. Blau, J. J. Boland, J. P. Hamilton and J. N. Coleman, Adv. Mater., 2008, 20, 1876–1881 CrossRef CAS.
- J. Vatamanu, L. Cao, O. Borodin, D. Bedrov and G. D. Smith, J. Phys. Chem. Lett., 2011, 2, 2267–2272 CrossRef CAS.
- Y. Hernandez, M. Lotya, D. Rickard, S. D. Bergin and J. N. Coleman, Langmuir, 2009, 26, 3208–3213 CrossRef PubMed.
- W. Lu, S. Liu, X. Qin, L. Wang, J. Tian, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, J. Mater. Chem., 2012, 22, 8775–8777 RSC.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- A. A. Green and M. C. Hersam, J. Phys. Chem. Lett., 2010, 1, 544–549 CrossRef CAS PubMed.
- C. Y. Peñalber and S. Baldelli, J. Phys. Chem. Lett., 2012, 3, 844–847 CrossRef.
- S. Baldelli, J. Phys. Chem. Lett., 2012, 4, 244–252 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |