Iron-promoted C–C bond formation in the total synthesis of natural products and drugs
Julien
Legros
*a and
Bruno
Figadère
*b aCOBRA UMR 6014 Normandie Univ, Univ Rouen, INSA Rouen and CNRS 1 rue Lucien Tesnière, 76821 Mont-Saint-Aignan, France. E-mail: julien.legros@univ-rouen.fr bCNRS, BioCIS UMR 8076 Labex LERMIT, Univ Paris Sud, and CNRS 5 rue J. B. Clément, 92296 Châtenay-Malabry, France. E-mail: bruno.figadere@u-psud.fr
Received
13th May 2015
First published on 23rd September 2015
Abstract
Covering: up to 2015
Iron salts are inexpensive and almost innocuous; they are thus the promoters of choice, even in stoichiometric amounts, for the formation of carbon–carbon bonds in the backbone of complex molecules. This review encompasses the key role of iron complexes in the total synthesis of some natural products or pharmacologically important compounds.
Julien Legros
Julien Legros was born in 1974 and educated at the Universities Paris East, and then Paris South-Faculty of Pharmacy where he received his PhD under the guidance of Dr D. Bonnet-Delpon and J.-P. Bégué (2002). After an Alexander von Humboldt post-doctoral position on iron catalysis with Prof. C. Bolm at the RWTH University (Aachen, Germany), he got a permanent position at the CNRS in 2004 (Univ Paris South-Faculty of Pharmacy) and then moved to the University of Rouen in 2011. Julien Legros is involved in synthetic methodology in non-conventional media (flow microreactors, hyperbaric conditions, fluorous solvents).
Bruno Figadère
Bruno Figadère, born in 1960, was educated in Paris (University Pierre et Marie Curie) where he received his PhD under Dr G. Cahiez leadership. After a post-doctoral position at the University of California at Riverside with Prof W. H. Okamura (1988–1990) he got a permanent position at the CNRS and since then works at University of Paris Sud-Faculty of Pharmacy. He is currently director of the BioCIS laboratory. Bruno Figadère is involved in the chemistry of natural products, especially in total synthesis, pharmacomodulation and characterisation of new bioactive compounds from different origins.
1. Introduction
Iron (Fe) is the fourth most abundant element in the Earth's crust, and most of this iron is found as iron oxides, such as the minerals hematite (Fe2O3) or magnetite (Fe3O4).1,2 For the biologist, iron is an essential element to most living beings, from single-celled microorganisms to humans, as illustrated for example by the Fe-containing protein hemoglobin.3 However, for the synthetic chemist, iron salts are often intimately connected to the centenarian Friedel–Crafts reaction, an electrophilic aromatic substitution promoted by metal salts, often including FeX3 (X = Br, Cl).4 Recently, iron complexes have found increasing application in organic synthesis, especially as catalysts. Indeed, metal-catalysed reactions are essential utensils in the toolbox of the synthetic chemist, and only those combining criteria of efficiency, selectivity and reliability with cost effectiveness and low toxicity shall hold a prominent position in the future.5–8 In this demanding context, iron occupies a place that probably no other transition metal can dispute: it is truly inexpensive and most of its salts exhibit low toxicity and they are generally environmentally benign. A comparison between iron salts (Fe(acac)3, FeCl2,3), and some commonly used metal salts (AlCl3, CuCl2, NiCl2, CoCl2, PdCl2) is given in Table 1.
aAverage range price for metal complexes with purity ≥ 99.9% taken from different providers.bLethal dose.cLethal concentration.
Fe(acac)3
3–5
No significant toxicity reported
1872
Unknown
FeCl3
18–28
No significant toxicity reported
1350
21.84
FeCl2
14–19
No significant toxicity reported
450
4
AlCl3
12–20
3450
36.6
CuCl2
15–22
584
0.23
NiCl2
37–46
186
Unknown
CoCl2
28–36
418
0.33
PdCl2
58–106
200
Unknown
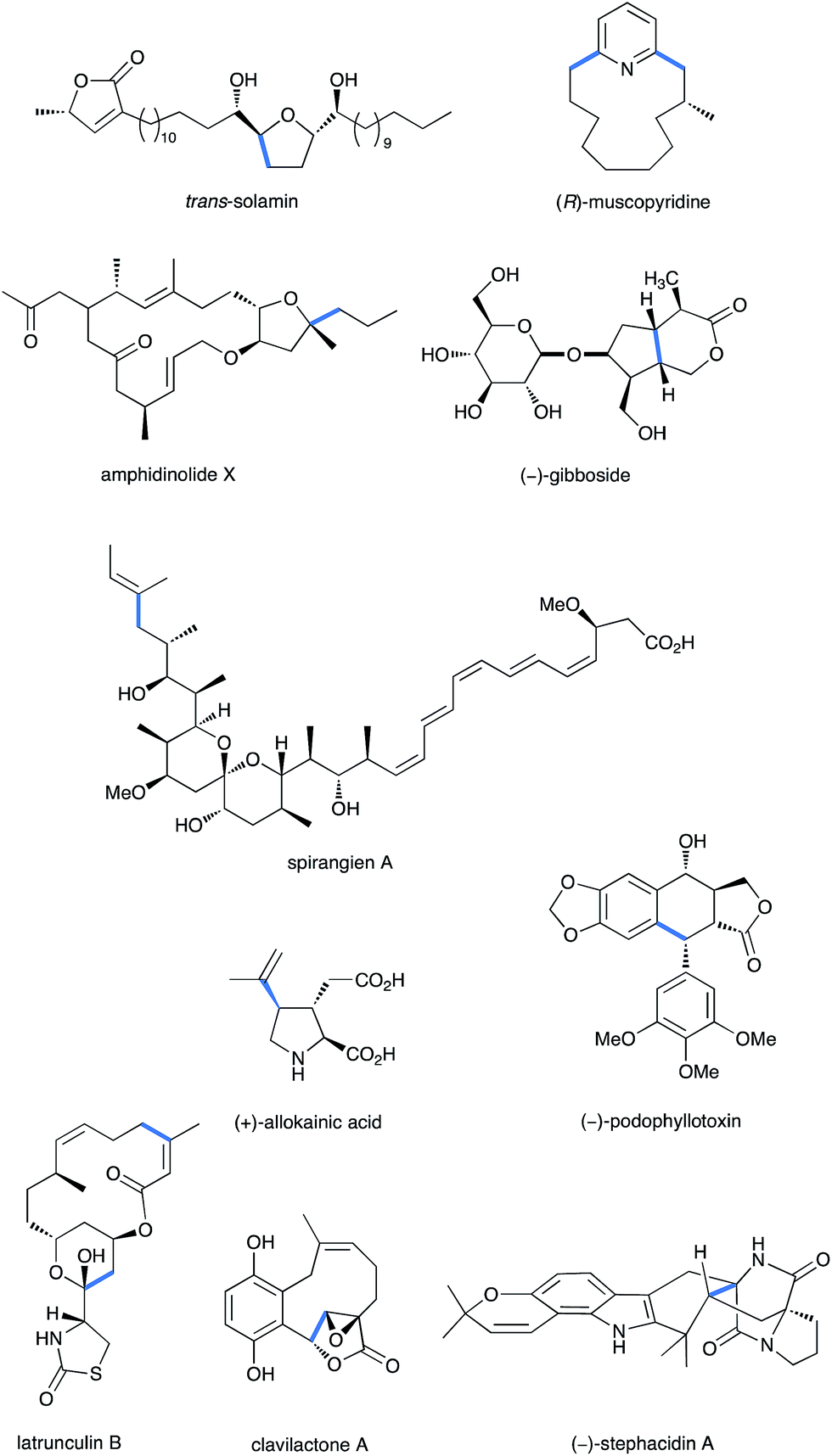
Thus, iron complexes are by far the least expensive ones, with poor toxicity, environment poisoning or CMR classification (carcinogenic, mutagenic or toxic for reproduction). Only aluminium chloride is cheaper with higher LD50 and LC50 but it exhibits teratogenicity effects. Therefore, even when used in stoichiometric amounts, iron salts remain cheaper and safer that many other catalytic metal complexes, such as the palladium ones, broadly used in cross-coupling reactions. These decisive advantages make iron complexes the promoters of choice (under catalytic or even stoichiometric amounts) for elegant C–C bond formation, as required to build the carbon backbone of poly-functionalized molecules in the total synthesis of natural products and bioactive ingredients. This review thus aims at illustrating how iron salts play a key role in carbon skeleton buildings in the total synthesis of natural products or pharmacologically important compounds (Fig. 1).
Fig. 1 Some relevant examples of natural products synthesized through a Fe-promoted key step (highlighted in blue).
2. Cross-coupling reactions of organometallic reagents
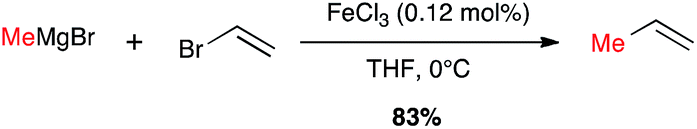
Metal-catalysed cross-coupling reactions constitute one of the most straightforward and strongest methodologies to form C–C bonds.9 In 1971 Tamura and Kochi reported for the first time successful and stereoselective examples of formal nucleophilic substitution between alkyl magnesium bromides and vinyl bromides in presence of very low catalytic loadings of FeCl3 in THF (Scheme 1).10,11 However, one year later Kumada and Corriu independently reported the cross-coupling reaction between Grignard reagents and organic halides by means of nickel catalysts, later extended to palladium complexes, and these transformations were then extensively studied leading eventually to the Nobel prize in 2010 to Negishi, Heck and Suzuki. The potential of Fe-catalysed cross-couplings remained in the shadow of palladium catalysis for many years and did not undergo much studies until Cahiez reported in 1998 a spectacular improvement allowing poorly reactive substrates, such as alkenyl chlorides, to be efficiently converted by using Fe(acac)3 as catalyst and N-methylpyrrolidinone (NMP) as co-solvent; the yield jumped then from 5% in THF to 85% in this solvent mixture (Scheme 2).12
Scheme 1 First Fe-catalysed cross-coupling reaction with a Grignard reagent by Tamura and Kochi (1971).
Scheme 2 Improved conditions for Fe-catalysed cross-coupling with a Grignard reagent by Cahiez (1998).
Therefore, methodology in Fe-catalysed cross-coupling reactions has met outstanding improvements, notably by Fürstner and Nakamura. Thus, the reaction scope has been extended to various Grignard reagents and numerous electrophilic partners such as vinyl-, aryl- and alkyl (pseudo)halides. Moreover, many efforts have also been devoted to the comprehension of the reaction mechanism. Whereas Kochi early suggested the formation of an active “reduced form of soluble iron” generated from organomagnesium reagents and iron chloride,11 recent experimental and theoretical investigations suggest that the nature of the active iron species generated from either Fe(III) or Fe(II) and Grignard reagents depends on the nature of the latter (e.g. Ar- or alkyl-MgBr, presence of a hydrogen atom in the β-position).13–18 To briefly summarize, two main pathways are generally considered: double-electron transfer or single-electron transfer mechanisms.6,19
Iron-catalysed coupling reactions with organometallics, such as Grignard reagents, have shown their great compatibility with various functional groups and it is then not surprising that they have then been frequently applied as a key step in total syntheses of natural products and/or compounds of pharmacological interest, due to the ease, chemo- and stereoselectivity of the processes.5,8,20,21
2.1 With alkenyl and alkynyl (pseudo)halides
Aryl and alkyl Grignard reagents have been involved with various alkenyl and alkynyl electrophilic partners.
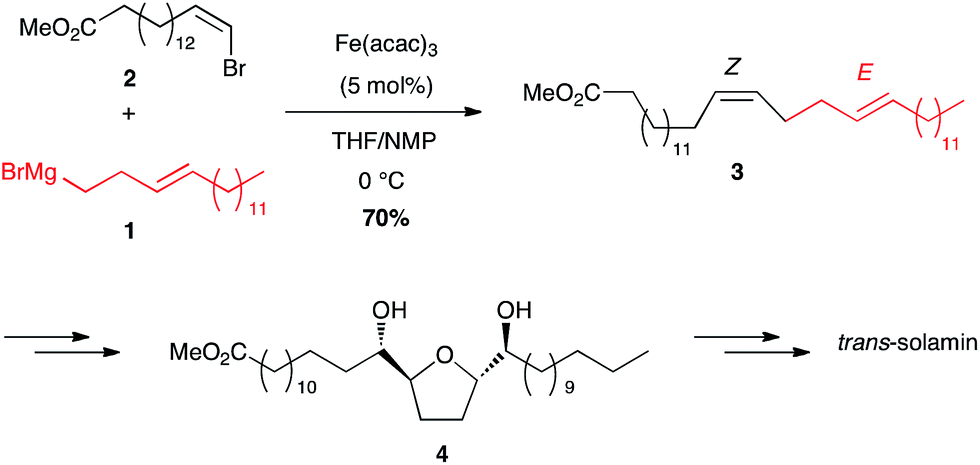
Alkenyl and alkynyl halides. Since the pioneering work of Kochi, vinyl halides have been widely used. For example, iron-catalysed coupling reaction of an homoallylic Grignard reagent 1 with a (Z)-bromoalkene 2 proceeded with complete maintaining of the geometry and led to a key diene 3 of defined Z,E configuration (70% yield), which after chemo- and enantioselective dihydroxylation will lead to cis and trans solamins, depending on the dihydroxylation procedures, two annonaceous acetogenins with cytotoxicity activity (Scheme 3).22
Scheme 3 Fe-catalysed cross-coupling of homoallylic magnesium bromide with a (Z)-bromoolefin for the synthesis of trans-solamin.
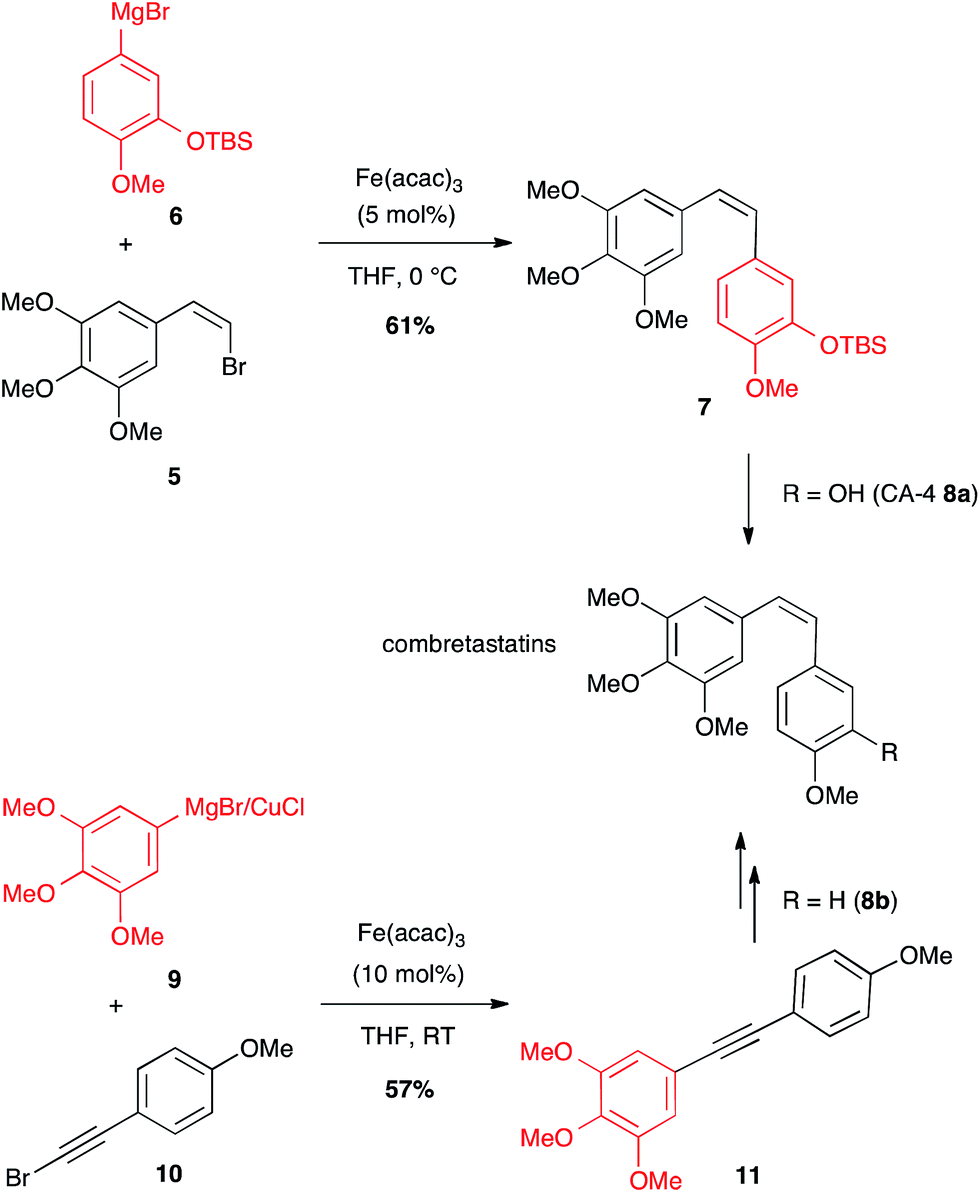
A (Z)-bromoalkene 5 has also been coupled under iron catalysis to an aryl Grignard reagent 6 to afford combretastatin-A4 (CA-4) protected as TBDMS ether 7 (61% yield), which after deprotection led to the desired anticancer compound 8a (Scheme 4).23 Later on, another Fe-catalysed path to access combretastatins has been described. This latter does not involve any olefin as coupling partner with the arene reagent, but a bromoalkyne: bimetallic aryl magnesium/copper halide 9 reacted with bromoarylacetylene 10 under iron catalysis to afford the corresponding bi(aryl)acetylene 11 (57% yield) that can be reduced to yield analogues of CA-4 8b (Scheme 4).24 It is worth noting that this is the single known example of Fe-catalysed coupling between a Grignard reagent and a bromalkyne.
Scheme 4 Fe-catalysed cross-coupling for the synthesis of combretastatins.
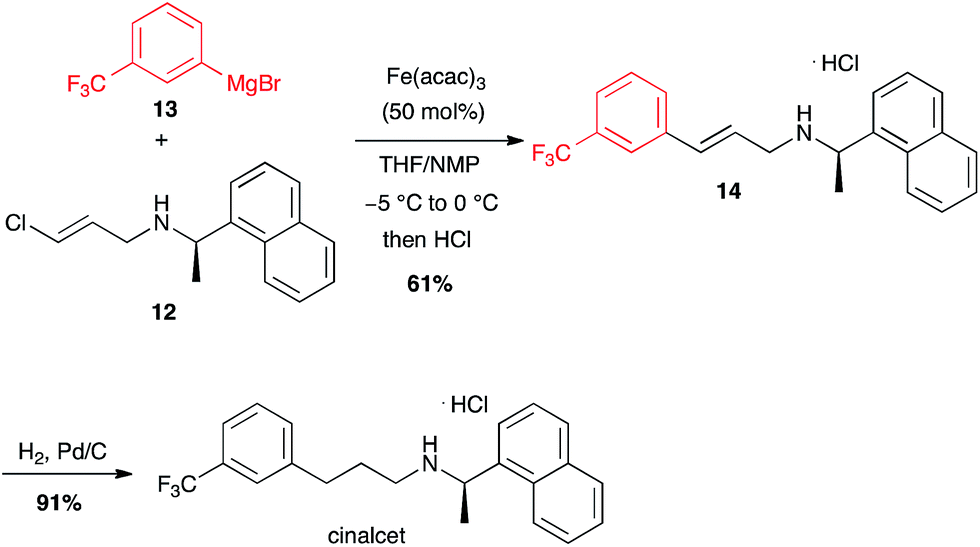
Interestingly, less reactive (E)-chloroolefins are also good substrates for iron-catalysed coupling with aryl Grignard reagents: cinacalcet, a drug used for the treatment of secondary hyperparathyroidism, was prepared on a kilogram scale using this reaction between 12 and 13 as a key step (Scheme 5).25
Scheme 5 Fe-catalysed coupling of aryl magnesium bromide with a (E)-chloroolefin for the synthesis of cinacalcet.
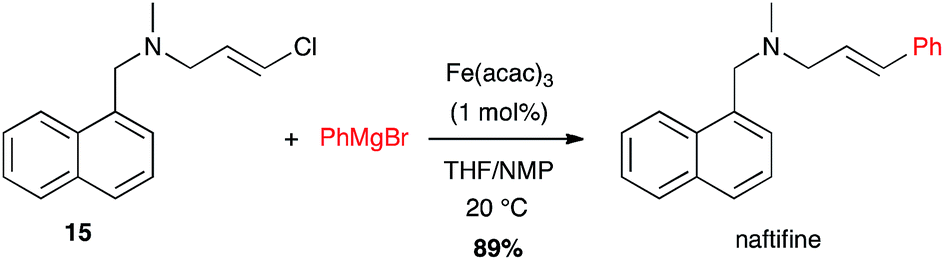
Another amine-containing molecule, naftifine, an antifungal drug, was prepared by an iron-catalysed coupling of a (E)-chloroolefin 15 and phenyl Grignard reagent in high yield (89%) and excellent stereoselectivity (Scheme 6).26
Scheme 6 Fe-catalysed coupling of phenyl magnesium bromide with a (E)-chloroolefin for the synthesis of naftifine.
Vinyl (pseudo)halides. However greatest applications with vinyl electrophiles in total synthesis have been found with vinyl triflates, as established by Fürstner.27 One of the major interest of these electrophiles is that enol triflates are readily accessible from the corresponding ketones (albeit with possible regioisomeric issues). Moreover, these triflates can in some cases be generated and used in cross-couplings without intermediate isolation.
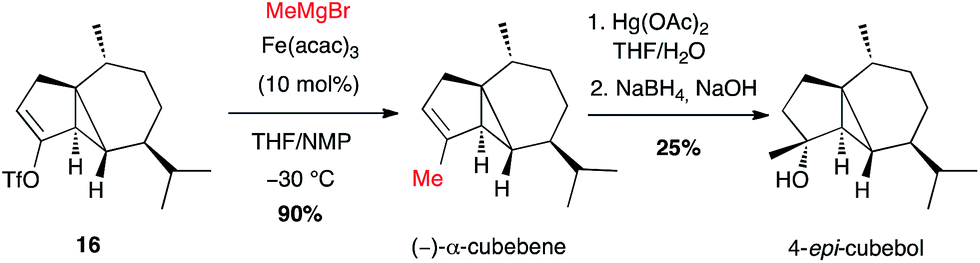
This method has been mainly used to introduce a methyl group. Thus, the natural sesquiterpene (−)-α-cubebene has been prepared in 2006 by Fürstner involving, at a late stage, coupling of methylmagnesium bromide with the isolated alkenyl triflate 16 in the presence of Fe(acac)3 (10 mol%) in a THF/NMP mixture, in high yield (90%). 4-epi-Cubebol has then been further obtained (Scheme 7).28
Scheme 7 Fe-catalysed coupling of methyl Grignard reagent with a vinyl triflate for the synthesis of (−)-α-cubebene and 4-epi-cubebol.
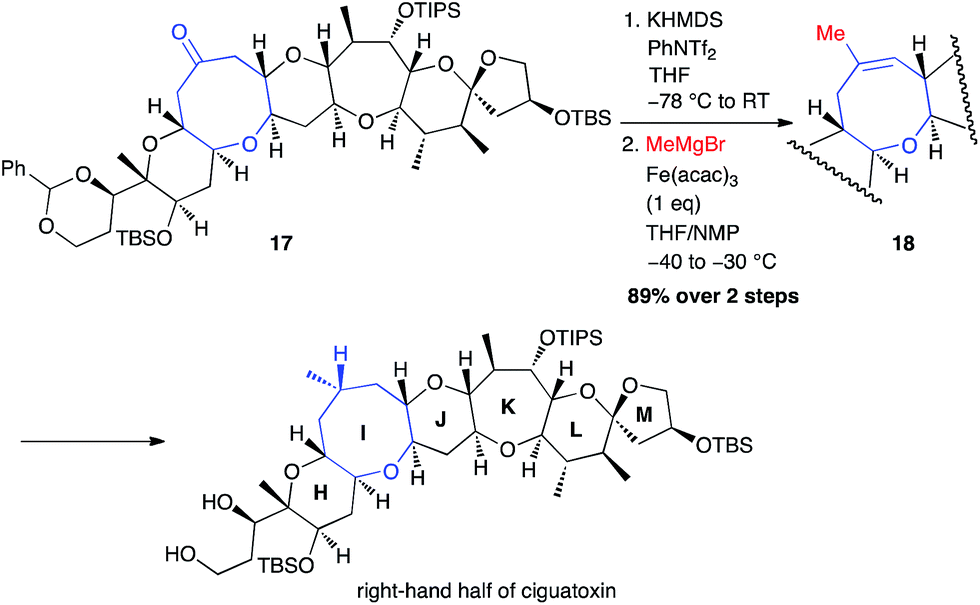
The same year, Isobe used this methodology to perform the synthesis of the right-hand segment of ciguatoxin.29 A methyl group was then successfully introduced from an isolated triflate generated from PhNTf2, using a stoichiometric amount of Fe(acac)3 (89% from ketone 17; Scheme 8).
Scheme 8 Fe-catalysed coupling of methyl Grignard reagent with a vinyl triflate for the synthesis of the right-hand segment of ciguatoxin.
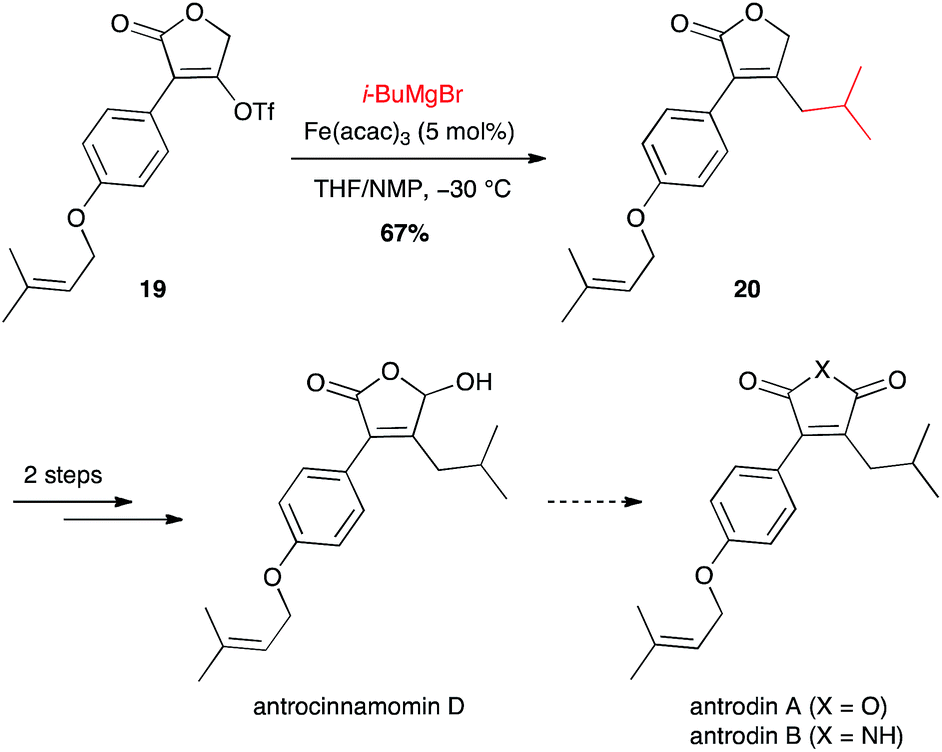
i-Butyl moiety has also been introduced for the synthesis of maleic anhydrides and derivatives, constituents of Antrodia camphorata: antrocinnamomin D and antrodins (Scheme 9).30 Interestingly the coupling between i-BuMgBr and the vinyl triflate 19 with the inexpensive Fe(acac)3 (5 mol%) was more efficient than the Suzuki–Miyaura coupling from i-BuB(OH)2 and 10 mol% Pd(dppf)Cl2 (67% vs. 55% yield for compound 20).
Scheme 9 Fe-catalysed coupling of methyl Grignard reagent with a vinyl triflate for the synthesis of antrocinnamomin D and antrodins.
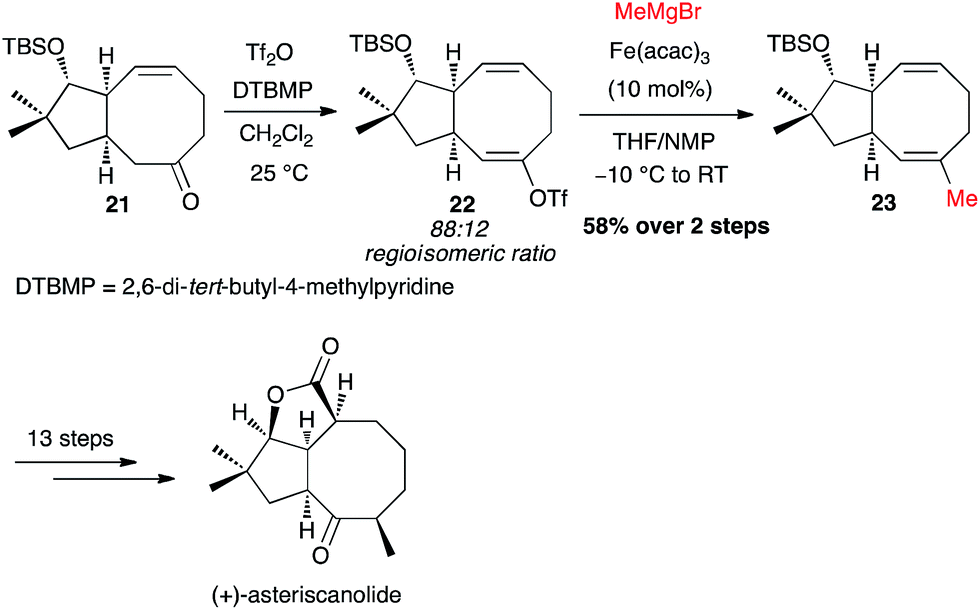
Yu and Nicolaou independently investigated the Fe-catalysed cross-coupling with MeMgBr directly on a crude triflate. Thus, Yu's synthesis of (+)-asteriscanolide involved the coupling of triflate 22 at an intermediate stage, generated with a 88:12 regioisomeric ratio, affording the methylated adduct 23 in 58% yield from the ketone 21 (Scheme 10).31
Scheme 10 Fe-catalysed coupling of methyl Grignard with a vinyl triflate for the synthesis of (+)-asteriscanolide.
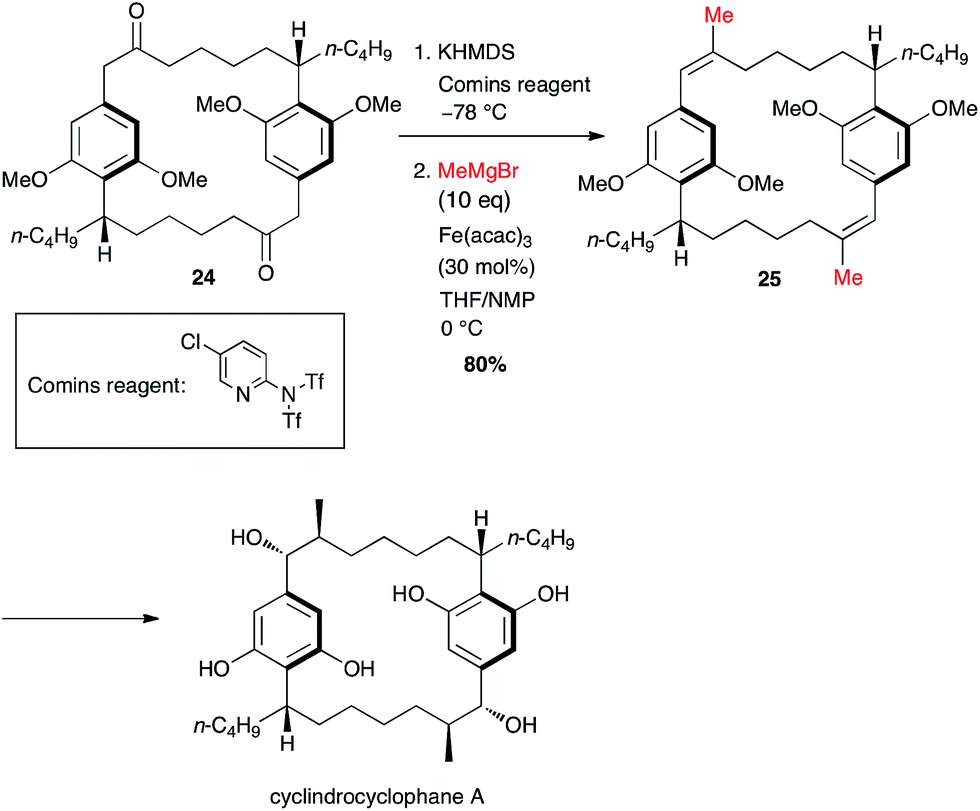
In his synthesis of cylindrocyclophane by Nicolaou, two methyl groups were introduced simultaneously and the generation of the bis-triflate did not undergo any isomeric issues since it took place at a benzylic position, and the coupling occurred smoothly to afford the double methylated product 25 in 80% yield over two steps from 24 (Scheme 11).32
Scheme 11 Fe-catalysed coupling of methyl Grignard with a bis-vinyltriflate for the synthesis of cylindrocyclophane A.
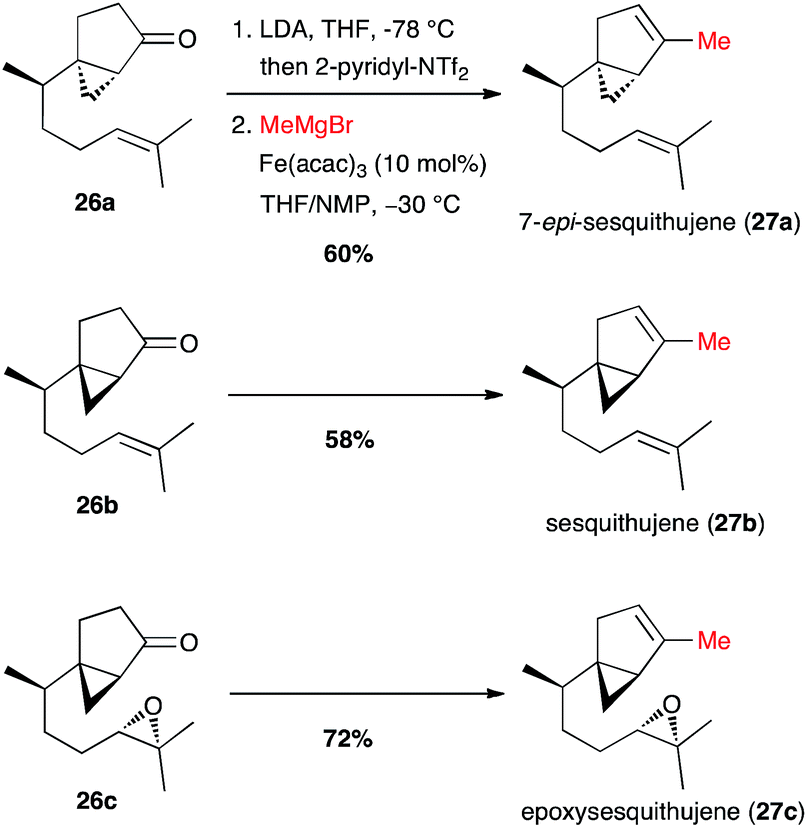
Finally, Fürstner reported the full one-pot triflate formation/cross-coupling at the last step of the synthesis of sesquithujene natural products (Scheme 12).33 Vinyl triflates were generated from 26a–c under kinetic conditions with 2-pyridinyl-NTf2, a Comins' reagent analogue, leaving unaffected the chiral cyclopropyl moieties. Then the reagents for the cross-coupling were added (MeMgBr, [Fe] cat. and NMP co-solvent), and the coupling products 27a–c were obtained in 60–72% yield. Interestingly, under these conditions the coupling with the Grignard reagent reacted selectively with the vinyl triflate and the epoxide moiety remained unchanged in the synthesis of epoxysesquithujene 27c.
Scheme 12 Fe-catalysed coupling of methyl Grignard with a vinyl triflate for the synthesis of three members of the sesquithujene family.
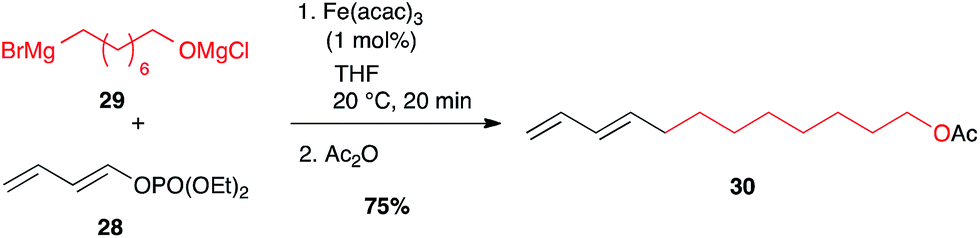
Besides classical leaving groups met in cross-couplings (I, Br, OTf,…), phosphates can also be used on the electrophilic partner. Thus, dienol phosphate 28 reacted very well with alkyl Grignard reagent 29 at low iron catalyst loading (1 mol%) while maintaining the double bond geometry.34,35 This strategy has been successfully used to access the Red Bollworm Moth (Diparopsis castanea) pheromone 30 (75% over two steps, Scheme 13).35
Scheme 13 Fe-catalysed coupling of an alkyl Grignard with a (E)-dienol phosphate for the synthesis of the Diparopsis castanea pheromone.
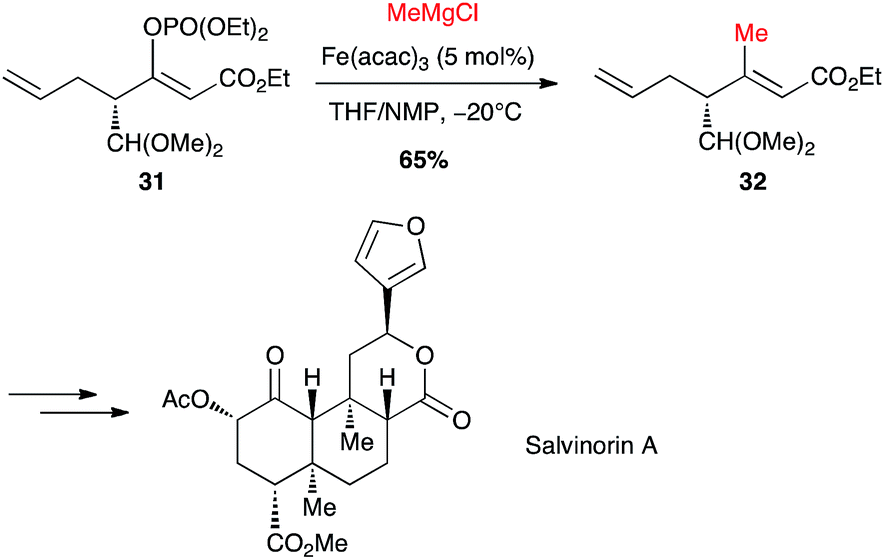
Selective formation of (Z)-enol phosphate 31 from a keto-ester, followed by an iron-catalysed coupling with methylmagnesium chloride, allowed Evans to prepare a key trisubstituted olefin 32, en route to the total synthesis of salvinorin A, a potent κ opioid receptor agonist (Scheme 14).36
Scheme 14 Fe-catalysed coupling of methyl Grignard with a (Z)-enol phosphate for the synthesis of salvinorin A.
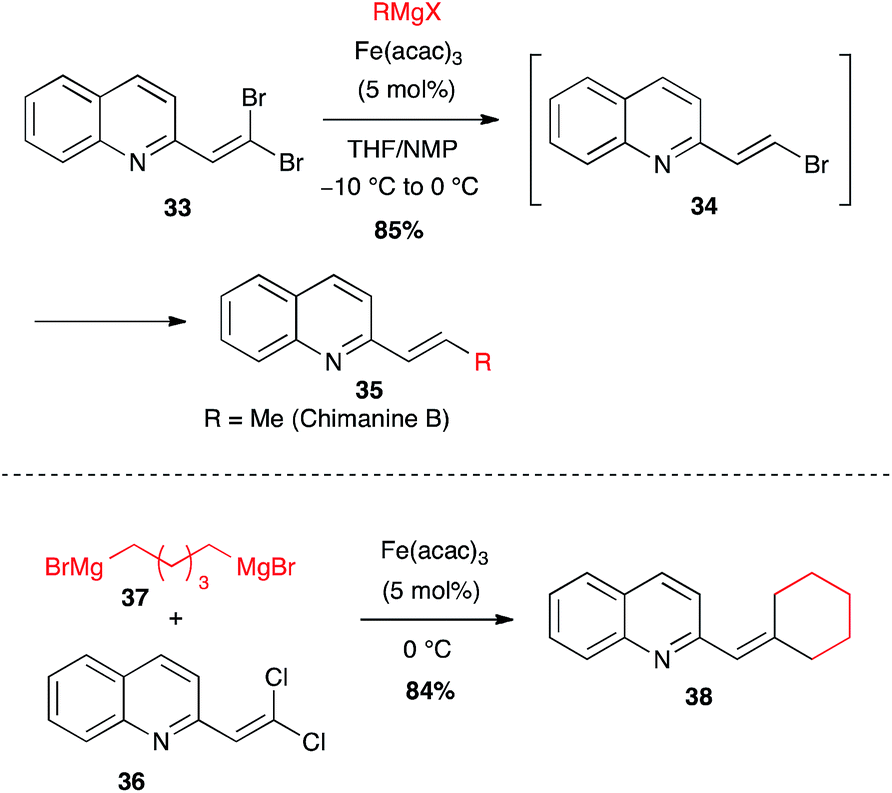
gem-Dihalogenoalkenes. gem-Dihalogenoalkenes constitute an interesting and specific class of electrophiles with a particular behavior towards organo-magnesium and -lithium reagents in Fe-catalysed cross-couplings, which met application in the synthesis of natural products. Thus, bioactive compounds derived from the chimanines, natural 2-substituted quinolines isolated from a Rutaceae, were prepared from quinolyl-gem-dihalogenoalkenes and organomagnesium reagents in presence of Fe(acac)3. While dibromo-compound 33 undergoes reduction to selectively afford the (E)-bromoalkene 34 (able to react by cross-coupling with a further equivalent of RMgX to afford 35),37 in contrast, 1,1-dichloroalkenes react rapidly to afford the disubstituted product, even if less than 2 equiv. of RMgX are used.38 Thus, gem-dichlorovinyl quinoline 36 has been engaged in cross-coupling with bis-Grignard reagent 37 to perform an intermolecular followed by an intramolecular cross-coupling leading thus to an original cyclohexylidene moiety on this family of bioactive compounds (84% for 38, Scheme 15).38
Scheme 15 Fe-catalysed coupling of an alkyl Grignard with gem-dihaloalkenes for the synthesis of chimanine derivatives.
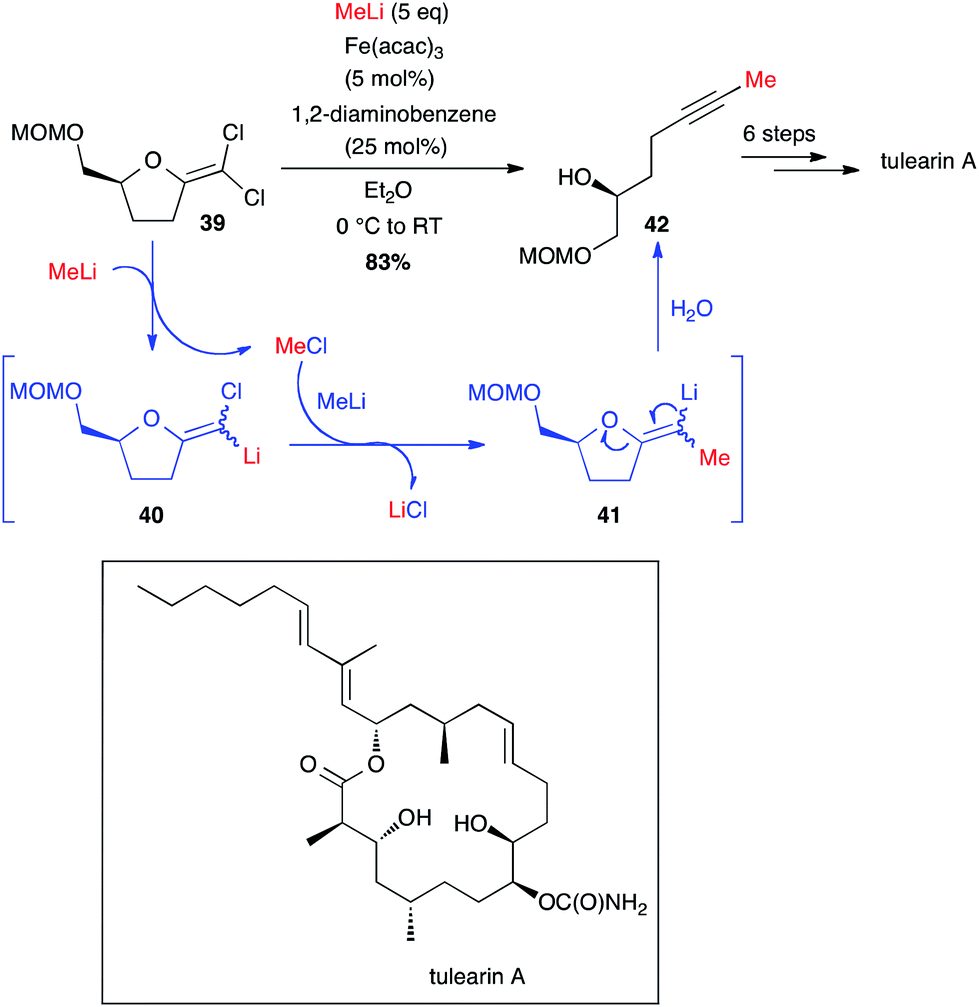
Very recently, Fürstner developed a brilliant methodology to access methyl-terminated alkynes from MeLi and gem-dichloro-olefins obtained from lactones. These non-terminal alkyne moieties are then involved in ring closing alkyne metathesis (RCAM) and tulearin A and brefeldin A have been prepared through this pathway (Schemes 16 and 17).39,40 The reaction between MeLi and gem-dichloroolefin 39, affording the methyl-capped acetylenic compound 42, is not stricto sensu a cross-coupling: a first Cl–Li exchange occurs and the MeCl formed is trapped by compound 40 and the rearrangement takes place to yield the target product 42 in 83% yield (Scheme 16). Even if this reaction is possible without catalyst, the presence of Fe(acac)3 and 1,2-diaminobenzene (5 and 25 mol%, respectively) significantly enhanced the course of the reaction (2 h vs. 48 h reaction time without [Fe]). Thus, starting from 39, tulearin A was obtained in 6 steps.39
Scheme 16 Fe-catalysed coupling of methyl lithium with a gem-dichloroalkene for the synthesis of tulearin A.
Scheme 17 Fe-catalysed coupling of methyl lithium with gem-dichloroalkene for the synthesis brefeldin A.
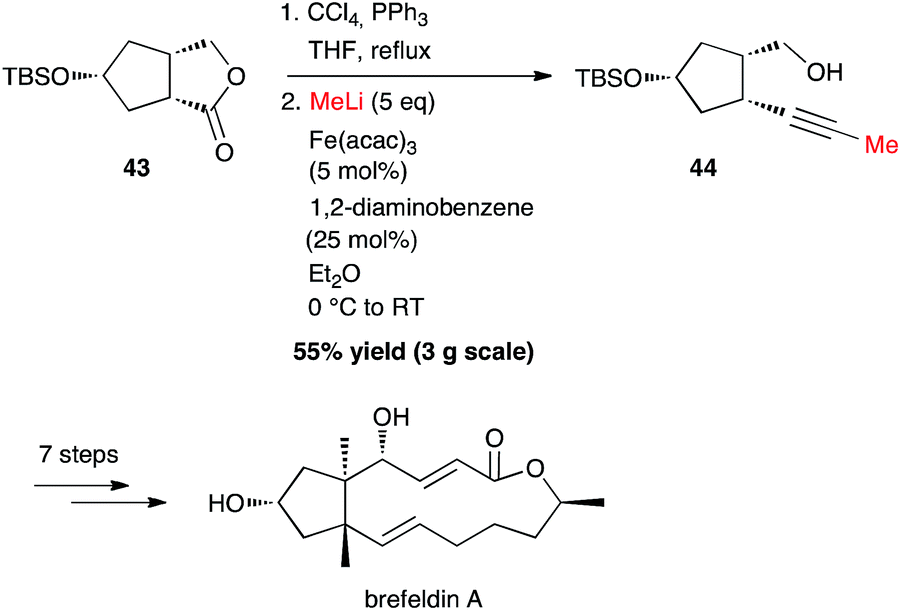
Interestingly, starting from 3 g of lactone 43, it was possible to synthesize the methyl-capped alkyne 44 in 55% yield, as an intermediate for the preparation of brefeldin A, a most studied biologically active macrolide from Penicillium decumbens (Scheme 17).40
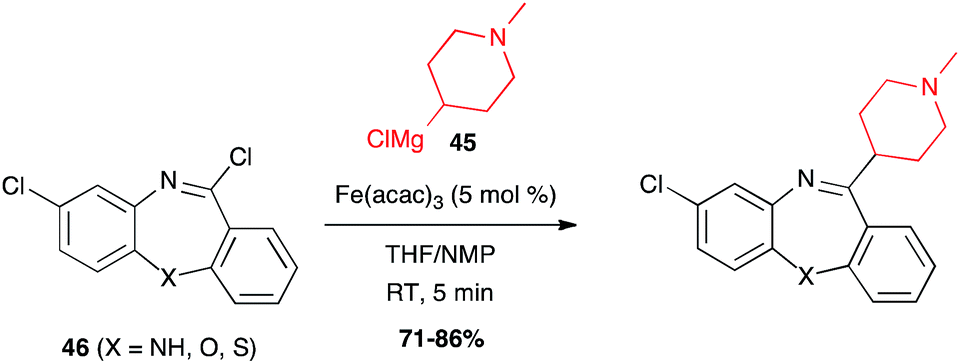
Imidoyl chloride. Olsson demonstrated the possibility to introduce various moieties on an imidoyl chloride with organomagnesium reagents to access benzodiazepine analogues (clozapines). Interestingly, the Grignard reagent 45 selectively reacted with the imidoyl chlorides 46 without affecting the chlorine atom connected to the arene (Scheme 18).41
Scheme 18 Fe-catalysed coupling of a Grignard reagent with imidoyl chlorides to synthesize clozapines.
2.2 With aryl (pseudo)halides
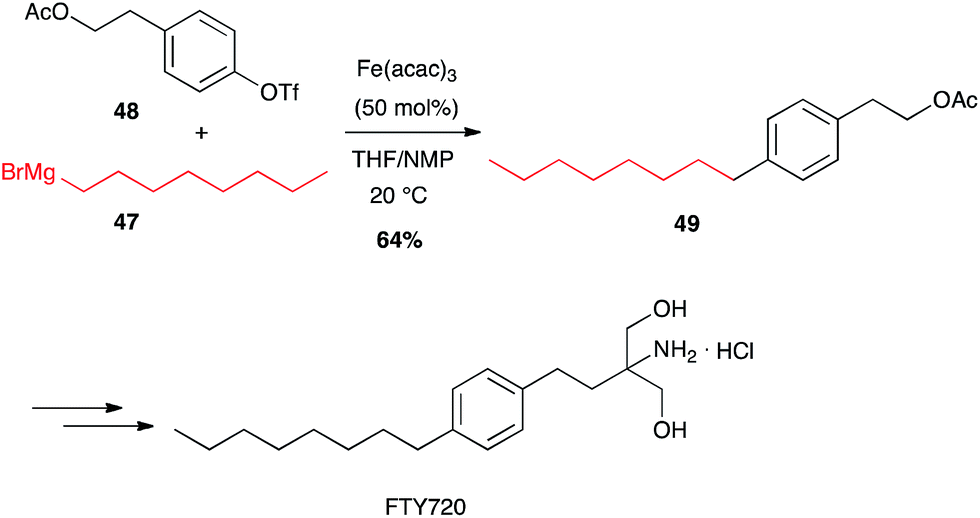
In contrast with palladium-catalysed cross-couplings, aryl-chlorides, -tosylates and -triflates have been shown to be the best partners for iron-catalysed reactions with Grignard reagents, bromo-/iodoarenes being poorer reagents.42 Fürstner has developed many iron-catalysed coupling reactions between organomagnesium reagents and various Ar-X electrophilic partners, and showed that alkyl Grignard reagents react very well with aromatic and heteroaromatic triflates in high yields. FTY720, an immunosuppressive agent was thus prepared in a multigram scale by Fürstner through Fe-catalysed coupling of an alkyl Grignard reagent 47 and an aryl triflate 48 (64%, Scheme 19).43
Scheme 19 Fe-catalysed coupling of octyl magnesium bromide with an aryl triflate for the synthesis of FTY720.
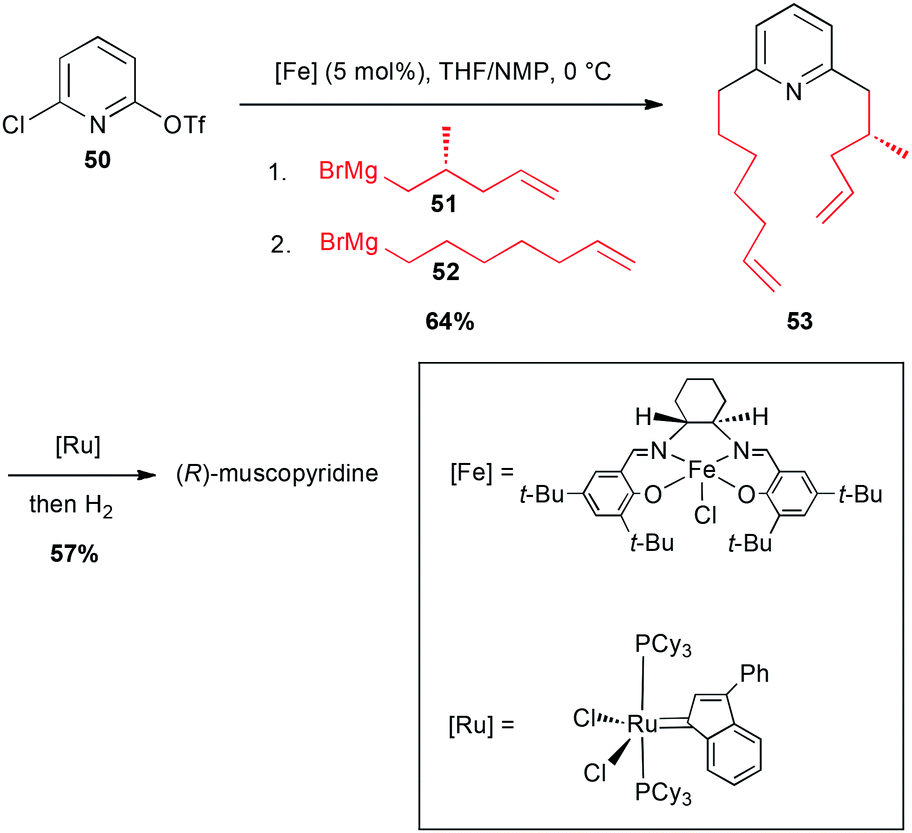
In 2003, these coupling reactions have been applied to an elegant one-pot synthesis of an odoriferous alkaloid from animal origin, (R)-muscopyridine. Indeed, mixing 2-chloro-6-triflate-pyridine 50 with an enantiomerically pure alkyl Grignard reagent 51 in the presence of an iron-salen salt (5 mol%) in a THF/NMP mixture resulted in a selective reaction with the–OTf group. Then, addition of 6-heptenylmagnesium bromide 52 (reacting on the C–Cl site), afforded the expected double adduct 53 (64%). This latter then underwent a ring-closing metathesis, and a hydrogenation to afford the expected natural product in 57% yield (Scheme 20).44
Scheme 20 Fe-catalysed coupling of alkyl Grignard reagents with a chloropyridinyl triflate for the synthesis of (R)-muscopyridine.
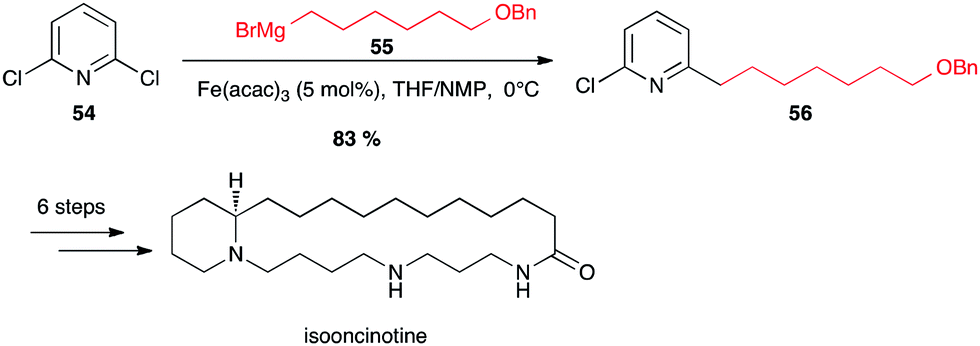
This group confirmed that halopyridines are indeed excellent substrates for iron-catalyzed Grignard couplings. Thus isooncinotine was prepared using as a key step the coupling of 2,6-dichloropyridine 54 with a functionalized alkyl Grignard reagent 55 in the presence of Fe(acac)3 in the THF/NMP solvent mixture at 0 °C (83% yield of 56; Scheme 21).45
Scheme 21 Fe-catalysed coupling of alkyl Grignard with a dichloropyridine for the synthesis of isooncinotine.
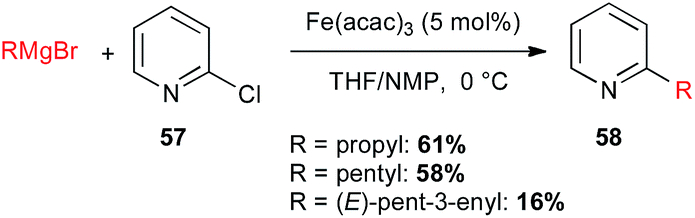
In 2014, Schulz exemplified the coupling of 2-chloropyridine 57 with alkyl Grignard reagents under iron catalysis for the preparation of volatile pyridine alkaloids 58 produced by Streptomyces sp. (Scheme 22).46
Scheme 22 Fe-catalysed coupling of alkyl Grignard reagents with 2-chloropyridine for the synthesis of streptopyridines.
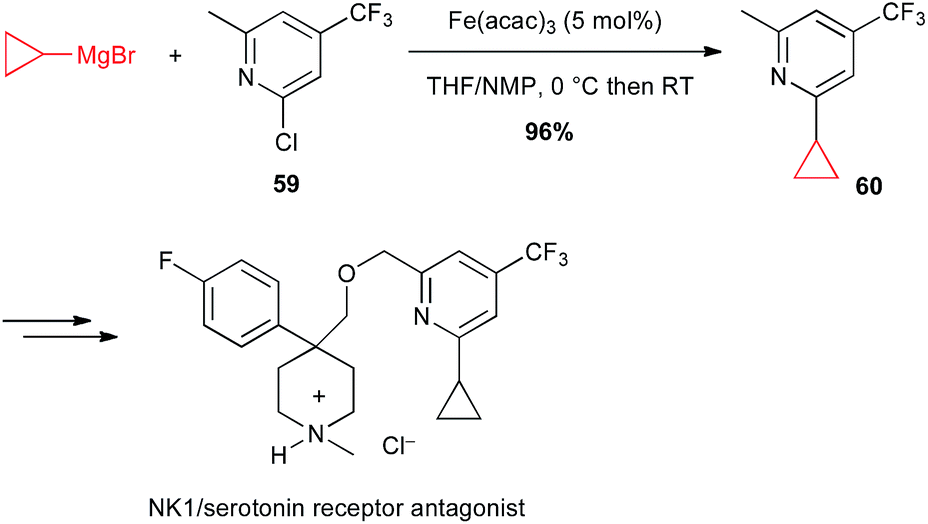
Moreover, Risatti performed a high yielding Fe(acac)3-catalysed coupling between a functionalized chloropyridine 59 and cyclopropylmagnesium bromide in their route to the synthesis of a dual NK-1/serotonin receptor antagonist (Scheme 23).47
Scheme 23 Fe-catalysed coupling of cyclopropylmagnesium bromide with a chloropyridine derivative for the synthesis of a NK1/serotonin receptor antagonist.
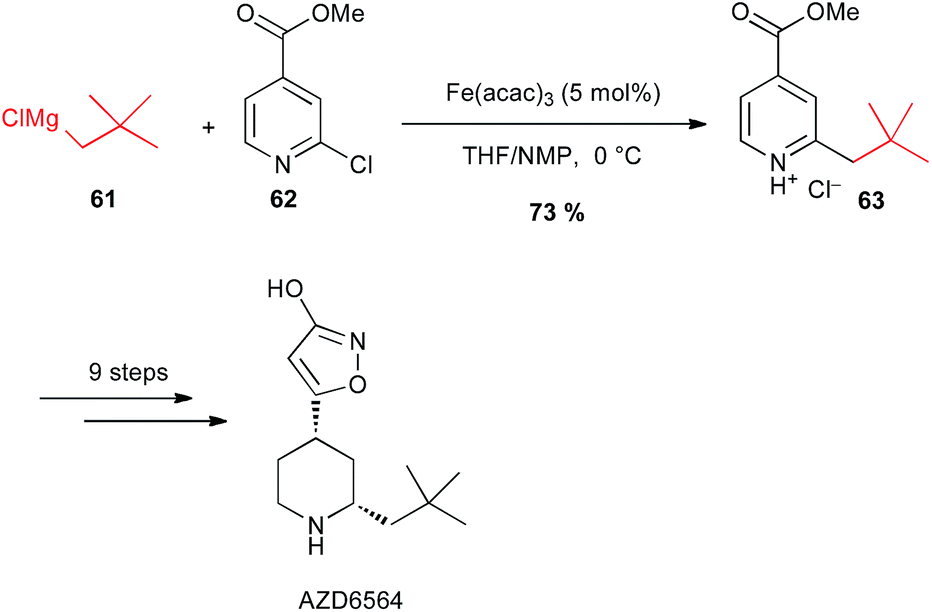
It is worth noting that Sörensen has used a coupling with alkyl Grignard reagent 61 with the 2-chloropyridine 62 under iron catalysis for a large scale preparation (2 kg) of the 3-isoxazolol fibrinolysis inhibitor AZD6564 (Scheme 24).48
Scheme 24 Fe-catalysed coupling of alkyl Grignard with a 2-chloropyridine derivative for the synthesis of fibrinolysis inhibitor AZD6564.
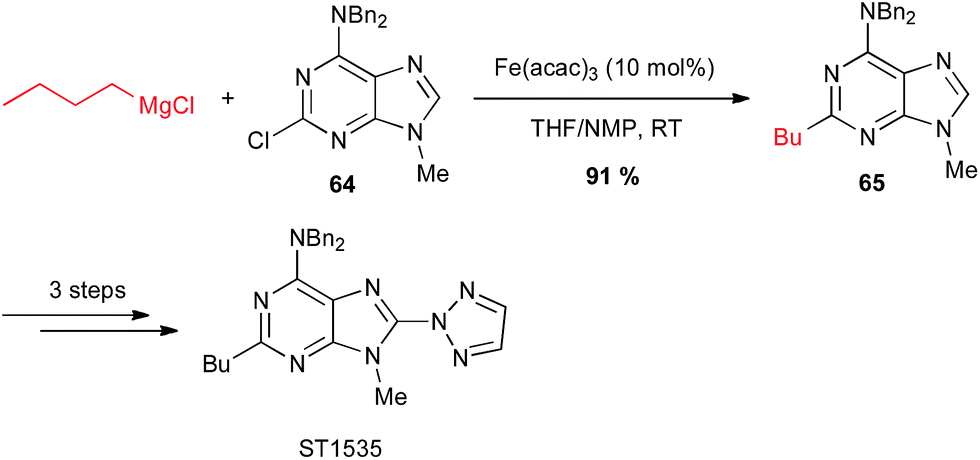
Iron-catalysed alkyl Grignard couplings can be applied to other heteroaryl halides. In particular, Cabri showed that a 2-chloropurine 64 can be coupled with n-butyl Grignard reagent in the presence of Fe(acac)3 in the THF/NMP solvent mixture at 0 °C, to afford a key intermediate 65 in 91% yield in the total synthesis of the adenosine A2A receptor antagonist ST1535 (Scheme 25).49
Scheme 25 Fe-catalysed coupling of alkyl Grignard with a 2-chloropurine derivative for the synthesis of adenosine A2A receptor antagonist ST1535.
2.3 With alkyl (pseudo)halides
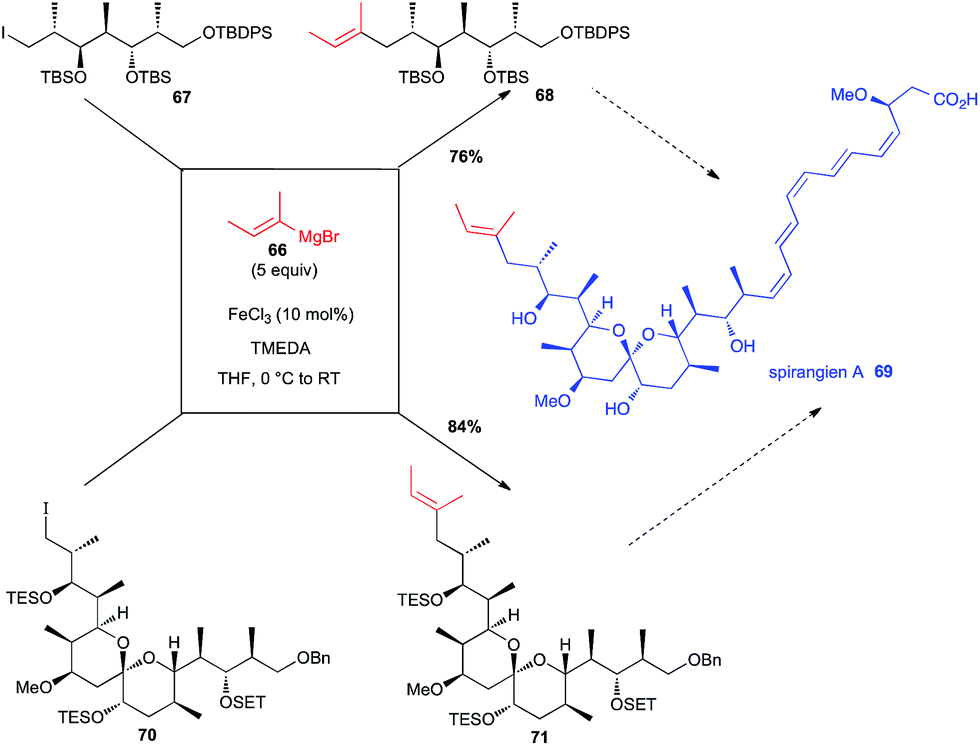
Total synthesis of spirangien A has been in the focus of two different investigations with a Fe-catalysed step to introduce the terminal 2-butenyl fragment. Thus, Cossy has used an iron-catalysed coupling between an alkenyl Grignard reagent 66 and a primary alkyl iodide 67 in an early step of her synthesis of the spiroketal core of spirangien A (76% yield for 68, Scheme 26).50 It is worth noting that this reaction was much more efficient than the Negishi cross-coupling reaction. More recently, Rizzacasa used the same iron-catalysed coupling between 66 and a primary alkyl iodide 70, but in a later step of their formal synthesis of spirangien A, still in good yield (84% yield for 71, Scheme 26).51
Scheme 26 Fe-catalysed coupling of a vinyl Grignard with a primary alkyl iodide for the synthesis of spirangien A.
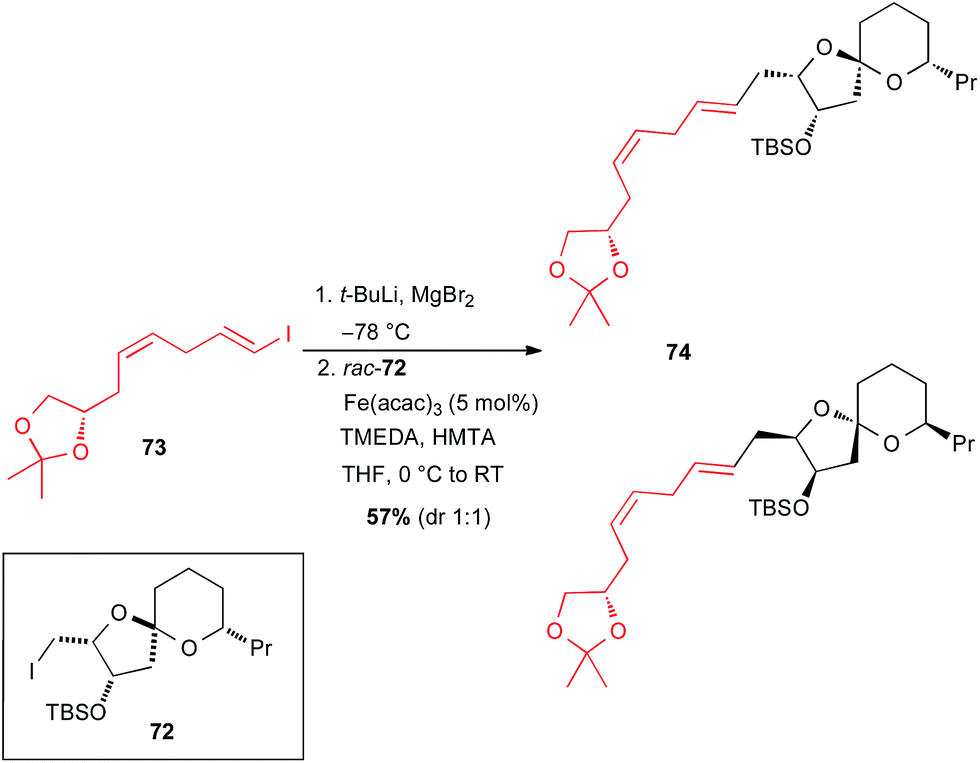
In 2015, Totah and Lam reported the synthesis of a model fragment 74 of spirastrellolide A through an iron catalysed cross-coupling from rac-72 (Scheme 27).52 The vinyl Grignard reagent was generated in situ by I–Li exchange from enantiopure 1-iodo-1,4-diene 73, followed by transmetallation with MgBr2 without any issues, and the Fe-catalysed coupling with rac-72 occurred to afford the target molecule 74 as a mixture of two diastereomers.
Scheme 27 Fe-catalysed coupling of a vinyl Grignard with a primary alkyl iodide for the synthesis of a model fragment of spirastrellolide A.
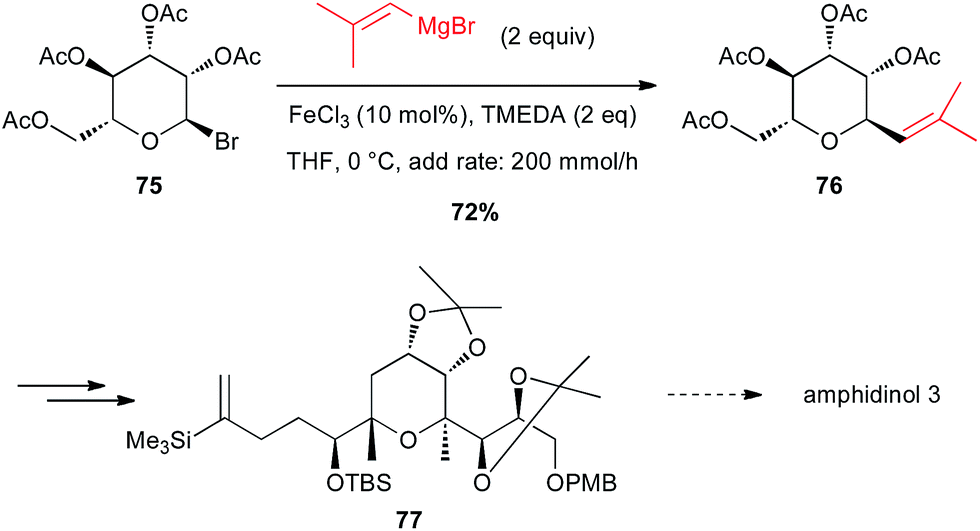
Secondary aliphatic bromides are also good substrates for iron-catalysed couplings with Grignard reagents. In this line, Cossy reported a coupling of a vinyl Grignard with a C-bromopyranoside 75 under Fe-catalysed conditions to prepare a key tetrahydropyrane 76 en route to the total synthesis of amphidinol 3. This cross-coupling exhibits excellent chemo- and diastereoselectivity since it tolerates the presence of acetoxy moieties and the product is obtained in 72% yield with dr = 9:1 (Scheme 28).53
Scheme 28 Fe-catalysed coupling of a vinyl Grignard with a secondary alkyl bromide for the synthesis of a tetrahydropyrane core of amphidinol 3.
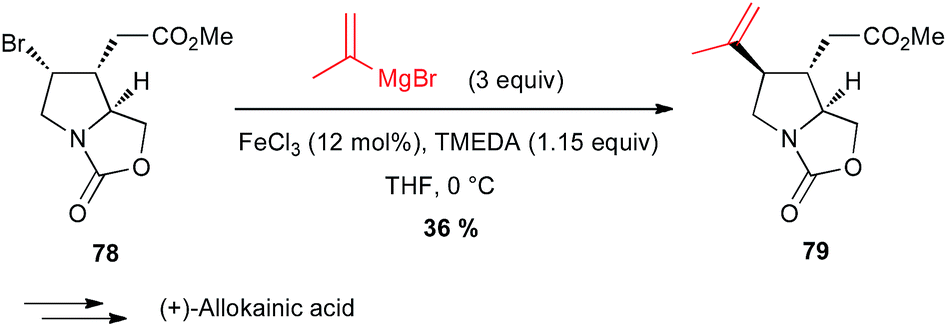
Yamada also reported in their formal total synthesis of (+)-allokainic acid the use of an iron-catalysed coupling between a vinyl Grignard and a secondary bromoalkyl derivative 78, although the yield of 79 remained low (36%) due to competitive opening of the pyrrolidine ring (Scheme 29).54
Scheme 29 Fe-catalysed coupling of a vinyl Grignard with a secondary alkyl bromide for the synthesis of (+)-allokainic acid.
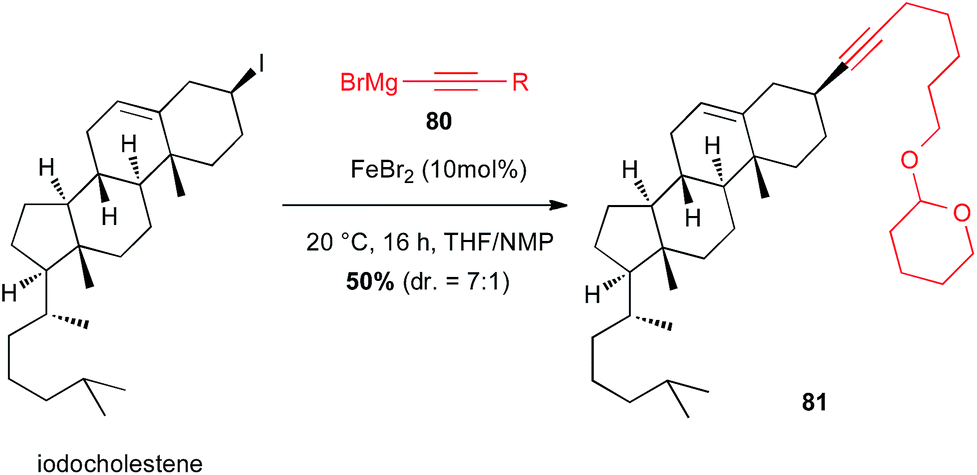
Iodocholestene was successfully reacted with an alkynyl Grignard reagent 80 under iron(II)-catalysis to afford the natural product derived compound 81 in moderate yield (50%) and selectivity favouring retention of configuration (dr = 7:1) (Scheme 30).55
Scheme 30 Fe-catalysed coupling of an alkynyl Grignard with a secondary alkyl iodide for the synthesis of cholestene derivative.
In 2015, an important breakthrough was reported by Nakamura who performed a Fe-catalysed enantioselective cross-coupling of racemic α-chloroesters with aryl Grignard reagents in presence of a C2-symmetric bisphosphine ligand.56 Moreover, this methodology allowed the preparation of the chiral non-racemic NSAID naproxen and dexibuprofen (Scheme 31). The coupling between chloropropionate 82 and the corresponding aryl magnesium bromides afforded the esters 83a and 83b with the same 74% ee for naproxen and dexibuprofen esters, respectively. However, saponification and crystallization with octyl amine allowed significant enantioenrichment to 86% and 98% ee, respectively. On a mechanistic standpoint, a catalytic cycle involving a radical intermediate formed by halogen abstraction and Fe(I) species with a chiral ligand has been proposed.
Scheme 31 Fe-catalysed enantioselective cross-coupling of racemic α-chloroesters with aryl Grignard reagents to prepare naproxen and dexibuprofen.
2.4 With acyl chlorides
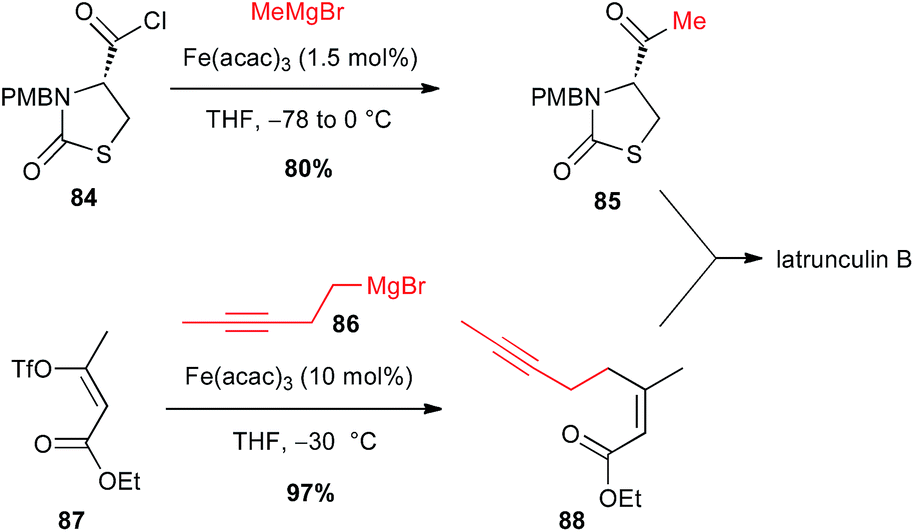
Iron-catalysed processes are also powerful tools to access ketones: Grignard reagents in presence of Fe(acac)3 catalyst were able to perform the formal nucleophilic substitution onto acyl chlorides and thioesters, without further addition to generate the tertiary alcohol, as demonstrated by Marchese.57 In 2003, Fürstner extended this methodology to functionalized sophisticate molecules such as an enantiopure thiazolidinone 84 bearing an acyl chloride moiety, in his synthesis of latrunculin B. The reaction proceeds cleanly, affecting only the acyl group and affording thus the ketone 85 in good 80% yield. Moreover, a second Fe-catalysed step was also involved in this total synthesis between a homopropagylic Grignard reagent 86 and a vinyl triflate 87 (Scheme 32).58
Scheme 32 Fe-catalysed acylation of an alkyl Grignard with an acyl chloride for the synthesis of latrunculin B.
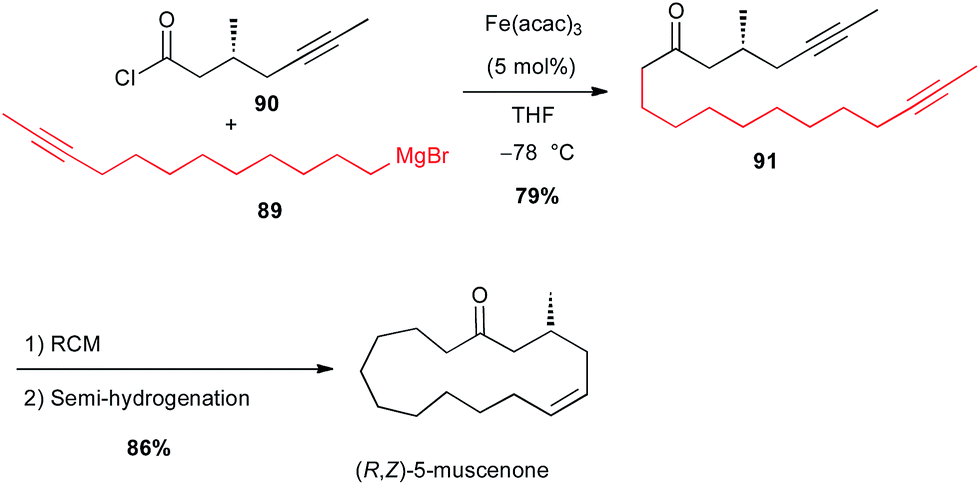
More recently, the same group applied this reaction between a Grignard reagent 89 terminated with an acetylenic moiety and an acyl chloride 90 to prepare a precursor 91 of (R,Z)-muscenone (Scheme 33).59
Scheme 33 Fe-catalysed acylation of an alkyl Grignard with an acyl chloride for the synthesis of (R,Z)-muscenone.
2.5 Ring opening
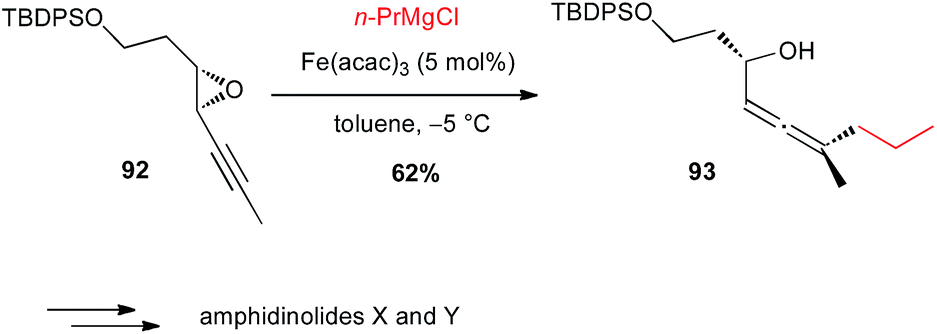
Propargyl epoxides. The addition of Grignard reagents onto enantiopure propargyl epoxides has also been shown by Fürstner to be a very elegant manner to obtain optically active allenols though SN2′ process. Thus, this Fe(acac)3-catalysed reaction proceeded with good C1 to axial chirality transfer (62% yield, dr > 8:1) for the preparation of a syn allenol 93, which was a key intermediate in the total synthesis of amphidinolides X and Y, with regards to the oxygenated ring systems (Scheme 34 and Fig. 1).60,61
Scheme 34 Fe-catalysed coupling of propyl magnesium chloride with a propargyl epoxide for the synthesis of amphidinolides X and Y.
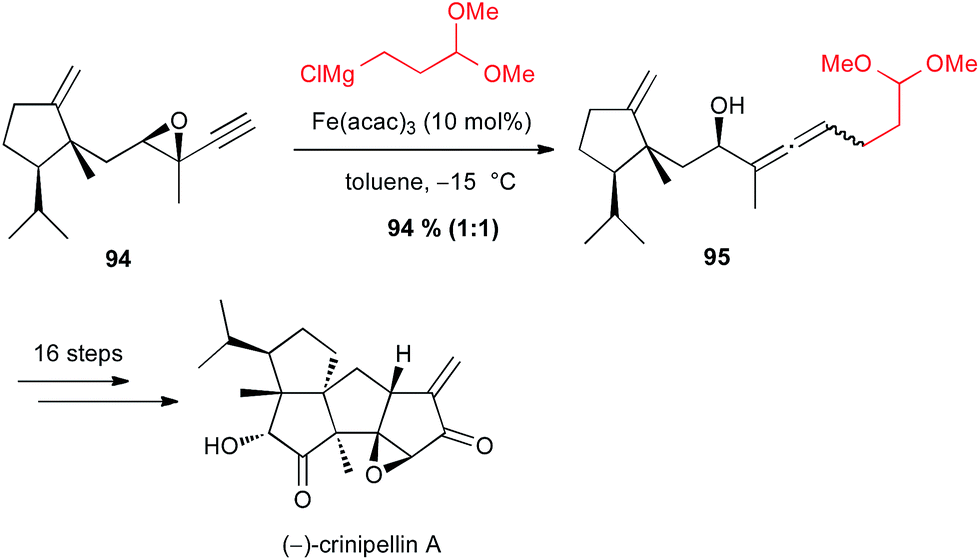
On the contrary, Lee found that iron-catalyzed SN2′ opening of an intermediate epoxyalkyne 94 in their synthesis of (−)-crinipellin A was not diastereoselective, since 95 was obtained in 94% yield as a 1:1 mixture of two diastereomers (Scheme 35).62
Scheme 35 Fe-catalysed coupling of a Grignard reagent with a propargyl epoxide for the synthesis of (−)-crinipellin A.
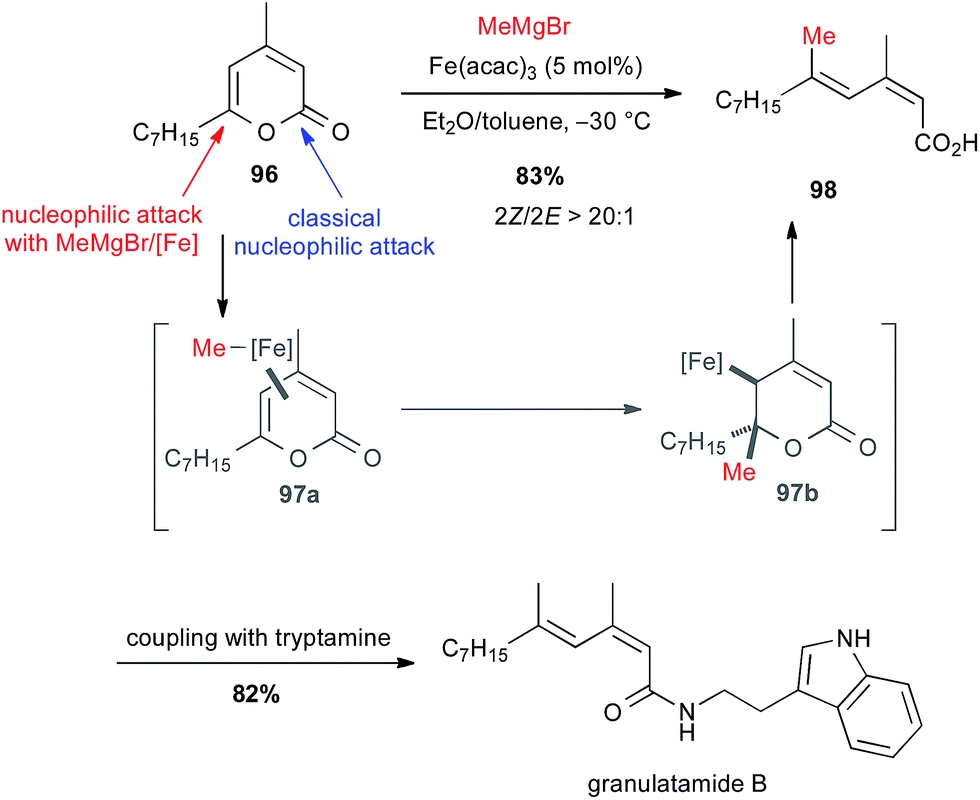
2-Pyrones. The ring opening of 2-pyrones with nucleophiles classically occurs on the carbonyl group. In contrast, the reaction between 2-pyrones and Grignard reagents with Fe(acac)3 in toluene or ether solvent (or a mixture of both) leads to the formation of 2,4-dienoic acids with selective 2Z configuration.63 Thus, pyrone 96 reacted under these conditions to give dienoic acid 98 (2Z/2E > 20:1) in 83% yield. This latter, after peptidic coupling with tryptamine, leads to granulatamide B, a cytotoxic compound found in Eunicella granulate (Scheme 36).
Scheme 36 Fe-catalysed ring-opening/cross-coupling with methyl Grignard and 2-pyrones for the synthesis of granulatamide B.
The mechanism of this cross-coupling is supposed to go through a π-complex 97a followed by conjugate addition of methyl iron on the γ,δ-olefin leading to intermediate 97b, which affords by β-elimination the target compound 98 with desired geometry.
3. Lewis acid catalysis
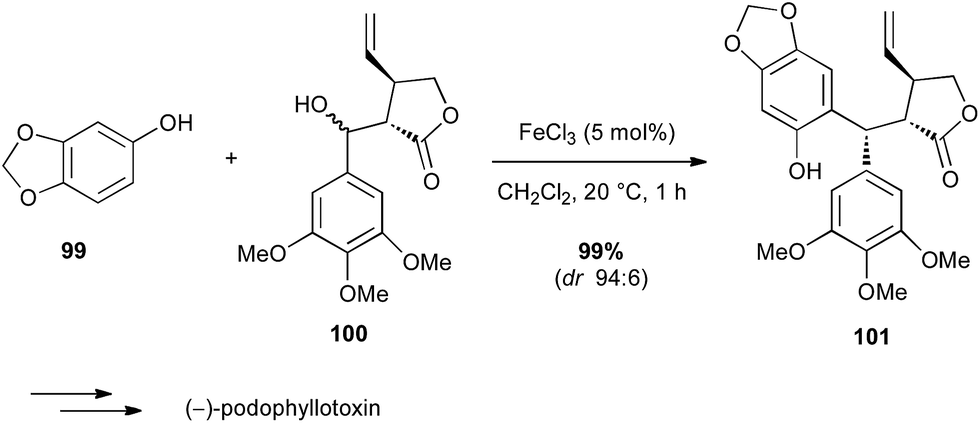
Iron salts are much known as one of the first Lewis acids used in the Friedel–Crafts reaction. Bach used FeCl3 in an intermolecular Friedel–Crafts reaction between a phenol derivative 99 and a functionalized benzyl alcohol 100 in the total synthesis of (−)-podophyllotoxin (Scheme 37).64 Interestingly, FeCl3 was by far the best catalyst for conversion and diastereoselectivity among several acids assessed (HBF4, AuCl3,…) leading to the target compound in almost quantitative yield and excellent dr.
Scheme 37 Fe-catalysed Friedel–Crafts reaction with a benzyl alcohol for the synthesis of (−)-podophyllotoxin.
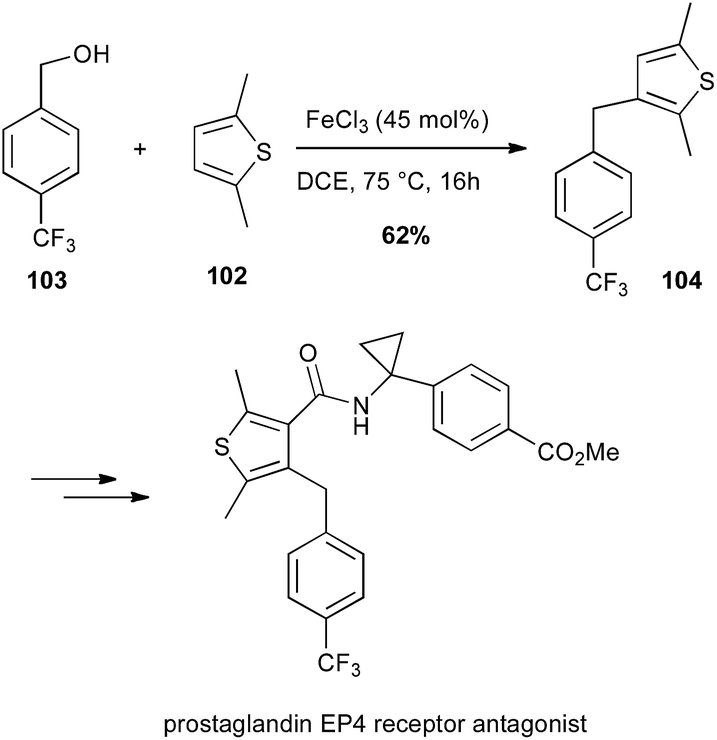
The same iron catalyst was also successfully used in the coupling of 2,5-dimethylthiophene 102 with benzylic alcohol derivative 103 for the total synthesis of a prostaglandin EP4 receptor antagonist (Scheme 38).65
Scheme 38 Fe-catalysed coupling of 2,5-dimethylthiophene with benzyl alcohol for the synthesis of a prostaglandin EP4 receptor antagonist.
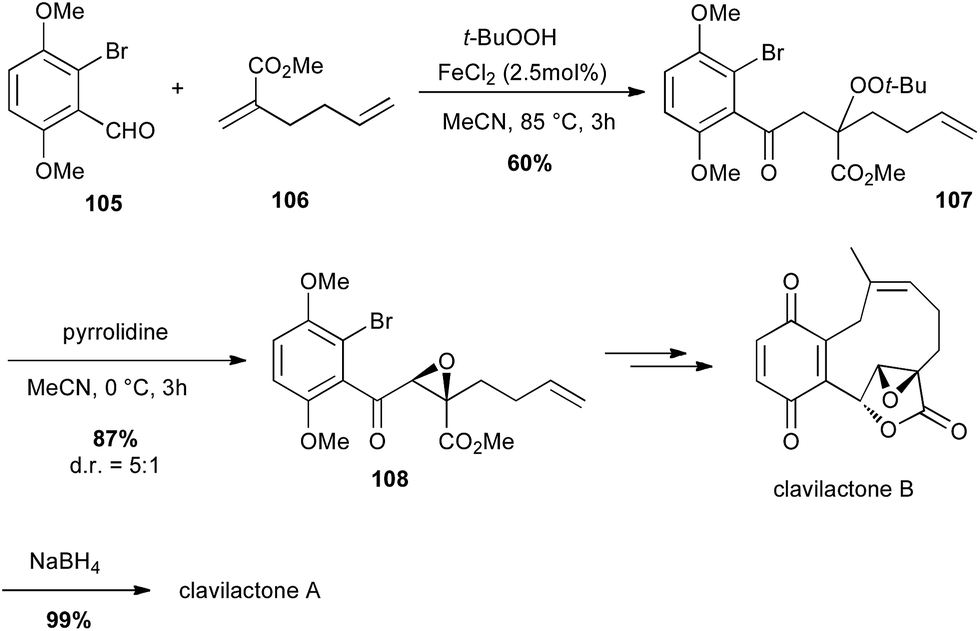
In 2010, Li described an interesting carbonylation–peroxidation of alkenes with aldehydes and hydroperoxides under 3-component conditions. The obtained β-peroxyketones can then offer an easy access to α-keto epoxides under basic conditions.66 The same authors used this method to prepare a key intermediate in the total synthesis of racemic clavilactones A and B: the β-peroxyketone 107 was prepared (60%) and pyrrolidine treatment led to a syn epoxide 108 in very good yield (87%) and dr (5:1), which allowed them to obtain the key lactone fragment of clavilactones (Scheme 39).67
Scheme 39 Fe-catalysed carbonylation–peroxidation of an olefin for the synthesis of (+/−)-clavilactones A and B.
4. Carbocyclisation
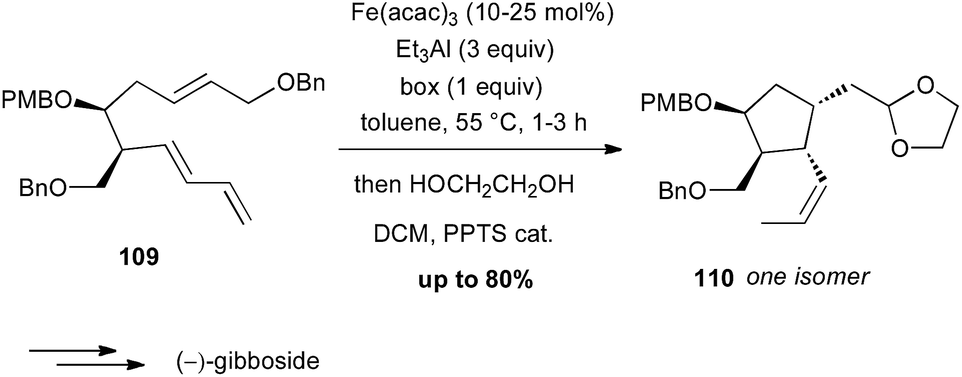
During the 1980's, Takacs reported a powerful carbocyclisation proceeding through an intramolecular Fe(0)-catalysed ene reaction between allylic ethers and diene moieties.68 In these reactions, Fe(0) is generated in situ from Fe(acac)3, Et3Al as a reductant and a ligand, such as bipyridine (bipy) or a bisoxazoline (box). In 2002, this methodology was successfully applied to the diastereoselective preparation of the key cyclopentane core of (−)-gibboside in the total synthesis of this natural iridoid glucoside isolated from Patrinia gibbosa (Scheme 40).69 The reaction is very sensitive to impurities and the yield varied significantly (40–80%), but always affording 110 as a single isomer.
Scheme 40 Fe-catalysed enediene cyclisation for the synthesis of (−)-gibboside.
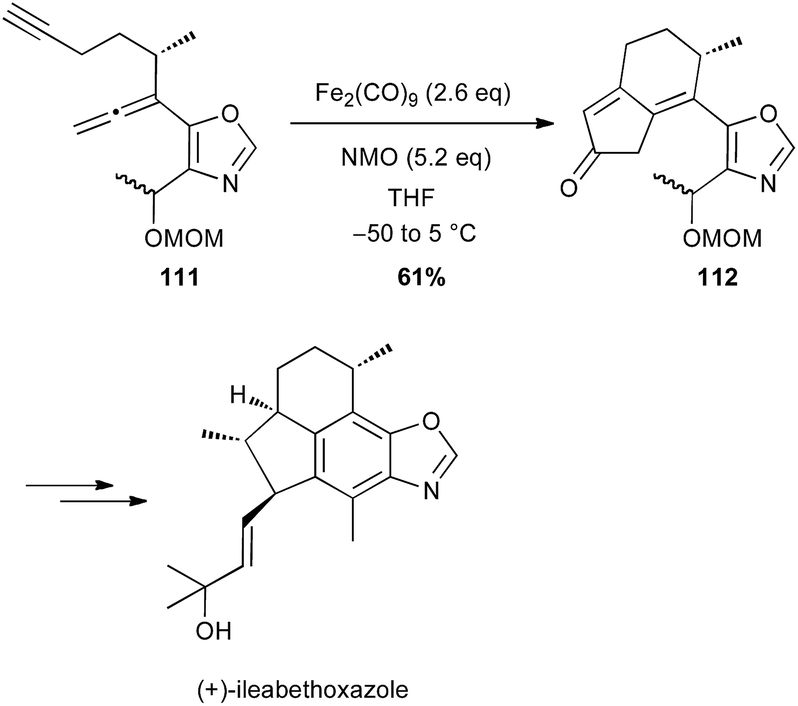
The Pauson–Khand reaction (PK) is also a powerful tool for synthesizing cyclopentenone systems.70 While this [2 + 2 + 1] carbocyclisation is generally performed with Co2(CO)8 or Mo(CO)6 in stoichiometric amounts or as catalysts under CO atmosphere, some limitations are observed with highly functionalised substrates. In contrast, Williams and Baik have shown that Fe2(CO)9 (2.6 equiv.) is an efficient promoter for PK reaction between allenes and alkynes, which tolerates various functional groups.71 Shah and Williams have thus been able to perform the PK reaction on a substrate 111 bearing an oxazole moiety (61% yield), affording a precursor 112 of (+)-ileabethoxazole (Scheme 41).72 Interestingly only the metal complex Fe2(CO)9 was able to promote the carbocyclisation with this substrate.
Scheme 41 Fe-promoted Pauson–Khand reaction for the synthesis of (+)-ileabethoxazole.
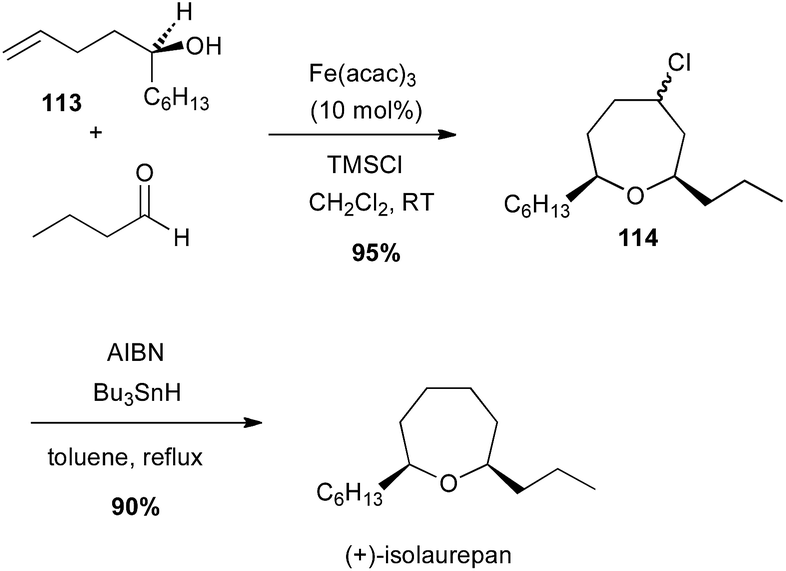
Prins-type cyclisation generally occurs between homoallylic alcohols and an aldehyde under Brønsted or Lewis acidic conditions. In 2009, Padrón and Martín reported a catalytic Prins cyclisation from homoallyl amines or alcohols with catalytic iron(III) salt and stoichiometric TMSX leading to haloheterocycles.73 Thus, an enantiopure tertiary bis-homoallylic alcohol 113 reacted with butyraldehyde in presence of Fe(acac)3 (10 mol%) and TMSCl (likely generating FeCl3in situ) to afford the corresponding chlorooxepan 114 (95% yield), which after reduction led to (+)-isolaurepan (Scheme 42).74
Scheme 42 Fe-catalysed Prins cyclisation in the synthesis of (+)-isolaurepan.
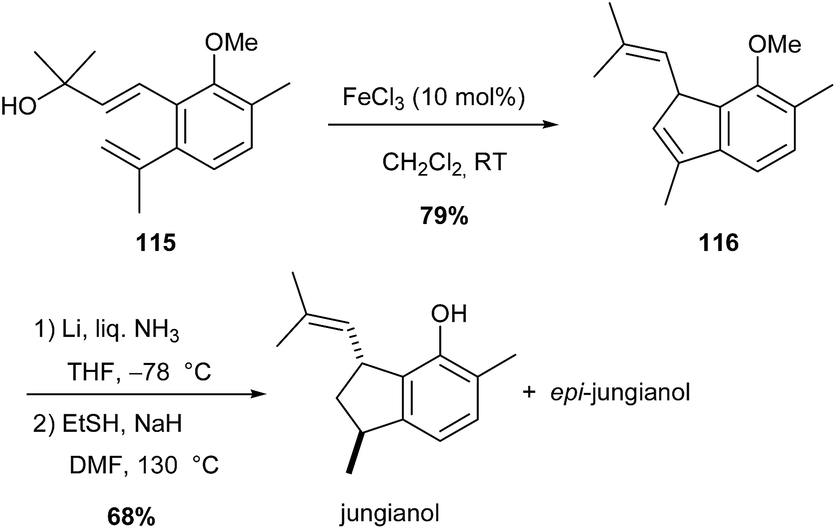
Analogous iron(III)-catalysed Prins-type cyclisation was also used to access an indene molecule 116 (79% yield), which after demethylation and reduction afforded jungianol and its epimer (Scheme 43).75
Scheme 43 Fe-catalysed Prins cyclisation in the synthesis of jungianol.
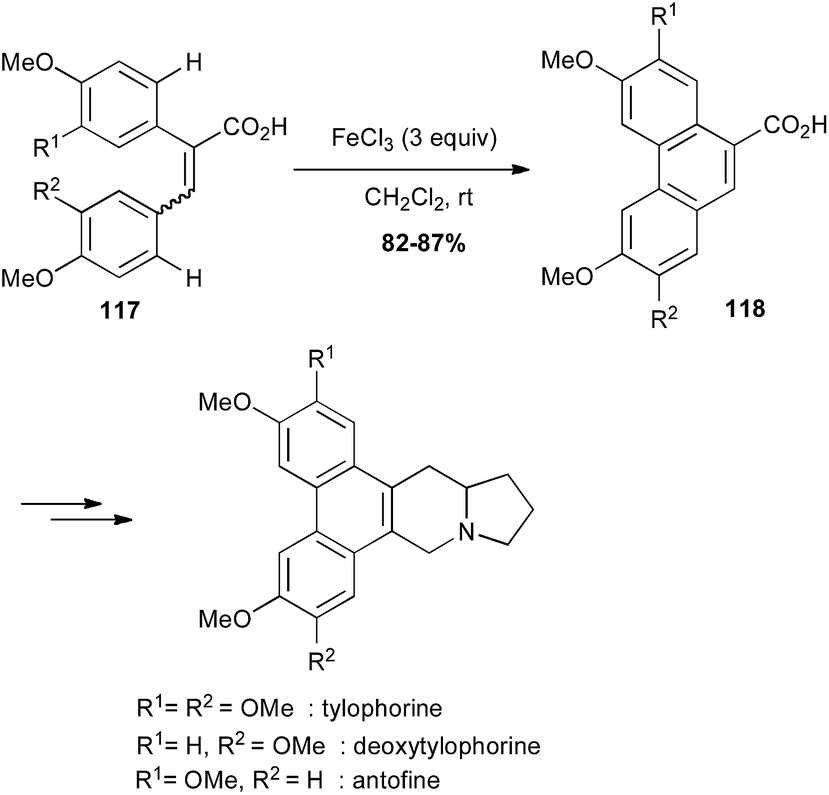
Oxidative coupling promoted by iron(III) chloride is a powerful method for C–C bond formation between two arene moieties.76 Thus, iron-mediated oxidative cyclisation of bi-aryl compounds 117 has been used for the preparation of the phenanthrene core of several alkaloids, which were prepared in a few more steps (Scheme 44).77
Scheme 44 Fe-catalysed oxidative cyclisation of a biaryl for the synthesis of tylophorine, deoxytylophorine and antofine.
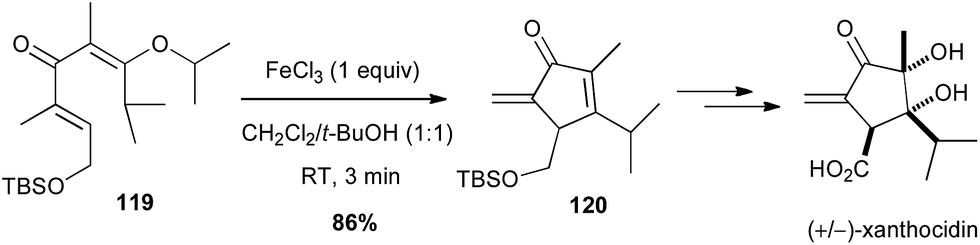
A modified Nazarov reaction on a divinyl ketone 119 promoted by iron trichloride (1 equiv.) allowed Shindo to obtain the key α-exo-methylene cyclopentadienone 120 in high yield (86%), en route to the total synthesis of (+/−)-xanthocidin, an antibiotic isolated from Streptomyces xanthocidicus (Scheme 45).78
Scheme 45 Fe-promoted Nazarov cyclisation of a divinyl ketone for the synthesis of (+/−)-xanthocidin.
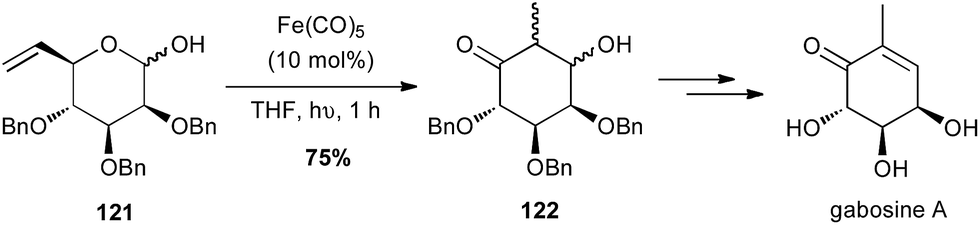
Grée reported in 2011 that an intramolecular tandem Fe(CO)5-catalysed isomerisation–aldolisation reaction starting from vinyl pyranose 121 allowed a short and elegant synthesis of natural products from the gabosine family (Scheme 46).79
Scheme 46 Fe-promoted isomerisation–aldolisation of a vinyl pyranose for the synthesis of gabosine A.
5. C–H activation
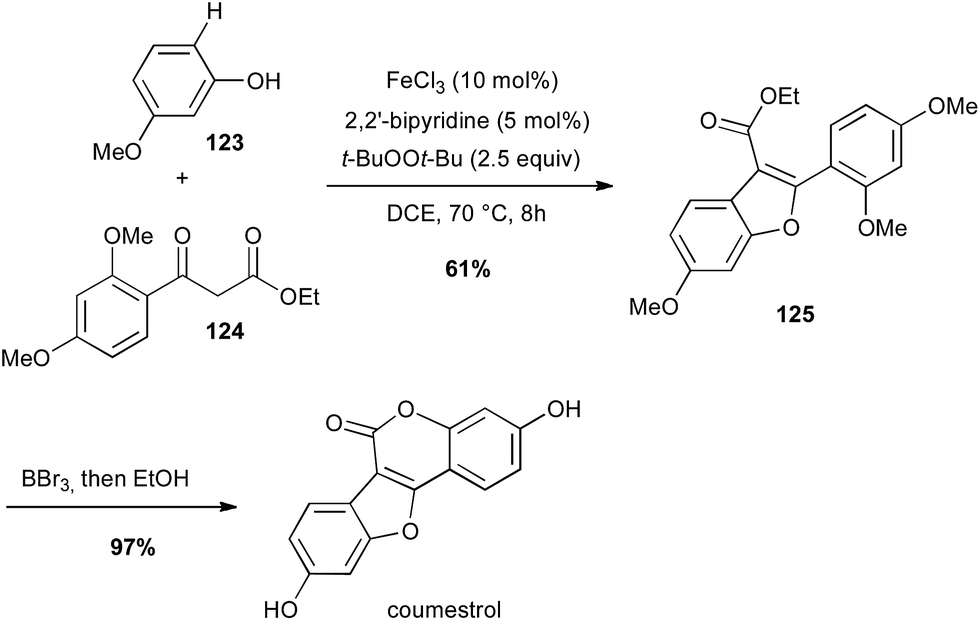
Iron has also been used for direct C–H activation. For instance, cross-dehydrogenative coupling (CDC) between sp2 C–H bond of a phenol 123 in the presence of a β-keto ester 124 and FeCl3 allowed the preparation in a gram-scale of the benzofuran core of coumestrol, a natural estrogenic analogue, in good yield (Scheme 47).80
Scheme 47 Fe-catalysed coupling of a phenol with β-keto ester for the synthesis of coumestrol.
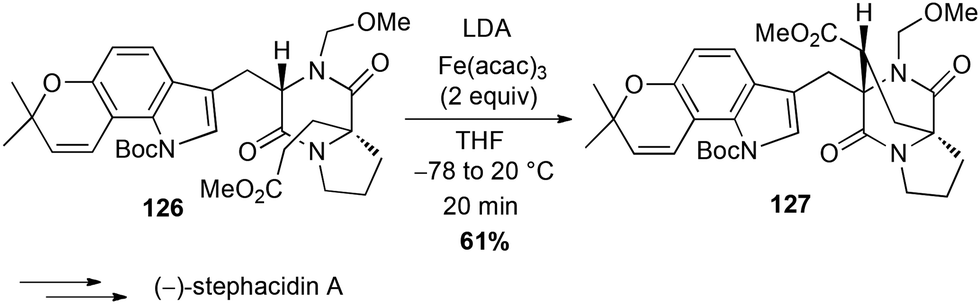
In 2006, Baran reported a formal intramolecular coupling between two sp3 C–H bonds albeit the reaction goes through oxidative heterocoupling of two types of enolates. Enolates are generated in presence of LDA and the coupling takes place in presence of an oxidant, among which Fe(acac)3 (2 equiv.) proved to be very efficient (61% yield of 127). Thus, this method allowed the preparation in a gram-scale of the polycyclic core of (−)-stephacidin A, a natural estrogenic analogue (Scheme 48).81
Scheme 48 Fe-catalysed oxidative coupling for the synthesis of (−)-stephacidin A.
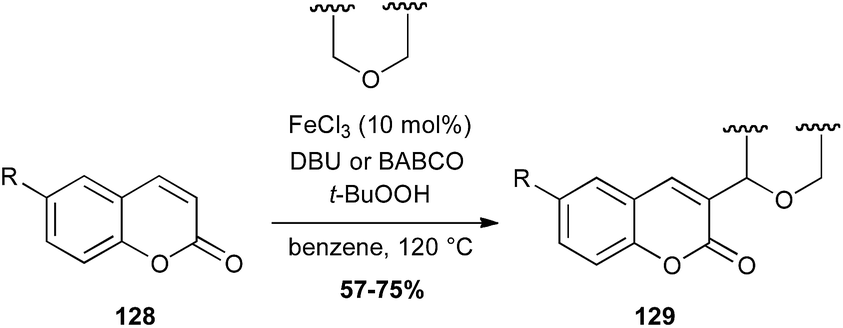
Coumarin is a fragrant compound found in many plants. In 2015, an interesting radical coupling between coumarins 128 and ethers catalyzed by iron chloride in presence of TBHP has been described. This reaction allows the formation of a C–C bond between an sp2 carbon of the coumarin and the carbon in α-position of the ether (Scheme 49).82
Scheme 49 Fe-catalysed coupling of coumarins with ethers.
6. Conclusions
Iron-catalysed reactions are nowadays-key tools in the total synthesis of natural products and pharmacologically important chemical entities. While the cross-coupling reaction between Grignard reagents and numerous electrophiles is well documented and constitutes the most visible facet, iron catalysis is only at the dawn of its development. Innovative and powerful methodology, such as Fe-catalysed C–H activation, is still an under-explored field, which will become shortly a source of new applications, especially for the large-scale preparation of highly functionalised molecules. Moreover, the number of enantioselective transformations with iron is still low compared to other metals and such methodologies are definitely required in total synthesis. Attractiveness of iron salts relies for sure on their cost and their non-toxic property, but efficiency and selectivity observed in these chemical transformations are also the driving force of the efforts brought in these studies.
7. Acknowledgements
J. L. is grateful to Labex SynOrg (ANR-11-LABX-0029), the Région Haute-Normandie and the European France (Manche) – England cross-border cooperation program INTERREG IV A “AI-CHEM CHANNEL” and co-financed by ERDF for financial support. Labex LERMIT (ANR-10-LABX-33) is acknowledged for financial support (BF).
8. References
P. Enghag, Encyclopedia of the elements technical data, history, processing, applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
J. W. Morgan and E. Anders, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 6973–6977 CrossRefCAS.
G. M. Yee and W. B. Tolman, in Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases, ed. P. M. H. Kroneck and M. E. S. Torres, Springer International Publishing, 2015, pp. 131–204 Search PubMed.
M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed.
C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRefCASPubMed.
I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRefCASPubMed.
B. Plietker, Iron catalysis in organic chemistry: reactions and applications, Wiley-VCH, Weinheim, 2008 Search PubMed.
A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 1364–1367 CrossRefPubMed.
Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2004 Search PubMed.
M. Tamura and J. Kochi, Synthesis, 1971, 303–305 CrossRefCAS.
M. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487–1489 CrossRefCAS.
G. Cahiez and H. Avedissian, Synthesis, 1998, 1199–1205 CrossRefCAS.
B. Bogdanović and M. Schwickardi, Angew. Chem., Int. Ed., 2000, 39, 4610–4612 CrossRef.
A. Fürstner, H. Krause and C. W. Lehmann, Angew. Chem., Int. Ed., 2006, 45, 440–444 CrossRefPubMed.
A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773–8787 CrossRefPubMed.
Q. Ren, S. Guan, F. Jiang and J. Fang, J. Phys. Chem. A, 2013, 117, 756–764 CrossRefCASPubMed.
R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804–1808 CrossRefCASPubMed.
A. Hedström, Z. Izakian, I. Vreto, C.-J. Wallentin and P.-O. Norrby, Chem.–Eur. J., 2015, 21, 5946–5953 CrossRefPubMed.
R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485–1493 CrossRefCASPubMed.
B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRefCASPubMed.
W. M. Czaplik, M. Mayer, S. Grupe and A. J. von Wangelin, Pure Appl. Chem., 2010, 82, 1545–1553 CrossRefCAS.
J. Defretin, C. Gleye, D. Cortes, X. Franck, R. Hocquemiller and B. Figadere, Lett. Org. Chem., 2004, 1, 316–322 CrossRefCAS.
A. A. Camacho-Davila, Synth. Commun., 2008, 38, 3823–3833 CrossRefCASPubMed.
D. Castagnolo and M. Botta, Eur. J. Org. Chem., 2010, 2010, 3224–3228 CrossRefPubMed.
N. Tewari, N. Maheshwari, R. Medhane, H. Nizar and M. Prasad, Org. Process Res. Dev., 2012, 16, 1566–1568 CrossRefCAS.
R. N. Shakhmaev, A. S. Sunagatullina and V. V. Zorin, Russ. J. Org. Chem., 2014, 50, 322–331 CrossRefCAS.
B. Scheiper, M. Bonnekessel, H. Krause and A. Fürstner, J. Org. Chem., 2004, 69, 3943–3949 CrossRefCASPubMed.
A. Fürstner and P. Hannen, Chem.–Eur. J., 2006, 12, 3006–3019 CrossRefPubMed.
A. Hamajima and M. Isobe, Org. Lett., 2006, 8, 1205–1208 CrossRefCASPubMed.
J. Boukouvalas, V. Albert, R. P. Loach and R. Lafleur-Lambert, Tetrahedron, 2012, 68, 9592–9597 CrossRefCASPubMed.
Y. Liang, X. Jiang and Z.-X. Yu, Chem. Commun., 2011, 47, 6659–6661 RSC.
K. C. Nicolaou, Y.-P. Sun, H. Korman and D. Sarlah, Angew. Chem., Int. Ed., 2010, 49, 5875–5878 CrossRefCASPubMed.
A. Fürstner and A. Schlecker, Chem.–Eur. J., 2008, 14, 9181–9191 CrossRefPubMed.
G. Cahiez, O. Gager and V. Habiak, Synthesis, 2008, 2636–2644 CrossRefCAS.
G. Cahiez, V. Habiak and O. Gager, Org. Lett., 2008, 10, 2389–2392 CrossRefCASPubMed.
J. R. Scheerer, J. F. Lawrence, G. C. Wang and D. A. Evans, J. Am. Chem. Soc., 2007, 129, 8968–8969 CrossRefCASPubMed.
M. A. Fakhfakh, X. Franck, R. Hocquemiller and B. Figadère, J. Organomet. Chem., 2001, 624, 131–135 CrossRefCAS.
M. Dos Santos, X. Franck, R. Hocquemiller, B. Figadere, J.-F. Peyrat, O. Provot, J.-D. Brion and M. Alami, Synlett, 2004, 2697–2700 CAS.
K. Lehr, S. Schulthoff, Y. Ueda, R. Mariz, L. Leseurre, B. Gabor and A. Fürstner, Chem.–Eur. J., 2015, 21, 219–227 CrossRefCASPubMed.
M. Fuchs and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 3978–3982 CrossRefCASPubMed.
L. K. Ottesen, F. Ek and R. Olsson, Org. Lett., 2006, 8, 1771–1773 CrossRefCASPubMed.
A. Fürstner and A. Leitner, Angew. Chem., Int. Ed., 2002, 41, 609–612 CrossRef.
G. Seidel, D. Laurich and A. Fürstner, J. Org. Chem., 2004, 69, 3950–3952 CrossRefCASPubMed.
A. Fürstner and A. Leitner, Angew. Chem., Int. Ed., 2003, 42, 308–311 CrossRefPubMed.
B. Scheiper, F. Glorius, A. Leitner and A. Fürstner, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11960–11965 CrossRefCASPubMed.
U. Groenhagen, M. Maczka, J. S. Dickschat and S. Schulz, Beilstein J. Org. Chem., 2014, 10, 1421–1432 CrossRefCASPubMed.
C. Risatti, K. J. Natalie, Z. Shi and D. A. Conlon, Org. Process Res. Dev., 2013, 17, 257–264 CrossRefCAS.
S. M. Andersen, M. Bollmark, R. Berg, C. Fredriksson, S. Karlsson, C. Liljeholm and H. Sörensen, Org. Process Res. Dev., 2014, 18, 952–959 CrossRefCAS.
F. Bartoccini, G. Piersanti, S. Armaroli, A. Cerri and W. Cabri, Tetrahedron Lett., 2014, 55, 1376–1378 CrossRefCASPubMed.
A. Guerinot, G. Lepesqueux, S. Sable, S. Reymond and J. Cossy, J. Org. Chem., 2010, 75, 5151–5163 CrossRefCASPubMed.
C. Gregg, C. Gunawan, A. W. Y. Ng, S. Wimala, S. Wickremasinghe and M. A. Rizzacasa, Org. Lett., 2013, 15, 516–519 CrossRefCASPubMed.
T. Lam and N. I. Totah, Tetrahedron Lett., 2015, 56, 3349–3352 CrossRefCASPubMed.
C. Bensoussan, N. Rival, G. Hanquet, F. Colobert, S. Reymond and J. Cossy, Tetrahedron, 2013, 69, 7759–7770 CrossRefCASPubMed.
K. Yamada, T. Sato, M. Hosoi, Y. Yamamoto and K. Tomioka, Chem. Pharm. Bull., 2010, 58, 1511–1516 CrossRefCAS.
C. W. Cheung, P. Ren and X. Hu, Org. Lett., 2014, 16, 2566–2569 CrossRefCASPubMed.
M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128–7134 CrossRefCASPubMed.
V. Fiandanese, G. Marchese, V. Martina and L. Ronzini, Tetrahedron Lett., 1984, 25, 4805–4808 CrossRefCAS.
A. Fürstner, D. De Souza, L. Parra-Rapado and J. T. Jensen, Angew. Chem., Int. Ed., 2003, 42, 5358–5360 CrossRefPubMed.
K. Lehr and A. Fürstner, Tetrahedron, 2012, 68, 7695–7700 CrossRefCASPubMed.
O. Lepage, E. Kattnig and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 15970–15971 CrossRefCASPubMed.
A. Fürstner, E. Kattnig and O. Lepage, J. Am. Chem. Soc., 2006, 128, 9194–9204 CrossRefPubMed.
T. Kang, S. B. Song, W.-Y. Kim, B. G. Kim and H.-Y. Lee, J. Am. Chem. Soc., 2014, 136, 10274–10276 CrossRefCASPubMed.
C.-L. Sun and A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 13071–13075 CrossRefCASPubMed.
D. Stadler and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 7557–7559 CrossRefCASPubMed.
D. Gauvreau, S. J. Dolman, G. Hughes, P. D. O'Shea and I. W. Davies, J. Org. Chem., 2010, 75, 4078–4085 CrossRefCASPubMed.
W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRefCASPubMed.
L. Lv, B. Shen and Z. Li, Angew. Chem., Int. Ed., 2014, 53, 4164–4167 CrossRefCASPubMed.
J. M. Takacs and L. G. Anderson, J. Am. Chem. Soc., 1987, 109, 2200–2202 CrossRefCAS.
J. M. Takacs, S. Vayalakkada, S. J. Mehrman and C. L. Kingsbury, Tetrahedron Lett., 2002, 43, 8417–8420 CrossRefCAS.
K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263–3283 CrossRefCAS.
D. R. Williams, A. A. Shah, S. Mazumder and M.-H. Baik, Chem. Sci., 2013, 4, 238–247 RSC.
D. R. Williams and A. A. Shah, J. Am. Chem. Soc., 2014, 136, 8829–8836 CrossRefCASPubMed.
P. O. Miranda, R. M. Carballo, V. S. Martín and J. I. Padrón, Org. Lett., 2009, 11, 357–360 CrossRefCASPubMed.
M. A. Purino, M. A. Ramírez, A. H. Daranas, V. S. Martín and J. I. Padrón, Org. Lett., 2012, 14, 5904–5907 CrossRefCASPubMed.
D. H. Dethe and G. Murhade, Org. Lett., 2013, 15, 429–431 CrossRefCASPubMed.
A. A. O. Sarhan and C. Bolm, Chem. Soc.
Rev., 2009, 38, 2730 RSC.
K.-L. Wang, M.-Y. Lü, Q.-M. Wang and R.-Q. Huang, Tetrahedron, 2008, 64, 7504–7510 CrossRefCASPubMed.
K. Yaji and M. Shindo, Tetrahedron, 2010, 66, 9808–9813 CrossRefCASPubMed.
D. H. Mac, R. Samineni, A. Sattar, S. Chandrasekhar, J. S. Yadav and R. Grée, Tetrahedron, 2011, 67, 9305–9310 CrossRefCASPubMed.
U. A. Kshirsagar, R. Parnes, H. Goldshtein, R. Ofir, R. Zarivach and D. Pappo, Chem.–Eur. J., 2013, 19, 13575–13583 CrossRefCASPubMed.
P. S. Baran, B. D. Hafensteiner, N. B. Ambhaikar, C. A. Guerrero and J. D. Gallagher, J. Am. Chem. Soc., 2006, 128, 8678–8693 CrossRefCASPubMed.
B. Niu, W. Zhao, Y. Ding, Z. Bian, C. U. Pittman, A. Zhou and H. Ge, J. Org. Chem., 2015, 80, 7251–7257 CrossRefCASPubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 






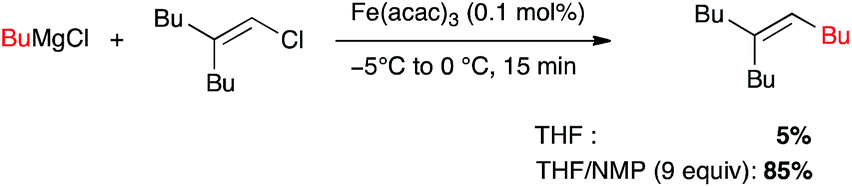
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: