
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlanar and distorted indigo as the core motif in novel chromophoric liquid crystals†
Jan H.
Porada
ab,
Jörg-M.
Neudörfl
a and
Dirk
Blunk
*a
aUniversität zu Köln, Institut für Organische Chemie, Greinstr. 4, 50939 Köln, Germany. E-mail: d.blunk@uni-koeln.de; Web: http://www.oc.uni-koeln.de/blunk Fax: +49 221 470 3064
bUniversität Stuttgart, Institut für Physikalische Chemie, Pfaffenwaldring 55, 70569 Stuttgart, Germany
First published on 31st July 2015
Abstract
Symmetrically bis-substituted indigo derivatives with long peripheral alkyl chains were synthesised by the reductive condensation of corresponding isatin derivatives. Their thermotropic mesomorphism was investigated with respect to different substitution patterns, which include the position and lateral modifications of the substituents. A systematic investigation of structure–property relationships revealed that only substitution at the 6 and 6′ positions affords the calamitic shape necessary to form smectic or nematic liquid crystalline phases. This finding is rationalised on the basis of a structural analysis of N,N′-diacetyl indigo and the consequences for 5,5′- and 6,6′-bis-substituted derivatives are discussed. Some of the liquid crystalline substances exhibit dichroism, which is especially pronounced in highly ordered phases. In addition, the 6,6′ substitution leads to significantly enhanced activity with respect to the photochemical trans–cis isomerization of N,N′-diacetylated indigo derivatives.
Introduction
The spatial anisotropic alignment of chromophores is an essential requirement for many high-tech applications, in which a macroscopic directed effect is generated by the interaction of light and matter. Examples of this can be found in the field of non-linear optics,1 organic photovoltaics,2 laser technology3 or supramolecular photo-chemistry.4 Due to the rather small HOMO–LUMO gap, which causes the colour of the chromophore, further applications in the field of molecular electronics are possible.5 The common methods for approaching the challenge of molecular alignment are shown in Fig. 1 and are described below. They are as follows: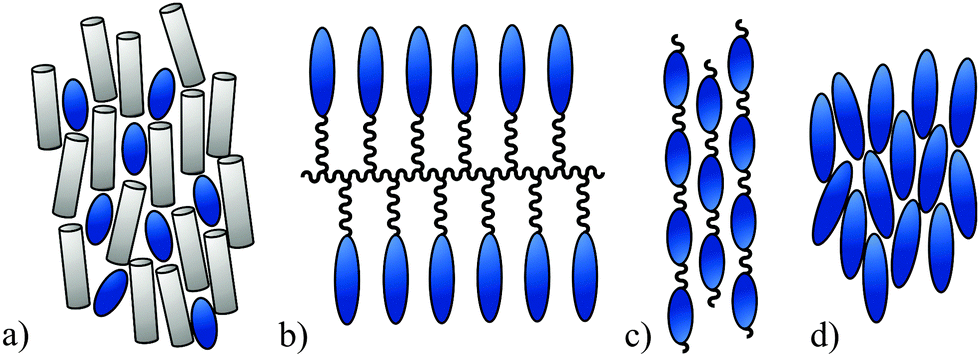 | ||
| Fig. 1 Possible methods used to align chromophores. | ||
(a) Embedding a chromophoric guest in an anisotropic host matrix. Such a matrix usually consists of low molecular weight or polymeric liquid crystals or polymers that have been aligned by stretching or even oriented in a polar order by various poling techniques.1b,c Field induced switching of the chromophore orientation can be achieved by choosing a polar matrix6 or an appropriate LC cell geometry.7
(b) Covalent integration of a chromophoric substructure into the side-chains of a polymer backbone (side-chain polymers).1b,c,8 Such side-chain polymers are often designed to have liquid-crystalline properties.1a
(c) Polymerization of chromophoric monomers to a main-chain polymer or copolymer.1b,9
(d) Structural integration of the chromophore into a molecular framework of a low molecular weight anisometric molecule, which is capable of forming a liquid crystalline phase.10 This method is by far the least common but nevertheless a very promising approach.
The latter method (d) leads to materials with a significantly higher chromophore density than host–guest-systems (a) without the possibility of phase separation or physical demixing. In contrast to polymers (b) or (c), the product can be synthesized and purified in a precise manner. The presumably higher solubility of such chromophoric mesogens compared to polymeric systems would be advantageous for technical applications.
In this context, indigo appears to be a suitable chromophoric molecular core as a starting point for the design of colored mesogens. This is due not only to its rather small chromogenic motif and high thermal and photochemical stability, but also to its C2h-symmetry. Due to the latter, indigo can be easily substituted in a symmetric way to generate rod-like molecules. Furthermore, many synthetic approaches towards indigo and its analogues—including donors such as selenium, sulphur or oxygen—are known, which will give broad access to a wide structural diversity of suitable molecules. Once the basic structure–property relations are studied, desymmetrisation by the corresponding synthetic strategies can broaden the structural diversity and give access to polar mesophases.
Despite the fact that indigo was intensely studied since the start of the 19th century, the reason for its remarkably low frequency absorption (indicating its small HOMO–LUMO gap) was inscrutable for decades. It was only in the 1960s that quantum mechanical calculations revealed that the biggest contribution to its chromophoric properties originates from its double-crossed donor–acceptor substructure, which is formed by vinylogous amides. The aromatic rings contribute to the chromophoric system mainly by stabilizing the coplanar conformation of the molecule and only marginally by mesomeric effects.11 The position of the absorption maximum can be fine-tuned by the type of the peripheral substituents.12
Herein, we present our results concerning the synthesis and thermotropic properties of a variety of symmetrically bis-substituted indigo derivatives.
Results and discussion
Synthesis
The structural requirements for mesogenic behaviour were established by the introduction of pendant groups with long aliphatic chains at the lateral positions of the phenyl rings of the indigo unit. Although numerous synthetic approaches to indigo are known, most of them are inadequate for the synthesis of complex derivatives due to their aqueous reaction conditions, which are incompatible with the extended aliphatic moieties of the pendant groups. An exception, which employs organic solvents, is the reaction of isatin (1) with phosphorus pentachloride in benzene and the subsequent reductive condensation of the resulting 2-chloro-3H-indole-3-one (2), and finally using zinc dust or hydrogen iodide as the reducing agent to yield indigo (3) with varying amounts of indirubin as the by-product. Actually, this procedure was one of the first indigo syntheses and was described by Baeyer in 1879.13 The formation of indirubin can be avoided by applying refined reductive conditions, i.e. using thiophenol as the reducing agent in benzene (Scheme 1) under an inert atmosphere, as published by Katritzky et al.14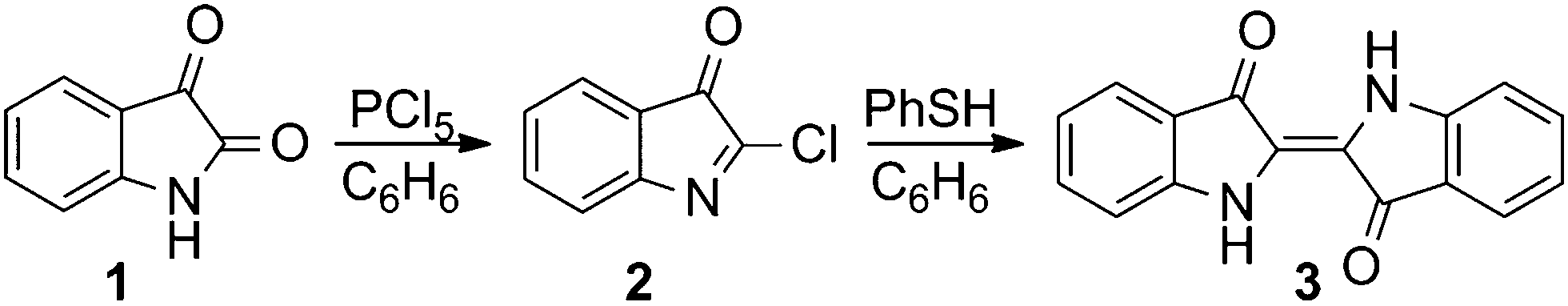 | ||
| Scheme 1 Synthesis of indigo (3) with thiophenol as a mild reducing agent performed in an organic solvent, as published by Katritzky et al.14 | ||
A modified one pot methodology was developed using the less toxic toluene as the solvent to transform substituted isatin derivatives of type B into the corresponding indigo derivatives of type C (Scheme 2). This procedure was also applied successfully with respect to the synthesis of analogue structures in our previous publication on phasmidic indigo derivatives.15 Pendant groups were introduced via Suzuki–Miyaura coupling of the bromoisatines of type A (Scheme 2) with the corresponding boronic acid of the pendant groups 4–7 (Fig. 2).16
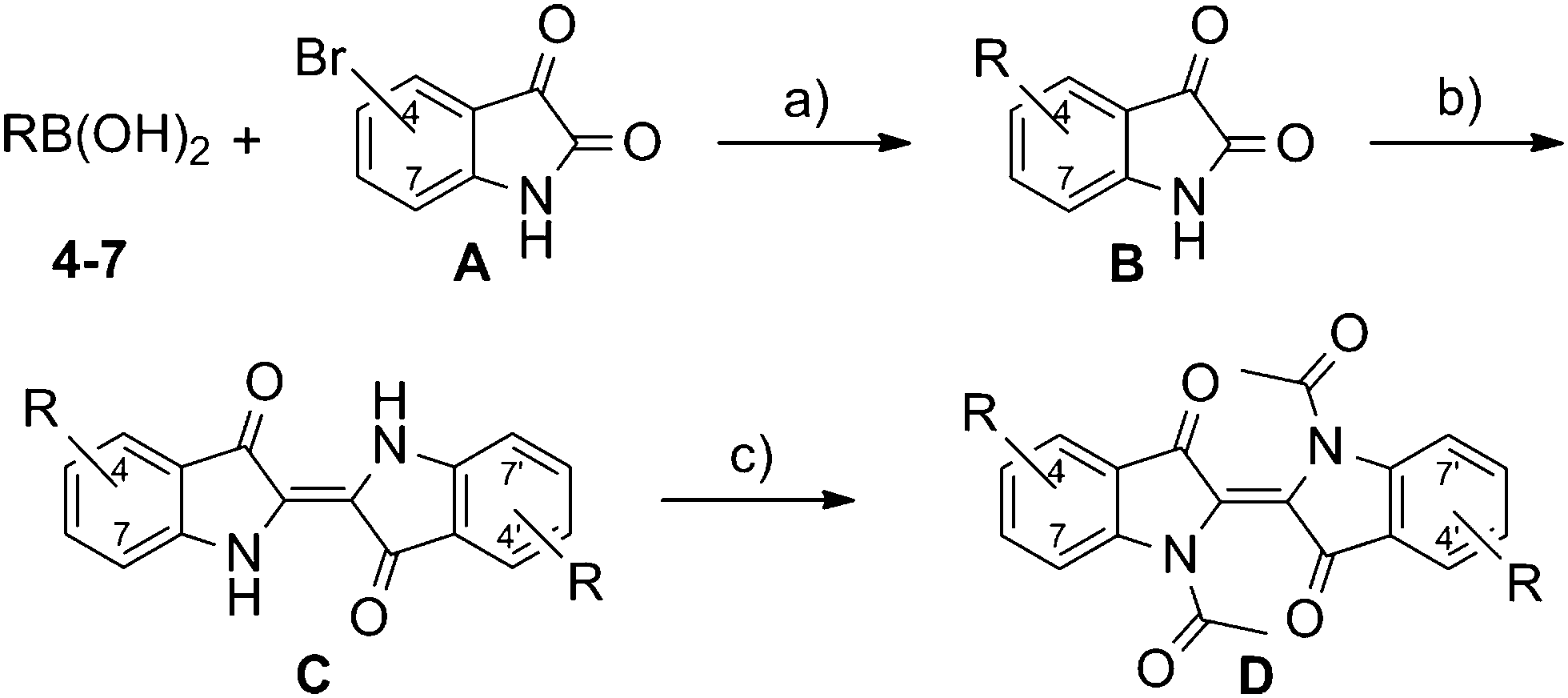 | ||
| Scheme 2 General synthetic route to the symmetrically bis-substituted indigo derivatives of type C and D. (a) K3PO4 (aq., 2 eq.), Pd(PPh3)4 (2 mol%), DME, 90 °C; (b) 1. PCl5 (1.05 eq.), 2. PhSH (2.2 eq.), toluene, 100 °C; (c) AcCl/Ac2O, NMP, 90 °C. | ||
For further diversification and to suppress the formation of hydrogen bonds, which are known to cause the high melting temperature of pure indigo,17 the vinylogous amide functions were masked by acetylation, thus affording the indigo derivatives of type D (Scheme 2). The acetylation method published by Liebermann and Dickhut using acetic anhydride as the solvent,18 though reactive towards indigo, did not result in any conversion. Moreover, when using polar aprotic solvents, conversion could be observed in NMP, but not in DMF or DMSO. The origin of this effect is not completely understood yet. The whole sequence, starting from the boronic acids 4–7 and bromoisatines A, is shown in Scheme 2.
To evaluate the influence of the pendant groups on mesomorphic behaviour, four boronic acids 4–7, which are shown in Fig. 2, were employed. These differ in the chain type, which are alkyl-(4) or alkoxy-chain (5–7) or in the lateral substitution of the substituent with a fluorine atom (6) or a methyl group (7).
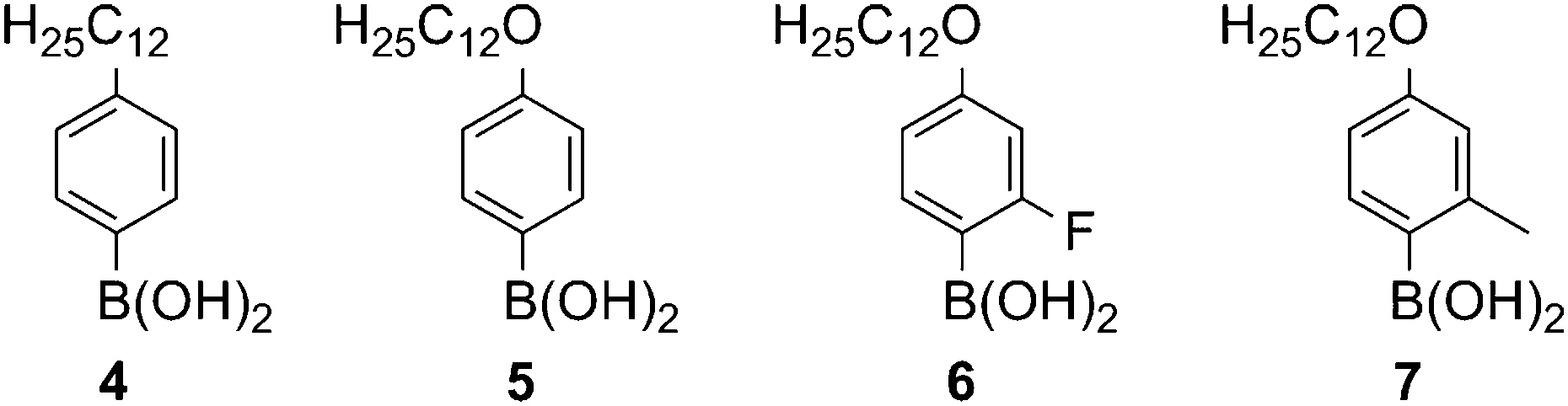 | ||
| Fig. 2 The boronic acids of the employed pendant groups 4–7. | ||
4–6 were synthesised according to published procedures,15,19 while 4-dodecyloxy-2-methyl-phenyl boronic acid (7) was obtained by the selective bromination of the 4-position of m-cresol in acetonitrile,20 followed by etherification with bromododecane, treatment with n-butyl lithium and trimethylborate and subsequent acidic workup.
Mesomorphic properties
The symmetrically substituted compounds of type C were investigated to observe the effect of shape anisotropy on the mesogenic properties of bis-substituted indigo derivatives.The four resulting regioisomeric 4-dodecyloxyphenyl-bis-substituted indigo derivatives 8–11 are shown in Fig. 3. Their thermotropic behaviour was investigated using polarization microscopy (PM) and differential scanning calorimetry (DSC) and the results are summarized in Table 1.
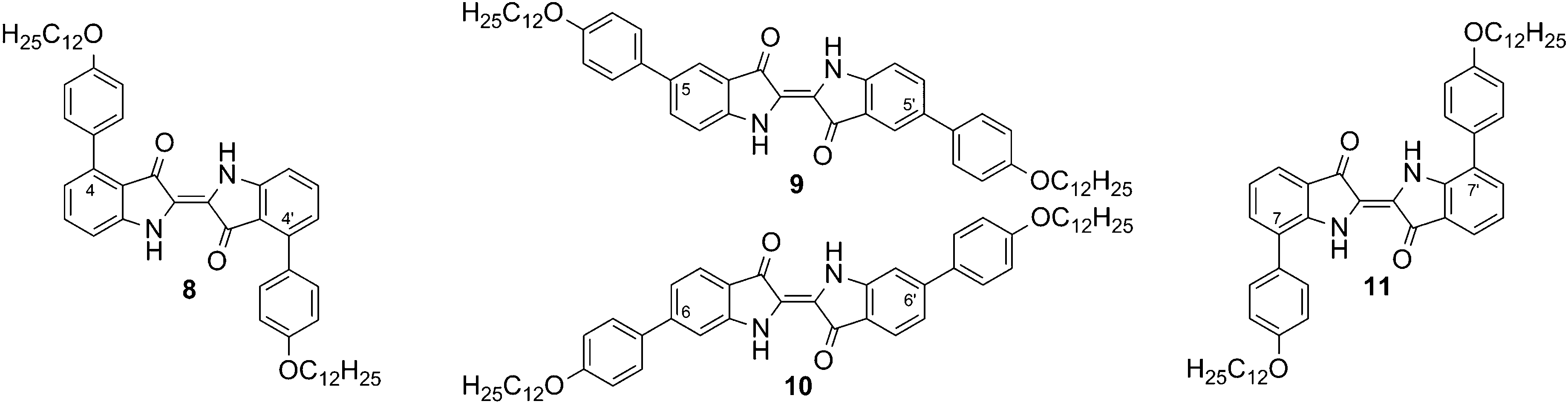 | ||
| Fig. 3 The four symmetrically 4-dodecyloxyphenyl-bis-substituted indigo derivatives 8–11. | ||
| Cr | • | I | |||
|---|---|---|---|---|---|
| a Form of designation: PM/DSC [°C] (ΔH [kJ mol−1]), SmX: smectic phase of unknown type. | |||||
| 8 | • | —/— | — | 253/251.1 (37.6) | • |
| 9 | • | 350/348 | SmX | Decomposition | — |
| 10 | • | 325/320 | — | Decomposition | — |
| 11 | • | —/— | — | 217/218.2 (30.1) | • |
As seen from Table 1, compounds 8 and 11 show a direct transition from crystal to isotropic liquid, whereas compounds 9 and 10 melt at considerably higher temperatures while undergoing decomposition. Shortly before the decomposition of compound 9, a fan-like texture, indicating a smectic phase, was observed by PM. Further characterization of the phase was not possible due to decomposition. The different melting behaviours of these four compounds can be rationalized with regard to the role of hydrogen bonds. It can be assumed that 9 and 10 form a hydrogen bond network, similar to indigo,17 which leads to high melting temperatures. In compounds 8 and 11, these intermolecular H-bonds are sterically hindered by 4-dodecyloxy substituents, which leads to a significant reduction of the melting temperatures. This interpretation is supported by the fact that 8 and 11 exhibit unusual high solubility in solvents such as chloroform, whereas 9 and 10 only form colloidal suspensions in chloroform.
Due to their rod-like molecular shape, the indigo derivatives 9 and 10 appeared to have the most promising structural motifs for designing liquid crystalline compounds. To overcome the high melting temperatures, the vinylogous amide functions were masked by acetylation, which led to the diacetylated compounds 12 and 13 shown in Fig. 4. The introduction of methyl groups as a possible alternative was not performed, because the dimethyl indigo is known to exhibit low thermal stability and readily oxidizes to methyl isatin.21
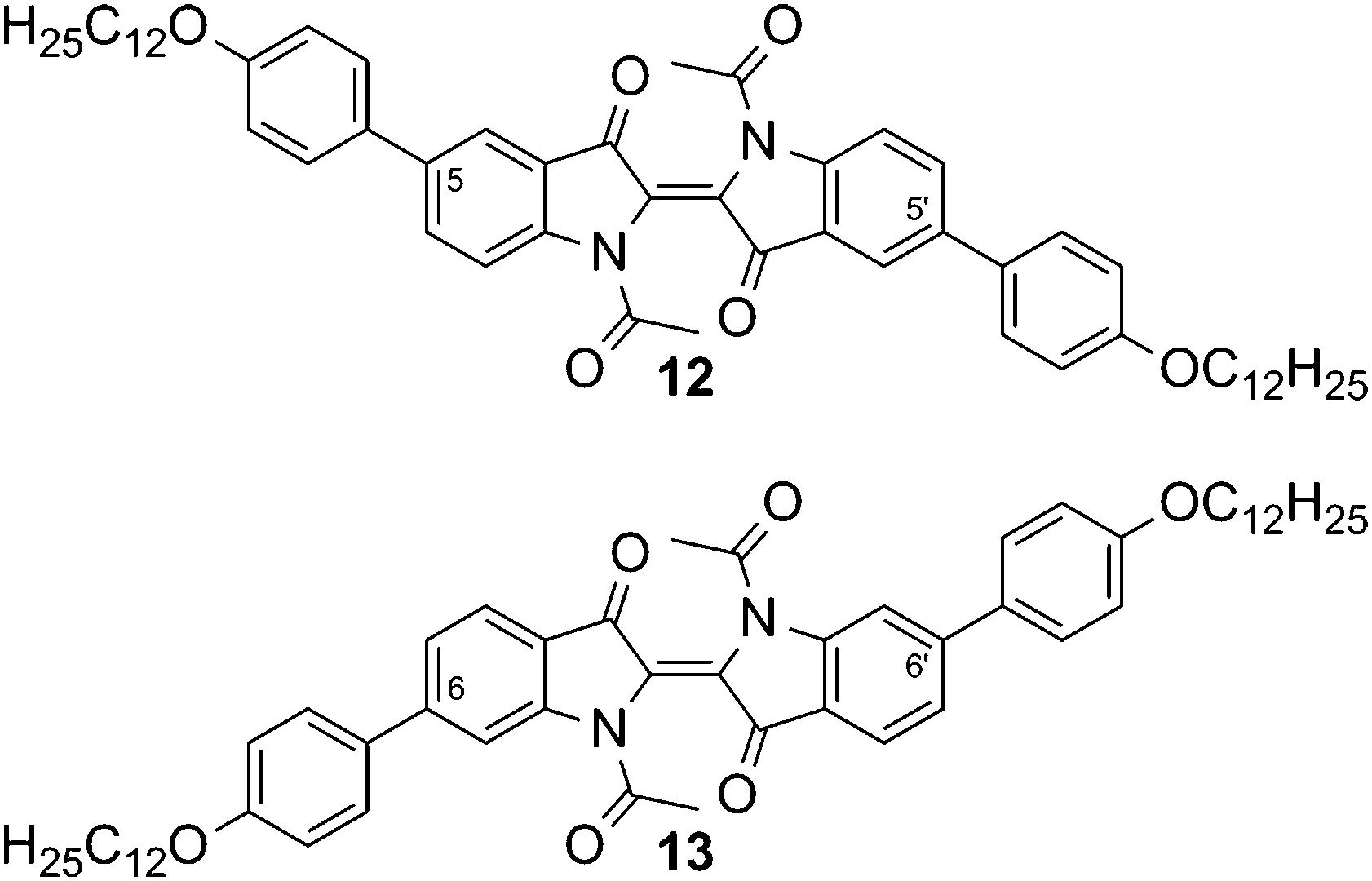 | ||
| Fig. 4 N,N′-Diacetylated indigo derivatives with 4-dodecyloxyphenyl pendant groups in positions 5,5′ (12) or 6,6′ (13). | ||
A dramatic reduction of melting temperatures, accompanied by an increase in solubility was achieved for compounds 12 and 13 with respect to their parent compounds 9 and 10, respectively (Table 2).
Upon heating, 12 undergoes a phase transition at 124.8 °C into a soft crystalline phase SC, which could only be observed on first heating. At 160.5 °C, this SC phase melts into an isotropic liquid. Compound 13 shows a phase transition at 180.6 °C into the SmA phase, as determined by PM. The phase grows from the isotropic melt as bâtonnets, which finally converge into a fan-like texture. Upon shearing, homeotropic alignment could be achieved. Using conoscopy, a uniaxial, optical positive character was identified, which is in accordance with the orientation of the molecular long axis perpendicular to the layers and parallel to the viewing axis.
In addition to our previously published phasmidic indigo derivatives, 13 now represents the first example of a calamitic liquid crystal that possesses an indigo core. To gain insight into the effect of different pendant groups on mesomorphic behaviour, the structures summarized in Fig. 5 were examined. The influence of the oxygen atom in the alkoxy chain was evaluated by comparison to analogue alkyl derivatives bearing no oxygen. Then, the effect of lateral substituents on the pendant groups was investigated. A differentiation between electronic and steric effects was achieved using fluorine or methyl groups as the lateral substituent. The thermal data of these compounds are presented in Table 3. The influence of the four different parameters, i.e. the substitution positions, the N-substituent R1, the chain type R2 and the lateral substituent X (see Fig. 5) will be discussed in the following section. In this discussion, the main subdivisions are initially given by the substitution positions and subsequently by the N-substituent R1.
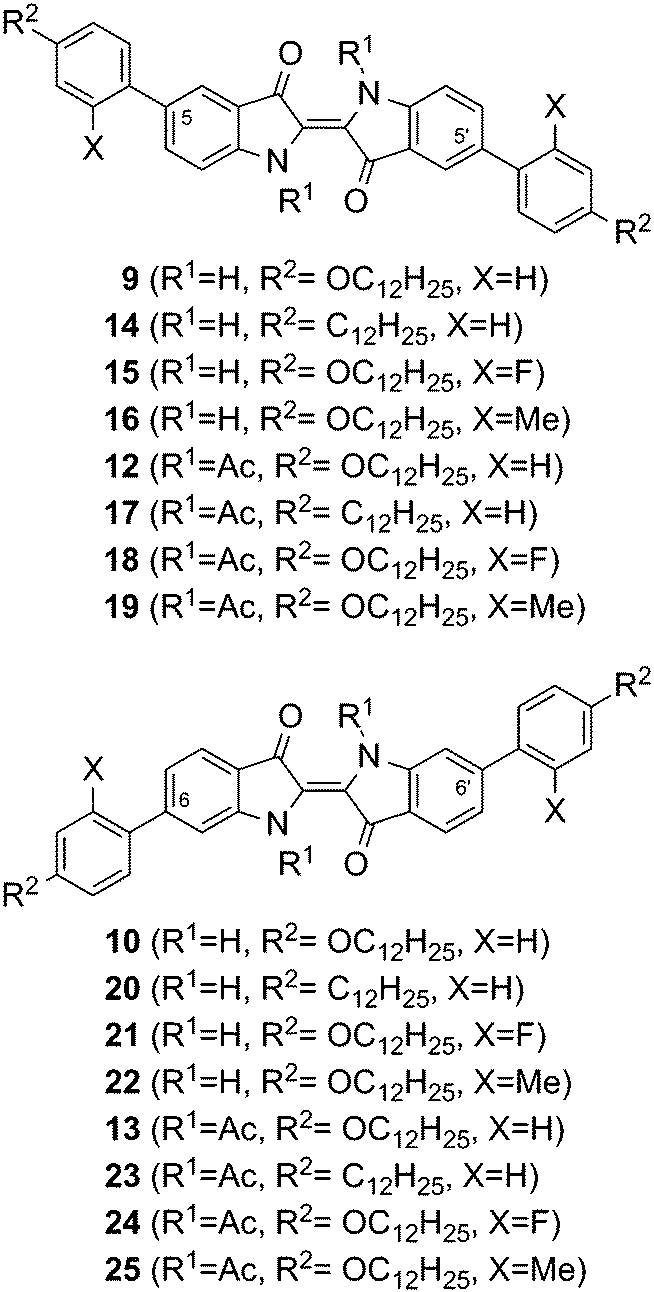 | ||
| Fig. 5 Compilation of the substitution patterns of the synthesised indigo derivatives with pendant groups in the positions 5,5′ or 6,6′ and N,N′-disubstitution. | ||
| Cr | • | • | I | ||||
|---|---|---|---|---|---|---|---|
| a Form of designation: PM/DSC [°C] (ΔH [kJ mol−1]). b Could only be observed in the first heating cycle. c Solidifies as amorphous glass. d Values from the cooling cycle, Cr: crystalline phase, SmX: smectic phase of unknown type, SC: soft crystalline phase, SmF/I: smectic F or smectic I phase, SmA: smectic A phase, N: nematic phase. | |||||||
| 14 | • | —/— | — | 345/341 | SmX | Decomposition | — |
| 15 | • | —/116.6 (38.3) | Cr2 | 320/322.9 (36.5) | SmX | Decomposition | — |
| 16 | • | —/— | — | —/241.4 (4.4) | SC | 266/268.6 (56.6) | • |
| 17 | • | —/— | — | —/— | — | 108/106.8 (56.2)b,c | • |
| 18 | • | —/— | — | —/— | — | 169/170.9 (32.6) | • |
| 19 | • | —/— | — | —/— | — | 110/110.6 (47.2) | • |
| 20 | • | —/— | — | 288/— | — | Decomposition | — |
| 21 | • | —/165.8 (13.7) | Cr2 | —/239.2 (7.9) | SmF/I | 295/296.8 (56.1) | • |
| 22 | • | —/— | — | —/141.0 (20.5) | SmF/I | 261/263.8 (44.2) | • |
| 23 | • | —/99.2 (18.8) | SC2 | 172/168.1 (7.6) | SmA | 176/178.0 (6.9)d | • |
| 24 | • | 141/140.7 (15.5) | SmA | 145/145.9 (1.3) | N | 149/149.3 (1.9) | • |
| 25 | • | —/— | — | —/— | — | 199/198.0 (50.3) | • |
For the 5,5′-bis-substituted indigo derivatives, a direct comparison between the N,N′-unsubstituted compounds 9 and 14 that differ only by an oxygen atom in the chain reveals a minor stabilization of the crystalline phase, which is caused by the alkoxy oxygen. Upon the introduction of lateral fluorinated pendant groups of compound 15, the formation of a second crystalline phase Cr2 was observed at 116.8 °C, which decomposes at 320 °C, forming a smectic phase while decomposing; this was also observed in compound 9. The laterally methylated pendant groups of 16 reduce the stability of the crystalline phase of the N,N′-unsubstituted compound enough to melt, without decomposition, into an isotropic phase after transitioning through a soft crystalline phase SC.
The mesomorphic behaviour of N,N′-diacetylated compounds 12 and 17 differ clearly with respect to each other. The soft crystalline phase SC, which was found in 12 was not detected in compound 17. Furthermore, its melting point is reduced by 53 °C with respect to 12 and it solidifies as an isotropic glass. For the corresponding laterally fluorinated compound 18, stabilization of the crystalline phase by 10 °C could be observed compared to 12.
The laterally methylated analogue 19, much like 17, shows significant destabilization of the crystalline phase by approximately 50 °C with respect to 12. None of the 5,5′-bis-substituted, N,N′-diacetylated compounds possess liquid crystalline phases.
The 6,6′-bis-substituted indigo derivatives already show clear differences at the N,N′-unsubstituted stage, in contrast to the 5,5′-bis-substituted derivatives. Compared to the bis-alkoxyphenyl substituted compound 10, the bis-alkylphenyl substituted compound 20 shows a reduction in the melting temperature by approx. 30 °C, but decomposition still occurs. For both, the N,N′-unsubstituted laterally fluorinated compound 21 and especially the lateral methylated compound 22, a decrease in the stability of the crystalline phase in comparison to 10 and the formation of a high order liquid crystalline phase were observed. The decrease of the crystal phase stability is approximately 80 °C for the fluorinated compound 21, while it is 180 °C for the methylated analogue 22. Both compounds enter the isotropic phase without decomposition. The liquid crystalline phase found for 21 and 22 was assigned either to be of SmI or SmF type, based on the following observations.
Shearing is possible, but high resistance indicates a high degree of molecular order. Furthermore, the high transition enthalpies of the liquid crystal to isotropic transition support this assumption.
The phase growth from the isotropic melt is dendritic, as is typically observed for highly ordered phases.
A smectic phase is more reasonable than a columnar phase, due to the particular molecular shape and non-curved polar/apolar-interface of these compounds.
Conoscopy revealed an optical biaxial character in each domain. An orientation of the bisectrix parallel or orthogonal to the viewing axis could be excluded. Such an orientation cannot be explained without assuming an intrinsic tilt of the molecules with respect to the layer normal. Therefore an SmB-phase can be excluded.
Evidently, bis-substitution in positions 6 and 6′ has a greater influence on phase behaviour compared to bis-substitution in positions 5 and 5′. Depending on the 6,6′-bis-substituent, the stability of the crystal phase can be reduced tremendously. An explanation for this could be that the sterically demanding substituents in these positions cause a change in the H-bond network. The formation of a three dimensional H-bond network, known from indigo,17 in which each molecule is connected with each one H-bond to four other molecules oriented perpendicular is sterically hindered. Instead one dimensional H-bond strands are formed in which each molecule is connected by two H-bonds to two others in parallel orientation. As a result of such uniform alignment of light absorbing molecules, a pronounced dichroism should be observed, which was found for the 6,6′-bis-substituted indigo derivatives 21 and 22 in the LC phase. Fig. 6 shows the UV-Vis-absorption spectrum of 22, which has two maxima in the visible range. The absorption at 604 nm causes a blue visual impression, whereas the absorption at 398 nm relates to a yellow colour. Indeed, as a consequence of the superposition, the substance appears green.
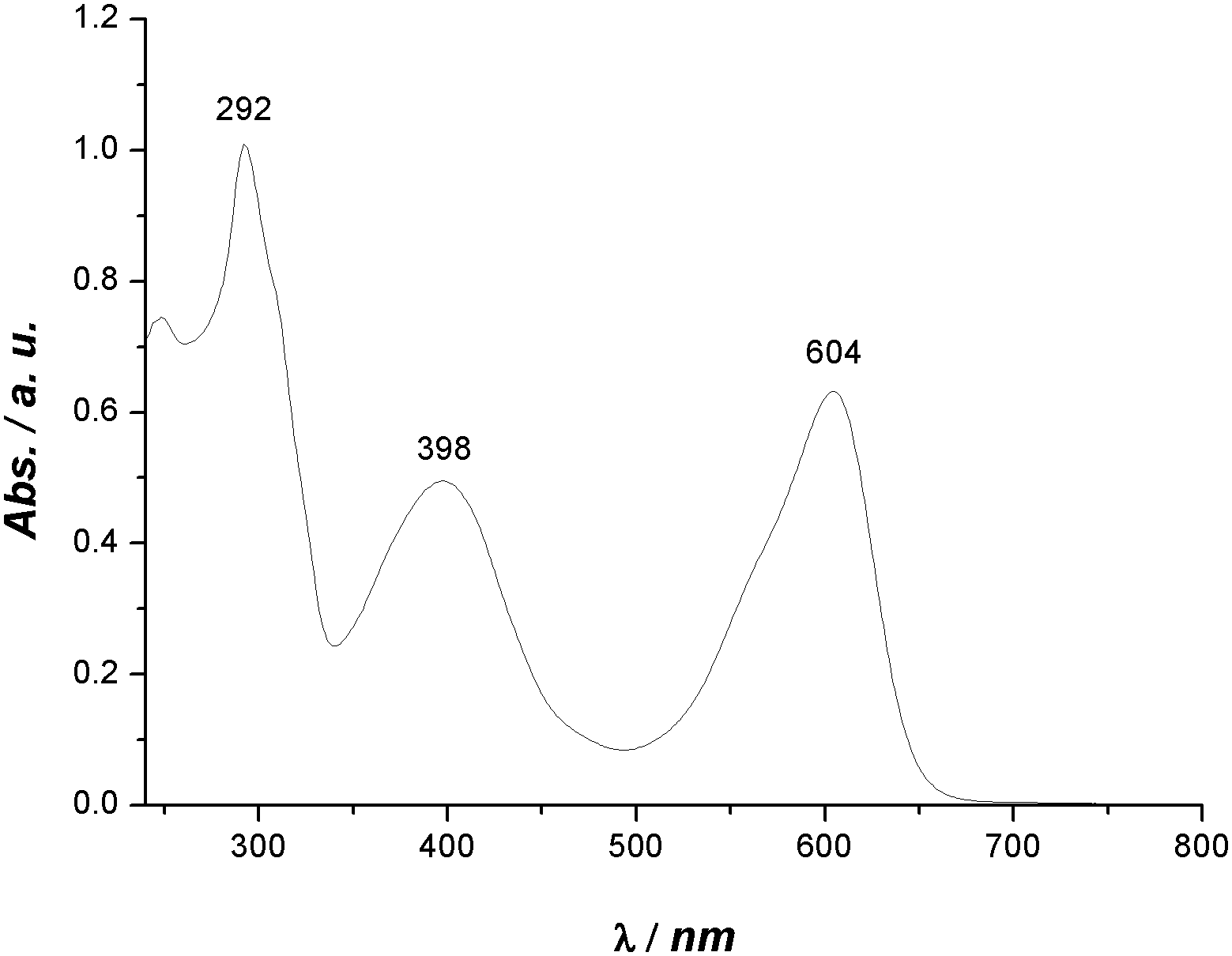 | ||
| Fig. 6 UV-Vis spectrum of 22 (20 mg L−1 in NMP). | ||
Under a polarization microscope, when only one polarizer is applied, domains ranging from blue to yellow can be observed. Upon rotating the polarizer by 90°, the colour appearance of the blue and yellow domains are inverted (Fig. 7), indicating that the absorption maxima belong to electronic excitation with the vectors of the transition dipole moments being oriented orthogonal. Dichroism in indigo derivatives has already been reported for 6,6′-dichloroindigo in the crystalline form.22 Large and homogeneous oriented domains can be easily generated within the LC phase by preparation on treated substrates and reach dimensions not easily accessible with crystalline materials. The orientation can be frozen by rapid cooling; thus, the appearance of dichroism in the LC phase might also be valuable for technical applications. The UV-Vis spectra of 8–13 are presented in the ESI.†
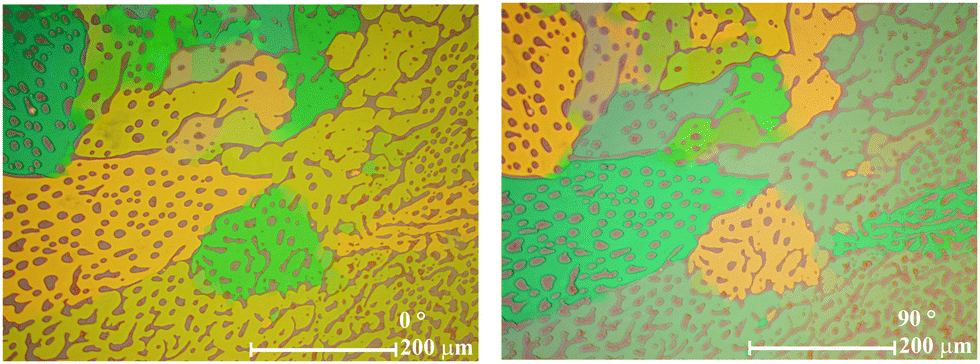 | ||
| Fig. 7 The liquid crystalline phase of 22 at 260 °C with only one polarizer horizontal (left) and vertical (right) exhibits strong dichroism. | ||
The N,N′-diacetylated, 6,6′-bis-alkylphenyl substituted 23 exhibits a different phase sequence as well as a lower clearing temperature, compared to its alkoxy-analogue 13. At 99.2 °C, phase transition into a soft crystalline phase SC2 was detected, which manifests in a paramorphic “quasi fingerprint texture” when cooling from the upper SmA phase. Such a texture is known from the soft crystalline E phase; however, at this stage of investigation, a definite assignment cannot be given. The SmA phase of 23 was identified using PM and conoscopy, which was observed to have an identical appearance as the SmA phase of 13.
The corresponding laterally fluorinated or methylated compounds 24 and 25 exhibit complementary properties not only with respect to their N,N′-unsubstituted analogues, but also with respect to each other. The crystalline phase of the lateral methylated compound 25 shows a much higher stability compared to the laterally fluorinated compound 24, which is the opposite case, as observed for the N,N′-unsubstituted analogues. The methylated indigo derivative 25 melts directly into the isotropic phase at 198.0 °C without formation of a liquid crystalline phase. The lateral methyl group even stabilizes the crystalline phase by approximately 20 °C compared to compound 13 which has no lateral substituent.
The fluorinated compound 24 shows a phase transition at 140.7 °C into the SmA phase followed by a transition at 145.9 °C into the nematic phase, which clears at 149.3 °C. Both phase types were identified by the characteristic textures observed by PM. The SmA phase shows a fan-like texture and has an identical appearance as described for 13. The nematic phase exhibits a schlieren texture, which includes singularities with the disclination strength of s = ±½ and s = ±1. The phase assignments were confirmed by wide angle X-ray scattering (WAXS). In addition, the order parameter S for each phase type could be obtained from the intensity profile of the wide angle area, according to the method of Davidson et al. (see ESI†).23
The nematic phase of 24 possesses a value of S = 0.47 at 148 °C, which is within the typical range of the nematic phase. The determined order parameter for the SmA-phase is S = 0.55 at 144 °C, which is unusually low.24 This is most likely a result of the bent shape of the distorted N,N′-diacetyl indigo core, which is discussed in more detail below.
Conformational aspects
For the examined N,N′-diacetylated compounds, liquid crystalline behaviour was exclusively found with the substitution pattern in the 6 and 6′ positions. To gain insight into the origins of this observation, the inherent molecular distortions of N,N′-diacetyl indigo in general were analysed and at a later step compared to those of the peripherally bis-substituted N,N′-diacetyl indigos 12 and 23.When the planar indigo molecule is N,N′-disubstituted, the ring systems, which are connected by the central double bond, experience rotational distortions in all three spatial dimensions with respect to the double bond, as illustrated in Fig. 8.
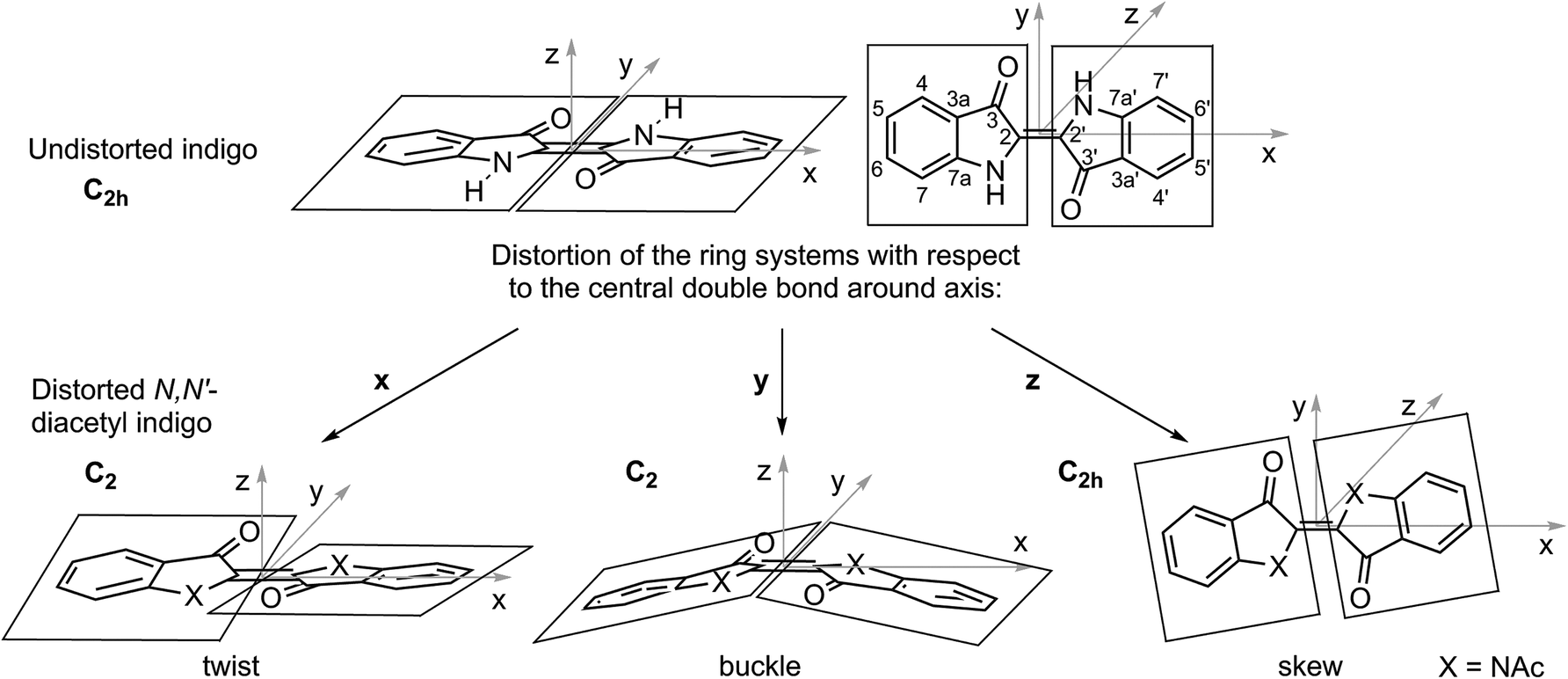 | ||
| Fig. 8 Rotational distortions of the ring systems with respect to the central double bond induced by N,N′-disubstitution of the indigo motive. When the molecule is positioned in the centre of the xy-plane with the double bond along the x-axis, a rotational distortion around the x-axis causes a twist (left), that around the y-axis causes a buckle and that around the z-axis causes a skew (right). For the twist and the buckle, the symmetry point group is reduced from C2h to C2, whereas it is retained C2h for the skew because this distortion is along the C2-axis of the molecule. | ||
It is known that the N,N′-disubstitution of indigo leads to the buckling and twisting of the central double bond. This phenomena is caused by the steric demand of the substituent and was first observed for N,N′-dimethyl indigo,25 but appears in a more pronounced form in N,N′-diacetyl indigo. The buckling is a direct result of the twist of the central π-bond, because the sp2 orbitals of the bonded atoms rehybridise with a higher p-character (spn, with 2 < n < 3), which leads to the pyramidalisation of the bond geometry. The origins of the skew are the different bond lengths and pyramidalisations for different atoms within each ring system.
Because buckling and twisting are interrelated to each other, it is nontrivial to separately describe these distortions as required for a quantitative comparison between different N,N′-diacetylated indigos. A powerful tool was found in the p-orbital axis vector (POAV) analysis, as introduced by Haddon et al.26 In the POAV1 theory, the p-orbital axis vector is defined to have equal angles Θσπ ≥ 90° to all three σ-bonds. Θ = Θσπ − 90° precisely quantifies the pyramidalisation from which the smp hybridization and the average spn hybridization may be calculated. The degree of pyramidalisation of the C2-atom ΘC2 = Θσπ(C2) − 90° is a direct measure for the buckling of the molecule.
However, in the POAV1 theory, equal σ-bond hybridization in the C3v symmetry is assumed, which may lead to significant deviations of the actual hybridization, when the bond angles Θij, of the surrounding atoms substantially differ. This case is given for the indigoid structures examined here, and a more precise description of the axis vector and the smp-hybridisation is then obtained from the POAV2 theory, which treats σ-bond hybridizations individually.27 The dihedral angle, Θππ, between two POAVs accurately designates the π-orbital alignment and the actual twist within the molecule and is separated from the buckling distortion. The torsion angle, Θ(C2–C2′), precisely quantifies the twist of the indigo motif. Details on the performed calculations of the POAVs are given in the ESI.† The angle, σ is defined as the angle between two lines, one passing through the centre of C5/C6 and the centre of C5′/C6′ and the other one passing through C2 and C2′. This angle σ specifies the lateral skew of ring systems with respect the central double bond. It is defined to be σ > 0 when the ring systems are shifted in a direction in which the C5 and C5′ atoms are more in line with the central double bonds than C6 and C6′.
In addition, four further characteristic angles were determined. The effective ring twist, α describes the torsion between the ring systems and is defined as the dihedral angle of C5–C6–C6′–C5′. This twist originates from the torsion Θ(C2–C2′) of the central double bond but may be altered in magnitude by compensation or amplification due to further internal twists within the ring systems. βC5 is defined as the angles between two lines, one passing through the C5 carbon atom and the opposing C7a atom in the same phenyl ring, and the other one passing through the C5′ and the C7a′ of the phenyl ring on the other side of the central double bond. βC6 is correspondingly defined for lines passing through C6 and C3a on one side and C6′ and C3a′ on the other side. These angles can have values from 0° for parallel alignment to 90° for orthogonal alignment and serve as a measure of the bent of the indigoid structure with respect to the corresponding substitution position 5 and 6. The overall bent γ is defined as the sum of two angles each between two lines. The central line passes through C2 and C2′. The plane lines connect the centre of C5/C6 and centre of C3a/C7a on one ring system and the centre of C5′/C6′ and centre of C3a′/C7a′ on the other ring system. The sum of the angle between the central line and each plane line describes the position unspecific bent of the molecule. The plane lines are on the same plane as the lines used to define βC5 and βC6, which allows the direct comparison of these different measures for the bent.
Computational results
The molecular geometry of N,N′-diacetyl indigo, which was obtained from X-ray diffraction, was discussed and compared to quantum mechanical calculations on the AM1-SCF-level by Grimme et al.28 However, in this discussion, different angle definitions were applied. The C2-symmetric molecular shape of this calculated structure is in accordance with the overall molecular geometry found from X-ray diffraction, but shows strong deviations with respect to the magnitude of the angles. Therefore, we decided to recalculate the molecular geometry using the more recent B3LYP density functional29 and the 6-311G(d)-basis set, as implemented in the Gaussian 0330 package (see ESI†). Although in the crystal structure only the C2-symmetric species was found, we also considered an alternative Ci-symmetric molecular geometry in our calculations, which also minimizes steric strain and, in principle, may exist in solution or fluid phases. In such Ci-symmetric conformation, the pyramidalisation, ΘC2, is of opposing algebraic sign for C2 and C2′, thus the angle of torsion, Θ(C2–C2′), of the ring systems is 180°, which leads to a step-like arrangement. Both possible geometries were found as local energetic minima and are shown in Fig. 9. However, the difference of the sum of the electronic and zero point energies of both the C2- and Ci-symmetric conformations was computed to be ΔH = 32.1 kJ mol−1, favouring the C2-symmetric conformer. Thus, the existence of the Ci-symmetric conformer in solution or a fluid phase can be neglected. | ||
| Fig. 9 Calculated geometries of N,N′-diacetyl indigo in the actual C2-symmetry (left) and the hypothetical Ci-symmetry (right). | ||
The calculated and experimental results are summarized in Table 4. The stepwise arrangement of the two ring systems in the Ci-symmetric conformer only allows the relief of the steric strain of the N-acetyl bond by an extremely high torsion of Θ(N–C8) = 38.4°. As a result, herein, the most dominant pyramidalisations are found for N and C8, whereas the pyramidalisations for C2 and C3 are significantly lower than in the C2-symmetric conformation. The inversion symmetry of the Ci-conformer implies opposing algebraic signs for each ring system, hence an effective ring twist α and an overall bent γ of zero. Consequently, β5 and β6 are zero, indicating a parallel alignment for each set of lines. The seemingly high skew of σ = 11.63° is an artefact caused by its definition, because for a stepwise alignment it determines the magnitude of the step and not the lateral skew. Other possible definitions, for instance the dihedral angle between the centre of C5/C6, the centre of C5′/C6′, C2′ and C2, indicate a skew of 0°. The situation is very different for the C2-symmetric conformer, which results from the combination of the twist, buckle and skew distortions in all three spatial dimensions. In this conformer, the ring systems of the indigo structure are distorted in a disrotatory way by the twist and buckle deformation, but in a conrotatory way for the skew deformation.
| C i-symmetry (DFT calc.) | C 2-symmetry (DFT calc.) | N,N′-Diacetyl indigo (exp.)28 | ||
|---|---|---|---|---|
| Θ X denotes the angle of pyramidalisation of the atom X, m is the degree of s-character of the smp hybrid orbital, Θ(X–Y) is the dihedral angle between the smp-orbitals of the two neighbouring atoms X and Y and dX–Y is the distance between X and Y. Pointy brackets indicate the average value from both sides of the central double bond. Ln denotes a line, which is defined to pass through the specified atoms. The angle between the lines was determined using the Diamond 3 software from Crystal Impact. The notation X/Y indicates that a dummy atom was used at the centre between atom X and Y.a Obtained from POAV1 analysis.b Obtained from POAV2 analysis.c Significant asymmetry. A detailed list of all the calculated parameters is given in the ESI. | ||||
| 〈ΘC2〉a [°]/mb | ±1.71/0.002 | 5.44/0.017 | 5.90/0.020 |
|
| 〈ΘN〉a [°]/mb | ±7.07/0.029 | 3.91/0.009 | 5.99/0.021 | |
| 〈ΘC3〉a [°]/mb | ±0.23/0.000 | 1.35/0.001 | 1.72/0.002c | |
| 〈ΘC8〉a [°]/mb | ±2.77/0.005 | 1.85/0.002 | 1.36/0.001 | |
| 〈Θ(C2–C2′)〉b [°]/dC2–C2′ [Å] | 180.00/1.362 | −24.15/1.364 | −19.37/1.349 | |
| 〈Θ(C2–C3)〉b [°]/〈dC2–C3〉 [Å] | ±11.64/1.514 | 12.44/1.509 | 13.87c/1.501 | |
| 〈Θ(C2–N)〉b [°]/〈dC2–N〉 [Å] | ±159.12/1.413 | 162.06/1.407 | 157.67c/1.414 | |
| 〈Θ(N–C8)〉b [°]/〈dN–C8〉 [Å] | ±38.40/1.438 | −21.02/1.424 | −20.49c/1.411 | |
| σ Ln (5/6–5′/6′) to Ln (2–2′) [°] | 11.63 | 4.28 | 7.37 | |
| α (C5–C6–C6′–C5′) [°] | 0.00 | −24.67 | −19.17 | |
| β C5 = Ln (5–7a) to Ln (5′–7a′) [°] | 0.00 | 49.14 | 52.90 | |
| β C6 = Ln (3a–6) to Ln (3a′–6′) [°] | 0.00 | 23.45 | 32.44 | |
| γ = Ln (5/6–3a/7a) to Ln (2–2′) + Ln (5′/6′–3a′/7a′) to Ln (2–2′) [°] | 0.00 | 43.65 | 51.44c | |
For the sake of clarity, it is helpful to first describe the effect of each distortion separately. The torsion of the central double bond by φ just by itself twists the ring planes such that φ = α, because the defined peripheral bond vectors of the dihedral angle are perpendicular to the rotational axis. In this case, the 5 and 5′ positions migrate to the opposite face of the central double bond compared to the 6 and 6′ positions.
The magnitude of β5 and β6 is identical, but smaller than φ, because the defining lines are not perpendicular to the axis of rotation. The overall bent γ would remain zero for only the twist deformation because all the lines are along the rotational axis. The buckling of the C2-symmetric conformer is a result of the pyramidalisation of C-2 and C-2′ by ΘC2 and ΘC2′, respectively, which have identical algebraic signs. Therefore, the buckling without twist or skew would lead to the bending of the central double bond, thus leaving the 5, 5′, 6 and 6′ positions in the same plane, parallel to the double bond, and resulting in α = 0 and βC5 = βC6. The overall bent γ would be more acute than βC5 and βC6 as the line between the centres of C5/C6 and C3a/C7a runs perfectly perpendicular to the axis of distortion, while the lines defining the angles β5 and β6 are not. The skew deformation by itself leaves both ring systems on the same plane, thus the effective ring twist α remains zero. Because the ring systems are rotated conrotatory, the defining lines for βC5 and βC6 each stay parallel; hence, βC5 = βC6 = 0. The defining angles for the overall bent γ are of the same magnitude, but of opposing algebraic signs and will cancel out, resulting in γ = 0.
As indicated in Fig. 8, the twist and buckle deformations, by themselves, each result in a chiral C2-symmetric conformer. By inverting the algebraic sign to either φ or ΘC2 or ΘC2′, the enantiomer of the respective conformation is formed. Because enantiomers are energetically degenerated, there is no directional preference for the bending or the torsion of the double bond.
The conformers that result when twisting and buckling occur together still retain C2-symmetry because the C2-axis for both singly distorted conformers are identical; however, but now they are diastereomeric. The buckling in one direction energetically discriminates the direction of the twist and vice versa. In the preferred conformation, the acetyl groups are on the convex face of the molecule, due to the reduction of steric strain. Thus, the 5 and 5′ positions, which are para relative to the amide groups, are pushed towards the concave molecular face. The degrees of the effective ring twist α and the overall bent γ remain nearly unaffected by the combination of these distortions; however, now there is a substantial difference between βC5 and βC6, because the angle between the lines defining βC5 are now more acute than the overall bent γ and the lines defining βC6 are now less acute. Just considering the twist and the buckle deformation, the difference between γ and each β angle should be identical. However, the positive skew rotates the ring systems in such a way that the 5 and 5′ positions are more in line with the central double bond, which shifts the ring systems up on their planes and decreases both βC5 and βC6, while leaving the overall bent γ nearly unaffected. Thus, the position specific molecular bending βC5 is now only slightly higher than γ, whereas the position specific molecular bending βC6 is significantly reduced compared to γ. As a result, 5,5′-bis-substituted N,N′-diacetyl indigo derivatives show high molecular bents and are not suitable for the formation of mesophases, whereas the 6,6′-bis-substituted N,N′-diacetyl indigo derivatives are considerably less bent. The molecular shape of the latter derivative is still not ideally rod-like, but within the window of calamitic mesogens. Deviations in magnitude from this general trend are caused by further internal distortions within each ring system.
Although the DFT-calculation of the C2-symmetric conformer gives much better results than that obtained by the AM1-SCF-method, it can be seen that the pyramidalisation, Θσπ, and the s-character of the smp hybrid orbital of the atoms C2, C3 and especially N are generally underestimated when compared to the crystal structure, whereas ΘC8 is slightly overestimated. On the other hand, the torsion Θ(C2–C2′) of the central π-bond was predicted to be 4.8° higher than actually found, which is also reflected in the difference of the bond lengths, whereas the average torsion 〈Θ(C2–N)〉 was computed to be smaller by 4.4°. It should be noted that due to crystal packing effects, these average torsion angles show significant asymmetric deviations of 7–12°; however, the more consistent bond lengths substantiate this trend.
The most significant deviations were found in connection to the nitrogen atom. It appears that the steric strain, which is induced by the acetyl groups, is stronger compensated by the N pyramidalisation ΘN and the torsion Θ(C2–N) than predicted. This allows a less twisted central π-bond and a higher degree of conjugation between the two ring systems. On the other hand, due to the higher distortion of the nitrogen bonds, the skew angle σ is significantly increased by 3.1°, when compared to the computed gas phase structure. For both cases, the computed and the measured structure, the effective ring twist α very accurately reflects the torsion Θ(C2–C2′) of the central π-bond. The overall bent γ, and thus βC5 and βC6, was found to be substantially higher for the crystal structure than predicted for the gas phase. However, in the crystal structure, the two angles contributing to γ, one for each ring system, largely differ (34.5° and 16.9°), indicating strong crystal forces. The higher value was found for the ring system with the stronger pyramidalisation of atoms.
Crystal structures
For the peripherally bis-substituted compounds 12 and 23, it was possible to obtain crystals that were suitable for single crystal X-ray diffraction. Fig. 10 and 11 show two projections of the molecular structures of 12 and 23, respectively.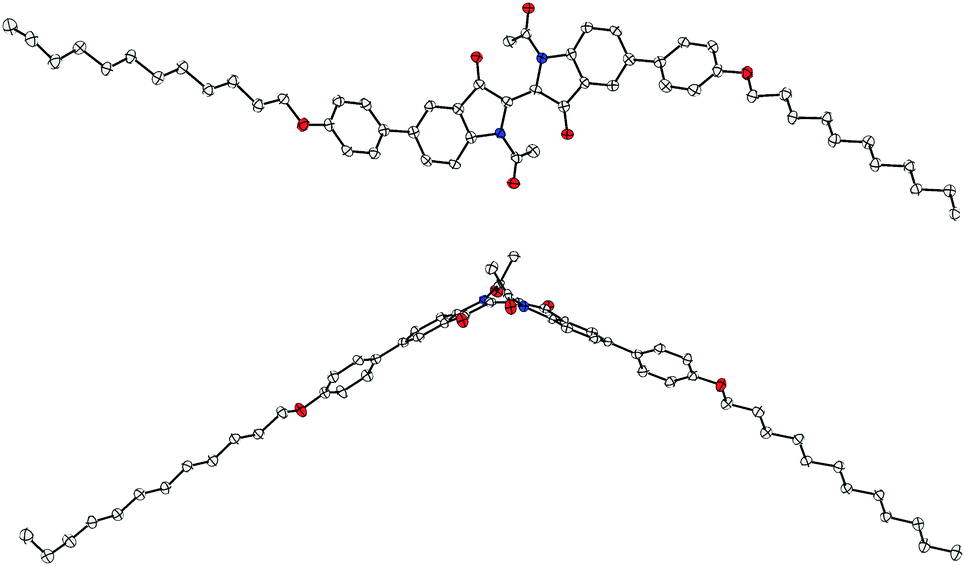 | ||
| Fig. 10 Structure of the 5,5′-bis-substituted compound 12 in the solid state, which is shown in two projections. The anisotropic displacement parameters are drawn at the 50% probability level. Oxygen atoms are drawn in red; nitrogen atoms are blue. Only one crystallographic type of molecule was observed. | ||
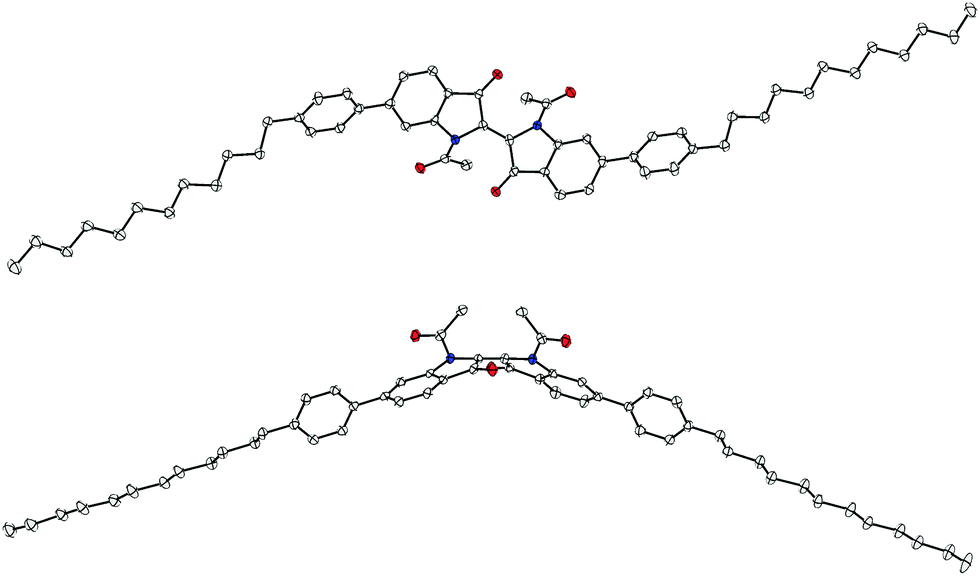 | ||
| Fig. 11 Molecular structure of the 6,6′-bis-substituted compound 23 as found in the crystalline state, which is shown in two projections. The anisotropic displacement parameters are drawn at the 50% probability level. Oxygen atoms are drawn in red; nitrogen atoms in blue. Only one crystallographic type of molecule was observed. | ||
Similar to the unit cell of the parent structure N,N′-diacetyl indigo, only one crystallographic type of molecule was found as a racemate of conformers in both crystal structures. However, 23 was crystallized from NMP and its crystal structure contains two molecules of NMP per molecule 23 with one of them being disordered.
It is known for anilines that electron withdrawing substituents decrease the pyramidalisation of the nitrogen due to the increased donor–acceptor conjugation between the nitrogen lone pair and the substituent. This effect is strongest for para-substitution, but prevails to a lesser extent for meta-substituents.31 Electron-pushing substituents, such as 4-alkylphenyl or 4-alkoxyphenyl, show the opposite effect. To estimate if the different distortions of 12 and 23 found in their crystal structures originate from the altered substitution pattern or from crystal packing effects, the geometries of a set of correspondingly substituted N,N′-diacetyl indigo structures were computed using the same DFT-method as described above (see ESI†). With regard to the computational effort, the chain length of the substituent was reduced to 4-ethylphenyl (PhEt) and 4-methoxyphenyl (PhOMe).
In Table 5, the experimentally determined molecular geometries of 12 and 23 are compared to the computed structures. The C2 pyramidalisation, ΘC2, is in good accordance for all the structures, and slightly increased for 12. The torsion, Θ(C2–C2′), of the central double bond shows high conformity in all experimentally determined geometries, but appears to be systematically overestimated by 4°–5° in the computed structures. Substantial differences were found for the parameters, which are associated with the nitrogen atom and the acetyl group. Compared to the unsubstituted N,N′-diacetyl indigo structure, the pyramidalisation, ΘN, of the nitrogen atom is strongly decreased for the 5,5′-bis-substituted 12, whereas it is significantly increased for the 6,6′-bis-substituted 23, which is also indicated by the higher torsion Θ(C2–N) between the nitrogen and the neighbouring carbon of the central π-bond. This finding could be explained by the conjugation between the electron-pushing substituent at position 5 with the N-acetyl group in the same ring system as well as the N-acetyl and the carbonyl group of the opposing one, all of which involve nitrogen atoms. Conjugation for the substituent in position 6 only occurs with the carbonyl group of the same ring system, thus leaving the nitrogen atom unaffected. The computed geometries of the corresponding substituted molecules confirm this trend but to a diminished extent, indicating strong crystal forces. In 12, the steric strain induced by the acetyl groups is mainly compensated by the high torsion Θ(N–C8) between the nitrogen atom and the carbon of the acetyl group, whereas 23 shows considerably lower values for these torsions. The position specific bent angles βC5 and βC6 of 12 are in good accordance with those of the computed structures. In 23, on the other hand, the strain appears to be compensated to a higher extent by internal distortions of the ring systems, causing them to be less twisted against each other but generally more bent than 12, as indicated by the values for α and γ. Both βC5 and especially βC6 of compound 23 are higher than that predicted computationally. All the computed structures show, independent of their substitution pattern, only little deviation among each other for all the determined parameters. Thus, it is very likely that the strong distortions found in 23 originate from crystal effects rather than from its substitution pattern. The value for βC6 of 23 in the fluid phase may therefore be expected to be lower than found in the crystal structure.
| Space group | 12 (5,5′) | 23 (6,6′) | 5,5′-DiPhEt | 5,5′-DiPhOMe | 6,6′-DiPhEt | 6,6′-DiPhOMe |
|---|---|---|---|---|---|---|
| Exp. | Exp. | Calc. | Calc. | Calc. | Calc. | |
| P21/n | P21/c | |||||
| Θ X denotes the angle of pyramidalisation of the atom X, m is the degree of s-character of the smp hybrid orbital, Θ(X–Y) is the dihedral angle between the smp-orbitals of the two neighbouring atoms X and Y and dX–Y is the distance between X and Y. Pointy brackets indicate the average value from both sides of the central double bond. Ln denotes a line that was defined to pass through the specified atoms. The angle between the lines was determined using the Diamond 3 software from Crystal Impact. The notation X/Y indicates that a dummy atom at the centre between atom X and Y was used.a Obtained from POAV1 analysis.b Obtained from POAV2 analysis.c Significant asymmetry. A detailed list of all the calculated parameters is given in the ESI. | ||||||
| 〈ΘC2〉a [°]/mb | 6.18/0.022 | 5.57/0.018 | 5.52/0.017 | 5.44/0.017 | 5.43/0.017 | 5.37/0.016 |
| 〈ΘN〉a [°]/mb | 2.72/0.004c | 8.83/0.047 | 3.84/0.008 | 3.89/0.009 | 4.22/0.010 | 4.26/0.010 |
| 〈ΘC3〉a [°]/mb | 1.62/0.001 | 1.58/0.001 | 1.33/0.001 | 1.37/0.001 | 1.38/0.001 | 1.37/0.001 |
| 〈ΘC8〉a [°]/mb | 2.34/0.003 | 1.33/0.001c | 1.83/0.002 | 1.75/0.002 | 1.73/0.002 | 1.78/0.002 |
| 〈Θ(C2–C2′)〉b [°]/dC2–C2′ [Å] | −19.46/1.362 | −19.71/1.351 | −24.60/1.364 | −24.53/1.364 | −24.30/1.363 | −23.69/1.363 |
| 〈Θ(C2–C3)〉b [°]/〈dC2–C3〉 [Å] | 12.44/1.499 | 15.21/1.500 | 12.44/1.510 | 12.11/1.510 | 12.66/1.510 | 12.47/1.510 |
| 〈Θ(C2–N)〉 b [°]/〈dC2–N〉 [Å] | 163.80/1.413 | 153.82/1.418 | 162.10/1.407 | 162.33/1.407 | 161.55/1.409 | 161.57/1.409 |
| 〈Θ(N–C8)〉 b [°]/〈dN–C8〉 [Å] | −31.52/1.419 | −17.18/1.412 | −20.48/1.423 | −20.75/1.422 | −20.06/1.422 | −20.55/1.422 |
| σ = Ln (5/6–5′/6′) to Ln (2–2′) [°] | 3.91 | 5.86 | 4.20 | 4.06 | 4.69 | 4.59 |
| α (C5–C6–C6′–C5′) [°] | −26.05 | −18.68 | −26.08 | −25.80 | −23.95 | −22.59 |
| β C5 = Ln (5–7a) to Ln (5′–7a′) [°] | 50.60 | 54.29 | 49.97 | 48.54 | 50.07 | 49.01 |
| β C6 = Ln (3a–6) to Ln (3a′–6′) [°] | 26.11 | 32.41 | 23.83 | 22.74 | 24.04 | 24.13 |
| γ = Ln (5/6–3a/7a) to Ln (2–2′) + | ||||||
| Ln (5′/6′–3a′/7a′) to Ln (2–2′) [°] | 45.71 | 52.90 | 44.29 | 42.75 | 44.79 | 44.20 |
The observed bent angle of approximately 130° for the 5,5′-bis-substituted indigo derivatives is within the suitable range for bent-core mesogens.32 However, these phases were not observed for the compounds presented in this article. The 6,6′-bis-substituted indigo derivatives have a more obtuse bent angle of approximately 150°. Bent angles around 150° are considered to be on the border between calamitic and bent core mesogens.
In this context, the unusual low order parameter found for the SmA phase of 24 can be understood as the higher degree of disorder caused by this bending. Such low bent angle mesogens have attracted considerable attention recently, due to their high tendency to form uncommon phases types such as SmAPA33 or SmAPR.34 By the introduction of flexible linking groups such as esters or imines, bent core liquid crystalline phases may be induced. Such sterically deformed π-system could serve as an unusual and unique bent unit and will be the subject of further investigation.
The peripheral substitution pattern also has a tremendous influence on the kinetics of the trans–cis-photoisomerization of the N,N′-diacetylated indigo derivatives. Substitution in the 5 and 5′ positions leads to a decrease in the rate of isomerization compared to N,N′-diacetyl indigo. The cis-product could not be observed, even under long and intense exposure to light. In contrast, substitution in the 6 and 6′ positions leads to a considerable increase in the isomerization rate. The cis-product was observed after a short-time exposure to sunlight, indicating an increase in the quantum yield of the isomerization process compared to N,N′-diacetyl indigo. This phenomenon could be exploited to generate new photoresponsive materials.
Conclusion
We presented an efficient synthesis method for a wide spectrum of peripherally substituted indigo derivatives. Substitution at the 4,4′ and 7,7′ positions leads to a considerable increase in solubility, which presumably originates from the steric hindrance of intermolecular H-bonding.The N,N′-unsubstituted compounds, bearing pendant groups in the 5,5′ and 6,6′ positions possess an approximately rod-like molecular shape. Low solubility and high melting temperature, which are caused by H-bonding, dominate their thermotropic behaviour which is characteristic for indigo. The 5,5′-bis-substituted derivatives exhibit liquid crystalline phases only shortly before or during decomposition. Stable, highly ordered smectic phases were found for the derivatives substituted with laterally fluorinated or methylated pendant groups at the 6 and 6′ positions. For these phases pronounced dichroism was observed.
Upon acetylation of the vinylogous amide groups the solubility of all indigo derivatives was increased and the melting temperatures were reduced. LC phases, however, were exclusively found for the compounds substituted at the 6 and 6′ positions. Based on the structural analysis of N,N′-diacetyl indigo, it is supposed that the combination of buckling and twisting of the central double bond leads to molecular bent, which is specific for the substitution position. For 6,6′-bis-substituted N,N′-diacetyl indigos, the molecular bent is reduced compared to the bent of the core system, whereas it is amplified for the substitution positions 5 and 5′.
Acknowledgements
We gratefully acknowledge Prof. F. Gießelmann for allowing us the access to his X-ray facilities for diffraction on LCs, Prof. Schwarz for respective devices for single crystal X-ray measurements, and M. Moran and R. Callahan for fruitful discussions.Notes and references
- (a) O. Ostroverkhova and W. E. Moerner, Chem. Rev., 2004, 104, 3267–3314 CrossRef CAS PubMed; (b) D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31–75 CrossRef CAS; (c) Polymers for Photonics Applications II, ed. F. Kajzar, K.-S. Lee and A. K.-Y. Jen, Springer, Berlin, 2003 Search PubMed.
- (a) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. Mackenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed; (b) S.-S. Sun and N. S. Sariciftci, Organic Photovoltaics – Mechanism, Materials and Devices, CRC Press, Boca Raton, FL, 2005 Search PubMed; (c) L. Li, S. W. Kang, J. Harden, Q. Sun, X. Zhou, L. Dai, A. Jakli, S. Kumar and Q. Li, Liq. Cryst., 2008, 35, 233–239 CrossRef CAS PubMed.
- (a) H. P. Preiswerk, M. Lubanski and F. K. Kneubühl, Appl. Phys. B: Lasers Opt., 1984, 33, 115–131 CrossRef; (b) S. M. Morris, M. M. Qasim, D. J. Gardiner, P. J. W. Hands, F. Castles, G. Tu, W. T. S. Huck, R. H. Friend and H. J. Coles, Opt. Mater., 2013, 35, 837–842 CrossRef CAS PubMed.
- (a) K. Ariga and T. Kunitake, Supramolecular Chemistry – Fundamentals and Applications, Springer Verlag, Berlin Heidelberg New York, 2006 Search PubMed; (b) V. Balzani, Tetrahedron, 1992, 48, 10443–10514 CrossRef CAS; (c) H. K. Bisoyi and Q. Li, Acc. Chem. Res., 2014, 47, 3184–3195 CrossRef CAS PubMed.
- C. A. Mirkin and M. A. Ratner, Annu. Rev. Phys. Chem., 1992, 43, 719–754 CrossRef CAS.
- S. Kundu, T. Ray, S. K. Roy and S. S. Roy, Jpn. J. Appl. Phys., Part 1, 2004, 43, 249–255 CrossRef CAS.
- S. J. Cowling, C. Ellis and J. W. Goodby, Liq. Cryst., 2011, 38, 1683–1698 CrossRef CAS PubMed.
- H. Kang, J. Hong and D. Kang, Mol. Cryst. Liq. Cryst., 2014, 605, 103–116 CrossRef CAS PubMed.
- H. Zhang, J. Zhang and B. Tieke, Polym. Chem., 2014, 5, 646–652 RSC.
- (a) Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem. – Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed; (b) X. Li, A. Liu, S. Xun, W. Qiao, X. Wan and Z. Y. Wang, Org. Lett., 2008, 10, 3786–3787 Search PubMed; (c) L. Marin, A. Zabulica and M. Sava, Soft Mater., 2013, 11, 32–39 CrossRef CAS PubMed.
- M. Klessinger and W. Lüttke, Tetrahedron, 1963, 19(suppl. 2), 315–335 CrossRef CAS.
- (a) R. Wizinger, Chimia, 1961, 15, 89–105 CAS; (b) R. Grinter and E. Heilbronner, Helv. Chim. Acta, 1962, 45, 2496 CrossRef CAS PubMed; (c) H. Labhart and G. Wagnière, Helv. Chim. Acta, 1963, 46, 1314–1326 CrossRef CAS PubMed; (d) W. Lüttke and M. Klessinger, Chem. Ber., 1964, 104, 2342–2357 CrossRef PubMed.
- (a) A. v. Baeyer, Ber. Dtsch. Chem. Ges., 1878, 11, 1296–1297 CrossRef PubMed; (b) A. v. Baeyer, Ber. Dtsch. Chem. Ges., 1879, 12, 456–461 CrossRef PubMed.
- A. R. Katritzky, W.-Q. Fan, A. E. Koziol and G. J. Palenik, J. Heterocycl. Chem., 1989, 26, 821–828 CrossRef CAS PubMed.
- J. H. Porada and D. Blunk, J. Mater. Chem., 2010, 20, 2956–2958 RSC.
- D. Blunk and J. H. Porada, ChemPhysChem, 2009, 10, 3260–3264 CrossRef CAS PubMed.
- P. Süsse, M. Steins and V. Kupcik, Z. Kristallogr., 1988, 184, 269 CrossRef.
- C. Liebermann and F. Dickhuth, Chem. Ber., 1891, 24, 4130–4136 CrossRef PubMed.
- (a) J. E. Klare, G. S. Tulevski, K. Sugo, A. de Picciotto, K. A. White and C. Nuckolls, J. Am. Chem. Soc., 2003, 125, 6030–6031 CrossRef CAS PubMed; (b) N. Lindner, M. Kölbel, C. Sauer, S. Diele, J. Jokiranta and C. Tschierske, J. Phys. Chem. B, 1998, 102, 5261–5273 CrossRef CAS; (c) G. W. Gray, C. Hogg and D. Lacey, Mol. Cryst. Liq. Cryst., 1981, 67, 1–23 CrossRef CAS PubMed.
- M. C. Carreño, J. L. García Ruano, G. Sanz, M. A. Toledo and A. Urbano, Synlett, 1997, 1241–1242 CrossRef.
- (a) G. Miehe, P. Süsse, V. Kupcik, E. Egert, M. Nieger, G. Kunz, R. Gerke, B. Knieriem, M. Niemeyer and W. Lüttke, Angew. Chem., Int. Ed., 1991, 30, 964–967 CrossRef PubMed; (b) G. A. Russell and G. Kaupp, J. Am. Chem. Soc., 1969, 91, 3851–3859 CrossRef CAS.
- P. Süsse and R. Wäsche, Naturwissenschaften, 1978, 65, 157 CrossRef.
- P. Davidson, D. Petermann and A. M. Levelut, J. Phys. II, 1995, 5, 113–131 CrossRef CAS.
- J. W. Goodby, D. Demus, J. Goodby, G. W. Gray, H. W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH Verlag GmbH, 2008, pp. 3–21 Search PubMed.
- G. Miehe, P. Süsse, V. Kupcik, E. Egert, M. Nieger, G. Kunz, R. Gerke, B. Knieriem, M. Niemeyer and W. Lüttke, Angew. Chem., Int. Ed., 1991, 30, 964–967 CrossRef PubMed.
- R. C. Haddon and L. T. Scott, Pure Appl. Chem., 1986, 58, 137–142 CAS.
- (a) R. C. Haddon, Chem. Phys. Lett., 1986, 125, 231–234 CrossRef CAS; (b) R. C. Haddon, J. Am. Chem. Soc., 1986, 108, 2837–2842 CrossRef CAS.
- G. Grimme, S. Grimme, P. G. Jones and P. Boldt, Chem. Ber., 1993, 126, 1015–1021 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- I. V. Alabugin, M. Manoharan, M. Buck and R. J. Clark, J. Mol. Struct., 2007, 813, 21–27 CrossRef CAS PubMed.
- R. A. Reddy and C. Tschierske, J. Mater. Chem., 2006, 16, 907–961 RSC.
- I. Wirth, S. Diele, A. Eremin, G. Pelzl, S. Grande, L. Kovalenko, N. Pancenko and W. Weissflog, J. Mater. Chem., 2001, 11, 1642–1650 RSC.
- (a) D. Pociecha, M. Čepič, E. Gorecka and J. Mieczkowski, Phys. Rev. Lett., 2003, 91, 185501 CrossRef; (b) Y. Shimbo, E. Gorecka, D. Pociecha, F. Araoka, M. Goto, Y. Takanishi, K. Ishikawa, J. Mieczkowski, K. Gomola and H. Takezoe, Phys. Rev. Lett., 2006, 97, 113901 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: A description of the experimental details, analytical data, UV-Vis spectra, calculations of the order parameters, π-orbital axis vectors and DFT calculations are given. CCDC 1033834 (12) and 811039 (23). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01594d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |