
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biology of 1,4-benzodioxane lignan natural products
Lisa I.
Pilkington
and
David
Barker
*
School of Chemical Sciences, University of Auckland, New Zealand. E-mail: d.barker@auckland.ac.nz
First published on 7th July 2015
Abstract
Covering: up to 2015
Lignan-derived 1,4-benzodioxane natural products have been shown to exhibit a diverse array of biological activities, which has lent them to be the focus of a wealth of synthetic attention. Herein we review the background, bioactivities, biosynthesis and synthetic approaches to the 1,4-benzodioxane lignan scaffold, with an emphasis on 1,4-benzodioxane oxyneolignans.
 Lisa I. Pilkington | Lisa I. Pilkington was born in Auckland, New Zealand. She graduated in 2010 from the University of Auckland with a BA/BSc conjoint degree majoring in Chemistry, Statistics and German. Lisa then went on to graduate with a BSc (Honours, First Class) in 2011, followed by a PhD in 2015 from the same university under the supervision of Dr David Barker, working on the asymmetric synthesis of 1,4-benzodioxane lignans. She is currently undertaking postdoctoral research with the Auckland Cancer Society Research Centre and the School of Chemical Sciences at the University of Auckland. |
 David Barker | David Barker was born in Altrincham, UK. After moving to Australia, he graduated from the University of Sydney with a BSc degree (Honours, First Class) and then completed his PhD in 2002 at the same university. After post-doctoral research at the School of Medical Sciences at the University of New South Wales working with Prof. Larry Wakelin, in 2004 he joined the University of Auckland as a lecturer. He is currently Senior Lecturer in Organic and Medicinal Chemistry and he has a diverse range of synthetic interests including biologically active natural products, drug discovery and development of novel polymeric scaffolds. |
1 Introduction
1,4-Benzodioxane lignans are a group of natural products that exhibit a wide range of interesting biological activities which has resulted in them receiving much synthetic attention over the years. The intent of this review is to provide detailed analysis of the background, biological activity, biosynthesis and the various methods developed towards the synthesis of these compounds. There has previously been limited discussion, and review, of these natural products,1,2 as such, it was decided to produce a comprehensive review on these previous syntheses of 1,4-benzodioxane lignans.The syntheses of 1,4-benzodioxane compounds can be categorised into groups subject to their general approach to construct the 1,4-benzodioxane structure. The aim of the synthetic section of this review is to provide an overview of these approaches to the synthesis of 1,4-benzodioxanes, showing the main methodologies to form these compounds, as well as other novel approaches that have been attempted. The intent is to particularly highlight the synthesis of natural 1,4-benzodioxane oxyneolignans and analogues thereof. There is reference, however to studies towards other 1,4-benzodioxane-containing natural products where appropriate.
1.1 Nomenclature and numbering
1,4-Benzodioxanes are subject to two different numbering systems mandated by traditional and lignan IUPAC nomenclature (Fig. 1). In this review, 1,4-benzodioxane compounds that do not have carbon sidechains representing C-7′ to C-9′ (e.g.1), are numbered in accordance to traditional IUPAC conventions. Compounds that do have a three carbon sidechain (e.g. eusiderin A (2)) are classified as lignan compounds and as such, each lignan fragment is numbered from C-1 to C-9.3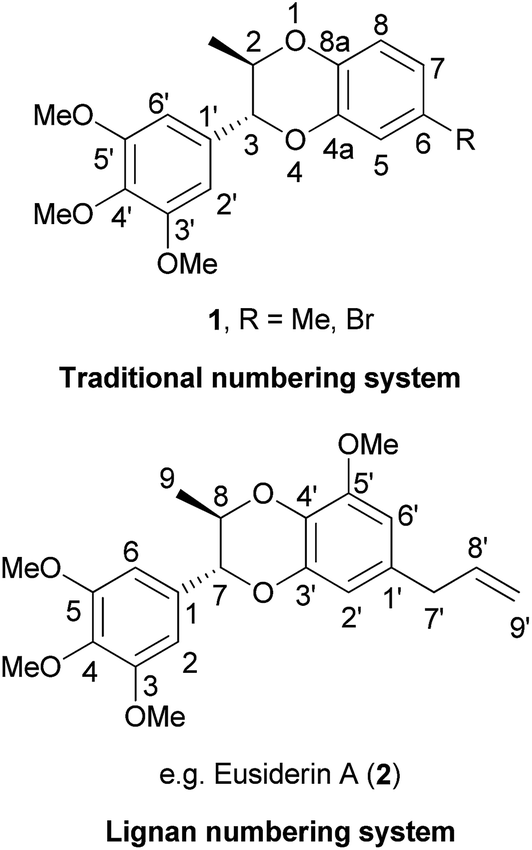 | ||
| Fig. 1 The two systems used in numbering 1,4-benzodioxanes. | ||
This review also refers to 2-aryl- and 3-aryl-1,4-benzodioxanes, numbered using the traditional IUPAC system (Fig. 2). 3-Aryl-1,4-benzodioxanes (e.g.1 and oxyneolignan isoamericanin A (3)) are the most abundant type of 1,4-benzodioxane lignan found in nature; 2-aryl-1,4-benzodioxanes (e.g.4) are significantly less common, but some examples have been isolated, one of the most notable of which being oxyneolignan, americanin A (5).
 | ||
| Fig. 2 The differing structures of 2-aryl- and 3-aryl-1,4-benzodioxanes. | ||
1.2 1,4-Benzodioxanes – oxyneolignans, flavonolignans, coumarinolignans, stillbenolignans
Naturally occurring, lignan-derived (containing a characteristic phenylpropanoid, C-6 to C-3, unit) 1,4-benzodioxane lignans can be categorised into different groups, based on the partner subclass to which their structure belongs. | ||
| Fig. 3 Biosynthesis of flavonolignans and highlighted structural terms; the flavone nucleus is in the blue rectangle, while the chromanone is in the red rectangle. | ||
 | ||
| Scheme 1 Proposed biosynthesis of 1,4-benzodioxanes. | ||
Flavonolignans were first discovered in the seeds of Silybum marianum, commonly known as milk thistle. The original flavonolignan to be discovered was silybin 8 (Fig. 3), and to this day it remains by far the most researched flavonolignan and 1,4-benzodioxane lignan.4,6–8 For more information on silybin, see the recent review by Biedermann et al.4
 | ||
| Fig. 4 Biosynthesis of comarinolignans. | ||
Cleomiscosin A (9), isolated from the seeds of Cleome viscosa and its regioisomer cleomiscosin B (10) were the first regioisomeric pair of 1,4-benzodioxane coumarinolignans to be found.9,11,12 They are both formed by the oxidative coupling of coniferyl alcohol 7 and coumarin 11, the orientation of the dimerization determining which regioisomer is formed (Fig. 4).
 | ||
| Fig. 5 Biosynthesis of aiphanol 12. | ||
2 Bioactivities of 1,4-benzodioxane-containing natural products and MOA studies
Of all 1,4-benzodioxane lignans, the biological activities of 1,4-benzodioxane flavonolignans have been most extensively investigated. 1,4-Benzodioxane flavonolignans, particularly silybin 8 display a range of activities that are exploited through the use of milk thistle extract, readily available to millions worldwide. There are a number of comprehensive reviews that summarise the several hundred studies that have been reported on these activities that include hepaprotective, anticancer and antioxidant activities, along with various others that offer silybin 8 and related flavonolignans the distinction of being one of the most sought-after compounds for potential pharmaceutical applications.4,5,16–182.1 Hepaprotective activities
The hepaprotective properties of silybin 8 and associated flavonolignans are well-documented, and up until recently, this biological property has been thought to be present by virtue of the chromanone functionality present in these compounds (see Fig. 3).19 HCV (Hepatitis C Virus) is a worldwide cause of chronic liver disease, with over 70% of HCV-infected patients developing a chronic condition.20 This has resulted in silybin being used to treat HCV-induced liver disease. Earlier this year, it was reported that several 1,4-benzodioxane oxyneolignans – members of the rodgersinine family (compounds 13–16) were found to exhibit anti-HCV activity, by analysing the reduction in expression of HCV non-structural proteins NS3 and NS5A (Fig. 6).21 This activity was independent of cytotoxicity. | ||
| Fig. 6 Structures of natural 1,4-benzodioxane lignans. | ||
2.2 Prostaglandin I2 inducer activity
Isoamericanin A (3) was first isolated in 1987 by Hasegawa et al.22 from Phytolacca americana and was found to be a prostaglandin I2 inducer – increasing the release of prostaglandin I2 from rat aorta by up to 149.8% at 10−5 M concentrations.222.3 Neurotrophic (ChAT) activity
The structurally similar isoamericanol A (17) was isolated five years later from the same plant by the same group.23 Both of these compounds 3 and 17, were isolated with their associated regioisomers americanin A (5) and americanol A (18) respectively, and have since been isolated from an array of trees and plants.24–26 Americanol A (18) and isoamericanol A (17) were discovered (along with isoamericanin A (3)) to exhibit interesting neurotrophic properties; they promoted the neurite outgrowth and enhancement of choline acetyltransferase (ChAT) activity in the primary neuronal cell cultures of fetal rat cerebral hemisphere.23 These compounds significantly enhanced the neurite sprouting morphologically and increased the ChAT activity at 10−5 M concentrations. When compounds that are considered the monomeric units of these active 1,4-benzodioxane lignans were tested for their activity, they failed to enhance ChAT activity at the same concentration, and in fact cinnamic acid decreased ChAT activity. This indicated that a dimeric structure, namely the 1,4-benzodioxane moiety, is required for this neurotrophic activity.2.4 Insecticidal activity
Haedoxan A (19), isolated from an extract of Haedokusou (Phryma leptostachya L.) shows remarkably high toxicity against houseflies (LD50 0.25 ng by topical administration).27,28 In light of this potent activity, a number of SAR studies have identified structural features that are critical for the insecticidal activity of haedoxan A (19). Alteration of the 1,4-benzodioxane moiety in haedoxan A (19) resulted in complete loss or significant decrease in activity.29,30 Replacement of the 3′′-aryl group with methyl and butyl groups also lowered the activity of the analogues, indicating that an aromatic substituent at the 3′′ position may be necessary for insecticidal activity.282.5 COX-1 and COX-2 inhibition
(−)-Aiphanol (12) shows potent inhibition of the cyclooxygenase enzymes, COX-1 and COX-2 with IC50 values of 1.9 and 9.9 μM, respectively.31 In contrast, the racemic mixture of (±)-aiphanol (12) exhibits a strong inhibition of COX-2 (IC50 0.17 μM) but only a modest inhibitor of COX-1 (IC50 7.3 μM), which suggests that (+)-aiphanol (12) could be a more potent COX-2 inhibitor.322.6 Anti-angiogenic activity
The anti-angiogenic activity of (±)-aiphanol (12) has also been studied, using an in vitro angiogenesis assay. The results showed that (±)-aiphanol (12) completely inhibited blood vessel growth at 100 μg mL−1 and 42% inhibition at 10 μg mL−1.32 It was also noted that (±)-aiphanol (12) was more active than PI-88, an oligosaccharide which exhibits anti-angiogenic properties and was in clinical development as an agent for the treatment of certain cancers.2.7 MDR pump inhibition activity
Flavonolignan (±)-5-methoxyhydnocarpin D (20), isolated from Berberis fremontii is a potent NorA MDR (Multi Drug Resistance) pump inhibitor.33 While (±)-5-methoxyhydnocarpin D (20) alone does not exhibit antibiotic activity, when it was used with subinhibitory concentrations of berberine it completely inhibited the growth of S. aureus. Silybin 8 was also found to act as a synergist with berberine, however it was not as active as (±)-5-methoxyhydnocarpin D (20) (10 μg mL−1vs. 1.2 μg mL−1).33 (±)-5-Methoxyhydnocarpin D (20) was also shown to enhance the activity of a range of antibiotic compounds including norfloxacin (2-fold) and pentamidine (>12-fold), against S. aureus.342.8 Anti-cancer activity
Hydnocarpin (21) and sinaicitin (22) (the 3′′-demethoxy derivative of hydnocarpin (21)), flavonolignans isolated from sinaiticum leaves found in the Sinai region of Egypt, have shown to have significant inhibitory activities against the murine lymphocytic leukaemia P-388 cell line (ED50 = 1.2 and 7.7 μg mL−1).35 Hydnocarpin (21) has also been shown to have significant activity against six human cancer cell lines and the murine cell line, L-1210. Cleomiscosin A (9) has also been shown to be active against the P-388 cell line (ED50 = 0.4 μg mL−1),36 and other 1,4-benzodioxane coumarinolignans have shown additional anti-proliferative activities in carcinoma, CNS and breast cancer cell lines.37,382.9 Anti-oxidant activity
Natural products cleomiscosin C (23) and jatrocin B (24) showed lipid peroxidation inhibitory activity in rat liver microsomes (IC50 = 0.7 and 1.4 μg mL−1, respectively) – values that are comparable to vitamin E (IC50 = 0.8 μg mL−1).39 Additionally, cleomiscosins A (9) and C (23) have been tested for their inhibition of LDL (low-density lipoprotein) oxidation.40 Cleomiscosin C (23) was found to dose-dependently inhibit LDL oxidation by either catalytic copper ions or free radicals generated by APPH. Electrophoretic analysis determined that cleomiscosin C (23) protects apolipoprotein B-100 against Cu2+-induced fragmentation (63.5% inhibition at 5.0 pM) and fluorescence analysis indicated that both cleomiscosin A (9) and C (23) protect apolipoprotein B-100 against oxidative modification by either Cu2+ and HOCl. This suggests that these natural products could be beneficial in preventing LDL oxidation in atherosclerotic lesions.40–422.10 Additional activities
An aiphanol derivative has been shown to have inhibitory activity against α-glucosidase and has potential for the development of hypoglycaemic treatments.14 Other 1,4-benzodioxane compounds have shown a range of biological properties. Compounds containing the 1,4-benzodioxane moiety have demonstrated activity as selective α1D-AR (adrenoreceptor) antagonists indicating the potential to act as anti-hypertensive agents. They have also been reported to act as 5-HT1A receptor agonists which may have use as antidepressant and neuroprotective agents.43–54 Additionally, 1,4-benzodioxanes have been found to inhibit 5-lipoxygenase, an activity which presents these compounds as potential anti-inflammatory agents, with use in the treatment of asthma, rheumatoid arthritis and inflammatory bowel disease.55 A close analogue of natural 1,4-benzodioxane oxyneolignans showed anti-Leishmania donovani promastigote activity, indicating possible activity against Leishmaniasis.56 Pyrrolidynylbenzodioxanes have been shown to have affinities for α4β2 and α7 central nicotinic receptors which play a key role in a wide range of CNS functions,57 and some 1,4-benzodioxanes have been shown to exhibit antiplatelet aggregation and organ-protective activities.583 Biosynthesis of 1,4-benzodioxane oxyneolignans
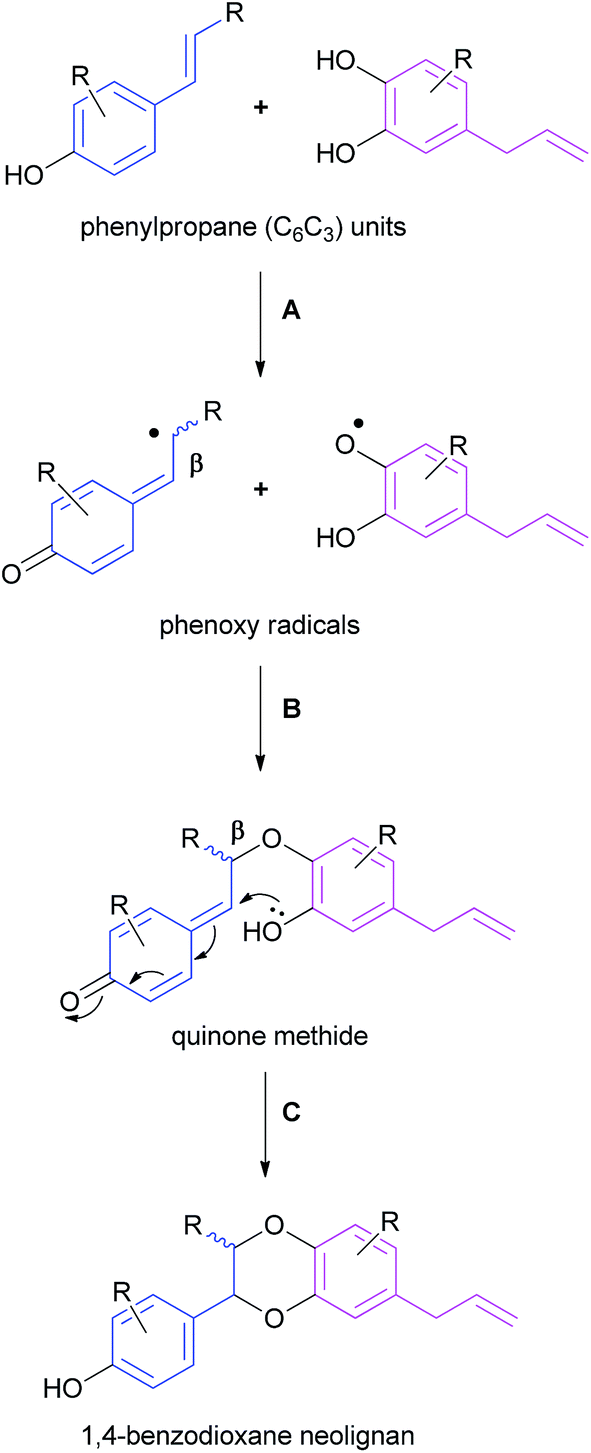
3.1 Proposed mechanism of biosynthesis
1,4-Benzodioxane oxyneolignans are believed to be formed by the phenolic coupling of two C6C3 units through a three-step process – (A) enzymatically-promoted phenol oxidation of the two phenylpropane units, (B) O-β coupling of the resulting phenoxy radicals and (C) cyclisation of the quinone methide system (Scheme 1).59–611,4-Benzodioxane lignan natural products are usually found as racemic mixtures; steps B and C may not be enzymatically controlled and therefore for the most part a mixture of stereo- and regioisomers are formed.62 There is, however an important collective of 1,4-benzodioxanes, most notably the eusiderin and rodgersinine families that are found as single enantiomers in nature – resulting from a stereoselective coupling in step B. Most natural products have a trans configuration across the 1,4-benzodioxane (as displayed in eusiderin A (2) and isoamericanol A (17)) although some cis natural products (e.g. cis-rodgersinine B (16)) have been isolated – an observation that is consistent with the thermodynamic control of step C.59
3.2 Initial investigation into the biomimetic synthesis
The synthesis of (±)-eusiderin A (2) by Merlini and Zanarotti in their efforts to test the postulated biosynthetic mechanism by which these compounds were hypothesised to form, constituted the first synthesis of a 1,4-benzodioxane lignan (Scheme 2).63 | ||
| Scheme 2 First synthesis of a 1,4-benzodioxane lignan, by Merlini and Zanarotti.63 | ||
This straightforward method, used silver(I) oxide in benzene to couple phenols 25 and 26, giving 1,4-benzodioxane (±)-27, provided the basis for a simple method by which a large number of products could be produced.
3.3 Further developments and application of biomimetic methods
Following the pioneering study by Merlini and Zanarotti, a number of investigations were conducted to determine the regioselectivity of these cross-coupling reaction to form either 2- or 3-aryl-1,4-benzodioxanes (Scheme 3).64,65 | ||
| Scheme 3 The effect of substituents on the regioselectivity of oxidative dimerisation to form 1,4-benzodioxane products.65 | ||
It was revealed that the substituents on the catechol fragment (i.e.28 in Scheme 3) have a strong effect on the regioselectivity of the reaction; electron donating substituents, like methyl as in 28, strongly favour the formation of 3-aryl products (e.g. (±)-29), which are by far the most commonly found in nature. The reaction is less selective and lower yielding when electron-withdrawing substituents (e.g. a formyl group) are present, giving both 2- and 3-aryl products, as demonstrated by the reaction of 7 and 30 to give both the 3-aryl (±)-31 and 2-aryl (±)-32 1,4-benzodioxanes in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ratio.
:7 ratio.
Oxidative dimerisation has been the prevailing method in synthesis of 2- or 3-hydroxymethyl-1,4-benzodioxanes, particularly that of members of the (iso)americanin family; isoamericanin A (3) and isoamericanol A (17) (3-aryl-1,4-benzodioxanes) and their corresponding regioisomers, americanin A (5) and americanol A (18) (2-aryl-1,4-benzodioxanes). Americanin A (5)66 was the first of the americanin family to be isolated. Initially, it was given the structure now attributed to isoamericanin A (3), however this was later revised.67 The 2- and 3-aryl regioselectivity of conditions and starting materials have been investigated and exploited to obtain desired products (Scheme 4).68–70
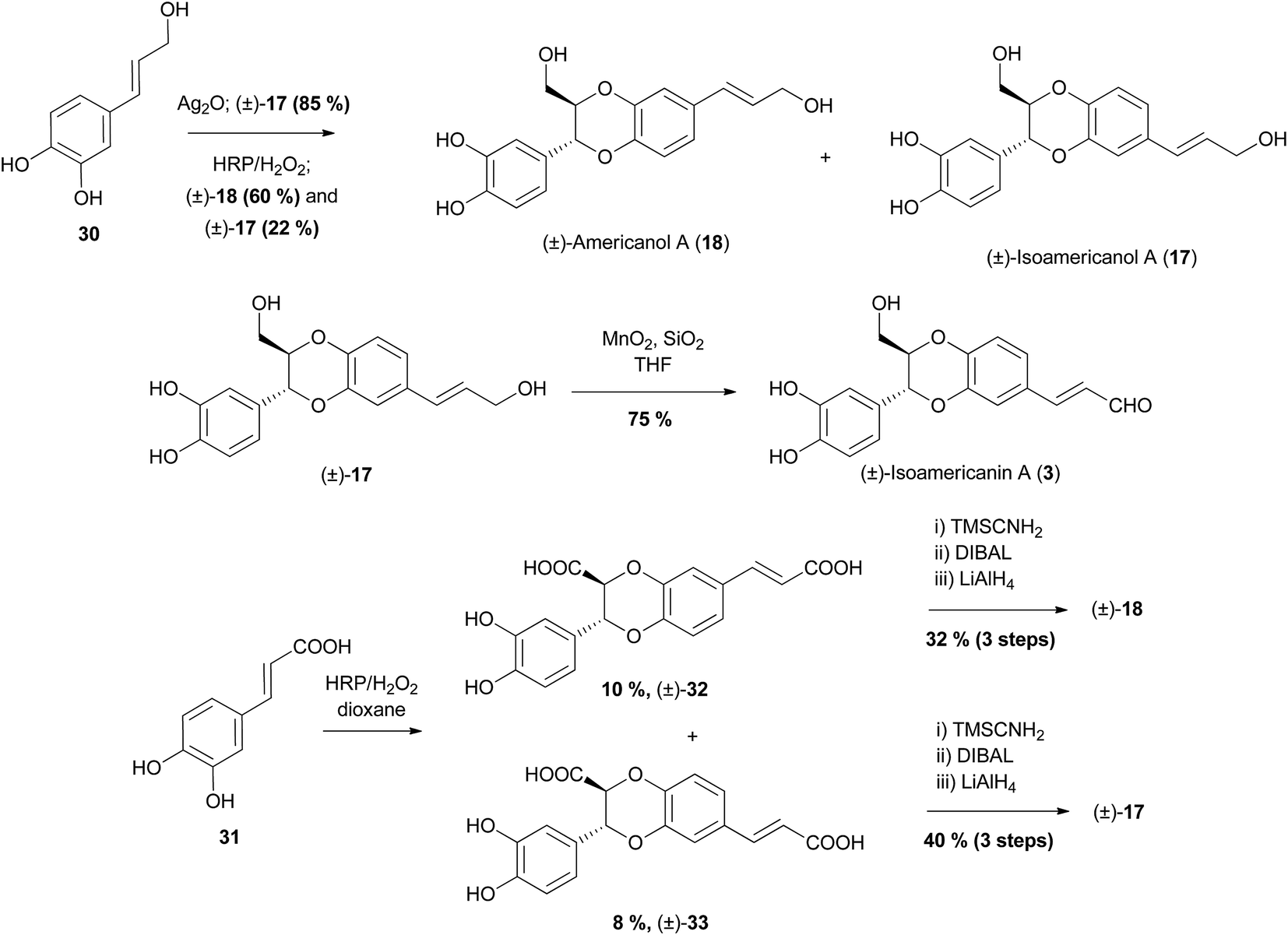 | ||
| Scheme 4 Biomimetic syntheses of (±)-isoamericanol A (17), (±)-isoamericanin A (3) and (±)-americanol A (18).68–70 | ||
The oxidative homocoupling of alcohol 30 has been effected by two oxidising agents with differing results. Initially, She et al. effected the coupling using Ag2O, obtaining only (±)-isoamericanol A (17) in 85% yield, which they were able to oxidise to (±)-isoamericanin A (3), using MnO2 in the presence of silica gel.69 Following this, Takahashi et al. used an alternative oxidant, horseradish peroxidase (HRP)/H2O2 to give a mixture of (±)-18 and (±)-17 in an approximate 3:1 ratio in 82% yield.68 When caffeic acid 31 was coupled with HRP/H2O2 a mixture of regioisomers (±)-32 and (±)-33 was isolated (among many other products). These were able to be converted to (±)-americanol A (18) and (±)-isoamericanol A (17) through conversion to the corresponding methyl diesters, followed by reduction.
As demonstrated in the above syntheses, the products of each reaction not only depend on the substituents on the aromatic ring, but also on the oxidative agents used in the coupling. This is particularly highlighted by She and co-workers in their synthesis of methyl ether analogues of members of the (iso)americanin families (Scheme 5).71,72
 | ||
| Scheme 5 Biomimetic synthesis of methyl ethers of the (iso)americanin family.71,72 | ||
By changing oxidising agents to couple phenylpropene 7 and catechol 34, different regioselectivity of products was achieved. Using silver carbonate as the oxidising agent produced 3-aryl-1,4-benzodioxane (±)-33 in high regioselectivity of ∼30:1 over the regioisomer (±)-34; this was in higher selectivity than the 20:1 mixture reported by Merlini et al.65,71 In contrast, when the same coupling reaction was performed using potassium hexacyanoferrate(III), the 2-aryl-1,4-benzodioxane (±)-34 was produced exclusively, albeit in a mixture of trans and cis isomers.72 This mixture was stirred with K2CO3 in DMF to effect isomerisation, giving the pure trans over the two steps. Reduction of esters (±)-33 and (±)-34 with LiAlH4 to give (±)-35 and (±)-36, followed by oxidation with MnO2 produced (±)-5-O-methyl-isoamericanin A (37) and (±)-5-O-methyl americanin A (38) respectively.
There was no explanation given by She et al. for the contrasting results achieved when using silver and iron salts, however Sefkow proposes an explanation for the difference.2 Sefkow postulates that the basic silver salts slowly deprotonate the phenol groups on the catechol (e.g.34), with the more acidic phenol at C-4 deprotonating faster than the less acidic phenol at C-3, to give more phenoxy radical 39 than phenoxy radical 40 (Scheme 6).
 | ||
| Scheme 6 Regioselectivity of oxidative dimerisation when using silver salts. | ||
In the same fashion as the biosynthesis (Section 3.1, Scheme 1), both radicals attack the double bond of the phenylpropene at C-8, eventually resulting in a mixture of 3-aryl- and 2-aryl-1,4-benzodioxanes. As more of phenoxy radical 39 would have been produced when using silver salts, this favours the formation of a 3-aryl-1,4-benzodioxane, as was observed.
It is well established that Fe(III) salts form strong complexes with catechols.73 Sefkow suggests that an Fe(III) complex 41 is an intermediate in the coupling reaction (Fig. 7).
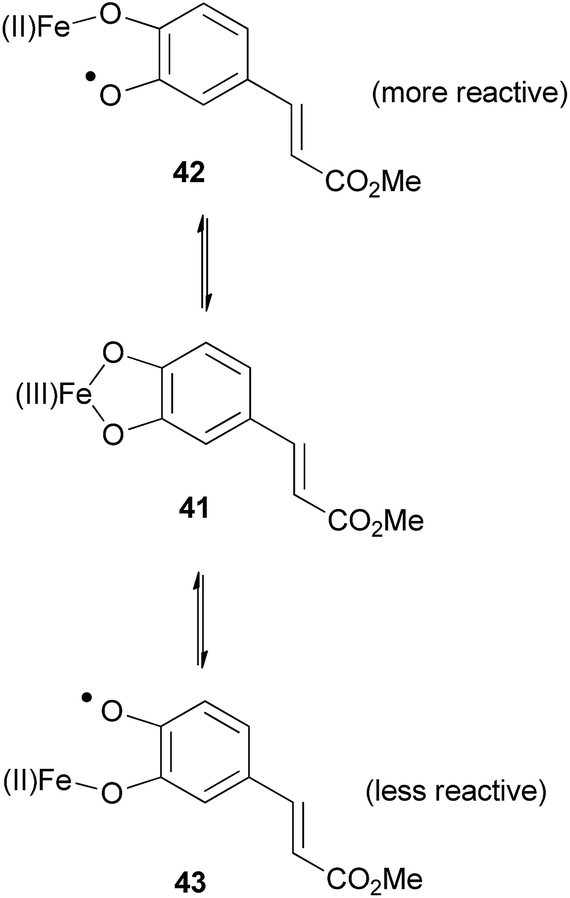 | ||
| Fig. 7 Two regioisomeric radicals formed using iron salts.2 | ||
The similar oxidation potential of Fe(II)/Fe(III) and of O˙/O− in catechols means the two radicals 42 and 43 can equilibrate via complex 41. Radical 42 is less stabilised and therefore more reactive than radical 43, therefore it primarily couples with the phenylpropene (e.g.7), favouring the formation of 2-aryl-1,4-benzodioxanes, for example (±)-34.
The high selectivity of iron and silver salts has been further exploited by the same group in the synthesis of (±)-sinaiticin 22 and (±)-aiphanol 12.74,75
4 Synthetic methods to 1,4-benzodioxane oxyneolignans
4.1 Biomimetic approaches and oxidative dimerization
Indisputably the most well-researched route to synthesise 1,4-benzodioxane lignans is the biomimetic synthesis involving the oxidative dimerisation of phenoxy units to give racemic products (introduced in Sections 3.2 and 3.3).Subsequent efforts by Jing and co-workers have expanded on the preliminary report by Merlini and Zanarotti, with several members of the eusiderin family synthesised by changing the C6C3 units involved in the oxidative coupling using silver oxide in benzene (Scheme 7).76,77
 | ||
| Scheme 7 Synthesis of five members of the eusiderin family by Jing et al.76,77 | ||
All the eusiderins that were synthesised were done so using phenylpropene 44 as a coupling partner. Phenylpropene 44 was prepared by global methylation of pyrogallol 45, followed by regioselective demethylation to give phenol 46. Allylation of 46 followed by a high-yielding Claisen rearrangement afforded an alkene that underwent PdCl2-catalysed migration of the double bond to form phenylpropene 44. Three different o-diphenol fragments: 25, 26 and 47, were then prepared to account for the different sidechains on eusiderins E (48) and F (49) compared to K (50) and A (2) and also G (51). The synthesis of these phenylpropene fragments also began with pyrogallol 45 which was selectively methylated to give 52 followed by allylation to give a substrate that underwent a Claisen rearrangement to give diphenol 26. Double-bond transposition gave phenol 25, the allyl group of which was then oxidised to give 47. Oxidative cross-coupling between phenylpropene 44 and either diphenol 25, 26, or 47 was mediated by Ag2O to give (±)-eusiderin E (48), (±)-eusiderin K (50) and (±)-53 respectively; all with the desired trans isomers as major products and in moderate yields circa 40%. Methylation of the 1,4-benzodioxane products with iodomethane gave pure trans isomers of (±)-eusiderin F (49), (±)-eusiderin A (2) and (±)-eusiderin G (51).
Chand and Banwell78 in their biomimetic synthesis of (±)-aiphanol 12, used slightly different starting materials in their oxidative coupling – instead of a phenylpropene, they coupled piceatannol 54 with sinapyl alcohol 53, which resulted in (±)-aiphanol 12. Three aiphanol isomers; cis-(±)-55, trans-(±)-56 and cis-(±)-57 were also procured directly from the coupling reaction (Scheme 8). The products were separated using semi-preparative HPLC.
 | ||
| Scheme 8 Synthesis of (±)-aiphanol 12 by Chand and Banwell.78 | ||
This method only produces racemic products, and while some 1,4-benzodioxanes are found as racemic mixtures in nature, non-racemic compounds make up a significant portion of isolated natural products. Although a certain degree of regioselectivity is achieved through altering conditions and starting materials, most of these reactions do give both regioisomers and many possible coupling products often isolated. In addition, the respective components of the oxidative dimerisation have to be assembled separately and then combined in the last or penultimate steps of the synthesis.
4.2 Phenolate substitution of a bromine in an α-bromophenone
An effective technique to form a 1,4-benzodioxane hinges on the condensation of an α-bromophenone (e.g.58) and a phenol (e.g.59) as the pivotal synthetic step (Scheme 9). | ||
| Scheme 9 Synthesis of (±)-silybin A (8).79 | ||
This approach was first reported in relation to synthesis of the 1,4-benzodioxane fragment of silybin A (8),6,7,79 followed by other compounds including insecticidal sesquilignans (e.g. haedoxan A (19),27Scheme 10).80–82 The phenolic component requires a second hydroxyl group, that can be protected or unprotected, ortho to the phenol substituent, for formation of the 1,4-benzodioxane later in the synthesis.
 | ||
| Scheme 10 Synthesis of (±)-haedoxan A (19).81 | ||
In this approach, the crucial step involved the reaction of racemic 2-bromo-3-oxypropionates 58 and 60 with phenols 59 and 61 to give keto-esters 63 and 64. Reduction of the ketone, followed by manipulation of protecting groups and acid-catalysed cyclisation gave the 1,4-benzodioxane structures (±)-65 and (±)-66. In all the acidic cyclisation conditions attempted, 1,4-benzodioxane (±)-66 was isolated as a mixture of trans and cis isomers.6 However, with the slightly different substrate, Tanaka et al. obtained only the trans-1,4-benzodioxane (±)-65 using similar conditions to those previously reported.79
While 2-bromo-3-oxypropionates are required for the synthesis of 1,4-benzodioxanes with a 9-hydroxymethyl (i.e. isoamericanin-type), α-bromopropiophenones (e.g.67) are required for the synthesis of 1,4-benzodioxanes with a methyl group (i.e. eusiderin-type). This route has been applied to the synthesis of members of the eusiderin and rodgersinine families.83–85
Miao and coworkers reported the racemic synthesis of members of the rodgersinine family using this method (Scheme 11).
 | ||
| Scheme 11 Synthesis of members of the rodgersinine family by Miao et al.83 | ||
This synthesis began with 2,4-dihydroxypropiophenone 68 which was benzyl-protected followed by a bromination step to give α-bromopropiophenone 67. The bromine in α-bromopropiophenone 67 was then displaced by the phenoxide of 3,4-dihydroxybenzaldehyde 30, to give ether 69. Global reduction using NaBH4, followed by acetic acid-mediated cyclisation gave 1,4-benzodioxane 70 in a 2:1 ratio of trans to cis isomers with concomitant acetylation of the benzylic alcohol. The cis/trans mixture of isomers was not separated and the remaining synthesis was performed using a cis/trans mixture. Next was the conversion of the benzylic ester to an alcohol and a series of deprotection/reprotection steps followed by oxidation with PCC resulted in aldehyde 71. To synthesise (±)-trans-rodgersinine A (13) and (±)-cis-rodgersinine A (15), aldehyde 71 underwent a Wittig reaction, to give a mixture of E and Z isomers which were isomerised to give exclusively the E isomer 72, which was then deprotected to give the natural products (±)-13 and (±)-15 in 11 steps with an overall yield of 7.1%. To introduce the alkyne functionality, aldehyde 71 underwent a Corey–Fuchs reaction. Subsequent deprotection of the MOM groups of 73 yielded (±)-trans-rodgersinine B (14) and (±)-cis-rodgersinine B (16) in 11 steps with an overall yield of 8.2%.
The main advantage of this method over the oxidative dimerisation method (Section 4.1), is control over which regioisomer (2-aryl or 3-aryl) is produced in the synthesis, rather than a mixture as was seen for the other method. However, like the preceding coupling method, this approach is both racemic and as stated above does not easily allow for the synthesis of a wide range of analogues and similar final products. As shown above in the synthesis of members of the rodgersinine family, the sidechain can be manipulated given that there is an appropriate substituent. The substitution on the non-1,4-benzodioxane aromatic ring, however, cannot be easily changed in the synthesis. When some members of the eusiderin family were synthesised using this approach, different α-bromopropiophenones were prepared and a parallel reaction sequence was required for the formation of analogous 1,4-benzodioxanes (Scheme 12).
 | ||
| Scheme 12 Synthesis of members of the eusiderin family and analogues.84,85 | ||
4.3 Condensation of an epoxide precursor
The first enantioselective synthesis of a 1,4-benzodioxane was that of a model 1,4-benzodioxane by Arnoldi et al. (Scheme 13).59 They used the enantiomerically pure (+)-epoxide 74, derived from ephedrine. Firstly, (+)-74 was heated at reflux with catechol 75 and NaH in DMF, giving separable diols 76 and 77 in a 1:2 ratio. Diol 76 was then cyclised with Amberlyst® 15 in toluene to give trans-benzodioxane 78 and cis-benzodioxane 79 in a 3:2 ratio.
 | ||
| Scheme 13 First enantioselective synthesis of a 1,4-benzodioxane structure.59 | ||
This pioneering work highlighted epoxides as an attractive option for the synthesis of 1,4-benzodioxanes, and even though the asymmetric version has not be applied to the synthesis of any eusiderin-type natural products, it has since been further developed by several groups to synthesise isoamericanin-type natural products.
The first synthesis of 1,4-benzodioxane natural products using epoxides was in 1987 by Tanaka et al. (Scheme 14).86 The method was carried out using achiral epoxide 80, and as such gave racemic products. Nevertheless, it provided a novel way to synthesise this type of naturally occurring compound.
 | ||
| Scheme 14 Synthesis of (±)-isoamericanin A (3) and (±)-americanin A (5) by Tanaka et al.86 | ||
The synthesis by Tanaka et al. began from caffeic acid 31, which was esterified, MOM protected and then reduced, to give allylic alcohol 81. Alkene 81 then underwent epoxidation with tert-butyl hydroperoxide (TBHP) in the presence of vanadyl acetylacetonate to give epoxide 80. Condensation of 80 with either phenol 82, or its regioisomer 83, in the presence of sodium hydroxide afforded the respective diols 84 and 85 as the erythro products. Mesylation of the primary alcohol in 84 and 85 followed by treatment with base, gave epoxides 86 and 87, respectively. Removal of the benzyl protecting groups, followed by cyclisation in basic conditions gave trans-1,4-benzodioxanes (±)-88 and (±)-89. Horner–Wadsworth–Emmons of aldehydes (±)-88 and (±)-89 with phosphonate 90 followed by treatment with acid afforded the natural products (±)-americanin A (5) and (±)-isoamericanin A (3).
Gu et al. were able to adapt this method, which previously gave racemic products, to an enantioselective synthesis through an asymmetric dihydroxylation and were able to use this to synthesise the 1,4-benzodioxane fragment of silybin A (8) (Scheme 15).87,88
 | ||
| Scheme 15 Enantioselective synthesis of the 1,4-benzodioxane fragment of silybin A (8).87,88 | ||
Starting from ferulic acid 91, a series of esterification, benzyl protection and reduction steps yielded cinnamyl alcohol 92. This underwent a Sharpless asymmetric dihydroxylation to give triol 93 with a 92% ee. Triol 93 was then converted to epoxyalcohol 94 which then underwent a Mitsunobu reaction with phenol 82 to yield benzyl aryl ether 95. Removal of the benzyl protecting groups followed by base-mediated cyclisation of the resultant diol produced 1,4-benzodioxane carbaldehyde (2R,3R)-31 as the trans isomer. Using AD-mix-β as opposed to AD-mix-α in the asymmetric dihydroxylation step provided triol ent-93, which was then transformed into the enantiomer of 31.
This method was applied to the synthesis of isoamericanin A (3) and isoamericanol A (17) by the same group.89 Banwell and co-workers also used this method to prepare (−)-aiphanol 12 and (+)-aiphanol 12 using AD-mix-β and AD-mix-α, respectively.31
The route was abridged by Ganesh et al.90 in their racemic synthesis involving the base mediated one-pot condensation-cyclisation step between bromoepoxide 96 and catechol 30 to give model (±)-trans-1,4-benzodioxane 97 (Scheme 16). The stereochemistry of benzodioxane 97 in this reaction is the result of initial attack of the more reactive 3-phenolate, over the resonance-stabilised 4-phenolate, to the benzylic bromide. Subsequent ring-opening of the epoxide results in the trans-1,4-benzodioxane.
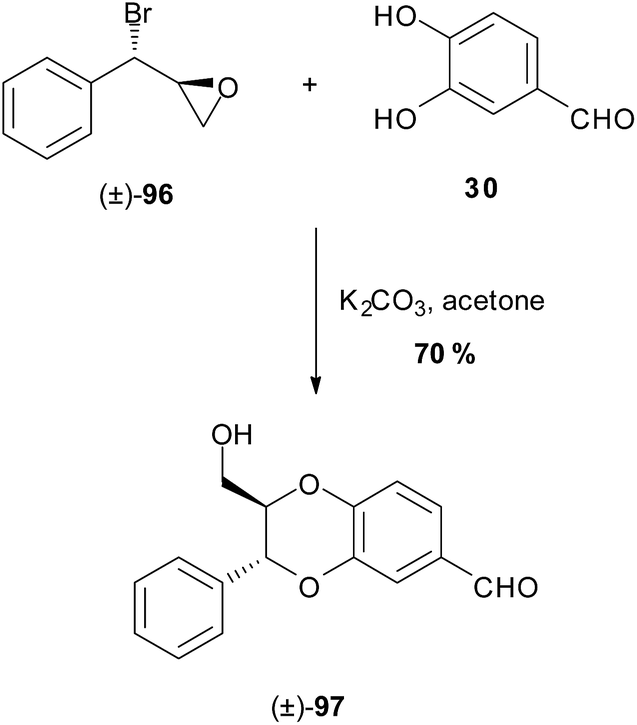 | ||
| Scheme 16 One pot cyclisation of the model 1,4-benzodioxane (±)-97 by Ganesh et al.90 | ||
This overall synthetic approach offered the first successful asymmetric synthesis of a 1,4-benzodioxane. Depending on the epoxide, the method can produce either racemic or enantioenriched products. Unfortunately, the asymmetric version has only been applicable to isoamericanin-type 1,4-benzodioxanes and not eusiderin-type. This method also offers regioselectivity of final products, best demonstrated by the separate synthesis of (±)-isoamericanin A (3) and (±)-americanin A (5) by Tanaka et al. (Scheme 14). Once again, synthesis of similar compounds using this method is not trivial, especially with compounds that differ in their substitution on the non-1,4-benzodioxane aromatic ring.
4.4 Transition metal assisted methods
In conjunction with the rise in popularity of transition-metal based chemistry in recent years, strategies to synthesise 1,4-benzodioxanes using transition-metals have been developed and expanded upon. | ||
| Scheme 17 Intramolecular palladium-catalysed etherification of aryl halides.91 | ||
It was found that the reactions with bromides 98–100 in toluene with Pd(OAc)2 and Cs2CO3 successfully gave 1,4-benzodioxanes 101–103, although the reactions were highly ligand dependent. Cyclisation of optically active alcohols, such as 104 is also possible with the enantiopurity of product 105 being achieved in 95% ee and a yield of 80%.
The reaction can also be performed intermolecularly by coupling a diol (e.g.106) and a o-dihaloaryl (e.g.107) species (Scheme 18).92
 | ||
| Scheme 18 Coupling of diols 106 and o-dihaloaryls 107 to give 1,4-benzodioxanes.92 | ||
It was found that the reaction did not proceed readily without addition of PPh3, however when a catalytic amount was introduced, the yield was greatly improved. Following these model reactions, the concept was applied to the synthesis of (±)-isoamericanin A (3) and (±)-isoamericanol A (17) (Scheme 19).
 | ||
| Scheme 19 Synthesis of (±)-isoamericanol A (17) and (±)-isoamericanin A (3) by Jing et al.92 | ||
Diol 108 was coupled with dibromide 109, using the optimised conditions to give (±)-110. Reduction of the aldehyde and removal of the acetyl protecting groups with LiAlH4 gave (±)-isoamericanol A (17) which was oxidised to (±)-isoamericanin A (3). Unfortunately, the regioselectivity or trans/cis isomerism of the coupled product was not reported.
Recently, the coupling of a chiral dibromide and a catechol was carried out by Rouf et al. to give an enantiopure 1,4-benzodioxane which was ultimately converted to (R)-doxazosin, an antidepressant drug.93
An enantioselective variant of this reaction has also been reported, forming an asymmetric 1,4-benzodioxane product 111 with 96% ee, In this case, the intramolecular desymmetrization of the 1,3-diol unit in 112 in the presence of a chiral ligand gave the desired chiral product 111 (Scheme 20).94
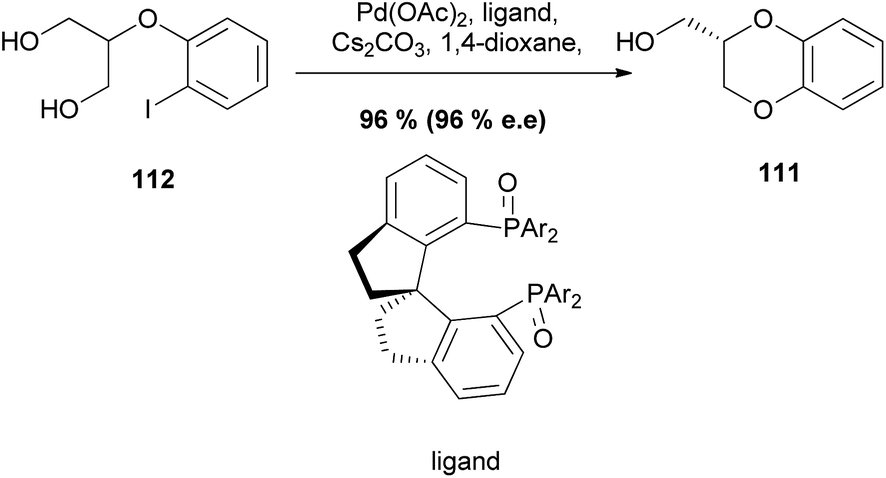 | ||
| Scheme 20 Asymmetric synthesis of 1,4-benzodioxane 111 from 112. | ||
 | ||
| Scheme 21 Palladium-catalysed coupling of catechol 75 with allylic biscarbonates.95 | ||
Interestingly all of the allyl biscarbonates 113–115 that were trialled gave 1,4-benzodioxane (±)-116 in a 60% yield and an ee of 37%, when ligand (R)-BINAP was used, this implies all three starting materials 113–115 form a common intermediate en route to benzodioxane 116.
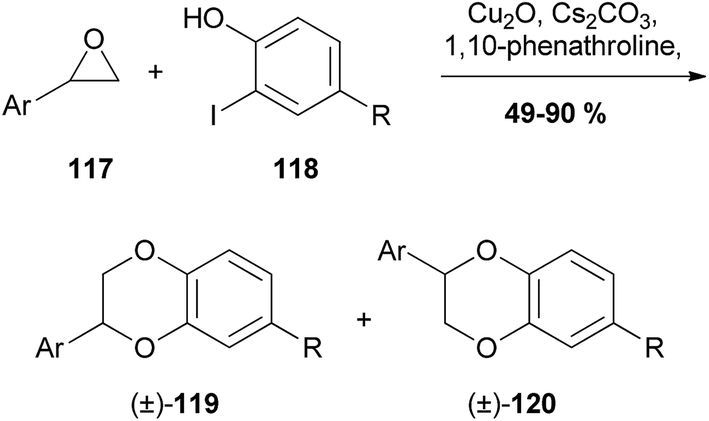 | ||
| Scheme 22 Copper-catalysed synthesis of 1,4-benzodioxanes by Bao et al.96 | ||
It was found that the 3-aryl-1,4-benzodioxane (±)-119 was formed preferentially over its 2-aryl regioisomer (±)-120. Unfortunately, only terminal epoxides were reacted, and therefore there were no 1,4-benzodioxanes with the potential for trans/cis isomers produced and the selectivity cannot be commented on.
While these new approaches to 1,4-benzodioxanes are interesting alternatives to earlier methods, their processes do not address many of the drawbacks of the existing methods. Once again, in all the discussed reactions, the same issue with flexibility is left unchallenged – as with the oxidative dimerisation method, components are assembled first and the 1,4-benzodioxane formed subsequently. Also, with the exception of the example described in the palladium catalysed etherification of aryl halides, all syntheses were racemic and have not been applied to the synthesis of natural products.
4.5 Mitsunobu coupling of phenols and chiral alcohols
A number of 1,4-benzodioxane oxyneolignans have been reported where the pivotal step was the Mitsunobu coupling of a phenol with a chiral alcohol.This approach was used for the first asymmetric synthesis of six members of the eusiderin family; eusiderins A (2), B (121), C (122), G (51), L (123) and M (124), as well as various analogues.97 This was achieved using a divergent synthesis, all from the same chiral aldehyde 125, derived from the Mitsunobu coupling of (S)-ethyl lactate 126 and phenol 127, itself synthesised from o-vanillin 128 (Scheme 23).
 | ||
| Scheme 23 Synthesis of six eusiderin natural products.97 | ||
Following the synthesis of aldehyde 125, addition of the appropriate aryl Grignard reagents, deprotection of the benzyl ether and cyclisation yielded separable trans/cis mixtures of the cyclised bromides 126a, 126b and 127. Installation of sidechains through either Suzuki cross-coupling reactions (to give 2, 121 and 122), or installation of the formyl group in 123 and subsequent manipulation (to give 51 and 124) provided the final products in a highly efficient synthesis. A subsequent ECD (Electronic Circular Dichroism) study confirmed the absolute stereochemistry of eusiderins A (2) and C (122) from different plant sources (Fig. 8). There was insufficient chiroptical data reported in the original isolation papers to confirm the stereochemistry of the remaining natural products.
 | ||
| Fig. 8 Absolute stereochemistry of eusiderin A (2) and eusiderin C (122) from different natural sources. | ||
This approach was recently applied to the asymmetric synthesis of members of the rodgersinine family of 1,4-benzodioxane oxyneolignans.21 Rather than the aryl-Grignard reagents used in the synthesis of the eusiderin family, this synthesis relied on the addition of the aryl lithiate 128 to aldehyde 129, providing ethers 130a and 130b. This mixture of diastereomers underwent hydrogenolysis and concomitant cyclisation in the acidic conditions, to give a trans/cis mixture of 1,4-benzodioxanes 131a and 131b, which were then converted to the natural products through installation and side-chain manipulation. This asymmetric synthesis was used to correct the stereochemical assignment of the natural products (Scheme 24).
 | ||
| Scheme 24 Asymmetric total synthesis of trans- and cis-rodgersinine A (13) and (15) and B (14) and (16).21 The upper blue percentages are the yields for the shown compounds and while the corresponding enantiomers are not drawn, their yields are indicated as the lower red percentages. | ||
This route has also been applied to the synthesis of 2-hydroxymethyl-1,4-benzodioxane oxyneolignans through the synthesis of members of the isoamericanin family, isoamericanin A (3) and isoamericanol A (17).98 Instead of using a lactate in the Mitsunobu coupling with a phenol (in this case 132), a glycerol derivative 133 was used to afford the 2-hydroxymethyl structural feature found in the final products (Scheme 25).
 | ||
| Scheme 25 Mitsunobu coupling of chiral alcohol 133 and phenol 132en route to the synthesis of 2-hydroxymethyl oxyneolignans (−)-isoamericanin A (3) and (+)-isoamericanol A (17).98 | ||
These methods offer a flexible approach to the synthesis of 1,4-benzodioxanes, particularly as both aryl groups are easily modified. The separate aryl units are added sequentially to a three carbon unit (either lactate of glycerol derived) which accounts for the remaining atoms of the 1,4-benzodioxane.
4.6 Alternative syntheses of the 1,4-benzodioxane structure
In addition to the general routes presented above, there are other ways in which 1,4-benzodioxane oxyneolignans, or analogues, have been synthesised. | ||
| Scheme 26 Asymmetric synthesis of americanin-type 1,4-benzodioxane (2R,3R)-32 by Chen et al.99 | ||
Instead of converting triol 93 (see Section 4.3, Scheme 15) to the corresponding epoxide, benzaldehyde dimethylacetal was used to protect the 1,3-diol to yield benzylidene 134. A Mitsunobu reaction added the next fragment, phenol 82, to give 135 and the cyclisation was effected by acid-mediated deprotection of acetal and benzyl protecting groups to give 1,4-benzodioxane (2R,3R)-32.
The same concept was also used in the 1,4-benzodioxane component of haedoxan A (19), albeit using an acetonide group as an alternative to a benzylidene (Scheme 27).100
 | ||
| Scheme 27 Asymmetric synthesis of 1,4-benzodioxane (2R,3R)-66 by Nakamura et al.100 | ||
Instead of an asymmetric dihydroxylation to install the stereochemistry of alcohol 136, an Evan's aldol condensation was used. Oxazolidinone 137101 underwent an aldol reaction with benzaldehyde 138 to give syn aldol adduct 139 in >98% de. Reduction of the aldol provided diol 136 which underwent protection to give acetonide 140. Hydrogenolysis of the benzyl group with Pearlman's catalyst provided alcohol 141, which was coupled with phenol 61 under Mitsunobu conditions to give ether 142. Deprotection and cyclisation steps produced (2R,3R)-66.
This method offers many of the advantages and disadvantages presented by the epoxide-based method. Although only applicable to the synthesis of isoamericanin-type 1,4-benzodioxanes, it does produce chiral products with in-built regioselectivity. While the 1,4-benzodioxane aromatic ring is added later in the synthesis and thus can be changed to allow for analogues, the non-benzodioxane aromatic ring is present from the beginning of the synthesis and as such is not easily altered to provide analogues.
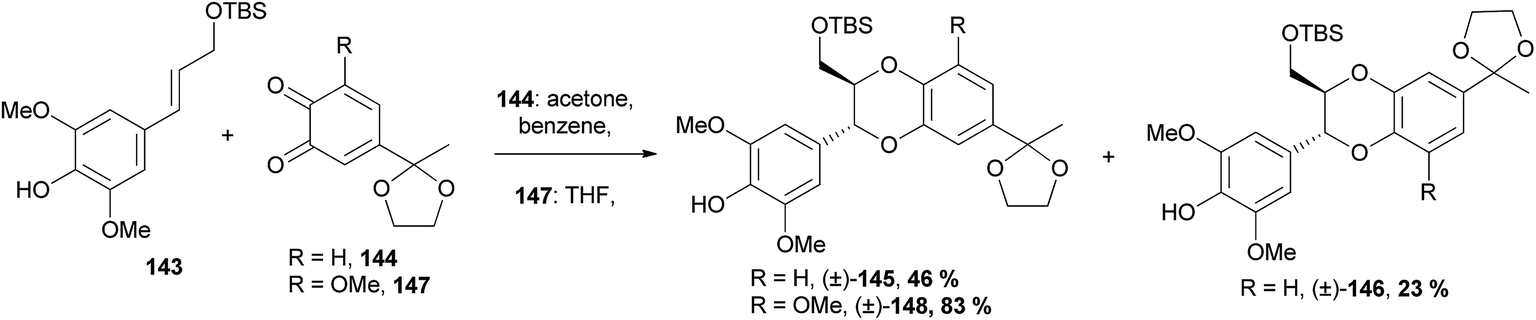 | ||
| Scheme 28 Synthesis of 1,4-benzodioxanes (±)-145, (±)-146 and (±)-148 through a [4 + 2] cycloaddition.102,103 | ||
It was found that the free phenol and a protected primary alcohol (in this case with TBS) on cinnamyl alcohol 143 were required for a successful cycloaddition. Interestingly, when a methoxy substituent was added to the o-quinone, as in 147, the selectivity of the reaction was greatly increased, with only the isoamericanin-type 3-aryl-1,4-benzodioxane (±)-148 produced, as opposed to a 2:1 mixture of 3-aryl- and 2-aryl-1,4-benzodioxanes (±)-145 and (±)-146 as was found when reacting the o-quinone 144.103 1,4-Benzodioxanes (±)-145 and (±)-148 were further reacted to procure natural products (±)-aiphanol 12 and (±)-nitidanin, respectively.
Similar to oxidative dimerisation, this route provides racemic products, and the regioselectivity can be somewhat controlled.
 | ||
| Scheme 29 Synthesis of (±)-5′-methoxyhydnocarpin-D (20) by Chan et al.34 | ||
Selective protection of the primary alcohol in diol 149, followed by tosylation of the secondary alcohol gave ether 150. The acetal protecting group was removed by stirring with acid, exposing the phenol for the base-mediated intramolecular nucleophilic substitution to give the trans isomer of desired 1,4-benzodioxane (±)-151 which was then converted to (±)-5′-methoxyhydnocarpin-D (20).
This example of a racemic synthesis involving the intramolecular cyclisation of tosylates demonstrates another synthesis that can produce isoamericanin-type 1,4-benzodioxanes with regioselectivity. Once again, as is the case with all the aforementioned syntheses, this method is not particularly flexible or able to produce a large number of analogues with variations on both aromatic rings.
5 Summary and conclusions
1,4-Benzodioxane lignans, isolated from a wide range of sources, are a promising sub-class of compounds with a broad range of bioactivities which lend them to be desirable synthetic targets. Their structures are such that a number of synthetic routes, involving a wide range of known and new reactions have been used and developed for their synthesis. A study of the synthesis toward these natural products highlights the evolution in the way that groups approach these targets. Initially, syntheses were mainly racemic with little stereochemical control, but they explored many aspects of these structures, including their formation and characterisation. Regioselective and enantioselective syntheses have since been developed which allowed for the confirmation of natural product stereochemistry. More recently the rise in use of transition-metal chemistry in organic synthesis has translated to the use of these versatile reactions to provide the 1,4-benzodioxane scaffold.6 References
- Y. Yang, T. Wu and X. Pan, Tianran Chanwu Yanjiu Yu Kaifa, 2003, 15, 61–65 CAS.
- M. Sefkow, Synthesis, 2003, 2595–2625 CrossRef CAS.
- G. P. Moss, Pure Appl. Chem., 2000, 72, 1493–1523 CrossRef CAS.
- D. Biedermann, E. Vavrikova, L. Cvak and V. Kren, Nat. Prod. Rep., 2014, 31, 1138–1157 RSC.
- S. AbouZid and O. M. Ahmed, in Studies in Natural Products Chemistry, Elsevier, 2013, vol. 40, pp. 469–484 Search PubMed.
- R. Hänsel, T.-L. Su and J. Schulz, Chem. Ber., 1977, 110, 3664–3671 CrossRef PubMed.
- R. Hänsel, J. Schulz, A. Pelter, H. Rimpler and A. F. Rizk, Tetrahedron Lett., 1969, 10, 4417–4420 CrossRef.
- A. Pelter and R. Hänsel, Tetrahedron Lett., 1968, 9, 2911–2916 CrossRef.
- S. A. Begum, M. Sahai and A. B. Ray, Prog. Chem. Org. Nat. Prod., 2010, 93, 1–70 CAS.
- F. Bourgaud, A. Hehn, R. Larbat, S. Doerper, E. Gontier, S. Kellner and U. Matern, Phytochem. Rev., 2006, 5, 293–308 CrossRef CAS PubMed.
- A. B. Ray, S. K. Chattopadhyay, C. Konno and H. Hikino, Tetrahedron Lett., 1980, 21, 4477–4480 CrossRef CAS.
- A. B. Ray, S. K. Chattopadhyay, C. Konno and H. Hikino, Heterocycles, 1982, 19, 19 CrossRef CAS.
- D. Lee, M. Cuendet, J. S. Vigo, J. G. Graham, F. Cabieses, H. H. S. Fong, J. M. Pezzuto and A. D. Kinghorn, Org. Lett., 2001, 3, 2169–2171 CrossRef CAS PubMed.
- S.-H. Lam and S.-S. Lee, Phytochemistry, 2010, 71, 792–797 CrossRef CAS PubMed.
- Y. Li, C.-L. Wang, S.-X. Guo, J.-S. Yang and P.-G. Xiao, Chem. Pharm. Bull., 2008, 56, 1477–1479 CrossRef CAS.
- V. Kren and D. Walterova, Biomed. Pap., 2005, 149, 29–41 CrossRef CAS.
- R. Gazak, D. Walterova and V. Kren, Curr. Med. Chem., 2007, 14, 315–338 CrossRef CAS.
- S. AbouZid, in Phytochemicals – A Global Perspective of Their Role in Nutrition and Health, ed. V. Rao, InTech, 2012 Search PubMed.
- B. Ahmed, S. A. Khan and T. Alam, Pharmazie, 2003, 58, 173–176 CAS.
- S. J. Polyak, C. Morishima, M. C. Shuhart, C. C. Wang, Y. Liu and D. Y.-W. Lee, Gastroenterology, 2007, 132, 1925–1936 CrossRef CAS PubMed.
- L. I. Pilkington, J. Wagoner, S. J. Polyak and D. Barker, Org. Lett., 2015, 17, 1046–1049 CrossRef CAS PubMed.
- T. Hasegawa, Y. Fukuyama, K. Koshino, K. Nakagawa, M. Tori and Y. Asakawa, Chem. Lett., 1987, 16, 329–332 CrossRef.
- Y. Fukuyama, T. Hasegawa, M. Toda, M. Kodama and H. Okazaki, Chem. Pharm. Bull., 1992, 40, 252–254 CrossRef CAS.
- R. Waibel, G. Benirschke, M. Benirschke and H. Achenbach, Phytochemistry, 2003, 62, 805–811 CrossRef CAS.
- P. Luecha, K. Umehara, T. Miyase and H. Noguchi, J. Nat. Prod., 2009, 72, 1954–1959 CrossRef CAS PubMed.
- K. H. Kim, E. Moon, S. Y. Kim and K. R. Lee, J. Agric. Food Chem., 2010, 58, 4779–4785 CrossRef CAS PubMed.
- E. Taniguchi, K. Imamura, F. Ishibashi, T. Matsui and A. Nishio, Agric. Biol. Chem., 1989, 53, 631–643 CrossRef CAS.
- S. Yamauchi and E. Taniguchi, Biosci., Biotechnol., Biochem., 1992, 56, 1744–1750 CrossRef CAS.
- S. Yamauchi, S. Nagata and E. Taniguchi, Biosci., Biotechnol., Biochem., 1992, 56, 1193–1197 CrossRef CAS.
- S. Yamauchi, F. Ishibashi and E. Taniguchi, Biosci., Biotechnol., Biochem., 1992, 56, 1760–1768 CrossRef CAS.
- M. G. Banwell, S. Chand and G. P. Savage, Tetrahedron: Asymmetry, 2005, 16, 1645–1654 CrossRef CAS PubMed.
- M. G. Banwell, A. Bezos, S. Chand, G. Dannhardt, W. Kiefer, U. Nowe, C. R. Parish, G. P. Savage and H. Ulbrich, Org. Biomol. Chem., 2003, 1, 2427–2429 CAS.
- F. R. Stermitz, J. Tawara-Matsuda, P. Lorenz, P. Mueller, L. Zenewicz and K. Lewis, J. Nat. Prod., 2000, 63, 1146–1149 CrossRef CAS PubMed.
- K.-F. Chan, Y. Zhao, L. M. C. Chow and T. H. Chan, Tetrahedron, 2005, 61, 4149–4156 CrossRef CAS PubMed.
- M. S. A. Afifi, M. M. Ahmed, J. M. Pezzuto and A. Douglas Kinghornt, Phytochemistry, 1993, 34, 839–841 CrossRef CAS.
- K.-H. Lee, N. Hayashi, M. Okano, H. Nozaki and M. Ju-ichi, J. Nat. Prod., 1984, 47, 550–551 CrossRef CAS.
- L.-G. Zhuang, O. Seligmann and H. Wagner, Phytochemistry, 1983, 22, 617–619 CrossRef.
- Y.-C. Chen, M.-J. Cheng, S.-J. Lee, A. K. Dixit, T. Ishikawa, I.-L. Tsai and I.-S. Chen, Helv. Chim. Acta, 2004, 87, 2805–2811 CrossRef CAS PubMed.
- B.-S. Yun, I.-K. Lee, I.-J. Ryoo and I.-D. Yoo, J. Nat. Prod., 2001, 64, 1238–1240 CrossRef CAS PubMed.
- W. Jin, P. T. Thuong, N. D. Su, B. S. Min, K. H. Son, H. W. Chang, H. P. Kim, S. S. Kang, D. E. Sok and K. Bae, Arch. Pharmacal Res., 2007, 30, 275–281 CrossRef CAS.
- J. A. Berliner and J. W. Heinecke, Free Radical Biol. Med., 1996, 20, 707–727 CrossRef CAS.
- R. Stocker and J. F. Keaney, Physiol. Rev., 2004, 84, 1381–1478 CrossRef CAS PubMed.
- W. Quaglia, A. Piergentili, F. Del Bello, Y. Farande, M. Giannella, M. Pigini, G. Rafaiani, A. Carrieri, C. Amantini, R. Lucciarini, G. Santoni, E. Poggesi and A. Leonardi, J. Med. Chem., 2008, 51, 6359–6370 CrossRef CAS PubMed.
- D. Giardina, R. Bertini, E. Brancia, L. Brasili and C. Melchiorre, J. Med. Chem., 1985, 28, 1354–1357 CrossRef CAS.
- W. L. Nelson, J. E. Wennerstrom, D. C. Dyer and M. Engel, J. Med. Chem., 1977, 20, 880–885 CrossRef CAS.
- G. Marciniak, A. Delgado, G. Leclerc, J. Velly, N. Decker and J. Schwartz, J. Med. Chem., 1989, 32, 1402–1407 CrossRef CAS.
- W. Quaglia, M. Pigini, S. K. Tayebati, A. Piergentili, M. Giannella, A. Leonardi, C. Taddei and C. Melchiorre, J. Med. Chem., 1996, 39, 2253–2258 CrossRef CAS PubMed.
- W. Quaglia, M. Pigini, S. K. Tayebati, A. Piergentili, M. Giannella, G. Marucci and C. Melchiorre, J. Med. Chem., 1993, 36, 1520–1528 CrossRef CAS.
- L. M. Gaster, A. J. Jennings, G. F. Joiner, F. D. King, K. R. Mulholland, S. K. Rahman, S. Starr, P. A. Wyman and K. A. Wardle, J. Med. Chem., 1993, 36, 4121–4123 CrossRef CAS.
- W. Quaglia, M. Pigini, M. Giannella and C. Melchiorre, J. Med. Chem., 1990, 33, 2946–2948 CrossRef CAS.
- A. P. Welbourn, C. B. Chapleo, A. C. Lane, P. L. Myers, A. G. Roach, C. F. C. Smith, M. R. Stillings and I. F. Tulloch, J. Med. Chem., 1986, 29, 2000–2003 CrossRef CAS.
- S. F. Campbell, M. J. Davey, J. D. Hardstone, B. N. Lewis and M. J. Palmer, J. Med. Chem., 1987, 30, 49–57 CrossRef CAS.
- M. F. Hibert, M. W. Gittos, D. N. Middlemiss, A. K. Mir and J. R. Fozard, J. Med. Chem., 1988, 31, 1087–1093 CrossRef CAS.
- A. K. Mir, M. Hibert, M. D. Tricklebank, D. N. Middlemiss, E. J. Kidd and J. R. Fozard, Eur. J. Pharmacol., 1988, 149, 107–120 CrossRef CAS.
- Y. Satoh, C. Powers, L. M. Toledo, T. J. Kowalski, P. A. Peters and E. F. Kimble, J. Med. Chem., 1995, 38, 68–75 CrossRef CAS.
- L. E. S. Barata, L. S. Santos, P. H. Ferri, J. D. Phillipson, A. Paine and S. L. Croft, Phytochemistry, 2000, 55, 589–595 CrossRef CAS.
- C. Bolchi, C. Gotti, M. Binda, L. Fumagalli, L. Pucci, F. Pistillo, G. Vistoli, E. Valoti and M. Pallavicini, J. Med. Chem., 2011, 54, 7588–7601 CrossRef CAS PubMed.
- T. Tomiyama, S. Wakabayashi and M. Yokota, J. Med. Chem., 1989, 32, 1988–1996 CrossRef CAS.
- A. Arnoldi and L. Merlini, J. Chem. Soc., Perkin Trans. 1, 1985, 2555–2557 RSC.
- H. Erdtman, Justus Liebigs Ann. Chem., 1933, 503, 283–294 CrossRef CAS PubMed.
- O. R. Gottlieb, Phytochemistry, 1972, 11, 1537–1570 CrossRef CAS.
- A. Arnone, L. Merlini and A. Zanarotti, J. Chem. Soc., Chem. Commun., 1979, 696–697 RSC.
- L. Merlini and A. Zanarotti, Tetrahedron Lett., 1975, 16, 3621–3622 CrossRef.
- S. Antus, Á. Gottsegen, E. Baitz-Gács, R. Bauer, O. Seligmann and H. Wagner, Liebigs Ann. Chem., 1989, 1989, 1147–1151 CrossRef PubMed.
- L. Merlini, A. Zanarotti, A. Pelter, M. P. Rochefort and R. Hänsel, J. Chem. Soc., Perkin Trans. 1, 1980, 775–778 RSC.
- W. S. Woo, S. S. Kang, H. Wagner and V. M. Chari, Tetrahedron Lett., 1978, 19, 3239–3242 CrossRef.
- S. Antus, O. Seligmann and H. Wagner, Liebigs Ann. Chem., 1986, 647–654 CrossRef CAS PubMed.
- Y. Fukuyama, H. Takahashi, K. Matsumoto, M. Ueda and Y. Miyake, Heterocycles, 2002, 56, 245 CrossRef PubMed.
- X. She, W. Gu, T. Wu and X. Pan, Synth. Commun., 1999, 29, 2625–2628 CrossRef CAS PubMed.
- K. Matsumoto, H. Takahashi, Y. Miyake and Y. Fukuyama, Tetrahedron Lett., 1999, 40, 3185–3186 CrossRef CAS.
- X. She, W. Gu, T. Wu and X. Pan, J. Chem. Res., Synop., 1999, 100–101 RSC.
- X. She, S. Qi, W. Gu and X. Pan, J. Chem. Res., Synop., 1998, 436–437 RSC.
- R. Yamahara, S. Ogo, H. Masuda and Y. Watanabe, J. Inorg. Biochem., 2002, 88, 284–294 CrossRef CAS.
- X. She, X. Jing, X. Pan, A. S. C. Chan and T.-K. Yang, Tetrahedron Lett., 1999, 40, 4567–4570 CrossRef CAS.
- X. L. Wang, J. P. Feng, X.-G. Xie, X. P. Cao and X. Pan, Chin. Chem. Lett., 2004, 15, 1036–1038 CAS.
- X. Jing, W. Gu, P. Bie, X. Ren and X. Pan, Synth. Commun., 2001, 31, 861–867 CrossRef CAS.
- X.-B. Jing, L. Wang, Y. Han, Y.-C. Shi, Y.-H. Liu and J. Sun, J. Chin. Chem. Soc., 2004, 51, 1001–1004 CrossRef CAS PubMed.
- S. Chand and M. G. Banwell, Aust. J. Chem., 2007, 60, 243–250 CrossRef CAS.
- H. Tanaka, M. Shibata, K. Ohira and K. Ito, Chem. Pharm. Bull., 1985, 33, 1419–1423 CrossRef CAS.
- S. Yamauchi and E. Taniguchi, Biosci., Biotechnol., Biochem., 1992, 56, 1751–1759 CrossRef CAS.
- F. Ishibashi and E. Taniguchi, Agric. Biol. Chem., 1989, 53, 1557–1563 CrossRef CAS.
- F. Ishibashi and E. Taniguchi, Agric. Biol. Chem., 1989, 53, 1565–1573 CrossRef CAS.
- Q. Miao, X.-G. Xie, J.-Y. Zhang, X.-G. She and X. Pan, J. Chin. Pharm. Sci., 2007, 16, 41–42 CAS.
- S. Goyal, P. K. Narula, P. Sharma and M. R. Parthasarathy, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1993, 32, 435–439 Search PubMed.
- S. Goyal, P. K. Mohakhud, J. A. Ray, V. K. Rastogi and M. R. Parthasarathy, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1995, 34, 87–92 Search PubMed.
- H. Tanaka, I. Kato and K. Ito, Chem. Pharm. Bull., 1987, 35, 3603–3608 CrossRef CAS.
- W. Gu, X. Jing, X. Pan, A. S. Chan and T.-K. Yang, Tetrahedron Lett., 2000, 41, 6079–6082 CrossRef CAS.
- W. Gu, X. Chen, X. Pan, A. S. C. Chan and T.-K. Yang, Tetrahedron: Asymmetry, 2000, 11, 2801–2807 CrossRef CAS.
- W. Gu, A. X. Wu, X.-G. She and X. Pan, Chin. Chem. Lett., 2001, 12, 485–486 CAS.
- T. Ganesh, K. K. Sharma and G. L. D. Krupadanam, Bull. Chem. Soc. Jpn., 2001, 74, 2397–2399 CrossRef CAS.
- S. Kuwabe, K. E. Torraca and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 12202–12206 CrossRef CAS PubMed.
- X.-B. Jing, C. G. Yan, J. Sun, L. Wang and L. An, Chin. Chem. Lett., 2004, 15, 1392–1394 CAS.
- A. Rouf, M. A. Aga, B. Kumar and S. C. Taneja, Tetrahedron Lett., 2013, 54, 6420–6422 CrossRef CAS PubMed.
- J. Shi, T. Wang, Y. Huang, X. Zhang, Y.-D. Wu and Q. Cai, Org. Lett., 2015, 17, 840–843 CrossRef CAS PubMed.
- M. Massacret, R. Lakhmiri, P. Lhoste, C. Nguefack, F. B. Ben Abdelouahab, R. Fadel and D. Sinou, Tetrahedron: Asymmetry, 2000, 11, 3561–3568 CrossRef CAS.
- W. Bao, Y. Liu, X. Lv and W. Qian, Org. Lett., 2008, 10, 3899–3902 CrossRef CAS PubMed.
- L. I. Pilkington and D. Barker, J. Org. Chem., 2012, 77, 8156–8166 CrossRef CAS PubMed.
- L. I. Pilkington and D. Barker, Eur. J. Org. Chem., 2014, 2014, 1037–1046 CrossRef CAS PubMed.
- X. Chen, X. Ren, K. Peng, X. Pan, A. S. C. Chan and T.-K. Yang, Tetrahedron: Asymmetry, 2003, 14, 701–704 CrossRef CAS.
- Y. Nakamura, M. Hirata, E. Kuwano and E. Taniguchi, Biosci., Biotechnol., Biochem., 1998, 62, 1550–1554 CrossRef CAS.
- J. R. Gage and D. A. Evans, Org. Synth., 1990, 68, 77 CrossRef CAS.
- A. Kuboki, T. Yamamoto and S. Ohira, Chem. Lett., 2003, 32, 420–421 CrossRef CAS.
- A. Kuboki, T. Yamamoto, M. Taira, T. Arishige and S. Ohira, Tetrahedron Lett., 2007, 48, 771–774 CrossRef CAS PubMed.
- E. Valoti, M. Pallavicini, L. Villa and D. Pezzetta, J. Org. Chem., 2001, 66, 1018–1025 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |