Mechanistic studies on the indole prenyltransferases
Martin E.
Tanner
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, V6T 1Z1, British Columbia, Canada. E-mail: mtanner@chem.ubc.ca
Received
26th July 2014
First published on 1st October 2014
Abstract
Covering: up to 2014
Prenylated indole alkaloids comprise a large and structurally diverse family of natural products that often display potent biological activities. In recent years a large family of prenyltransferases that install prenyl groups onto the indole core have been discovered. While the vast majority of these enzymes are evolutionarily related and share a common protein fold, they are remarkably versatile in their ability to catalyze reverse and normal prenylations at all positions on the indole ring. This highlight article will focus on recent studies of the mechanisms utilized by indole prenyltransferases. While all of the prenylation reactions may follow a direct electrophilic aromatic substitution mechanism, studies of structure and reactivity suggest that in some cases prenylation may first occur at the nucleophilic C-3 position, and subsequent rearrangements then generate the final product.
Martin E. Tanner
Martin Tanner received his Ph.D. at UCLA working with Prof. Donald Cram in the area of molecular recognition. He then completed postdoctoral studies on mechanistic enzymology at Harvard University with Prof. Jeremy Knowles. He is currently a Professor of Chemistry at the University of British Columbia where he works on enzyme mechanism, biosynthetic pathways, and inhibitor design. He has interests in epimerases/racemases, sugar nucleotide biosynthesis, peptidoglycan biosynthesis/modification, menaquinone biosynthesis, and alkaloid biosynthesis.
1 Prenylated indole alkaloids and indole prenyltransferases
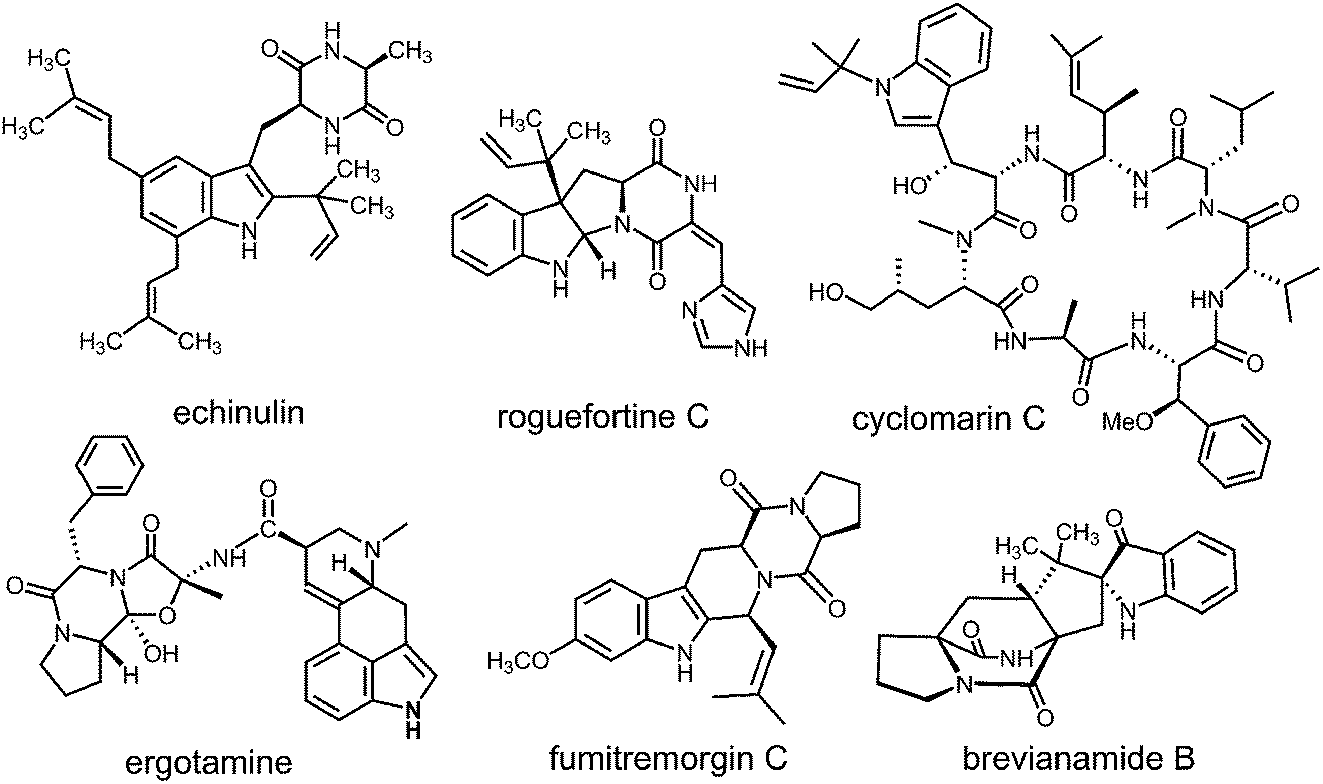
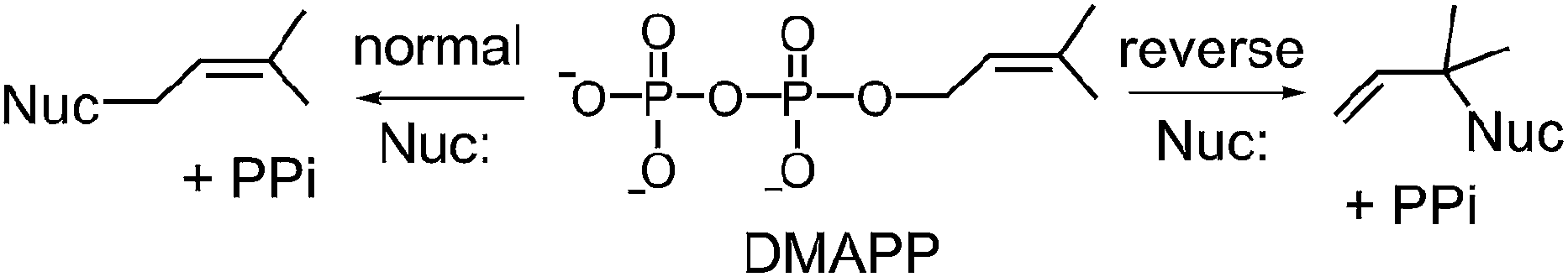
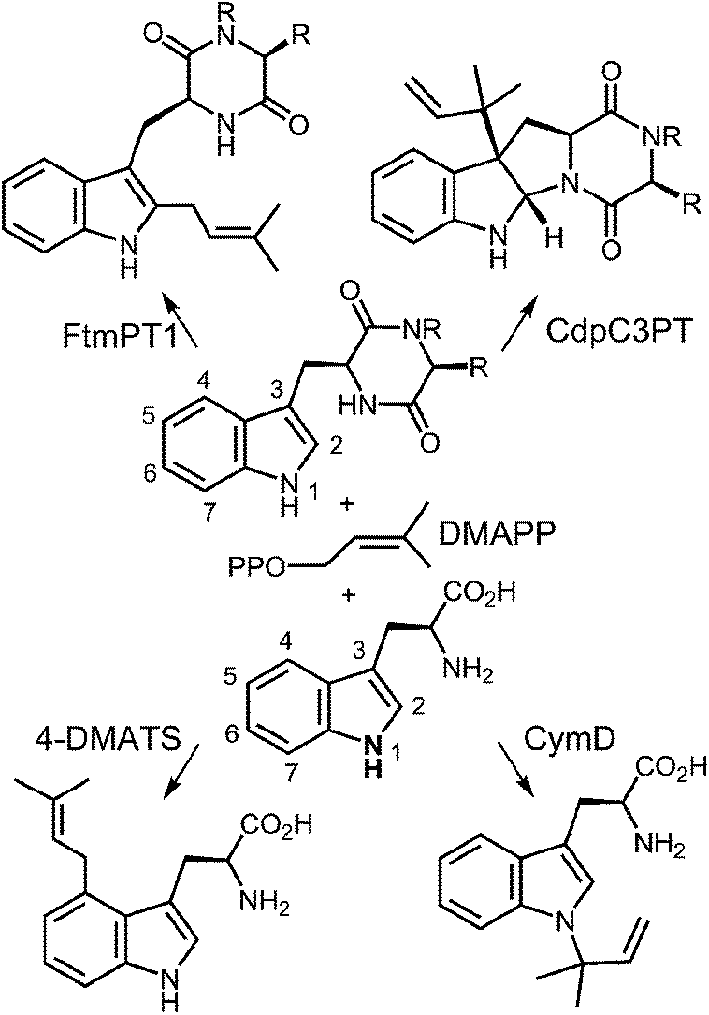
Prenylated indole alkaloids comprise a large family of biologically active natural products produced by plants, fungi, and bacteria.1–3 The indole core in these compounds is typically derived from L-tryptophan, or less commonly, from indole-3-glycerol phosphate. The prenyl carbons may reside on the periphery (such as with echinulin or roguefortine C), or may ultimately be embedded within the core of, the final alkaloid product (such as with ergotamine or brevianamide B) (Fig. 1). Thus, they may serve either to increase the lipophilicity of the alkaloids or to provide a carbon skeleton that is integral to the final structure. In most cases, prenylation involves the addition of a five-carbon dimethylallyl moiety; however, the addition of larger prenyl groups is known. For example, the addition of a ten-carbon geranyl group is involved in the biosynthesis of the lyngbyatoxins, and that of a twenty-carbon geranylgeranyl group is involved in the biosynthesis of the indole diterpenes such as paxilline.4,5 The prenylations may take place in a “normal” sense where the primary carbon of the allylic moiety adds to the indole ring, or in a “reverse” sense where the tertiary carbon adds (Fig. 2).
Fig. 1 Representative structures of prenylated indole alkaloids.
Fig. 2 Normal and reverse prenylation with dimethylallyl diphosphate (DMAPP). Nuc: represents a nucleophilic nitrogen or carbon of the indole ring.
In more structurally complex alkaloids, the prenylation of the indole ring often occurs early in the biosynthetic sequence and common substrates are either free tryptophan or cyclic dipeptides/benzodiazopinediones bearing a tryptophan subunit. The prenyl donors are allylic diphosphates and the reactions are catalyzed by indole prenyltransferases.2,6–8 In the past ten years, an explosion in the number of identified indole prenyltransferases has occurred. Enzymes that prenylate at all possible positions of the indole ring (N-1,9,10 C-2,11–13 C-3,14–16 C-4,17–19 C-5,20,21 C-6,22,23 and C-711,24–26) have been identified (Fig. 3). Most of these enzymes utilize dimethylallyl diphosphate (DMAPP) as a substrate and transfer a five-carbon prenyl group. They are all members of the soluble indole prenyltransferase superfamily that share a common structural motif known as the “ABBA” fold.27 Unlike other families of prenyltransferases, these enzymes act in a metal-independent fashion. The majority of studies on this new family of prenyltransferases have focused on identification of the enzymes and an examination of their substrate scope. What is clear from this work is that many of the enzymes are somewhat promiscuous and will accept a variety of alternate indole- or phenol-containing substrates.14,28–30 In addition, with alternate substrates, products containing prenylation at unexpected sites are often observed.25,31–33 While fewer studies have focused on mechanism, recent work in this area is addressing the way in which enzymes with similar active sites catalyze both normal and reverse prenylations at a variety of positions on the indole ring. This highlight article will focus on recent mechanistic and structural studies that have been performed on the indole prenyltransferases.
Fig. 3 Representative examples of normal and reverse prenylations of cyclic dipeptides and tryptophan.
2 4-Dimethylallyltryptophan synthase (4-DMATS or FgaPT2)
The first identified, and most thoroughly studied, of the soluble indole prenyltransferases is 4-dimethylallyltryptophan synthase (4-DMATS or FgaPT2).18,19,34,35 This enzyme catalyzes a normal C-4 prenylation of L-Trp with dimethylallyl diphosphate (DMAPP) in the first committed step of ergot alkaloid biosynthesis (Fig. 4).36 The ergot alkaloids are well known for their potent biological properties.37 In the middle ages, the ingestion of rye infected with the ergot fungus led to Saint Anthony's fire (ergotism) that results in convulsive and gangrenous symptoms ultimately causing death. With more controlled doses, ergot alkaloids have been used as medicines. For example, ergotamine has been used to treat migraine headaches and ergometrine has been used to prevent post-natal bleeding. Finally, lysergic acid diethylamide (LSD) is a semi-synthetic psychedelic drug used both recreationally and in psychedelic therapy.
Fig. 4 The reaction catalyzed by 4-DMATS in the biosynthesis of the ergot alkaloids. Atoms bearing an asterisk convey the stereochemical outcomes reported in ref. 38.
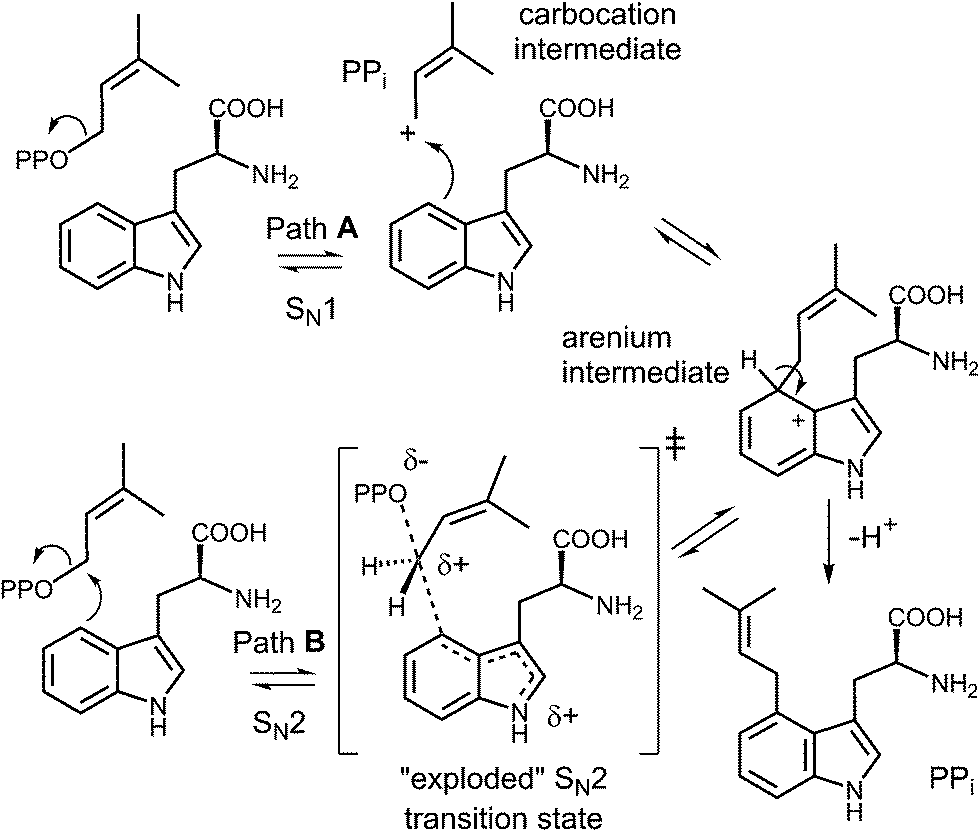
Early mechanistic studies on 4-DMATS were performed by Floss and co-workers.38 Using isotopically labeled substrates, they established that the prenyl group of DMAPP is transferred onto the indole ring with inversion of configuration at the C-2 methylene position and that no scrambling of the methyl groups occurred during this process (Fig. 4). Subsequent studies by Poulter and co-workers examined the effects of substituting electron-withdrawing groups onto either the DMAPP methyl groups or the L-Trp indole ring (C-7 position).39 In both cases the reaction was slowed considerably by electron-withdrawing substituents, consistent with an electrophilic addition mechanism involving the development of cationic character in both the prenyl and indole groups. The magnitude of the effects were somewhat lower than seen in non-enzymatic model reactions, suggesting that there is less development of positive charge in the transition states for the enzymatic reaction. These observations can be explained by either a dissociative or associative electrophilic aromatic substitution mechanism (Fig. 5).40 In the dissociative mechanism an initial ionization of DMAPP forms a dimethylallyl cation-pyrophosphate ion pair in an SN1-like fashion (Path A). The indole then attacks the allylic cation intermediate to form an arenium ion intermediate and a final deprotonation generates 4-dimethylallyltryptophan (4-DMAT). The associative mechanism invokes an SN2-like attack in which the indole directly displaces the pyrophosphate leaving group in a single step to form the arenium ion intermediate (Path B). In this case, a discrete dimethylallyl carbocation intermediate is not formed, but the displacement occurs via an “exploded” transition state with considerable carbocation character.41
Fig. 5 Dissociative (SN1, Path A) and associative (SN2, Path B) mechanisms for the reaction catalyzed by 4-DMATS. PP represents a diphosphate moiety.
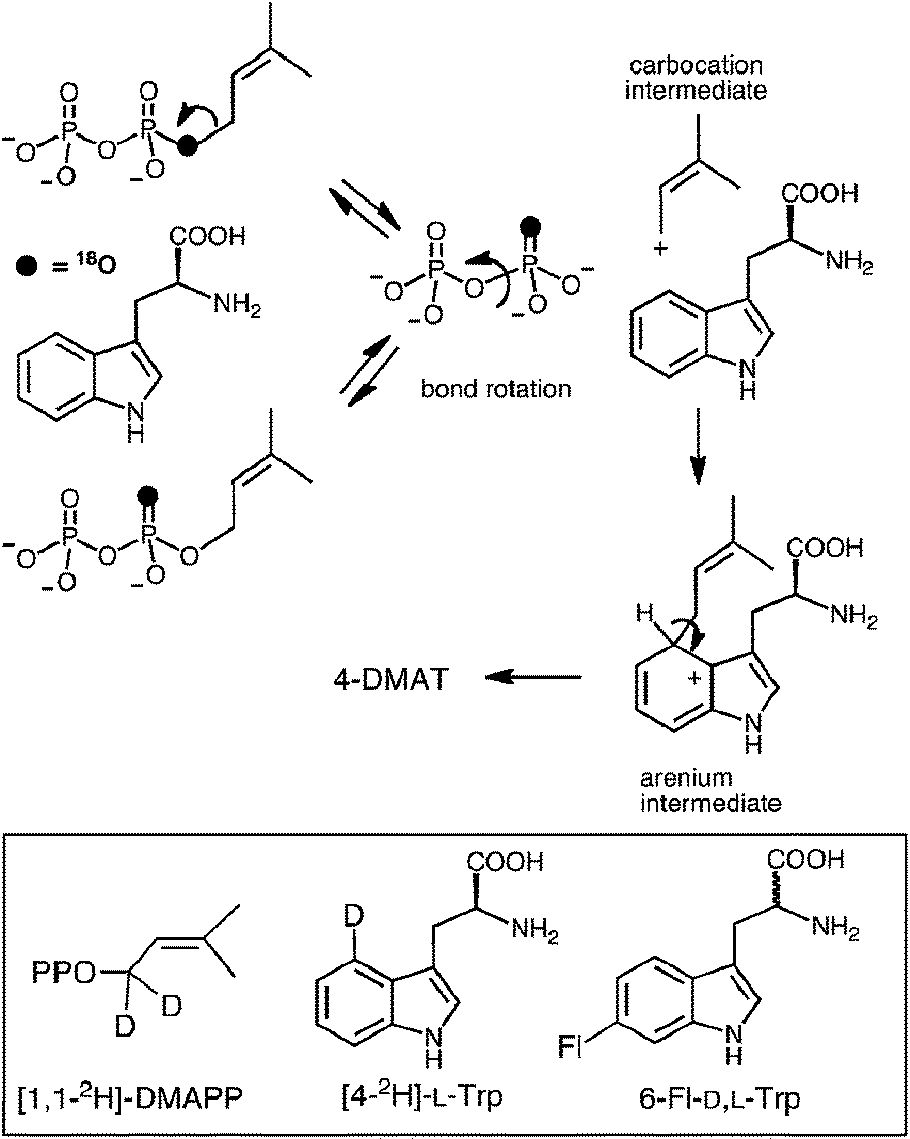
In 2007, 4-DMATS from Aspergillus fumigatus was overproduced in Escherichia coli, greatly simplifying further mechanistic and structural studies on this enzyme.42 Shortly thereafter, a study aimed at discerning between the dissociative and associative mechanistic possibilities was reported.40 A positional isotope exchange (PIX) experiment was used to probe for the existence of a carbocation intermediate in the 4-DMATS reaction. DMAPP was synthesized bearing an 18O-label in the position bridging the dimethylallyl and pyrophosphate groups (Fig. 6, see darkened atoms). The labeled compound was partially converted to product in an enzymatic reaction with 4-DMATS (57% completion) and the remaining starting material was analyzed for isotopic scrambling using 31P NMR spectroscopy. The observation that 15% of the recovered DMAPP contained 18O-label that had scrambled into a non-bridging position indicates that the C–O bond cleavage step is a reversible process. This could occur via a dissociative (SN1) mechanism in which the dimethylallyl carbocation could partition either forward to the arenium ion or back towards DMAPP. If the lifetime of the cation is long enough for P–O bond rotation to occur in the enzyme-bound pyrophosphate, isotopic scrambling would be observed in the pool of recovered starting material. Alternatively, the scrambling could be explained by a reversible associative (SN2) process. This would require a rate-determining step to exist subsequent to arenium ion formation. In order to distinguish between these possibilities, kinetic isotope effect (KIE) experiments were performed. When [1,1-2H]-DMAPP (see Fig. 6, inset) was used as a substrate, a normal secondary KIE of kH/kD = 1.16 was observed. This is consistent with a partially rate-limiting C–O bond cleavage step in which the hybridization at the C-1 carbon of DMAPP changes from sp3 to sp2. When [4-2H]-L-Trp was used as substrate, an inverse secondary isotope effect of kH/kD = 0.81 was observed. This is consistent with a partially rate limiting step involving attack of the indole C-4 onto the dimethylallyl carbocation and a change of hybridization from sp2 to sp3. The isotope effects rule out a rate-limiting step involving deprotonation of the arenium ion, and when taken together with the observation of PIX, strongly support a stepwise dissociative mechanism involving a discrete carbocation intermediate (Fig. 5, Path A).
Fig. 6 The positional isotope exchange (PIX) experiment with 4-DMAT synthase. Inset shows structures of isotopically-labeled and fluorinated substrates used in this study.
To further probe this notion, 6-fluorotryptophan (6F-D,L-Trp, Fig. 6 inset) was used in the PIX experiment.40 The electron withdrawing nature of the substituent should decrease the ability of the indole ring to compete with the pyrophosphate in nucleophilically attacking the carbocation intermediate, and the extent of PIX should increase. When 6F-D,L-Trp was tested as a substrate, no products were observed under standard incubation conditions. This could either mean that the initial ionization to form a carbocation can no longer take place with this alternate substrate, or that the partitioning of the carbocation lies strongly in favor of starting materials. The latter scenario proved to be the case as complete PIX was observed using this substrate with a 2:1 ratio of non-bridging:bridging isotopic label in the recovered pool of unreacted starting material. This provided further support for the existence of a dimethylallyl carbocation intermediate in the 4-DMATS reaction.
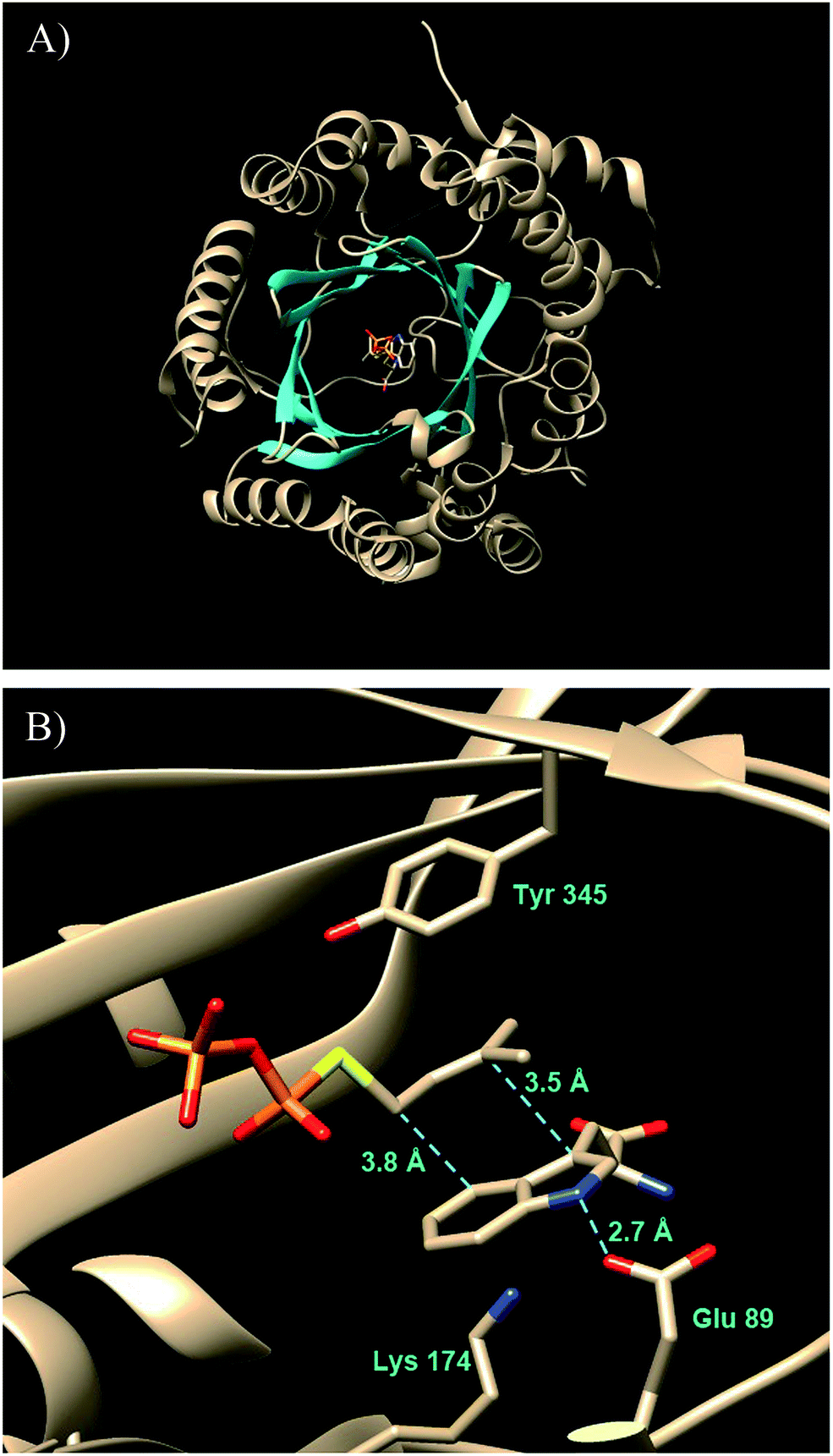
A significant advance in our understanding of the indole prenyltransferases came in 2009 went the first structure of a member of this family was solved.43 4-DMATS from A. fumigatus was crystallized in the presence of L-Trp and an unreactive analog of DMAPP (dimethylallyl S-thiolodiphosphate, DMSPP). The enzyme was found to adopt an unusual ABBA fold with a central barrel that is formed by 10 antiparallel β-strands surrounded by 10 α-helixes (Fig. 7a).27
Fig. 7 The structure of FtmPT1 in complex with L-Trp and DMSPP. (A) (upper panel) The ABBA fold with active site at the center. (B) (lower panel) The active site with key amino acid residues displayed.
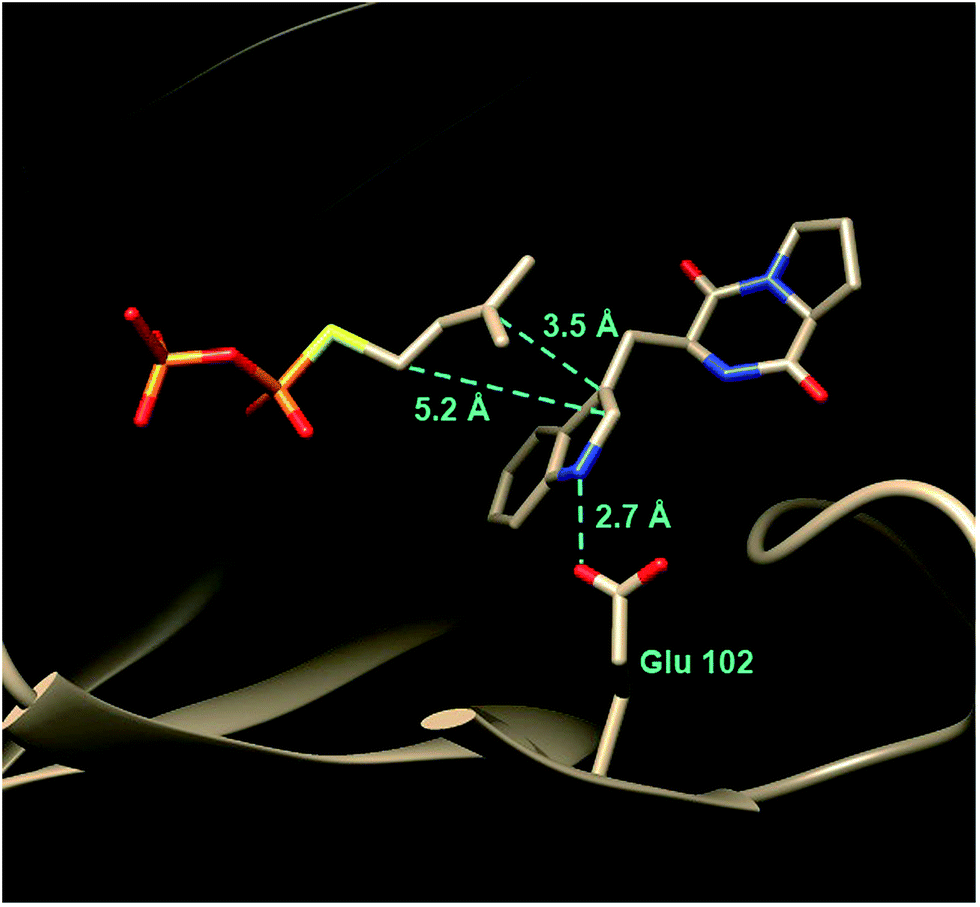
This fold was first seen in the prenyltransferase NphB that acts on a phenolic substrate, and has also been termed the prenyltransferase-fold (PT-fold).44 The active site is positioned in the center of the barrel and the absence of any metal ion in this location is consistent with the lack of requirement of divalent metals for activity. The presence of the bound substrate/analog provides a beautiful picture of the Michaelis complex and allows one to view the active site arrangement immediately prior to catalysis (Fig. 7b). The dimethylallyl group of DMAPP is coplanar with the indole ring of the L-Trp substrate, and the C-4 of the indole is located 3.8 Å away from the C-1 of DMAPP, as expected for the proposed mechanism (Fig. 5, Path A). It should be noted however, that the C-3 of the indole is located 3.5 Å away from the C-3 of DMAPP, as will be discussed later. Recognition of the diphosphate group is achieved by electrostatic interactions with three arginine residues and two lysine residues in addition to multiple H-bonds with active site tyrosine residues (not shown). The dimethylallyl group of DMAPP is sandwiched between the substrate indole ring and a phenolic ring of Tyr345. These interactions facilitate the initial ionization step by stabilizing the carbocation intermediate via π–cation interactions. Glu89 and Lys174 presumably act as key acid–base residues during catalysis. Glu89 is H-bonded to the NH of the indole and likely assists it in acting as a nucleophile via deprotonation or the formation of a charged H-bond (Fig. 8). Lys174 is positioned appropriately to carry out the final deprotonation step that rearomatizes the indole ring to give product.
Fig. 8 A direct alkylation mechanism for the 4-DMATS reaction showing the putative roles of Glu89 and Lys174 in catalysis.
The structure of 4-DMATS identified the active site residues and allowed for mutagenesis studies to determine their importance in catalysis.43,45 Mutation of the positively charged residues that interact with the diphosphate greatly impaired catalysis. Glu89, that forms an H-bond with the indole NH, was also crucial for catalysis and the Glu89Ala mutant was essentially devoid of activity. Lys174 was less important for catalysis; the value of kcat for the Lys174Gln mutant was decreased by 20% and that of Lys174Ala was decreased by 95%. From its position in the active site, it appears likely that Lys174 does play the role of a base in the final deprotonation step, but given the low pKa of the hydrogen being abstracted, other weak bases (such as a Gln) may compensate in mutant enzymes. PIX experiments with both Glu89 and Lys174 mutants showed that the extent of isotopic scrambling increased when compared to the wild type reaction run under similar conditions.45 This is consistent with the notion that these residues are required for catalytic steps that occur subsequent to the initial ionization event.
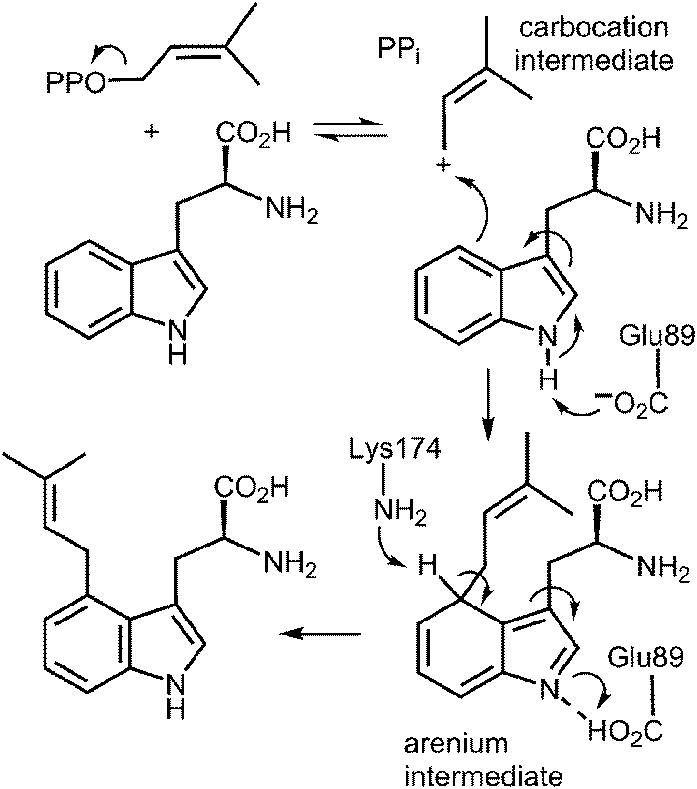
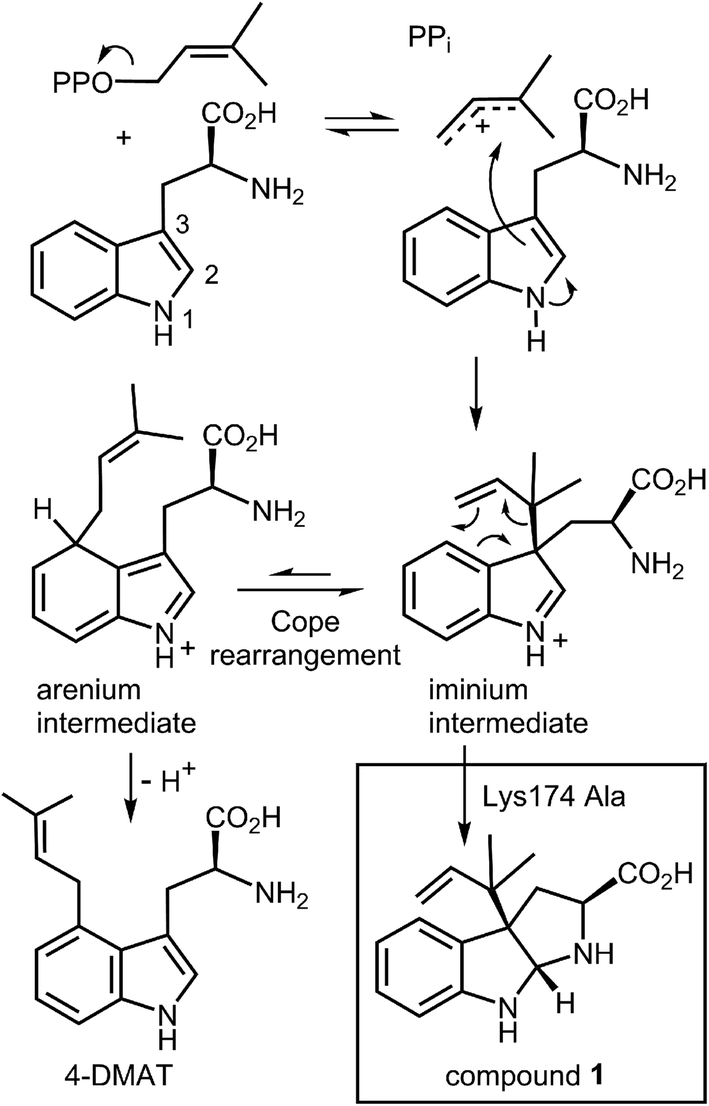
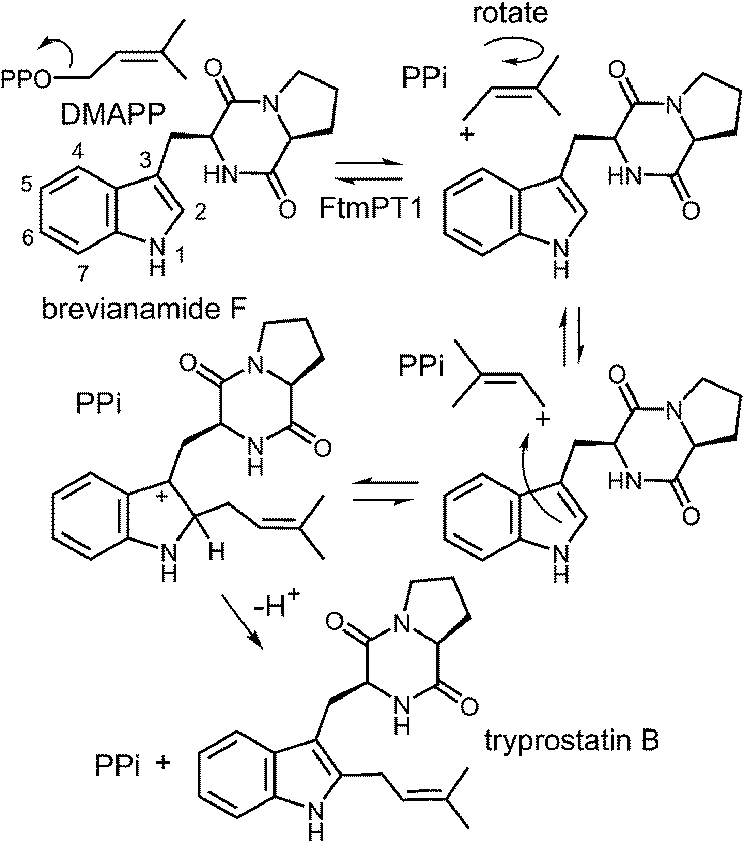
An interesting observation was made when a product analysis was performed on the reaction catalyzed by the Lys174Ala mutant.45 It was found that the major product (90%) was not 4-DMAT, but instead it was a reverse prenylated species. Ultimately it was possible to show that this compound was reverse prenylated at C-3 and a ring closure event had formed the hexahydropyrroloindole compound 1 (Fig. 9). This observation prompted the notion that 4-DMATS may employ a mechanism involving a Cope rearrangement as a key step in catalysis. This mechanism had previously been considered as a way to explain how prenylation is directed to the poorly nucleophilic position of the indole ring (C-4), but was eventually discounted due to lack of non-enzymatic precedence for the key rearrangement.46–48 This mechanism involves an initial reverse prenylation at the C-3 position of the indole (Fig. 9). This step is strongly supported by the structural data, as the C-3 of the indole is located 3.5 Å away from the C-3 of DMAPP.43 Furthermore, the C-3 position of the indole is much more nucleophilic than the C-4 position, and therefore one would expect such an attack to predominate. The C-3 reverse prenylated iminium intermediate would then undergo a reversible Cope rearrangement that would convert it into a C-4 normal prenylated arenium ion. The enzyme would catalyze this step largely by holding the prenyl group in a position that resembles the transition state for the rearrangement. This is quite reasonable as the crystal structure of 4-DMATS shows that the prenyl group has a coplanar relationship with the indole ring and is positioned such that its C-1 and C-3 carbons reside directly above the C-4 and C-3 carbons of the indole ring, respectively. In the final step of catalysis, Lys174 would deprotonate the arenium ion, driving the formation of 4-DMAT. It was argued that in the absence of Lys174, the more stable C-3 reverse prenylated intermediate would simply undergo a ring closure involving the α-amino group of L-Trp to give the observed product of the mutant reaction. The Cope mechanism is also consistent with the observed KIE's if one assumes that the ionization step ([1,1-2H]-DMAPP gives a normal secondary KIE) and the Cope rearrangement step ([4-2H]-L-Trp gives an inverse secondary KIE) are both partially rate-limiting during catalysis.
Fig. 9 A Cope rearrangement mechanism for the reaction catalyzed by 4-DMAT synthase. Inset shows the formation of compound 1 with the Lys174Ala mutant.
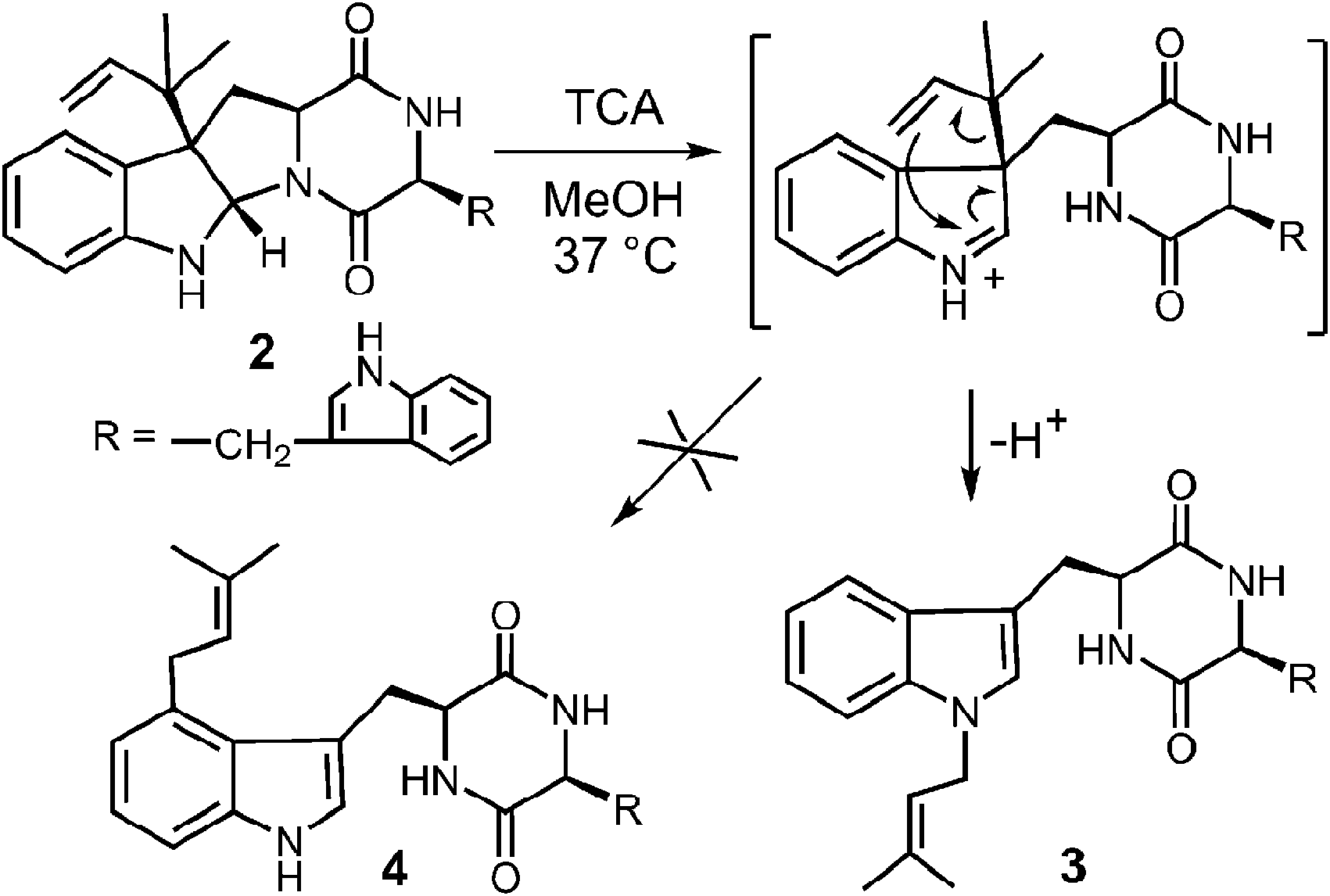
While it is quite difficult to experimentally distinguish between a direct electrophilic aromatic substitution mechanism (Fig. 5, Path A) and a Cope mechanism (Fig. 9), a key question remains as to the intrinsic barrier for the rearrangement in the latter case. Enzymes that catalyze pericyclic reactions often do so largely by conformational control as there is no extensive charge development in the transition state.49 Therefore, the observed rate accelerations are often not large and the non-enzymatic reaction often proceeds at measurable rates. The observation of a non-enzymatic Cope rearrangement on a C-3 reverse prenylated indole would therefore provide strong support for the latter mechanism. To investigate this possibility, the reverse C-3 prenylated compound 2 was incubated in the methanol containing 100 mM trichloroacetic acid at 37 °C (Fig. 10).31,50 The major product formed in this reaction was found to be the normal N-1 prenylated compound 3, and no trace of the normal C-4 prenylated product 4 could be detected. Presumably, the acid facilitated ring opening and the formation of an iminium ion, and an aza-Cope rearrangement occurred to give compound 3.51 This demonstrates that a Cope rearrangement is feasible at ambient temperatures in these systems, but that in the absence of enzyme, the preference is for the prenyl group to migrate to N-1 instead of C-4.
Fig. 10 The non-enzymatic aza-Cope rearrangement of compound 2.
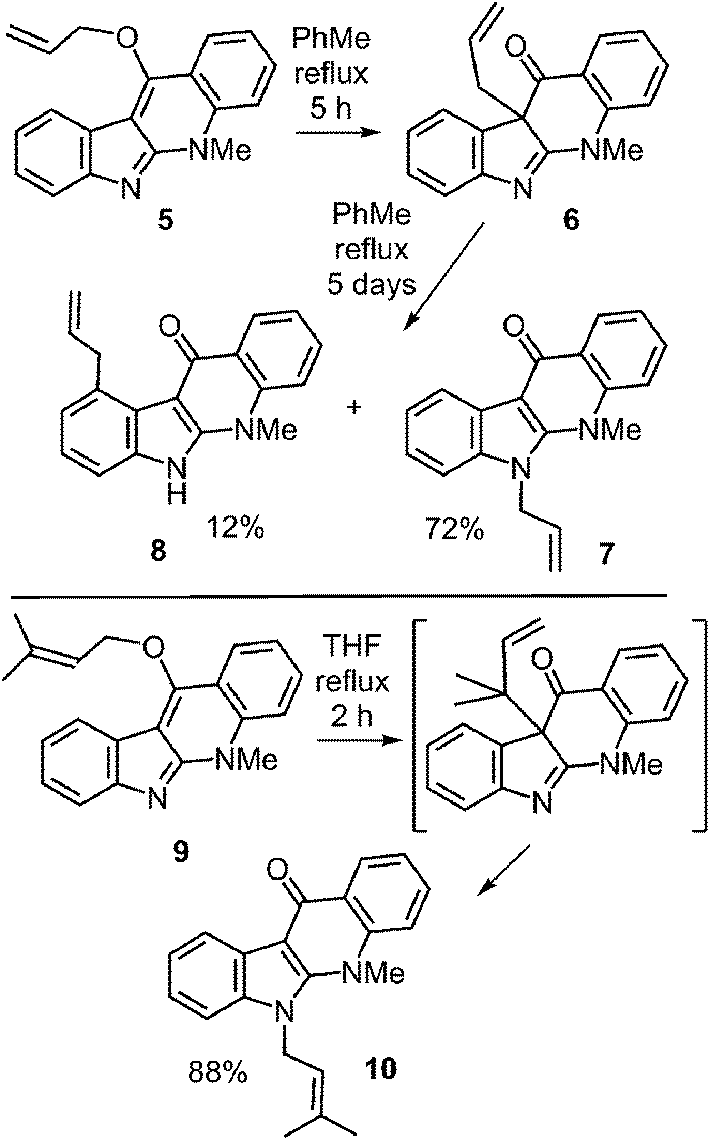
Literature precedent for competing N-1 and C-4 migrations can be found in a somewhat similar system (Fig. 11).52 When compound 5 was refluxed in toluene it was gradually converted into compound 6via a Claisen rearrangement. Continued refluxing produced a mixture of the N-1 prenylated compound 7 (72%, aza-Cope product) and the C-4 prenylated compound 8 (12%, Cope product). This demonstrates that the two rearrangements do not have vastly different energy barriers, and that an enzyme may be able to dictate the regioselectivity via conformational control. This paper also demonstrated that the migration of a dimethylallyl group was markedly faster than that of an allyl group due to the gem-dimethyl effect.53 Compound 9 was converted in the aza-Cope product 10 after two hours of reflux in THF.
Fig. 11 Non-enzymatic Cope and aza-Cope rearrangements of C-3 reverse prenylated indole derivatives.
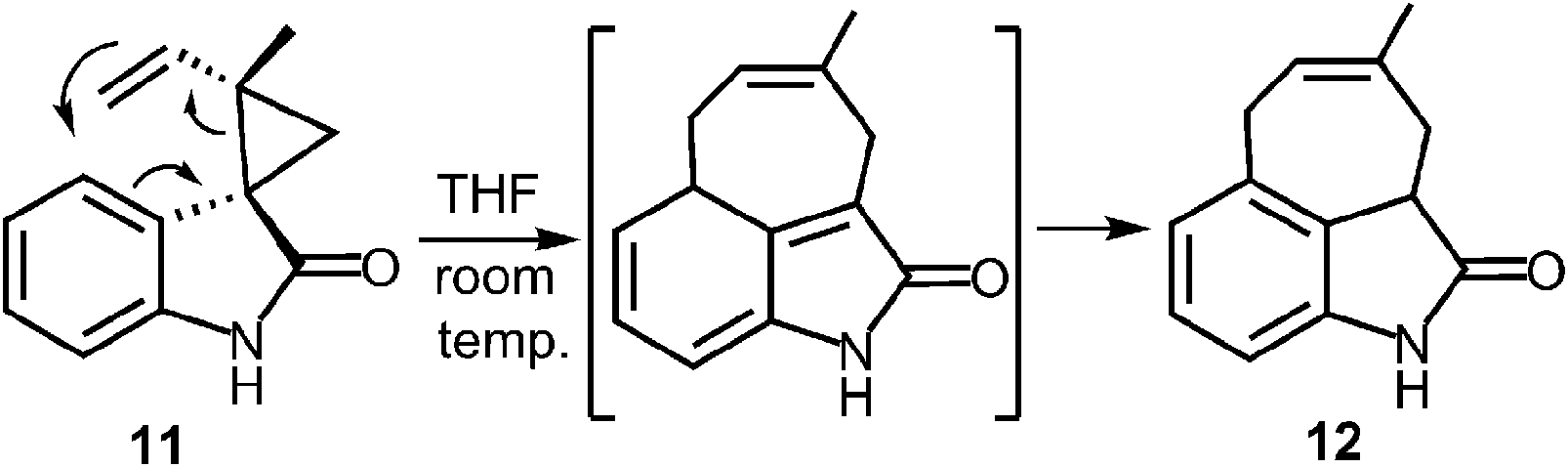
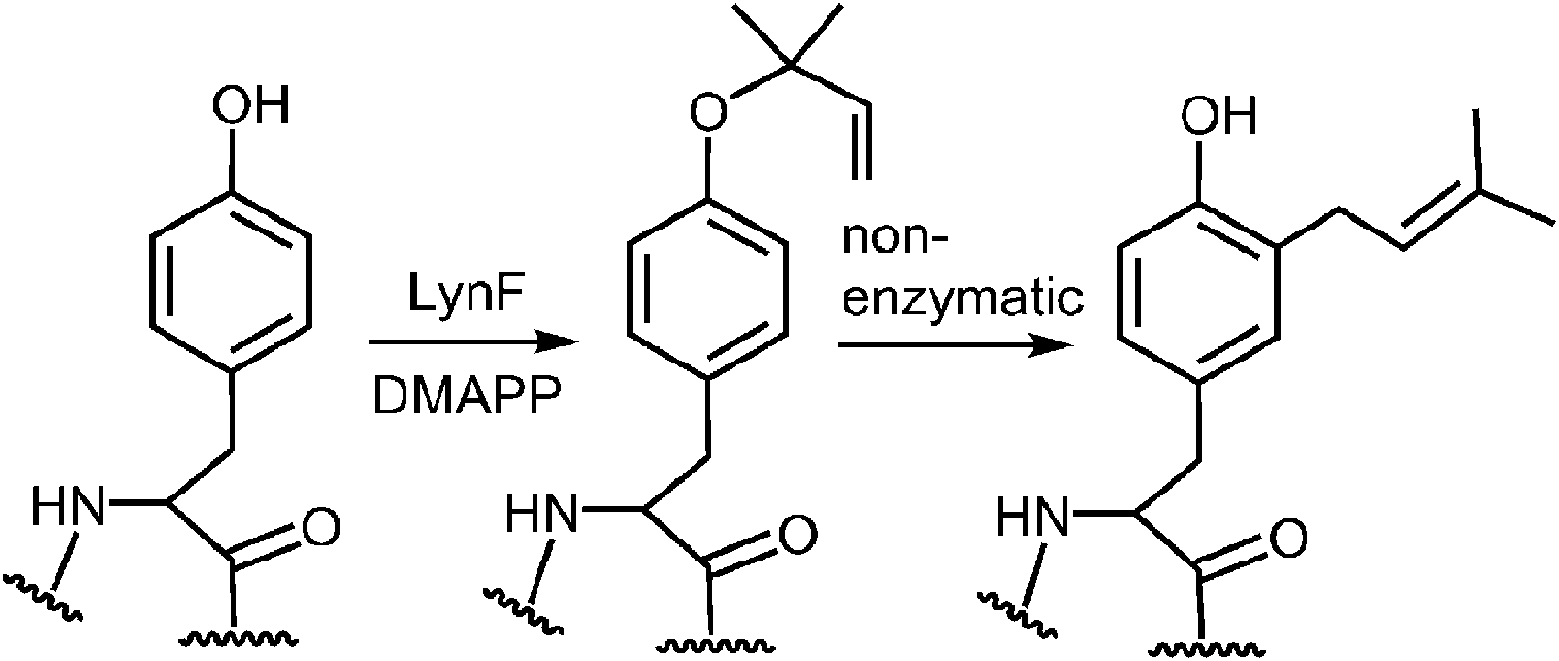
A more recent study has described a Cope rearrangement that produces a C-4 prenylated indole at room temperature (Fig. 12).54 Compound 11 was shown to convert into compound 12 after 10 hours of incubation in THF. Of course, this rearrangement is accelerated by the cleavage of a strained cyclopropane ring, so it may not represent a truly accurate model of the proposed 4-DMATS mechanism. While exact precedence for the Cope rearrangement remains to be found, it should be noted that non-enzymatic oxy-Cope and aza-Cope rearrangements of allyl phenols and anilines are well known, and sometimes occur under mild conditions.31,55,56 Of particular relevance to this study is the action of the enzyme LynF that is responsible for the normal ortho-prenylation of a tyrosine phenol (Fig. 13). It has recently been shown that the enzyme actually catalyzes the formation of the reverse O-prenylated phenol, and that a non-enzymatic oxy-Cope rearrangement gradually converts it into product.57 This scenario has striking similarities to the proposed Cope rearrangement for DMATS catalysis.
Fig. 12 A non-enzymatic Cope rearrangement as a mimic of the proposed 4-DMATS mechanism.
Fig. 13 Reverse prenylation of tyrosine by LynF followed by a non-enzymatic Claisen rearrangement.
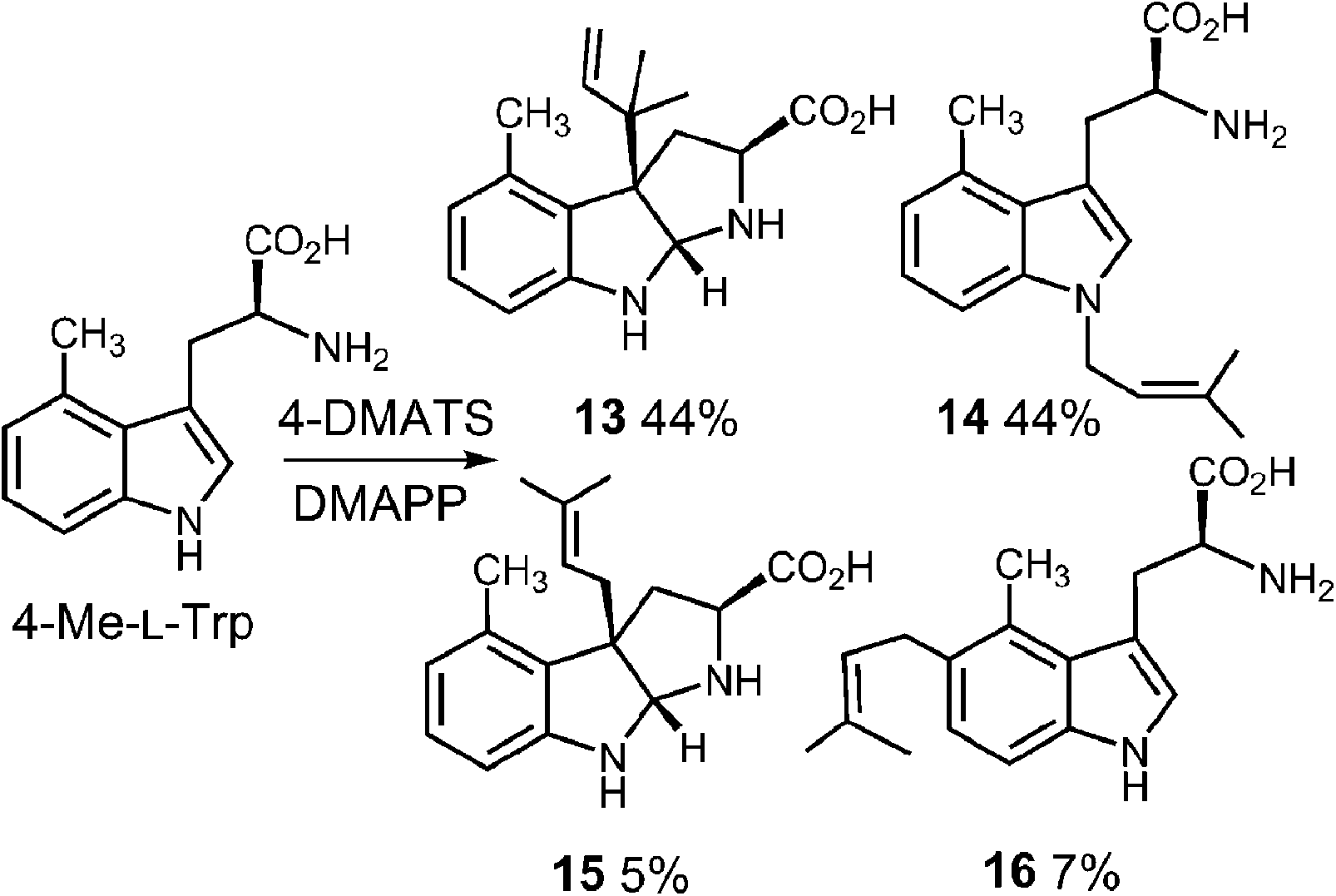
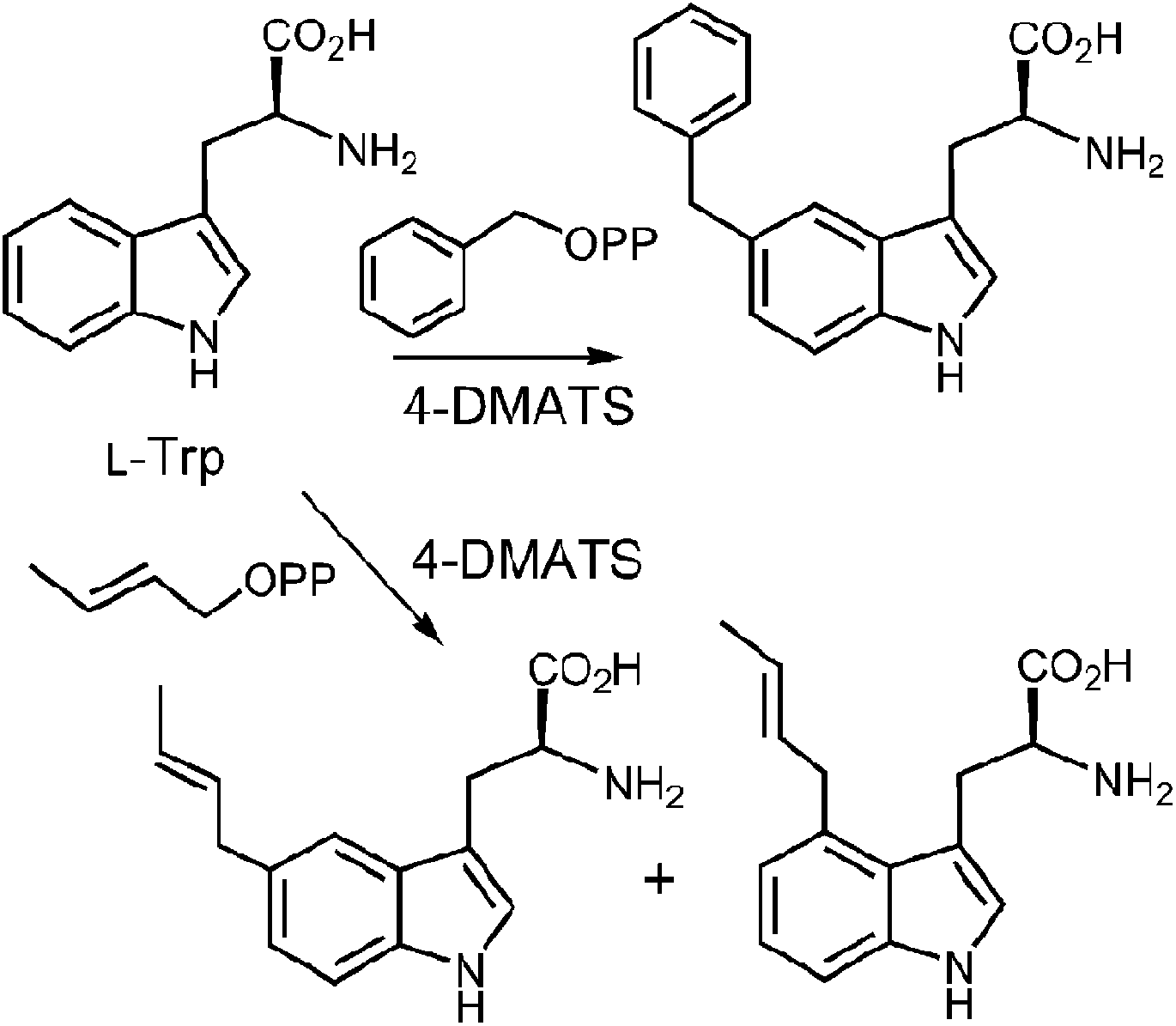
In a very recent study, 4-substituted tryptophans were tested as substrates with 4-DMATS (Fig. 14).32 These compounds have the normal site of substitution blocked yet were found to serve as slowly reacting alternate substrates that gave mixtures of products. In the case of 4-methyltryptophan, the major products were the reverse C-3 prenylated compound 13 (44%) and the N-1 normal prenylated compound 14 (44%). Minor products included the normal C-3 prenylated compound 15 (5%) and the normal C-5 prenylated compound 16 (7%, assignment tentative). 4-Methoxytryptophan and 4-aminotryptophan also gave mixtures of products with the major species being normal prenylated at positions ortho or para to the substituent (C-5 or C-7, not shown). The authors suggest that the observation of multisite prenylation favors a simple electrophilic addition mechanism for this family of enzymes, and that a Cope rearrangement does not need to be invoked for reaction at weakly nucleophilic positions. Instead, the enzyme very specifically localizes the highly reactive carbocation to the desired site of substitution. Any modifications made to the substrate (or active site residues), could lead to loss of control of the cation and a variety of alternate products. Similar unexpected prenylation products have been observed when DMAPP analogs are used in the 4-DMATS reaction (Fig. 15).58,59 With benzyl diphosphate, benzylation occurs primarily at C-5, and with E-2-butenyl diphosphate, alkylation occurs at both C-4 and C-5. Thus it is clear that a direct addition mechanism is possible at indole ring positions of lower nucleophilicity.
Fig. 14 Multisite prenylation of 4-methyltryptophan by 4-DMATS.
Fig. 15 Alkylation at C-4 and C-5 with unnatural diphosphates.
In summary, direct alkylation is the simpler of the two mechanisms proposed for the 4-DMATS reaction and is chemically reasonable. Nevertheless, the structural analysis of the pseudo-Michaelis complex, combined with the intrinsic reactivity of the C-3 position, provide compelling evidence to consider the Cope rearrangement mechanism for this enzyme.
3 C-2 prenylation: normal and reverse
Tryprostatin B synthase, or FtmPT1, catalyzes the normal C-2 prenylation of the cyclic dipeptide, brevianamide F (cyclo-L-Trp-L-Pro) to give tryprostatin B (Fig. 16).13 Tryprostatin B is a cytotoxic agent that causes cell cycle arrest in the G2/M phase and is also a precursor to the fumitremorgin-type alkaloids.60,61 FtmPT1 serves as an interesting test case for understanding the mechanisms of the indole prenyltransferases as it is the second example to have been structurally characterized.
Fig. 16 A direct alkylation mechanism for the reaction catalyzed by FtmPT1.
In 2010, the structure of FtmPT1 was solved in complex with the unreactive DMAPP analog, DMSPP, and brevianamide F (Fig. 17).62 This structure was remarkably similar to that of 4-DMATS both in overall fold and in active site architecture. The binding site for DMAPP is well conserved and the larger uncharged peptide substrate is accommodated into the active site via a replacement of threonine with a glycine (Gly115) and an arginine with a histidine (His279). A glutamate residue (E102) is H-bonded to the indole NH and mutation of this residue to a glutamine abolished activity. The relative orientation of the prenyl group and the indole ring is remarkably similar to that observed in 4-DMATS, although the plane of the indole is shifted by 1 Å and tilted by 18°. This similarity is quite surprising as FtmPT1 must prenylate the indole at C-2, and this carbon is 5.2 Å away from the C-1 of the DMAPP analog. Instead, the structure seems to suggest a C-3 reverse prenylation would be preferred as the indole C-3 position is located 3.5 Å away from the C-3 position of the DMAPP analog. To explain this unexpected positioning, the authors invoked a mechanism involving a rotation of the free carbocation subsequent to ionization (Fig. 16).62 This reorientation would position the primary carbon of the allylic carbocation in the vicinity of the indole C-2 so that proper alkylation could occur. The key residue E102 serves to activate the indole for nucleophilic attack, and may also act as a base in the final deprotonation step.
Fig. 17 The active site structure of FtmPT1 complexed with DMSPP and brevianamide F.
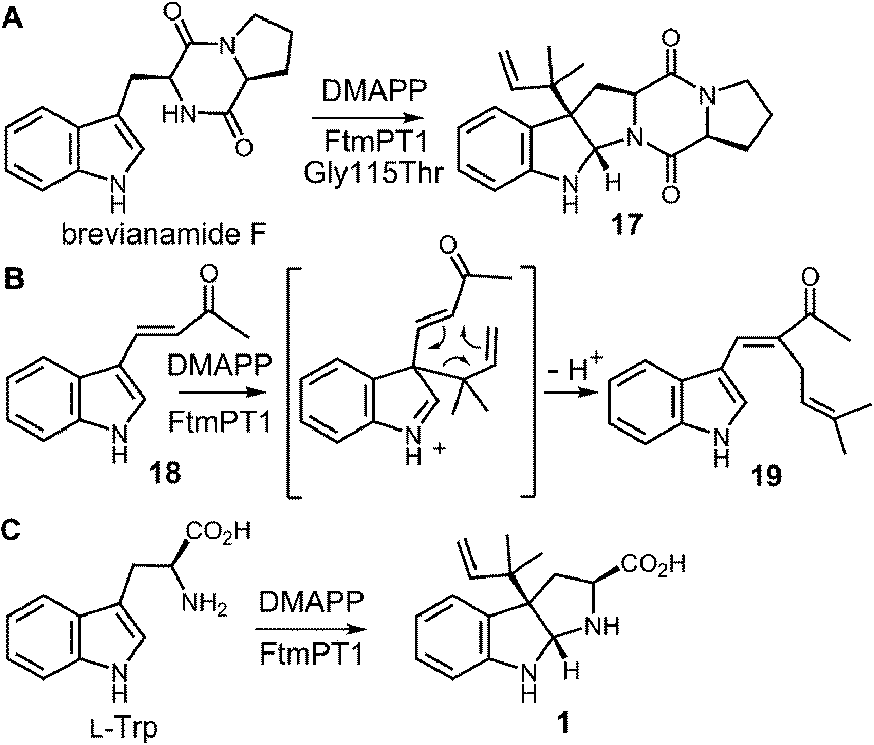
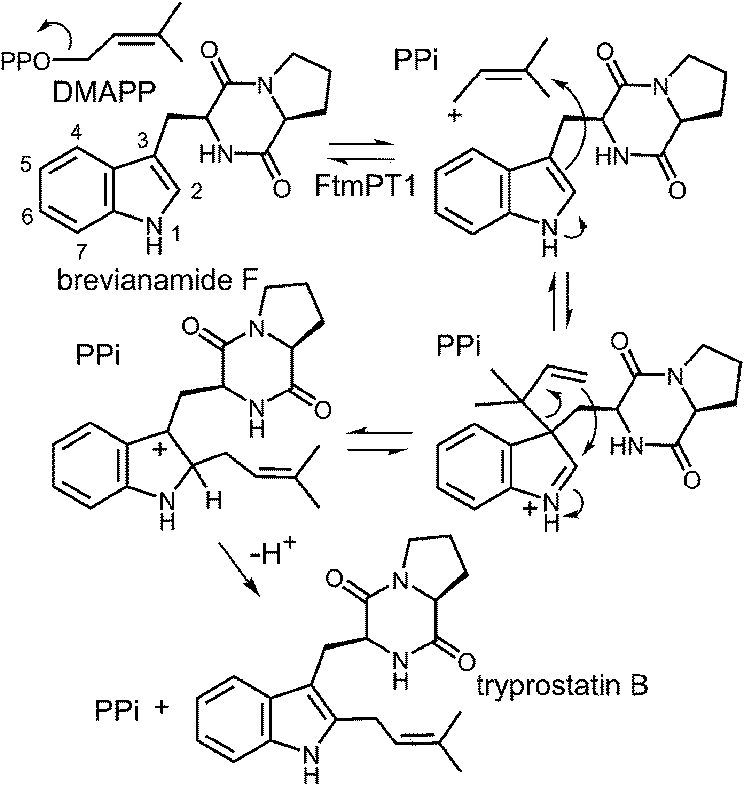
While the indole C-2 position is fairly nucleophile and direct alkylation at this position is chemically reasonable, the structure of the pseudo-Michaelis complex does not position the DMAPP appropriately for such an attack. Apart from the structural analysis, several observations have led to the speculation that a C-3 prenylation occurs first, and a rearrangement subsequently produces tryprostatin B.31,63 The first such observation, involved the mutation of Gly115 into a threonine in an attempt to revert the active site structure closer to that of 4-DMATS.62 Surprisingly, the Gly115Thr mutant produced the reverse C-3 prenylated species 17 (Fig. 18a), which has an analogous structure to that of compound 1 formed by the 4-DMATS Lys174Ala mutant (Fig. 9). Either the mutation prevents the proposed rotation of the cation from occurring and C-3 attack intercepts, or the enzyme normally catalyzes reverse C-3 prenylation as the first step of catalysis and compound 17 represents an intermediate that has been released into solution and cyclized. A second observation involved the use of indolylbutenone 18 as an alternate substrate in the FtmPt1 reaction (Fig. 18b).64 Surprisingly, the observed product 19 was found to be normal prenylated at a non-aromatic alkene carbon. While this could conceivably occur by direct prenylation at this remote position, it is perhaps more likely that a reverse C-3 prenylation occurs, and a Cope rearrangement migrates the prenyl group out onto the butenone side chain (shown in Fig. 18b). Finally, when free L-tryptophan is used as a substrate in the FtmPT1 reaction, the product is the same reverse C-3 prenylated compound 1 that is formed by the 4-DMATS Lys174Ala mutant (Fig. 18c).31 All three of these observations, combined with the structural analysis, suggest a mechanism may be operative in which a reverse C-3 prenylation is the first step in catalysis (Fig. 19). The resulting intermediate must then undergo a rearrangement to produce a C-2 normal prenylated arenium ion. This rearrangement is formally a [3,5]-sigmatropic shift and is therefore symmetry forbidden to occur as a concerted process.65,66 Instead it would likely proceed in a stepwise fashion via a secondary carbocation (not shown).
Fig. 18 Products formed from FtmPT1 that indicate a reverse C-3 prenylation is possible.
Fig. 19 A reverse C-3 prenylation mechanism for the reaction catalyzed by FtmPT1.
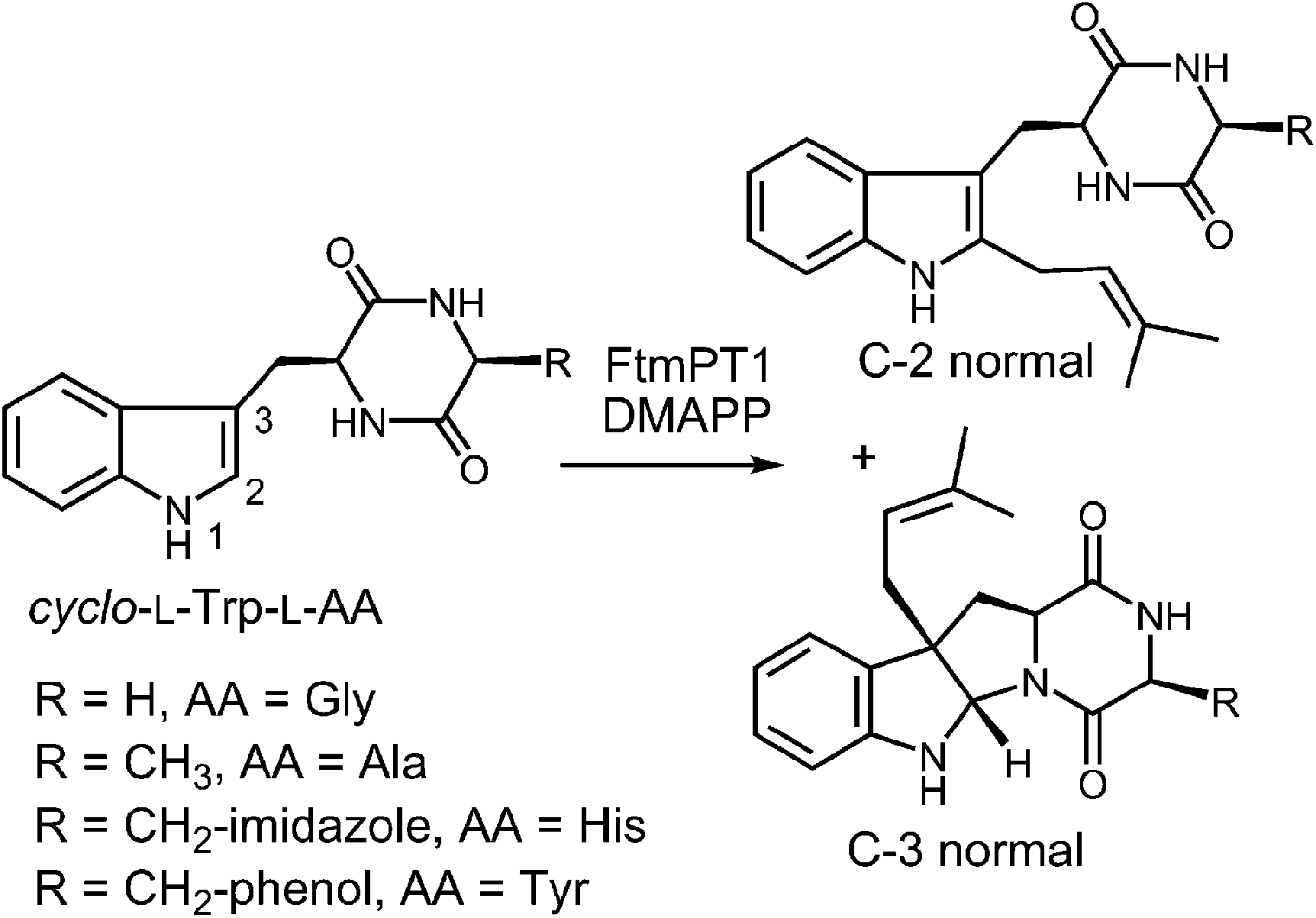
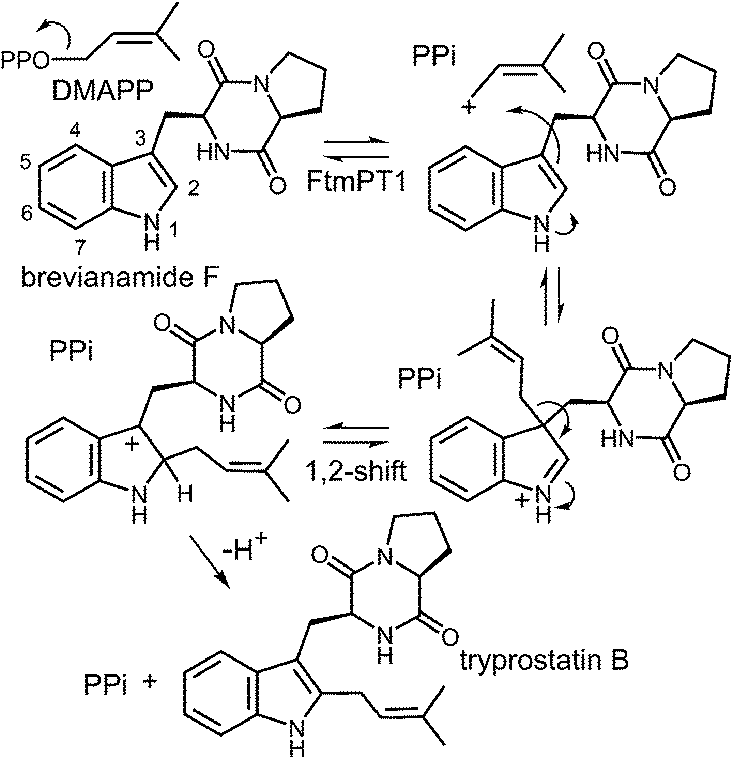
Another series of studies utilized a series of cyclic dipeptides in which the L-Pro residue of brevianamide F is exchanged for other amino acids such as Gly, L-Ala, L-Tyr and L-His (Fig. 20).33 These compounds were accepted as alternate substrates for FtmPT1 and primarily gave products that were normal C-2 prenylated, as expected. However, in each case, a side product was produced that was normal C-3 prenylated and the amount of this species could account for as much as 30% of the product formed. The authors speculate that the structural changes imparted by the replacement of L-Pro with a different amino acid relax the enzyme's control over the carbocation and allow both C-2 and C-3 attacks to occur. An alternate explanation is that the enzyme may normally utilize a mechanism involving a C-3 normal prenylation as the first step of catalysis and these side products represent released reaction intermediates that cyclize in solution (Fig. 21). In this scenario, the C-3 normal prenylated intermediate would have to undergo a 1,2-alkyl shift to generate the C-2 prenylated intermediate and a final deprotonation would give product. Such 1,2-alkyl shifts are well known in indole chemistry and have been called Plancher rearrangements.67–69 It is notable that this exact rearrangement has been used in the chemical synthesis of tryprostatin B.70
Fig. 20 Products formed from FtmPT1 and a series of cyclic dipeptides.
Fig. 21 A normal C-3 prenylation mechanism for the reaction catalyzed by FtmPT1.
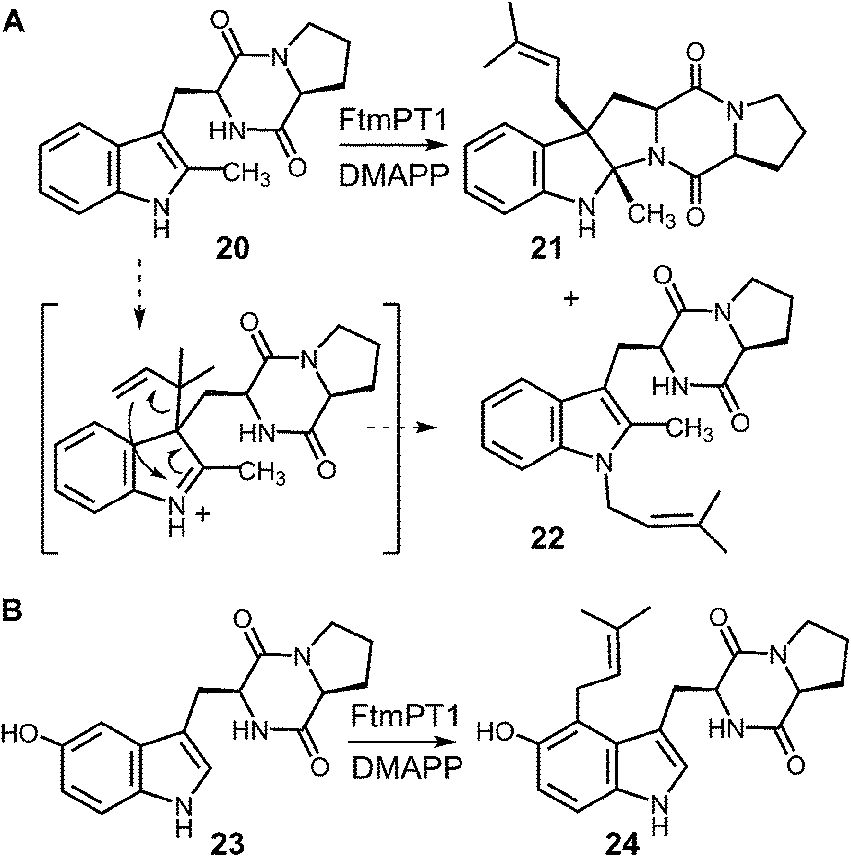
In a recent study, an attempt was made to discern between these potential mechanisms using 2-methyl-brevianamide F 20 (Fig. 22a).31 This compound retains both the L-Trp and the L-Pro, but the normal site of attachment is blocked. If either C-3 reverse or C-3 normal prenylation was the first step of catalysis, one might expect the corresponding products to predominate from a reaction with compound 20. Incubation of compound 20 with DMAPP and FtmPT1 lead to the formation of two products, 21 and 22. Compound 21 was the major product and was normal C-3 prenylated, consistent with the mechanism shown in Fig. 21. Surprisingly, the minor product 22 was found to be N-1 normal prenylated. While this could be due to a direct addition to this position, the indole nitrogen is very poorly nucleophilic when compared to either C-2 or C-3 positions. Alternatively, this product could have arisen from a reverse C-3 prenylation followed by an aza-Cope rearrangement (shown in Fig. 22a). Such a rearrangement is feasible as discussed previously (Fig. 10). Thus, the observed products could be formed via either an initial reverse, or normal, C-3 prenylation. In this study, 5-hydroxy-brevianamide F 23 was also tested as a substrate in order to probe whether the structural analysis of FtmPT1 accurately reflected the orientation of the reactants following the ionization step (Fig. 22b). Despite the fact that the C-2 position is available for substitution in this compound, only the normal C-4 prenylated product 24 was formed. This could occur via a direct attack from C-4 onto the dimethylallyl cation as this position is highly activated due to the presence of the hydroxyl functionality. Alternatively, it could be formed by a reverse C-3 prenylation, followed by a Cope rearrangement. The C-5 hydroxyl group should accelerate such a rearrangement as it is conjugated to the π-system of the resulting intermediate. In either case, the exclusive formation of compound 24 indicates that the relative geometry observed in the structure of the FtmPT1 pseudo-Michaelis complex is relevant to catalysis.
Fig. 22 Reactions of 2-methyl- and 6-hydroxybrevianamide F with FtmPT1.
In summary, the direct C-2 alkylation mechanism remains a viable possibility for the FtmPT1 reaction. However, the structural analysis indicates that the relative orientation of the substrates is incorrect for such an attack and demands that a reorientation of the dimethylallyl cation occur prior to alkylation. The observation of C-3 prenylated products with a variety of alternate substrates or mutant enzyme lends support to alternate mechanisms involving C-3 prenylation (either normal or reverse) as the first step of catalysis. However, it is difficult to make solid conclusions from these studies as it has become clear that structural modifications to the active site or substrate can lead to a variety of unexpected products in certain cases.
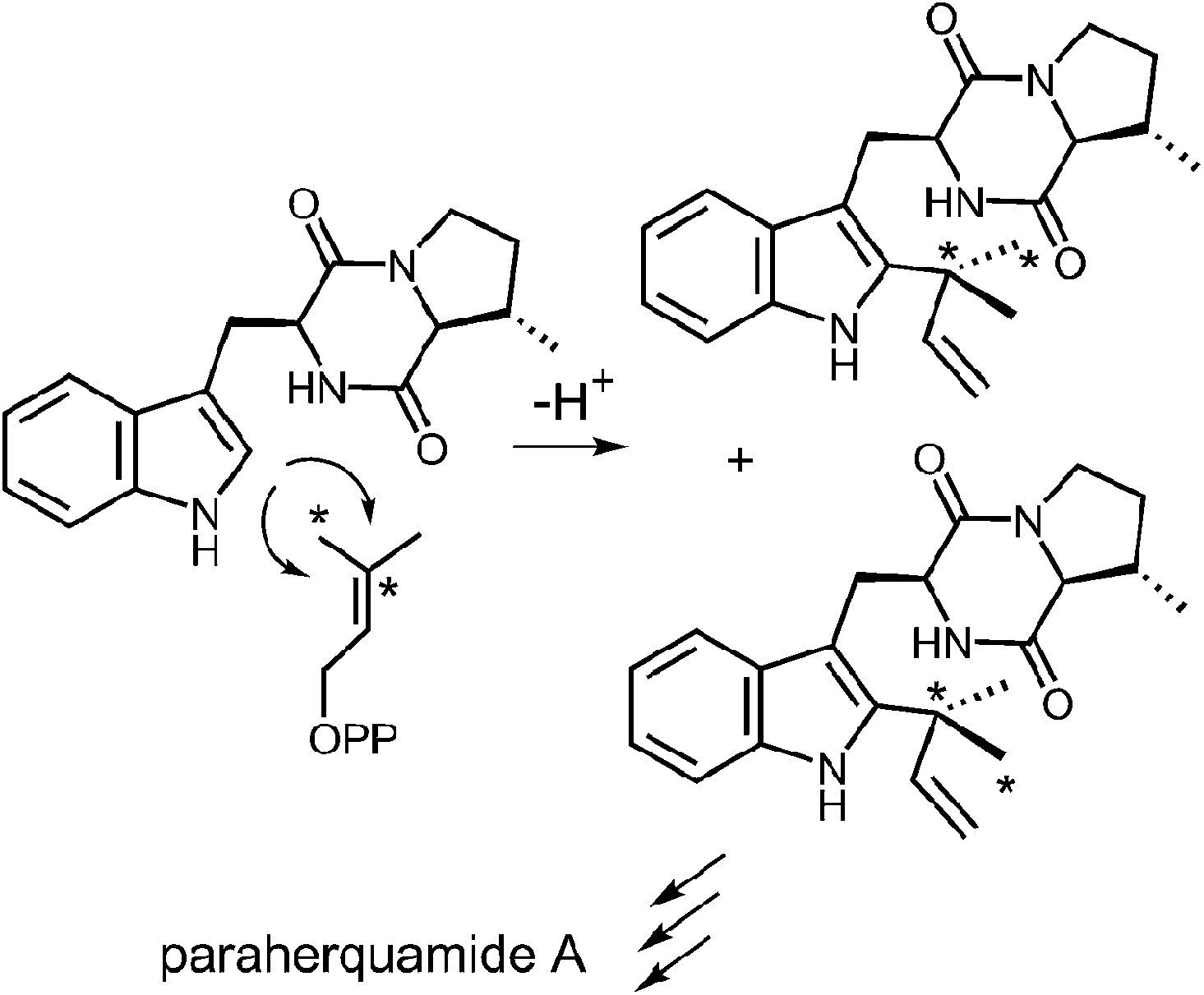
Reverse C-2 prenylation also occurs in the biosynthesis of several indole alkaloids such as the notoamides, the paraherquamides, the malbrancheamides, and fumigaclavine C. In vitro activity has been demonstrated in some cases, however, mechanistic studies with purified enzymes are largely absent.11,12 One notable in vivo study involved a feeding experiment in which [13C2]-labeled acetate was used in the biosynthesis of paraherquamide A, brevianamide A, and austamide.71,72 In each case, the resulting labeling pattern in the natural products indicated that the methyl groups of DMAPP became equivalent during the prenylation process (Fig. 23). The authors conclude that the prenyltransferase exhibits poor facial selectivity and may present either face of the dimethylallyl moiety to the indole ring. This physical basis behind this unexpected lack of selectivity presents an interesting study for further in vitro experiments.
Fig. 23 A lapse in facial selectivity during reverse C-2 prenylation.
4 Reverse C-3 prenyltransferases (CdpNPT, AnaPT, and CdpC3PT)
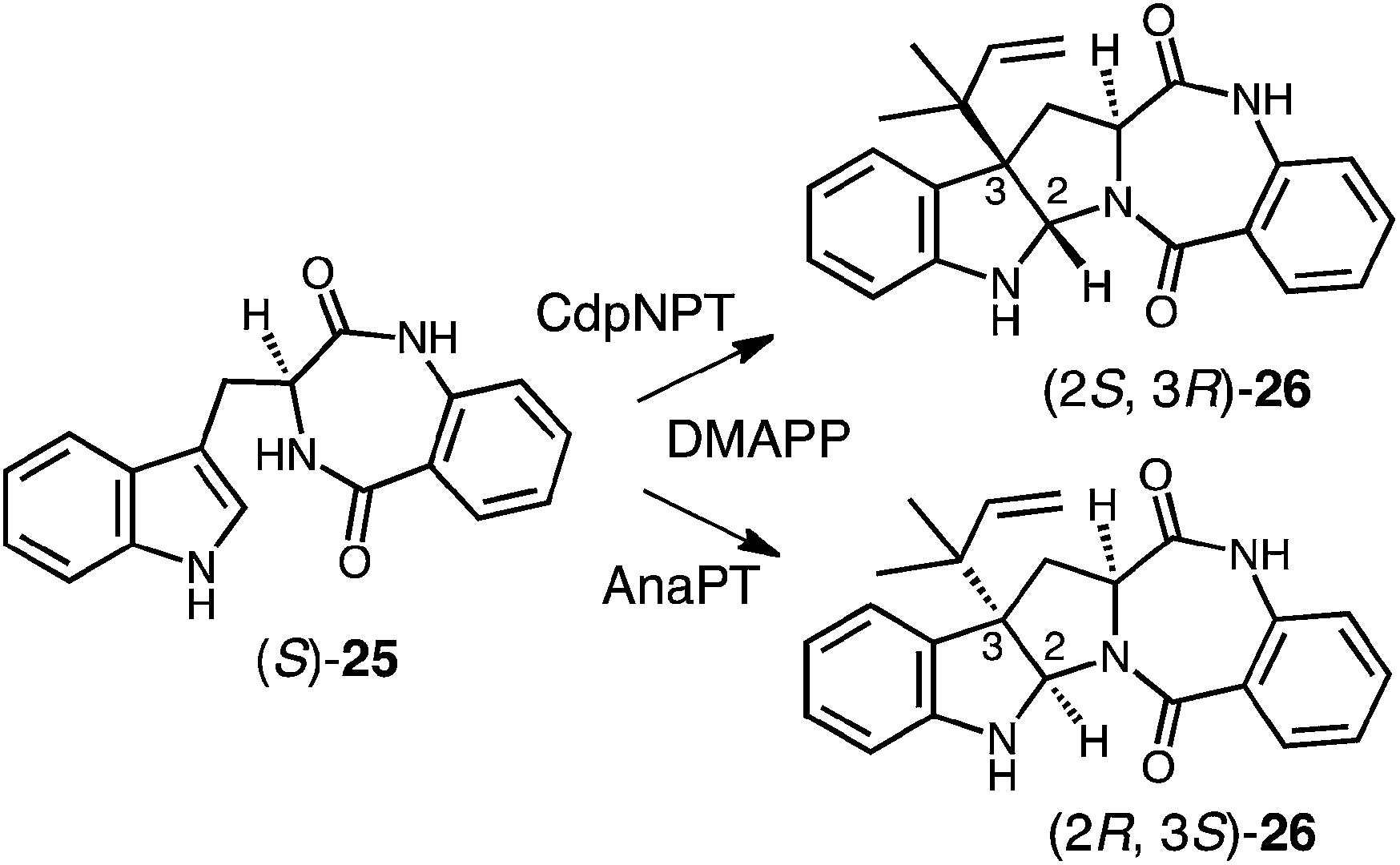
Genomic mining of indole prenyltransferase genes from fungi led to the discovery of three new enzymes that catalyze a reverse C-3 prenylation on cyclic dipeptide/benzodiazepinedione substrates.14–16,73 The best understood of these is CdpNPT whose exact substrate is unknown and which has an extremely broad substrate specificity.14 With (S)-benzodiazepinedione 25 it generates exclusively the (2S, 3R) product 26 (Fig. 24). A structure of CdpNPT has been solved in complex with both (S)-25 and thiolodiphosphate (SPP, the hydrolyzed version of DMSPP). The missing prenyl moiety was modeled into the active site using the structure of FtmPT1 as a guide. The resulting active site had a very similar geometry to those of 4-DMATS and FtmPT1 (not shown). The DMAPP binding site was highly conserved and the indole substrate binding pocket was enlarged somewhat to accept the larger substrate. The C-3 position of the indole appears to sit directly adjacent to the C-3 position of DMAPP as expected for a direct reverse C-3 prenylation mechanism. In addition, the conserved glutamate residue E116 forms an H-bond with the indole NH, and is crucial for catalysis. CdpNPT also accepts the (R)-enantiomer of 25 as well as a variety of cyclic dipeptides as substrates.73,74 A curious observation is that the stereoselectivity of prenyl addition lapses as the non-indole-containing amino acid decreases in size and rigidity. For example, with cyclo-L-Trp-L-Ala, a mixture of the (2S, 3R) and (2R, 3S) products are obtained. This is a surprising observation as it means that opposite faces of the indole ring must have access to the dimethylallyl cation. This would require a rotation within the active site that would presumably disrupt the interaction with the key glutamate residue E116.
Fig. 24 Reverse C-3 prenylations catalyzed by CdpNPT and AnaPT.
Two other enzymes that catalyze C-3 reverse prenylation are known and also accept a wide variety of substrates. AnaPT will prenylate (S)-benzodiazepinedione 25 to give product 26 with the alternate (2R, 3S) configuration (Fig. 24).73,74 Unlike CdpNPT, this enzyme is stereoselective for a wide variety of cyclic dipeptide substrates and the stereochemistry of the products is dictated by the stereochemistry of the tryptophan. With L-Trp-containing substrates the (2R, 3S) products are formed, and with D-Trp-containing substrates the (2S, 3R) products are formed. A structure of AnaPT is available but it only contains SPP bound in its active site and thus the relative orientation the prenyl and indole moieties is not known.73 Finally, a third member, CdpC3PT, shows high stereoselectively but in an opposite sense from AnaPT.15,73 With L-Trp-containing substrates the (2S, 3R) products are formed, and with D-Trp-containing substrates the (2R, 3S) products are formed. The mechanistic basis behind this fascinating stereochemical outcome is not yet known and structures of ternary complexes will help to elucidate its origins.
5 Reverse N-1 prenyltransferase (CymD)
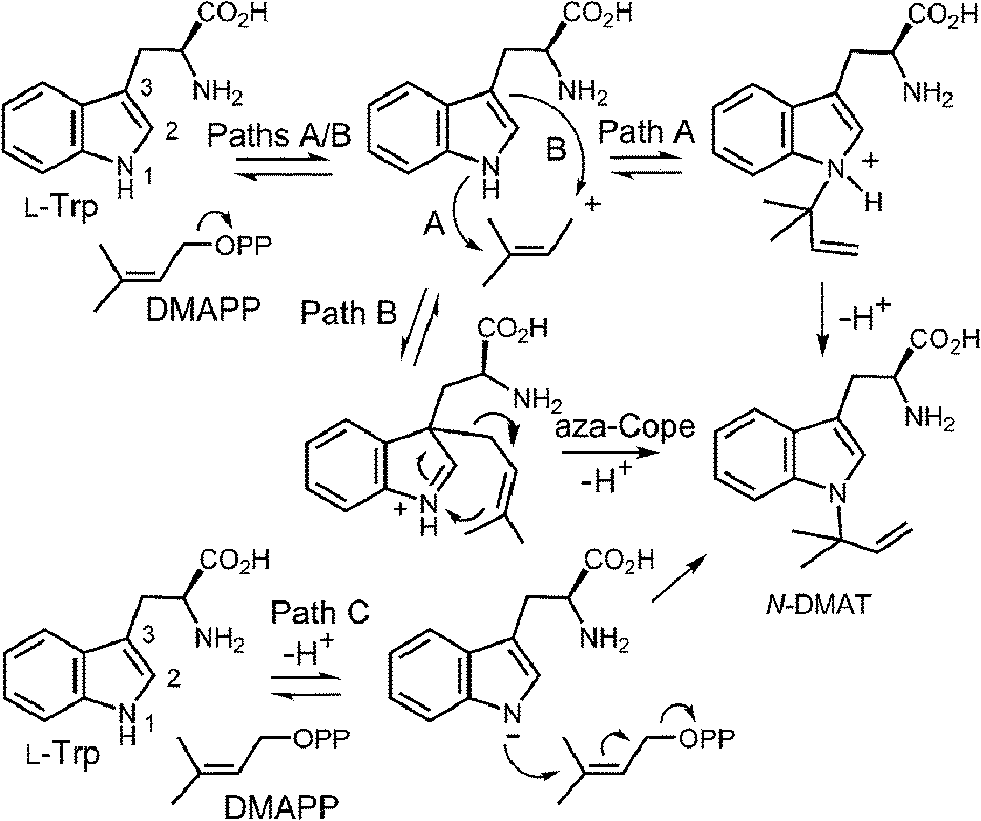
CymD is an unusual indole prenyltransferase as it catalyzes the reverse prenylation of tryptophan at the N-1 position to produce N-dimethylallyltryptophan (N-DMAT) (Fig. 25).10,75,76 This is the first step in the biosynthesis of the cyclic peptides, cyclomarin and cyclomarazine, that are produced in the marine actinobacterium Salinispora arenicola. This reaction is of interest since the indole nitrogen is poorly nucleophilic due to the participation of its lone pair in the aromaticity of the indole ring. Three mechanisms were initially considered for the CymD reaction (Fig. 25).75 The first involves an initial ionization of DMAPP to produce the dimethylallyl cation, followed by a direct attack of the indole amine (Path A). Deprotonation would then generate N-DMAT. The second also involves carbocation formation, followed by a normal C-3 prenylation (Path B). The resulting iminium ion would lose a proton and undergo an aza-Cope rearrangement (or vice versa) to give N-DMAT. This mechanism has the advantage of utilizing a better nucleophile (C-3), and there is precedence for an analogous aza-Cope rearrangement proceeding at ambient temperature (Fig. 10).31 The third mechanism involves an initial deprotonation of the imine NH to give an anion, followed by an SN2′ attack onto DMAPP (Path C). This is reasonable as the pKa of the indole amine is approximately 17 in water, and associative mechanisms (SN2) are known for prenyltransferases that alkylate good nucleophiles such as cysteine.77–79
Fig. 25 Potential mechanisms for the reaction catalyzed by CymD.
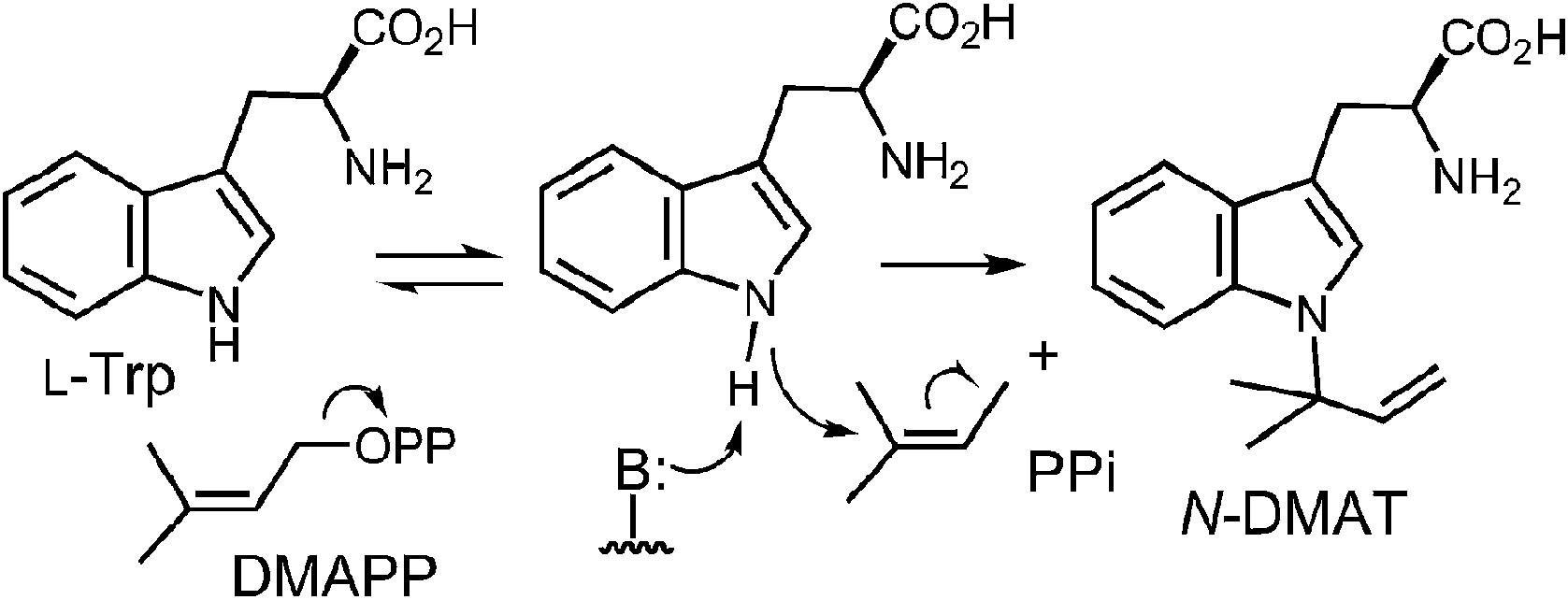
Initial studies on CymD involved the use of fluorinated substrates to analyze the effects of electron withdrawing groups on catalysis.75 Surprisingly, when 4-fluorotryptophan and 6-fluorotryptophan were tested as substrates, the reduction in the rate of catalysis was modest (relative kcat values of 30% and 10%, respectively). This is unlike the effects seen previously with 4-DMATS, and suggests that the amount of positive charge development on the indole ring is minimal. When E-4-fluoro-DMAPP was tested as a substrate, the maximal rate decreased to 1% of that with DMAPP, indicating that there is greater positive charge accumulation on the prenyl moiety during catalysis. When a PIX experiment was performed with tryptophan as substrate (see Fig. 6 for PIX with 4-DMATS), no isotopic scrambling was observed. When the same experiment was performed with 4-fluorotryptophan, however, 44% of the isotopic label had scrambled into a non-bridging position after 59% conversion to product, indicating that a dimethylallyl carbocation had reversibly formed. Presumably, with the electron poor indole, collapse of the ion pair back to DMAPP became competitive with the forward reaction. Given the similarity in the overall rate of catalysis between 4-fluorotrytophan and tryptophan, it is quite likely that the same mechanism is employed in each case. Therefore, the PIX result indicates that CymD uses a dissociative mechanism involving a carbocation intermediate, ruling out Path C (Fig. 25). In a final experiment, the rate of the CymD reaction was monitored in both H2O and D2O to look for a solvent KIE during catalysis. A value of kH2O/kD2O = 2.3 was measured, indicating that the transfer of a solvent-exchangeable proton occurred during a rate limiting step of catalysis. This can presumably be attributed to the removal of the indole NH proton as it is the only requisite proton transfer step in the CymD reaction. Taken together a mechanism was proposed which is a hybrid of Paths A and C (Fig. 26). The reaction is initiated by the dissociation of DMAPP to form a dimethylallyl cation. In a second step, deprotonation of the indole NH proton accompanies, or precedes, the attack of the indole N-1 onto the carbocation to directly give N-DMAT. This explains the observed solvent KIE (rate-limiting NH deprotonation) and the modest effect of indole fluorination on catalysis (negligible positive charge build-up on indole ring). While it is not possible to rule out a similar hybrid mechanism involving normal C-3 prenylation followed by an aza-Cope rearrangement from these studies, one might expect that the rearrangement would be rate limiting and mask the solvent KIE.
Fig. 26 Proposed mechanism for the reaction catalyzed by CymD. B: represents an enzymic base.
6 Geranyl and geranylgeranyl prenyltransferases (LtxC/TleC, PaxC)
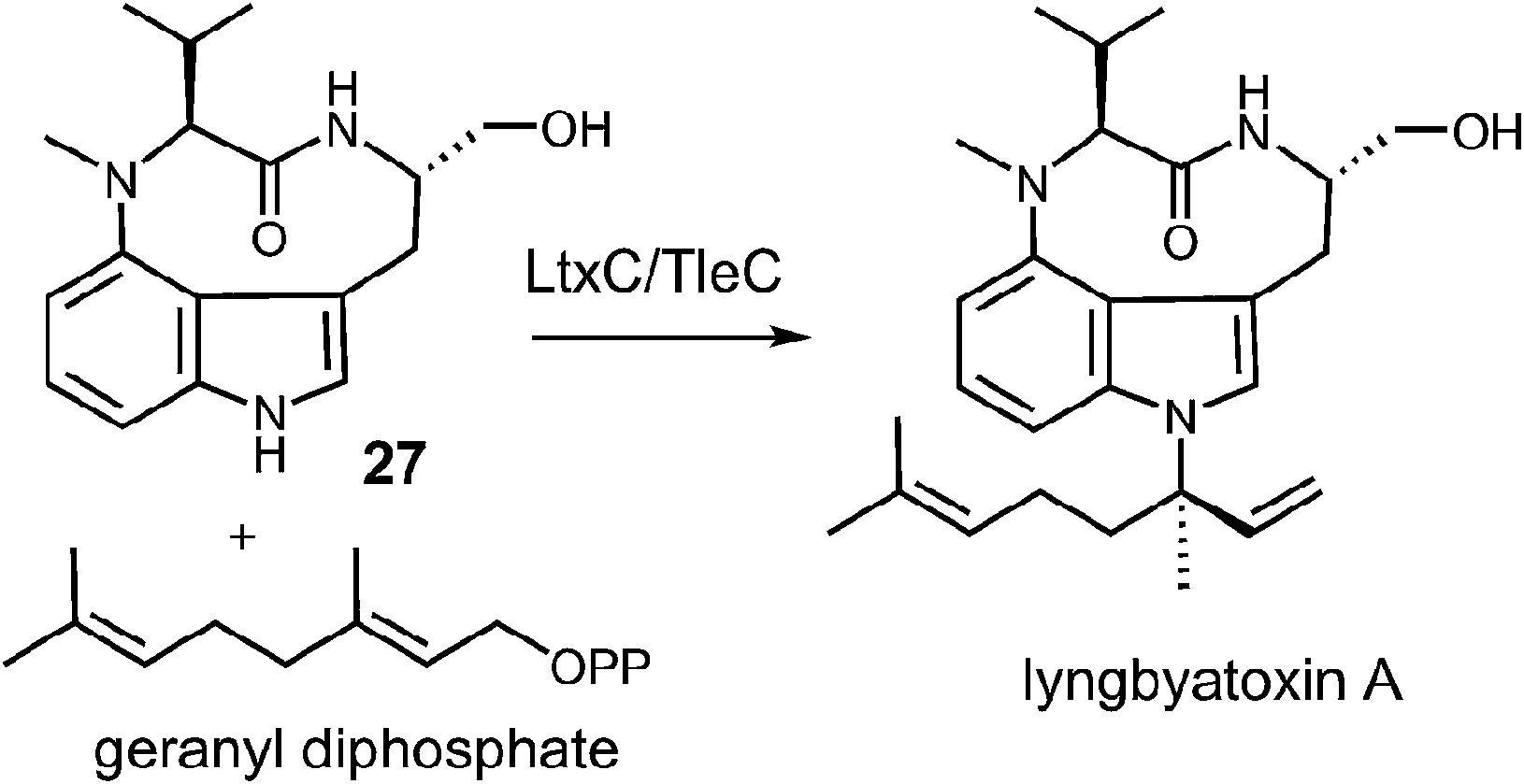
Examples of enzymes that transfer larger prenyl moieties onto an indole nucleus have also been identified, although mechanistic studies on these systems are limited. One of the first such enzymes to be identified is LtxC which prenylates indolactam 27 using geranyl diphosphate in the biosynthesis of lyngbyatoxin A (Fig. 27).5 This activity has also recently been identified in the enzyme TleC, which is involved in the biosynthesis of the teleocidins.80 LtxC catalyzes a reverse prenylation of the C-7 position on an indole ring. This is likely done by a direct electrophilic substitution mechanism as the indole also bears an amino substituent at C-4 and therefore the C-7 position is quite nucleophilic. The possibility of a normal N-1 prenylation followed by an aza-Cope rearrangement has been suggested as an alternate mechanism, but no studies have been performed to address this scenario. LtxC also appears to act in a metal-independent fashion, however, shows no sequence homology to enzymes such as 4-DMATS.
Fig. 27 A reverse C-7 geranylation catalyzed by LtxC or TleC.
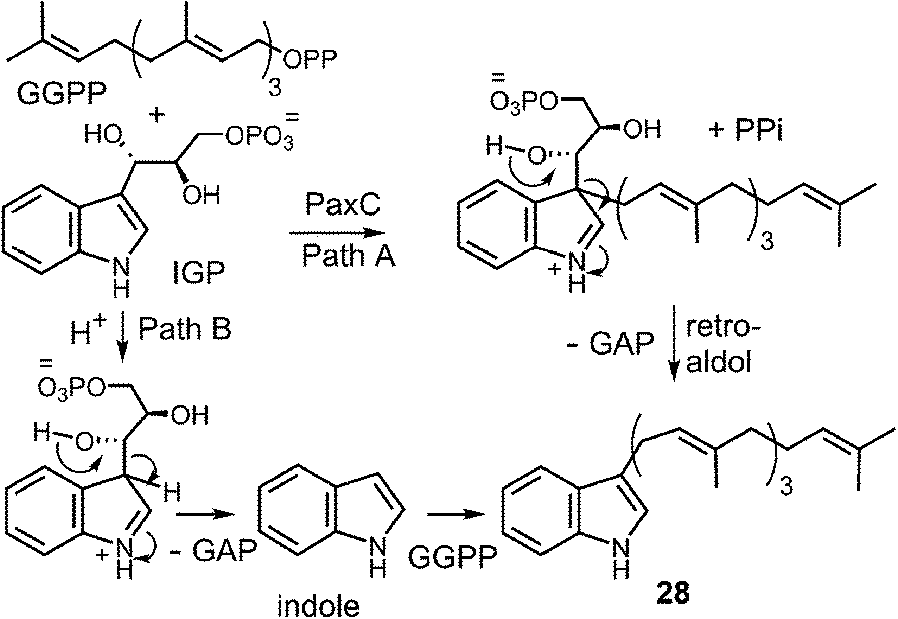
The transfer of a geranylgeranyl group is required in the biosynthesis of the indole diterpene alkaloids. Recently, the enzyme PaxC was shown to catalyze such a reaction using geranylgeranyl diphosphate (GGPP) and indole-3-glycerol phosphate (IGP) to give compound 28 during the biosynthesis of paxilline (Fig. 28).4 This enzyme is quite different from the soluble indole prenyltransferases discussed thus far as it shares sequence homology with the metal-dependent polyprenyl synthases, and since glyceraldehyde 3-phosphate (GAP) is extruded during catalysis. Two mechanisms can be invoked for this reaction. The first involves an initial ionization to form the allylic cation, followed by the direct addition to the C-3 indole position of IGP (Fig. 28, Path A). A retroaldol reaction of the resulting iminium ion would produce the geranylgeranylated indole product 28 and GAP. Precedence for this mechanism is found with the MenA prenyltransferase of menaquinone biosynthesis that catalyzes both decarboxylation and prenylation at a single position of a hydroquinone substrate.81 An alternate mechanism involves protonation of IGP and a retroaldol reaction to generate free indole and GAP (Fig. 28, Path B). The indole then adds to the geranylgeranyl cation and a deprotonation gives product. Precedence for the latter mechanism is found in the reaction catalyzed by tryptophan synthase in which free indole is formed from IGP prior to a condensation with a pyridoxal phosphate bound amino acrylate.82 While mechanistic studies have yet to distinguish between these two possibilities, it is interesting to note that indole itself may replace IGP in this reaction, albeit at a somewhat slower rate. This seems to indirectly support the latter mechanism where indole is an actual intermediate of catalysis, however, it is also possible that it simply acts as an alternate substrate and intercepts the allylic carbocation.
Fig. 28 Two potential mechanisms for the geranylgeranylation catalyzed by PaxC.
A recent report also indicates that the previously discussed enzyme AnaPT is capable of geranylating a variety of cyclic dipeptides and benzodiazepinediones.83 With the normal substrate DMAPP, this enzyme catalyzes a reverse C-3 prenylation, however, with GPP it catalyzes a normal C-6 or C-7 geranylation. This surprising switch in regiochemistry is not well understood, and provides a remarkable example of how altered substitution patterns may arise with unnatural substrates. Finally, a 6-DMATS has recently been shown to accept GPP in place of DMAPP, but prenylation still occurs at the C-6 position.22
7 Conclusions
The indole prenyltransferases represent a newly discovered family of soluble metal-independent enzymes that show an amazing versatility in prenylating all possible sites of the indole ring in either a normal or reverse fashion. Despite this scope of reactivity, the enzymes all appear to share remarkably similar structural folds with highly conserved allylic diphosphate binding sites and similar indole binding pockets. In all cases, it is possible that an electrophilic aromatic substitution mechanism occurs directly at the site of substitution. However, in some cases it has been suggested that prenylation occurs first at the reactive C-3 position and subsequent rearrangements produce differently substituted products. These suggestions are supported by structural analyses and studies with mutant enzymes or alternate substrates. Complicating factors in such experiments are that the crystal structures represent static pictures of the Michaelis complex and may not reflect on the true positioning of the carbocation formed upon ionization. Furthermore, it is now clear that minor modifications to either the substrate structure or the enzyme active site can result in loss of control of the carbocation intermediate and the formation of products with differing substitution patterns. Therefore, in order to discriminate between direct addition and rearrangement mechanisms, it may be necessary to employ rapid quench techniques with the normal substrates and wild type enzymes in an effort to trap intermediates prior to rearrangement. Alternatively, the use of 13C kinetic isotope effects could provide evidence for such intermediates. Finally, further structural studies with bound intermediate or transition state analogs will help to more clearly delineate the mechanistic details of these remarkable enzymes.
8 Acknowledgements
The author would like to acknowledge Qi Qian for assistance in making Fig. 7 and 17.
9 References
T. Lindel, N. Marsch and S. K. Adla, Top. Curr. Chem., 2011, 309, 67–130 CrossRef.
R. M. Williams, E. M. Stocking and J. F. Sanz-Cervera, Top. Curr. Chem., 2000, 209, 97–173 CrossRefCAS.
K. Tagami, C. Liu, A. Minami, M. Noike, T. Isaka, S. Fueki, Y. Shichijo, H. Toshima, K. Gomi, T. Dairi and H. Oikawa, J. Am. Chem. Soc., 2013, 135, 1260–1263 CrossRefCASPubMed.
D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRefCASPubMed.
T. Bonitz, V. Alva, O. Saleh, A. N. Lupas and L. Heide, PLoS One, 2011, 6, e27336 CAS.
L. Heide, Curr. Opin. Chem. Biol., 2009, 13, 171–179 CrossRefCASPubMed.
A. Grundmann, T. Kuznetsova, S. S. Afiyatullov and S.-M. Li, ChemBioChem, 2008, 9, 2059–2063 CrossRefCASPubMed.
A. W. Schultz, C. A. Lewis, M. R. Luzung, P. S. Baran and B. S. Moore, J. Nat. Prod., 2010, 73, 373–377 CrossRefCASPubMed.
Y. Ding, J. R. de Wet, J. Cavalcoli, S. Li, T. J. Greshock, K. A. Miller, J. M. Finefield, J. D. Sunderhaus, T. J. McAfoos, S. Tsukamoto, R. M. Williams and D. H. Sherman, J. Am. Chem. Soc., 2010, 132, 12733–12740 CrossRefCASPubMed.
I. Unsold and S.-M. Li, ChemBioChem, 2006, 7, 158–164 CrossRefPubMed.
A. Grundmann and S.-M. Li, Microbiology, 2005, 151, 2199–2207 CrossRefCASPubMed.
J. M. Schuller, G. Zocher, M. Liebhold, X. Xie, M. Stahl, S.-M. Li and T. Stehle, J. Mol. Biol., 2012, 422, 87–99 CrossRefCASPubMed.
H. Zou, X. Zheng and S.-M. Li, J. Nat. Prod., 2009, 72, 44–52 CrossRefCASPubMed.
N. Mahmoodi and M. E. Tanner, ChemBioChem, 2013, 14, 2029–2037 CrossRefCASPubMed.
J. D. Rudolf, H. Wang and C. D. Poulter, J. Am. Chem. Soc., 2013, 135, 1895–1902 CrossRefCASPubMed.
B. Wollinsky, L. Ludwig, X. Xie and S.-M. Li, Org. Biomol. Chem., 2012, 10, 9262–9270 CAS.
H.-F. Tsai, H. Wang, J. C. Gebler, C. D. Poulter and C. L. Schardl, Biochem. Biophys. Res. Commun., 1995, 216, 119–125 CrossRefCASPubMed.
S.-L. Lee, H. G. Floss and P. Heinstein, Arch. Biochem. Biophys., 1976, 177, 84–94 CrossRefCAS.
C. Wallwey and S.-M. Li, Nat. Prod. Rep., 2011, 28, 496–510 RSC.
T. Haarmann, Y. Rolke, S. Giesbert and P. Tudzynski, Mol. Plant Pathol., 2009, 10, 563–577 CrossRefCASPubMed.
M. Shibuya, H.-M. Chou, M. Fountoulakis, S. Hassam, S.-U. Kim, K. Kobayashi, H. Otsuka, E. Rogalska, J. M. Cassady and H. G. Floss, J. Am. Chem. Soc., 1990, 112, 297–304 CrossRefCAS.
J. C. Gebler, A. B. Woodside and C. D. Poulter, J. Am. Chem. Soc., 1992, 114, 7354–7360 CrossRefCAS.
L. Y. P. Luk and M. E. Tanner, J. Am. Chem. Soc., 2009, 131, 13932–13933 CrossRefCASPubMed.
J. P. Richard and W. P. Jencks, J. Am. Chem. Soc., 1984, 106, 1383–1396 CrossRefCAS.
N. Steffan, I. A. Unsold and S.-M. Li, ChemBioChem, 2007, 8, 1298–1307 CrossRefCASPubMed.
U. Metzger, C. Schall, G. Zocher, I. Unsold, E. Stec, S.-M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRefCASPubMed.
T. Kuzuyama, J. P. Noel and S. B. Richard, Nature, 2005, 435, 983–987 CrossRefCASPubMed.
L. Y. P. Luk, Q. Qian and M. E. Tanner, J. Am. Chem. Soc., 2011, 133, 12342–12345 CrossRefCASPubMed.
E. Wenkert and H. Sliwa, Bioorg. Chem., 1977, 6, 443–452 CrossRefCAS.
H. G. Floss, Tetrahedron, 1976, 32, 873–912 CrossRefCAS.
M.-P. Seiler, Ph.D. Dissertation No. 4574, ETH Zürich, 1970.
S. Zhao, K. S. Smith, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, T. L. Macdonald and J. M. Cook, J. Med. Chem., 2002, 45, 1559–1562 CrossRefCASPubMed.
M. Jost, G. Zocher, S. Tarcz, M. Matuschek, X. Xie, S.-M. Li and T. Stehle, J. Am. Chem. Soc., 2010, 132, 17849–17858 CrossRefCASPubMed.
N. Mahmoodi, O. Qian, L. Y. P. Luk and M. E. Tanner, Pure Appl. Chem., 2013, 85, 1935–1948 CrossRefCAS.
J. Chen, H. Morita, T. Wakimoto, T. Mori, H. Noguchi and I. Abe, Org. Lett., 2012, 14, 3080–3083 CrossRefCASPubMed.
A. S. P. Cardoso, M. M. B. Marques, N. Srinivasan, S. Prabhakar, A. M. Lobo and H. S. Rzepa, Org. Biomol. Chem., 2006, 4, 3966–3972 CAS.
A. G. Leach, S. Catak and K. N. Houk, Chem.–Eur. J., 2002, 8, 1290–1299 CrossRefCAS.
C. C. J. Loh, G. Raabe and D. Enders, Chem.–Eur. J., 2012, 18, 13250–13254 CrossRefCASPubMed.
Y. Kanaoka, K. Miyashita and O. Yonemitsu, J. Chem. Soc., Chem. Commun., 1969, 1365 RSC.
M. Nakazaki, K. Yamamoto and K. Yamagami, Bull. Chem. Soc. Jpn., 1960, 33, 466–472 CrossRefCAS.
E. Caballero, C. Avendano and J. C. Menendez, J. Org. Chem., 2003, 68, 6944–6951 CrossRefCASPubMed.
E. M. Stocking, R. M. Williams and J. F. Sanz-Cervera, J. Am. Chem. Soc., 2000, 122, 9089–9098 CrossRefCAS.
E. M. Stocking, J. F. Sanz-Cervera and R. M. Williams, Angew. Chem., Int. Ed., 1999, 38, 786–789 CrossRefCAS.
X. Yu, G. Zocher, X. Xie, M. Liebhold, S. Schutz, T. Stehle and S.-M. Li, Chem. Biol., 2013, 20, 1492–1501 CrossRefCASPubMed.
W.-B. Yin, J. Cheng and S.-M. Li, Org. Biomol. Chem., 2009, 7, 2202–2207 CAS.
Q. Qian, A. W. Schultz, B. S. Moore and M. E. Tanner, Biochemistry, 2012, 51, 7733–7739 CrossRefCASPubMed.
A. W. Schultz, D.-C. Oh, J. R. Carney, T. Williamson, D. W. Udwary, P. R. Jensen, S. J. Gould, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2008, 130, 4507–4516 CrossRefCASPubMed.
C.-C. Huang, K. E. Hightower and C. A. Fierke, Biochemistry, 2000, 39, 2593–2602 CrossRefCASPubMed.
C. M. Harris and C. D. Poulter, Nat. Prod. Rep., 2000, 17, 137–144 RSC.
V. A. Weller and M. D. Distefano, J. Am. Chem. Soc., 1998, 120, 7975–7976 CrossRefCAS.
T. Awakawa, L. Zhang, T. Wakimoto, S. Hoshino, T. Mori, T. Ito, J. Ishikawa, M. E. Tanner and I. Abe, J. Am. Chem. Soc., 2014, 136, 9910–9913 CrossRefCASPubMed.
K. Suvama, D. Stevenson, R. Meganathan and M. E. S. Hudspeth, J. Bacteriol., 1998, 180, 2782–2787 Search PubMed.
S. Raboni, S. Bettati and A. Mozzarelli, Cell. Mol. Life Sci., 2009, 66, 2391–2403 CrossRefCASPubMed.
D. Pockrandt and S.-M. Li, ChemBioChem, 2013, 14, 2023–2028 CrossRefCASPubMed.



![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: