Generation, structure and reactivity of tertiary organolithium reagents
Matthew A.
Perry
*a and
Scott D.
Rychnovsky
b aAnacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA, USA. E-mail: mperry@anacor.com bDepartment of Chemistry, University of California, 1102 Natural Sciences 2, Irvine, CA, USA. E-mail: srychnov@uci.edu
Received
29th September 2014
First published on 5th December 2014
Abstract
Covering: up to 2014
Tertiary alkyllithium reagents are very useful intermediates in synthesis. Alkyllithium reagents with adjacent heteroatoms may be formed stereoselectively or may react stereoselectively, and have been used in the synthesis of alkaloids, C-glycosides and spirocycles. An overview of the generation, reactivity and stereochemistry of tertiary alkyllithium reagents will be presented, as well as examples of their use in organic synthesis. The discussion will be focused on a conceptual understanding of the generation and reactivity of these intermediates. The reactions described herein generate fully substituted carbon atoms, and the forces driving stereoselectivity will be discussed in detail. Where appropriate, computational results will be introduced to provide a better understanding for the structure and reactivity of tertiary alkyllithium reagents.
Matthew A. Perry
Matthew A. Perry is a native of Seattle Washington and received his BS in chemistry from the University of Washington in 2006. After two years of research with professor D. Scott Wilbur (UW), he joined the research group of Professor Scott D. Rychnovsky at University of California, Irvine in 2008. There, his research focused on the reductive generation and utility of tertiary organolithiums in the synthesis of alkaloid natural products and spirocyclic heterocycles. After receiving his PhD in 2013, he began his industrial career at Anacor Pharmaceuticals (Palo Alto, Ca) as a Research Scientist developing Boron based small molecule antibiotics.
Scott D. Rychnovsky
Professor Rychnovsky earned his Bachelor's degree from UC Berkeley in 1981 and his PhD from Columbia University in 1986 with Gilbert Stork. After postdoctoral work at Harvard and Yale, he began his academic career at the University of Minnesota in 1988. Professor Rychnovsky moved to UC Irvine as a full Professor in 1995. His research has focused on the synthesis and structural assignment of natural products, as well as the development of new methods for organic synthesis. The generation and reactivity of complex alkyllithium reagents has been a longstanding area of interest.
Preparation of organolithium reagents
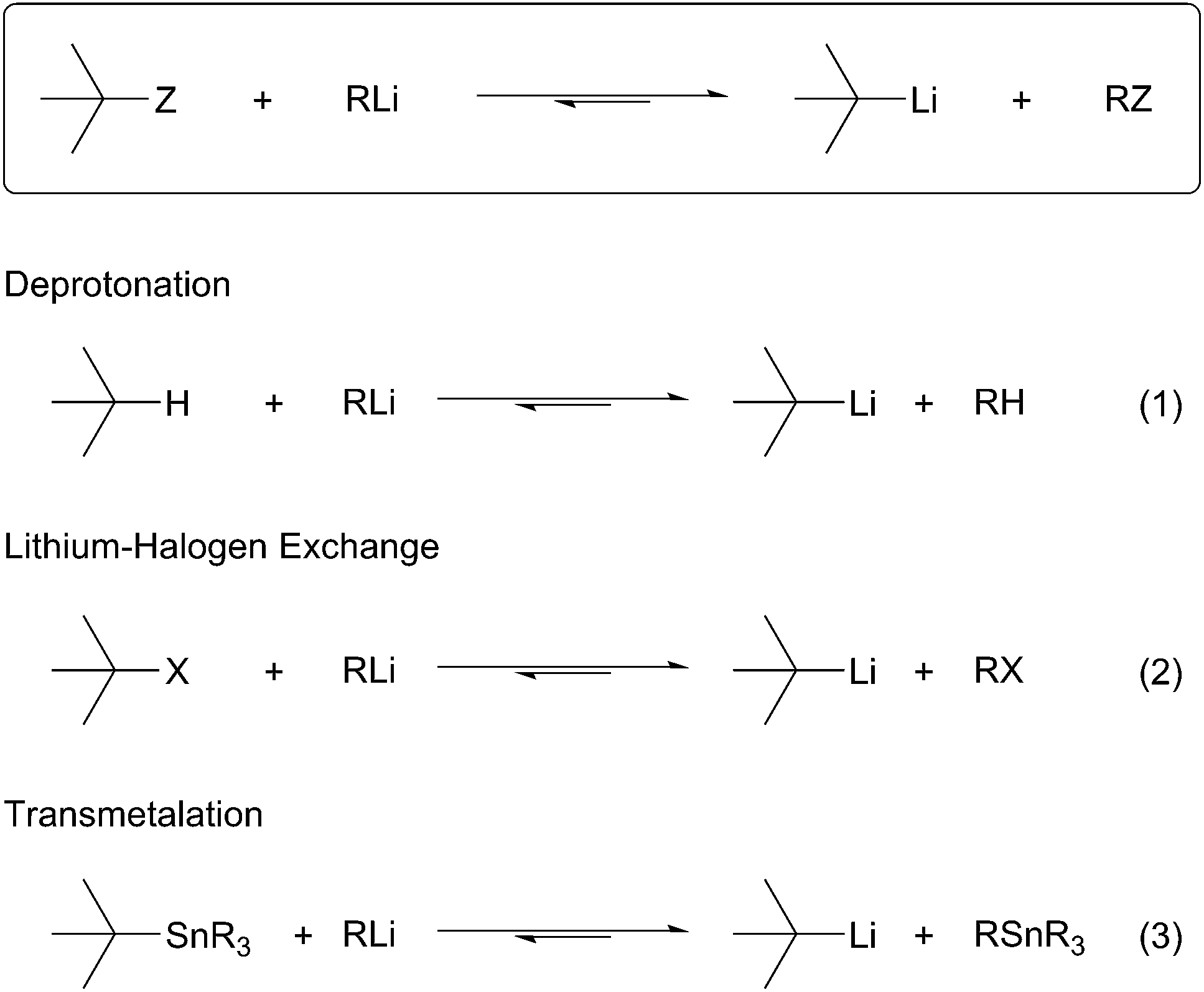
All organolithium species are prepared ultimately from the obvious precursor, lithium–metal. The limited compatibility of lithium–metal with organic substrates has resulted in the development and use of more general methods for the selective introduction of C–Li bonds. Schlosser describes two main methods to generate organolithium species.1 The first method, termed permutational interconversion, involves the displacement of a substituent Z with subsequent transfer of the electrofugal group to the original metal carrier (Scheme 1). This method may be further divided into three classes based on the mechanism of the transformation; (1) deprotonation, (2) transmetalation, and (3) lithium–halogen exchange. Deprotonation is the most direct method, but is limited by the low acidities of C–H bonds, poor regioselectivity, and limited substrate scope (Scheme 1, eqn (1)). Lithium–halogen exchange may be the most common method to selectively introduce a C–Li bond (Scheme 1, eqn (2)). Early work performed by Gilman determined lithium–halogen exchange to be an equilibrium process favoring the formation of the less basic, more stable organolithium.2 Transmetalation occurs through formation of a lithium–metal ate complex followed by lithium–metal exchange:3 the most applicable method involves Sn–Li exchange (Scheme 1, eqn (3)). Organolithiums tend to react rapidly and reversibly with stannanes under thermodynamic control to produce the more stable organolithium. To a large extent the current knowledge regarding reactivity and configurational stability of organolithiums was elucidated by their preparation from organostannanes. In general, permutational interconversion is the most mild and substrate compatible method for the generation of functionalized organolithium species.
Scheme 1 Permutational interconversion methods.
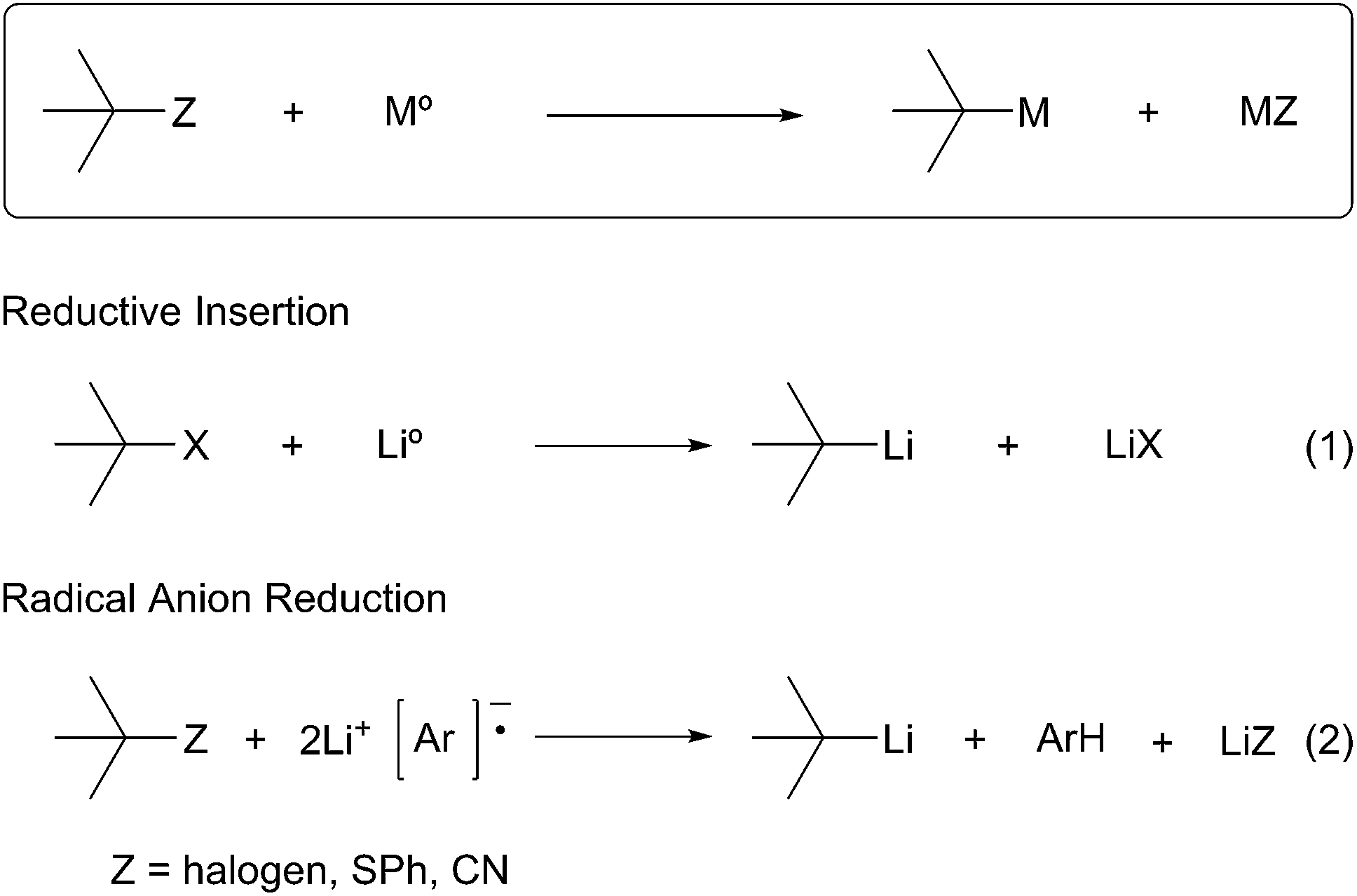
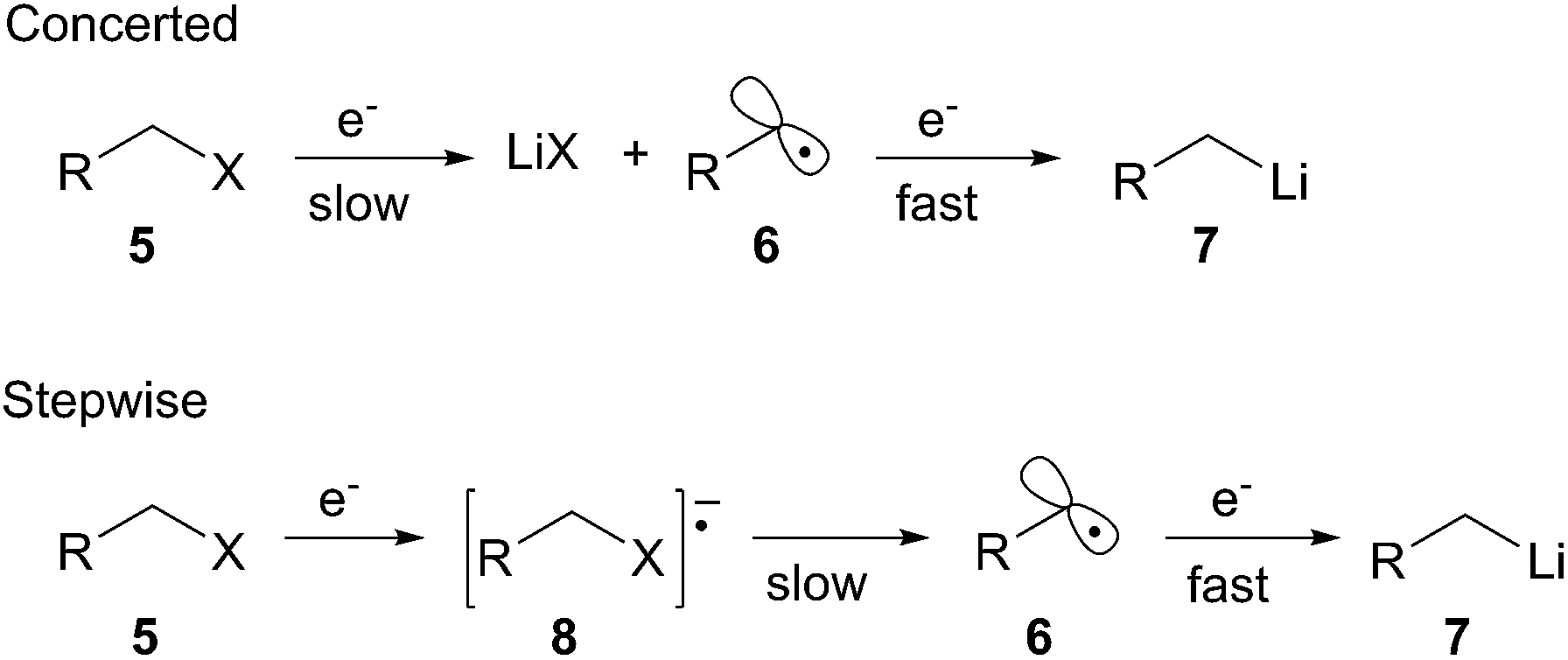
The second general method for preparing organolithium species is reductive insertion. This process involves insertion of lithium–metal into an activated C–Z bond (Z = I, Br, Cl, CN, etc.) with concomitant release of Z as a nucleofugal group (Scheme 2). The common commercially available unfunctionalized organolithium reagents (n-, sec-, tert-butyllithium) are prepared by reductive lithiation of alkyl chlorides and lithium–metal.4 In alkyl halides, an electron from a lithium atom enters the C–X σ* orbital with concomitant C–X sigma bond cleavage to the alkyl radical and halide anion in a concerted process.5 The radical intermediate is then reduced by a second lithium atom to provide the organolithium (Scheme 2, eqn (1)). Since the reduction is a radical process, the relative rate of formation is contingent upon the stability of the radical intermediate. Complementary to deprotonation, reductive lithiation is fastest for more substituted alkyllithiums and slowest for aryllithiums (Fig. 2).
Scheme 2 Reductive insertion methods.
Arene radical anion reducing reagents
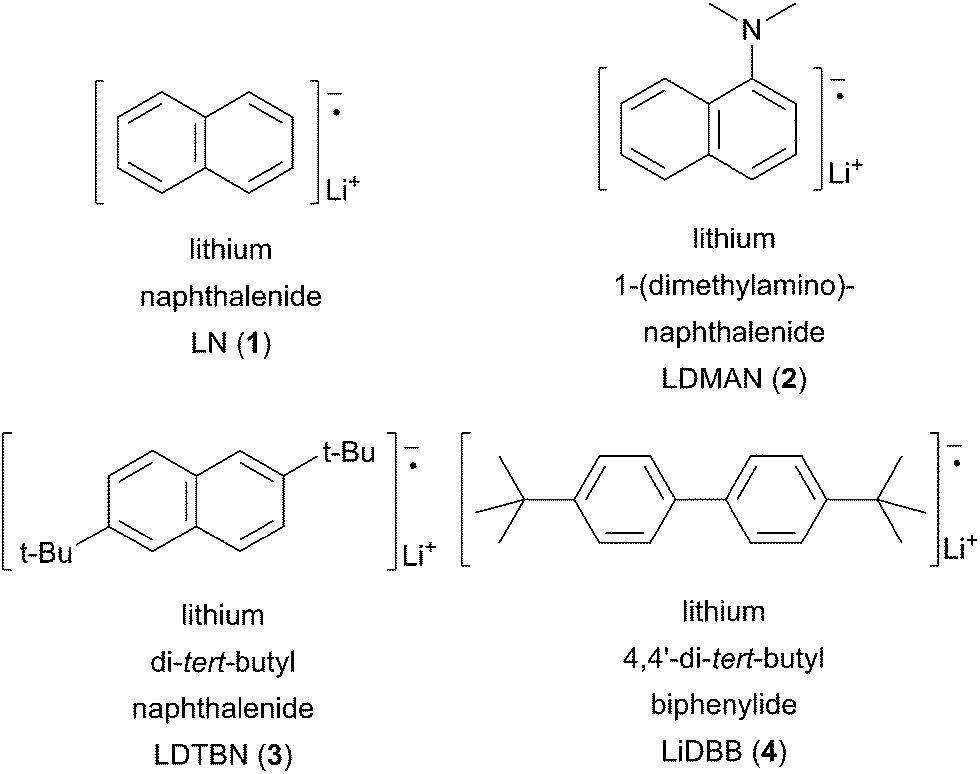
More recently, arene radical anion reductions have been developed to circumvent the variability and undesired reactivity of lithium–metal reductions (Scheme 2, eqn (2)). Polynuclear aromatics accept electrons from lithium–metal to form radical anion intermediates that act as single electron transfer (SET) reagents between the metal and the reducible functionality (Fig. 1). The “dissolving metal” conditions provided by arene radical anions are homogeneous and therefore provide efficient and rapid electron transfer under a wide range of temperatures. In addition, the stoichiometry between the arene radical species and the compound to be reduced is precisely controlled. One limitation to dissolving metal conditions involves the nucleophilic addition of the intermediate arene radical/organolithium to an electrophile on the substrate being reduced. Another drawback is the radical anion solution must be freshly prepared before use; their highly reducing nature results in a short shelf life. This method is complementary to traditional dissolving metal conditions (i.e. Li/NH3 or Na/NH3) in which the anion generated upon reductive insertion of lithium or sodium is rapidly protonated by the solvent ammonia.
Fig. 1 Common aromatic radical anion carriers.
Reductive lithiation proceeds by the same general mechanism regardless of the radical anion carrier used. The first step involves a SET to the LUMO of the nucleofugal substituent of the substrate being reduced. The loss of the nucleofuge to form the intermediate alkyl radical may proceed through either a stepwise or concerted process (Scheme 3). A stepwise process is possible when the nucleofuge is able to accept an electron into a low-lying empty d-orbital or π*-orbital. A concerted process occurs when the nucleofuge is unable to accommodate an additional electron into a low energy molecular orbital prior to bond cleavage. The first SET to form the alkyl radical intermediate is the rate-limiting step in the reduction to an organolithium. The second SET from a second equivalent of radical anion carrier forms the organolithium intermediate.
Scheme 3 General mechanism for reductive lithiation.
The rate of reduction for the first SET is dictated by the relative stability of the resultant radical intermediate. Therefore, the reduction is fastest for tertiary alkyllithiums and slowest for aryllithiums (Fig. 2).
Fig. 2 Relative rate of formation by reductive lithiation.
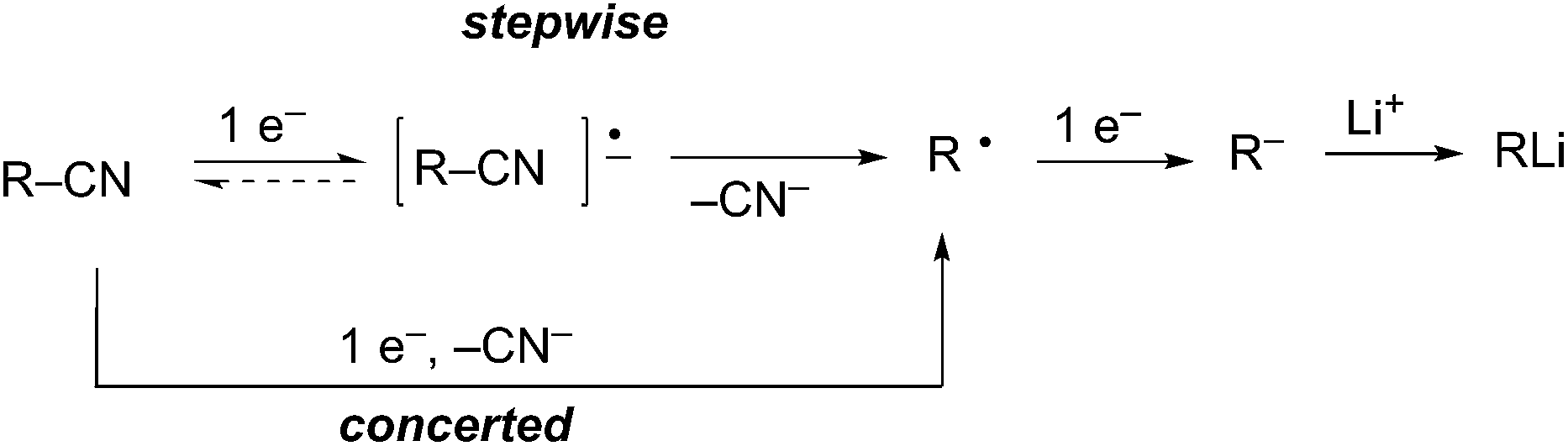
Reductive decyanation is mechanistically related to reductive desulfurization6 and dehalogenation.7 Initial single electron transfer (SET) to the alkyl nitrile, a potentially reversible process, leads to a transient radical anion that fragments to generate an alkyl radical and cyanide (Scheme 4). Alternately, a concerted single electron transfer/cleavage sequence can also lead directly to the alkyl radical. A second single electron transfer to the alkyl radical produces a carbanion that is protonated in protic media (e.g. NH3) or trapped by an electrophile in aprotic media (e.g. THF, ether).
Scheme 4 Generalized mechanism for reductive decyanation.
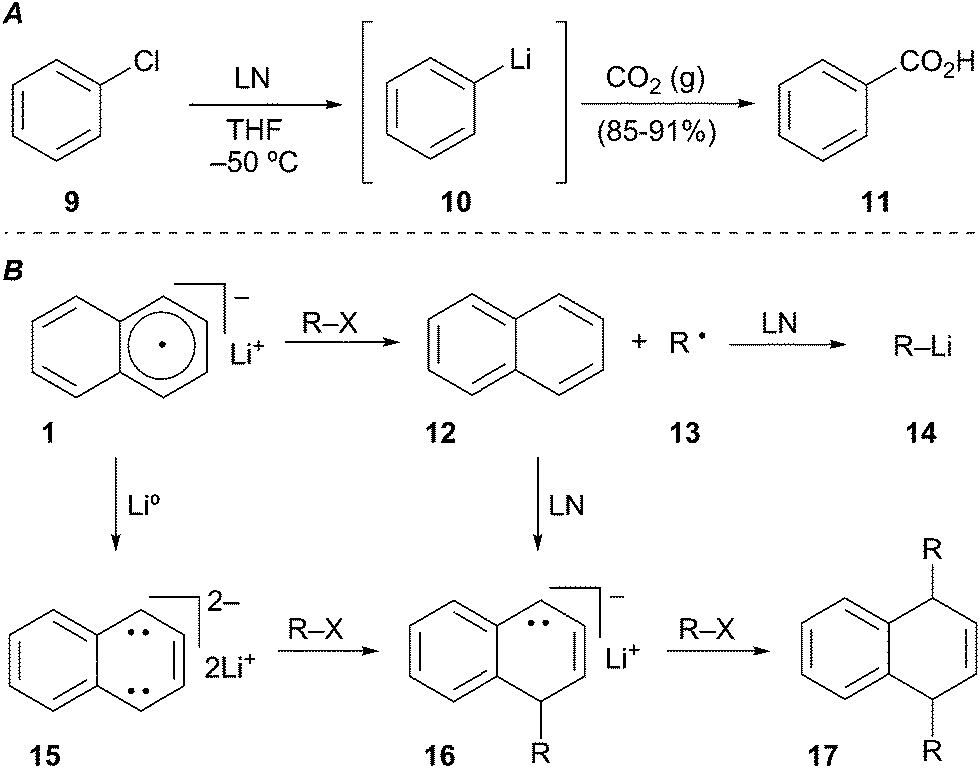
Screttas was the first to describe the use of a polynuclear aromatic radical carrier for reductive lithiation.8 In this case, naphthalene was used with lithium–metal to mediate the reduction of chlorobenzene (9) to phenyllithium (10). Electrophilic trapping of the organolithium with carbon dioxide gas efficiently provided benzoic acid (11, Scheme 5A). The limited utility of LN (1) arises from the propensity of naphthalene to undergo deleterious alkylation or dimerization (Scheme 5B). In the desired pathway, reduction of naphthalene by lithium–metal generates radical anion 1 that mediates two sequential SET processes with RX to provide the alkyllithium 14. Unproductive pathways stem from the Birch reduction of LN to the aromatic dianion 15. Coupling of the dianion with either RX or the alkyl radical 13 afford alkylated anion 16. The resulting anion 16 may undergo a second alkylation reaction in the presence of primary or secondary alkyl halides (RX) to generate 17. To avoid the undesired by-product reactivity associated with lithium naphthalenide (LN) and chromatographic separation of naphthalene, other arene radical anion carriers were developed.
Scheme 5 Potential reaction pathways of lithium naphthalenide.
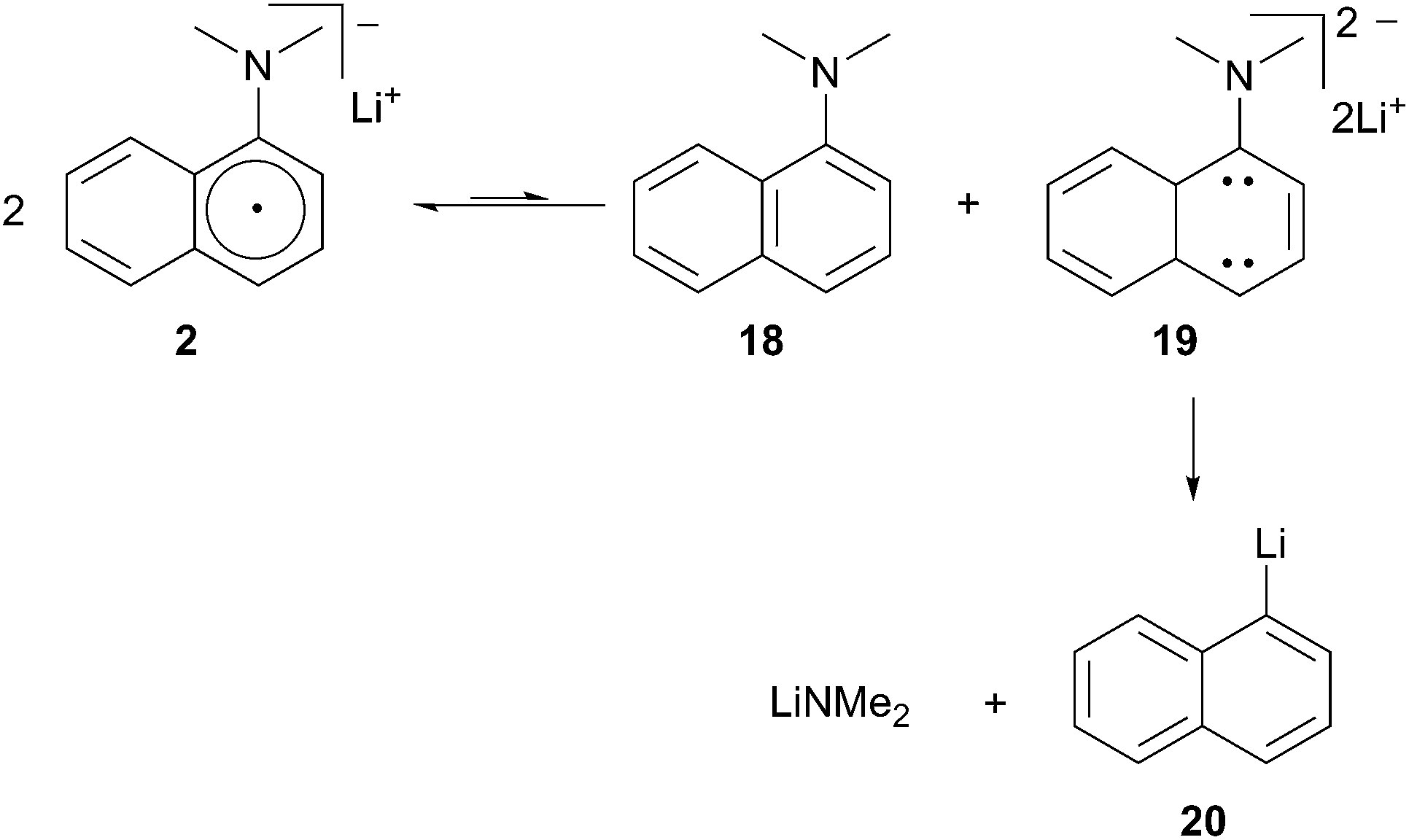
Cohen described the use of aqueous acid soluble 1-dimethylaminonaphthalene (DMAN) as an alternative solution to the issue of purification.9 The generation and use of lithium 1-(dimethylamino)-naphthalenide (2) requires that the reaction take place at −45 °C or below to avoid degradation of the radical anion to 1-lithionaphthalene.10 Cohen postulated a decomposition pathway involving an unfavorable equilibrium between two equivalents of LDMAN (2) to afford DMAN and dianion 19 (Scheme 6). The dianion further decomposes to the more stable lithium dimethyl amide and 1-lithionaphthalene 20.
Scheme 6 Decomposition pathway of LDMAN.
In an effort to prevent the undesired alkylation problems, Freeman reported on the use of lithium 4,4′-di-tert-butylbiphenylide (LiDBB, 4) as an arene radical carrier.11 Freeman's reagent has the advantage of minimizing deleterious alkylation side reactions based on steric hindrance. The steric bulk of the tert-butyl substituents inhibit bond formation, which require distances of <2.0 Å. Fortunately, electron transfers can occur between 7–9 Å allowing for single electron reduction to occur.12 In addition to providing beneficial steric interactions, the tert-butyl groups also infer a higher reduction potential (Table 1).
Table 1Reduction potentials of arene radical carriers
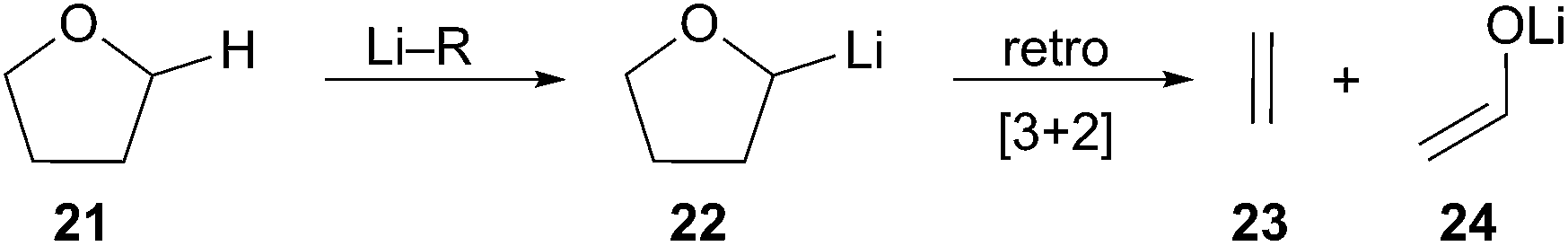
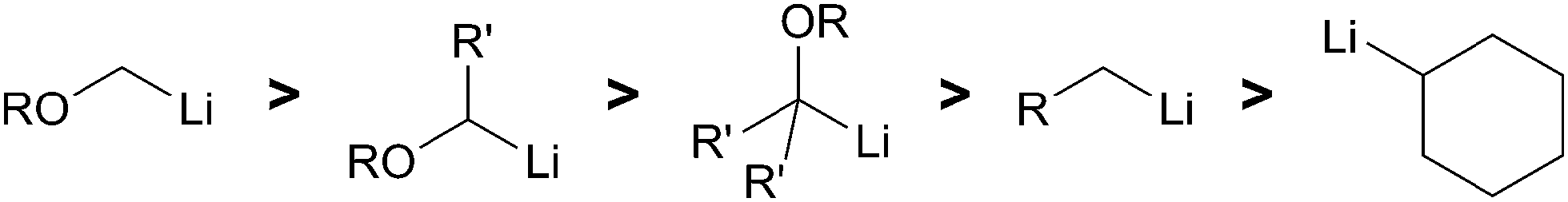
Solvent effects play a key role in the generation of organolithiums. The solubility and stability of an arene radical species dictates which solvents are appropriate. The extremely basic nature of organolithiums can result in deprotonation of ethereal solvents such as THF. Both LN and LiDBB have limited solubility in solvents other than THF as described by Cohen.13 At times, THF has been detrimental to the optimum utilization of reductive lithiation. The ability of organolithiums to remove a proton from the 2-position of THF can be a major problem chiefly for the more basic organolithiums or at temperatures above 0 °C.14 Deprotonation of THF produces lithiated intermediate 22 that subsequently undergoes a facile retro-[3 + 2] rearrangement to afford acetaldehyde enolate 24 and ethene gas (Scheme 7).15 The instability of highly basic tertiary organolithiums is typified by the relatively short 5.6 hour half-life of tert-BuLi in THF at −40 °C and 0.7 hour half-life at −20 °C.16 A decrease in the rate of electrophilic trapping of the tertiary organolithium–may result in greater competitive deprotonation of solvent. This decomposition pathway has become a fundamental limitation for reductive lithiation. The restrictions imposed by the solubility of the radical anion reagent and the high basicity of the organolithiums produced leave few alternative solvents that are compatible with the reaction conditions.
Scheme 7 Decomposition of THF.
Tertiary α-alkoxyorganolithiums
Generation, structure and reactivity
McGarvey et al. demonstrated that α-alkoxy substituted lithium species were stabilized compared to the alkyllithium counterparts.17 The relative stability of such α-alkoxyorganolithiums and alkyllithiums was determined through competitive exchange experiments (Fig. 3). Both primary and secondary α-alkoxyorganolithiums are more stable than methyllithium while the tertiary α-alkoxyorganolithium are more stable than primary alkyllithiums. In most cases, the stereospecific transformations of transmetalation from tin and subsequent electrophilic trapping allow for stereoselective C–C bond formation.
Fig. 3 Relative stability of α-alkoxyorganolithium and alkyllithium species.
Generation by permutational interconversion
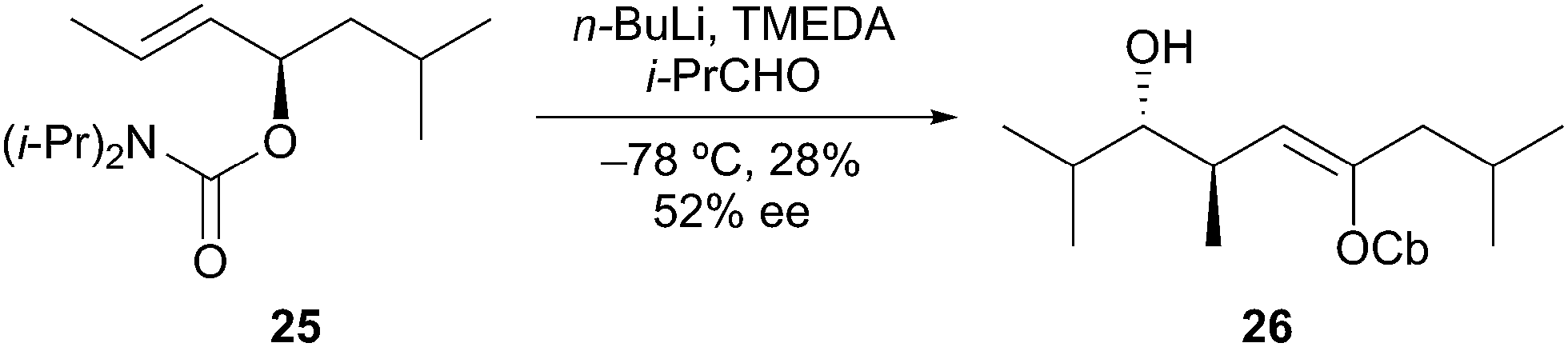
Deprotonation. Hoppe has studied extensively the lithiation of carbamates alpha to oxygen by deprotonation in racemic and enantioselective processes.18 Unfortunately, the substrate features and reaction conditions needed for appropriate reactivity tend to work against configurational stability. Polar solvents (ether, THF, TMEDA) required for lithium–halogen exchange reactions generally make the lithiated intermediates configurationally unstable. In some cases, racemization can be decreased with the presence of a stabilizing α-heteroatom or small ring. The same types of issues arise for the formation of α-alkoxyorganolithium species by deprotonation. The functionality needed to acidify the alkoxy proton(s) and favor deprotonation also favor racemization of the organolithium species formed. Hoppe overcame these issues by using a carbamate group to stabilize the lithiated intermediate. Tertiary allylic alkoxyorganolithium derived from 25, formed by deprotonation with n-butyllithium in a mixture of ether/hexane/TMEDA, was configurationally stable over a period of a few hours (t1/2 = 3 h at −78 °C). Electrophilic quenching of the organolithium with isobutyraldehyde provided the enantiomerically enriched (52% ee) alcohol 26 in low yield (Scheme 8). Hoppe later reported increased configurational stability when ether was excluded from the reaction conditions.19 More recent examples demonstrating the formation of tertiary α-alkoxyorganolithiums by deprotonation of epoxides20 and tetrahyrdofurans21 were described in the literature. Aggarwal and co-workers have also described an efficient approach for the enantioselective synthesis of tertiary boronic esters from the corresponding secondary benzylic alcohols by deprotonation and stereospecific 1,2-boronate rearrangement.22
Scheme 8 Stabilized α-alkoxyorganolithium by deprotonation.
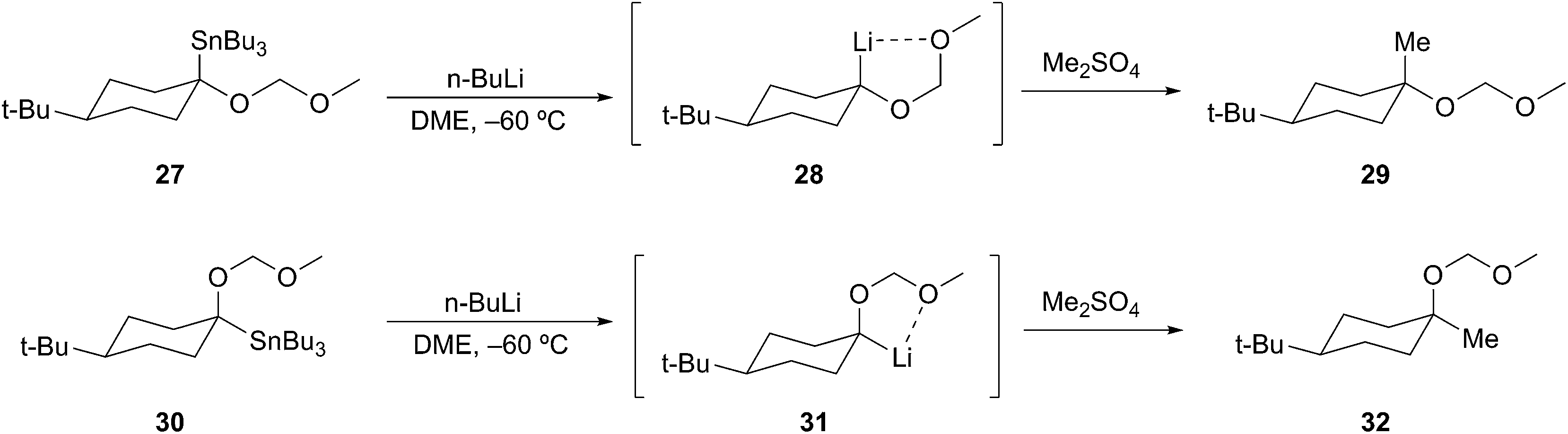
Transmetalation. Stille was the first to recognize the synthetic potential of unstabilized α-alkoxyorganolithium reagents.23 Prepared from their corresponding stannanes through tin–lithium exchange, the resulting organolithium species were configurationally stable at low temperature (Scheme 9). McGarvey and co-workers subsequently demonstrated the stereospecificity of the transformation using diastereomeric tertiary-α-alkoxystannanes 27 and 30 to undergo Sn–Li exchange to tertiary organolithiums with retention of configuration. The α-alkoxyorganolithiums 28 and 31 were found to be configurationally stable (−60 °C), and could be alkylated with retention of configuration to provide 29 and 32, respectively. These studies demonstrate the enhanced stability of α-alkoxyorganolithiums.
Scheme 9 Tertiary α-alkoxyorganolithiums generated by Sn–Li exchange.
Generation by reductive insertion
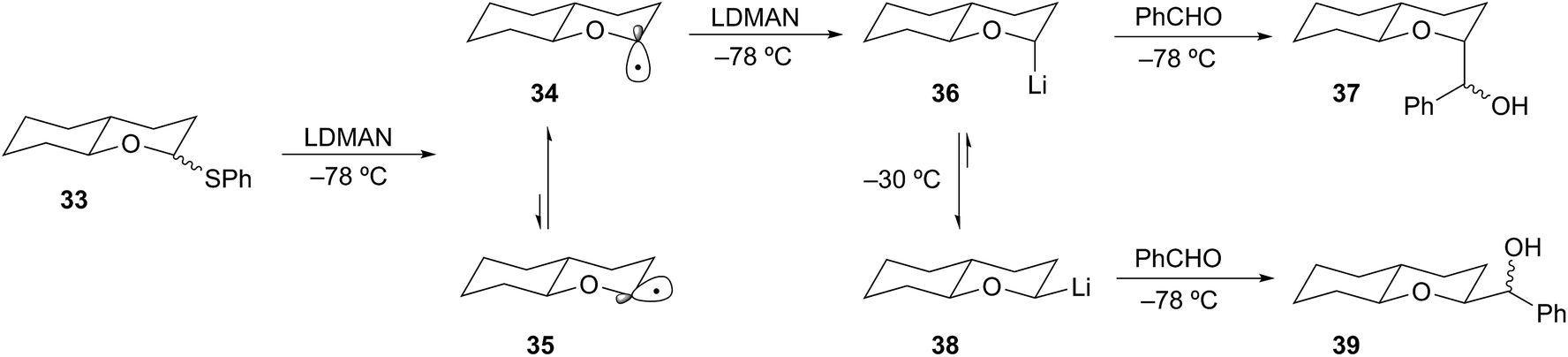
Complementary to the permutational interconversion approach is the reductive insertion method described earlier. Cohen demonstrated that formation of α-alkoxyorganolithium intermediates by reductive lithiation of thiophenyl ethers is a general and reliable method.24 These studies also show that stereoelectronic effects strongly influence the stereochemical disposition of the lithium species. For example, reductive lithiation (LDMAN) of thioether 33 gave a mixture of radicals 34 and 35. Rapid equilibration of 34 and 35 through a small energy barrier (<0.5 kcal mol−1)25 affords the thermodynamically preferred axial lithium 36 (Scheme 10).26 A favorable HOMO–SOMO anomeric stabilization involving overlap of the oxygen lone pair with the SOMO of the radical can rationalize the preferential formation of the axial radical 34.27 In this way, the configuration of thioether 33 is inconsequential to the stereochemical outcome of the reduction. Facile reduction of the radical 34 by a second equivalent of LDMAN (kred = 1 × 109 M s−1) provided the axial organolithium. Consequently, this organolithium is destabilized by an anti-anomeric (HOMO–HOMO) interaction.28 Organolithium 36 is configurationally stable at −78 °C and can be efficiently trapped to provide 37 in good yield and high diastereoselectivity (ax/eqn (95):5). Epimerization of 36 can be accomplished by raising the temperature of the reaction to −30 °C to provide the lower energy isomer 38. Subsequent cooling to −78 °C and trapping with benzaldehyde provided 39 in 87:13 dr. While this example does not involve a tertiary α-alkoxyorganolithium, the stereochemical stability and lability of the secondary case described is an important feature of α-alkoxyorganolithium chemistry.
Scheme 10 Cohen's reductive lithiation to equilibrating α-alkoxyorganolithiums.
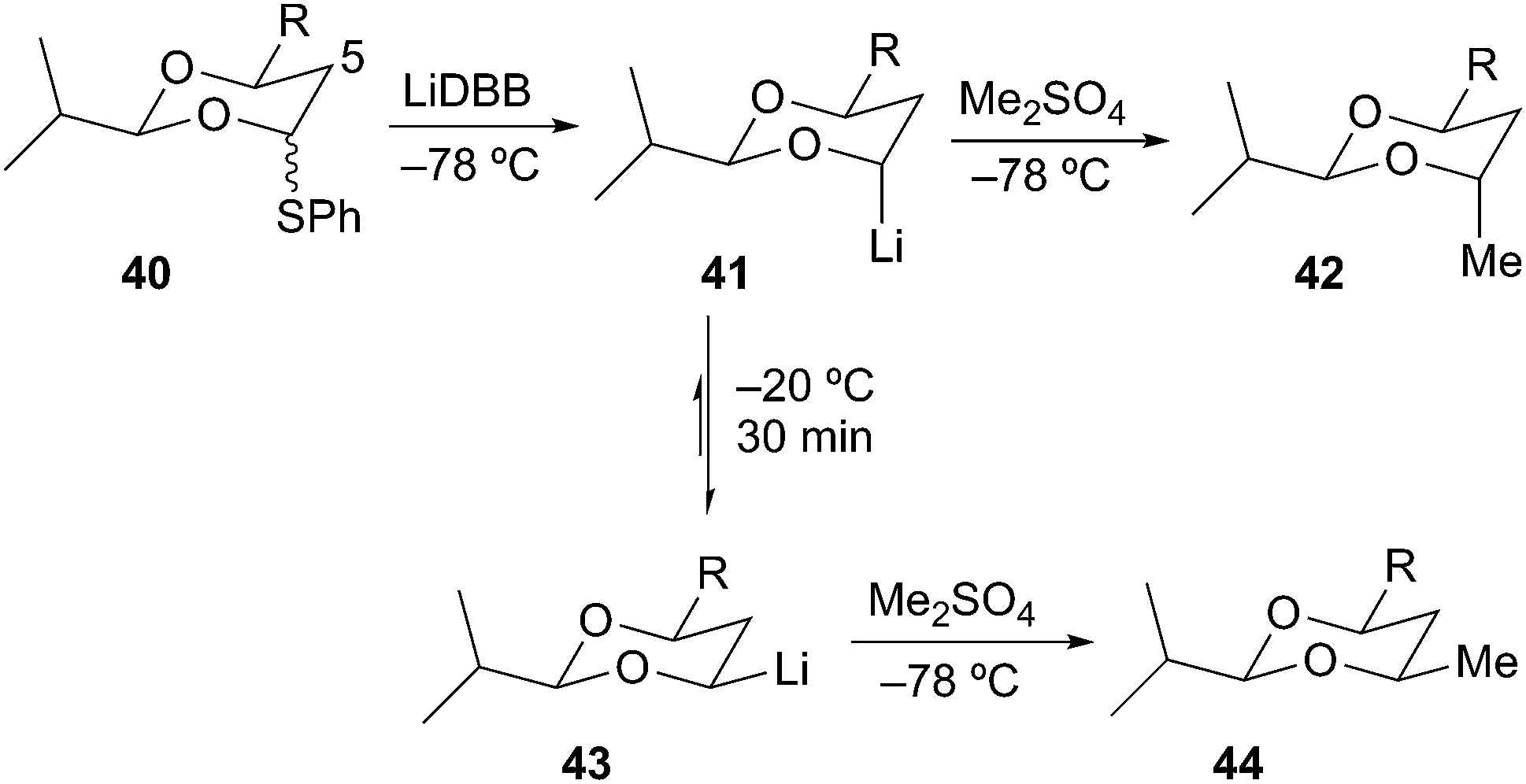
Rychnovsky and Skalitzky later demonstrated high levels of selectivity in the reductive lithiation of 4-phenythio-1,3-dioxanes to provide stereochemically defined 1,3 diols. Reductive lithiation of 40 with LiDBB at −78 °C gave the axial alkyllithium 41, which upon alkylation with dimethyl sulfate afforded the protected anti-1,3-diol 42 in 79% yield and >99:1 selectivity (Scheme 11).29 The equatorial alkyllithium 43 was prepared by equilibration of axial organolithium 41 at −20 °C for 30 min followed by alkylation at −78 °C to give the protected syn-1,3-diol 44 in 79% yield and >99:1 dr. Beak has described these epimerizing alkyllithium systems as Dynamic Thermodynamic Resolutions in which the reactive intermediate is equilibrated prior to reaction with the next reagent.30
Scheme 11 Rychnovsky's reductive lithiation and equilibration of acetonides.
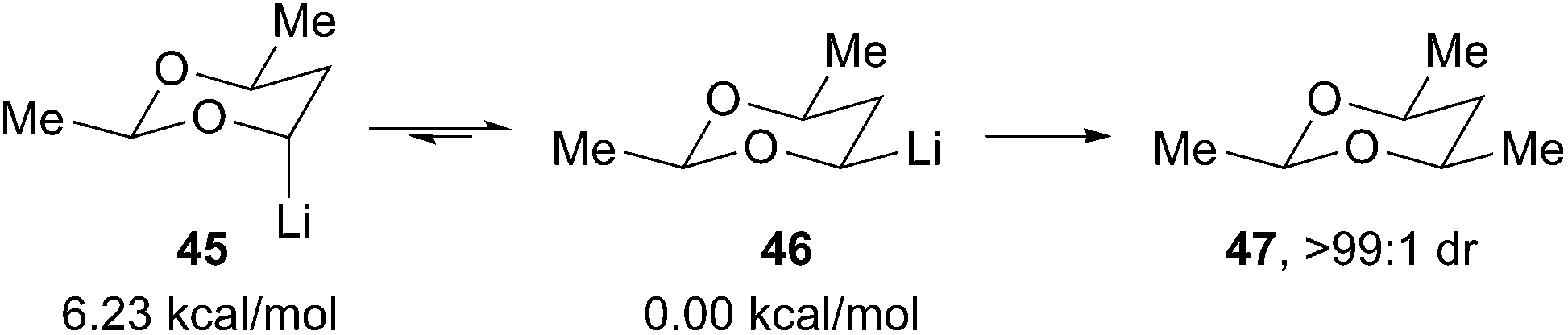
The equatorial alkyllithium 43 is more stable than the axial isomer 41 by at least 2.4 kcal mol−1 based on the equilibration experiment in Scheme 11. Rychnovsky and Skalitzky reported computational studies to evaluate the thermodynamic preference for equatorial over axial disposition.31 However, the studies were carried out many years ago using only modest levels of theory (B3LYP/6-31G(d), but larger systems were evaluated at the HF/3-21G minima) without explicit solvation. The calculations showed a strong preference for the equatorial isomer, varying from 5.3 to 6.2 kcal mol−1 depending upon the substitution pattern around the 4-lithio-1,3-dioxane ring (Scheme 12). Experimentally, the equilibration of systems with C-5 substitution equilibrated more slowly, but the calculated preference for the equatorial isomer was essentially unchanged. This conclusion is consistent with experiments where the equilibrium was approached from both directions. Although all of the 4-lithio-1,3-dioxane systems studied showed a large thermodynamic preference for the equatorial isomer, the equilibration strategy was only practical for the less substituted systems as shown in Scheme 11.
Scheme 12 Calculated energies for equilibrating acetonide organolithiums.
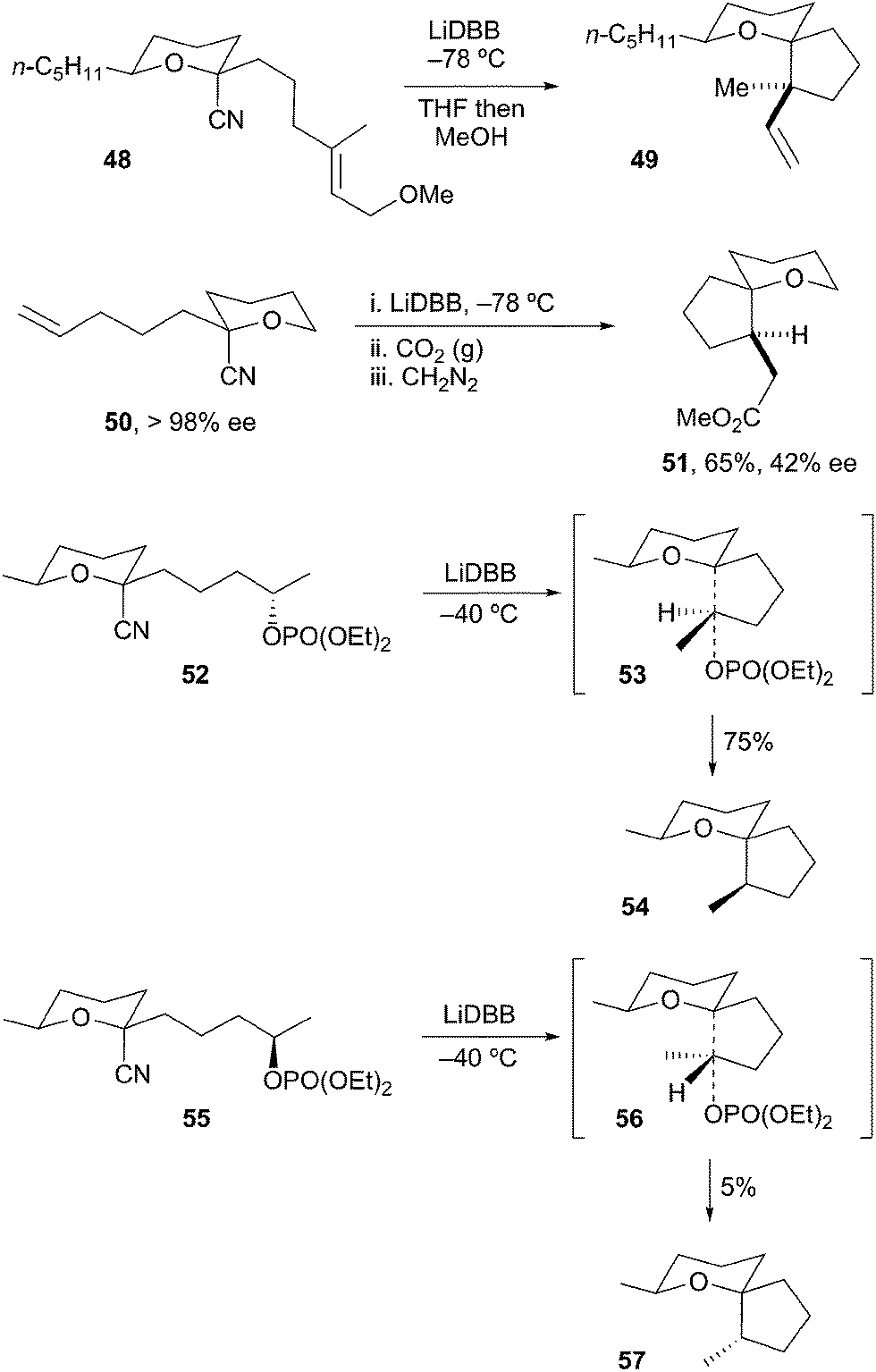
The generation and spiroannulation of tertiary α-alkoxyorganolithiums has also been described. Rychnovsky and Takaoka reported on the annulation of lithiated THP 48 onto tethered alkenes to afford spirocycle 49 (Scheme 13).32 In a similar fashion, Rychnovsky et al. cyclized lithiated THP 50 by a carbolithiation/carboxylation strategy to provide spiro-THP 51 in good yield and high diastereoselectivity. The optical purity of the reductive cyclization product 51 (42% ee) indicated a lifetime for the radical intermediate (ca. 2.4 × 10−7 s) that was too brief for a radical cyclization to take place. Thus, the reduction of 50 to 51 proceeds via cyclization of the alkyllithium. Rychnovsky and Morin later described the stereoselective spiroannulation of lithiated THP 52 onto resolved secondary tethered electrophiles to provide spirocycles such as 54.33 As might be expected based on transition states 53 and 56, the rate of cyclization is strongly dependent on the configuration of the electrophilic carbon. In all instances, the tertiary α-alkoxyorganolithium intermediate was configurationally stable at −78 °C and reacted with retention of configuration.
Scheme 13 Reductive spiroannulation of tertiary α-alkoxyorganolithiums.
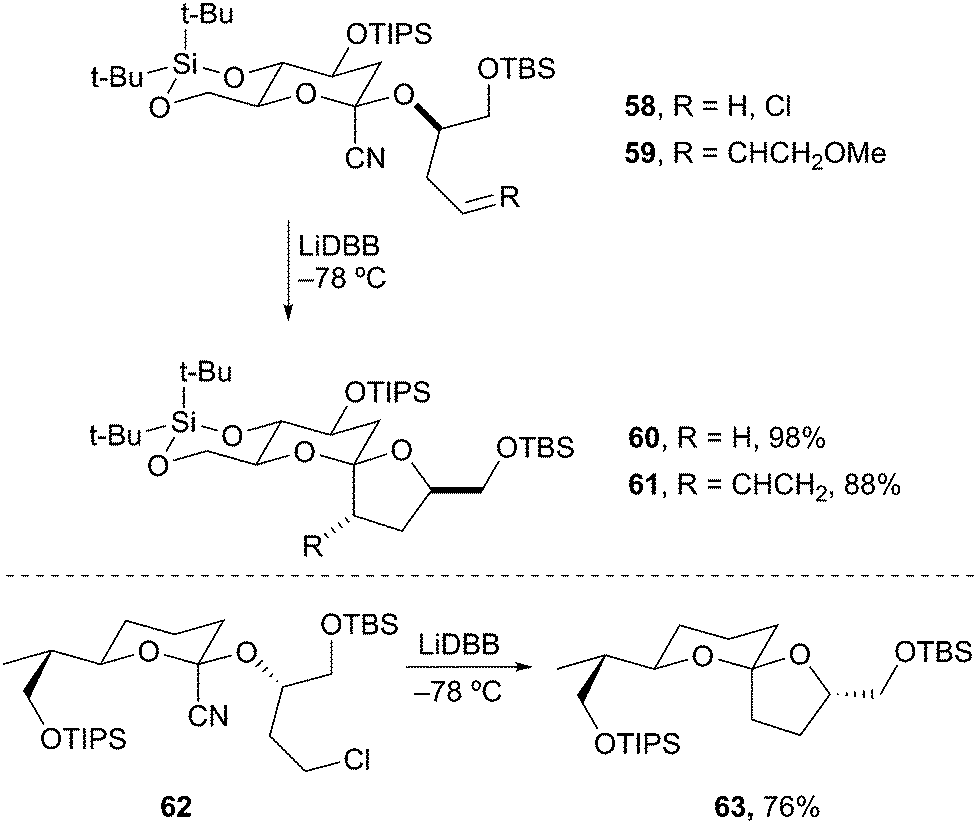
Spiroacetal motifs are common in natural products and several methods have been developed for their synthesis.34 Most methods rely on acid-catalyzed cyclization, which lead to a thermodynamic mixture of spiroacetals. The most stable configuration usually positions both oxygen atoms in a spatial arrangement favoring double anomeric stabilization.35 A number of natural products contain contra-thermodynamic spiroacetals (only a single anomeric stabilization) and have been more difficult to access. Rychnovsky and coworkers accomplished the construction of contra-thermodynamic spiroacetals by reductive lithiation and intramolecular trapping of 2-cyanotetrahydropyranyl acetals. Specifically, reductive lithiation of 2-cyano-THP 58 produced an axially disposed 2-lithio-THP that cyclized with retention to give spiroacetal 60 in 98% yield as a single diastereomer (Scheme 14). The cyclization also proceeds via an SN2′-type mechanism to provide 4-alkenyl tetrahydrofuran spiroacetal 61. Rychnovsky and Vellucci subsequently applied this approach to the synthesis of the A/B non-anomeric spiroacetal of pectenotoxin 2.36 The reductive cyclization of cyano acetal 62 provided the non-anomeric spiroacetal 63 in 76% yield as a single diastereomer. This approach constitutes the first highly diastereoselective approach to the less stable spiroacetal of the pectenotoxin natural products.
Scheme 14 Reductive spiroannulation to contra-thermodynamic spiroacetals.
Tertiary α-aminoorganolithiums
Generation, structure and reactivity
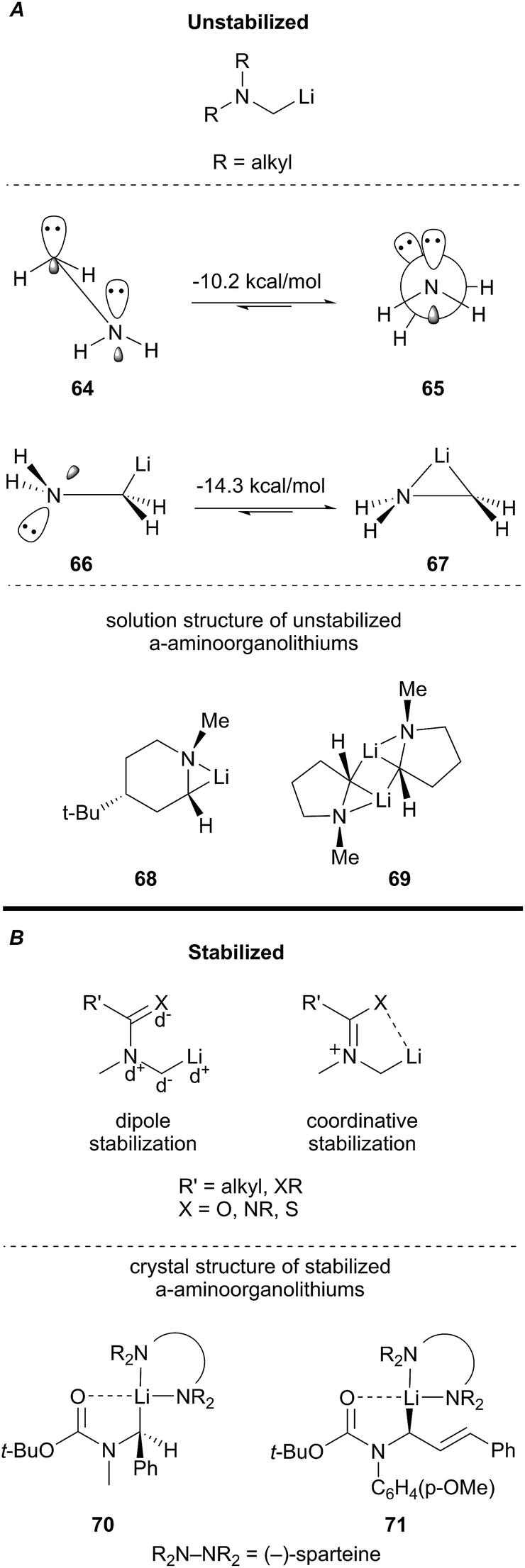
The reactivity of a α-aminoorganolithium depends on the nature of substitution at nitrogen. Broadly, there are two classes of α-aminoorganolithiums: unstabilized and stabilized α-aminoorganolithiums (Fig. 4).37 Unstabilized α-aminoorganolithiums possess simple alkyl substituents that infer little to no stereoelectronic or coordinative stabilization of the C–Li bond. Computational studies indicate that N–Li bridging favors the syn conformation (67) in α-aminoorganolithiums. The solution state structure of 68 and 69 support that hypothesis (Fig. 4A).38 The analogous syn conformation of the α-aminocarbanion (64) is 10.2 kcal mol−1 higher in energy and prefers to adopt a staggered geometry (65). However, nitrogen–lithium bridging plays a negligible role in the case of stabilized α-aminoorganolithiums (Fig. 4B). Stabilized α-aminoorganolithiums benefit from a combination of a favorable dipole arrangement and heteroatom–Li coordination to form a carbon centered anion.39 This effect has been unequivocally demonstrated in the crystal structures of N-acyl α-aminoorganolithiums 70 and 71.40
Fig. 4 Geometry and stabilization of α-aminoorganolithiums.
Generation by permutational interconversion
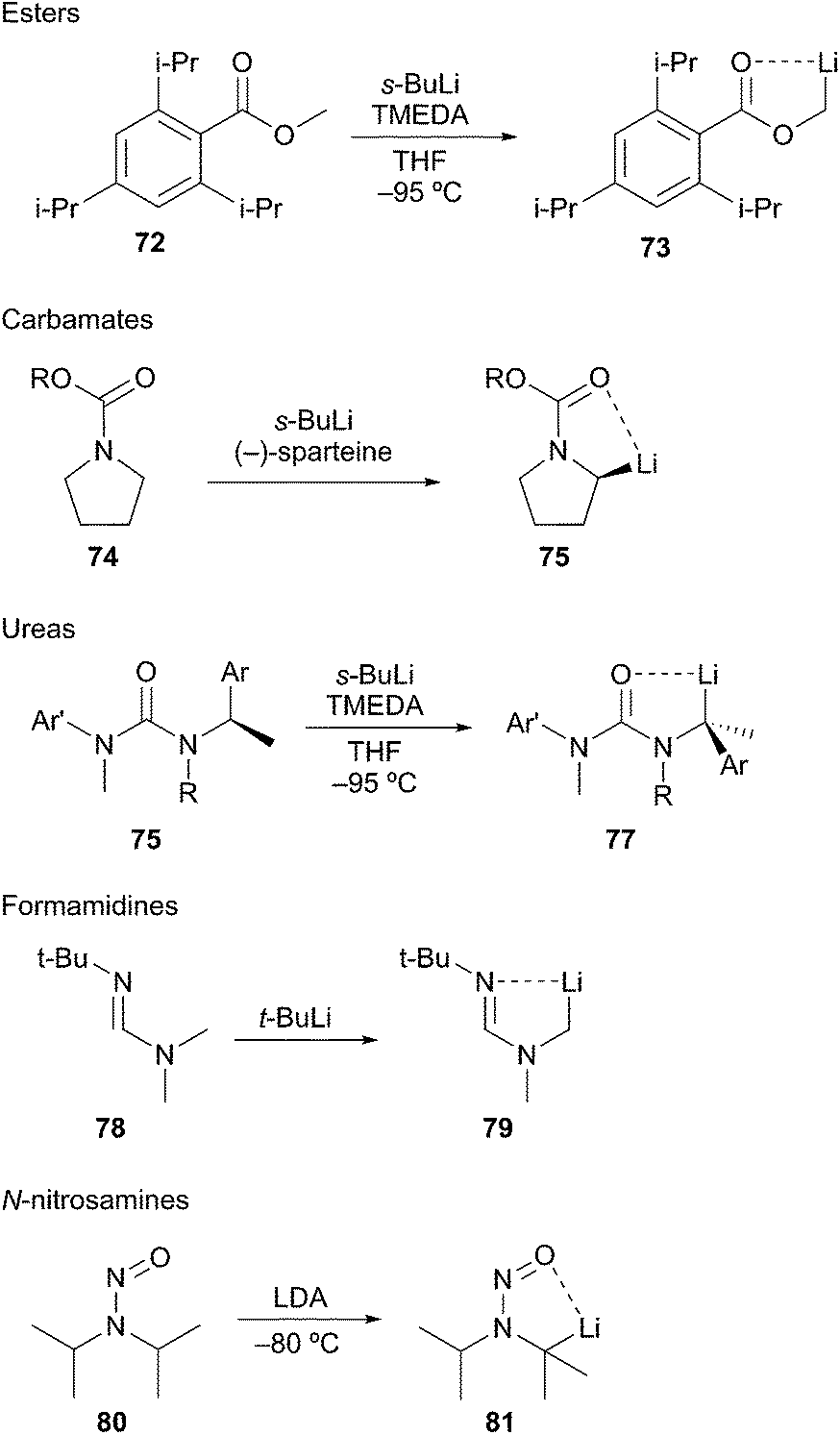
Deprotonation. Permutational interconversion methods for generation of α-aminoorganolithiums mostly rely on deprotonation and tin–lithium exchange. Simple alkyl amine substrates generated by deprotonation with strong organolithium bases exhibit poor regioselectivity and have a high kinetic barrier.41 Efforts to devise direct alkylamine metalation procedures have not resulted in generally applicable methods. One strategy with limited success involves the introduction of an electron-withdrawing group (i.e. nitrosyl,42 carbamate,43 formamidine,44 urea45) on nitrogen to increase the acidity of the adjacent protons to make deprotonation achievable (Scheme 15). The most reliable methods use benzylic or cyclic amines where the amine contains a substituent capable of coordinating the organolithium.46 Generating α-aminoorganolithiums by lithium–halogen exchange is challenging due to the instability and synthetic intractability of α-haloamines. Tin–lithium transmetalation has proven to be a successful approach into these highly substituted compounds.63–67
Scheme 15 Deprotonation to stabilized α-heteroatomorganolithiums.
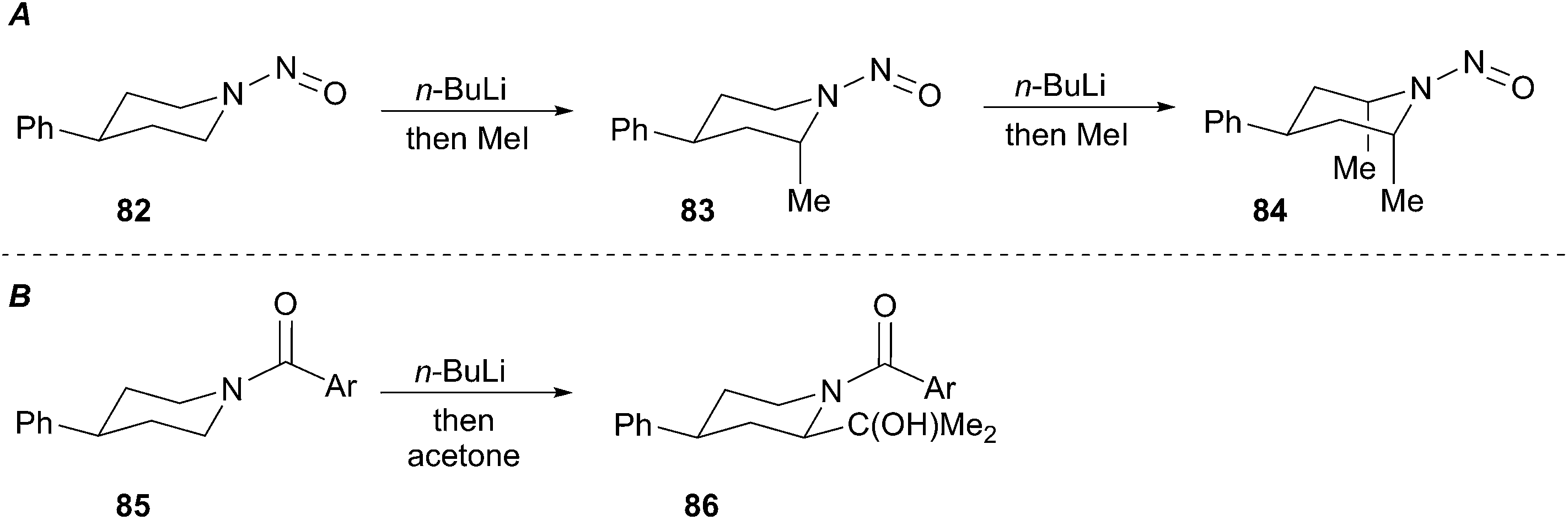
Seebach developed efficient low-temperature lithiation procedures of N-nitrosamines using lithium diisopropylamide (LDA).47 The resulting lithiated species react in good yields with a wide range of electrophiles. A noteworthy application of this method involves the lithiation and alkylation of tertiary α-amino positions as illustrated in Scheme 15 (i.e.81). Further developments revealed a highly diastereoselective alkylation of cyclohexyl nitrosamine 82 to give exclusively axially alkylated products 83 and 84 (Scheme 16A). The major drawback of N-nitrosamines is attributed to their mutagenic and carcinogenic properties.48 Extreme care to minimize exposure is necessary when using any N-nitrosamine reagents. Based on work conducted by Fraser,49 Seebach showed that 4-phenylpiperidine-derived amide 85 undergoes lithiation and alkylation at the equatorial position to give 2,4-cis-piperidine 86 (Scheme 16B).50 The stereochemical disparity in the alkylation of the N-nitroso and N-acyl examples suggested the organolithium intermediates were configurationally stable. Few examples of discrete tertiary organolithiums generated by deprotonation have been reported. As such, the description of configurationally stable secondary α-aminoorganolithiums will be discussed as a means of exemplifying the stereoelectronic and steric factors that govern stereoselectivity and reactivity.
Scheme 16 Seebach's lithiation and alkylation of protected piperidines.
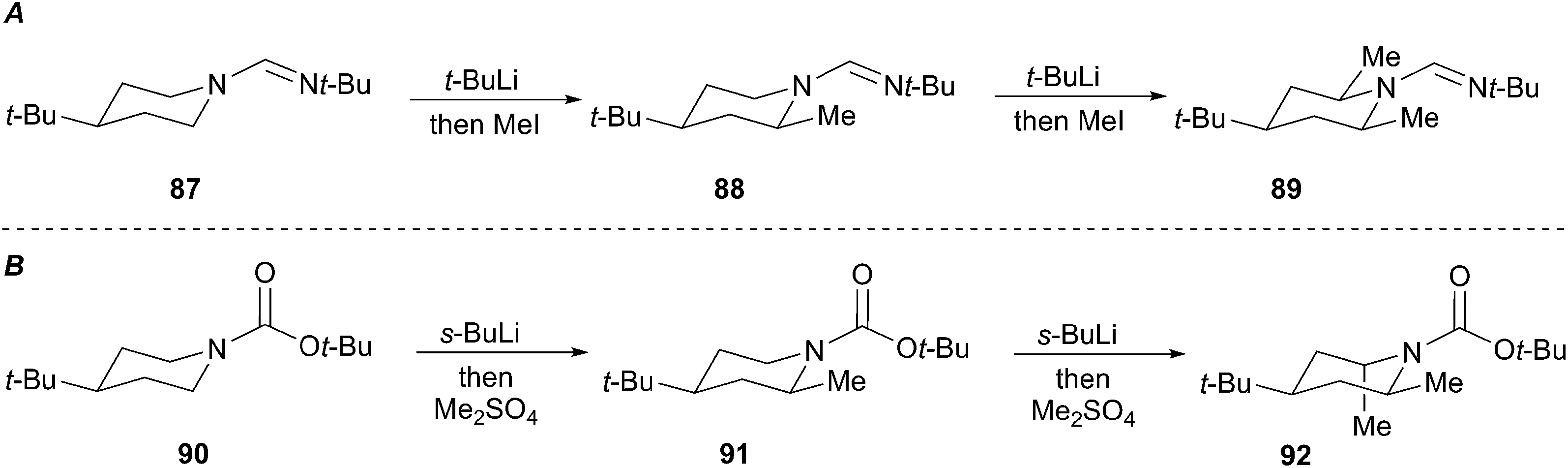
Both Meyers and Beak investigated the use of activating groups of greater synthetic utility. Meyers focused on N-formamidinyl activating groups and Beak employed the N-Boc group. Meyers devised a sequence involving successive equatorial lithiation/alkylation to give exclusively the 2,4,6-cis derivative 89 (Scheme 17A).51 Beak's lithiation/alkylation sequence proceeds first with equatorial alkylation then with axial lithiation/alkylation to provide the 2,4-cis-6-trans piperidine 92 as a single diastereomer (Scheme 17B).52
Scheme 17 Meyer's and Beak's directed lithiation of piperidines.
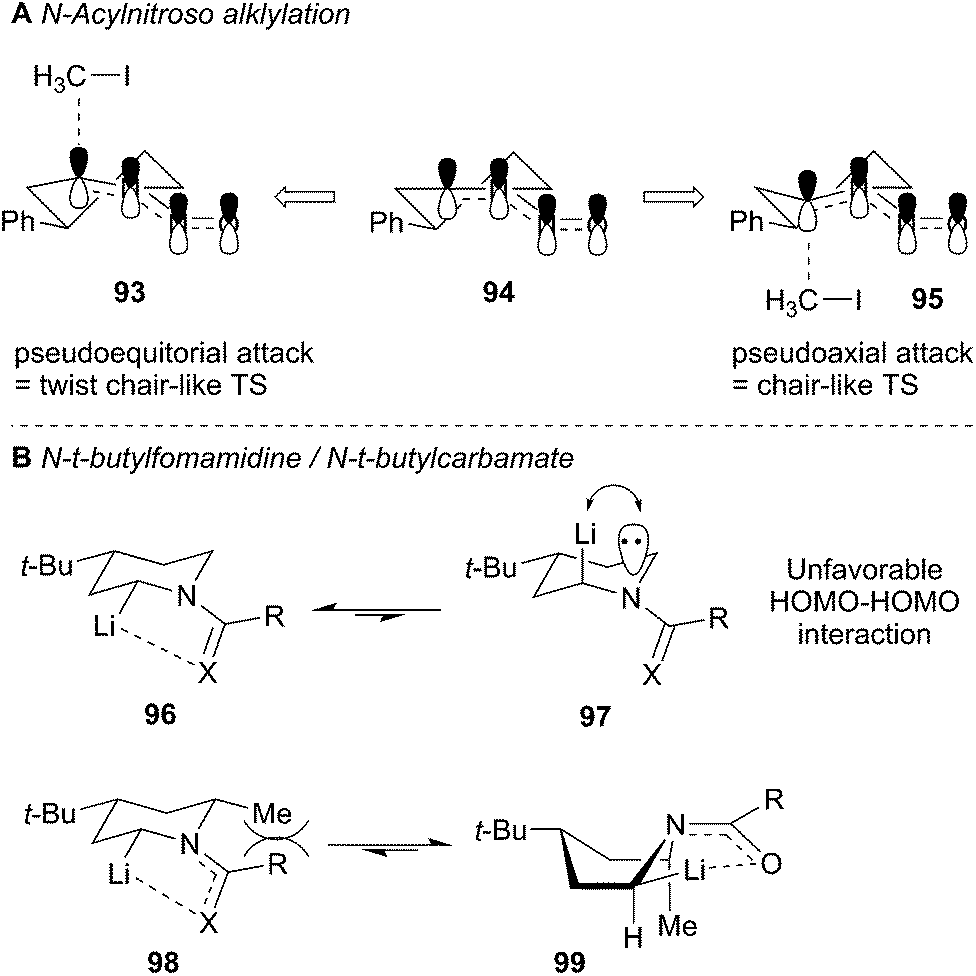
The proposed rationalization for axial-selective N-nitroso alkylation is based on the strong electron-withdrawing capability of the nitroso group. The anion is largely delocalized in a coplanar C–N–N–O π-system, which favors axial alkylation over equatorial alkylation for stereoelectronic purposes. Specifically, a lower energy chair-like transition state (95) that leads to axial alkylation is preferred over a higher energy twist-boat-like transition state (93) that leads to equatorial alkylation (Fig. 5A). For N-tert-butylformamidinyl and N-Boc lithiation and alkylations the delocalization of the nitrogen lone pair into the carbonyl π-bond is less significant than the N-nitroso systems. As a result, the C–Li bond prefers to adopt an orthogonal orientation to the axial nitrogen lone pair (96) to avoid the high energy (∼17 kcal mol−1) HOMO–HOMO interaction (97) (Fig. 5B). Efficient O–Li coordination also stabilizes this geometry. Subsequent alkylation with retention of configuration provides the equatorially substituted product 91. For the N-t-butylformamidinyl case, the second alkylation sequence occurs as above to provide the 2,4,6-cis product 89. In contrast, the second alkylation of N-Boc piperidines gives rise to a 2,4-cis-6-trans product 92. Resonance induced planarization of the N–C–O bonds creates A1,3-strain between the Boc group and the equatorial substituent (98). Conformational adjustment to the lower energy twist-boat-like conformation (99) allows lithiation to occur at a pseudo-equatorial position to give a 2,6-trans relationship.
Fig. 5 Proposed rationale for alkylation stereoselectivity.
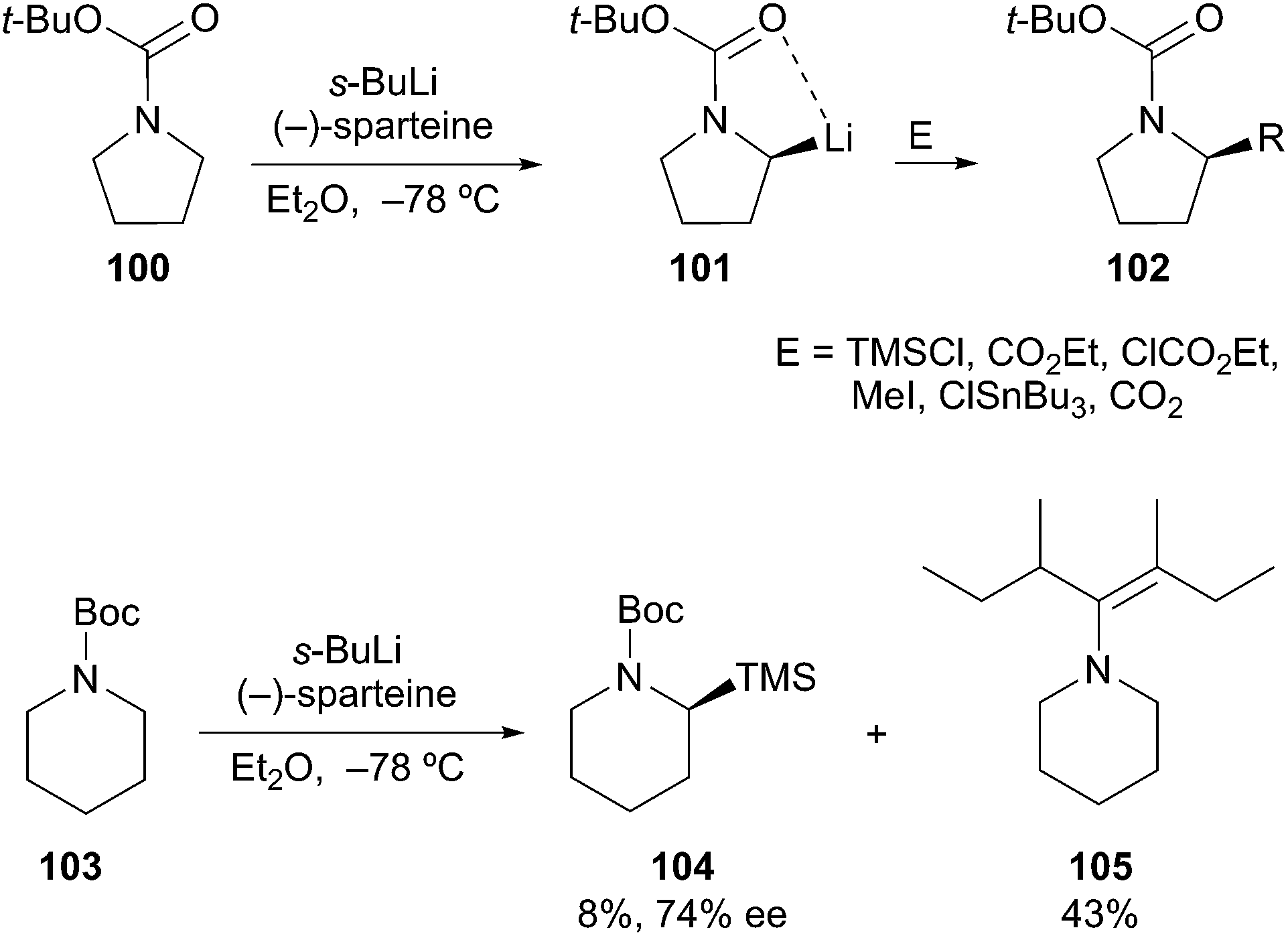
Following the work of Nozaki53 and Hoppe,54 Beak described the asymmetric deprotonation of N-Boc-directed pyrrolidine 100 by a (−)-sparteine·s-BuLi complex (Scheme 18). Subsequent electrophilic alkylation provided the substituted pyrrolidine 102 in good yield and high enantioselectivity. Since the first report in 1991,55 considerable attention has been focused on the mechanistic and synthetic utility of this method.56 In simple cases, sparteine and related amines are effective chiral ligands. One notable limitation is the use of Beak's protocol in the deprotonation of N-Boc piperidine 103. Lithiation followed by silylation provided adduct 104 in low yield and with moderate enantioselectivity. Unfortunately, the favored course of the reaction involved sec-BuLi addition into the Boc carbonyl to give 105.57 Interestingly, O'Brien and co-workers reported on the asymmetric deprotonation and electrophilic trapping of piperidine 103 with s-BuLi and a (+)-sparteine surrogate in moderate to high yield and moderate enantioselectivity (88:12 er, 76% ee).58 Gawley and Beng have also reported on the catalytic dynamic resolution of rac-2-lithio-N-Boc-piperidine using a chiral ligand in the presence of TMEDA to afford various 2-substituted piperidines with high enantioselectivity (96:4 to >99:1 er).59
Scheme 18 α-Aminoorganolithiums by asymmetric deprotonation.
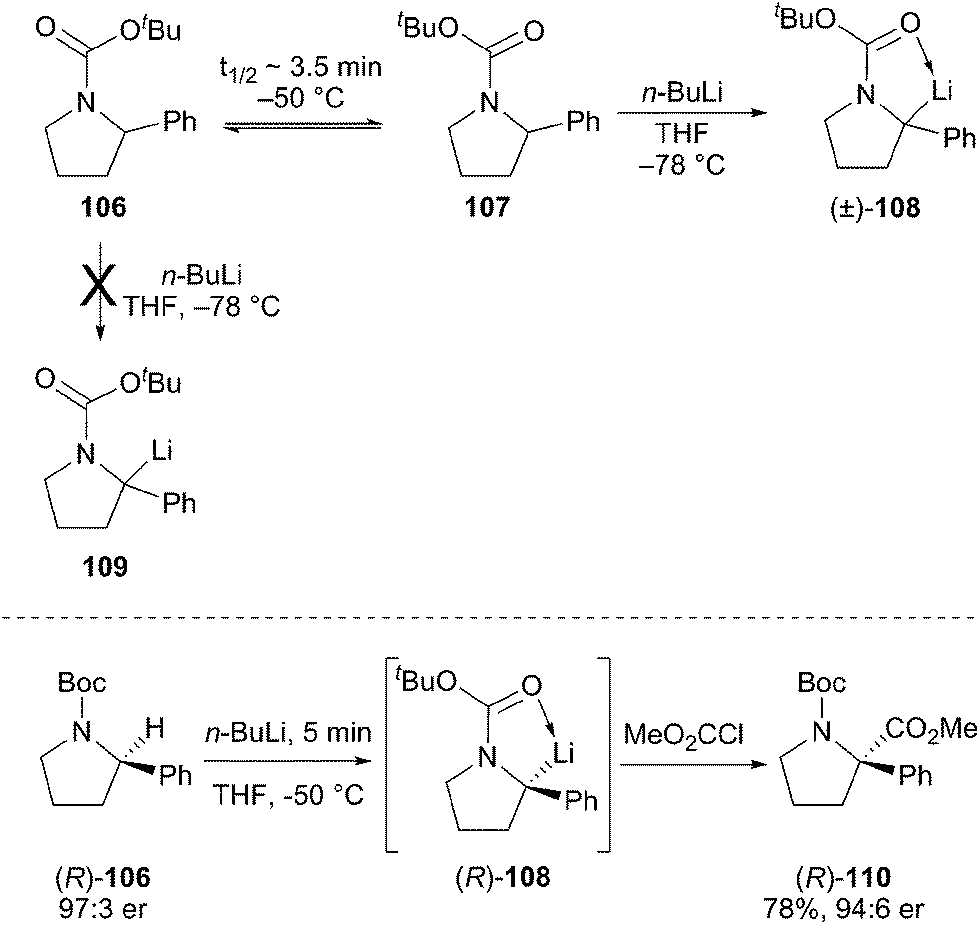
Recent work has shown that configurationally stable alkyllithium reagents can be generated by direct deprotonation of N-Boc amides with an activating group such as a phenyl ring. O'Brien and Coldham studied the deprotonation reaction using React-IR and found that the two rotamers of the Boc carbamate show very different reactivity (Scheme 19).60 The example of N-Boc-2-phenylpyrrolidine is particularly demanding. Deprotonation of rotamer 107 at −78 °C takes place rapidly, but the other rotamer 106 does not react at low temperatures. The interconversion of rotamers 106 and 107 has a measured barrier of ca. 15.4 kcal mol−1, which leads to a half-life of rotation of ∼10 h at −78 °C and ∼3.5 min at −50 °C. Rotamer interconversion is thus the slow step in the deprotonation reaction at −78 °C, but warming to 0 °C leads to complete and efficient deprotonation. The corresponding N-Boc-2-phenylpiperidine rotamers interconvert much more rapidly and can be efficiently deprotonated at low temperature.
Optically pure N-Boc-2-phenylpyrrolidine (R)-106 can be deprotonated and alkylated with a high degree of retention (Scheme 19).54 Under carefully optimized conditions, the deprotonation of pyrrolidine (R)-106 at −50 °C for 5 min generates the tertiary alkyllithium (R)-108, which slowly epimerizes at this temperature. By keeping the reaction times short and using reactive electrophiles, this alkyllithium reagent can be trapped with overall retention of configuration in good yield. The method works well with the analogous piperidine, and provides a very nice strategy to prepare optically pure, fully substituted centers adjacent to nitrogen in these important ring systems. In 2014, Coldham et al. reported on the kinetic resolution of rac-N-Boc-2-phenylpiperidines with n-BuLi/(−)-sparteine or n-BuLi/(+)-sparteine in moderate yield and high enantioselectivity to afford 2,2-disubstituted piperidines.61
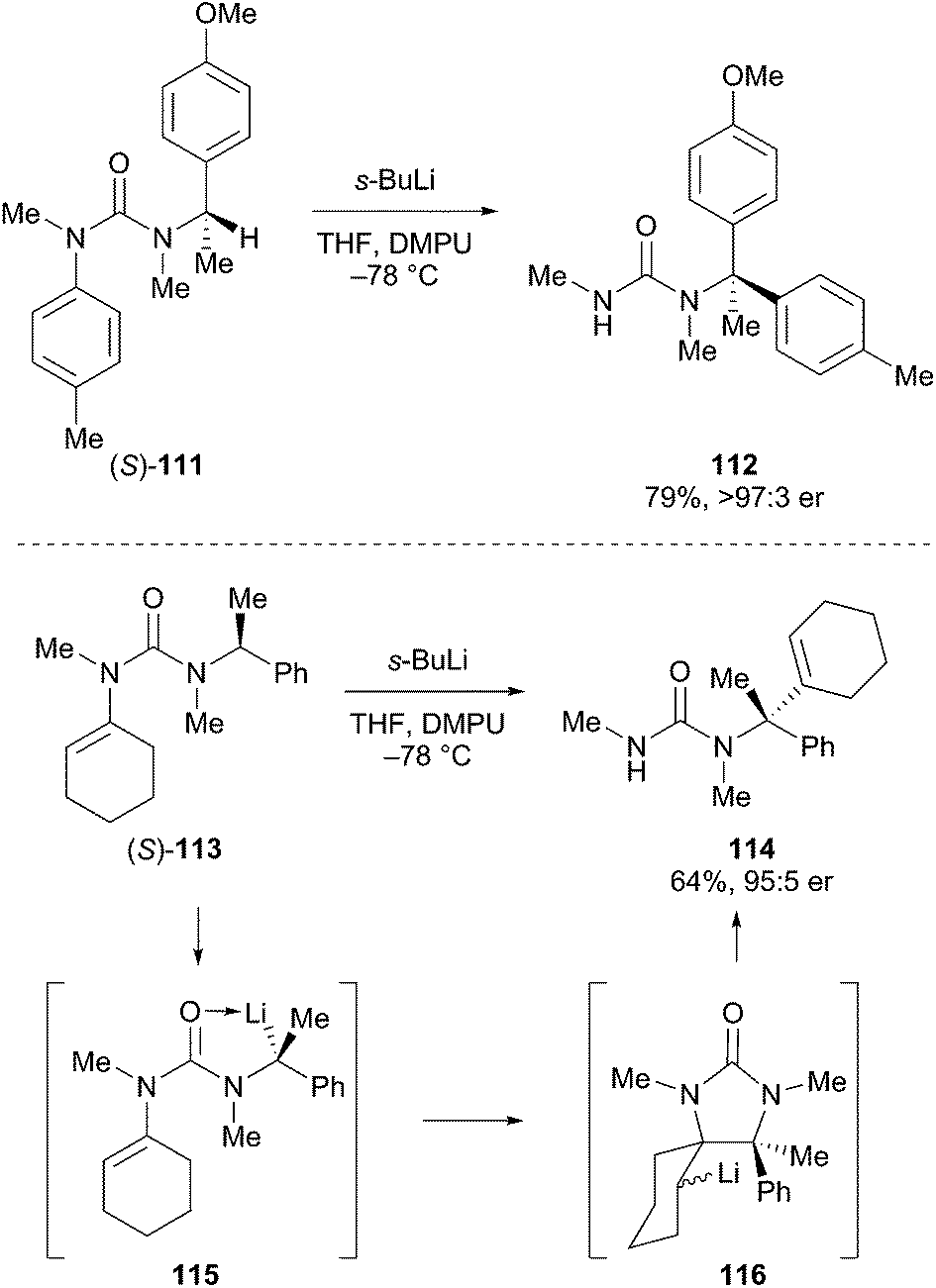
An interesting method to prepare optically enriched fully substituted centers adjacent to nitrogen atoms was developed by Clayden.62 For example, the reaction is initiated by deprotonation of optically pure benzylic urea (S)-111 (Scheme 20). Intramolecular migration of the aryl group took place with retention of configuration to give urea 112 in good overall yield. Similarly, alkene migration was demonstrated in the transformation of 113 to 114 with good levels of selectivity. The proposed mechanism involved deprotonation with retention of configuration to produce the alkyllithium 115, which is configurationally stable under the reaction conditions. The migration proceeds by cyclization to 116 and ring opening to produce the fully substituted carbon atom adjacent to nitrogen. These transformations are effective in the absence of any obvious anion-stabilizing group on the aryl ring or alkene, and led to optically active amines that would be difficult to access through other methods.
Scheme 20 Deprotonation and migration adjacent to benzylic ureas.
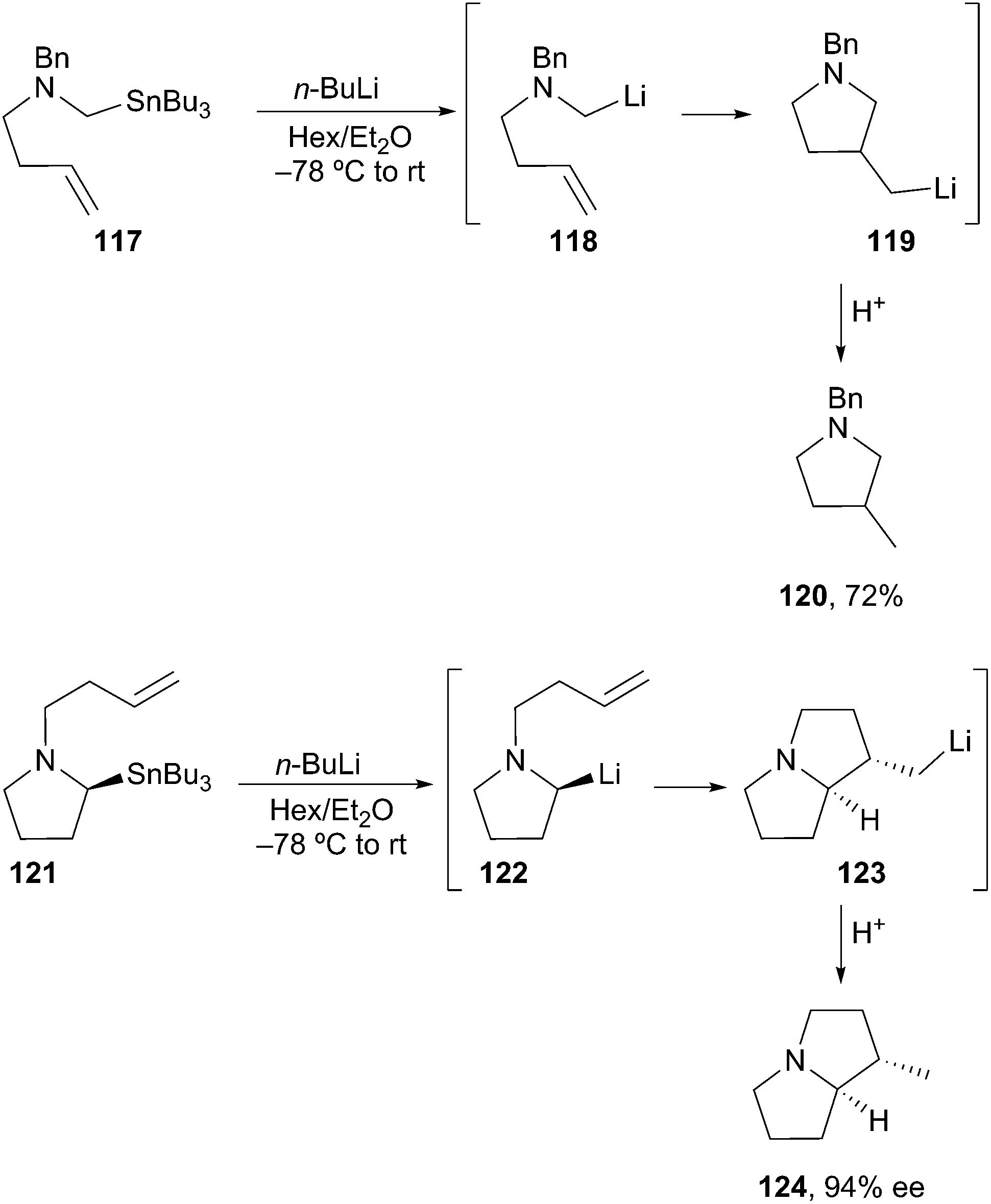
Transmetalation. Access to unstabilized α-aminoorganolithiums by Sn–Li exchange is well documented. Coldham demonstrated that transmetalation of stannane 117 afforded the unstabilized α-aminoorganolithium 118. The organolithium then underwent intramolecular carbolithiation on the pendant alkene to afford the more stable primary organolithium 119. Subsequent addition of methanol produced exclusively pyrrolidine 120 (Scheme 21).63 In an enantioselective fashion, transmetalation/carbolithiation of enantioenriched stannane 121 (94% ee) produced pyrrolizidine 124 (94% ee) with no loss of enantiomeric purity. This method accesses α-aminoorganolithiums (e.g.122 and 118) that are inaccessible by deprotonation.64
Scheme 21 Unstabilized α-aminoorganolithiums by Sn–Li exchange.
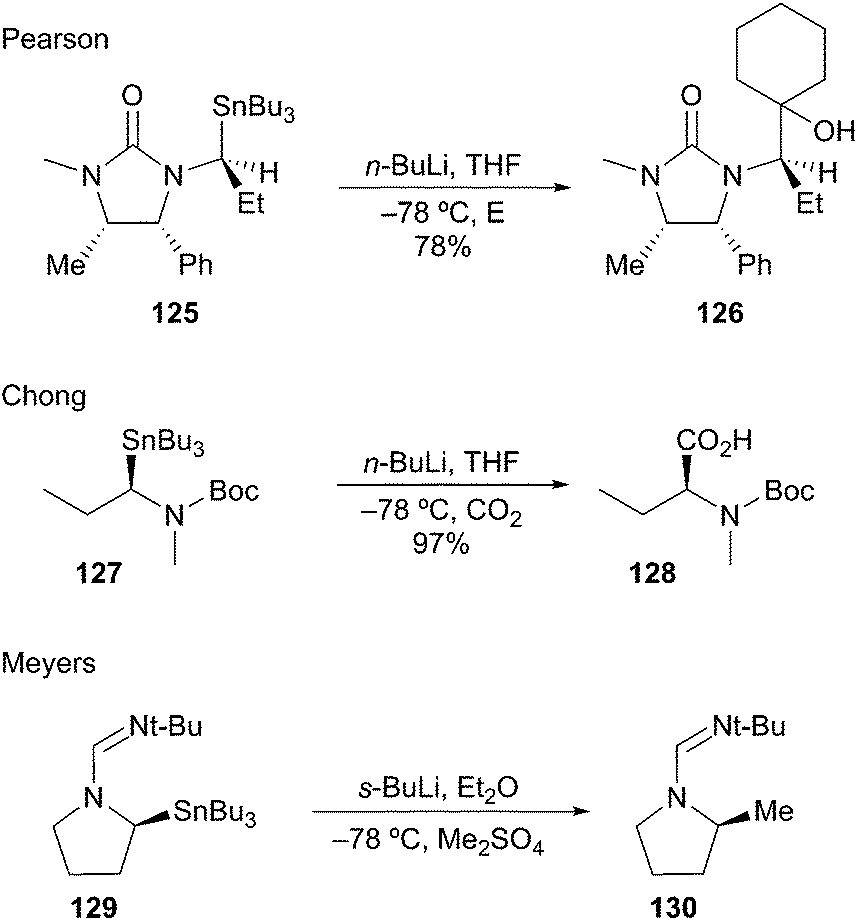
Pearson was the first to demonstrate Sn–Li exchange to generate stabilized α-aminoorganolithiums.65 Transmetalation of 125 at −78 °C afforded the organolithium with retention, which was trapped with cyclohexanone to afford 126 in good yield (Scheme 22). Chong and co-workers also demonstrated the generation and electrophilic trapping of an α-aminoorganolithium from enantioenriched stannane 127 to afford acid 128 with high stereofidelity.66 Meyers later demonstrated the configurational stability of formamide-stabilized α-aminoorganolithium at −78 °C from stannane 129; subsequent methylation provided 130.67
Scheme 22 Stabilized α-aminoorganolithiums by Sn–Li exchange.
Generation by reductive insertion
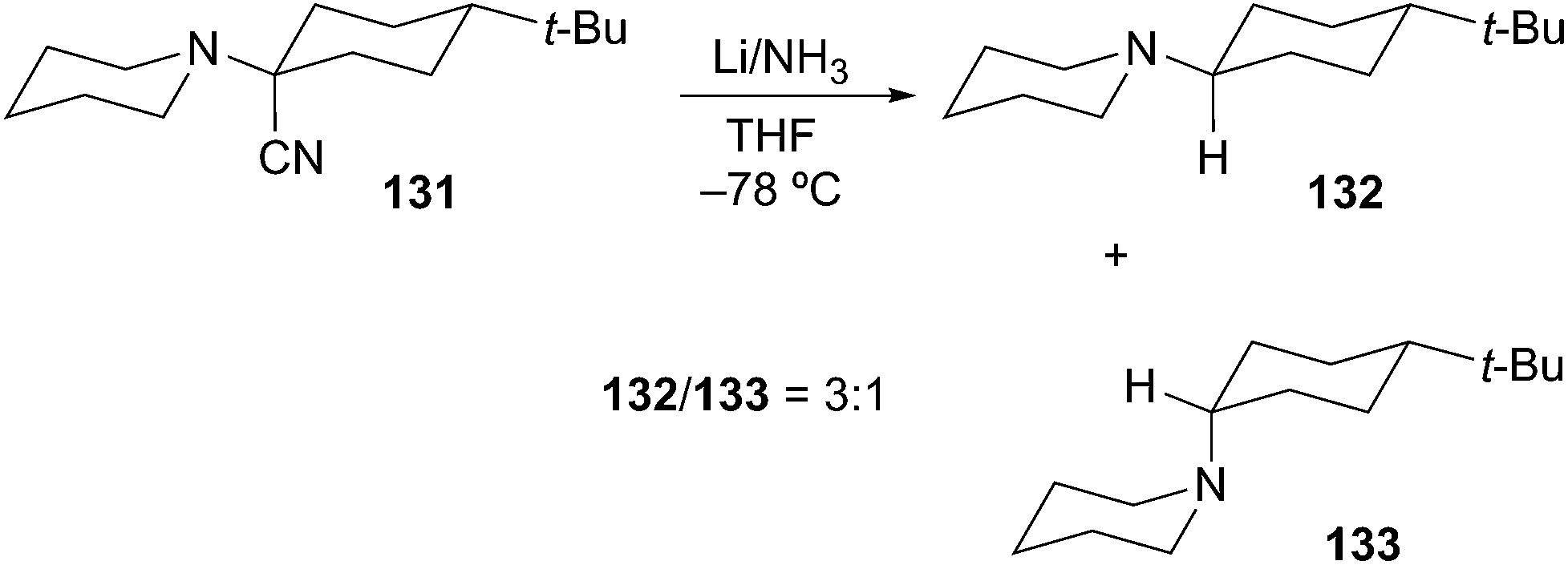
The last, and perhaps, most efficient method for the generation of α-aminoorganometallics is reductive decyanation/metalation of suitable functional groups by low-valent metals (e.g. zero-valent Group 1 metals: Li, Na, and K). In 1970, Welvart described the reduction of a tertiary α-aminonitrile precursor under dissolving metal conditions (Li/NH3 or Na/NH3) to effect replacement of the nitrile group by a hydrogen atom (Scheme 23).68
Scheme 23 Welvart reductive decyanation.
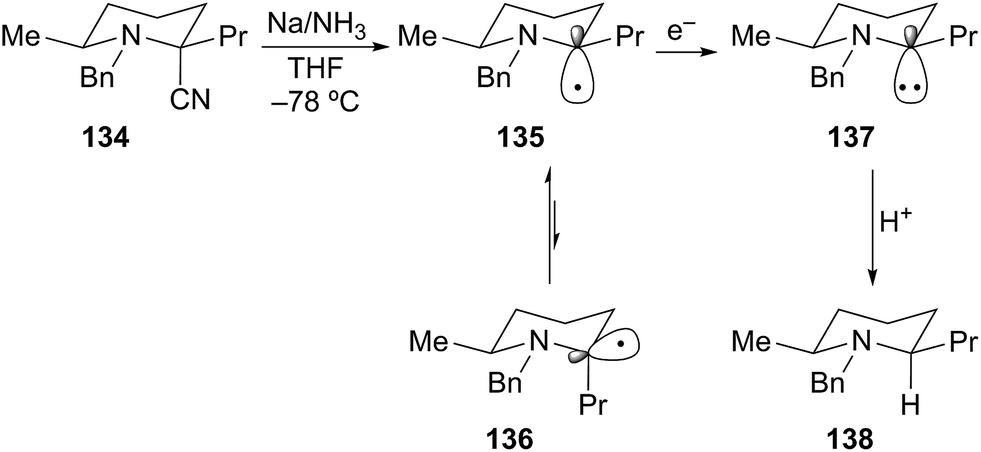
Husson later described the stereoselective reductive decyanation of 2-cyanopiperidines under dissolving metal conditions.69 Reductive decyanation (Na/NH3) of 134 to afforded piperidine 138 as a single diastereomer with 2,6-cis relative stereochemistry (Scheme 24). The selectivity in this process is rationalized by an anomeric-type effect as described earlier for the case of α-alkoxyorganolithium generation. Initial SET produces a rapidly equilibrating mixture of anomeric radicals 135 and 136 where equilibrium lies toward the pseudo-axial radical 135 due to a stabilizing HOMO–SOMO interaction. A second SET produces the axial carbanion 137. Rapid protonation of the highly basic anion proceeds with retention of configuration to generate the cis product 138. As noted above, this process proceeds first through the thermodynamically preferred axial radical configuration and then through the thermodynamically disfavored axial anion configuration (HOMO–HOMO interaction). On this basis, the equatorial anion should be preferred as in the N-acyl/formamidinyl piperidine lithiations shown in Schemes 16 and 17.
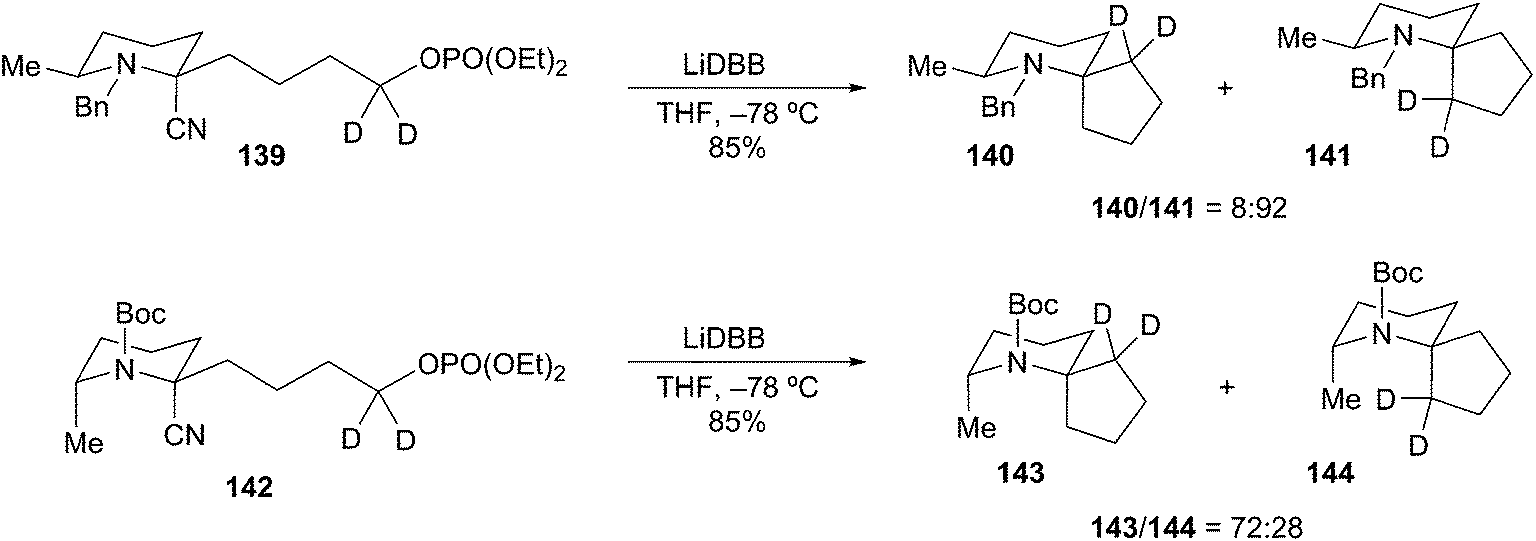
Rychnovsky and Wolckenhauer have also reported several examples for the generation of tertiary α-aminoorganolithiums by reductive decyanation.70 Their study examined the roles of protecting group and tether length with regards to cyclization of substituted 2-cyanopiperidines. As with examples described above, the use of unstabilized (N-Bn) versus stabilized (N-Boc) organolithiums had a noticeable influence on the course of the reductive cyclization. Reductive decyanation of N-Bn protected cyanopiperidine 139 (LiDBB, −78 °C) afforded cyclized diastereomers 141 and 140 in a 92:8 ratio (Scheme 25). The major cyclization product 141 arises from cyclization with retention from an axial organolithium. The configuration of the organolithium is assumed based on analogy to the deuterium-trapped alkyllithium of a similar piperidine (Scheme 27). In contrast, subjecting N-Boc cyanopiperidine 142 to the same reductive cyclization conditions produced diastereomeric products 143 and 144 in a 72:28 ratio. Trapping studies of a model system with trimethyl phosphate indicated that the equatorial organolithium was the major species in solution. These results exemplify the disparate yet complementary nature of seemingly similar reactions where only the protecting group dictates the course of the reaction.
Scheme 25 Complementary reductive cyclization of protected piperidines.
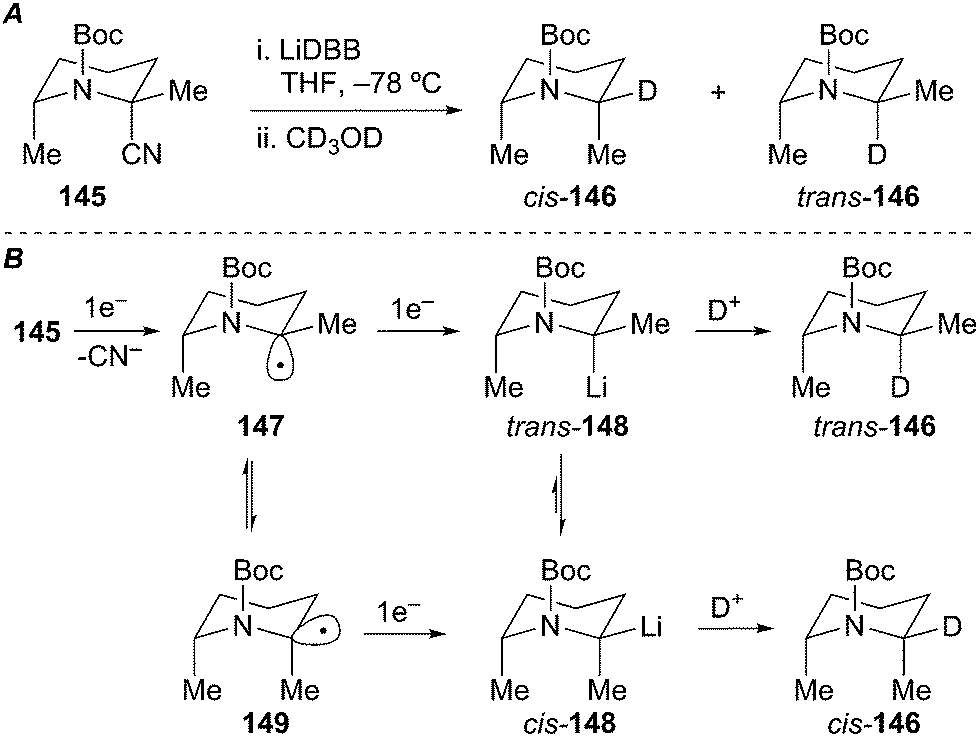
Wolckenhauer and Rychnovsky also explored the stereoselectivity in the reductive decyanation of simple N-benzyl 2-cyanopiperidine and N-Boc 2-cyanopiperidine. Rapid (<1 h) CD3OD quenches afforded low ratios of diastereomeric products cis/trans-146 with the cis-146 predominating (Scheme 26A). Slow equilibration of the intermediate diastereomeric organolithiums was observed at low temperature (−78 °C, t = 1 h gave 62:38 cis/trans versus t = 12.5 h, 78:22 cis/trans) over extended time periods. Conducting the reductive lithiation at −40 °C provided higher diastereomer ratios (94:6 cis/trans) with lower yield and deuterium incorporation. The observed selectivity can be rationalized by considering the stability of the intermediate radical and organolithium intermediates. Single-electron transfer to produce an axially disposed anomerically stabilized radical (147) would be expected. In the N-Boc piperidine case, the anomeric stabilization is diminished due to delocalization of the nitrogen lone pair into the carbonyl moiety. Additionally, A1,3-strain between the Boc group and the equatorial methyl group results in isomerization to the equatorially disposed radical intermediate 149. Further single-electron reduction of the diastereomeric radical intermediates leads to α-aminoorganolithiums trans/cis-148. An anti-anomeric (HOMO–HOMO) effect of the C–Li bond and the nitrogen lone pair electrons, and A1,3-strain arising from interactions between the Boc group and the equatorial methyl group favor formation of the equatorial organolithium intermediate. Coordinative stabilization between the Boc group and the equatorial lithium may also favor equilibration to the equatorial organolithium cis-148. The combination of these effects overrides the 1,3-diaxial (Me–Me) interactions in the cis-isomer. Deuterium incorporation with retention of configuration produces a mixture of the 2,6-trans and 2,6-cis piperidines. Assuming a thermodynamic preference for formation of cis-148, the slow equilibration at −78 °C indicates a substantial barrier to inversion. Computational work by Gawley et al. suggests an inversion barrier of ∼16 kcal mol−1 for related N-Boc 2-lithiopyrrolidines.71
Scheme 26 Reductive lithiation to cis and trans-N-Boc-2-deuteropiperidines.
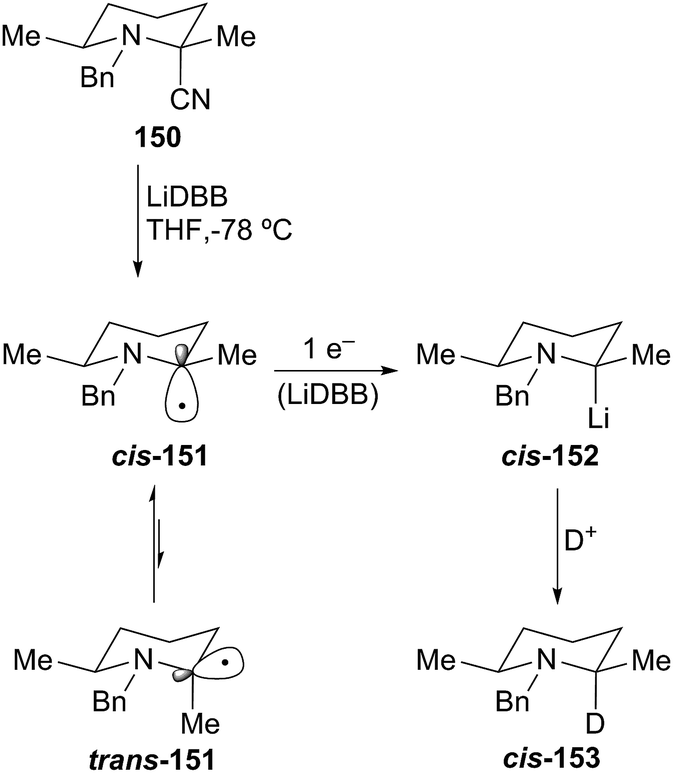
The analogous N-benzyl 2-cyanopiperidine gave complementary diastereoselectivity compared to the N-Boc 2-cyanopiperidine case. Reductive lithiation of 150 proceeded with high stereoselectivity (>95:5 cis/trans) to produce the cis-lithiopiperidine 152, which appears to be configurationally stable at −78 °C for at least one hour (Scheme 27). The axial incorporation of the proton can be explained by carbon–CN bond cleavage by single-electron transfer to predominantly generate the anomerically-stabilized axial radical cis-151. The subsequent single-electron transfer occurs with retention of configuration to form the axial 2-lithiopiperidine cis-152, which reacts with retention of configuration to produce cis-N-benzyl piperidine 153.
Scheme 27 Reductive lithiation to cis and trans-N-Bn-2-deuteropiperidines.
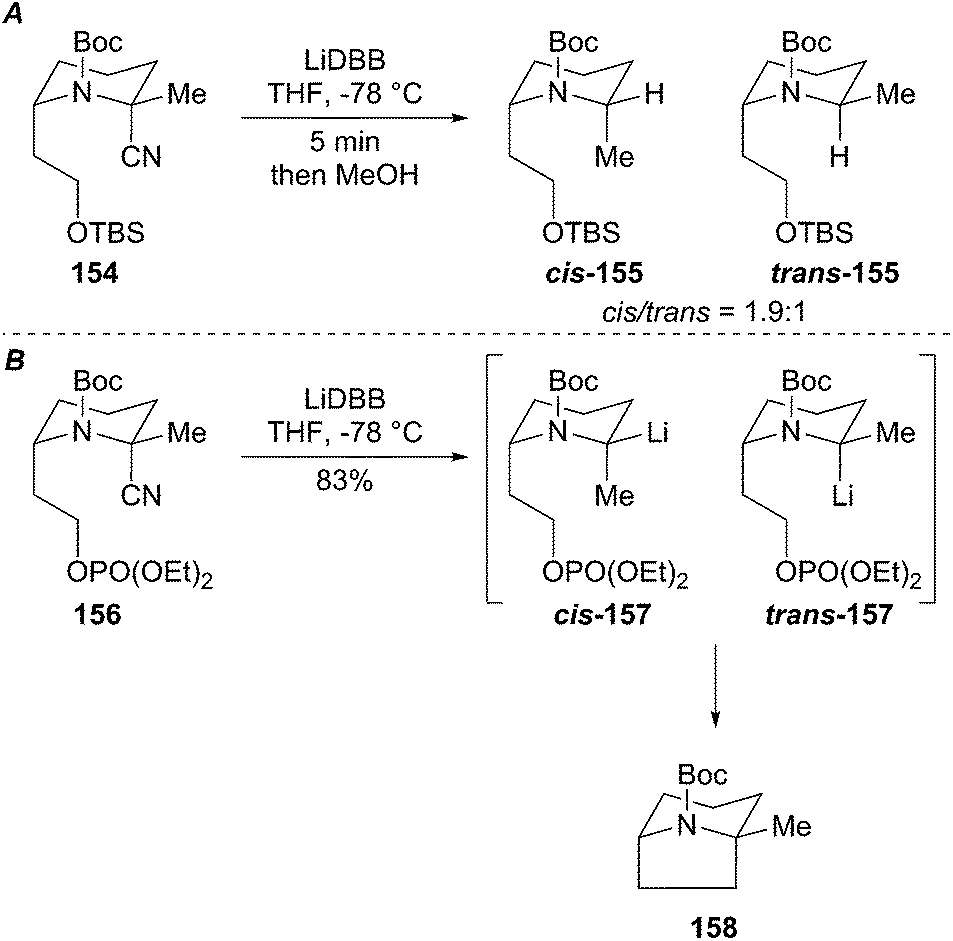
As shown above, the reductive lithiation of N-Boc 2-cyanopiperidines produces organolithium diastereomers with less than 2:1 ratio at −78 °C, which slowly equilibrate over several hours. Reductive lithiation of analog 154 also produced a mixture of organolithium diastereomers cis/trans-155 (Scheme 28A). By proper positioning of the tethered leaving group on the N-Boc piperidine, one organolithium diastereomer is forced to react with retention of configuration and the other with inversion. Alternatively, the organolithium diastereomer may be non-reactive towards cyclization with inversion and may be protonated upon quenching the reaction. A reasonable mechanism based on the reductive decyanation product ratio of cis/trans-155 involves non-stereoselective reductive lithiation to generate a pair of diastereomeric organolithiums (cis/trans-157) where one organolithium diastereomer reacts via a SE2ret pathway while the other diastereomer reacts via SE2inv. The hypothesis was confirmed by reducing the 2-cyanopiperidine phosphate 156 to give the bridged bicycle 158 in 83% yield as a single diastereomer (Scheme 28B). This example demonstrates that a stereogenic organolithium can be forced to proceed through a non-preferred electrophilic substitution pathway by conformational constraint.
Scheme 28 Reductive cyclization of cis- and trans-2-lithiopiperidines.
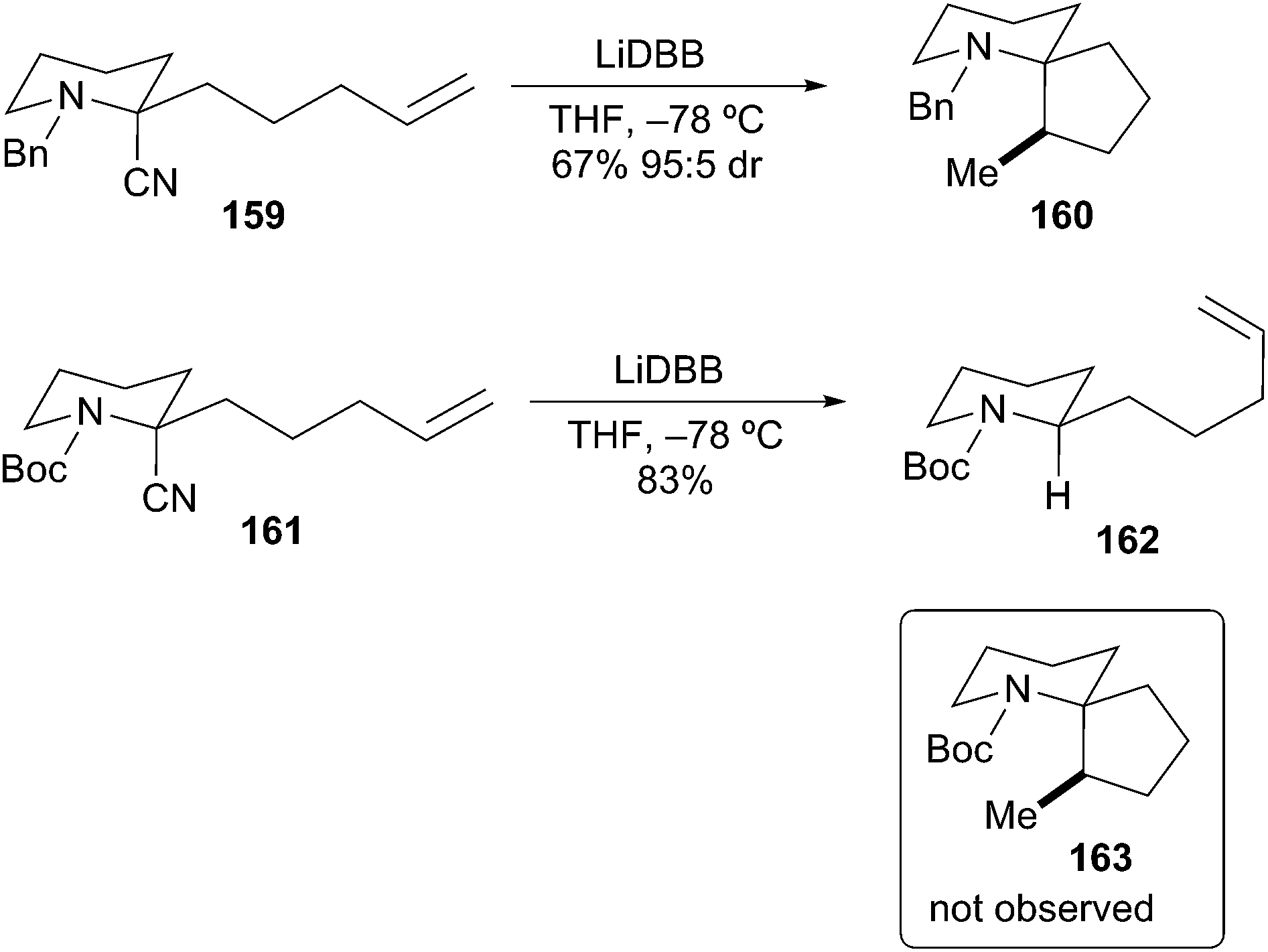
Bahde and Rychnovsky also described the different reactivity of N-Boc and N-Bn protected α-aminoorganolithiums.72 Reductive lithiation of N-Bn-2-cyanopiperidine 159 followed by carbolithiation cyclization produced spiropiperidine 160 in good yield with excellent diastereoselectivity (Scheme 29). In an analogous experiment, reductive lithiation of the N-Boc-2-cyanopiperidine 161 produced an α-aminoorganolithium species that did not undergo cyclization to 163; only reduced starting material 162 was isolated. Coldham et al. have reported a similar lack of cyclization for N-Boc alkenyl stannanes.73 These examples further demonstrate the reactivity differences between unstabilized and stabilized α-aminoorganolithium species.
Scheme 29 Reductive lithiation of N-Boc- and N-Bn-2-cyanopiperidines.
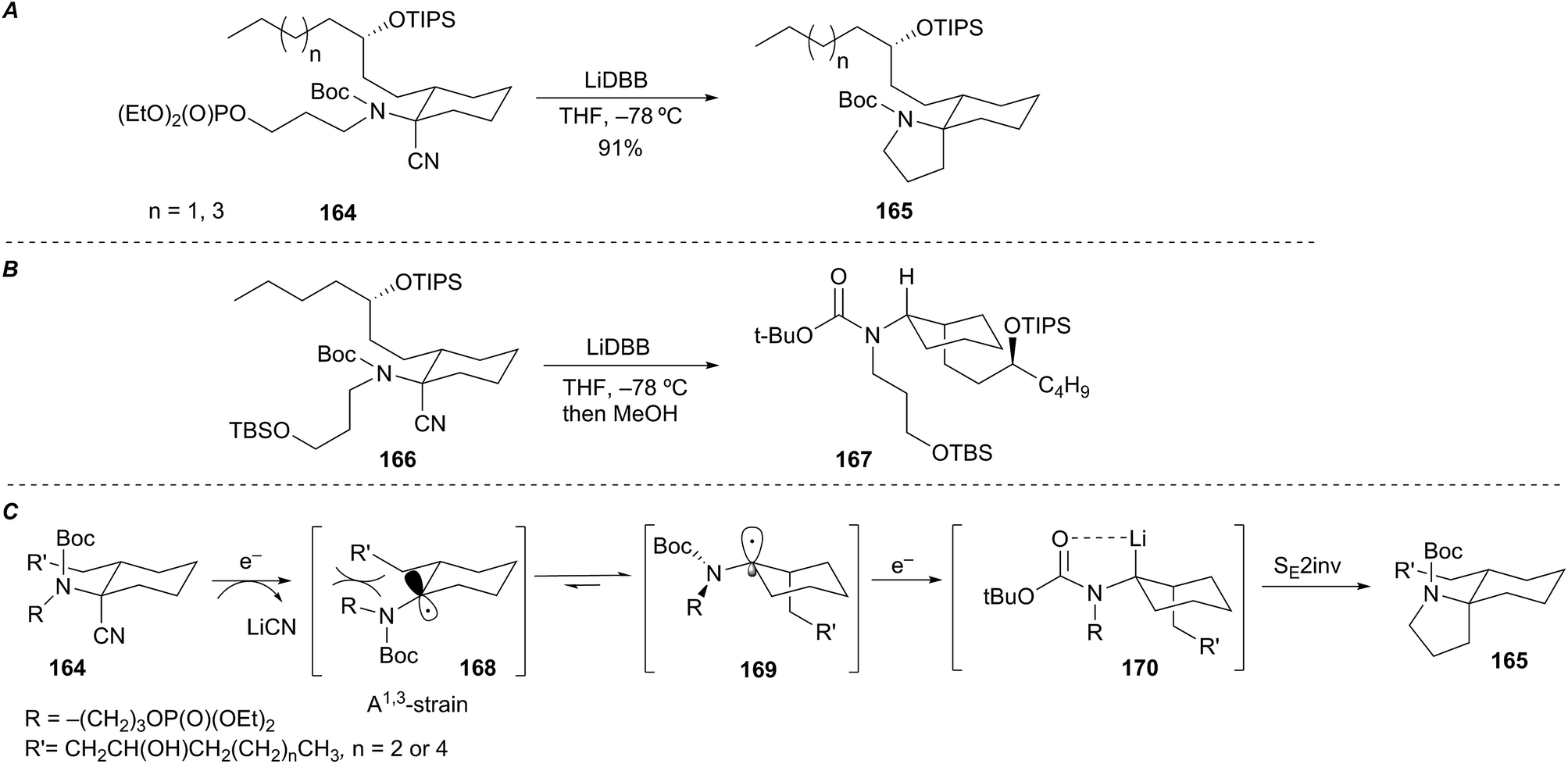
Rychnovsky et al. described an efficient and highly stereoselective reductive cyclization of an exocyclic α-aminoorganolithium to form the spiropyrrolidine ring system of the lepadiformine alkaloids (Scheme 30A).74 The mechanistic origin of selectivity was of interest and prompted an investigation aimed at identifying the configuration of the intermediate organolithium. Reductive lithiation of control substrate 166 followed by protonation of the resultant organolithium gave cis-disubstituted cyclohexane 167 (Scheme 30B). Assuming protonation proceeded with retention of configuration,75 the mechanism of the reductive cyclization may progress as shown in Scheme 30C. The radical intermediate 168, stabilized by interaction with the nitrogen electrons, creates a sterically encumbered environment between the equatorial R′ group and the nitrogen R group (or N-Boc group) resulting in a conformational isomerization to produce radical 169. Further reduction of the axially disposed radical 169 to alkyllithium 170 allows for cyclization via a SEinv pathway and leads to spiropyrrolidine 165. The overall stereochemical outcome of the event is cyclization with retention of configuration through a double inversion sequence.
Scheme 30 Reductive cyclization to the spiropyrrolidine of lepadiformine A–C.
Tertiary alkyl organolithiums
Generation, structure and reactivity
Reported concurrently by Wittig76 and Gilman,77 lithium halogen exchange has proven to be a useful method for the selective introduction of carbon–lithium bonds. This equilibrium process provides rapid and efficient preparation of primary and secondary alkyllithiums from alkyl bromides and iodides from less stable organolithiums (e.g. tert-butyllithium).78 Therefore, the generation of tertiary alkyllithiums by equilibration with another unstable tertiary organolithium is energetically and synthetically prohibitive. Similar to lithium–halogen exchange, tin–lithium exchange also relies on an equilibrium process that is limited by the unfavorable formation of less stable unfunctionalized tertiary alkyllithiums. Deprotonation is by far the least common method for their preparation. Lithiation by deprotonation to form unstabilized alkyllithiums is confounded by the very low acidity of the C–H bond, the lack of intramolecular coordination of the electron deficient lithium atom to a heteroatom, and/or the lack of an adjacent empty orbital or electron withdrawing group necessary for stabilization of the electron rich C–Li bond. The best method for generating tertiary unstabilized alkyllithiums is reductive insertion.
Generation by reductive insertion
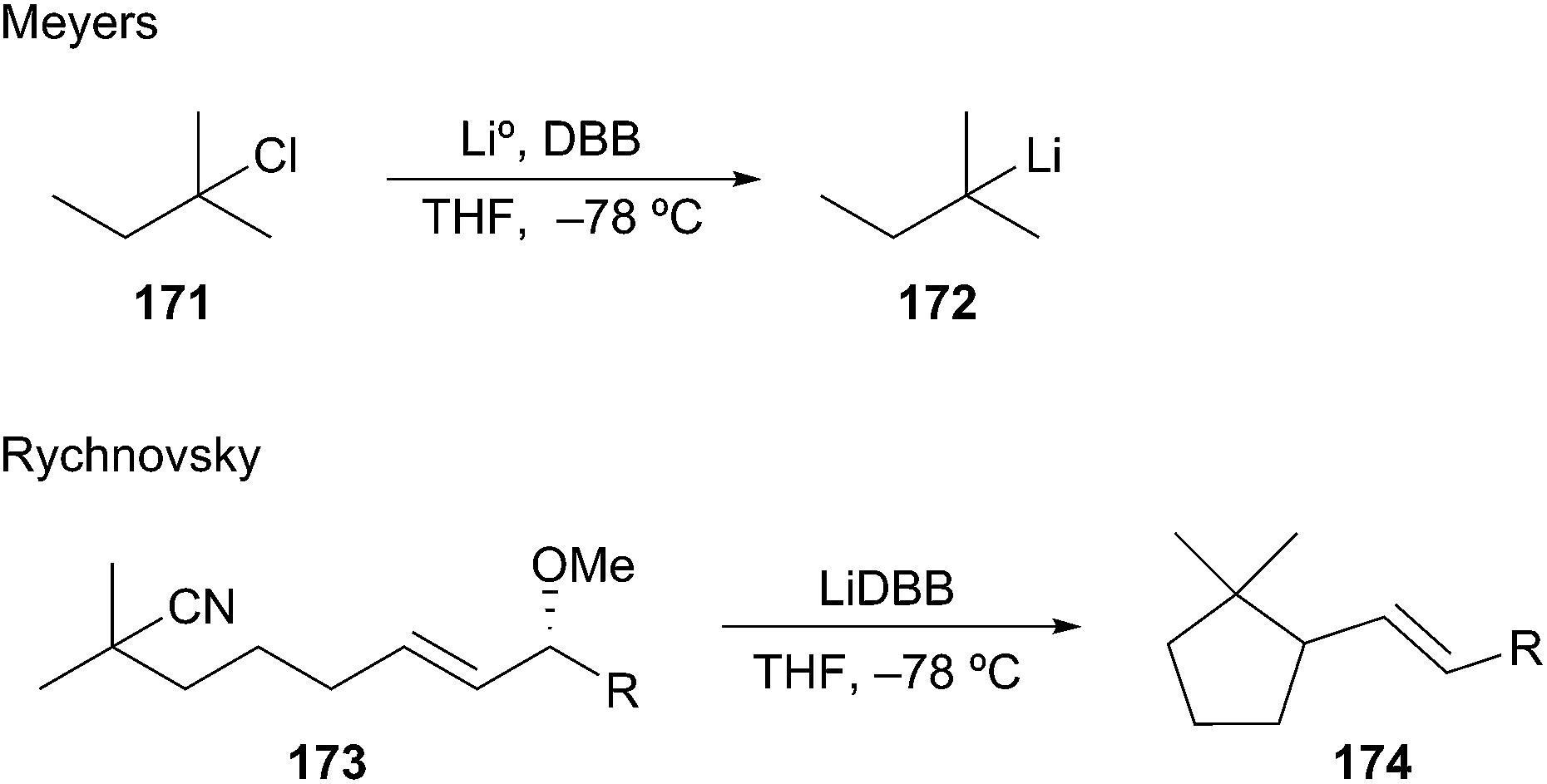
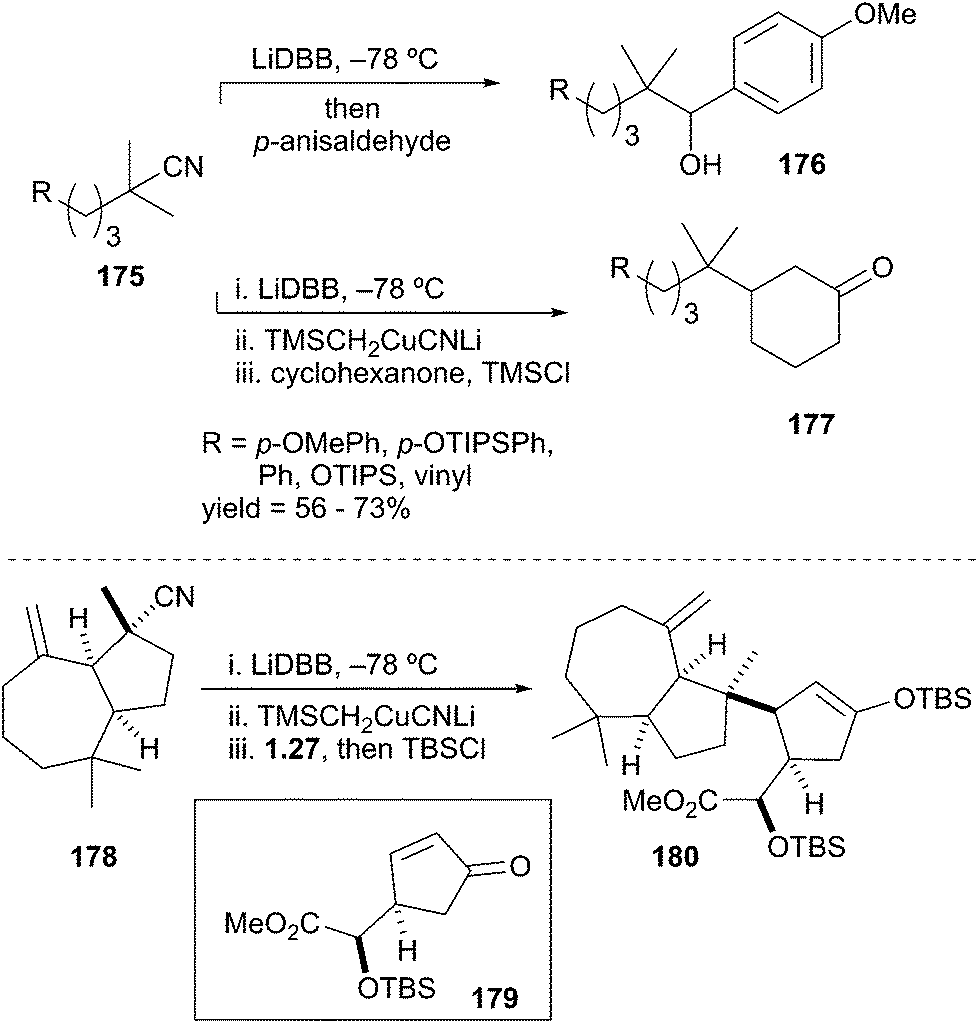
In contrast to deprotonation, the rate of reductive lithiation is fastest for substituted alkyllithiums (Fig. 2). This trend logically coincides with the relative stabilities of the intermediate radicals, which are generated in the rate-determining step of the reduction sequence. Meyers showed that the radical anion carrier DBB was very efficient at forming tert-amyl lithium 172 by reductive lithiation (Scheme 31).79 In 2004, Rychnovsky and La Cruz reported on the syn-SN2′ cyclization of a tertiary alkyllithium onto a tethered allylic methoxy ether to give cyclopentyl alkene 174 in 95:5 er.80 More recently, Overman et al. demonstrated the formation and reactivity of unstabilized tertiary alkyllithium reagents and their derived cuprates in bimolecular reactions with carbon-centered electrophiles.81 The reductive lithiation and electrophilic trapping of tertiary nitriles 175 with p-anisaldehyde gave neopentyl alcohols 176 in synthetically useful yields (Scheme 32). The approach was then extended to the formation of tertiary organocuprates by transmetalation. Generation of the organolithium from nitriles 175 followed by transmetalation and subsequent conjugate addition to the cyclohexenone afforded β-substituted cyclohexanone derivatives 177. The utility of this transformation was highlighted by the union of hydrazulene nitrile 178 to cyclopentenone 179 to fashion the C8–C14 bond and the C8 quaternary stereocenter of the rearranged spongian diterpene aplyviolene precursor 180.82 The above examples highlight the facile reduction of alkyl nitriles and halogens to provide synthetically useful tertiary alkyllithium intermediates.83 However, this area of research remains underdeveloped in part due to the highly basic and reactive nature of the generated alkyllithium species.
Scheme 31 Reductive lithiation to tertiary unstabilized alkyllithiums.
Scheme 32 Overman's reductive generation of unstabilized tertiary organolithiums.
Conclusion
Functionalized organometallic species are versatile reagents for carbon–carbon bond formation constituting a key step during the synthesis of many organic non-natural and natural products. Tertiary α-heteroatom organolithiums constitute a unique class due to their high reactivity and interesting modes of stereoselectivity. In general, the stereoselective generation of organolithiums by either permutational interconversion or direct insertion is difficult to predict without robust and abundant empirical data and analysis. The examples contained in this article highlight the unique nature of seemingly similar systems as it relates to changes in stereoselectivity and reactivity. The design and use of appropriately poised substrates as well as new investigations into the generation and selectivity of organolithium species will continue to provide the synthetic chemist a powerful tool in the construction of ever increasingly complex molecules.
Notes and references
M. Schlosser, in Organometallics in Synthesis: A Manual, ed. M. Schlosser, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2nd edn, ch. 1, pp. 1–352 Search PubMed.
H. Gilman and R. G. Jones, Org. React., 1951, 6, 339 Search PubMed; R. G. Jones and H. Gilman, Chem. Rev., 1954, 54, 835 CrossRefCAS.
W. C. Still, J. Am. Chem. Soc., 1978, 100, 1481 CrossRefCAS; N. Meyer and D. Seebach, Chem. Ber., 1980, 113, 1290 CrossRef.
B. J. Wakefield, Organolithium Methods, Academic Press, London, 1988 Search PubMed.
J. F. Garst, Acc. Chem. Res., 1974, 4, 400 CrossRef.
T. Cohen and M. Bhupathy, Acc. Chem. Res., 1989, 22, 152 CrossRefCAS.
C. P. Andrieux, I. Gallardo, J.-M. Saveant and K.-B. Su, J. Am. Chem. Soc., 1986, 108, 638 CrossRefCAS.
C. G. Screttas, J. Chem. Soc., Chem. Commun., 1972, 752 RSC.
T. Cohen and J. R. Matz, Synth. Commun., 1980, 10, 371 CrossRef.
T. Cohen, J. P. Sherbine, R. R. Hutchins and M.-T. Lin, Organomet. Synth., 1986, 3, 361 Search PubMed.
(a) P. K. Freeman and L. L. Hutchinson, Tetrahedron Lett., 1976, 22, 1849 CrossRef;
(b) P. K. Freeman and L. L. Hutchinson, J. Org. Chem., 1980, 45, 1924 CrossRefCAS.
K. Shimada and M. Szwarc, J. Am. Chem. Soc., 1975, 97, 3313 CrossRefCAS.
T. Cohen, T. Kreethadumrongdat, X. Liu and V. Kulkarni, J. Am. Chem. Soc., 2001, 123, 3478 CrossRefCASPubMed.
(a) S. D. Rychnovsky, J. Org. Chem., 1989, 54, 4982 CrossRefCAS;
(b) M. E. Jung and R. B. Blum, Tetrahedron Lett., 1977, 3791 CrossRefCAS;
(c) A. Krief and P. Barbeaux, Synlett, 1990, 511 CrossRefCAS;
(d) F. Chen, B. Mudryk and T. Cohen, Tetrahedron, 1994, 50, 12793 CrossRefCAS;
(e) D. Cheng, S. Zhu, X. Liu, S. H. Norton and T. Cohen, J. Am. Chem. Soc., 1999, 121, 10241 CrossRefCAS;
(f) B. Mudryk and T. Cohen, J. Am. Chem. Soc., 1993, 115, 3855 CrossRefCAS.
(a) R. B. Bates, L. M. Kroposki and D. E. Potter, J. Org. Chem., 1972, 37, 560 CrossRefCAS;
(b) J. Clayden and S. S. Yasin, New J. Chem., 2002, 26, 191 RSC.
P. Stanetty and M. D. Mihovilovic, J. Org. Chem., 1997, 62, 1514 CrossRefCAS.
(a) J. S. Sawyer, T. L. Macdonald and G. J. McGarvey, J. Am. Chem. Soc., 1984, 106, 3376 CrossRefCAS;
(b) J. S. Sawyer, A. Kucerovy, T. L. Macdonald and G. J. McGarvey, J. Am. Chem. Soc., 1988, 110, 842 CrossRefCAS.
(a) D. Hoppe and T. Krämer, Angew. Chem., Int. Ed. Engl., 1986, 98, 171 CrossRefCAS;
(b) D. Hoppe, F. Hintze and P. Tebben, Angew. Chem., Int. Ed., 1990, 29, 1422 CrossRef;
(c) P. Sommerfeld and D. Hoppe, Synlett, 1992, 764 CrossRefCASPubMed;
(d) F. Hintze and D. Hoppe, Synthesis, 1992, 1216 CrossRefCAS.
T. Kramer and D. Hoppe, Tetrahedron Lett., 1987, 28, 5149 CrossRef.
R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2013, 49, 4911 RSC.
R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Commun., 2013, 49, 10160 RSC.
(a) H. K. Scott and V. K. Aggarwal, Chem.–Eur. J., 2011, 17, 13124 CrossRefCASPubMed;
(b) A. P. Pulis and V. K. Aggarwal, J. Am. Chem. Soc., 2012, 134, 7570 CrossRefCASPubMed;
(c) A. P. Pulis, P. Fackler and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 4382 CrossRefCASPubMed;
(d) A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054 CrossRefCASPubMed;
(e) K. R. Fandrick, N. D. Patel, J. A. Mulder, J. Gao, M. Konrad, E. Archer, F. G. Buono, A. Duran, R. Schmid, J. Daeubler, D. R. Fandrick, S. Ma, N. Grinberg, H. Lee, C. A. Busacca, J. J. Song, N. K. Yee and C. H. Senanayake, Org. Lett., 2014, 16, 4360 CrossRefCASPubMed.
(a) W. C. Still and C. Sreekumar, J. Am. Chem. Soc., 1980, 102, 1201 CrossRefCAS;
(b) W. C. Still, J. Am. Chem. Soc., 1978, 100, 1481 CrossRefCAS.
(a) T. Cohen and M.-T. Lin, J. Am. Chem. Soc., 1984, 106, 1130 CrossRefCAS;
(b) T. Cohen and M. Bhupathy, Acc. Chem. Res., 1989, 152 CrossRefCAS;
(c) T. Cohen and J. R. Matz, J. Am. Chem. Soc., 1980, 102, 6902 CrossRef.
D. Griller, K. U. Ingold, P. J. Krusic and H. Fischer, J. Am. Chem. Soc., 1978, 100, 6750 CrossRefCAS.
T. Cohen and M.-T. Lin, J. Am. Chem. Soc., 1984, 106, 1130 CrossRefCAS.
For computational studies of related 2-cyanotetrahydropyran reductive decyanations, see: S. D. Rychnovsky, J. P. Powers and T. J. LePage, J. Am. Chem. Soc., 1992, 114, 8375 CrossRefCAS.
A. I. Meyers, P. D. Edwards, W. F. Rieker and T. R. Bailey, J. Am. Chem. Soc., 1984, 106, 3270 CrossRefCAS.
S. D. Rychnovsky and D. J. Skalitzky, J. Org. Chem., 1992, 57, 4336 CrossRefCAS.
(a) P. Beak, D. R. Anderson and M. D. Curtis, Acc. Chem. Res., 2000, 33, 715 CrossRefCASPubMed;
(b) W. K. Lee, Y. S. Park and P. Beak, Acc. Chem. Res., 2009, 42, 224 CrossRefCASPubMed.
S. D. Rychnovsky, A. J. Buckmelter, V. H. Dahanukar and D. J. Skalitzky, J. Org. Chem., 1999, 64, 6849 CrossRefCASPubMed.
(a) L. R. Takaoka and S. D. Rychnovsky, Angew. Chem., Int. Ed., 2003, 42, 818 CrossRefPubMed;
(b) S. D. Rychnovsky, T. Hata, A. I. Kim and A. J. Buckmelter, Org. Lett., 2001, 3, 807 CrossRefCASPubMed.
M. D. Morin and S. D. Rychnovsky, Org. Lett., 2005, 7, 2051 CrossRefCASPubMed.
(a) K. T. Mead and B. N. Brewer, Curr. Org. Chem., 2003, 7, 227 CrossRefCAS;
(b) F. Perron and K. F. Albizati, Chem. Rev., 1989, 89, 1617 CrossRefCAS;
(c)
V. Vaillancourt, N. E. Pratt, F. Perron and K. F. Albizati, in Total Synthesis of Natural Products, ed. J. Apsimon, Wiley, New York, 1992, vol. 8, pp. 533–691 Search PubMed;
(d) T. L. B. Boivin, Tetrahedron, 1987, 43, 3309 CrossRefCAS.
F. Perron and K. F. Albizati, Chem. Rev., 1989, 89, 1617 CrossRefCAS.
D. Vellucci and S. D. Rychnovsky, Org. Lett., 2007, 9, 711 CrossRefCASPubMed.
P. Beak and D. B. Reitz, Chem. Rev., 1978, 78, 275 CrossRefCAS.
P. V. R. Schleyer, T. Clark, A. J. Kos, G. W. Spitznagel, C. Rohde, D. Arad, K. Houk and N. G. Rondan, J. Am. Chem. Soc., 1984, 106, 6467 CrossRefCAS.
P. Beak, W. J. Zajdel and D. B. Reitz, Chem. Rev., 1984, 84, 471 CrossRefCAS.
(a) D. J. Pippel, G. A. Weisenburger, S. R. Wilson and P. Beak, Angew. Chem., Int. Ed., 1998, 37, 2522 CrossRefCAS;
(b) G. Boche, M. Marsch, J. Harbach, K. Harms, B. Ledig, F. Schubert, J. C. W. Lohrenz and H. Ahlbrecht, Chem. Ber., 1993, 126, 1887 CrossRefCAS.
R. E. Gawley and Q. Zhang, Tetrahedron, 1994, 50, 6077 CrossRefCAS.
(a) R. R. Fraser, T. B. Grindley and S. Passannanti, Can. J. Chem., 1975, 53, 2473 CrossRefCASPubMed;
(b) D. Seebach and D. Enders, Angew. Chem., Int. Ed., 1972, 11, 1101 CrossRefCAS;
(c) D. Seebach and D. Enders, Angew. Chem., Int. Ed., 1975, 14, 15 CrossRef.
Y. S. Park, M. L. Boys and P. Beak, J. Am. Chem. Soc., 1996, 118, 3757 CrossRefCAS.
A. I. Meyers, B. Du and M. A. Gonzalez, J. Org. Chem., 1990, 55, 4218 CrossRefCAS.
J. Clayden, J. Dufour, D. M. Grainger and M. Helliwell, J. Am. Chem. Soc., 2007, 129, 7488 CrossRefCASPubMed.
(a)
P. Beak, T. A. Johnson, D. D. Kim and S. H. Lim, Topics in Organometallic Chemistry, in Organolithiums in Enantioselective Synthesis, ed. D.M. Hodgson, Springer-Verlag, Berlin, 2003, vol. 5, pp. 139–176 Search PubMed;
(b) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRefCASPubMed.
D. Seebach and D. Enders, Angew. Chem., Int. Ed., 1975, 14, 15 CrossRef.
National Toxicology Program, Reports on Carcinogens, National Institutes of Health, 2011, vol. 12, p. 302 Search PubMed.
R. R. Fraser, T. B. Grindley and S. Passannanti, Can. J. Chem., 1975, 53, 2473 CrossRefCASPubMed.
D. Seebach, W. Wykpiel, W. Lubosch and H.-O. Kalinowski, Helv. Chim. Acta, 1978, 61, 3100 CrossRefCAS.
(a) T. T. Shawe and A. I. Meyers, J. Org. Chem., 1991, 56, 2751 CrossRefCAS;
(b) A. I. Meyers, P. D. Edwards, W. F. Reiker and T. R. Bailey, J. Am. Chem. Soc., 1984, 106, 3270 CrossRefCAS;
(c) A. I. Meyers and G. Milot, J. Am. Chem. Soc., 1993, 115, 6652 CrossRefCAS.
(a) P. Beak and W. K. Lee, J. Org. Chem., 1990, 55, 2578 CrossRefCAS;
(b) P. Beak and W. K. Lee, J. Org. Chem., 1993, 58, 1109 CrossRefCAS.
H. Nozaki, T. Aratani, T. Toraya and R. Noyori, Tetrahedron, 1971, 27, 905 CrossRefCAS.
D. Hoppe, F. Hintze and P. Tebben, Angew. Chem., Int. Ed., 1990, 29, 1422 CrossRef.
S. T. Kerrick and P. Beak, J. Am. Chem. Soc., 1991, 113, 9708 CrossRefCAS.
P. O'Brien, K. B. Wiberg, W. F. Bailey, J.-P. R. Hermet and M. J. McGrath, J. Am. Chem. Soc., 2004, 126, 15480 CrossRefPubMed and references therein.
W. F. Bailey, P. Beak, S. T. Kerrick, S. Ma and K. B. Wiberg, J. Am. Chem. Soc., 2002, 124, 1889 CrossRefCASPubMed.
D. Stead, G. Carbone, P. O'Brien, K. R. Campos, I. Coldham and A. Sanderson, J. Am. Chem. Soc., 2010, 132, 7260 CrossRefCASPubMed.
(a) T. K. Beng and R. E. Gawley, J. Am. Chem. Soc., 2010, 132, 12216 CrossRefCASPubMed;
(b) T. K. Beng, W. S. Tyree, T. Parker, C. Su, P. G. Williard and R. E. Gawley, J. Am. Chem. Soc., 2012, 134, 16845 CrossRefCASPubMed.
(a) N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300 CrossRefCASPubMed;
(b) X. Li and I. Coldham, J. Am. Chem. Soc., 2014, 136, 5551 CrossRefCASPubMed;
(c) X. Li, D. Leonori, N. S. Sheikh and I. Coldham, Chem.–Eur. J., 2013, 19, 7724 CrossRefCASPubMed;
(d) T. K. Beng, J. S. Woo and R. E. Gawley, J. Am. Chem. Soc., 2012, 134, 14764 CrossRefCASPubMed.
E. J. Cochrane, D. Leonori, L. A. Hassall and I. Coldham, Chem. Commun., 2014, 50, 9910 RSC.
(a) J. Lefranc, A. M. Fournier, G. Mingat, S. Herbert, T. Marcelli and J. Clayden, J. Am. Chem. Soc., 2012, 134, 7286 CrossRefCASPubMed;
(b) A. M. Fournier, C. J. Nichols, M. A. Vincent, I. H. Hillier and J. Clayden, Chem.–Eur. J., 2012, 18, 16478 CrossRefCASPubMed;
(c) M. B. Tait, P. A. Ottersbach, D. J. Tetlow and J. Clayden, Org. Process Res. Dev., 2014, 18, 1245 CrossRefCAS.
I. Coldham, R. Hufton and D. J. Snowden, J. Am. Chem. Soc., 1996, 118, 5322 CrossRefCAS.
N. J. Ashweek, P. Brandt, I. Coldham, S. Dufour, R. E. Gawley, F. Haeffner, R. Klein and G. Sanchez-Jimenez, J. Am. Chem. Soc., 2005, 127, 449 CrossRefCASPubMed.
(a) W. H. Pearson and A. C. Lindbeck, J. Am. Chem. Soc., 1991, 113, 8546 CrossRefCAS;
(b) W. H. Pearson, A. C. Lindbeck and J. W. Kampf, J. Am. Chem. Soc., 1993, 115, 2622 CrossRefCAS.
J. M. Chong and S. B. Park, J. Org. Chem., 1992, 57, 2220 CrossRefCAS.
A. I. Meyers and T. R. Elworthy, Tetrahedron, 1994, 50, 6089 CrossRef.
C. Fabre and Z. Welvart, C. R. Acad. Sci. Ser., 1970, 270, 1887 CAS.
M. Bonin, J. R. Romero, D. S. Grierson and H.-P. Husson, Tetrahedron Lett., 1982, 23, 3369 CrossRefCAS.
S. A. Wolckenhauer and S. D. Rychnovsky, Tetrahedron, 2005, 61, 3371 CrossRefCASPubMed.
F. Haeffner, P. Brandt and R. E. Gawley, Org. Lett., 2002, 4, 2101 CrossRefCASPubMed.
R. J. Bahde and S. D. Rychnovsky, Org. Lett., 2008, 10, 4017 CrossRefCASPubMed.
(a) I. Coldham and R. Hufton, Tetrahedron, 1996, 52, 12541 CrossRefCAS;
(b) I. Coldham, J.-C. Fernàndez, K. N. Price and D. J. Snowden, J. Org. Chem., 2000, 65, 3788 CrossRefCASPubMed.
(a) M. A. Perry, M. D. Morin, B. W. Slafer, S. A. Wolckenhauer and S. D. Rychnovsky, J. Am. Chem. Soc., 2010, 132, 9591 CrossRefCASPubMed;
(b) M. A. Perry, M. D. Morin, B. W. Slafer and S. D. Rychnovsky, J. Org. Chem., 2012, 77, 3390 CrossRefCASPubMed.
N. C. Faibish, Y. S. Park, S. Lee and P. Beak, J. Am. Chem. Soc., 1997, 119, 11561 CrossRefCAS.
G. Wittig, U. Pockels and H. Dröge, Ber. Dtsch. Chem. Ges. A, 1938, 71, 1903 CrossRef.
H. Gilman, W. Langham and A. L. Jacoby, J. Am. Chem. Soc., 1939, 61, 106 CrossRefCAS.
W. F. Bailey and E. R. Punzalan, J. Org. Chem., 1990, 55, 5404 CrossRefCAS.
D. J. Rawson and A. I. Meyers, Tetrahedron Lett., 1991, 32, 2095 CrossRefCAS.
T. E. La Cruz and S. D. Rychnovsky, Chem. Commun., 2004, 168 RSC.
M. J. Schnermann, N. L. Untiedt, G. Jiménez-Osés, K. N. Houk and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9581 CrossRefCASPubMed.
M. J. Schnermann and L. E. Overman, Angew. Chem., Int. Ed., 2012, 51, 9576 CrossRefCASPubMed.
For additional examples of tertiary alkyllithiums see;
(a) W. F. Bailey and M. W. Carson, J. Org. Chem., 1998, 63, 9960 CrossRefCAS;
(b) A. Krief, B. Remacle and J. Mercier, Synlett, 2000, 2000, 1443 CrossRefPubMed;
(c) R. W. Hoffmann, R. Koberstein and K. Harms, J. Chem. Soc., Perkin Trans. 2, 1999, 183 RSC;
(d) M. Yus, R. Ortiz and F. F. Huerta, Tetrahedron, 2003, 59, 8525 CrossRefCASPubMed.



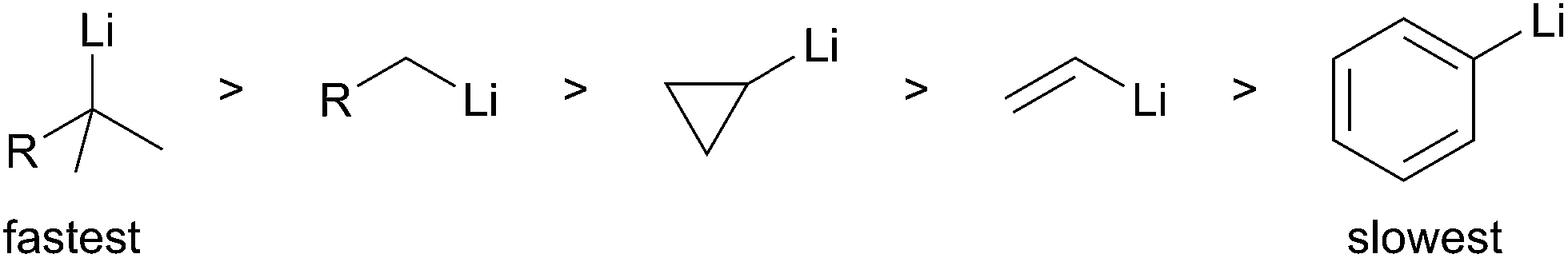
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: