
Syntheses of polycyclic aromatic diimides via intramolecular cyclization of maleic acid derivatives†
Ranran
Wang
,
Ke
Shi
,
Kang
Cai
,
Yikun
Guo
,
Xiao
Yang
,
Jie-Yu
Wang
,
Jian
Pei
* and
Dahui
Zhao
*
National Laboratory for Molecular Sciences, Centre for Soft Matter Science and Engineering and the Key Laboratory of Polymer Chemistry and Physics of the Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: dhzhao@pku.edu.cn; jianpei@pku.edu.cn
First published on 15th October 2015
Abstract
Using readily available aryl glyoxylic acids and arylene diacetic acids as starting materials, a series of polycyclic aromatic molecules bearing two phthalimide functional groups are synthesized via Perkin condensation followed by intramolecular cyclization reactions. Two different cyclization methods, photo-oxidation and Heck cross-coupling, are employed, both of which effectively accomplish the transformations from diaryl maleic anhydride or maleimide to polycyclic aromatic phthalimide functionality. The photocyclization protocol conveniently allows direct bridging of two plain aromatic C–H sites linked by a maleic anhydride group and uniquely produces the more twisted polycyclic framework as the major product, whereas the Heck coupling approach can typically afford more extended polycyclic skeletons. Thionation reactions are then carried out for the obtained polycyclic diimide molecules using Lawesson's reagent. For all isolated stable products, partial thionation occurs. The prepared polycyclic diimide compounds possess relatively low LUMO levels, and thionation further decreases the LUMO energy of the molecules by 0.2–0.3 eV. Electron-transporting properties are characterized by using solution-processed OFET devices, and an electron mobility of 0.054 cm2 V−1 s−1 is demonstrated by a selected compound. Such semiconducting performance promises great potentials of this class of compounds as useful electron-accepting and transporting building blocks in developing various new semiconductive materials.
Introduction
Polycyclic aromatic dicarboximide (PAI) compounds have demonstrated prominent potentials for applications in organic electronics.1 Amongst various PAI molecules, naphthalene and perylene diimides are the most intensively studied n-type semiconductors.2,3 Recently, impressive research progress has also been made with PAIs being used as electron-accepting materials in organic solar cells.4 Moreover, the unique self-assembly propensity and supramolecular architectures of PAIs have attracted great interest in the field of supramolecular chemistry. PAI derivatives are also utilized for developing novel low-band-gap dyes.5 Apparently, all these appealing applications of PAIs are dependent on the electron-deficient and low-LUMO (lowest unoccupied molecular orbital) attributes of the polycyclic aromatic systems installed with multiple electron-withdrawing dicarboximide groups. In view of such versatile and attractive properties, we deem that more diverse PAI structures should be designed and examined. Compared to PAI structures functionalized with 6-membered-ring dicarboximide units, analogous molecules featuring 5-membered-ring phthalimide-type groups are less reported and studied. Recently, a number of polycyclic aromatics bearing phthalimide moieties has been prepared and their optical properties are carefully investigated,6 but the semiconducting potentials of PAI systems still await exploration.Here, we report the syntheses and properties of a series of polycyclic aromatic molecules bearing phthalimide functional groups, which are potentially suitable building blocks of new electron-accepting or transporting materials. Specifically, eight different PAI compounds are obtained, each containing 5 to 7 fused benzene rings decorated with two phthalimide units (Chart 1). Among various available synthetic strategies,7 we select Perkin condensation to assemble a series of different diaryl maleic anhydrides, which serve as effectual precursors to the target PAI molecules. Great merits of Perkin condensation are its facile synthetic procedures and readily available substrates. The starting materials used in the current work are phenyl and naphthyl glyoxylic acids along with various arylene diacetic acids. Two annulation methods, the intramolecular Heck coupling8 and photo-oxidation cyclization,9–11 are employed to accomplish different polycyclic aromatic scaffolds (Scheme 1). In all the prepared PAIs, branched alkyl side groups are attached to the imide nitrogen atoms, which help in ensuring adequate solubility of the products in common organic solvents.3,4,12 Electrochemical characterization shows that these PAI molecules possess relatively low LUMO levels at −3.4 to −3.7 eV. As an effort to further lower the LUMO levels, thionation of the carbonyl groups is then carried out for the PAI compounds using Lawesson's reagent (Scheme 2). Interestingly, for all isolated stable products, partial yet regio-selective thionation occurs. That is, only one of the two carbonyl groups in every dicarboximide unit gets thionated (Chart 1). Compared to their oxo-counterparts, these thio-substituted PAI derivatives exhibit further decreased LUMO energy levels at −3.8 to −3.9 eV. Subsequently, for molecules suitable for fabricating solution-processed organic field effect transistors (OFETs), the electron-transporting ability is evaluated. An electron mobility of 0.054 cm2 V−1 s−1 is determined for one of the PAI molecules. Such semiconducting properties promise this class of compounds as potentially useful subunits for developing new electron-accepting and transporting (polymer) materials.
 | ||
| Chart 1 Synthesized polycyclic aromatic diimide compounds and their thionation derivatives. | ||
 | ||
| Scheme 1 Synthetic routes towards diimides 1–8. | ||
 | ||
| Scheme 2 Thionation reaction results. | ||
Results and discussion
PAI syntheses
Using phenylglyoxylic acid to react with 2,2′-(1,4-phenylene)- and 2,2′-(1,3-phenylene)diacetic acids, respectively, regio-isomeric molecules 9 and 10 of bis-maleic anhydride were obtained through Perkin condensation in neat acetic anhydride (Scheme 1). Subsequently, intramolecular cyclization was attempted under photo-oxidation conditions. The reaction mixture, containing the bis-maleic anhydride dissolved in mixed acetone and propylene oxide (PO) along with iodine, was irradiated using a high-pressure mercury lamp at room temperature under a nitrogen atmosphere.9,10 The resultant cyclized dianhydrides were poorly soluble in most organic solvents and thus directly applied to the subsequent imidization step. Upon reacting with 2-octyldodecan-1-amine in refluxing N-methyl-2-pyrrolidone (NMP), the dianhydrides were transformed into corresponding polycyclic aromatic diimides, showing reasonably good solubility.Upon further examination, the final PAI products derived from dianhydride 9 were a mixture of two different molecules, regio-isomers 1 and 2, which could be separated and characterized respectively. Although the 1H NMR spectra offered certain evidence for differentiating 1 and 2, unambiguous structural identifications were realized only after isomer 2 was independently prepared via a different synthetic route (Scheme 1). In this alternative pathway, 2,2′-(2,5-dibromo-1,4-phenylene)diacetic acid was subjected to Perkin condensation conditions and coupled with two equivalents of phenylglyoxylic acid, affording a dibromo-substituted bis-maleic anhydride. Imidization was then carried out and offered intermediate 11 with improved solubility, which allowed proper conduction of subsequent intramolecular Heck cross coupling,8 yielding exclusively diimide 2.
After identifying the structures of 2, the helicene-type isomer 1 was found to be the major product from the photo-oxidation reaction, along with byproduct diimide 2 having a relatively extended polycyclic skeleton formed in <5% yield. Such a regio-selectivity of the photocyclization protocol was consistent with the results previously reported by Frimer et al.10 On the other hand, with an overall yield of >40% over three steps, the route harnessing the Heck coupling was apparently a more favorable synthetic path for PAI 2, although it required the pre-installation of bromine atoms at specific positions. Nonetheless, the photocyclization method uniquely generated diimide 1 with a helicene-type scaffold, which was difficult to attain from alternative approaches.
When similar photocyclization procedures followed by an imidization process were applied to bis-maleic anhydride 10, again a pair of regio-isomer PAI products was produced. Distinct 1H NMR spectra clearly identified and differentiated 3 and 4, because 3 had an asymmetric polycyclic backbone, while 4 featured a mirror-plane symmetry. Moreover, PAI 3 was found to be the major component in the final product mixture, indicating that the main product yielded from photocyclization of 10 was the asymmetric dianhydride structure.9,10 This result was also in accordance with previous observations made with photocyclization of 1,3-distyrylbenzene derivatives.13 From the above results, it was clear that such photo-induced oxidative couplings preferentially happened to arene C–H sites with two ortho-substitutions.10,13
To further explore the substrate scope of the photocyclization protocol, we then used naphthylenediacetic acids as the central moiety to prepare PAI molecules with larger aromatic skeletons. When 2,2′-(naphthalene-2,6-diyl)diacetic acid was employed as a reactant to couple with phenylglyoxylic acid, only diimide 5 was isolated upon a series of reactions including Perkin condensation, photocyclization and imidization. No other regio-isomer involving photo-cyclization on the β-positions of naphthylene was detected. This result was not surprising since the α-position of naphthalene was commonly more reactive than the β-position. When phenylglyoxylic acid was subjected to reaction with 2,2′-(naphthalene-1,5-diyl)diacetic acid, PAI 6 was formed as the only product after photocyclization and imidization procedures. It is noteworthy that a prolonged irradiation time was necessary for the 1,5-naphthylene-derived substrate 13 than corresponding 2,6-substituted analogue 12. Presumably this was also because the reactivity of the β-positions of naphthalene was much lower compared to the α-positions in the photocyclization.
We then employed another dianhydride substrate 14, which was obtained from Perkin condensation between 1,4-phenylenediacetic acid and 1-naphthylglyoxylic acid. As expected, PAI 7 was formed as the major product, with a minimal byproduct of isomer 8. Evidently, the same regio-selectivity was manifested here as in the reaction generating PAI 1. Meanwhile, following the route of Perkin condensation, imidization, and Heck coupling, PAI 8 was obtainable in a more favorable yield from dianhydride intermediate 15, prepared from 2,5-dibromo-1,4-phenylenediacetic acid and 1-naphthylglyoxylic acid as the substrates.
Thionation reactions
As an effort to further lower the LUMO of the prepared PAIs,14 thionation reactions were carried out using Lawesson's reagent (Scheme 2). However, it was found that the thionation products from a number of studied PAI compounds were unstable and decomposed during the reaction. Stable thionation products were only achieved from compounds 6, 7 and 8, and these isolated thionation derivatives, 6-S, 7-S and 8-S, appeared as well sensitive to moisture and displayed a tendency to decompose gradually under ambient conditions. Additionally, the mass spectroscopy characterization revealed that 6-S, 7-S and 8-S all contained only two sulfur atoms, instead of four as expected. Furthermore, the 1H and 13C NMR spectra of these compounds indicated that such partial thionation products displayed similar molecular symmetry to that of their respective synthetic precursors of oxo-analogues. Hence, although we could not obtain unambiguous evidence to identify the exact structures of 6-S, 7-S and 8-S, regarding the specific locations of sulfur atoms, it was confirmable that one of the two carbonyl oxygen atoms in all dicarboximide functional groups was converted to a sulfur atom in a site-selective fashion (Chart 1).Photophysical and electrochemical characterization
Subsequently, all PAI molecules were subjected to electronic property characterization, with the absorption properties examined first. For PAIs 1–7, the main absorption bands occurred between 300 and 500 nm (Fig. 1), whereas compound 8 exhibited an additional peak of much longer wavelength at over 500 nm. Compared to their oxo-counterparts, the absorption of thionated compounds 6-S, 7-S and 8-S was considerably red-shifted (Fig. 2). Based on the onset of the absorption band, the optical band gap (Eg) of 6-S, 7-S and 8-S was narrowed by 0.58, 0.45 and 0.38 eV, respectively, relative to those of 6, 7 and 8 (Table 1). | ||
| Fig. 1 Absorption spectra of PAI 1–8 in CHCl3 (1 × 10−5 M). | ||
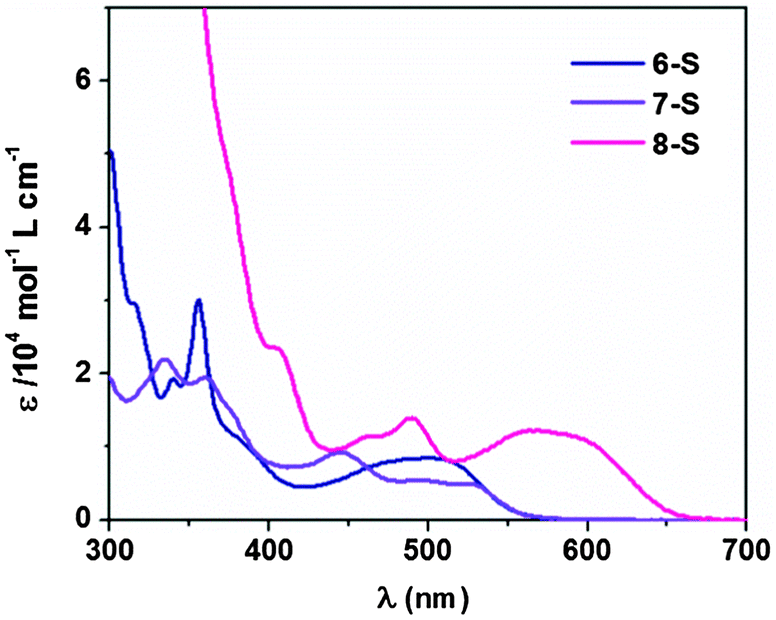 | ||
| Fig. 2 Absorption spectra of 6-S, 7-S and 8-S in CHCl3 (1 × 10−5 M). | ||
| Φ | E g /eV | LUMOc/eV | HOMOd/eV | |
|---|---|---|---|---|
| a Fluorescence quantum yields measured in chloroform. b Band gap values estimated from the absorption onset. c From the onset of the first reduction wave in CV. d Calculated from Eg and the LUMO. | ||||
| 1 | 0.10 | 2.64 | −3.51 | −6.15 |
| 2 | 0.21 | 2.50 | −3.56 | −6.06 |
| 3 | 0.01 | 2.66 | −3.41 | −6.07 |
| 4 | 0.25 | 2.63 | −3.49 | −6.12 |
| 5 | 0.15 | 2.51 | −3.41 | −5.92 |
| 6 | 0.21 | 2.58 | −3.40 | −5.98 |
| 7 | 0.15 | 2.44 | −3.54 | −5.98 |
| 8 | 0.14 | 2.23 | −3.67 | −5.90 |
| 6-S | <0.01 | 2.00 | −3.76 | −5.76 |
| 7-S | <0.01 | 1.99 | −3.83 | −5.82 |
| 8-S | <0.01 | 1.85 | −3.90 | −5.75 |
Most of the prepared PAI molecules were moderately fluorescent (Fig. 3), showing fluorescence quantum yields ranging from 0.10 to 0.25 (Table 1).15 Nonetheless, the Φ value of 3 (∼0.01) was significantly lower than those of other PAIs, which was likely a result from its twisted polycyclic backbone. The steric hindrance between the imide carbonyl and the aromatic backbone possibly caused additional non-radiative decay in the excited state of 3. All three thionated compounds 6-S, 7-S and 8-S were nearly non-fluorescent, reflecting a significant emission quenching effect of the sulfur atoms.
 | ||
| Fig. 3 Fluorescence emission spectra of PAI 1–8 (1 × 10−6 M, excited at 410 nm). | ||
In subsequent electrochemical characterization, PAIs 1–8 all showed two reversible reductive waves in their cyclic voltammograms (CVs) (Fig. 4).16 Estimated from the potential onset of the first reduction peaks, the LUMO energy levels of 1–7 were all in the range of −3.40 to −3.56 eV, while PAI 8 exhibited a lower LUMO at −3.67 eV (Table 1). Specifically, among regio-isomers 1–4, PAI 2 having an extended benzo[k]tetraphene-5,6,12,13-tetracarboxydiimide backbone displayed the lowest LUMO level and the narrowest band gap, whereas molecule 3 featuring an asymmetric benzo[c]chrysene-1,2,7,8-tetracarboxydiimide displayed the highest LUMO and the most widened band gap. Such electronic properties of 3 could as well be related to its twisted polycyclic framework, which frustrated electron delocalization over the conjugated aromatic skeleton. This assumption was confirmed by density functional theory (DFT) calculations, which revealed that molecule 3 had a non-planar polycyclic backbone due to the steric interference between one of the terminal benzene rings and a nearby imide carbonyl group (Fig. S1, ESI†). As a result, the LUMO of 3 was localized to part of the polycyclic backbone, covering only one of the two dicarboximide moieties. In contrast, the LUMOs of all other three isomers were delocalized over the entire polycyclic scaffolds.
 | ||
| Fig. 4 Cyclic voltammograms of 1–8, 6-S, 7-S and 8-S in CH2Cl2. | ||
With additional benzene rings fused to the polycyclic frameworks, the HOMO levels of 5–8 were noticeably boosted relative to those of 1–4 (Table 1). While regio-isomers 5 and 6 possessed similar LUMOs at −3.4 eV, much lowered LUMOs were detected with 7 and 8. Particularly, compound 8 manifested the lowest LUMO at −3.67 eV among all herein studied PAIs.
Partial thionation of the dicarboximide groups brought about conspicuous decrease in the LUMO levels of these polycyclic diimide molecules. According to the CV data, the LUMOs of 6-S, 7-S and 8-S were all lowered by 0.2 to 0.3 eV relative to their respective oxo- analogues 6–8. Such a pronounced effect of carbonyl thionation was also consistent with the DFT calculation results, which showed that thiocarbonyl moieties made substantial contributions to the LUMOs of the molecules (Fig. S2, ESI†).
OFET characterization
Subsequently, the semiconducting properties of these PAIs and their thionation derivatives were subjected to examination in OFET devices using the solution-processing technique. The devices were fabricated with the top-gate/bottom-contact (TG-BC) configuration, which has proven more suitable for characterizing solution-processed electron-transporting semiconductors. During the device fabrication, a CYTOP dielectric layer was necessarily introduced on the top of the semiconductive layer and was baked at 100 °C. Organic molecules having low melting points near or below 100 °C were thus not suitable for such device characterization. Unfortunately, most PAI molecules synthesized in this work exhibited relatively low melting points and hence could not be characterized, except for 2 and 8. It is particularly noteworthy that thionated derivatives 6-S, 7-S and 8-S surprisingly possessed lower melting points than their oxo-counterparts. We suspect that such properties were related to the steric characteristics of the molecules. That is, although sulfur atoms induce larger van der Waals interactions, the larger size of the atom entails greater distortion and non-planarity of the polycyclic skeletons. The DFT calculation results were consistent with such a proposition, showing nonplanar geometry of the optimized structures of 6-S, 7-S and 8-S (Fig. 5). Such nonplanar structures could apparently preclude very compact molecular packing and cause lowering of the melting points. | ||
| Fig. 5 Top- and side-views of optimized geometry of possible structures of 6-S, 7-S and 8-S. | ||
In addition to the melting points, the device performance of solution-processed semiconductors was also sensitive to the crystallinity of the active materials. Most of the prepared PAI molecules exhibited evident tendency to crystallize under solution processing conditions, rendering them unsuitable for solution-processed OFET characterization. An electron mobility of ∼10−3 cm2 V−1 s−1 was observed for PAI 2. Such a mobility value most likely suffered from the high tendency of compound 2 to crystallize under the device fabrication conditions, since discontinuous crystalline domains were perceivable with the semiconductive layer. Atomic force microscopy (AFM) and X-ray diffraction (XRD) provided supportive evidence for the high crystallinity of 2 in the thin-film state (Fig. S3 and S4, ESI†). In comparison, PAI 8 exhibiting a suitable melting point and more favorable film morphology displayed a much more desirable electron mobility of 0.054 cm2 V−1 s−1 under ambient conditions (Fig. 6).
 | ||
| Fig. 6 The transfer (a) and output (b) profiles of 8 measured in solution-processed OFET under ambient conditions. | ||
Conclusions
In summary, using readily available aryl glyoxylic and arylene diacetic acids as reactants, we synthesize eight different polycyclic aromatic tetracarboxydiimide derivatives via facile Perkin condensation followed by photo-oxidation or intramolecular Heck cross-coupling reactions. Each of these PAI molecules contains 5 to 7 fused benzene rings bearing two phthalimide-type functional groups at varied positions. The two employed cyclization methods, photo-oxidation and Heck cross-coupling, are both effective in accomplishing the functional group transformation from diaryl maleic anhydride to polycyclic phthalimide. The advantage of the photocyclization method lies in the fact that it conveniently allows direct bridging of two plain C–H sites on properly linked aromatic rings. Moreover, the photo-oxidation procedure uniquely produces the more twisted polycyclic skeletons as the major products, which are difficult to attain from alternative procedures such as Heck cross-coupling. On the other hand, the Heck protocol offers higher overall reaction yields and better control at the product structures. In light of such distinct regio-chemistry features, both approaches represent valuable synthetic methods that complement each other. Furthermore, using Lawesson's reagent thionation reactions are performed to obtain PAIs. For unclear reasons, some thionated products are unstable. For those isolated, stable products, partial thionation occurs, resulting in regio-selective substitution of one of the two oxygen atoms in every dicarboximide functionality.Owing to the electron-withdrawing effect of the dicarboximide groups, all the prepared polycyclic aromatic dicarboximide molecules show relatively low LUMOs at −3.4 to −3.7 eV, and the LUMOs of partially thionated molecules are typically further lowered by 0.2 to 0.3 eV relative to their respective oxo- analogues. An electron mobility of 0.054 cm2 V−1 s−1 is observed in a solution-processed field effect transistor with a selected structure 8 having suitable melting point and film morphology. Such semiconductive performance demonstrates great promise of this class of compounds as useful electron-accepting and transporting building blocks in developing new semiconductive materials. The thionated derivatives with lower LUMOs are especially attractive subunit structures.
Experimental section
Materials and methods
All chemicals were obtained from commercial sources and used as received unless otherwise indicated. 2,2′-(2,5-Dibromo-1,4-phenylene)diacetic acid,6 2,2′-(naphthalene-1,5-diyl)diacetic acid,17 2-(naphthalen-1-yl)-2-oxoacetic acid18 and compound 99,10 were prepared according to the reported methods, and their structures were confirmed with 1H NMR spectra. Photocyclization was carried out under a nitrogen atmosphere using a CEL-LAM500 high-pressure mercury lamp. 1H and 13C NMR spectra were recorded at 298 K on a Varian Mercury plus 300 (300 MHz) or a Bruker Avance 400 (400 MHz) in CDCl3 or otherwise noted solvent. Chemical shifts are reported in parts per million (ppm) with tetramethylsilane (TMS, 0 ppm) as the reference for 1H NMR spectra and CDCl3 (77.0 ppm) as the reference for 13C NMR spectra, unless noted otherwise. High-resolution ESI mass spectra (HR-ESI MS) were recorded on a Bruker Apex IV Fourier transformation mass spectrometer. Elemental analyses were performed using a German Vario EL CUBE elemental analyzer. Melting points were acquired through a Q100DSC modulation differential scanning calorimeter. The absorption spectra were recorded on a Hitachi U-4100 spectrophotometer using a 1 cm quartz cell at 298 K. The emission spectra were recorded on a Perkin Elmer LS55 Luminescence Spectrometer at 298 K. The emission quantum yields (Φ) were measured at 10−6 M in chloroform at 298 K using perylene as the standard for 1–7 and perylene orange as the standard for 8.15 Cyclic voltammetry (CV) was recorded using a BASI Epsilon workstation and the measurements were carried out at 298 K in CH2Cl2 containing 0.1 M n-Bu4NPF6 as the supporting electrolyte. A glassy carbon electrode was used as the working electrode, with a platinum sheet as the counter electrode. All potentials were recorded with Ag/AgCl as the reference electrode, at a scan speed of 50 mV s−1. Ferrocene/ferrocenium was employed as an external reference, the energy level of which is assumed to be −4.8 eV below the vacuum level.16 The LUMO levels were estimated from the onset potentials of the first reduction waves in CV.Device fabrication and characterization
Top-gate/bottom-contact FET devices were fabricated using n++-Si/SiO2 (300 nm) substrates. The gold source and drain bottom electrodes (with Ti as the adhesion layer) were patterned by photolithography on the SiO2 surface. The substrates were subjected to cleaning using ultrasonication in acetone, the cleaning agent, deionized water (twice), and isopropanol. The cleaned substrates were dried under vacuum at 80 °C for 2 h and then transferred into a glovebox. The semiconductive layer was deposited on the treated substrates by spin-coating trichloroethylene solutions of respective compounds (at 10 mg ml−1) at 1800 rpm for 60 s. A CYTOP solution (CTL809M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CT-solv180 = 3:1) was spin-coated onto the semiconducting layer at 2000 rpm for 60 s, resulting in a thick dielectric layer of 500 nm, which was then baked at 100 °C for 1 h. Gate electrodes of Al (50 nm thick) were then thermally evaporated through a shadow mask onto the dielectric layer. The OFET devices had a channel length (L) of 5 μm and a channel width (W) of 200 μm. The evaluations of the FETs were carried out under ambient conditions (humidity 50–60%) on a probe stage using a Keithley 4200 SCS as the parameter analyzer. The carrier mobility, μ, was calculated from the data in the saturated regime according to the equation ISD = (W/2L)Ciμ(VG − VT)2, where ISD is the drain current in the saturated regime. W and L are the semiconductor channel width and length, respectively. Ci (Ci = 3.7 nF) is the capacitance per unit area of the gate dielectric layer. VG and VT are the gate voltage and threshold voltage, respectively. VG − VT of the device was determined from the relationship between the square root of ISD and VG at the saturated regime.
:CT-solv180 = 3:1) was spin-coated onto the semiconducting layer at 2000 rpm for 60 s, resulting in a thick dielectric layer of 500 nm, which was then baked at 100 °C for 1 h. Gate electrodes of Al (50 nm thick) were then thermally evaporated through a shadow mask onto the dielectric layer. The OFET devices had a channel length (L) of 5 μm and a channel width (W) of 200 μm. The evaluations of the FETs were carried out under ambient conditions (humidity 50–60%) on a probe stage using a Keithley 4200 SCS as the parameter analyzer. The carrier mobility, μ, was calculated from the data in the saturated regime according to the equation ISD = (W/2L)Ciμ(VG − VT)2, where ISD is the drain current in the saturated regime. W and L are the semiconductor channel width and length, respectively. Ci (Ci = 3.7 nF) is the capacitance per unit area of the gate dielectric layer. VG and VT are the gate voltage and threshold voltage, respectively. VG − VT of the device was determined from the relationship between the square root of ISD and VG at the saturated regime.
General synthesis procedures for PAIs
1: 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 2H), 9.09 (m, 2H), 8.08 (m, 2H), 7.65 (m, 2H), 7.27 (m, 2H), 3.72 (d, J = 7.2 Hz, 4H), 1.99 (br, 2H), 1.41–1.22 (m, 64H), 0.86–0.83 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 169.7, 169.5, 133.7, 130.3, 129.5, 128.7, 128.5, 127.4, 127.1, 126.2, 125.4, 125.1, 42.4, 37.3, 31.9, 31.6, 30.05, 30.02, 29.68, 29.65, 29.60, 29.3, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C66H93N2O4 (M + H+): 977.7130; found: 977.7103. Elem. anal.: calcd for C66H92N2O4: C, 81.10%; H, 9.49%; N, 2.87%; found: C, 81.02%; H, 9.51%; N, 2.91%. m.p.: 109.6 °C.
2: 1H NMR (300 MHz, CDCl3) δ 10.10 (s, 2H), 9.04 (d, J = 7.5 Hz, 2H), 8.76 (d, J = 7.5 Hz, 2H), 7.85–7.76 (m, 4H), 3.60 (d, J = 7.5 Hz, 4H), 1.95 (br, 2H), 1.40–1.20 (m, 64H), 0.86–0.83 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 169.0, 168.9, 132.0, 130.1, 129.5, 128.5, 127.9, 125.9, 124.5, 123.6, 123.2, 119.4, 41.9, 37.4, 31.92, 31.89, 31.5, 30.1, 29.72, 29.67, 29.4, 26.4, 22.7, 22.6, 14.1. HR-ESI MS: calcd for C66H93N2O4 (M + H+): 977.7130; found: 977.7152. Elem. anal.: calcd for C66H92N2O4: C, 81.10%; H, 9.49%; N, 2.87%; found: C, 80.97%; H, 9.68%; N, 2.85%. m.p.: 137.0 °C.
3: 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 9.26 (s, 1H), 9.18 (d, J = 8.0 Hz, 1H), 8.80 (d, J = 9.2 Hz, 1H), 8.76 (m, 1H), 8.37 (d, J = 8.0 Hz, 1H), 7.91–7.87 (m, 2H), 7.76 (t, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 3.73 (d, J = 6.8 Hz, 2H), 3.56–3.42 (m, 2H), 1.97 (br, 1H), 1.86 (br, 1H), 1.43–1.22 (m, 64H), 0.89–0.83 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 170.0, 169.63, 169.60, 168.1, 135.3, 134.9, 132.9, 130.7, 130.3, 130.2, 129.8, 129.4, 128.7, 128.5, 127.9, 127.5, 126.22, 126.20, 126.1, 125.80, 125.76, 125.5, 125.3, 124.1, 123.4, 120.0, 116.4, 42.3, 37.34, 37.29 31.9, 31.6, 31.5, 30.1, 30.0, 29.63, 29.57, 29.3, 26.6, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C66H93N2O4 (M + H+): 977.7130; found: 977.7154. Calcd for C66H92N2O4: C, 81.10%; H, 9.49%; N, 2.87%; found: C, 81.05%; H, 9.62%; N, 2.90%. m.p.: 100.2 °C.
4: 1H NMR (300 MHz, CDCl3) δ 10.33 (s, 1H), 9.38 (s, 1H), 8.98 (d, J = 3.9 Hz, 2H), 8.61 (d, J = 3.9 Hz, 2H), 7.75–7.72 (m, 4H), 3.62 (d, J = 4.5 Hz, 4H), 1.97 (br, 2H), 1.48–1.22 (m, 64H), 0.86–0.81 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 169.6, 169.2, 132.4, 131.4, 129.4, 128.9, 128.1, 127.2, 126.8, 125.5, 124.1, 123.9, 123.0, 116.7, 42.4, 37.2, 31.9, 31.6, 30.1, 29.9, 29.7, 29.3, 26.5, 22.7, 14.1. HR-ESI MS: calcd for C66H93N2O4 (M + H+): 977.7130; found: 977.7152. Calcd for C66H92N2O4: C, 81.10%; H, 9.49%; N, 2.87%; found: C, 80.99%; H, 9.52%; N, 2.83%. m.p.: 86.6 °C.
5: 1H NMR (300 MHz, CDCl3) δ 9.13 (d, J = 8.1 Hz, 2H), 8.83 (d, J = 8.7 Hz, 2H), 8.66 (d, J = 8.7 Hz, 2H), 8.62 (d, J = 8.1 Hz, 2H), 7.77–7.66 (m, 4H), 3.56 (d, J = 7.2 Hz, 4H), 1.94 (br, 2H), 1.38–1.22 (m, 64H), 0.87–0.82 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 169.7, 169.4, 132.3, 131.2, 130.9, 129.0, 128.8, 128.2, 127.4, 126.6, 125.8, 124.5, 122.2, 42.1, 37.2, 31.9, 31.5, 30.0, 29.9, 29.63, 29.58, 29.5, 29.3, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C70H95N2O4 (M + H+): 1027.7286; found: 1027.7284. Elem. anal.: calcd for C70H94N2O4: C, 81.82%; H, 9.22%; N, 2.73%; found: C, 81.75%; H, 9.35%; N, 2.70%.
6: 1H NMR (300 MHz, CDCl3) δ 9.34 (d, J = 9.0 Hz, 2H), 9.30 (m, 2H), 8.75 (m, 2H), 8.71 (d, J = 9.0 Hz, 2H), 7.85–7.80 (m, 4H), 3.68 (d, J = 7.5 Hz, 4H), 1.99 (br, 2H), 1.46–1.13 (m, 64H), 0.88–0.82 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 169.69, 169.67, 133.6, 133.0, 131.8, 129.8, 129.7, 128.9, 128.8, 128.0, 126.3, 126.0, 123.6, 119.7, 42.6, 37.2, 31.9, 31.7, 30.1, 29.70, 29.65, 29.6, 29.4, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C70H95N2O4 (M + H+): 1027.7286; found: 1027.7274. Elem. anal.: calcd for C70H94N2O4: C, 81.82%; H, 9.22%; N, 2.73%; found: C, 81.65%; H, 9.21%; N, 2.55%. m.p.: 106.5 °C.
7: 1H NMR (400 MHz, CDCl3) δ 9.94 (d, J = 4.0 Hz, 2H), 9.47 (s, 2H), 7.87 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 8.0 Hz, 2H), 7.76 (t, J = 12.0 Hz, 2H), 7.54–7.46 (m, 4H), 3.75 (d, J = 8.0 Hz, 4H), 2.04 (br, 2H), 1.41–1.22 (m, 64H), 0.86–0.82 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 169.3, 169.1, 135.4, 132.7, 130.7, 130.2, 129.5, 129.0, 128.5, 127.7, 127.62, 127.59, 126.9, 126.8, 125.2, 42.9, 37.2, 31.9, 31.7, 30.1, 30.0, 29.7, 29.66, 29.60, 29.3, 26.5, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C74H97N2O4 (M + H+): 1077.7443; found: 1077.7445. Elem. anal.: calcd for C74H96N2O4: C, 82.48%; H, 8.98%; N, 2.60%; found: 82.24%; H, 8.86%; N, 2.54%. m.p.: 98 °C.
8: 1H NMR (400 MHz, CDCl3) δ 10.51 (s, 2H), 9.45 (d, J = 8.0 Hz, 2H), 8.86 (d, J = 8.0 Hz, 2H), 8.16 (d, J = 12.0 Hz, 2H), 8.00 (d, J = 8.0 Hz, 2H), 7.77–7.69 (m, 4H), 3.56 (d, J = 8.0 Hz, 4H), 1.97 (br, 2H), 1.39–1.18 (m, 64H), 0.84–0.79 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 169.0, 168.8, 133.14, 133.09, 131.3, 130.8, 129.9, 129.7, 128.8, 127.8, 127.7, 127.5, 126.4, 124.2, 123.8, 121.1, 120.7, 42.5, 37.1, 31.93, 31.91, 31.6, 30.2, 29.74,29.72, 29.68, 29.0, 29.37, 26.4, 22.7, 14.1. HR-ESI MS: calcd for C74H97N2O4 (M + H+): 1077.7443; found: 1077.7466. Elem. anal.: calcd for C74H96N2O4: C, 82.48%; H, 8.98%; N, 2.60%; found: C, 82.41%; H, 8.99%; N, 2.52%. m.p.: 195.6 °C.
6-S: 1H NMR (400 MHz, CDCl3) δ 10.13 (m, 2H), 9.21 (d, J = 8.0 Hz), 8.80 (m, 2H), 8.71 (d, J = 8.0 Hz, 2H), 7.85–7.79 (m, 4H), 4.05 (d, J = 8.0 Hz, 4H), 2.17 (br, 2H), 1.37–1.22 (m, 64H), 0.87–0.82 (m, 12H). 13C NMR (120 MHz, CDCl3) δ 197.9, 171.1, 134.0, 133.5, 132.1, 130.8, 130.1, 129.3, 129.0, 128.0, 127.0, 123.8, 122.9, 119.3, 100.0, 45.5, 36.5, 31.9, 31.8, 30.1, 29.7, 29.6, 29.6, 29.3, 26.4, 22.7, 14.1. Calcd for C70H95N2O2S2 (M + H+): 1059.6830; found: 1059.6839. m.p.: 75.0 °C.
7-S: 1H NMR (400 MHz, CDCl3) δ 9.97 (d, J = 2.8 Hz, 2H), 9.68 (d, J = 8.0 Hz, 2H), 7.78–7.67 (m, 6H), 7.34–7.24 (m, 4H), 4.03 (t, J = 8.0 Hz, 4H), 2.18–2.14 (m, 2H), 1.39–1.20 (m, 64H), 0.86–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 196.6, 170.4, 134.9, 132.8, 131.5, 130.4, 129.1, 129.1, 128.5, 128.3, 127.7, 127.7, 127.6, 126.6, 126.4, 125.2, 125.0, 45.7, 36.6, 31.95, 31.93, 31.7, 30.2, 30.1, 29.74, 29.73, 29.70, 29.68, 29.66, 29.40, 29.39, 26.4, 22.7, 14.2. Calcd for C74H97N2O2S2 (M + H+): 1109.6986; found: 1109.6952. Elem. anal.: calcd for C74H96N2O2S2: C, 80.09%; H, 8.72%; N, 2.52%; found: C, 79.94%; H, 8.87%; N, 2.41%. m.p.: 65.7 °C.
8-S: 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 2H), 8.93 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.95–7.67 (m, 4H), 7.60–7.54 (m, 4H), 3.54 (d, J = 8.0 Hz, 4H), 1.96 (br, 2H), 1.37–1.20 (m, 64H), 0.85–0.82 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 196.1, 170.3, 133.3, 133.0, 131.9, 131.2, 129.7, 129.3, 128.6, 127.7, 127.4, 125.9, 125.0, 124.1, 123.0, 122.9, 121.1, 45.1, 36.5, 32.0, 31.9, 31.7, 30.2, 29.78, 29.76, 29.7, 29.43, 29.40, 26.4, 22.72, 22.70, 14.1. Calcd for C74H97N2O2S2 (M + H+): 1109.6986; found: 1109.6949. Elem. anal.: calcd for C74H96N2O2S2: C, 80.09%; H, 8.72%; N, 2.52%; found: C, 80.12%; H, 8.87%; N, 2.51%. m.p.: 137.0 °C.
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (Projects 21174004, 21222403 and 51473003).Notes and references
- (a) B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed; (b) W. Yue, A. Lv, J. Gao, W. Jiang, L. Hao, C. Li, Y. Li, L. E. Polander, S. Barlow, W. Hu, S. Di Motta, F. Negri, S. R. Marder and Z. Wang, J. Am. Chem. Soc., 2012, 134, 5770–5773 CrossRef CAS PubMed; (c) A. R. Mohebbi, C. Munoz and F. Wudl, Org. Lett., 2011, 13, 2560–2563 CrossRef CAS PubMed; (d) T. V. Pho, F. M. Toma, M. L. Chabinyc and F. Wudl, Angew. Chem., Int. Ed., 2013, 52, 1446–1451 CrossRef CAS PubMed.
- (a) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed; (b) F. Würthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC; (c) L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- (a) Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed; (b) X. Gao, C. Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697–3699 CrossRef CAS PubMed; (c) D. K. Hwang, R. R. Dasari, M. Fenoll, V. Alain-Rizzo, A. Dindar, J. W. Shim, N. Deb, C. Fuentes-Hernandez, S. Barlow, D. G. Bucknall, P. Audebert, S. R. Marder and B. Kippelen, Adv. Mater., 2012, 24, 4445–4450 CrossRef CAS PubMed; (d) F. Zhang, Y. Hu, T. Schuettfort, C. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed; (e) X. Chen, Y. Guo, L. Tan, G. Yang, Y. Li, G. Zhang, Z. Liu, W. Xu and D. Zhang, J. Mater. Chem. C, 2013, 1, 1087–1092 RSC; (f) K. Cai, J. Xie, X. Yang and D. Zhao, Org. Lett., 2014, 16, 1852–1855 CrossRef CAS PubMed.
- (a) Q. Yan, Y. Zhou, Y. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389–4394 RSC; (b) X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed; (c) Y. Zhou, T. Kurosawa, W. Ma, Y. Guo, L. Fang, K. Vandewal, Y. Diao, C. Wang, Q. Yan, J. Reinspach, J. Mei, A. L. Appleton, G. I. Koleilat, Y. Gao, S. C. B. Mannsfeld, A. Salleo, H. Ade, D. Zhao and Z. Bao, Adv. Mater., 2014, 26, 3767–3772 CrossRef CAS PubMed; (d) Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed; (e) T. Earmme, Y. J. Hwang, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2014, 26, 6080–6085 CrossRef CAS PubMed; (f) Y. Zang, C.-Z. Li, C.-C. Chueh, S. T. Williams, W. Jiang, Z.-H. Wang, J.-S. Yu, A. K. Y. Jen and A. K. Y. Jen, Adv. Mater., 2014, 26, 5708–5714 CrossRef CAS PubMed.
- (a) K. Cai, J. Xie and D. Zhao, J. Am. Chem. Soc., 2014, 136, 28–31 CrossRef CAS PubMed; (b) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC; (c) C. Lin, M. Velusamy, H. Chou, J. Lin and P. Chou, Tetrahedron, 2010, 66, 8629–8634 CrossRef CAS; (d) J. Zheng, W. Qiao, X. Wan, J. Gao and Z. Wang, Chem. Mater., 2008, 20, 6163–6168 CrossRef CAS.
- (a) P. Sarkar, F. Durola and H. Bock, Chem. Commun., 2013, 49, 7552–7554 RSC; (b) H. Bock, D. Subervie, P. Mathey, A. Pradhan, P. Sarkar, P. Dechambenoit, E. A. Hillard and F. Durola, Org. Lett., 2014, 16, 1546–1549 CrossRef CAS PubMed; (c) H. Bock, P. Carre, E. A. Hillard and F. Durola, Eur. J. Org. Chem., 2015, 1028–1032 CrossRef CAS; (d) H. Bock, S. Huet, P. Dechambenoit, E. A. Hillard and F. Durola, Eur. J. Org. Chem., 2015, 1033–1039 CrossRef CAS; (e) L. Zou, X.-Y. Wang, X.-X. Zhang, Y.-Z. Dai, Y.-D. Wu, J.-Y. Wang and J. Pei, Chem. Commun., 2015, 51, 12585–12588 RSC.
- (a) L. Shan, Z. Liang, X. Xu, Q. Tang and Q. Miao, Chem. Sci., 2013, 4, 3294–3297 RSC; (b) K. V. Rao and S. J. George, Org. Lett., 2010, 12, 2656–2659 CrossRef CAS PubMed; (c) Z. Wang, C. Kim, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 13362–13363 CrossRef CAS PubMed.
- (a) L. Nassar-Hardy, C. Deraedt, E. Fouquet and F. X. Felpin, Eur. J. Org. Chem., 2011, 4616–4622 CrossRef CAS; (b) S. L. Suraru, C. Burschka and F. Würthner, J. Org. Chem., 2014, 79, 128–139 CrossRef CAS PubMed; (c) R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518–5526 CrossRef CAS.
- E. K. Fields, S. J. Behrend, S. Meyerson, M. L. Winzenburg, B. R. Ortega and H. K. Hall, J. Org. Chem., 1990, 55, 5165–5170 CrossRef CAS.
- A. A. Frimer, J. D. Kinder, W. J. Youngs and M. A. B. Meador, J. Org. Chem., 1995, 60, 1658–1664 CrossRef CAS.
- (a) F. B. Mallory and M. B. Baker, J. Org. Chem., 1984, 49, 1323–1326 CrossRef CAS; (b) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed; (c) M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC; (d) M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC; (e) G. Tsuji, K. Kawakami and S. Sasaki, Bioorg. Med. Chem., 2013, 21, 6063–6068 CrossRef CAS PubMed.
- (a) J. Li, J. Chang, H. Tan, H. Jiang, X. Chen, Z. Chen, J. Zhang and J. Wu, Chem. Sci., 2012, 3, 846–850 RSC; (b) H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513–5517 CrossRef CAS PubMed.
- (a) T. Toyoshima, S. Yoshida and S. Watanabe, Tetrahedron, 2013, 69, 1904–1911 CrossRef CAS; (b) K. Noller, F. Kosteyn and H. Meier, Chem. Ber., 1988, 121, 1609–1615 CrossRef CAS.
- (a) Y. Ie, S. Jinnai, M. Nitania and Y. Aso, J. Mater. Chem. C, 2013, 1, 5373–5380 RSC; (b) A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- (a) J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bassler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS; (b) Q. Sun, H. Wang, C. Yang and Y. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- W. Wang, S. V. Dandrea, J. P. Freeman and J. Szmuszkovicz, J. Org. Chem., 1991, 56, 2914–2915 CrossRef CAS.
- J. Zhuang, C. Q. Wang, F. Xie and W. B. Zhang, Tetrahedron, 2009, 65, 9797–9800 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete synthetic details, NMR spectra and DFT calculation results. See DOI: 10.1039/c5nj01849h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |