
Syntheses and biological studies of marine terpenoids derived from inorganic cyanide
Martin J.
Schnermann
*a and
Ryan A.
Shenvi
*b
aChemical Biology Laboratory, National Cancer Institute, Frederick, MD 21701, USA. E-mail: martin.schnermann@nih.gov
bDepartment of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: rshenvi@scripps.edu
First published on 16th December 2014
Abstract
Covering up to 2014
Isocyanoterpenes (ICTs) are marine natural products biosynthesized through an unusual pathway that adorns terpene scaffolds with nitrogenous functionality derived from cyanide. The appendage of nitrogen functional groups – isonitriles in particular – onto stereochemically-rich carbocyclic ring systems provides enigmatic, bioactive molecules that have required innovative chemical syntheses. This review discusses the challenges inherent to the synthesis of this diverse family and details the development of the field. We also present recent progress in isolation and discuss key aspects of the remarkable biological activity of these compounds.
 Martin J. Schnermann | Martin Schnermann is an investigator in the Chemical Biology Laboratory in the intramural program of the National Cancer Institute (NCI). He attended Colby College and graduated in 2002 with degrees in Chemistry (research with Prof. Dasan Thamattoor) and Physics. After a year at Pfizer, he was a graduate student with Prof. Dale Boger at the Scripps Research Institute and obtained his Ph.D. in 2008. He then completed an NIH-postdoctoral fellowship with Prof. Larry Overman at the University of California, Irvine. In 2012, Dr. Schnermann joined the NCI where his research focuses on the development of new chemical approaches for drug delivery and imaging in cancer, as well as the synthesis of natural products of relevance to cancer imaging and therapy. |
 Ryan A. Shenvi | Ryan Shenvi is an Associate Professor with tenure in the Department of Chemistry at The Scripps Research Institute. He earned his B.S. degree with honors and distinction in chemistry from Penn State University, where he was a research student in the laboratories of John Desjarlais and Ray Funk. He obtained his Ph.D. in 2008 as an NDSEG predoctoral fellow with Phil Baran at The Scripps Research Institute, and then joined the laboratory of E. J. Corey at Harvard University as an NIH-funded postdoctoral fellow. In 2010, Ryan returned to Scripps to start his own lab, which explores the chemical synthesis of secondary metabolites that are relevant to human health. These efforts frequently require the invention of new chemical reactions and also lead to discoveries in biology. |
1 Introduction
A variety of nitrogenous terpenes have been isolated from marine organisms – sponges and tunicates – that retain vestiges of a biogenesis involving inorganic cyanide. These compounds are defined by terpenoid carbon scaffolds decorated with functionality revealing their unusual biosynthesis. The isonitrile, formamide, isocyanate, or isothiocyanate functional groups embedded in these terpenes impart biological and chemical properties distinct from more common oxygenated terpenes. This class of molecules is significant and growing (>130 structures), and has been a source of innovation and discovery in chemistry and biology for over the past 4 decades.Various aspects of the chemistry and biology of these natural products has been reviewed previously.1–4 In particular, a 2004 review by Garson in this journal provided extensive discussion, particularly of biosynthetic considerations.5 The present review will provide an update of the field and provide expanded discussion in two areas: (1) an overview of their pharmacology with particular focus on recent biological disclosures and antimalarial activity; and (2) the chemical synthesis of complex isonitrile-containing or -derived natural products. Syntheses will be analyzed according to C–C framework construction and installation of the key C–N bonds – a didactic exercise meant to illustrate advances in synthesis and methods over the last 4 decades. The overall goal of this review is to provide an overview of modern aspects of the synthesis of these molecules and their potential biomedical applications, as well as to point out unaddressed questions for the future.
Shown in Fig. 1A are representative members of the key structural classes. The unusual biosynthetic nitrogen incorporation step is generally thought to involve an N-selective hydroisocyanation of unsaturated terpenes (ionization and capture of the corresponding alcohol is also a possibility).6 The nitrogen source can be either cyanide or thiocyanate ions, though these inorganic species can interconvert through enzymatic processes (Fig. 1B).7 The conversion of the initially formed isonitriles and isothiocyanates to formamides, amines, and isocyanates has not been studied in great detail and may arise, at least in part, through non-enzymatic reactions. Because the nitrogen functional groups of ICTs are not generally involved in gross, skeleton-forming biosynthetic steps, ICTs belong more within the terpene family of metabolite than with the alkaloids, even though the presence of basic amine groups within some of these molecules could place them in the latter class.
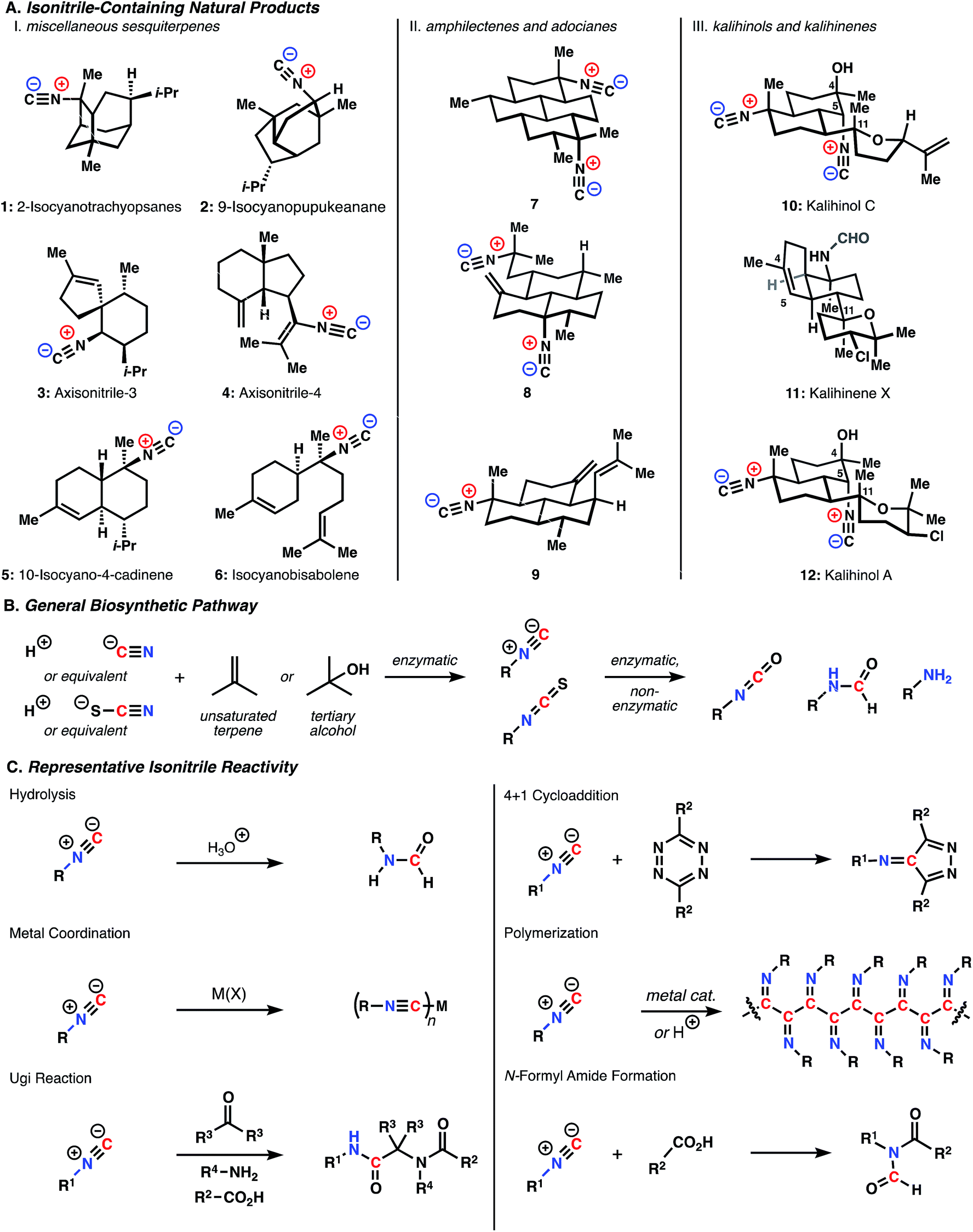 | ||
| Fig. 1 (A) Isonitrile-containing natural products (B) general biosynthetic scheme (C) representative isonitrile reactivity. | ||
The functional groups contained within these molecules immediately raise questions regarding their reactivity in the biological milieu. Isonitriles are of particular interest due to their extensive associated biological activity (discussed in detail below). As shown with key examples in Fig. 1C, isonitriles undergo a range of chemical transformations. These reactions find broad utility in a wide array of synthetic and biomedical applications, though our understanding of their role in the context of these natural products is still in its infancy. In addition to extensively studied reactions, such as protonation, Ugi and Passerini reactions, and metal coordination, recent reports have employed less well-known isonitrile reactivity for various applications. These efforts include the demonstration that isonitriles undergo a [4 + 1] cycloaddition with tetrazines in water with applications as a potentially bioorthogonal “click” bioconjugation approach.8 Isonitriles can polymerize to form helical structures in the presence of certain transition metals (e.g. Ni(II)) and were recently shown to form hydrogel structures that mimic the mechanical response of key cytoskeletal proteins.9,10 Finally, Danishefsky and coworkers recently showed that isonitriles and carboxylic acids react to form N-formyl amides, with application for the synthesis of complex peptidic structures.11
2 Recent progress in isolation
Since the most recent review, a number of new structures have been disclosed expanding the landscape of structural diversity of ICT and ICT-derived natural products. We particularly highlight studies where the reactivity patterns of the characteristic functional groups (i.e. isonitrile, isocyanate) are intimately involved in isolation and structural characterization efforts.A number of bisabolene sesquiterpenes have been isolated (Fig. 2). An isolation effort from “twilight sponges”, found at depths below 50 m, produced a number of active extracts and resulting active compounds validating this unusual source material.12 These studies identified a new bisabolene derivative theonellin isocyanate 13, as well the known isonitrile and isothiocyanate congeners. Other efforts obtained a variety of compounds in which the acyclic portion of the bisabolene scaffold is extensively oxidatively modified. These efforts include the isolation of epoxides, 14 and 15, as well as a large number of oxidatively modified structures in a single isolation study from Axinyssa sponges, termed the axinyssines A–L, 16 to 28.13,14 Relative and absolute assignments were obtained through mix of derivatization, NMR, MS, and CD experiments. In several instances the stereochemical assignment at C7, the position adjacent to the cyclohexane ring, remains to be defined.
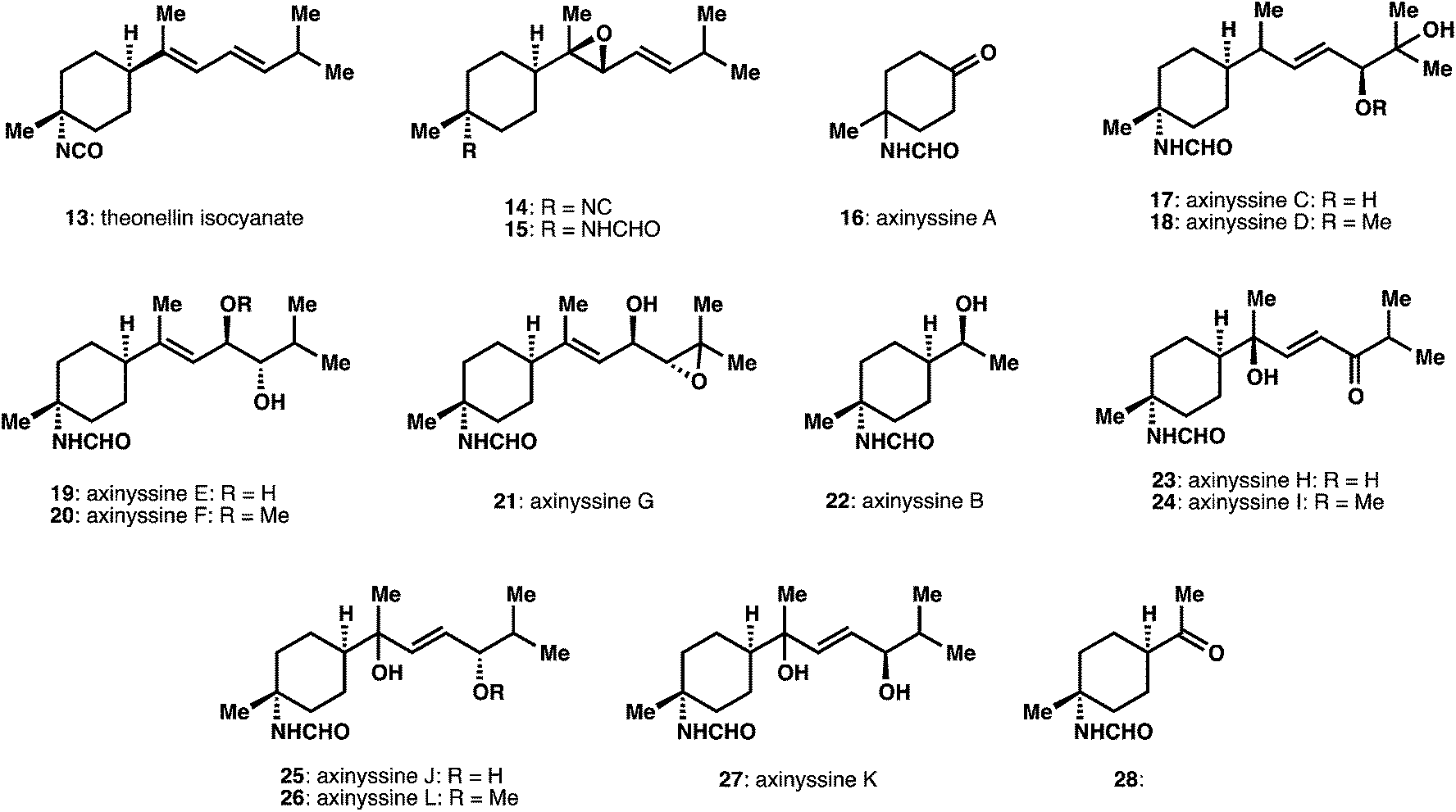 | ||
| Fig. 2 Bisabolene sesquiterpenes isolated from 2004–2014. | ||
A number of isolation efforts have identified additional sesquiterpene ICTs (Fig. 3). The unusual cyclopentenone-containing structure, 3-oxo-axisonitrile 29, was isolated from an Acanthella sponge.15 Zubía and coworkers disclosed a significant series of new bicyclic sesquiterpenes, the axinisothiocyanates, 31 to 44, from sponges of the Axinyssa genus.16,17 Among these is the nitrile-containing compound axinynitrile (30), the structure of which was verified by semi-synthesis. Notably, these highly substituted decalins are extensively oxygenated, including with peroxides, which is unusual among ICTs. Several other isolation efforts have provided isothiocyanates, formamides, and isocyanates, i.e.45 to 48, that complete the standard functional group tetrad (isonitrile, isothiocyanate, isocyanate and formamide) common with these compounds.18–20 One of these efforts appeared to isolate new diastereomers within the cadinene group of sesquiterpenes,20 however subsequent synthetic efforts failed to validate the proposed structure and further efforts appear to be required.21 Also quite significant is a systematic survey that compared the different constituent sesquiterpenes from various Acanthella cavernosa populations, which were taken from different isolation sites.22 These studies included critical efforts to clarify uncertainties in the absolute and relative structures of several previously proposed structures. Consequently, some caution should be taken in evaluating the isolation literature for these isocyanoterpenes, since assignment of stereochemistry in particular is challenged by overlapping signals in associated spectra.
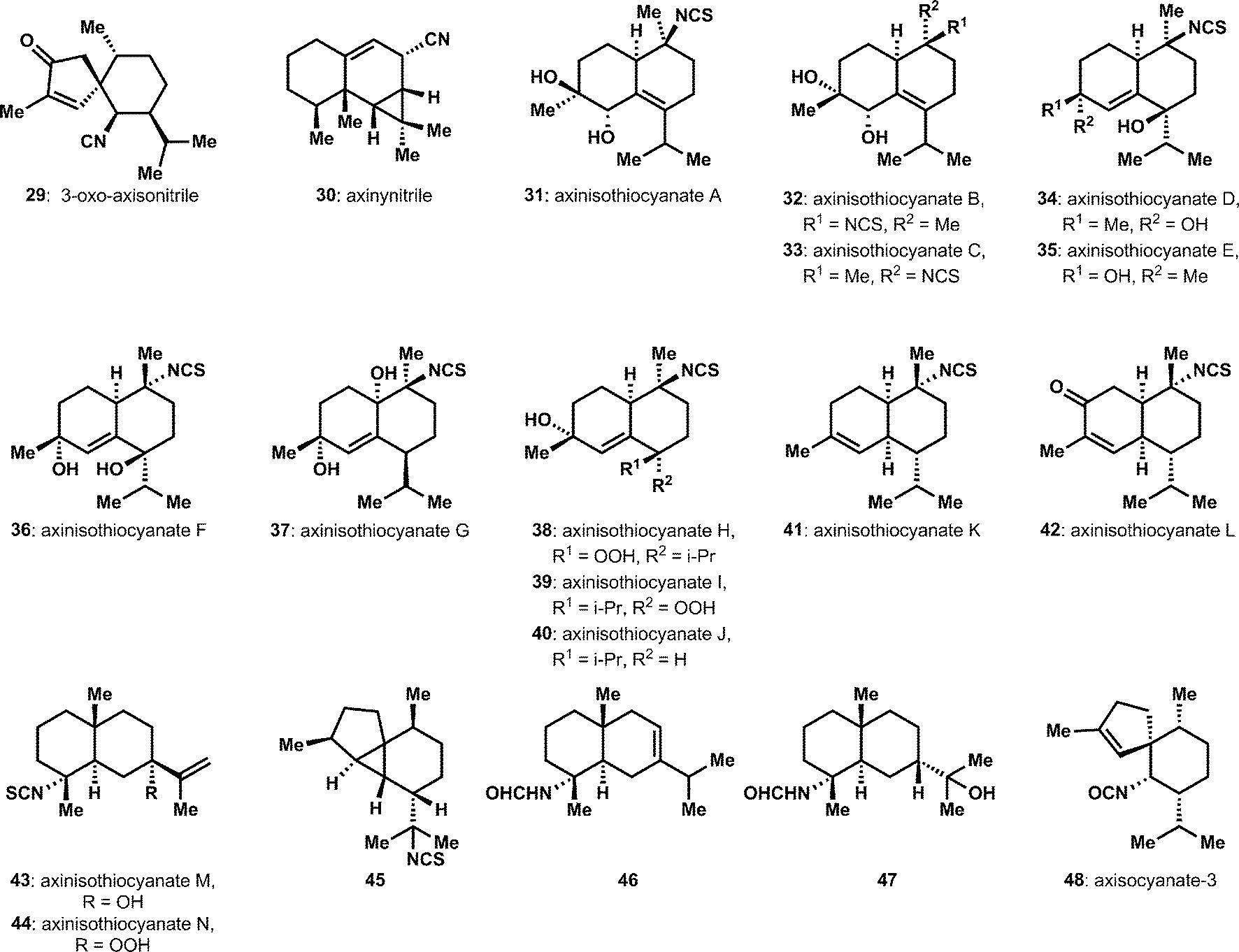 | ||
| Fig. 3 Sesquiterpenes isolated from 2004–2014. | ||
The diversity of diterpenes has also significantly expanded in recent years. The cavernene natural products, 49–52, as well as additions to the kalihinol and kalihinene families, 53–58, were reported through isolation efforts from Acanthella cavernosa (Fig. 4A).23–25 Notably, kalihinols M–T (59–66) were obtained from the South China Sea in a bioassay guided fraction effort targeted at biofouling. These compounds show a range of anti-biofouling activity, including several compounds in the submicromolar range.26 The variety of amphilectane structures has also grown, as shown in Fig. 4B.20,27–30 Of particular interest are the methylamine and formamide, 77 and 78, variants of the isoneoamphilectane scaffold, which were obtained from a Svenzea flava sponge and shown to display moderate antitubercular activity (MICs between 6 and 32 μg mL−1 against a M. tuberculosis H37RV).31 Also quite notable was a significant reassignment effort of the absolute and relative stereochemistry of several previously assigned32 compounds through a combination of X-ray and Mosher ester analysis.33
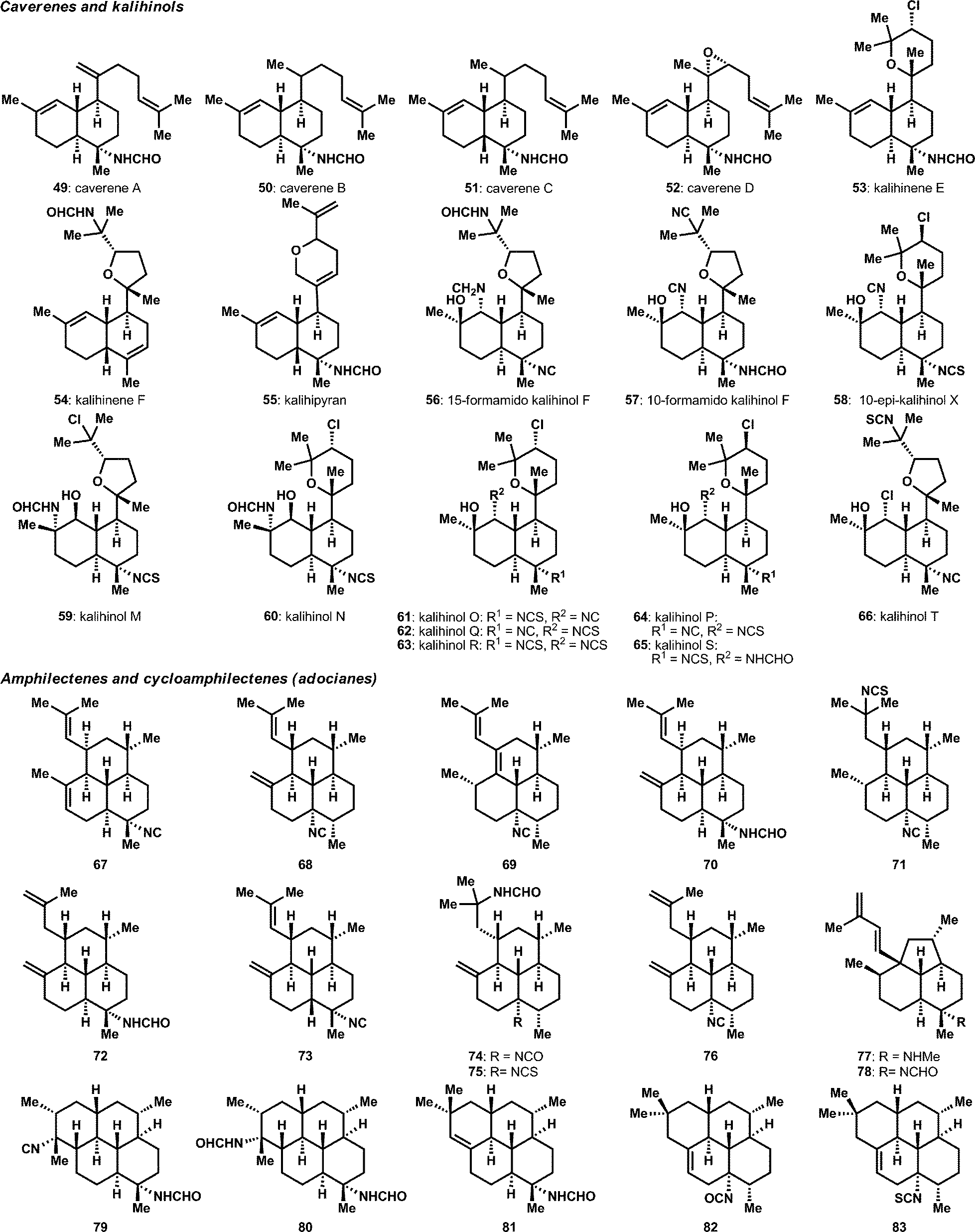 | ||
| Fig. 4 Diterpene natural products isolated from 2004–2014. | ||
Recent findings point out the role isonitrile reactivity can play in the biosynthesis, and isolation, of these molecules. Rodriguez and coworkers reported the isolation, structure, activity, and semisynthesis of the unusual β-lactam substituted amphilectene, monamphilectine A (84, Fig. 5a). The natural product 84 was prepared in a – presumably biosynthetic – single step from amphilectene 8 through Ugi reaction with formaldehyde and β-alanine. The anti-malarial activity of β-lactam 84 is somewhat diminished relative to 8 (0.60 vs. 0.04 μM against Plasmodium falciparum strain W2).34 Nevertheless, given the central role of β-lactam antibiotics in medicine, it is quite possible these new structures will ultimately be found to have useful activity.
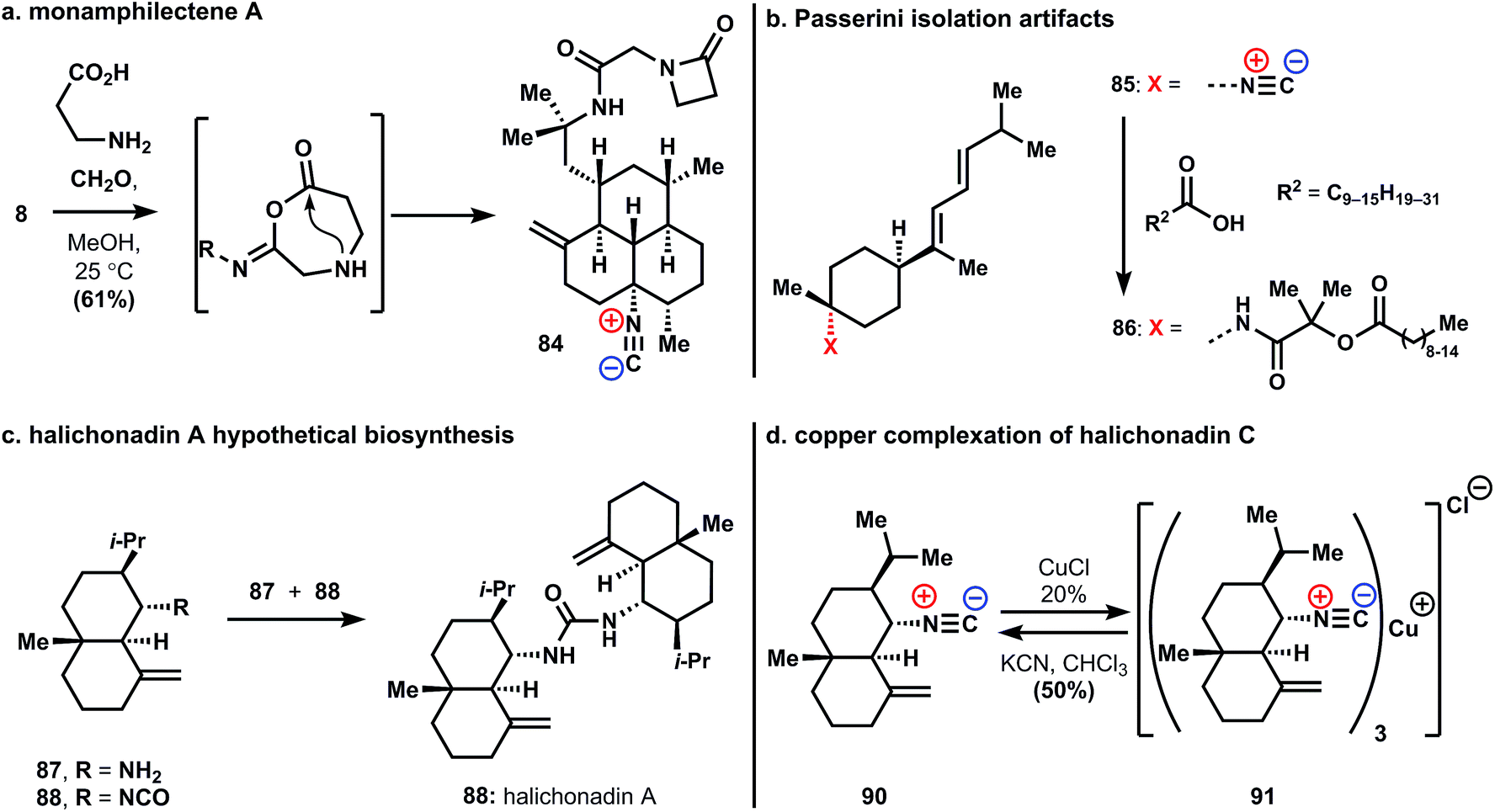 | ||
| Fig. 5 (a) Semisynthesis of monamphilectine A. (b) Passerini isolation artifacts. (c) Proposed biosynthesis of urea-linked sesquiterpenes. (d) Copper complexation and decomplexation of isocyanoterpenes. | ||
A recent result reveals the care required in the isolation of these potentially reactive natural products. Compound 86 was obtained from an Axinyssa sponge and ultimately shown to be an artifact of the isolation process (Fig. 5b). This compound results from a Passerini reaction occurring during isolation between known sesquiterpene 3-isocyanotheonellin, 85, acetone, and long-chain alkyl carboxylic acids.35
Garson and coworkers suggested an intriguing biosynthetic hypothesis regarding the origin of a significant, and growing, group of urea-linked sesquiterpenes.36–38 These ureas, exemplified in Fig. 5c with halichonadin A, 89, are isolated from marine sponges of the genus Halichondria. It is suggested that these arise through non-enzymatic addition of amines, formed from isonitriles through hydration and decarboxylation, to isocyanates. This suggestion is based on the isolation of an isocyanate and the isonitrile congener from the same natural source. Model studies verified the general underlying reactivity. These results suggest that halichonadin natural products are derived from ICTs and provide an example of non-enzymatic reactivity in biosynthesis.
Metal chelation represents another form of isonitrile reactivity that could conceivable play a key role in the biology of these compounds. In this context, a quite unusual copper(I) complex of halichonadin C (91) was isolated from the Halichondria sponge (Fig. 5d). The authors report the chemical interconversion of the demetalated parent natural product halichonadin C (90) and its copper complex.39
3 Chemical synthesis of isocyanoterpenes
3.1 Introduction
Retrosynthetic analysis of a molecule is guided by higher-level strategies that dictate the identity and order of transforms, intermediate targets en route, and occasionally the starting materials of the synthesis.40 For example, functional groups and their relative positions on the carbon skeleton of a molecular target provide useful ‘handles’ to guide the target's initial dissection.41 If the target is a naturally-occurring molecule, then biosynthesis – a known pathway,42 or a reasonable, hypothesized pathway – can inform the choices made in retrosynthesis. Within the alkaloid classes of natural products, biosynthesis-guided retrosynthesis is an established higher-level strategy that is easily recognized and employed, since the overwhelming majority of C–N bonds in these molecules are synthesized by imine/iminium chemistry, and this chemistry is historically simple to execute in the laboratory.43 Therefore the occurrence of isocyanoterpenes (ICTs) – nitrogenous, marine terpenoids whose C–N bonds are apparently derived from carbocations,44 not imines – present a significant challenge to synthesis.Apart from the challenges imposed by their aberrant biosynthesis, the problems encountered in ICT chemical synthesis diverge between the major subclasses. For instance, among the amphilectenes and adocianes (or cycloamphilectenes, 7–9, Fig. 1A-II) the absence of heteroatomic functional groups aside from widely spaced isonitriles limit the number of easily-identified polar disconnections.41 In contrast, the dense functional groups within the kalihinols (10–12, Fig. 1A-III) provide a range of viable retrosynthetic transforms but chemoselectivity then limits the availability of corresponding chemical reactions. Chemical synthesis of any subclass must address the challenge of installing the isonitrile functional group.
Almost without exception, the isonitrile function is installed at the end of the synthesis. Therefore, the tactics used for isonitrile installation determine significant components of the global strategy since downstream maneuvers are influenced by the isonitrile precursor functional groups. Inasmuch as these functional groups are the sole heteroatoms in the target, their efficient utilization without introduction of superfluous functionality (functional group addition, FGA) is crucial. Therefore, before discussing any specific syntheses, the general approaches to isonitrile installation will be summarized.
3.2 C–N bond formation
Most naturally occurring, marine ICTs contain stereogenic tert-alkyl isonitriles and their derivatives (amines, amides, isocyanates, isothiocyanates), although some contain sec-alkyl substitution. The sec-alkyl amino groups are generally and easily established on the carbon scaffold by reductive amination of the corresponding ketone (Fig. 6a),45,46 or SN2 displacement of a secondary leaving group with a nitrogenous nucleophile.47 However, introduction of stereogenic tert-alkyl isonitriles, amines, and amides is challenging and has required diverse strategies to achieve its efficient and stereoselective introduction (Fig. 6b). The Curtius rearrangement has been utilized by Piers,48–50 Miyaoka/Yamada,51 Asaoka,52 and Mander53 to introduce the C–N bond with the correct stereochemistry based on the high stereospecificity associated with this reaction. The stereochemistry of the product isocyanate is specified by the stereochemistry of the precursor carboxylic acid, which is easy to establish by stereoselective alkylation (see discussion below). However, this strategy works best when the carboxylate is retained throughout the synthesis; otherwise its installation from the corresponding dehomologated ketone consumes several steps. An alternative approach devised by Wood establishes the correct stereochemistry via reductive cleavage of an aziridine.54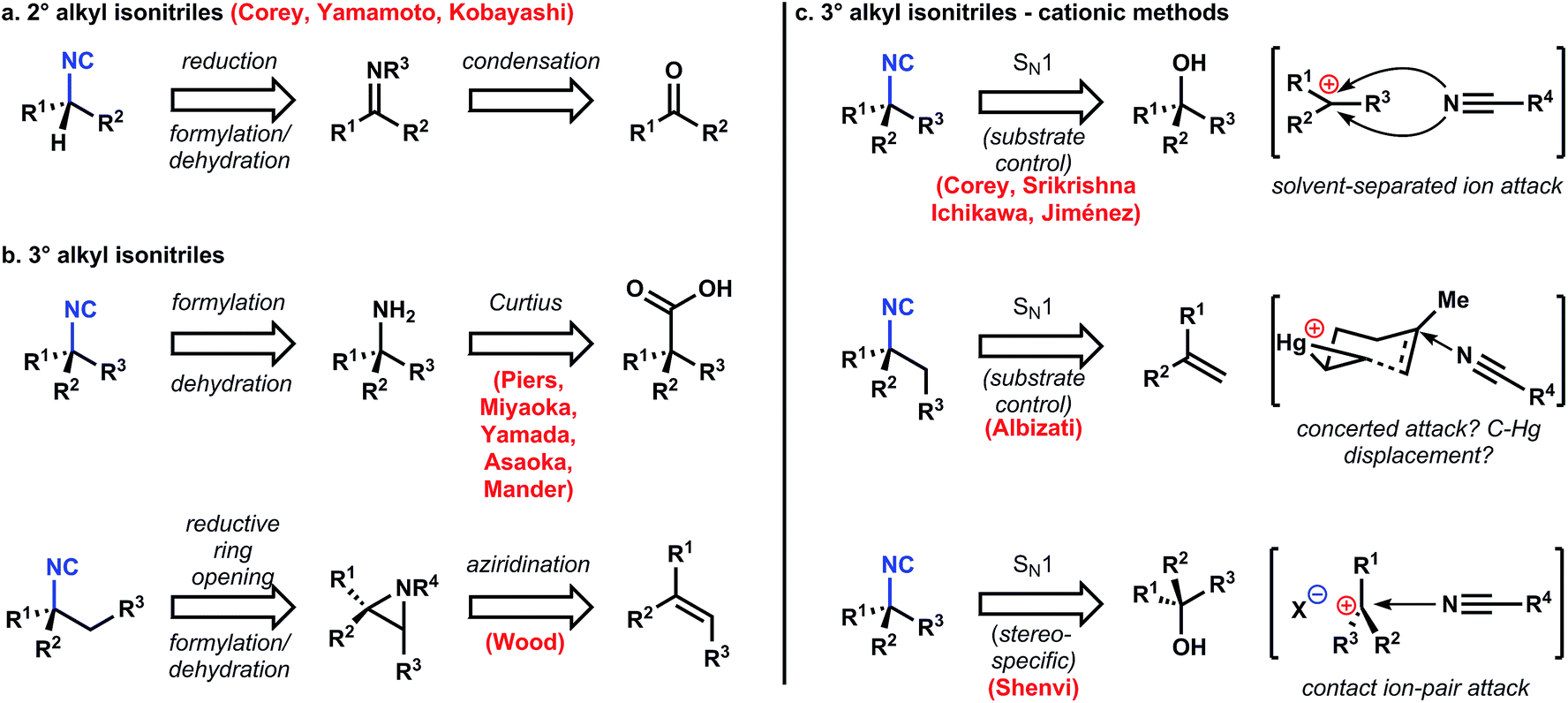 | ||
| Fig. 6 Retrosynthetic strategies to generate stereogenic sec- and tert-alkyl isonitriles. | ||
Variations on the Ritter reaction also quickly establish the tertiary stereogenic isonitriles by capture of carbocations derived either from alkenes or tertiary alcohols and their derivatives. Most reports utilize the attack of a solvent separated ion pair,55–57 which means that existing, proximal stereocenters are necessary to induce any stereoselectivity. There is a single and remarkable report by Albizati58 where the stereochemistry of the C–N bond is determined not by an ion pair, but by attack on a nascent carbocation (or by displacement of a C–Hg bond, vide infra). Finally, a recent report from Shenvi and coworkers describes a stereospecific tertiary alcohol displacement (via an intermediate perfluoroester) to form isonitriles.59
3.3 Substituted or embedded decalin motif
Treatments of conformational analysis by organic chemistry textbooks usually describe the lesser stability of cis- versus trans-decalin (92cvs.92t) due to more gauche butane interactions in the former (three) versus the latter (none).60,61 Occasionally, the corresponding ketone, 1-decalone (93c or 93t), is discussed also; this molecule prefers overwhelmingly the trans-ring fusion upon epimerization of the α-carbon.62 Therefore, the trans-decalin motifs embedded in the largest families of ICTs (amphilectenes/adocianes, kalihinols, miscellaneous sesquiterpenes) would appear upon cursory analysis to be simple, and equilibration to favor the trans-ring junction by wide margin to be a foregone conclusion. However, even slight variations from 1-decalone perturb the equilibrium to either inconvenient mixtures of stereoisomers (94c:94t, 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) or even towards a bias for cis (95c:95t, 4.6:1). Therefore, the planning phase for synthesis of polydecalin ICTs should include a careful conformational analysis of the thermodynamically preferred configuration at equilibrium. Some examples benefit from a preference for the targeted stereochemistry upon epimerization (96 → 97,5698 → 99,63 ratios are not given but high selectivity is implied in experimental details). Other cases suffer from the production of inconvenient mixtures (100 → 101,64102 → 103;54 in the latter case, Wood solves this problem in a subsequent step, vide infra). In a final example, Vanderwal attempts to form the all-trans tricyclic core of the amphilectenes and adocianes via a transannular Michael reaction,65 according to the report by Swaminathan that 105 is formed preferentially from 104.66 Vanderwal determined that 105 was misassigned, and the Michael reaction instead produces 106 and 107, which contain stable, mixed cis- and trans-decalin motifs, as proven by X-ray analysis. Furthermore, 106 and 107 are kinetic traps; their attempted equilibration does not lead to 105.
:1) or even towards a bias for cis (95c:95t, 4.6:1). Therefore, the planning phase for synthesis of polydecalin ICTs should include a careful conformational analysis of the thermodynamically preferred configuration at equilibrium. Some examples benefit from a preference for the targeted stereochemistry upon epimerization (96 → 97,5698 → 99,63 ratios are not given but high selectivity is implied in experimental details). Other cases suffer from the production of inconvenient mixtures (100 → 101,64102 → 103;54 in the latter case, Wood solves this problem in a subsequent step, vide infra). In a final example, Vanderwal attempts to form the all-trans tricyclic core of the amphilectenes and adocianes via a transannular Michael reaction,65 according to the report by Swaminathan that 105 is formed preferentially from 104.66 Vanderwal determined that 105 was misassigned, and the Michael reaction instead produces 106 and 107, which contain stable, mixed cis- and trans-decalin motifs, as proven by X-ray analysis. Furthermore, 106 and 107 are kinetic traps; their attempted equilibration does not lead to 105.
The point is that the all trans-ring junctures should not be dismissed offhand as trivial, nor are epimerizable stereocenters clearable40 at every stage of the retrosynthetic analysis. Instead, careful planning should identify at what stage the decalins might be equilibrated to favor the desired stereochemistry (Fig. 7).
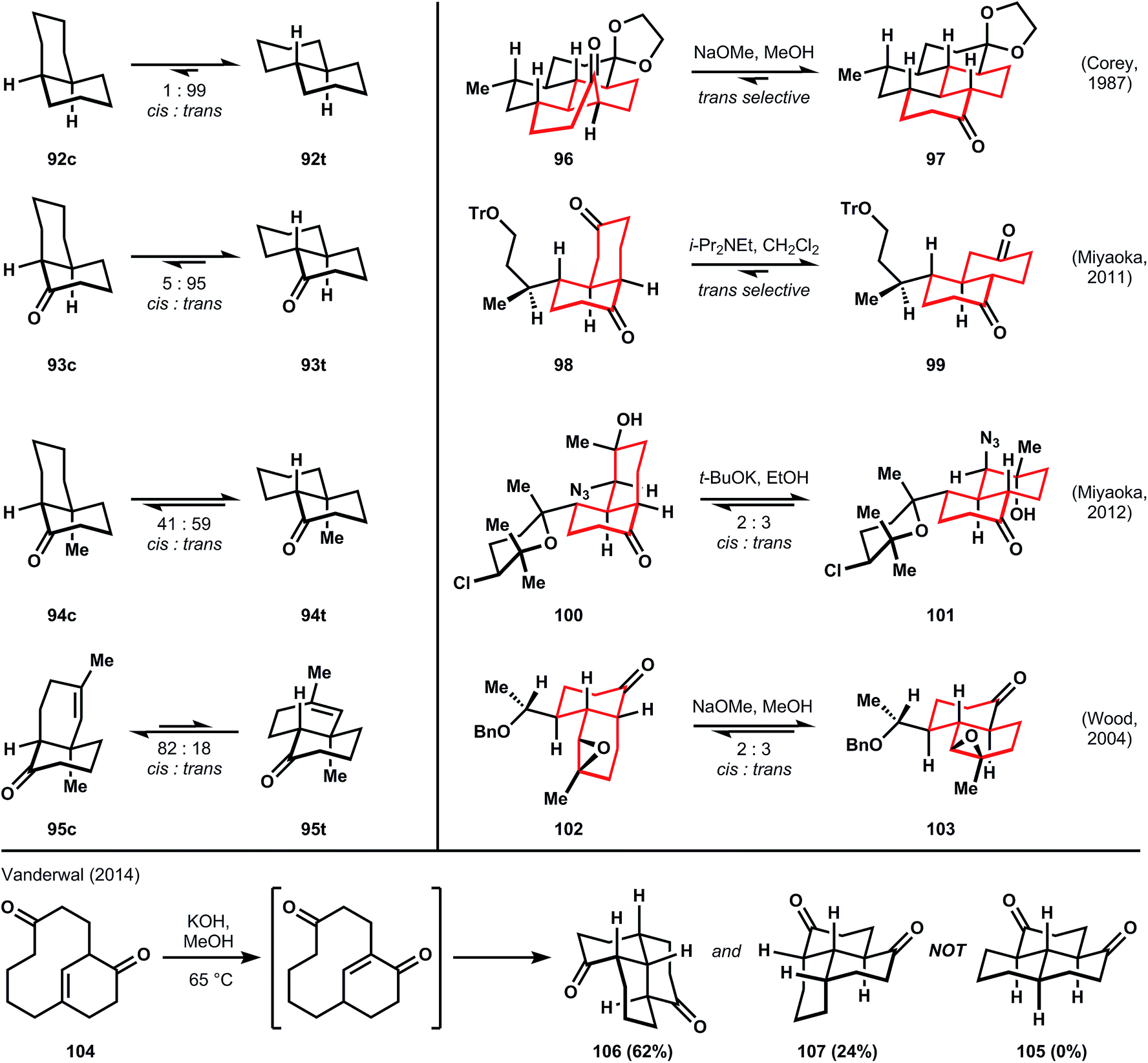 | ||
| Fig. 7 Cis-/trans-isomerism of substituted decalins. | ||
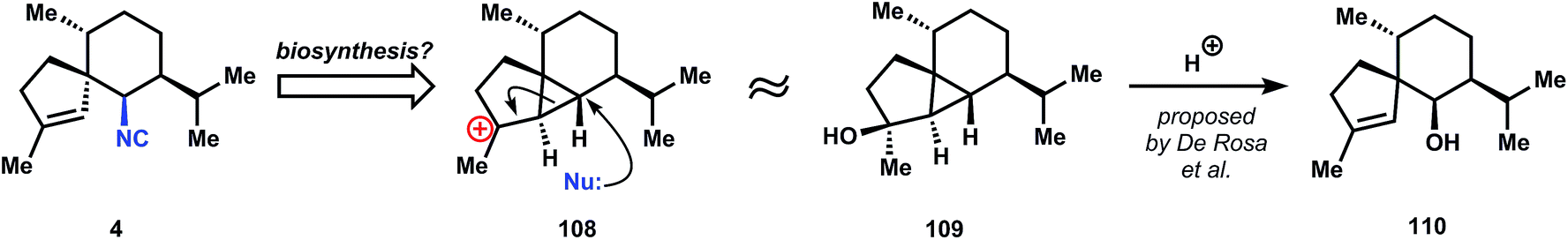 | ||
| Fig. 8 A hypothetical biosynthesis of 4 based on related terpene relationships. | ||
3.4 Overview
The differing strategies for isonitrile installation position the molecule for dissection along numerous possible retrosynthetic branches, depending on the functional group used as an isonitrile precursor. Below, we compare the different ways in which subclasses of these molecules have been analyzed, and then discuss the syntheses as actually executed. In most cases, we take the analysis at face-value, even though a chemical synthesis almost never proceeds exactly as designed (also, the assumption is made that a retrosynthetic analysis was utilized in all cases, which may be an incorrect framework imposed by the authors) (Fig. 9–11).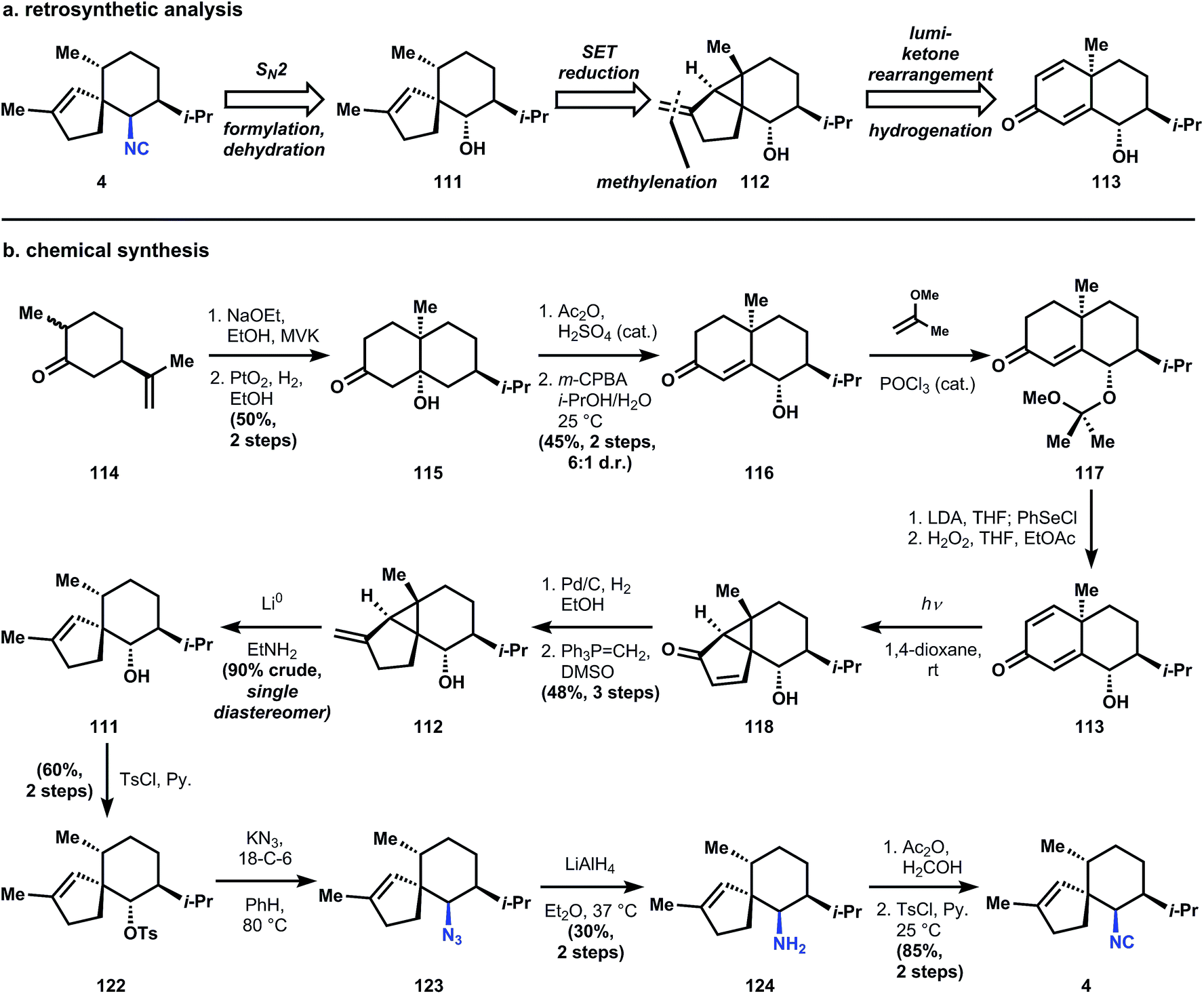 | ||
| Fig. 9 Caine's synthesis of (−)-axisonitrile-3. | ||
 | ||
| Fig. 10 Piers' studies on the stereochemistry of cyclopropyl ketone reductive cleavage. | ||
 | ||
| Fig. 11 Hypothesized biosynthesis of 2. | ||
The syntheses are organized roughly by subclass and within those groups presented mostly in chronological order. Understanding the chronology helps to properly assign credit to conceptual forerunners. In almost all cases, the targeted molecule is drawn initially in three dimensions to convey the actual geometry of the bonds and how this geometry keys certain retrosynthetic transforms (a symbol indicating rotation of Cartesian coordinates orients the reader, e.g. see Fig. 12, 2![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 2). Only in a few cases are multiple synthetic steps replaced with a simple step count and yield; instead, each synthesis is covered thoroughly. Even though this level of detail yields large schemes, the larger format allows for the laboriousness of a sequence to be appreciated and also serves an educational role: learning many reactions (and how to avoid using so many!). Not every synthesis is covered; the remaining syntheses and relevant synthesis studies may be found in ref. 67–83.
2). Only in a few cases are multiple synthetic steps replaced with a simple step count and yield; instead, each synthesis is covered thoroughly. Even though this level of detail yields large schemes, the larger format allows for the laboriousness of a sequence to be appreciated and also serves an educational role: learning many reactions (and how to avoid using so many!). Not every synthesis is covered; the remaining syntheses and relevant synthesis studies may be found in ref. 67–83.
 | ||
| Fig. 12 (a) Corey's analysis of 2; (b) Corey's chemical synthesis of 2. | ||
3.5 Miscellaneous sesquiterpenes
Although the targeted molecule contains a secondary isonitrile, its biosynthesis probably follows a variation of the Garson hypothesis (Fig. 8),44 since 4 can be imagined to arise from a cationic fragmentation and nucleophilic capture of cyclopropane 108. Indeed, De Rosa et al. have isolated (−)-cubebol (109) and axenol (110) from the marine alga Taonia atomaria and proposed their biosynthetic relationship via hydrolysis of 109.85 A related anionic fragmentation lies at the heart of Caine's synthesis of 4.
The Caine strategy to access 4 is unusually beautiful, and relies, surprisingly, on steroid chemistry.86 Specifically, the synthesis derives some inspiration from the work of Piers (another contributor to isocyanoterpene chemistry, see Section 3.6.2) who demonstrated the stereoselective reductive cleavage of lumiketones related to lumicholestenone.87 The contributions of Caine and Deutsch to this area include expansion of these reductive fragmentations to vinyl cyclopropanes and application to the synthesis of 4. Utilization of a vinyl cyclopropane rather than a cyclopropyl ketone is crucial, since the former leads directly to the correct trisubstituted alkene regioisomer (111), whereas derivatization from the corresponding carbonyl would require regiochemical control over enolization. The cyclopropane 112 would arise from the lumiketone rearrangement of 113, which can be easily derived from dihydrocarvone (see synthesis). A final note: the authors install the isonitrile function via an SN2 displacement of a sterically congested, secondary, neopentyl tosylate, which differs significantly from most strategies. Thirty years later, Kobayashi attempted to reproduce the reaction and failed, but discovered that a sodium cyanoborohydride reduction of the corresponding oxime established the same correct stereochemistry88 – details to keep in mind for future investigators.
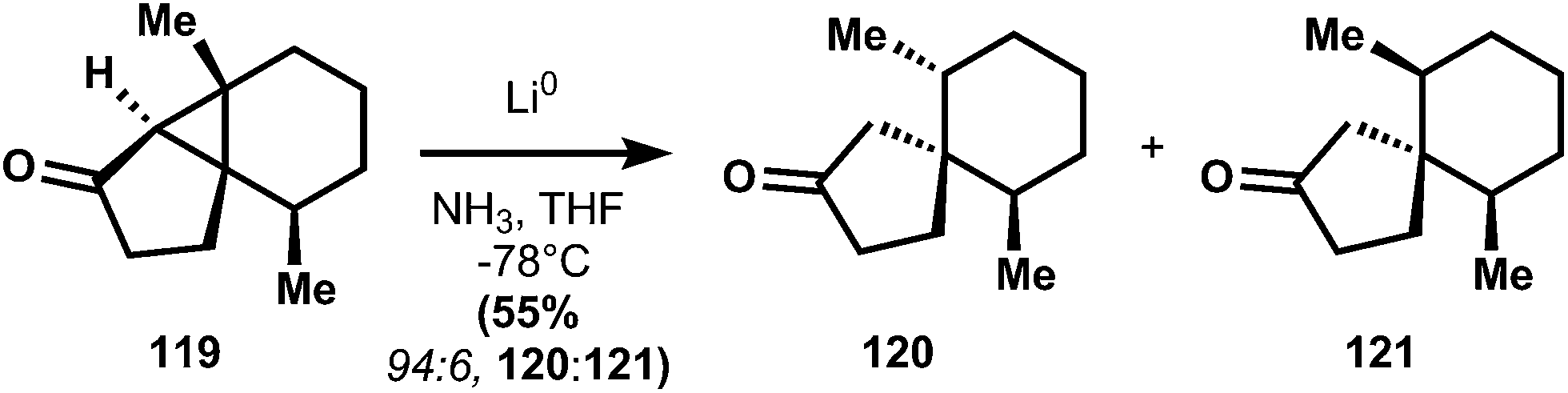
The synthesis begins with the isolation of the tertiary alcohol intermediate in the Robinson annulation of dihydrocarvone (114) with methyl vinyl ketone, followed by isopropenyl hydrogenation with Adam's catalyst.89 Treatment of 114 with catalytic sulfuric acid in acetic anhydride effects elimination of the hydroxyl and conversion of the nascent enone to the extended enol acetate. This intermediate is treated with m-CPBA under aqueous conditions to generate a labile epoxide that opens to form enone 116 with good (6:1) stereoselectivity. In order to access the cyclohexadienone substrate for rearrangement, the stereogenic alcohol is first protected as its methoxyisopropylidine ether (117). Unsaturation is carried out using selenation of the ketone enolate, followed by oxidative elimination to yield 113 (presumably the aqueous acidic workup90 to remove diisopropylamine after selenation also cleaves the methoxyisopropylidine ether; the authors do not comment). Irradiation of a dilute, room temperature dioxane solution of 113 with a low-pressure mercury lamp causes stereospecific rearrangement to 118, equivalent to the classical photochemical rearrangement of santonin.91 Catalytic hydrogenation and Wittig olefination of 118 provide 112, which is poised for the planned reductive fragmentation. Whereas carbonyls undergo single-electron reduction at cryogenic temperatures in liquid ammonia, alkene 112 requires more activation energy – a balmy 16 °C in ethylamine – to initiate reduction. Although this energy barrier usually limits functional group compatibility,92 there are no easily reducible groups in 112 and therefore the reaction proceeds cleanly to 111. It is noteworthy that the stereogenic methyl of 111 possesses the thermodynamically preferred equatorial stereochemistry, even though Piers had observed selectivity for the equatorial–axial dimethyl cyclohexane 120 (see 119 → 120 + 121).87 Both Caine's and Piers' systems yield inversion of stereochemistry, but the basis of this selectivity is unclear.
Regardless, 111 can be elaborated into the target structure first by tosylation to 122 and then SN2 displacement with potassium azide in the presence of 18-crown-6, optimized conditions that minimized elimination. Lithium aluminum hydride reduction produced the corresponding amine 124, which was converted to the isonitrile 4 using conditions developed by Corey93 – the most commonly applied method for this conversion (vide infra).
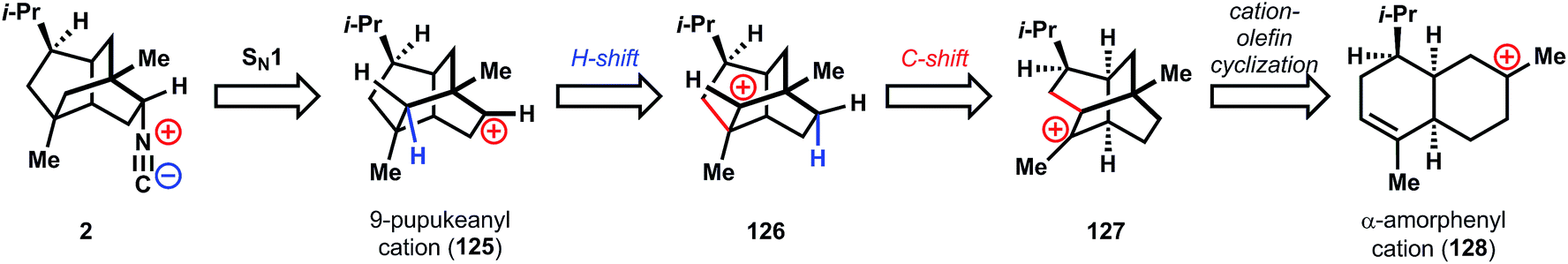
The biosynthesis of 2 is proposed94,95 to traverse several carbocationic intermediates by a series of hydrogen- and carbon-shifts. While these reaction cascades are not unusual in terpene biosynthesis, they are still remarkable given the high energy of carbocations (vide infra) and therefore remain a vibrant research area today.96,97 Formation of the isonitrile would proceed according to the Garson hypothesis44 from the 9-pupukeanyl cation (125), which is hypothesized to derive from isomeric cation 126via a hydrogen shift, in turn formed from twistane cation 127. Cation 127 could be formed by Markovnikov cation-olefin cyclization from α-amorphenyl cation 128, a commonly hypothesized cationic intermediate in the biosynthesis of diverse terpenes.98
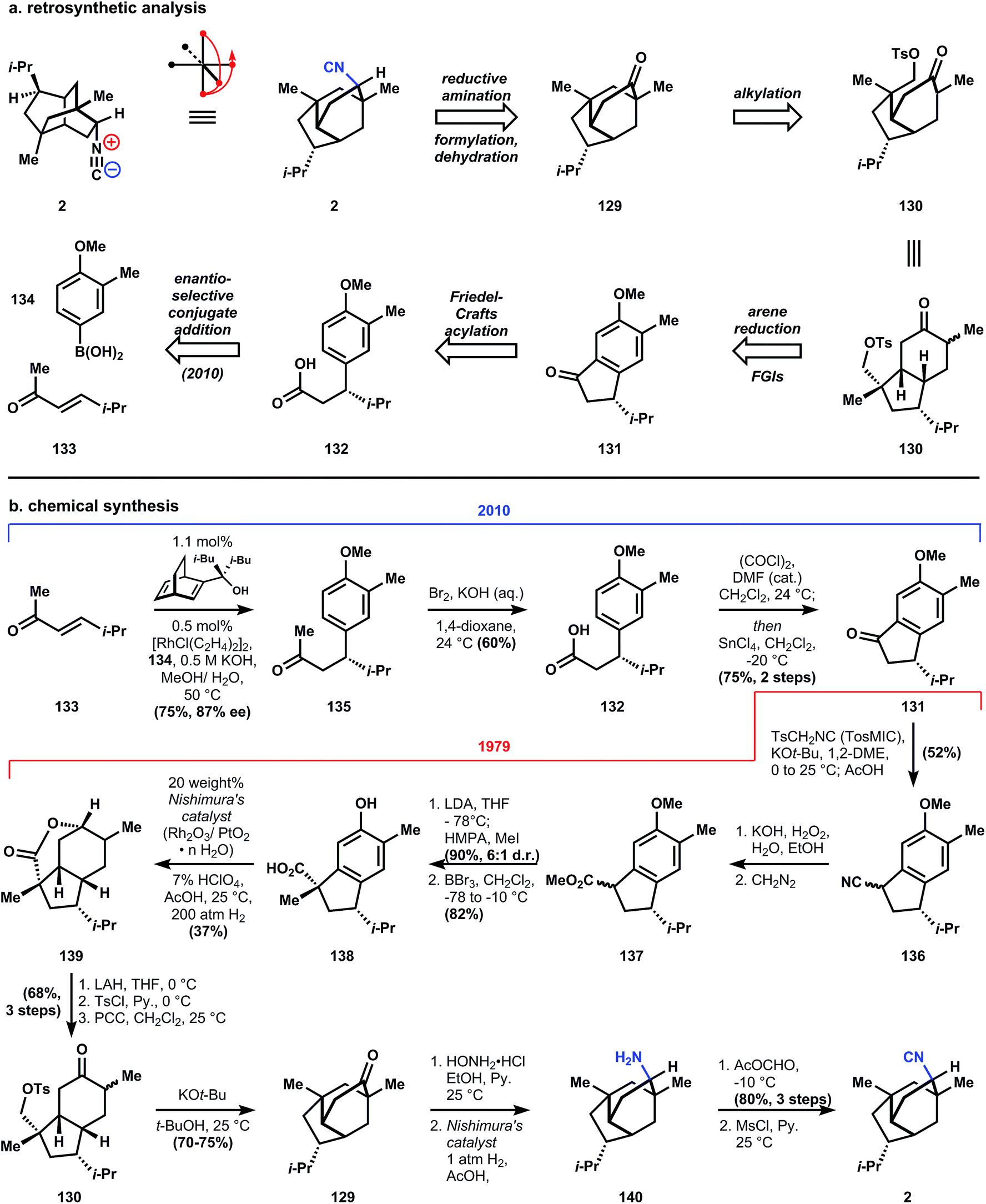
The evolving heuristics for retrosynthetic analysis being developed in the Corey laboratories at the time helped shape his strategy (originally racemic, vide infra) to access isocyanopupukeanane 2 (also evolving was the name for this approach for synthesis planning, which in this paper is called antithetic analysis, although retrosynthetic has since stuck).40 The first transform applied to 2, conversion of the isonitrile to the ketone, is a common and almost universally conserved strategic move within ICTs (vide infra) for three important reasons. First, a stereocenter is removed, thereby reducing the complexity of the molecule. Second, a challenging functional group is removed, which simplifies the practical aspects of applying chemoselective transforms downstream. And third, the ketone allows many more transforms to be directly applied since this functional group's chemistry is so well explored. Corey's strategy takes advantage of one such transform: the alkylation of ketone 129 with a pendant electrophilic carbon, which cleaves a ring of maximal bridging by breaking an exendo bond, a strategic, high priority bond according to his retrosynthetic analysis guidelines (for detailed description of these rules and nomenclature beyond the scope of this review, see ref. 40). This cleavage delivers a cis-fused 5–6 ring system 130 where the electrophilic methyltosylate is positioned endo and therefore proximal in space to the ketone's alpha-carbons. The saturated ring system is retrosynthetically unsaturated and the stereogenic quaternary carbon of 130 is simplified to the ketone of indanone 131. This broad stroke move is presumably based on the idea that the stereogenic isopropyl group would dominate other molecular features in controlling stereochemistry. Indanone 131 is derived from the arene 132, which was synthesized in racemic form in 1979, but rendered scalemic (87% ee) in 2010.99
Preparation of enantioenriched starting material is accomplished by the development of a new chiral diene ligand by Brown and Corey for the rhodium-catalyzed enantioselective conjugate addition of aryl- and alkenyl boronic acids to enones (133 + 132 → 135).99 Subjection of 135 to potassium hydroxide and bromine effects a haloform reaction to generate carboxylic acid 132, which is converted to its corresponding acid chloride and cyclized via a Friedel–Crafts acylation to provide indanone 131, thus intercepting the 1979 synthesis. The ketone can be homologated to 136 by the Van Leusen reaction with TosMIC;100 a similar homologation is utilized 30 years later in a related class of ICT (see Section 3.7.1). Saponification and diazomethane esterification of 136 provides methyl ester 137 as an inconsequential mixture of diastereomers. The stereochemistry of the alpha carbon is set by methylation with good stereoselectivity (6:1) due to delivery of the methyl group to the ester enolate face opposite to the isopropyl group. Boron tribromide is used to remove the methyl protecting group of the phenol, and concurrently demethylates the ester. Reduction of indane 138 to the perhydroindane is accomplished with Nishimura's catalyst101 – a mixed oxide of rhodium and platinum prepared in analogy to Adam's catalyst, and based on the observations that colloidal rhodium black is effective to reduce aromatic rings.102 Although a pressure of 200 atmospheres of hydrogen is required, the major product 139 (37% yield, no d.r. given) possesses the correct cis-hydrindane ring fusion and its alcohol has cyclized into a lactone, likely a result of the catalytic perchloric acid used. Reduction of the lactone provides a diol that is chemoselectively tosylated at the least hindered primary alcohol, which allows the remaining secondary alcohol to be oxidized with pyridinium chlorochromate (PCC, developed103 by Corey and Suggs in 1975), yielding ketone 130.
When 130 is deprotonated with LDA in THF, the kinetic enolate reacts to form a four-membered ring, which is presumably formed slower than the six-membered ring, but closes faster than proton exchange between the enolate and the product. Therefore, treatment of 130 with strong base under equilibrating conditions (t-BuOK, t-BuOH) produces 129 as the major isomer. Installation of the isonitrile with the correct stereochemistry involves a four – step process of: hydroxylamine condensation to an oxime; reduction with Nishimura's catalyst101 to cleave the N–O bond and reduce the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond; and finally N-formylation of amine 140 and formamide dehydration according to a procedure developed in the Corey labs93 (and utilized in nearly all of the ICT syntheses) to yield 9-isocyanopupukeanane (2).
N bond; and finally N-formylation of amine 140 and formamide dehydration according to a procedure developed in the Corey labs93 (and utilized in nearly all of the ICT syntheses) to yield 9-isocyanopupukeanane (2).
Important features of Corey's synthesis of 2 include the use of TosMIC to homologate a cyclic ketone, which finds use in subsequent ICT syntheses; the two step formylation/dehydration conversion of an amine to an isonitrile, which finds use in nearly every ICT synthesis; and the demonstrated ability of Corey's retrosynthetic analysis guidelines to provide workable and efficient (17 steps) laboratory syntheses. The unsolved problem of inducing asymmetry was met with a solution in 2010 by the use of a chiral diene ligand, thus completing a thirty year cycle of problem identification, solution, and application – recursive logic that appears frequently in the Corey oeuvre.
The chemical synthesis begins with DIBAL reduction of enone 144 to yield a secondary allylic alcohol, which is heated in the presence of ethyl vinyl ether and mercuric acetate to produce aldehyde 145. Addition of vinylmagnesium bromide to 145 produces an inconsequential mixture of 4 diastereomeric alcohols, which are protected as their tetrahydropyran ethers (yielding eight diastereomers!). Allylic oxidation of the cyclohexene is accomplished with chromium trioxide-pyridine complex, leading to eight enone diastereomers of 146. Deprotonation and silylation leads to enolsilane 142, the targeted diene for the intramolecular Diels–Alder cycloaddition, which occurs with heating in benzene in a sealed vessel at 160 °C. Subsequent treatment with aqueous acetic acid at elevated temperature removes both the trimethylsilyl and tetrahydropyran groups to produce tricycle 147. After ketalization of the ketone with ethylene glycol and Corey–Kim oxidation104 of the secondary alcohol, all superfluous stereocenters are removed and the eight diastereomers of 146 converge to a single diastereomer 148. The ketal is necessary to prevent subsequent addition of the isopropyl group to the other ketone. Thus, installation of the isopropyl group occurs over four steps: isopropenyl lithium addition to 148, deketalization to yield 149, alcohol elimination (to 150), and diene hydrogenation with iridium black to provide tricycle 129. Use of iridium led to a very high diastereomeric ratio (>98:2) favoring the correct geometry, whereas other catalysts led to lower stereoselectivity, likely a result of competitive alkene isomerization.105 Tricyclic ketone 129 is an intermediate in the Corey synthesis of 2, and conversion to the target structure follows a similar path with slightly different conditions. Specifically, oxime formation is conducted in the absence of pyridine and therefore requires heating to produce 151. Oxime reduction utilizes an iterative procedure of deoxygenation with low valent titanium, followed by imine reduction with DIBAL to amine 140. Conversion of this amine into 9-isocyanopupukeanane (2) utilizes the same conditions as described in Caine's and Corey's syntheses above.
 | ||
| Fig. 13 (a) Yamamoto's analysis of 2. (b) Yamamoto's chemical synthesis of 2. | ||
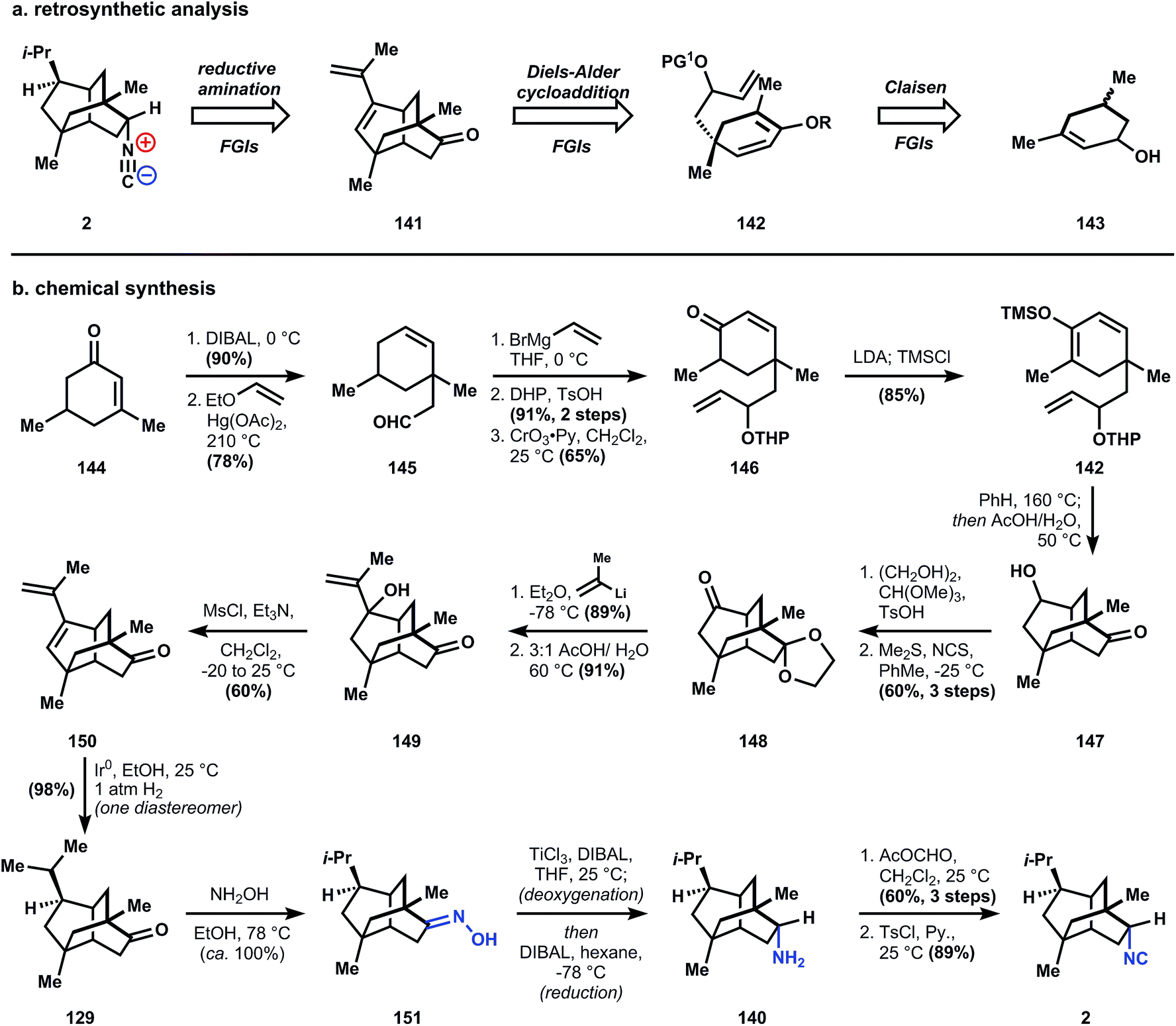
First, the isonitrile is excised through a cationic Ritter reaction, where stereochemistry is controlled by the concavity of tricycle 152. The alkene equilibrium is inconsequential to the erasure of any adjacent stereocenters because they are bridgehead carbons. The bond network of 152 is established quickly through another cationic reaction, a Wagner–Meerwein shift, which simplifies the ring system by embedding a [2.2.2]-bicyclic motif (153). This new tricycle is dissected using a rhodium-catalyzed C–H insertion to establish a dissonant relationship between the two oxygen functional groups, whereas the consonant relationships can be easily established using tandem Michael reactions, ultimately arriving at carvone 154. It should be noted that ent-1 and formamide 159 (see chemical synthesis, Fig. 14b) are both isolates of marine organisms, so a single synthetic route is bound to access the incorrect enantiomer of either 1 or 159. Fortunately carvone is commercially available as either enantiomer, so the desired target is specified by the starting material with little extra expense (Fig. 15).
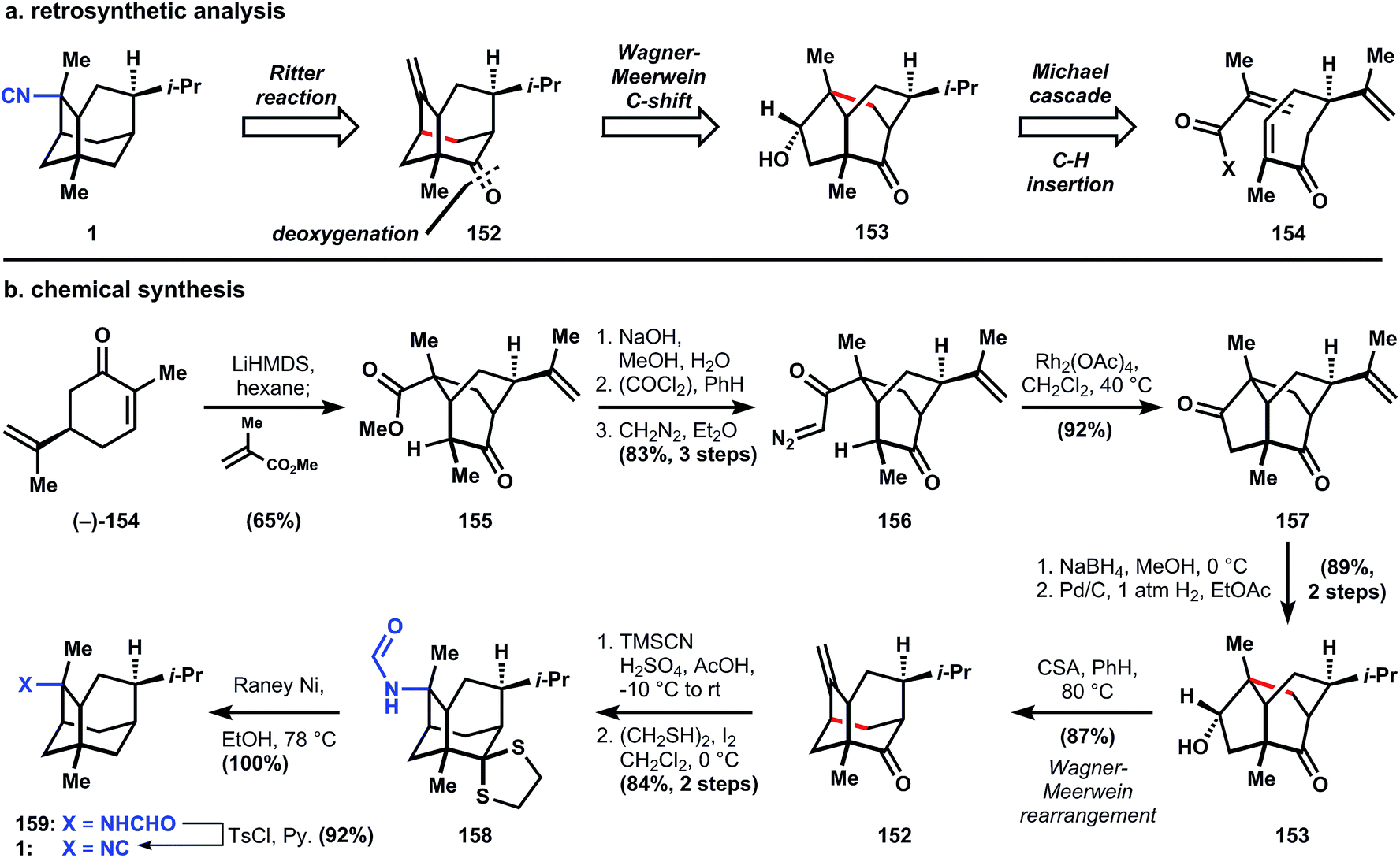 | ||
| Fig. 14 (a) Srikrishna's analysis of 1; (b) Srikrishna's chemical synthesis of 1. | ||
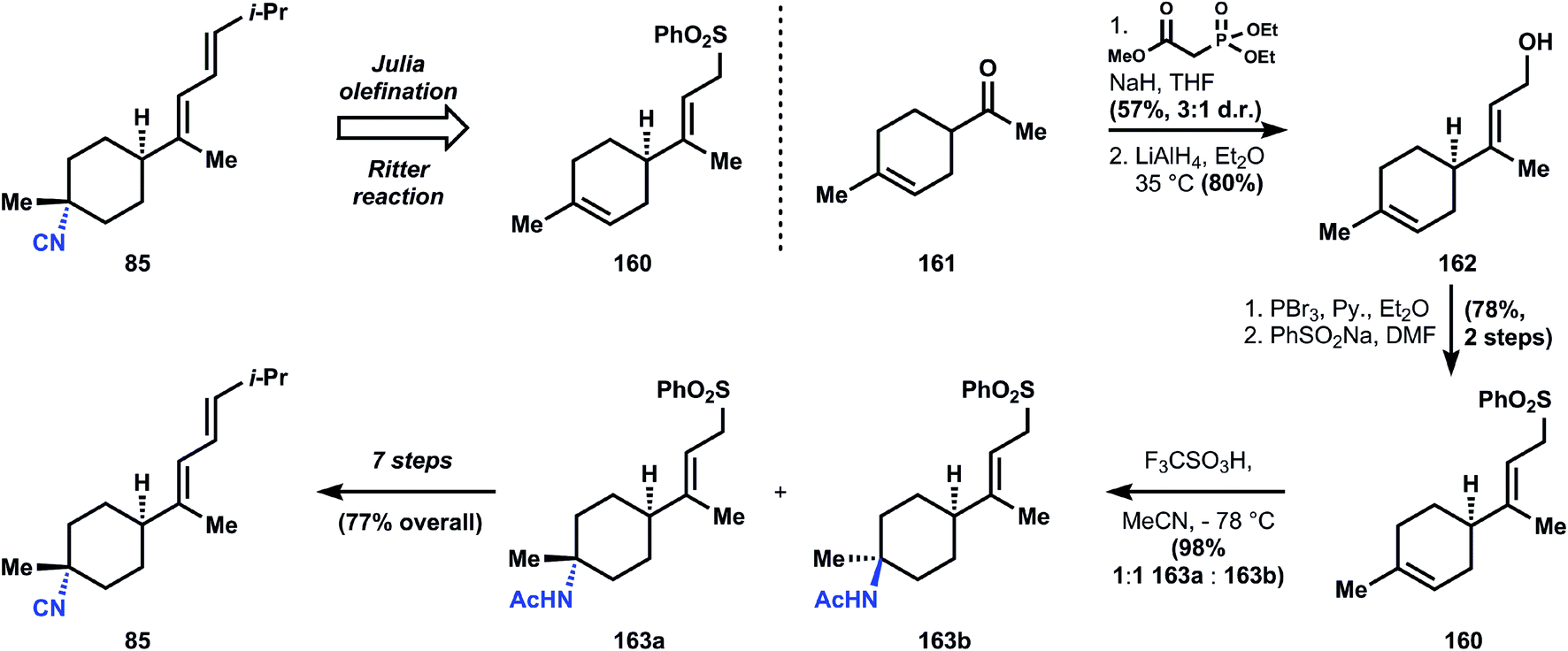 | ||
| Fig. 15 Ichikawa's analysis and synthesis of 85 using a Ritter reaction. | ||
The route to convert (−)-carvone (154) into the intermediate tricycle 157 (and (−)-9-pupukeanone)108 had previously been published by Srikrishna109 and traverses a remarkably concise sequence. First, the carvone enolate attacks methyl methacrolate in a Michael fashion from the face away from the isopropenyl group, and the newly formed ester enolate engages the enone in a second Michael reaction to generate bicycle 155. Saponification, acid chloride formation, and diazoketone installation proceed efficiently (82% overall) to yield 156, which is treated with catalytic rhodium(II) acetate (catalyst loading not given) in refluxing dichloromethane to produce diketone 157via C–H insertion. Borohydride reduction occurs selectively at the cyclopentanone on the convex face, followed by a palladium-catalyzed hydrogenation, which reduces the isopropenyl group to the stereogenic isopropyl found in 153 and 1. When 153 is heated with camphor sulphonic acid in refluxing benzene, the transient carbocation undergoes a Wagner–Meerwein carbon shift to the more thermodynamically-stable tricycle 152. A Ritter reaction with cyanotrimethylsilane (or trimethylsilyl cyanide, TMSCN) generates a formamide with stereochemical control of C–N bond formation caused by substrate bias (concave versus convex attack). Dithane formation is accomplished using ethanedithiol and iodine, which generates hydroiodic acid in situ and promotes nucleophilic attack and thionium formation. Finally, Raney nickel desulfurization generates (−)-2-(formylamino)trachyopsane 159, which is dehydrated to (−)-2-isocyano-trachyopsane 1, the enantiomer of the isolated material (Fig. 16).
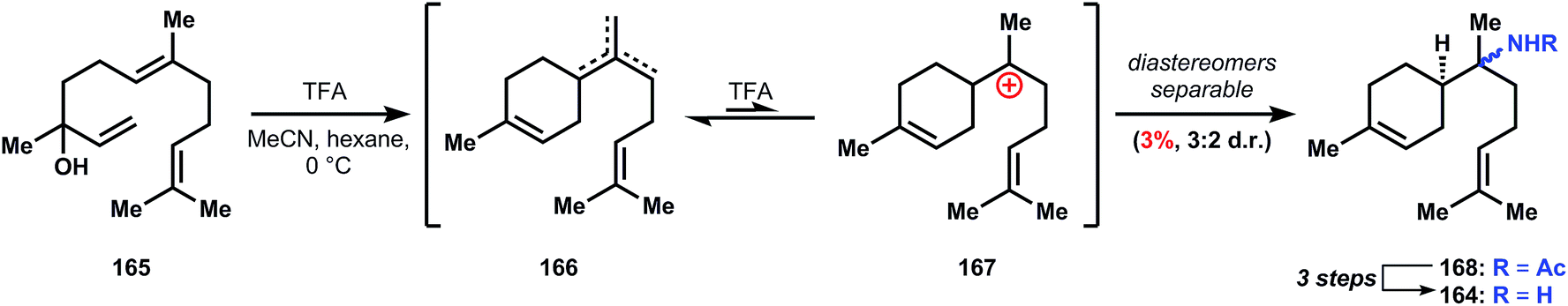 | ||
| Fig. 16 Ichikawa's polycyclization of nerolidol en route to bisabolyl amine 164. | ||
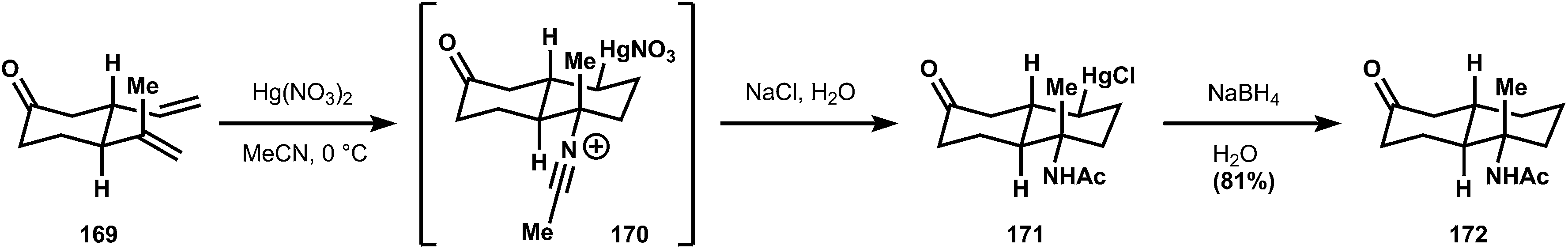 | ||
| Fig. 17 Albizati's mercury-mediated polyene cyclization and nitrile capture. | ||
The synthesis begins with ketone 161, the Diels–Alder adduct of methyl vinylketone and isoprene, which is converted to allyl alcohol 162via Horner–Wadsworth–Emmons olefination (3:1 stereoselectivity) and reduction. The alcohol is transformed into the corresponding allylic bromide, enabling alkylation of sodium phenylsulfinate to generate sulfone 160. At this point, the isobutylidene sidechain can be appended via Julia olefination, but a subsequent Ritter reaction is low yielding (38%, 63% based on recovered starting material) and more importantly the C–N bond formation favors the wrong diastereomer. The alternative Ritter addition to sulfone 160 offers a better yield (98%; fewer alkenes to protonate, electronic differentiation) and a marginal improvement in stereoselectivity, where now the two isomers 163a and 163b are produced in equimolar ratio. The isomers can be separated, and 163a carried forward to the natural product 85. Since, like most SN1 processes, the stereoselectivity of the Ritter reactions depends on direction from the substrate, no better solution is available to Ichikawa (Fig. 18).
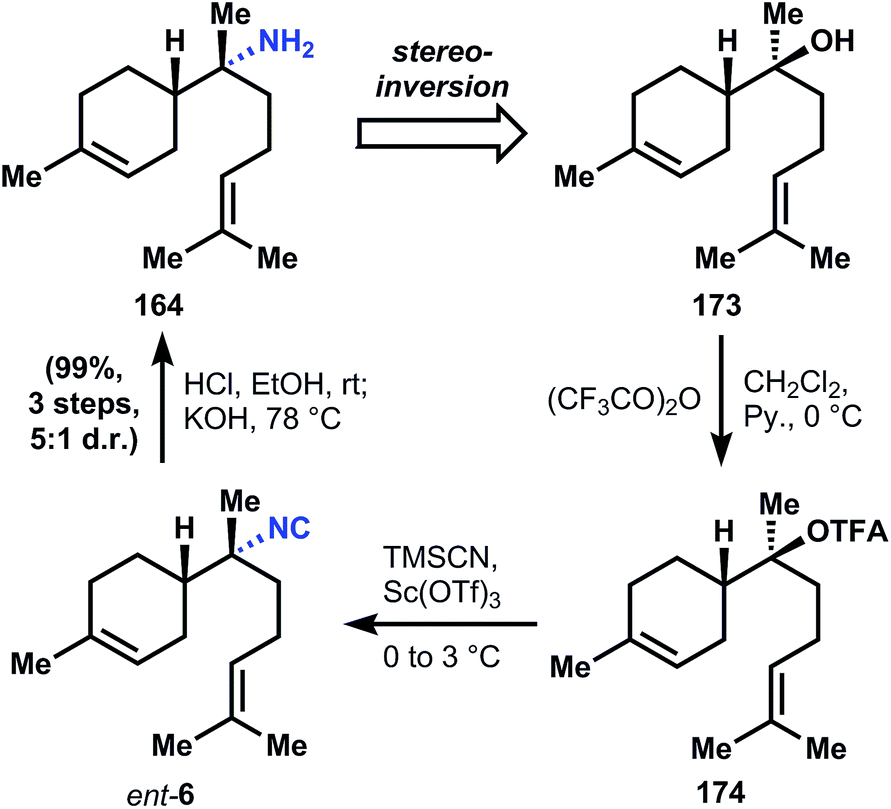 | ||
| Fig. 18 Shenvi's route to (−)-164 via stereoinversion of 173. | ||
:2 mixture of diastereomers. A three step hydrolysis procedure is known to convert the minor amide diastereomer into its corresponding α-bisabolyl amine (164).
Amidine 177 can be generated in good yield over 4 steps from orthoester 178 by first reaction with the Ellman auxiliary to provide imidate 179, whose corresponding iodide will couple with the reagent derived from isopropenylmagnesium bromide and cuprous iodide to give 180. Conversion of imidate 180 to amidine 177 is necessary for subsequent alkylation, since the electron withdrawing sulfinyl group renders the metalloenamide derived from 180 unreactive towards alkyl halides. Thus treatment of imidate 180 with morpholine in the presence of catalytic cyanide provides amidine 177, whose corresponding metalloenamide is efficiently alkylated by allyl bromide with perfect diastereocontrol; addition of methylcerium chloride yields sulfinimide 176. Ring closing metathesis using Grubbs' second generation catalyst produces cyclohexene 175, and addition of homoprenyl lithium provides sulfinamide 181, again with perfect stereocontrol for the correct diastereomer. Removal of the sulfinyl group is accomplished with hydrochloric acid and basification of the reaction delivers (+)-164.
These isocyano-sesquiterpenes (1, 2, 4, 85 and 164), although structurally disparate, possess similar challenges, mainly related to the problems of the nitrogen substitution patterns and stereochemistry imparted by their unusual biosynthesis. In contrast, the following sections analyze structurally similar isocyanoterpenes, which possess (by definition) similar challenges, but which are solved in very different ways, for better or worse (Fig. 19).
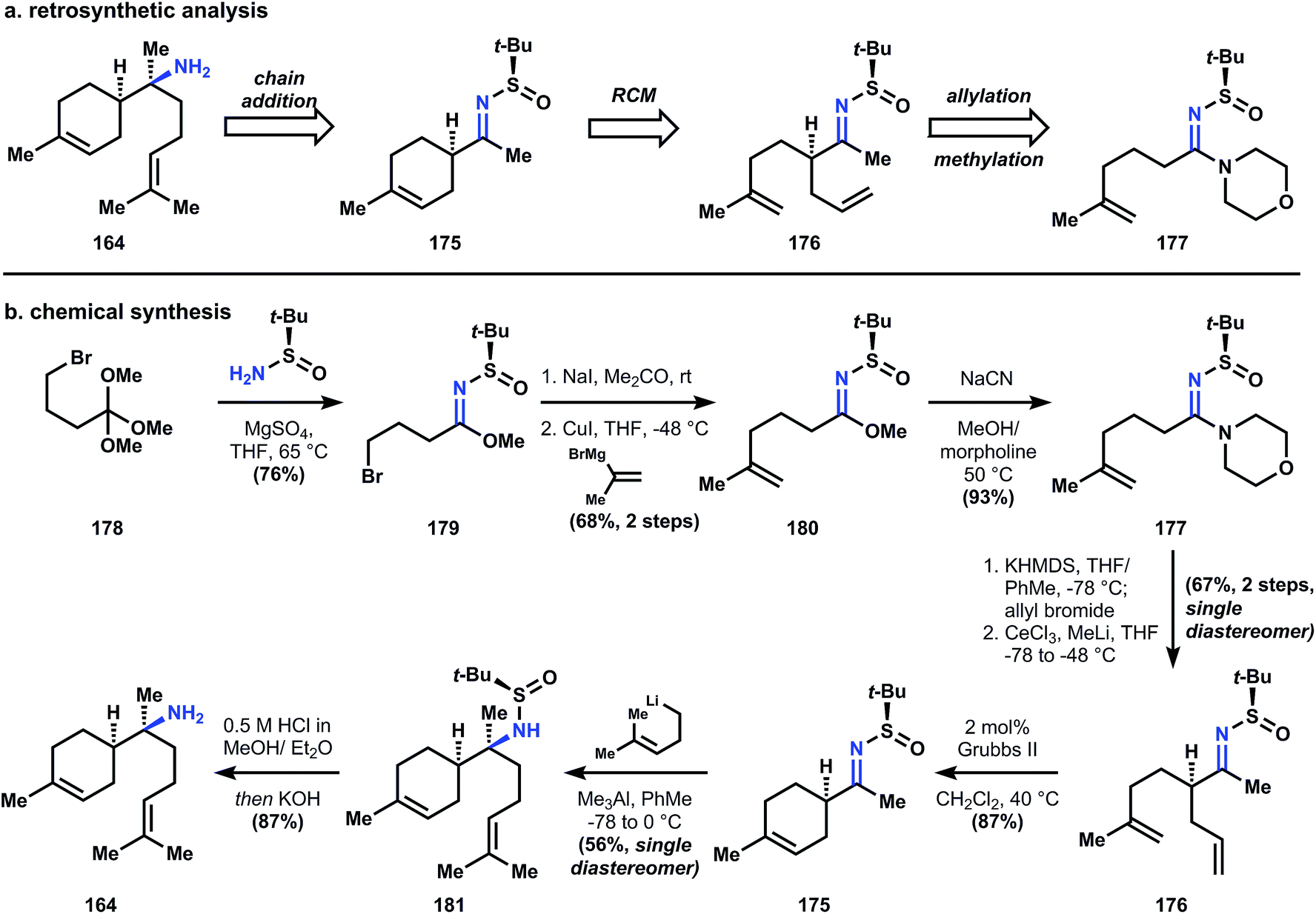 | ||
| Fig. 19 (a) Ellman's analysis and (b) synthesis of (+)-164 using his auxiliary. | ||
3.6 Amphilectenes and adocianes
The amphilectenes and adocianes, also called cycloamphilectenes (Fig. 20), are diterpenes (C20) composed of fused tri- and tetracyclic skeletons with isonitrile substitution conserved at C7 (occasionally at C8) and/or C20. There is minor variation of stereochemistry, alkene position, and methyl groups within this family, but generally its members are readily identifiable by their fused ring systems. The skeletons are stereochemically dense and usually consist of all-trans decalin motifs, though not exclusively. However, the all-trans stereochemistry is an unusual challenge since it limits the number of bonds susceptible to application of the Diels–Alder transform, which, when applied to cyclohexenes, gives rise to cis-stereochemistry, and therefore one newly formed center must be epimerized (see Fig. 21 below). Furthermore, the trans-decalins are not always thermodynamically favored versus cis-decalins (see Section 3.3) – this configurational preference is surprisingly sensitive to substitution patterns and attempted equilibrations of cis- to trans-decalins are often met with poor diastereoselectivity (Fig. 22).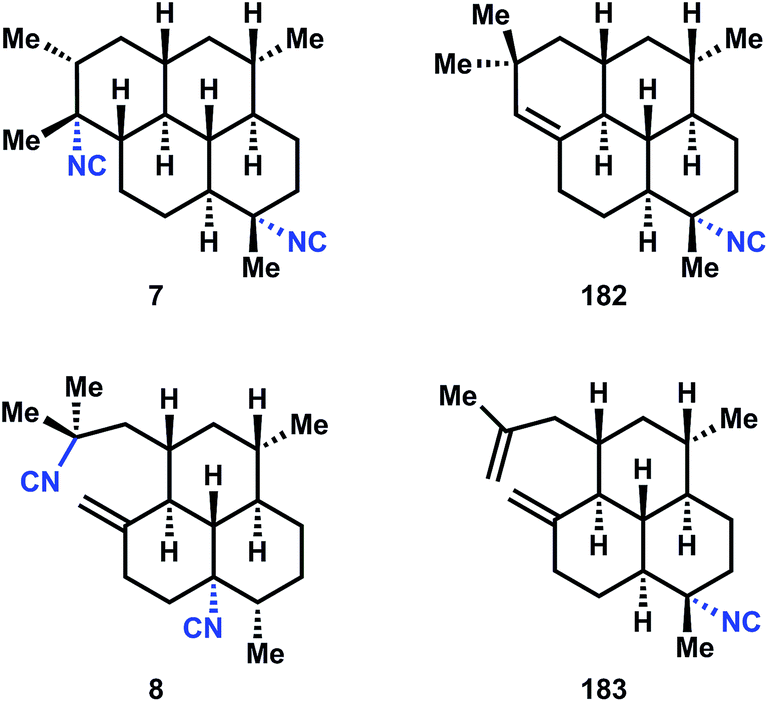 | ||
| Fig. 20 Amphilectenes and adocianes. | ||
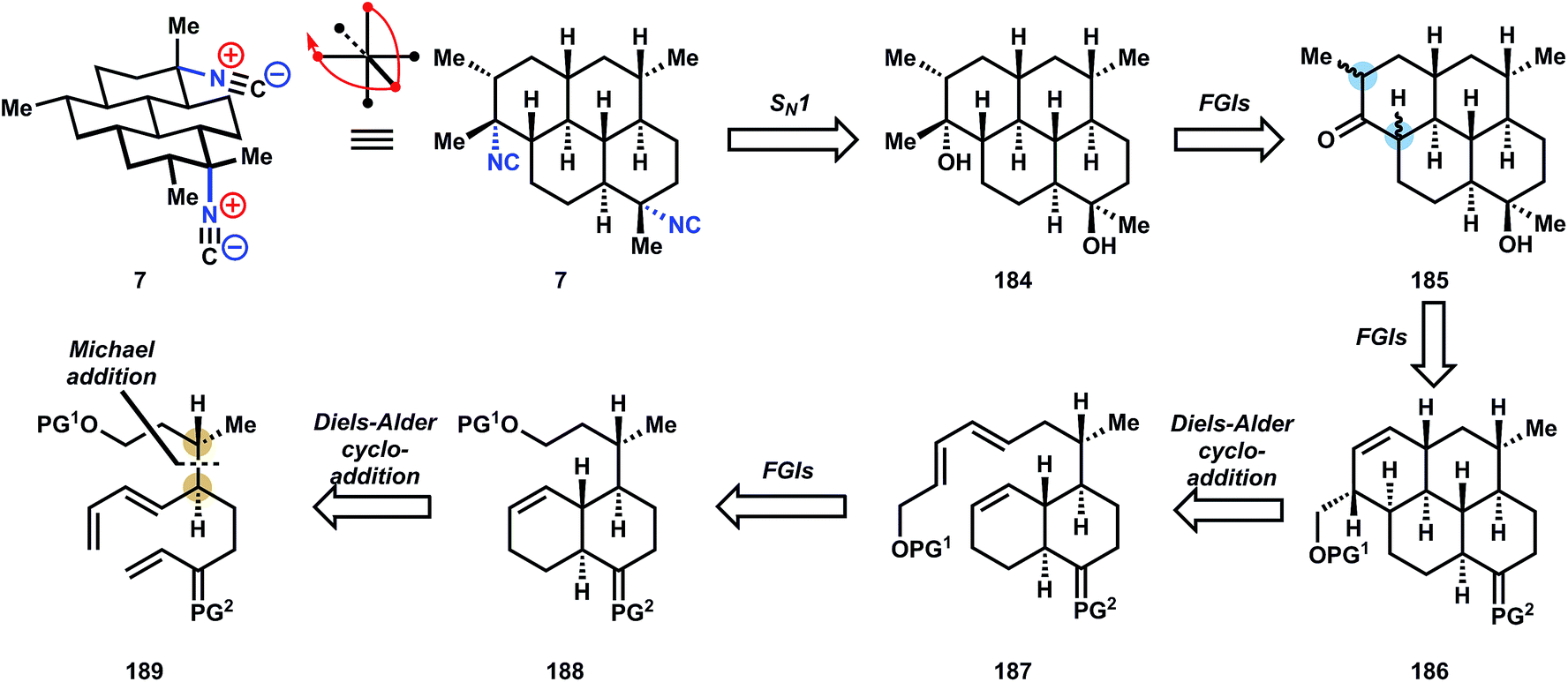 | ||
| Fig. 21 Corey's retrosynthetic analysis of (+)-7,20-diisocyanoadociane (7). | ||
 | ||
| Fig. 22 Corey's chemical synthesis of 7. | ||
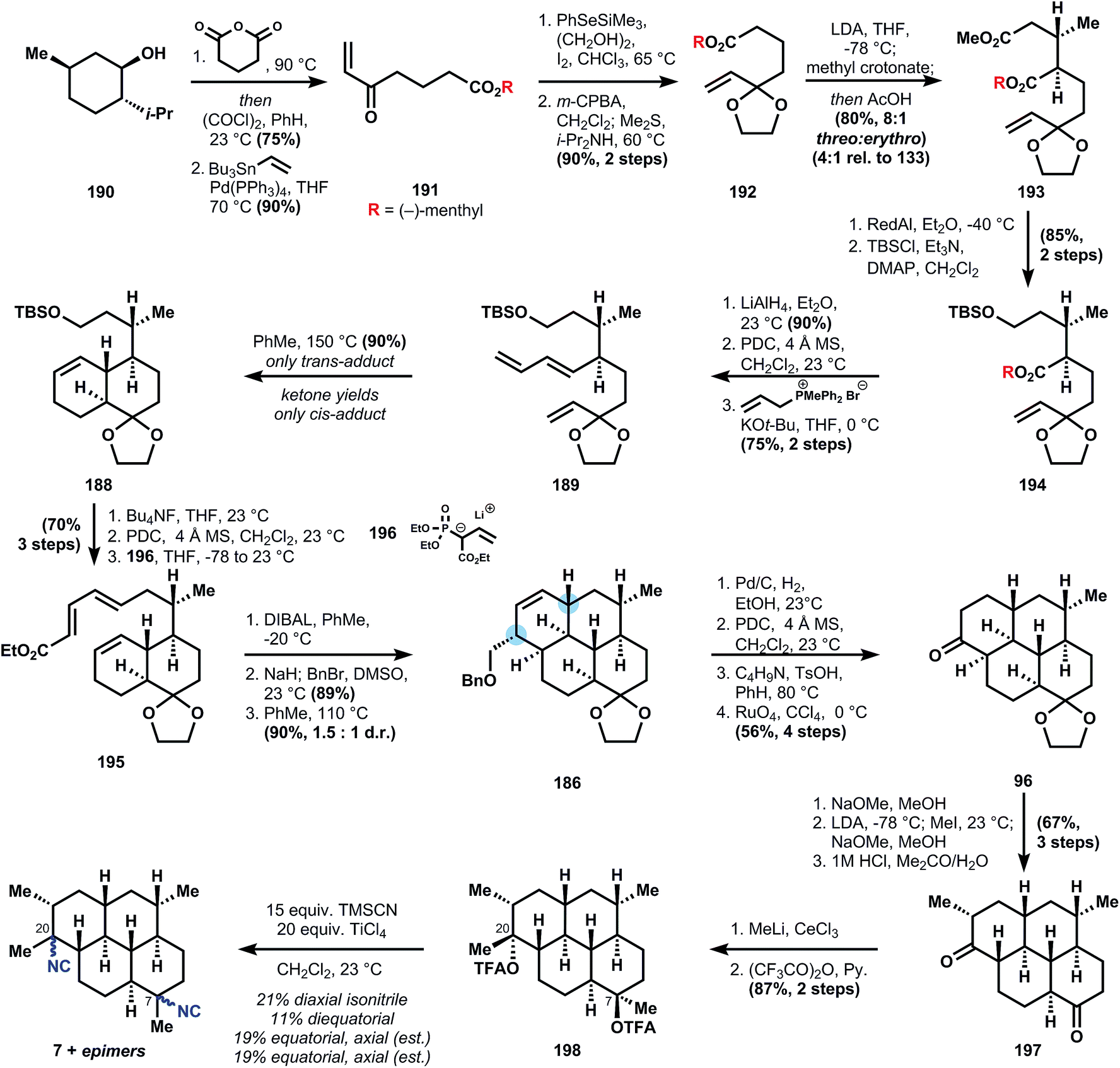
 | ||
| Fig. 23 Piers' analysis of 8 using difunctional reagent 204 as precursor to diene 201. | ||
The synthesis begins by appending a chiral auxiliary, (−)-menthol 190, onto what will become the first section of the carbon scaffold, glutaric anhydride, to give a carboxylic acid that is transformed into its acid chloride and Stille coupled to vinyltributyl stannane to provide enone 191. Standard ketalization of 191 is not possible, so a two-step workaround was devised whereby a combination of TMS phenyl selenide, iodine and ethylene glycol generate a β-phenylseleno ketal, whose selenide can be oxidized and eliminated to generated unsaturated ketal 192. Michael addition of the enolate derived from 192 into methyl crotonate provides 193 in good yield and acceptable diastereoselectivity (8:1 threo:erythro and 4:1 selectivity relative to (−)-menthol). Selective reduction of the less sterically-shielded methyl ester is achieved with RedAl (sodium bis(2-methoxyethoxy)aluminumhydride) and the resultant primary alcohol is protected as its silylether. The menthol auxiliary is removed by LAH reduction, the primary alcohol is oxidized with PDC, and the resulting aldehyde is subjected to Vedejs-Wittig olefination121 to yield the trans-butadiene 189. The Diels–Alder cycloaddition of ketal 189 occurs at 150 °C to provide the trans-decalin 188 selectively, whereas the Diels–Alder reaction of the corresponding ketone proceeds at room temperature122 but produces the cis-decalin, which cannot be quantitatively epimerized to trans- in related systems.122
Conversion of the primary silyl ether to the requisite diene 195 was accomplished by fluoride-mediated deprotection, another PDC oxidation, and E,E-selective Horner–Wadsworth–Emmons olefination with phosphonate anion 196. It was not reported whether the inverse-demand Diels–Alder cycloaddition of 195 was attempted, although an oblique reference to this possibility was made in the paper (changing the terminal substituent affects the stereoselectivity of the cycloaddition). Instead, DIBAL reduction of ester 195 and alkylation with benzyl bromide provides a terminal benzyl ether, which can be heated in toluene to effect the required cycloaddition, although the diastereoselectivity to produce 186 is modest because addition to the opposing cyclohexene face is competitive (1.5:1 d.r.) (Fig. 24).
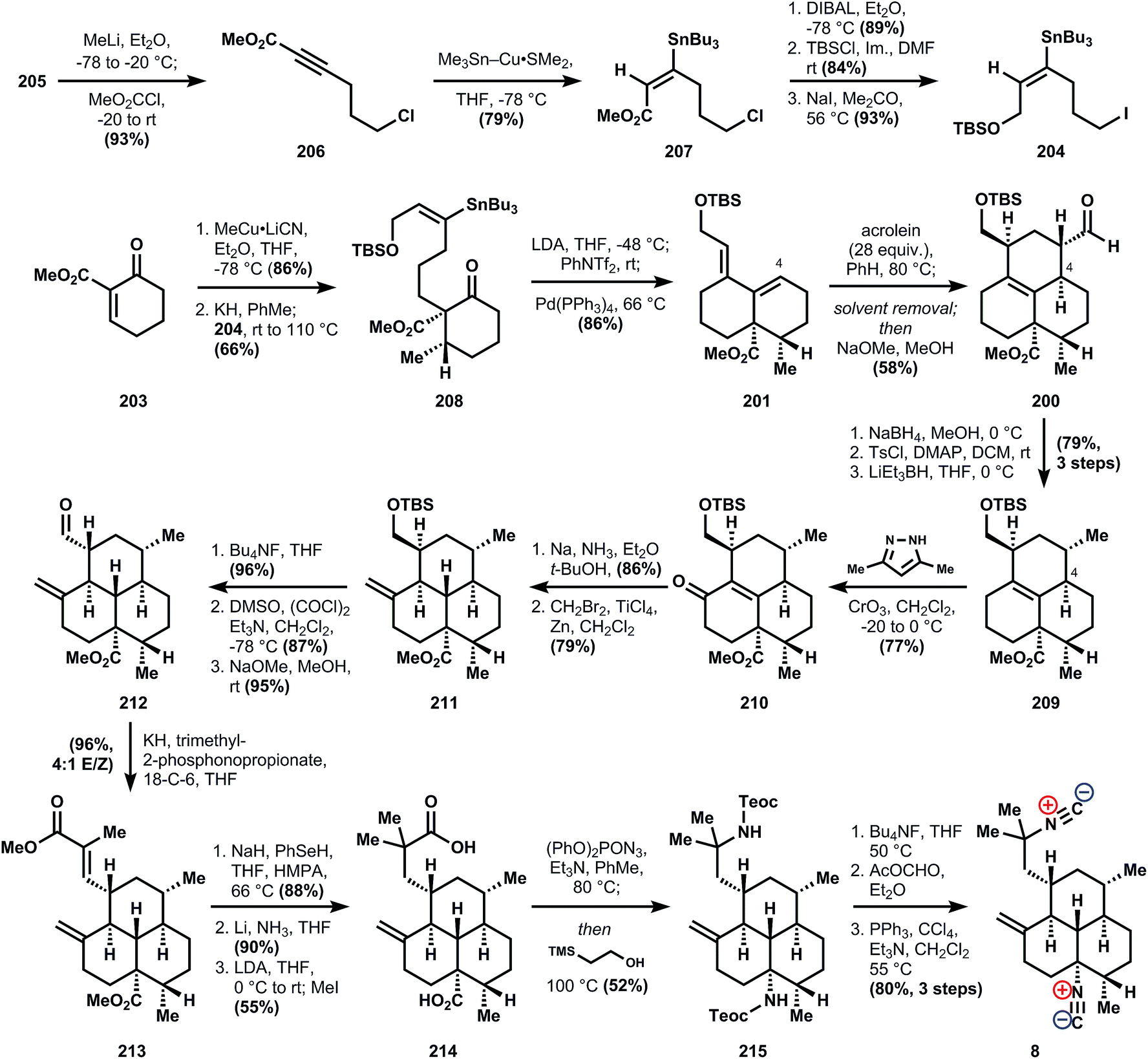 | ||
| Fig. 24 Piers' synthesis of 8. | ||
The next several steps dehomologate 186 to ketone 96via (1) benzyl ether hydrogenolysis, (2) alcohol oxidation to its aldehyde, (3) pyrrolidine enamine formation, and (4) oxidative cleavage of the enamine alkene with ruthenium tetroxide. At this point, the epimerizable locations adjacent to the western ketone can be used to advantage. Even though one cyclohexane ring is cis-fused, its equilibration to the trans-junction can be easily accomplished with sodium methoxide in methanol. Then, although methylation of the kinetic enolate preferentially forms the axial α-methyl ketone, an additional equilibration with sodium methoxide delivers the equatorial methyl. Finally, deketalization in the presence of acetone yields diketone 197.
The endgame of the synthesis involves double methyl addition using methylcerium chloride and trifluoroacetylation in the presence of pyridine to establish the two tertiary centers. Since equatorial attack is highly favored in methylation, the bis-axial alcohol is produced almost exclusively. Ironically, however, this inherent stereoselectivity of the substrate causes two problems: (1) addition of methyl nucleophiles to the corresponding imines would generate the wrong stereochemistry at C7 for the amphilectene and kalihinol classes (this problem is encountered in nearly every subsequent synthesis), and (2) whereas the C7 alcohol must be inverted, the stereochemistry at the C20 alcohol must be retained. In audacious fashion, the problems are tackled head-on by ionizing the trifluoroacetyl esters 198 with titanium tetrachloride in the presence of excess TMSCN. This reaction leads to a mixture of four diastereomers in nearly statistical mixture and 70% total yield that are separated by HPLC. Over one-half of the mixture corresponds to equatorial/axial isonitriles, and assuming neither stereocenter influences the other, 7 would then constitute about 19% of the reaction mixture. So, whereas this synthesis provides a valuable answer to the question of absolute stereochemistry of 7, further advances are needed to secure access to 7 efficiently.
Herewith, important features of this synthesis that significantly impacted future work: first, at a strategic level, the intermediacy of a branched polyene 189 has been chosen by multiple chemists working in the amphilectene/kalihinol family. Installation of this branch point prior to cycloaddition decisively places the burden of stereocontrol on the linear stereodiad of 189 and similar structures, and therefore the efficiency of the whole synthesis hangs on the accessibility of these branched diads. In later syntheses, we explore this access in more depth. Second, this synthesis is the first report of a Ritter-type reaction used in the synthesis of these marine isonitriles, with TMSCN in particular as the nitrogen source – an innovation that was further explored multiple times in the literature. The 42-step formal synthesis of 7 reported by Mander in 2006 (ref. 53) and the 32-step formal synthesis of 7 reported by Miyaoka in 2011 (ref. 123) should be examined by the interested reader (Fig. 25).
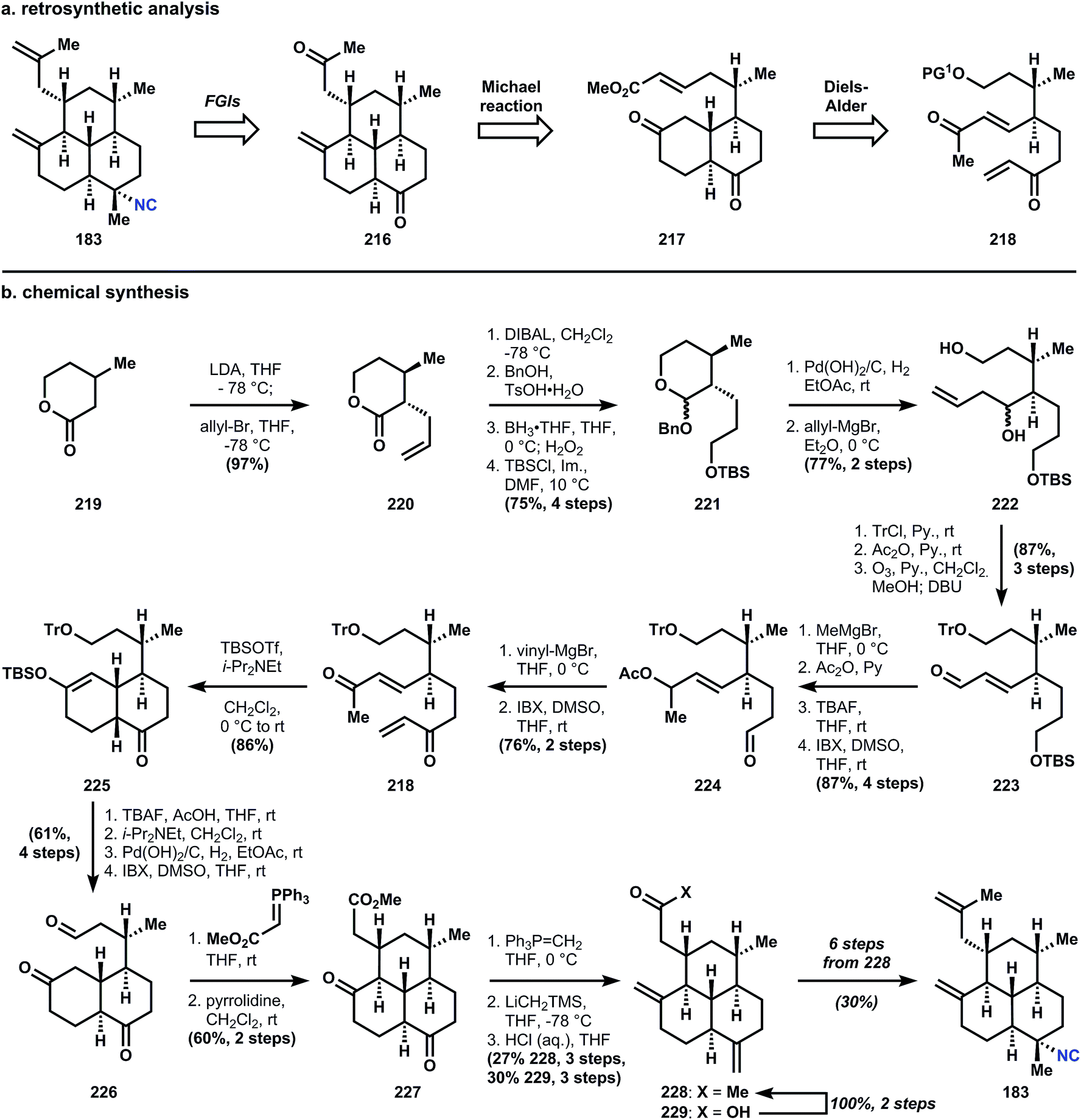 | ||
| Fig. 25 (a) Miyaoka's analysis and (b) synthesis of amphilectene 183. | ||
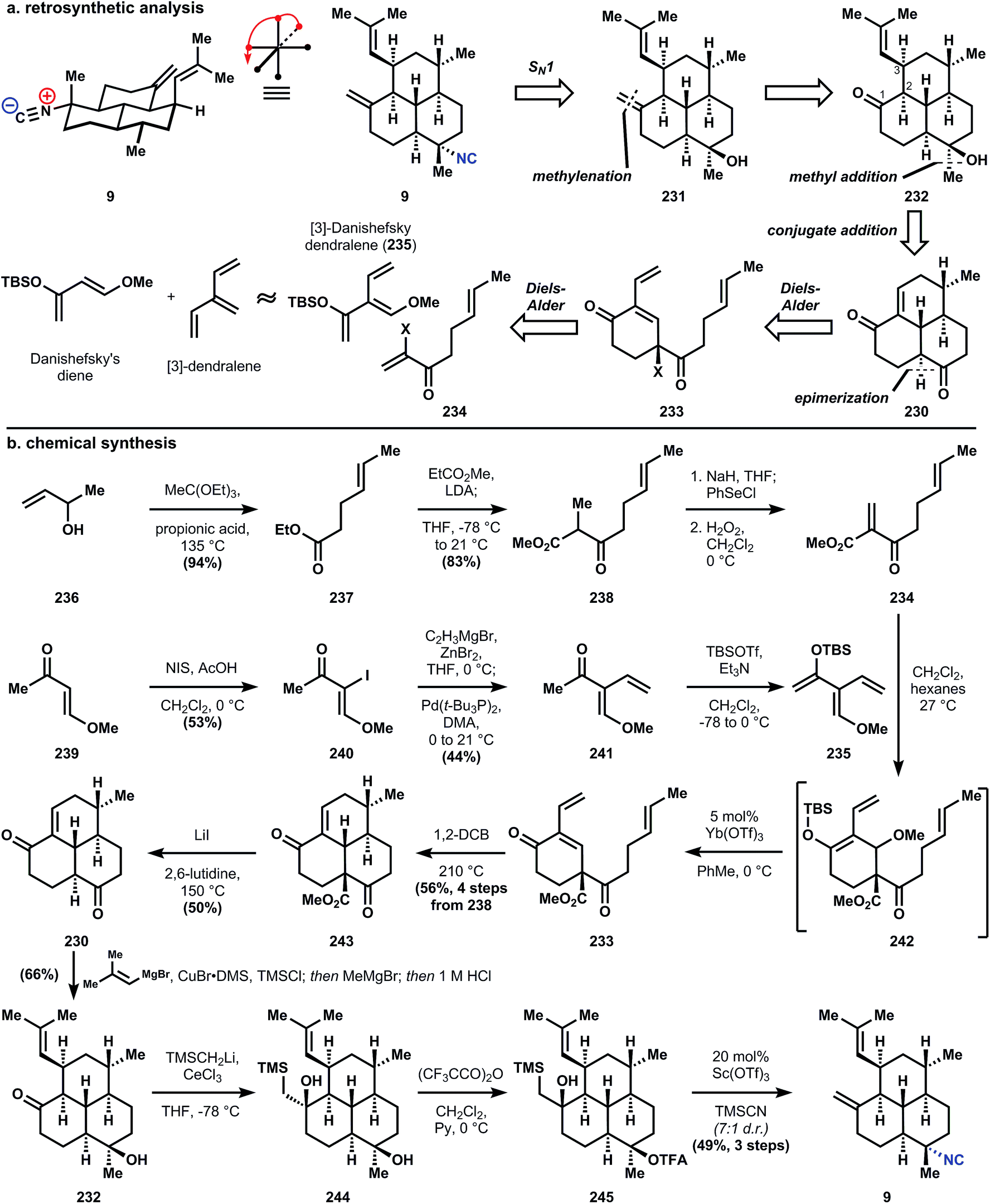 | ||
| Fig. 26 (a) Shenvi's analysis and (b) synthesis of amphilectene 9 using [3]-Danishefsky dendralene 235 and a stereospecific SN1 reaction. | ||
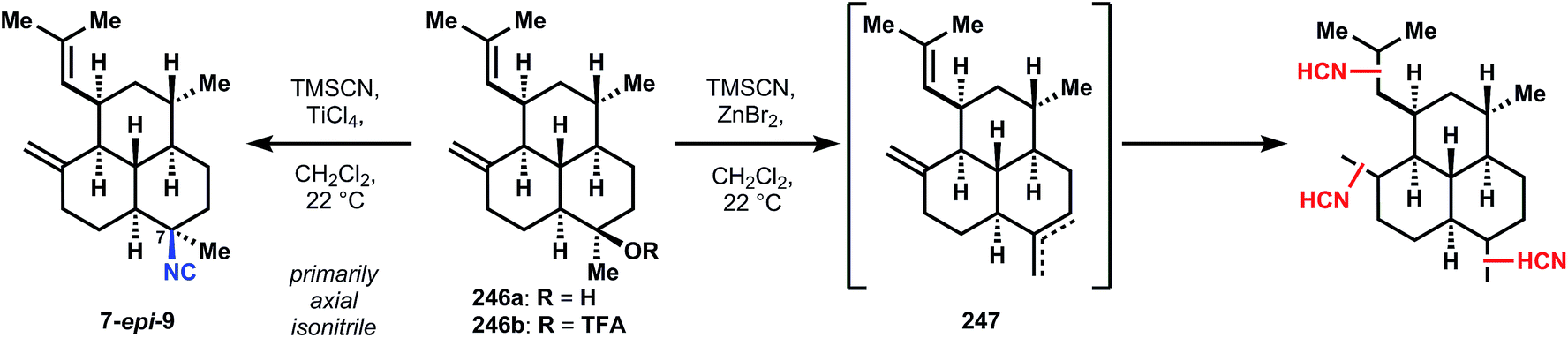 | ||
| Fig. 27 Pitfalls of other methods for isocyanation in the synthesis of 7. | ||
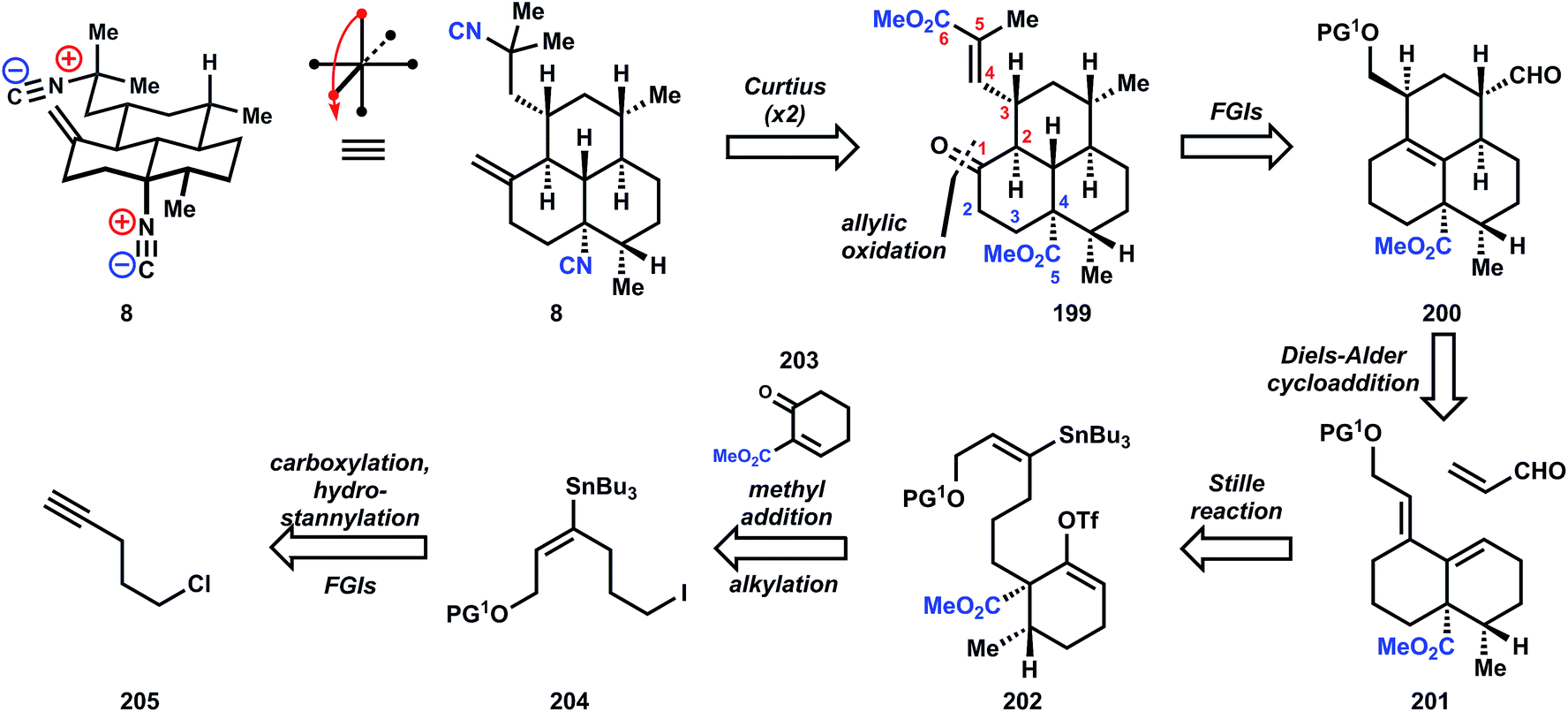
The synthesis begins with carboxylation of 1-chloro-4-pentyne (205) to ynoate 206, which is stereoselectively functionalized to stannane 207via conjugate addition using stannyl-copper. Ester reduction, followed by alcohol protection and Finkelstein halide exchange delivers iodide 204. Although alkylation of 204 by the enolate derived from organocopper addition to enone 203 is not efficient, the corresponding potassium enolate derived from deprotonation with KH cleanly generates the hindered quaternary center in 208. Formation of diene 201 is accomplished in one pot by addition of palladium tetrakistriphenylphosphine into the crude reaction mixture containing the vinyl triflate 202 generated from deprotonation of 208 and reaction with N-phenyltriflimide. Refluxing 201 in the presence of excess acrolein effects a Diels–Alder reaction, and while the diastereoselectivity is poor (ca. 2:1 at C4) even after equilibration of the aldehyde stereochemistry, the correct isomer 200 can still be isolated in 58% yield. Sodium borohydride reduction of the aldehyde, tosylation of the alcohol, and deoxygenation via SN2 displacement with Super-Hydride (lithium triethylborohydride) furnishes tricycle 209 in good overall yield. Allylic oxidation to install the conspicuously absent ketone is accomplished with the Corey–Fleet reagent,125 which was observed by Salmond and co-workers at UpJohn to significantly increase the rate of allylic oxidation in steroids.126 Dissolving metal reduction of 210 delivers the thermodynamically-favored trans-ring fusion selectively, in accordance with observations by Sarrett127 and Barton,128 and methylenation with Lombardo's reagent129 furnishes 211. Further FGIs – deprotection, Swern oxidation, epimerization to the equatorial aldehyde – transform the silylether into the corresponding aldehyde 212, which is engaged in a Horner–Wadsworth–Emmons olefination to supply ester 213. At this point, reduction of the unsaturated ester was unsuccessful (comments indicate poor selectivity for reducing this enoate versus the alkene or esters), so both esters are first demethylated with sodium benzeneselenoate. Dissolving metal reduction then chemoselectively reduces the unsaturated acid, and methylation of the trianion delivers diacid 214. The double Curtius reaction produces an intermediate bis-isocyanate, which is intercepted with 2-trimethylsilylethanol, presumably to aid in purification (the yield is only 52%). Removal of these newly formed Teoc protecting groups in 215 then provides the bis-amine which is converted into the bis-isonitrile 8 by a variation of the standard formylation/dehydration procedure, where a phosphorous(v) reagent replaces the commonly-used methanesulfonyl chloride (Fig. 28 and 29).
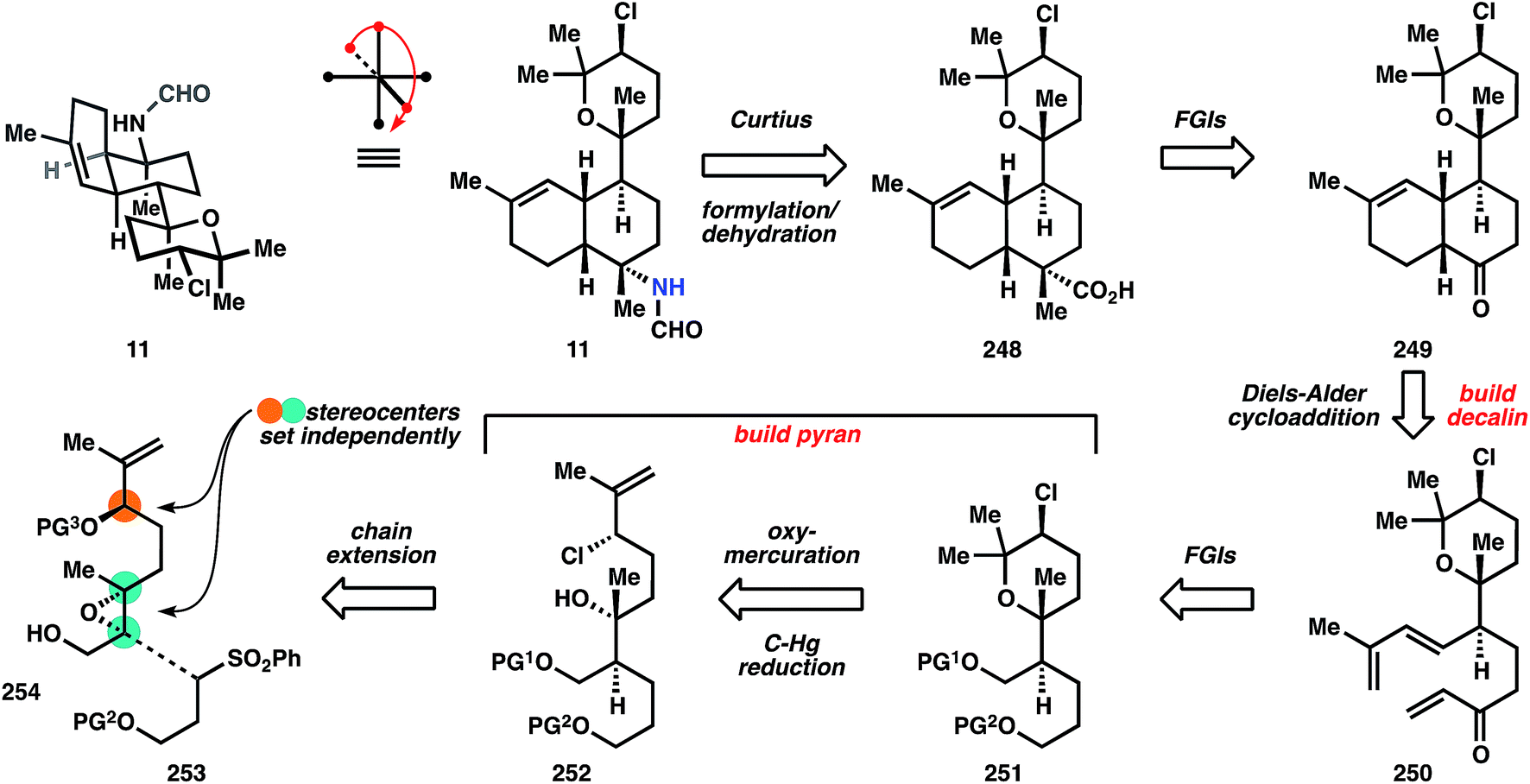 | ||
| Fig. 28 Retrosynthetic analysis of 11 according to Miyaoka and Yamada's synthesis. | ||
 | ||
| Fig. 29 Miyaoka's and Yamada's synthesis of 11. | ||
Not only was this synthesis the first of the tricyclic amphilectane class, but it also laid the ground work for extension to a closely related member (8-isocyano-10,14-amphilectadiene) from intermediate 210.130 Furthermore, Piers' work established the benchmark for efficiency in this class, which took 25 more years to improve upon.
The branching stereocenters are derived from stereogenic α- and β-carbons adjacent to an ester (in this case a lactone). In Miyaoka's synthesis, these centers are established via trans-selective allylation of β-methyl velero lactone (219). Multiple FGIs are then applied to advance intermediate 220 to the requisite Diels–Alder precursor 218. Of the 15 steps separating the two intermediates, 6 steps involve protecting group manipulation, and 4 cause oxidation state changes, leaving only 3 steps that build the carbon skeleton (2 other steps are also FGIs). The Diels–Alder reaction of 218 is accomplished upon silylation of the enone to provide 225 as the cis-decalin exclusively, in accordance with Taber's122 and Corey's observations (Corey used the corresponding ketal to alter the selectivity to favor the trans-decalin; it is likely that the presence of an alkene prevented thermodynamic equilibration of Corey's decalone;122 also see Section 3.7.1 below). Desilylation with tetrabutylammonium fluoride and epimerization using Hünig's base provides the trans-fused decalin diketone.
Removal of the trityl group is accomplished with hydrogenation over palladium hydroxide, and oxidation of the primary alcohol delivers aldehyde 226. Homologation with a stabilized ylide provides an unsaturated ester that undergoes pyrrolidine catalyzed Michael addition to yield tricycle 227. Wittig olefination occurs selectively at the southern ketone, and a Peterson olefination is required to install the western alkene. Fortuitously, attack also occurs on the ester and so methyl ketone 228 is also isolated along with carboxylic acid 229, which can be converted in two steps to 228via its Weinreb amide. From 228, six additional steps are required for conversion into 183. Miyaoka uses the Wood sequence54,131 (see Fig. 31) for introduction of the equatorial isonitrile, and for those reactions, we must visit the kalihinols. First, however, a final synthesis from the amphilectene family.
Synthesis of the dienophile 234 is accomplished in a short sequence from 3-buten-2-ol (236), which is heated with triethylorthoacetate in the presence of propionic acid to effect Claisen rearrangement and yield ester 237. A subsequent Claisen condensation with methyl propionate yields β-ketoester 238, which is unsaturated via selenation and oxidative elimination. It should be noted that 234 decomposes rapidly when stored neat, so this highly electrophilic dienophile is used directly without concentrating the reaction mixture. The [3]-Danishefsky dendralene 235 is synthesized in a three step sequence of iodination, Negishi coupling, and silylation; and like 234, 235 is not stable over long periods of time. Therefore, a crude dichloromethane solution of 234 and 235 is concentrated in vacuo, which effects immediate cycloaddition to form 242 as an inconsequential mixture of diastereomers. Redissolution of 242 in toluene and treatment at 0 °C with catalytic ytterbium triflate causes elimination of tert-butyldimethylsilyl methyl ether to yield enone 233. The second Diels–Alder is carried out at high temperature (likely a consequence of strain) in 1,2-dichlorobenzene to provide enone 243. The strain inherent to 243 complicates removal of the methyl carboxylate since hydroxide will add conjugatively to the enone, so a modified Krapcho decarboxylation136 is used to demethylate and decarboxylate 243 to parent tricycle 230 – the trans-decalin is formed exclusively.
Conjugate addition is accomplished with isobutenylmagnesium bromide in the presence of copper bromide and trimethylchlorosilane, which protects the western ketone as its enolsilane, allowing selective addition of methylmagnesium bromide into the southern ketone to supply intermediate 232, also as the trans-decalin. Notably, if conjugate addition is effected prior to decarboxylation, both fused decalin motifs contain cis-ring junctions. The methylene is installed in a two-step interrupted Peterson olefination first to silylhydrin 244; trifluoroacetylation is selective for the less hindered southern tertiary alcohol, providing 243. Treatment of 245 with a solution of catalytic scandium triflate in cyanotrimethylsilane (TMSCN) inverts the stereochemistry of 245 at the tertiary ester to provide the equatorial isonitrile of 9 preferentially (though unknown at the time, this reaction is stereospecific and can invert single isolated stereocenters). Elimination of the silylhydrin is also accomplished under these conditions. Previously reported procedures for SN1 isocyanation fail to convert 245 or 246a/b (Fig. 27) to 9. For instance, the procedure of Tada137 and Kitano138 proceeds through a mixture of alkenes 247 (Fig. 27) and Brønsted acid-mediated Ritter-type isocyanation does not differentiate between the electron-neutral olefins. The procedure of Corey56 that was successful to generate stereoisomeric mixtures of 7,20-diisocyanoadociane (7, Section 3.6.1) preferentially forms 7-epi-9 and in low yield; studies on a model decalin and related SN1 reactions suggest this stereochemical outcome derives from attack on a solvent separated ion pair, i.e. the substrates alone dictates the stereochemistry.59
This most recent synthesis of 9, an admittedly simple member of the amphilectene class, achieves the shortest route yet to these bioactive compounds. The true value of the work lies in the invention of a demonstrably useful functionalized dendralene, and the initial disclosure of a stereospecific tertiary alcohol inversion reaction. The amphilectenes (and adocianes), while stereochemically complex, are functionally sparse and therefore chemoselectivity is not a significant problem. In contrast, the functionally dense kalihinols are represent the pinnacle of complexity within the isocyanoterpene class and a major challenge for chemical synthesis.
3.7 Kalihinols
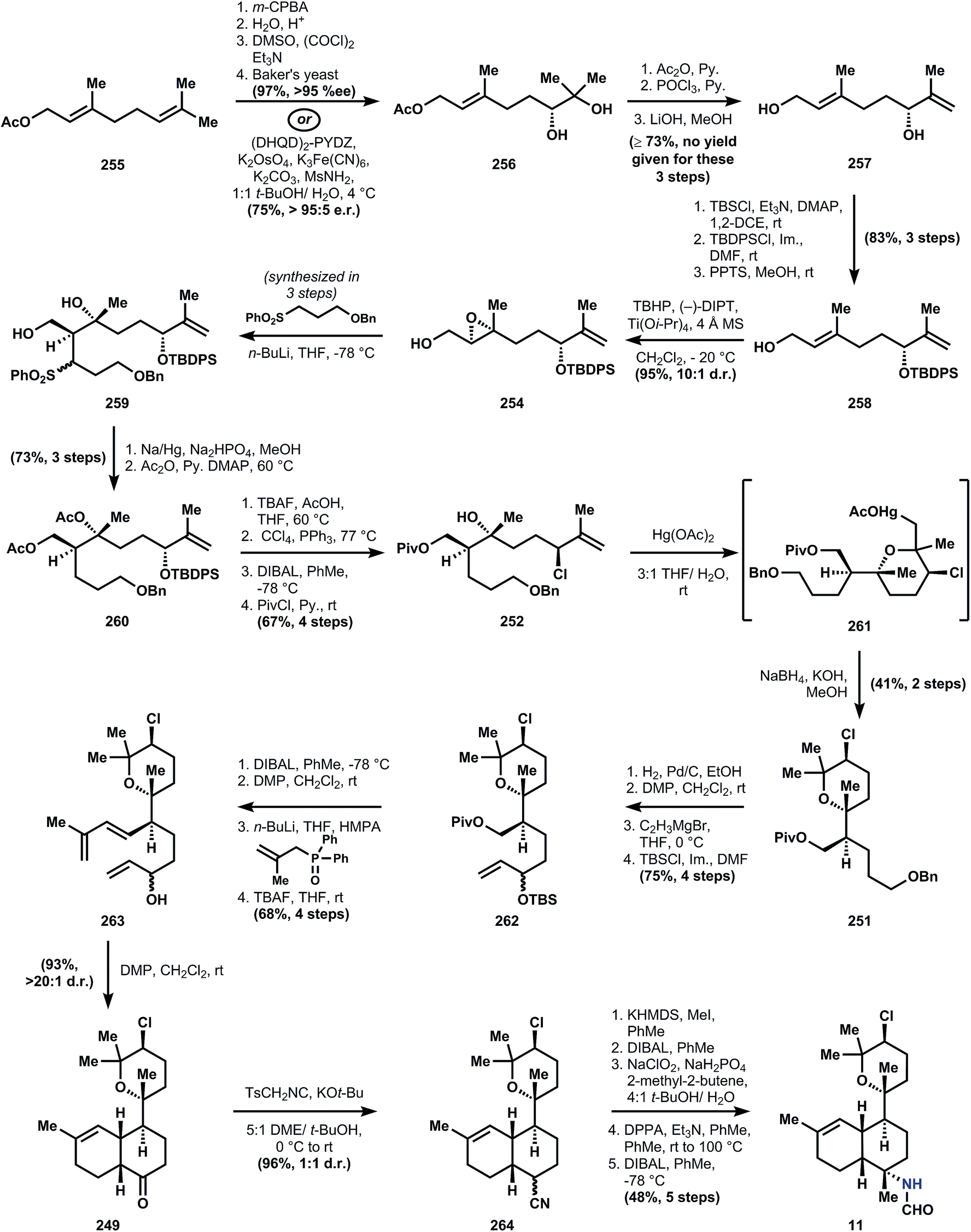
There are two common names given to members of this class: the kalihinols and the kalihinenes, corresponding to a presence or absence of hydroxylation at C4 or C5 (see Fig. 1A-III). The authors prefer the catch-all term ‘kalihinol’ for the entire family since the name (1) captures the unique C11-oxy-biflorin skeleton139 common to and distinctive of the class, and (2) likely reflects the biosynthetic origins of the heterocyclic ring as derived from an alcohol. The functional group-density of the kalihinols and the close proximity of their numerous branching carbons have confounded attempts at an efficient synthesis. In particular, the sterically-encumbered heterocycle directly joined to a polysubstituted decalin motif poses a dilemma for its installation: how to situate this key bond joining the two systems with stereocontrol and efficiency. Ideally, vicinal stereocenters should be directly cleared to two achiral carbons,40 but this feat has never been achieved in the kalihinols. Either the rings are built around pre-existing stereochemistry (Miyaoka and Yamada), or one stereocenter controls the stereochemistry of its neighbor (Wood); these strategies are described in the forthcoming synopses.Geranyl acetate 255 is processed to diol 256 using a Kodama procedure140 relying on enantioselective reduction of the intermediate hydroxy ketone corresponding to 256. The same result can be obtained in one step by Sharpless dihydroxylation,141 so the choice of the longer sequence is curious, but might have been due to financial constraints. Acetylation of the secondary alcohol of 256, elimination of the residual tertiary alcohol and global deacetylation provide alcohol 257. Selective silylation of the secondary allylic alcohol is not possible, so the primary alcohol is protected first as the TBS ether. Appendage of the TBDPS group onto the more sterically encumbered hydroxyl is followed by selective removal of the primary silyl ether to deliver alcohol 258. Sharpless epoxidation using (−)-DIPT provides epoxide 254, which upon alkylation by the anion derived from sulfone 253 yields 259 as a mixture of diastereomers. Desulfonylation with sodium amalgam and bisacetylation provide the polysubstituted 9-decenyl ether 260. The stereogenic chloride is installed upon removal of the tert-butyldiphenylsilyl group with TBAF and SN2 displacement using Appel conditions. In order to prepare the substrate for ring formation at the tertiary alcohol, the acetates are removed reductively with DIBAL, and the resultant primary alcohol is protected as a pivaloyl ester. At this point, 18 steps have been invested in the sequence to reach 252, with no rings yielded as dividend.
The first ring is formed by oxymercuration to provide organomercurial 261, and the carbon–mercury bond is reduced with sodium borohydride in the subsequence step to yield pyran 251. This move circumvents the challenge of stereocontrolled heterocycle formation, since the cyclization does not form permanent stereocenters; all requisite stereochemical information is contained in linear precursor 252. Elaboration of 251 to the intramolecular Diels–Alder substrate is conducted over 8 steps that contain two skeleton-building reactions. In accordance with Taber's122 and Corey's observations (ref. 56, footnote 11), the ketone derived from oxidation of alcohol 263 undergoes spontaneous cycloaddition at ambient temperature to provide primarily a cis-decalin, in this case 249. Whereas most members of the kalihinol family contain a trans-decalin, the cis-decalin is represented in a handful of metabolites, including formamide 11, which becomes the target of this synthesis and is now only 6 steps away. The ketone function in 249 again serves as a flexible synthon for the tert-alkyl amines, and thus is homologated to nitriles 264 using TosMIC100 (see also Section 3.5.2). The nitrile is alkylated on the convex face, corresponding to the correct stereochemistry for the targeted C–N bond. Notably, related studies suggest that analogous alkylation of the trans-decalin will not give the desired stereochemistry for the kalihinols.142 Reduction of the nitrile to the aldehyde and Pinnick oxidation to the carboxylic acid, followed by Curtius rearrangement and reduction of the resultant isocyanate with DIBAL produces the formamide, kalihinene X (11).
This first synthesis of a kalihinol exhibits several notable problems associated with the family. In concert with Wood's model study (see below) and Corey's synthesis of 7, this work by Miyaoka and Yamada illustrates the challenge associated with installation of the equatorial C–N bond: multiple steps are required and its success relies on the concavity of the cis-ring fusion. Furthermore, the synthesis highlights the challenge of generating the stereopentad that delineates the contours of the directly-joined rings and fused rings of the kalihinol skeleton. Such challenges prolong the synthesis to a total of 35-steps.
 | ||
| Fig. 30 Wood's analysis of 10 uses substrate control of most stereochemistry. | ||
 | ||
| Fig. 31 Wood's synthesis of kalihinol C (10). | ||
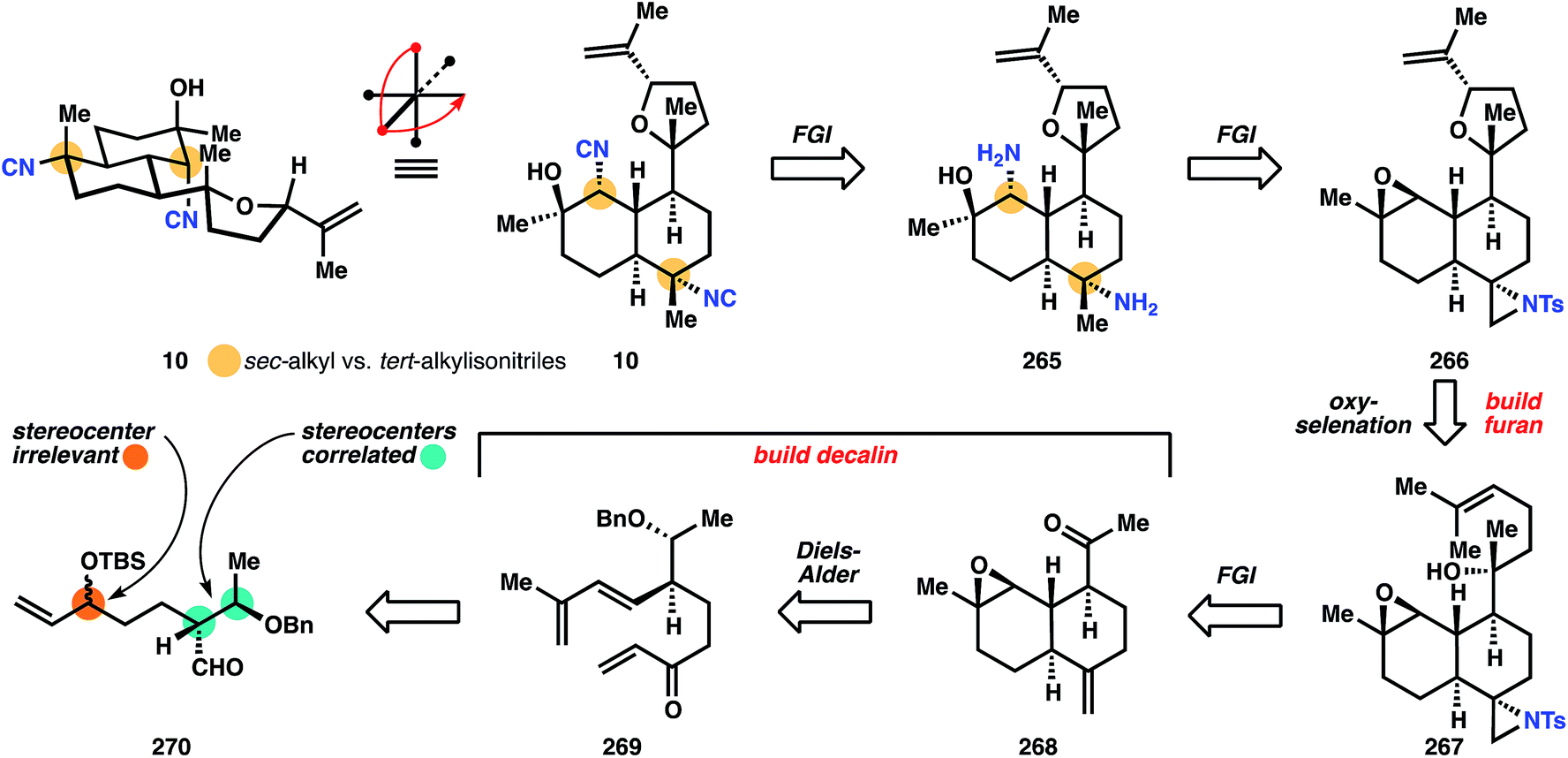
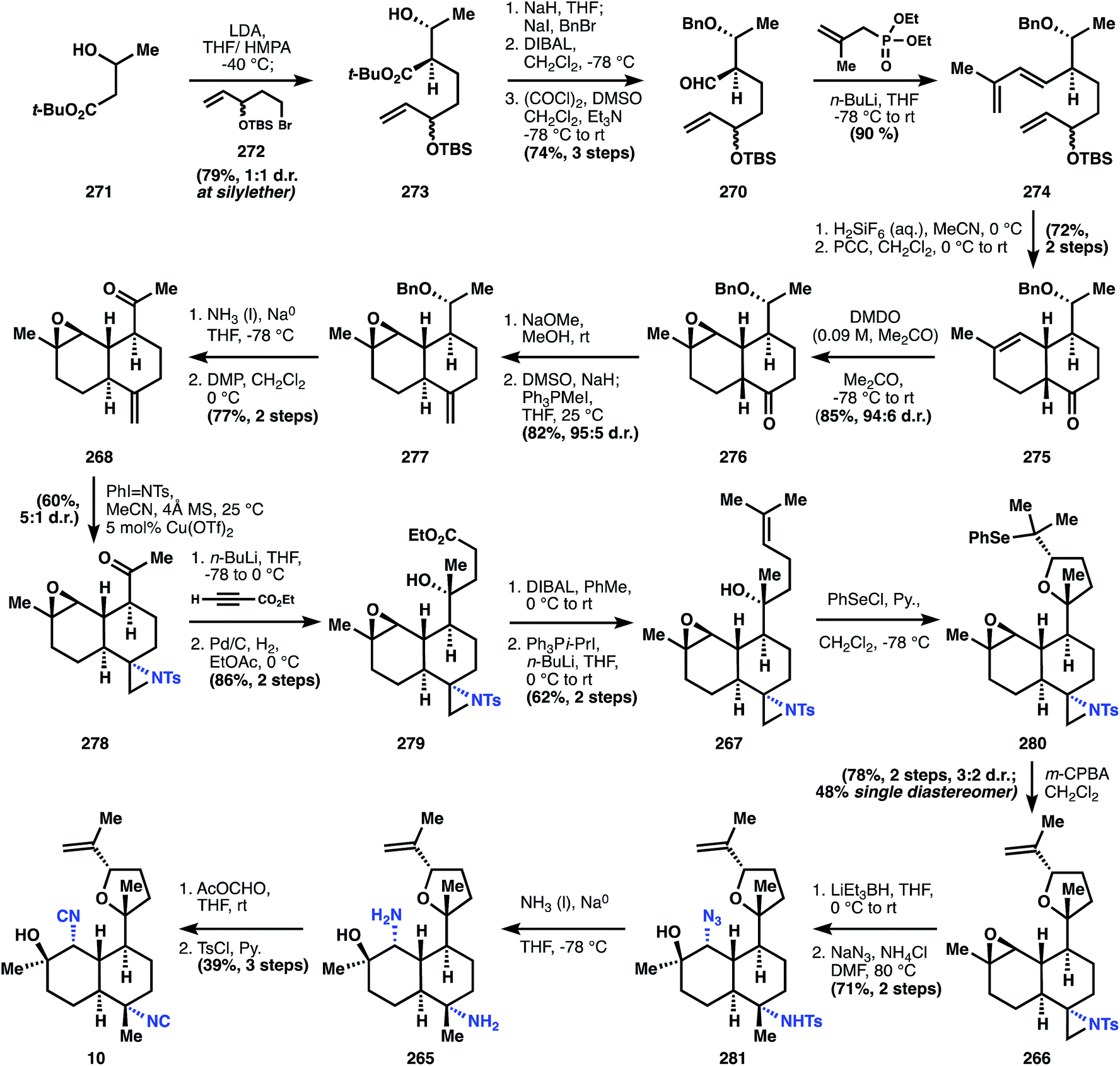
The synthesis of 10 commences with the alkylation of the dianion of 271 with alkyl bromide 272, itself synthesized in three steps. This mixture of diastereomers (273) is carried through five steps of mostly functional group interconversion, including a Horner–Wadsworth–Emmons olefination to form the diene cycloaddition partner 274. Deprotection of the TBS ether in 274 with fluorosilicic acid liberates the southern secondary alcohol for oxidation with PCC, initiating the Diels–Alder reaction at or below ambient temperatures to provide 275. The nascent cyclohexene is epoxidized with dimethyldioxirane with high facial selectivity to generate epoxide 276. Although the cis-ring junction mediates this high stereoselectivity, ultimately the trans-stereochemistry is preferred, since far more kalihinols possess this configuration. Therefore, 276 is partially epimerized with sodium methoxide to a 1.5:1 mixture with the trans-decalin epimer in excess, and this crude mixture is methylenated. Counterintuitively but conveniently, the trans-decalin 277 is produced almost exclusively from the epimerized mixture, whereas subjection of pure 276 to the same conditions provides ‘a variable ratio’ of cis- and trans-decalins.
One of the two stereocenters in the initially formed stereodiad is then erased to prepare for installation of the challenging tert-alcohol functionality. Dissolving metal reduction deprotects the benzyl ether and Dess–Martin periodane then oxidizes the secondary alcohol to the methyl ketone in 268. The Wood group then developed a useful strategy to install the correctly disposed C–N bond: aziridination of alkene 268, followed later in the sequence by reduction of the less-hindered C–N bond. It is noteworthy that the correct stereocontrol is achieved on this trans-decalin system, and telling that Miyaoka utilizes this sequence in a later synthesis of kalihinol A. Having established all the stereocenters in the decalin core, Wood now turns to the heterocyclic motif. Addition of the lithium acetylide derived from ethyl propiolate to methyl ketone 278 results in the anti-Felkin stereochemistry of the tert-alcohol with very high diastereoselectivity (98:2), and subsequent hydrogenation of the alkyne yields ester 279. Monoreduction of the ethyl ester with DIBAL provides a lactol, which is olefinated with isopropylidene phosphonium ylide to yield alkene 267. The tetrahydrofuran 280 is built using an oxyselenation that provides modest selectivity for the correct diastereomer (3:2). Elimination to 266 is effected upon m-CPBA oxidation.
At this point, both of the three-membered rings are opened to reveal the substitution patterns of the final target: the aziridine is reductively opened with Super-Hydride (LiEt3BH), and the epoxide is opened at the less hindered position with sodium azide to provide intermediate 281. Dissolving metal reduction then deprotects the tosyl amide and reduces the azide to the amine 265. Double formylation/dehydration then establishes the bis-isonitrile of kalihinol C (10). Notable advances in this synthesis include a substantially more efficient entry into the polysusbtituted kalihinol framework than previously established, a multistep but useful strategy for installation of the equatorial isonitrile motif, and reconnaissance data regarding substrate stereocontrol, especially formation of the O-heterocycle.
:2 margin upon epimerization) is accomplished using Wood's reconnaissance. The identical triene 250 as found in the synthesis of kalihinene X (11) is intercepted, and its synthesis relies again on pre-installation of all stereocenters on a linear chain 284, which is synthesized in a nearly identical manner as chain 252 in Section 3.7.1. It is instructive to point out that the biosynthetic pathway to the diterpenoid kalihinols also begins with a linear precursor, but with no stereocenters preinstalled. Instead, the stereocenters of the carbon framework form during the cyclization of achiral tetraene 285, geranylgeranyl pyrophosphate, once again highlighting the elegant reactivity that so eludes synthetic chemistry today (Fig. 32).
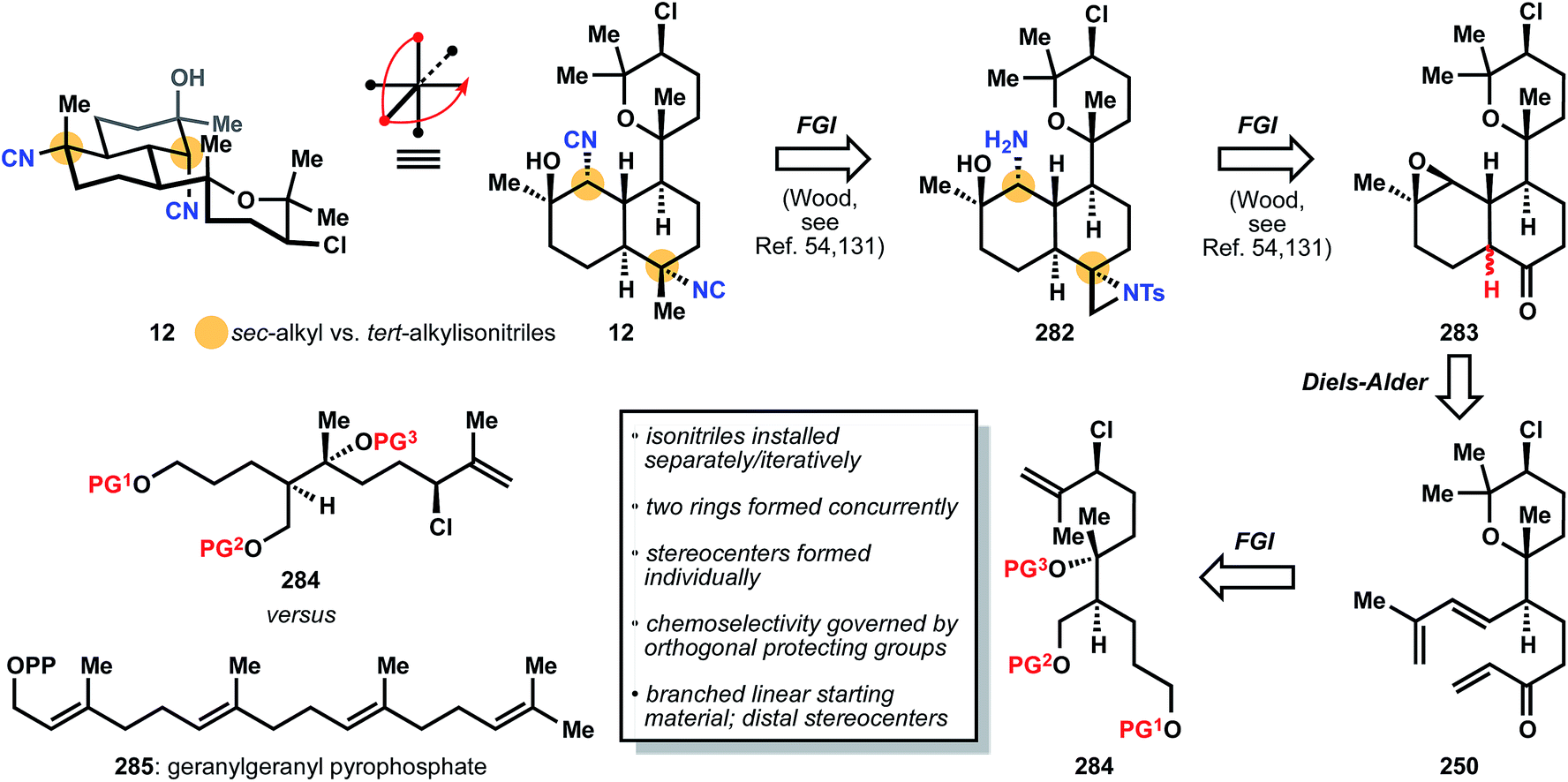 | ||
| Fig. 32 Miyaoka's analysis of 12 relies on preinstalled stereocenters (e.g.284) prior to ring building, in contrast to the hypothesized biosynthesis of 12 from 285. | ||
Conclusion of synthesis section. Having surveyed most syntheses of the isocyanoterpenes, this section aims to convey the difficulties, idiosyncracies, benefits and defects of current approaches. Clearly, an overriding theme in the field is the challenge of efficiency – both in synthetic steps and yield. For the kalihinols in particular, a unifying and divergent sequence that avoids the intermediacy of stereochemically complex, linear motifs would be ideal. The biosynthetically-aberrant isonitrile pharmacophore also remains a challenge. Although a biomimetic contact ion pair attack is advantageous in many cases, this approach may not be applicable to all substitution patterns, the isocyanohydrin motif of the kalihinols in particular. Furthermore, careful attention should be paid in the planning stage to points in the synthesis where the trans- or cis-decalin ring junctures might be obtained selectively through equilibrating epimerization. The impetus for all of this work and all of this thought is two-fold. First, synthetic chemistry is valuable as a central science that impacts the interfacing fields of biology, medicine, physics and polymers, and therefore the advances in chemistry that accrue from pure science endeavors in synthesis are inherently, if unpredictably, worthwhile. Second, the phenotypic effects of the isocyanoterpenes remain poorly understood, and therefore ‘on-demand’ access to these molecules and their analogs holds the potential to unlock the mysteries of their activity. This activity is the subject of the next section.
4 Biological activity
4.1 Cytotoxicity and antibacterial activity
Given their long history, a variety of phenotypic effects have been ascribed to the marine isocyanoterpenes. Notably, these compounds generally display only moderate to weak activity in mammalian cell cytotoxicity assays (10–100 μM against a range of cell lines). In related work, early toxicity studies found that several simple isonitriles show little toxicity, with oral and subcutaneous doses of up to 5 g kg−1 tolerated in rodent studies.145 Together these findings suggest that ICTs are generally tolerated by mammalian systems. In regard to potential therapeutic applications, early findings showed that kalihinols display moderate antibacterial activity,146 and it was recently demonstrated that several kalihinols inhibit bacterial folate biosynthesis and show potent growth inhibition against B. subtilis.147 Also adding to the range of associated biological activities, a recent report suggested that several amphilectane isonitriles have promising activity in an anti-inflammatory assay measuring thromboxane B2 and superoxide anion generation from LPS-activated rat brain microglia.1484.2 Kalihinol F copper chelation
The use of zebrafish embryos for drug screening is of some interest due to the significant body of literature linking phenotypic observations to genotypic information in this rapidly employed animal model. A recent screen using this approach identified kalihinol F, 286, as inducing a dramatic alteration in zebrafish development.149 The observed phenotype was similar to that observed with mutation of the copper transporter, atp7a – a loss of function event that induces copper deficiency. It is proposed that kalihinol F chelates copper through a bi- or tridentate chelation model (Fig. 33). Support for this hypothesis included NMR studies demonstrating copper binding by the isonitrile and abolition of its zebrafish developmental phenotype with additional copper(I). Copper chelation is useful in certain therapeutic contexts, such as Wilson's disease, which result from copper accumulation. The capacity of kalihinol F to alleviate the effects of copper toxicity was demonstrated in zebrafish and mammalian cell studies, with activity comparable to ethylenediaminetetraacetic acid (EDTA). The generality of this free-metal binding hypothesis is discussed below.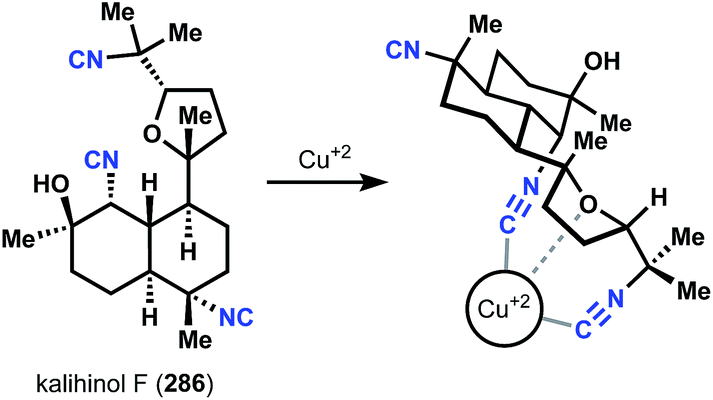 | ||
| Fig. 33 Kalihinol F copper chelation model. | ||
4.3 Anti-malarial activity
The distinguishing biological effects discovered for ICTs are their antimalarial activities. Approximately 50% of the world's population lives in regions affected by endemic malaria. Recently, malaria parasites resistant to the standard-of-care artemisinin combination therapy (ACT) have been found in the Thai-Cambodia border region, a common hotspot for the development and proliferation of drug-resistant parasite strains.150–153 The rise of artemisinin drug resistance points to significant need for novel chemotypes to act as lead compounds for further medicinal chemistry optimization, and has stimulated renewed interest in the isocyanoterpenes.59,65Here we attempt to provide perspective on the existing literature in this area, which has not been surveyed in a systematic fashion since an early review almost 20 years ago.154 In the mid 1990's, Wright and coworkers reported that several sesquiterpene and amphilectene natural products, including 4 and 7 (Fig. 1), display potent nanomolar activity against the causative agent of deadly forms of malaria Plasmodium falciparum. Here, as in most of these studies, activity was determined using a standard blood-borne parasite assay, [3H]-hypoxanthine incorporation, which measures parasitic nucleic acid synthesis.155,156 A subsequent report by Miyaoka disclosed similar activity for kalihinol A.157 In total, 9 reports have ascribed varying levels of anti-malarial activity – from weak to single-digit nanomolar – for isonitrile containing natural products and synthetic variants, clearly establishing the ICTs as intriguing antimalarial lead structures.30,143,158–162 A promising aspect of this collection of data is that several independent studies observed essentially equipotent activity between common chloroquine-sensitive (including D6 and HB3) and drug-resistant/drug-insensitive (including W2 and Dd2) strains of P. falciparum parasites.
4.4 Anti-malarial activity – mechanistic questions
While elucidating the mechanistic basis of many antimalarial drugs has proved to be a complex matter, small molecule–heme interactions are invoked with many of the most common antimalarial drugs, including chloroquine (289), mefloquine, and artemisinin (288).163 Parasites in the intraerythrocytic stage catabolize large quantities of hemoglobin within a specialized organelle, the digestive vacuole (DV). Hemoglobin degradation leads to release of the ferrous heme prosthetic group, which rapidly oxidizes to generate Fe(III) protoporphyrin IX (FPIX, 287).164 In addition to facilitating formation of reactive oxygen species, FPIX depletes the parasitic glutathione pool, interfering with redox homeostasis.165,166 Furthermore, the lipophilicity of FPIX is detrimental to lipid organization and membrane permeability and can destabilize interactions between cytoskeletal proteins and membranes.Malaria parasites lack heme catabolism mechanisms, such as the heme oxygenase pathway,167 and thus have developed alternative routes to counter toxic effects. The putative mechanism to mitigating heme toxicity is biocrystallization of FPIX to form hemozoin. The proposed mode of action for many antimalarial drugs is disruption of this natural detoxification pathway by forming stable complexes with one or more precrystalline forms of FPIX, thereby restoring the toxic effects described above.168 Prevailing theories are that these complexes block biocrystallization by keeping FPIX solubilized or by “capping” growing crystal faces, however certain mechanistic ambiguities remains.169,170 Nevertheless, inhibition of hemozoin formation in the presence of quinoline drugs has been visualized in live parasites171 and this has been correlated to inhibition of parasite growth.172
While details of the biological mechanism(s) remain to be fully determined, there has been significant effort characterizing small molecule/heme binding at the structural level. With respect to the quinolines, a diverse array of non-covalent heme–drug interactions have been suggested.163 An exemplar is the dative monomer complex (291), which is stabilized by coordination between the quinoline nitrogen and the iron center (Fig. 34). In the case of artemisinin, a heme C-alkylation process to form the heme complex 290 has been characterized in detail.173 These alkylated products occur through addition of carbon-centered radicals, which arise from reductive Fe(II) mediated cleavage of the peroxide functional group, to the accessible methine positions on the heme ring system. These heme alkylation products have been identified in living mice infected with malaria.174 It is important to note that radicals derived from artemisinin and other antimalarial peroxides have been proposed to have roles other then heme alkylation, and a recent review has covered these issues in detail.175,176
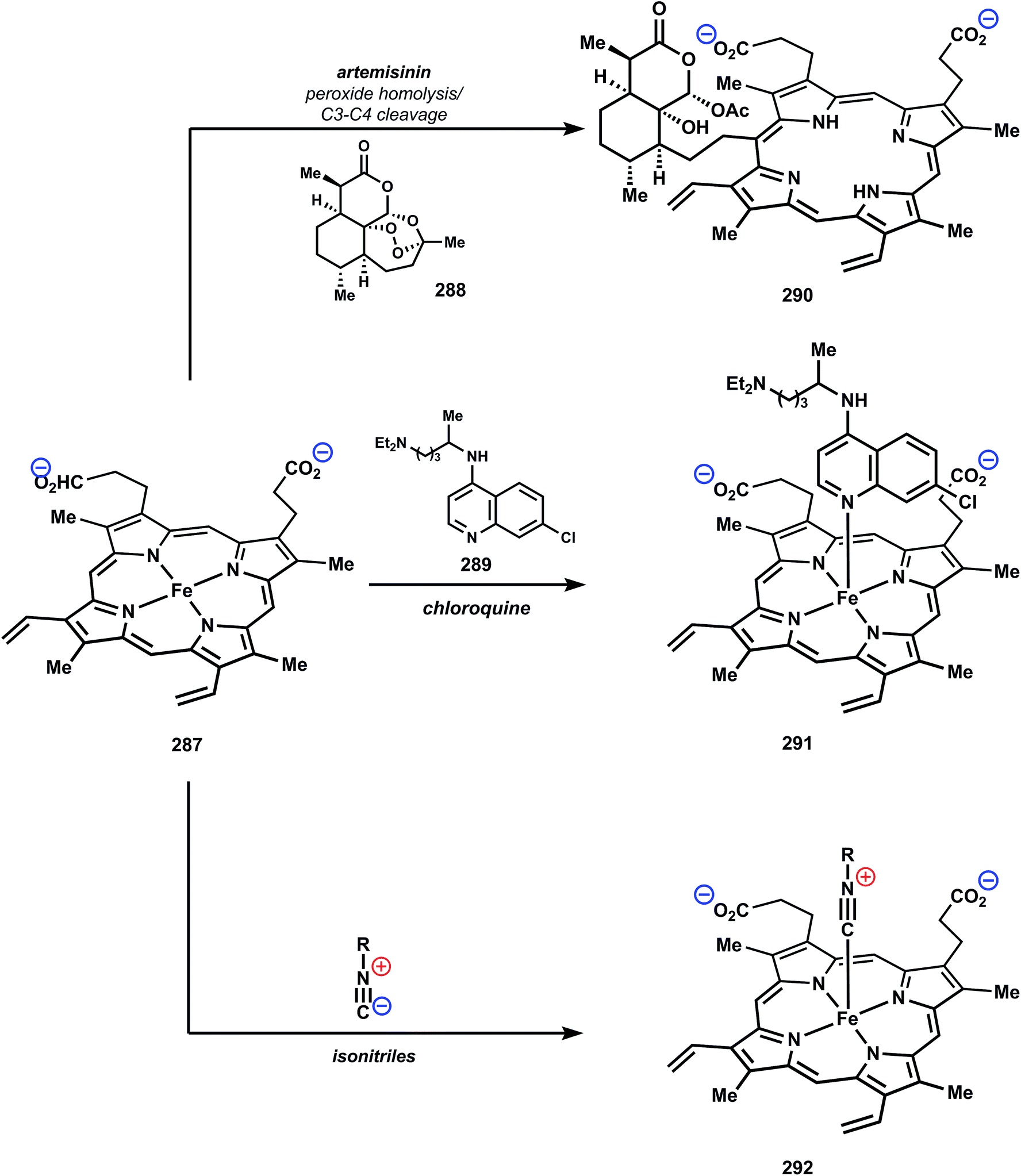 | ||
| Fig. 34 Small molecule/heme interactions. | ||
Heme/small molecule interactions are central to the current understanding of the ICT natural products discussed here. A variety of simple alkyl isonitriles bind the iron center of protein bound Fe(II)–heme, and starting with seminal studies by Pauling in 1956, these have served as probes of hemoglobin, myoglobin, and related proteins.177 Variations in the strength of isonitrile–heme binding interaction have been used to interrogate steric and electronic parameters of the heme-binding site, though advances in structural biology have largely supplanted such applications. More recently, Marletta and coworkers demonstrated a role for a nitric oxide/cysteine interaction in guanylate cyclase by using n-butyl isonitrile to block the heme-binding site.178
In 2001, Wright suggested that formation of isonitrile–heme(II) complex (292, Fig. 34) or the symmetrical binary complex lies at the heart of the antimalarial activity of these compounds. Isonitrile–heme binding was demonstrated using UV Stokes shift and ESI-MS. Several compounds, including 7, were shown to inhibit β-hematin formation and prevent the destruction of heme by peroxide and glutathione. In addition, a computational pseudoreceptor model was proposed.159 Similar to the proposed mechanism of action for quinoline and peroxide drugs, the suggestion is that these complexes disrupt hemozoin formation, leading to parasite death. This single binding interaction has been cited as the origin of the antimalarial activity of isonitrile natural products.179 While appealing, single-point binding of isonitriles to Fe(II)–heme does not offer a clear explanation for the complex SAR patterns highlighted above, nor does it explain the 200 nM activity of the formamide amphilectene (Fig. 34). In other words, the binding of heme by ICTs may be only one mechanism that contributes to their potency amidst possibly broad polypharmacology. In fact, recent studies examining chloroquine and related quinolines have suggested that targets other than heme are likely important for the cytocidal activity for these long-studied and extensively-used agents.180
Given the capacity of isonitriles to form transition metal complexes, a role for free metal binding is conceivable. The isolation the isonitrile–copper complex, 91 (Fig. 5d), and the zebrafish study by Ireland, both discussed above, provide support for this notion. Furthermore, anti-malarial activity has been reported for several copper and iron chelating scaffolds. In particular, iron chelators have been studied extensively, and desferrioxamine has undergone clinical studies to treat malaria with significant success.181 With regard to copper chelation, a cell permeable copper-chelating compound was shown to arrest parasite growth at the ring-to-trophozoite stage transition.182 The antimalarial activity of these agents is proposed to occur by two mechanisms: sequestration of free metal ions required for metabolic pathways and through the formation of toxic metal complexes.
The structure–activity relationships that can be defined from existing data suggest that ambiguities remain regarding the mechanism of action (Fig. 35 and 36). The following general observations can be made: (1) the presence of the isonitrile is required and, with one particularly notable exception (discussed below), only the isonitrile, not the corresponding isothiocyanate or formamide, display high activity. (2) The presence of an isonitrile alone is not sufficient for potent activity. For example, in a study examining the activity of 10 simple non-natural isonitriles, only 4 showed even modest activity (MIC = 3–14 μM). In these efforts, adamantyl isonitrile, 293, was the most active, (MIC = 2.5 μM) and was shown to display weak in vivo activity (2 of 5 mice survived to day 28 at 50 mg per kg per day administration) with poor therapeutic index (toxic at day 4 at 100 mg per kg per day).160 (3) In several cases, functionality distal to the isonitrile is critical to activity. Comparison of 294 and 9 suggests that a distal stereocenter affects the potency by over 20 fold, which is difficult to explain based on a non-specific (non-lock-and-key) binding event (Fig. 35). In a notable exception to these rules, the formamide of the mono-isonitrile amphilectane is reported to exhibit quite potent activity (200 nM, Fig. 36).162 In total, these results provide conflicting information about activity: if free-heme or free-metal binding is key, then why is stereochemistry important for potency? Alternatively, if the observations relate to drastic differences in pharmacokinetics (membrane permeability, efflux, metabolic degradation, etc.), why is formamide 295 active? In particular, protein active site binding is a distinct possibility that has not been examined in detail to date. A full understanding of the reported anti-malarial activity will certainly require further study.
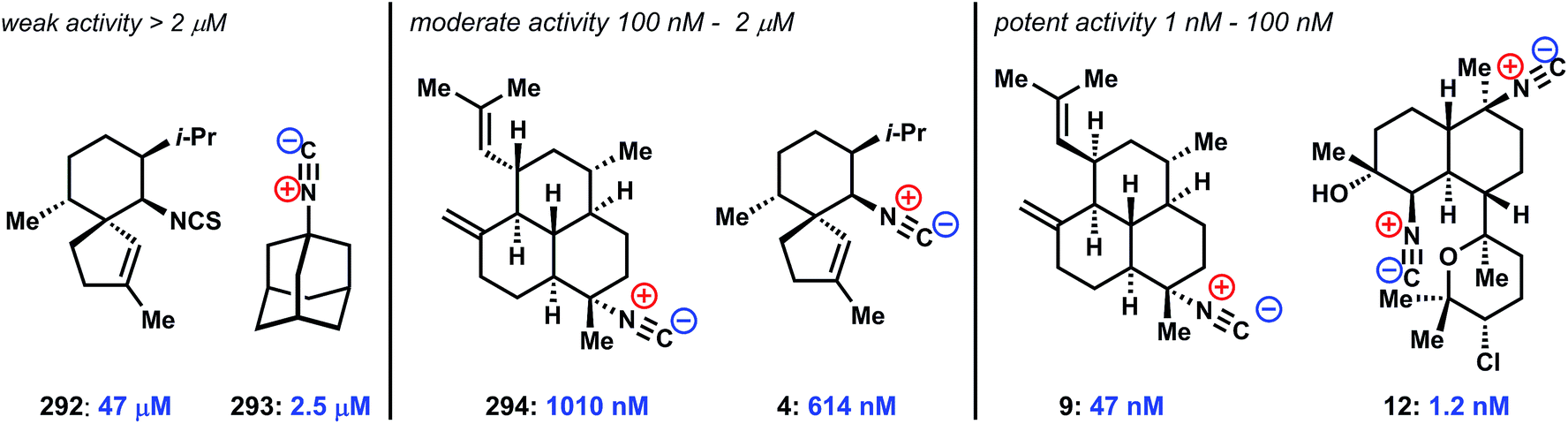 | ||
| Fig. 35 Representative anti-malarial (Plasmodium falciparum) activity.155,157,159,160 | ||
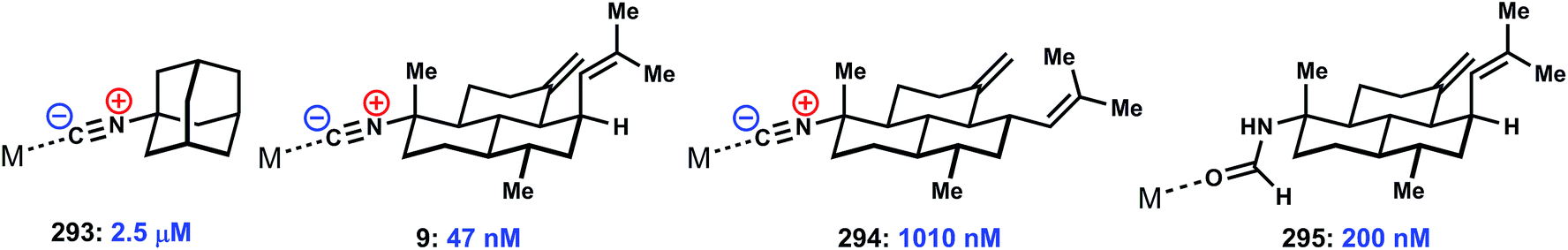 | ||
| Fig. 36 Metal binding alone does not explain SAR. | ||
5 Conclusion
We conclude with several points that emerge from the discussion above. First, although the isocyanoterpenes are recognizable by the substitution patterns imparted by their biosynthesis (Part 1), the structural diversity within this class has thwarted attempts to unify their chemical synthesis, particularly with a high level of efficiency (Part 3). Second, the value of synthetic chemistry research within this class is two-fold: as stimulus for the development of new chemical methods (Part 3), and as a means to procure material for biological study (Part 4). Third, we note that research efforts to broadly and rigorously address molecular interactions of this class in complex biological settings (Part 4) are noticeably absent, perhaps due to the assumption (correct or incorrect) that these molecules possess nonspecific mechanisms of action. The recent synthetic advances highlighted here, coupled with progress in chemoproteomic techniques suggest more rigorous evaluation of the ICTs as a potentially intriguing avenue for future work. The chemical reactivity of isonitriles (Part 1, Fig. 1C) may underpin target interactions and should be considered as these efforts proceed.6 Acknowledgements
M.J.S. is supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. R.A.S. acknowledges the generous support of the National Institutes of Health (GM105766), as well as the Alfred P. Sloan Foundation, Amgen, the Baxter Foundation, Boehringer-Ingelheim, and Eli Lilly. M.J.S. thanks Alexander P. Gorka for helpful suggestions. R.A.S. thanks Ted Molinski (UCSD) for helpful conversations, members of his lab for proofreading (Steven W. M. Crossley, Chris A. Reiher, Jeremy J. Roach, Kanny K. Wan), and Joseph Gassert for help with formatting.References
- P. J. Scheuer, Acc. Chem. Res., 1992, 25, 433–439 CrossRef CAS.
- M. S. Edenborough and R. B. Herbert, Nat. Prod. Rep., 1988, 5, 229–245 RSC.
- D. Laurent and F. Pietra, Mar. Biotechnol., 2006, 8, 433–447 CrossRef CAS PubMed.
- E. Fattorusso and O. Taglialatela-Scafati, Mar. Drugs, 2009, 7, 130–152 CrossRef CAS PubMed.
- M. J. Garson and J. S. Simpson, Nat. Prod. Rep., 2004, 21, 164–179 RSC.
- J. S. Simpson and M. J. Garson, Org. Biomol. Chem., 2004, 2, 939–948 CAS.
- J. S. Simpson and M. J. Garson, Tetrahedron Lett., 1998, 39, 5819–5822 CrossRef CAS.
- H. Stockmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Org. Biomol. Chem., 2011, 9, 7303–7305 Search PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- R. M. Wilson, J. L. Stockdill, X. Wu, X. Li, P. A. Vadola, P. K. Park, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 2834–2848 CrossRef CAS PubMed.
- A. D. Wright, P. J. Schupp, J. P. Schror, A. Engemann, S. Rohde, D. Kelman, N. J. de Voogd, A. Carroll and C. A. Motti, J. Nat. Prod., 2012, 75, 502–506 CrossRef CAS PubMed.
- J. Z. Sun, K. S. Chen, H. L. Liu, R. van Soest and Y. W. Guo, Helv. Chim. Acta, 2010, 93, 517–521 CrossRef CAS.
- W. Cheng, D. Liu, F. Y. Zhang, Q. Y. Zhang, P. Pedpradab, P. Proksch, H. Liang and W. H. Lin, Tetrahedron, 2014, 70, 3576–3583 CrossRef CAS PubMed.
- Y. N. Yan, X. Z. Zhu, J. L. Yu, D. Z. Jin, Y. W. Guo, E. Mullo and G. Cimino, J. Asian Nat. Prod. Res., 2006, 8, 579–584 CrossRef PubMed.
- E. Zubia, M. J. Ortega and J. L. Carballo, J. Nat. Prod., 2008, 71, 2004–2010 CrossRef CAS PubMed.
- E. Zubia, M. J. Ortega, C. J. Hernandez-Guerrero and J. L. Carballo, J. Nat. Prod., 2008, 71, 608–614 CrossRef CAS PubMed.
- W.-J. Lan, H.-P. Wan, G.-X. Li, H. J. Li, Y. Y. Chen, C. Z. Liao and J. W. Cai, Helv. Chim. Acta, 2008, 91, 426 CrossRef CAS.
- P. Jumaryatno, K. Rands-Trevor, J. T. Blanchfield and M. J. Garson, ARKIVOC, 2007, 7, 157–166 Search PubMed.
- H. Mitome, N. Shirato, H. Miyaoka, Y. Yamada and R. W. M. van Soest, J. Nat. Prod., 2004, 67, 833–837 CrossRef CAS PubMed.
- K. Nishikawa, T. Umezawa, M. J. Garson and F. Matsuda, J. Nat. Prod., 2012, 75, 2232–2235 CrossRef CAS PubMed.
- P. Jumaryatno, B. L. Stapleton, J. N. A. Hooper, D. J. Brecknell, J. T. Blanchfield and M. J. Garson, J. Nat. Prod., 2007, 70, 1725–1730 CrossRef CAS PubMed.
- T. S. Bugni, M. P. Singh, L. Chen, D. A. Arias, M. K. Harper, M. Greenstein, W. M. Maiese, G. P. Concepcion, G. C. Mangalindan and C. M. Ireland, Tetrahedron, 2004, 60, 6981–6988 CrossRef CAS PubMed.
- J. Z. Sun, K. S. Chen, L. G. Yao, H. L. Liu and Y. W. Guo, Arch. Pharmacal Res., 2009, 32, 1581–1584 CrossRef CAS PubMed.
- Y. Xu, J. H. Lang, W. H. Jiao, R. P. Wang, Y. Peng, S. J. Song, B. H. Zhang and H. W. Lin, Mar. Drugs, 2012, 10, 1445–1458 CrossRef CAS PubMed.
- Y. Xu, N. Li, W. H. Jiao, R. P. Wang, Y. Peng, S. H. Qi, S. J. Song, W. S. Chen and H. W. Lin, Tetrahedron, 2012, 68, 2876–2883 CrossRef CAS PubMed.
- A. D. Wright and N. Lang-Unnasch, J. Nat. Prod., 2009, 72, 492–495 CrossRef CAS PubMed.
- C. Wattanapiromsakul, N. Chanthathamrongsiri, S. Bussarawit, S. Yuenyongsawad, A. Plubrukarn and K. Suwanborirux, Can. J. Chem., 2009, 87, 612–618 CrossRef CAS.
- D. Lamoral-Theys, E. Fattorusso, A. Mangoni, C. Perinu, R. Kiss and V. Costantino, J. Nat. Prod., 2011, 74, 2299–2303 CrossRef CAS PubMed.
- N. Chanthathamrongsiri, S. Yuenyongsawad, C. Wattanapiromsakul and A. Plubrukarn, J. Nat. Prod., 2012, 75, 789–792 CrossRef CAS PubMed.
- E. Aviles, A. D. Rodriguez and J. Vicente, J. Org. Chem., 2013, 78, 11294–11301 CrossRef CAS PubMed.
- M. L. Ciavatta, A. Fontana, R. Puliti, G. Scognamiglio and G. Cimino, Tetrahedron, 1999, 55, 12629–12636 CrossRef CAS.
- M. L. Ciavatta, M. Gavagnin, E. Manzo, R. Puliti, C. A. Mattia, L. Mazzarella, G. Cimino, J. S. Simpson and M. J. Garson, Tetrahedron, 2005, 61, 8049–8053 CrossRef CAS PubMed.
- E. Aviles and A. D. Rodriguez, Org. Lett., 2010, 12, 5290–5293 CrossRef CAS PubMed.
- D. Q. Xue, E. Mollo, G. Cimino and Y. W. Guo, Helv. Chim. Acta, 2009, 92, 1428–1433 CrossRef CAS.
- H. Ishiyama, A. Hashimoto, J. Fromont, Y. Hoshino, Y. Mikami and J. Kobayashi, Tetrahedron, 2005, 61, 1101–1105 CrossRef CAS PubMed.
- S. Suto, N. Tanaka, J. Fromont and J. Kobayashi, Tetrahedron Lett., 2011, 52, 3470–3473 CrossRef CAS PubMed.
- N. Tanaka, S. Suto, H. Ishiyama, T. Kubota, A. Yamano, M. Shiro, J. Fromont and J. Kobayashi, Org. Lett., 2012, 14, 3498–3501 CrossRef CAS PubMed.
- H. Ishiyama, S. Kozawa, K. Aoyama, Y. Mikami, J. Fromont and J. Kobayashi, J. Nat. Prod., 2008, 71, 1301–1303 CrossRef CAS PubMed.
- E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, J. Wiley, New York, 1995 Search PubMed.
- D. A. Evans, An Organizational Format for the Classification of Functional Groups. Application to the Construction of Difunctional Relationships, Chemistry 206: Advanced Organic Chemistry, Handout 27A, Harvard University, 2001 Search PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, West Sussex, 2009 Search PubMed.
- Biomimetic Organic Synthesis, ed. E. Poupon and B. Nay, Wiley-VCH, Weinheim, 2011 Search PubMed.
- M. J. Garson, J. Chem. Soc., Chem. Commun., 1986, 35–36 RSC.
- E. J. Corey, M. Behforouz and M. Ishiguro, J. Am. Chem. Soc., 1979, 101, 1608 CrossRef CAS.
- H. Yamamoto and H. L. Sham, J. Am. Chem. Soc., 1979, 101, 1609 CrossRef CAS.
- D. Caine and H. Deutsch, J. Am. Chem. Soc., 1978, 100, 8030 CrossRef CAS.
- E. Piers and M. Llinas-Brunet, J. Org. Chem., 1989, 54, 1483–1484 CrossRef CAS.
- E. Piers, M. Llinas-Brunet and R. M. Oballa, Can. J. Chem., 1993, 71, 1486 Search PubMed.
- E. Piers and M. A. Romero, Tetrahedron, 1993, 49, 5791–5800 CrossRef CAS.
- H. Miyaoka, H. Shida, N. Yamada, H. Mitome and Y. Yamada, Tetrahedron Lett., 2002, 43, 2227–2230 CrossRef CAS.
- T. Ohkuba, H. Akino, M. Asaoka and H. Takei, Tetrahedron Lett., 1995, 36, 3365 CrossRef.
- K. A. Fairweather and L. N. Mander, Org. Lett., 2006, 8, 3395–3398 CrossRef CAS PubMed.
- R. D. White, G. F. Keaney, C. D. Slown and J. L. Wood, Org. Lett., 2004, 6, 1123–1126 CrossRef CAS PubMed.
- A. Srikrishna, G. Ravi and D. R. C. Subbaiah, Synlett, 2009, 1, 32–34 CrossRef PubMed.
- E. J. Corey and P. A. Magriotis, J. Am. Chem. Soc., 1987, 109, 287–289 CrossRef CAS.
- L. Castellanos, C. Duque, J. Rodríguez and C. Jiménez, Tetrahedron, 2007, 63, 1544–1552 CrossRef CAS PubMed.
- R. V. Stevens and K. F. Albizati, J. Org. Chem., 1985, 50, 632–640 CrossRef CAS.
- S. V. Pronin, C. A. Reiher and R. A. Shenvi, Nature, 2013, 501, 195–199 CrossRef CAS PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Springer, New York, 2007, p. 159 Search PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Chemistry, University Science Books, Sausalito, 2006, p. 108 Search PubMed.
- H. W. Thompson and D. J. Long, J. Org. Chem., 1988, 53, 4201–4209 CrossRef CAS.
- H. Miyaoka and Y. Okuba, Synlett, 2011, 4, 547–550 CrossRef PubMed.
- H. Miyaoka, Y. Abe, N. Sekiya, H. Mitome and E. Kawashima, Chem. Commun., 2012, 48, 901–903 RSC.
- P. C. Roosen and C. D. Vanderwal, Org. Lett., 2014, 16, 4368–4371 CrossRef CAS PubMed.
- C. S. S. Rao, G. Kumar, K. Rajagopalan and S. Swaminathan, Tetrahedron, 1982, 38, 2195–2202 CrossRef CAS.
- C. E. Dietinger, M. G. Banwell, M. J. Garson and A. C. Willis, Tetrahedron, 2010, 66, 5250–5261 CrossRef CAS PubMed.
- A. Srikrishna and S. J. Gharpure, J. Org. Chem., 2001, 66, 4379–4385 CrossRef CAS PubMed.
- A. Srikrishna and S. J. Gharpure, ARKIVOC, 2002, 7, 52–62 CrossRef.
- T. L. Ho, L. R. Kung and R. J. Chein, J. Org. Chem., 2000, 65, 5774–5779 CrossRef CAS PubMed.
- T. L. Ho and L. R. Kung, Org. Lett., 1999, 1, 1051–1052 CrossRef CAS.
- T. L. Ho and G. H. Jana, J. Org. Chem., 1999, 64, 8965–8967 CrossRef CAS.
- S. L. Hsieh, C. T. Chiu and N. C. Chang, J. Org. Chem., 1989, 54, 3820–3823 CrossRef CAS.
- E. J. Corey and M. Ishiguro, Tetrahedron Lett., 1979, 30, 2745–2748 CrossRef.
- K. Nishikawa, T. Umezawa, M. J. Garson and F. Matsuda, J. Nat. Prod., 2012, 75, 2232–2235 CrossRef CAS PubMed.
- F. Senejoux, L. Evanno and E. Poupon, Eur. J. Org. Chem., 2013, 2013, 453–455 CrossRef CAS.
- K. Saito, A. Nishimori, H. Kotsuki, K. Nakano and Y. Ichikawa, Synlett, 2013, 24, 757–761 CrossRef CAS PubMed.
- K. Nishikawa, N. Hiroshi, Y. Shirokura, Y. Nogata, E. Yoshimura, T. Umezawa, T. Okino and F. Matsuda, J. Org. Chem., 2011, 76, 6558–6573 CrossRef CAS PubMed.
- K. Nishikawa, H. Nakahara, Y. Shirokura, Y. Nogata, R. Yoshimura, T. Umezawa, T. Okino and F. Matsuda, Org. Lett., 2010, 12, 904–907 CrossRef CAS PubMed.
- C. M. de Oliveira, C. C. da Silva, C. H. Collins and A. J. Marsaioli, J. Braz. Chem. Soc., 2001, 12, 661–666 CrossRef CAS PubMed.
- O. Schwarz, R. Brun, J. W. Bats and H. G. Schmalz, Tetrahedron Lett., 2002, 43, 1009–1013 CrossRef CAS.
- Y. Kitano, A. Yokoyama, Y. Nogata, K. Shinshima, E. Yoshimura, K. Chiba, M. Tada and I. Sakaguchi, Biofouling, 2003, 19, 187–192 CrossRef CAS PubMed.
- Y. Ichikawa, Y. Matsuda, K. Okumura, M. Nakamura, T. Masuda, H. Kotsuki and K. Nakano, Org. Lett., 2011, 13, 2520–2523 CrossRef CAS PubMed.
- H. Prawat, C. Mahidol, S. Wittayalai, P. Intachote, T. Kanchanapoom and S. Ruchirawat, Tetrahedron, 2011, 67, 5651–5655 CrossRef CAS PubMed.
- S. De Rosa, A. De Giulio, C. Iodice and N. Zavodink, Phytochemistry, 1994, 5, 1327–1330 CrossRef.
- W. G. Dauben and E. J. Deviny, J. Org. Chem., 1966, 31, 3794–3798 CrossRef CAS.
- E. Piers and P. M. Worster, J. Am. Chem. Soc., 1972, 94, 2895 CrossRef CAS.
- K. Tamura, A. Nakazaki and S. Kobayashi, Synlett, 2009, 15, 2449–2452 Search PubMed.
- J. A. Marshall, W. I. Fanta and H. Roebke, J. Org. Chem., 1966, 31, 1016–1020 CrossRef CAS.
- D. Caine, A. A. Boucugnani and W. R. Pennington, J. Org. Chem., 1976, 41, 3632 CrossRef CAS.
- G. R. Clemo and R. D. Haworth, J. Chem. Soc., 1929, 1, 2369–2387 Search PubMed.
- K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. M. Crossley and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 1300–1303 CrossRef CAS PubMed.
- W. R. Hertler and E. J. Corey, J. Org. Chem., 1958, 23, 1221–1222 CrossRef CAS.
- P. Karuso, A. Poiner and P. J. Scheuer, J. Org. Chem., 1989, 54, 2095–2097 CrossRef CAS.
- N. Fusetani, H. J. Wolstenhohne, S. Matsunaga and H. Hirota, Tetrahedron Lett., 1991, 32, 7291–7294 CrossRef CAS.
- D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035–1053 RSC.
- D. W. Christianson, Chem. Rev., 2006, 106, 3412–3442 CrossRef CAS PubMed.
- S. Schulz and J. S. Dickschat, Nat. Prod. Rep., 2007, 24, 814–842 RSC.
- M. K. Brown and E. J. Corey, Org. Lett., 2010, 12, 172–175 CrossRef CAS PubMed.
- O. H. Oldenziel, D. Van Leusen and A. M. Van Leusen, J. Org. Chem., 1977, 42, 3114–3118 CrossRef CAS.
- S. Nishimura, Bull. Chem. Soc. Jpn., 1961, 34, 32–36 CrossRef CAS.
- L. B. Hunt, Platinum Met. Rev., 1962, 6, 150–152 Search PubMed.
- E. J. Corey and W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef.
- E. J. Corey and C. U. Kim, J. Am. Chem. Soc., 1972, 94, 7586–7587 CrossRef CAS.
- A. W. Weitkamp, J. Catal., 1966, 6, 431–457 CrossRef CAS.
- M. M. Toteva and J. P. Richard, J. Am. Chem. Soc., 1996, 118, 11434–11445 CrossRef CAS.
- S. V. Pronin and R. A. Shenvi, Nat. Chem., 2012, 4, 915–920 CrossRef CAS PubMed.
- A. Srikrishna and P. R. Kumar, Tetrahedron Lett., 2002, 43, 1109–1111 CrossRef CAS.
- A. Srikrishna and S. J. Gharpure, Chem. Commun., 1998, 1589–1590 RSC.
- Y. Ichikawa, Synlett, 1991, 715–716 CrossRef CAS PubMed.
- Y. Kitano, T. Ito, T. Suzuki, Y. Nogata, K. Shinshima, E. Yoshimura, K. Chiba, M. Tada and I. Sakaguchi, J. Chem. Soc., Perkin. Trans. 1, 2002, 2251–2255 RSC.
- Y. Ichikawa, Chem. Lett., 1990, 1347–1350 CrossRef CAS.
- N. H. Andersen and D. D. Syrdal, Tetrahedron Lett., 1972, 24, 2455–2458 CrossRef.
- Y. Ohta and Y. Hirose, Chem. Lett., 1972, 263–266 CrossRef CAS.
- I. Okada and Y. Kitano, Synthesis, 2011, 24, 3997–4002 Search PubMed.
- E. D. Hughes, C. K. Ingold, R. J. L. Martin and D. F. Meigh, Nature, 1950, 166, 679–680 CrossRef CAS.
- W. v. E. Doering and H. H. Zeiss, J. Am. Chem. Soc., 1953, 75, 4733–4738 CrossRef CAS.
- P. Müller and J.-C. Rossier, J. Chem. Soc., Perkin Trans. 2, 2000, 2, 2232–2237 RSC.
- T. Kochi and J. A. Ellman, J. Am. Chem. Soc., 2004, 126, 15652–15653 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- E. Vedejs and H. W. Fang, J. Org. Chem., 1984, 49, 210–212 CrossRef CAS.
- D. Taber and B. P. Gunn, J. Am. Chem. Soc., 1979, 101, 3992–3993 CrossRef CAS.
- H. Miyaoka, Y. Okubo, M. Muroi, H. Mitome and E. Kawashima, Chem. Lett., 2011, 40, 246–247 CrossRef CAS.
- E. Piers and R. W. Friesen, Can. J. Chem., 1987, 65, 1681–1683 CrossRef CAS.
- E. J. Corey and G. W. J. Fleet, Tetrahedron Lett., 1973, 14, 4499–4501 CrossRef.
- W. G. Salmond, M. A. Barta and J. L. Havens, J. Org. Chem., 1978, 43, 2057–2059 CrossRef CAS.
- L. H. Sarett, G. E. Arth, R. M. Lukes, R. E. Beyler, G. I. Poos, W. F. Johns and J. M. Constantin, J. Am. Chem. Soc., 1952, 74, 4974–4976 CrossRef CAS.
- D. H. R. Barton and C. H. Robinson, J. Chem. Soc., 1954, 3045–3051 RSC.
- L. Lombardo, Tetrahedron Lett., 1982, 23, 4293–4296 CrossRef CAS.
- E. Piers and M. A. Romero, Tetrahedron, 1993, 49, 5791–5800 CrossRef CAS.
- R. D. White and J. L. Wood, Org. Lett., 2001, 3, 1825–1827 CrossRef CAS PubMed.
- S. V. Pronin and R. A. Shenvi, J. Am. Chem. Soc., 2012, 134, 19604–19606 CrossRef CAS PubMed.
- H. Hopf and M. S. Sherburn, Angew. Chem., Int. Ed., 2012, 51, 2298–2338 CrossRef CAS PubMed.
- S. J. Danishefsky, Acc. Chem. Res., 1981, 14, 400–406 CrossRef CAS.
- S. J. Danishefsky, Aldrichimica Acta, 1986, 19, 59–69 CAS.
- I. Hayakawa, Y. Asuma, T. Ohyoshi, K. Aoki and H. Kigoshi, Tetrahedron Lett., 2007, 48, 6221–6224 CrossRef CAS PubMed.
- Y. Kitano, K. Chiba and M. Tada, Tetrahedron Lett., 1998, 39, 1911–1912 CrossRef CAS.
- I. Okada and Y. Kitano, Synthesis, 2011, 24, 3997–4002 Search PubMed.
- D. F. Wiemer, J. Meinwald, G. D. Prestwich, B. A. Solheim and J. J. Clardy, J. Org. Chem., 1980, 45, 191–192 CrossRef CAS.
- M. Kodama, H. Minami, Y. Mima and Y. Fukuyama, Tetrahedron Lett., 1990, 31, 4025–4026 CrossRef CAS.
- D. Xu, C. Y. Park and K. B. Sharpless, Tetrahedron Lett., 1994, 35, 2495–2498 CrossRef CAS.
- S. M. Abdur Rahman, H. Ohno, N. Maezaki, C. Iwata and T. Tanaka, Org. Lett., 2000, 2, 2893–2895 CrossRef PubMed.
- R. D. White, PhD Thesis, Yale University, 2003.
- G. F. Keaney, PhD Thesis, Yale University, 2005.
- I. Ugi, Isonitrile Chemistry, Academic Press, New York, 1971, pp. 1–278 Search PubMed.
- C. W. J. Chang, A. Patra, J. A. Baker and P. J. Scheuer, J. Am. Chem. Soc., 1987, 109, 6119–6123 CrossRef CAS.
- T. S. Bugni, M. P. Singh, L. Chen, D. A. Arias, M. K. Harper, M. Greenstein, W. M. Maiese, G. P. Concepcion, G. C. Mangalindan and C. M. Ireland, Tetrahedron, 2004, 60, 6981–6988 CrossRef CAS PubMed.
- A. M. Mayer, E. Aviles and A. D. Rodriguez, Bioorg. Med. Chem., 2012, 20, 279–282 CrossRef CAS PubMed.
- I. T. Sandoval, E. J. Manos, R. M. Van Wagoner, R. G. C. Delacruz, K. Edes, D. R. Winge, C. M. Ireland and D. A. Jones, Chem. Biol., 2013, 20, 753–763 CrossRef CAS PubMed.
- P. B. Bloland, Drug resistance in malaria, Document WHO/CDS/CSR/DRS/2001.4, World Health Organization, Geneva, 2001 Search PubMed.
- F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, M. Imwong, K. Chotivanich, P. Lim, T. Herdman, S. S. An, S. Yeung, P. Singhasivanon, N. P. Day, N. Lindegardh, D. Socheat and N. J. White, N. Engl. J. Med., 2009, 361, 455–467 CrossRef PubMed.
- M. Eisenstein, Nature, 2012, 484, S16–S18 CrossRef CAS PubMed.
- M. Enserink, Malaria's Drug Miracle in Danger, Science, 2010, 328, 844–846 CrossRef CAS PubMed.
- A. D. Wright, G. M. Konig, C. K. Angerhofer, P. Greenidge, A. Linden and R. Desqueyroux-Faundez, J. Nat. Prod., 1996, 59, 710–716 CrossRef CAS PubMed.
- C. K. Angerhofer, J. M. Pezzuto, G. M. Konig, A. D. Wright and O. Sticher, J. Nat. Prod., 1992, 55, 1787–1789 CrossRef CAS.
- G. M. Konig, A. D. Wright and C. K. Angerhofer, J. Org. Chem., 1996, 61, 3259–3267 CrossRef.
- H. Miyaoka, M. Shimomura, H. Kimura, Y. Yamada, H. S. Kim and Y. Wataya, Tetrahedron, 1998, 54, 13467–13474 CrossRef CAS.
- J. S. Simpson, M. J. Garson, J. N. A. Hooper, E. I. Cline and C. K. Angerhofer, Aust. J. Chem., 1997, 50, 1123–1127 CrossRef CAS.
- A. D. Wright, H. Q. Wang, M. Gurrath, G. M. Konig, G. Kocak, G. Neumann, P. Loria, M. Foley and L. Tilley, J. Med. Chem., 2001, 44, 873–885 CrossRef CAS PubMed.
- C. Singh, N. C. Srivastav and S. K. Puri, Bioorg. Med. Chem. Lett., 2002, 12, 2277–2279 CrossRef CAS.
- O. Schwarz, R. Brun, J. W. Bats and H. G. Schmalz, Tetrahedron Lett., 2002, 43, 1009–1013 CrossRef CAS.
- A. D. Wright, A. McCluskey, M. J. Robertson, K. A. MacGregor, C. P. Gordon and J. Guenther, Org. Biomol. Chem., 2011, 9, 400–407 CAS.
- A. P. Gorka, A. de Dios and P. D. Roepe, J. Med. Chem., 2013, 56, 5231–5246 CrossRef CAS PubMed.
- J. M. Rifkind, S. Ramasamy, P. T. Manoharan, E. Nagababu and J. G. Mohanty, Antioxid. Redox Signaling, 2004, 6, 657–666 CrossRef CAS PubMed.
- M. Goyal, A. Alam and U. Bandyopadhyay, Curr. Med. Chem., 2012, 19, 1475–1503 CrossRef CAS.
- S. E. Francis, D. J. Sullivan Jr and D. E. Goldberg, Annu. Rev. Microbiol., 1997, 51, 97–123 CrossRef CAS PubMed.
- P. A. Sigala, J. R. Crowley, S. Hsieh, J. P. Henderson and D. E. Goldberg, J. Biol. Chem., 2012, 287, 37793–37807 CrossRef CAS PubMed.
- T. J. Egan and H. M. Marques, Coord. Chem. Rev., 1999, 192, 493–517 CrossRef.
- M. Chugh, V. Sundararaman, S. Kumar, V. S. Reddy, W. A. Siddiqui, K. D. Stuart and P. Malhotra, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5392–5397 CrossRef CAS PubMed.
- D. E. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5283–5284 CrossRef CAS PubMed.
- B. Gligorijevic, R. McAllister, J. S. Urbach and P. D. Roepe, Biochemistry, 2006, 45, 12400–12410 CrossRef CAS PubMed.
- A. P. Gorka, J. N. Alumasa, K. S. Sherlach, L. M. Jacobs, K. B. Nickley, J. P. Brower, A. C. de Dios and P. D. Roepe, Antimicrob. Agents Chemother, 2013, 57, 356–364 CrossRef CAS PubMed.
- A. Robert, J. Cazelles and B. Meunier, Angew. Chem., Int. Ed., 2001, 40, 1954–1957 CrossRef CAS.
- A. Robert, F. Benoit-Vical, C. Claparols and B. Meunier, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13676–13680 CrossRef CAS PubMed.
- P. M. O'Neill, V. E. Barton and S. A. Ward, Molecules, 2010, 15, 1705–1721 CrossRef PubMed.
- F. Bousejra-El Garah, M. H. Wong, R. K. Amewu, S. Muangnoicharoen, J. L. Maggs, J. L. Stigliani, B. K. Park, J. Chadwick, S. A. Ward and P. M. O'Neill, J. Med. Chem., 2011, 54, 6443–6455 CrossRef CAS PubMed.
- R. C. St. George and L. Pauling, Science, 1951, 114, 629–634 CAS.
- E. R. Derbyshire and M. A. Marletta, J. Biol. Chem., 2007, 282, 35741–35748 CrossRef CAS PubMed.
- U. Bandyopadhyay, S. Kumar, M. Guha, V. Choubey and P. Maity, Life Sci., 2007, 80, 813–828 CrossRef PubMed.
- P. D. Roepe, Trends Parasitol., 2014, 30, 130–135 CrossRef CAS PubMed.
- G. F. Mabeza, M. Loyevsky, V. R. Gordeuk and G. Weiss, Pharmacol. Therapeut., 1999, 81, 53–75 CrossRef CAS.
- D. Rasoloson, L. Shi, C. R. Chong, B. F. Kafsack and D. J. Sullivan, Biochem. J., 2004, 381, 803–811 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |