
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-nuclear NMR of axially chiral biaryls in polypeptide orienting solvents: spectral discriminations and enantiorecognition mechanisms†
Philippe
Berdagué
a,
Jose-Enrique
Herbert-Pucheta
a,
Vishwajeet
Jha
b,
Armen
Panossian
b,
Frédéric R.
Leroux
 b and
Philippe
Lesot
*a
b and
Philippe
Lesot
*a
aLaboratoire de RMN en Milieu Orienté, Institut de Chimie Moléculaire et des Matériaux d'Orsay (ICMMO), UMR CNRS 8182, Université Paris-Sud, Université Paris-Saclay, 91405 Orsay, France. E-mail: philippe.lesot@u-psud.fr; Fax: +33 (0)1 69 15 81 05; Tel: +33 (0)1 69 15 47 59
bLaboratoire de Chimie Moléculaire, Université de Strasbourg, UMR CNRS 7509, ECPM, 25 Rue Becquerel, 67087 Strasbourg, France
First published on 11th September 2015
Abstract
Due to the importance of axially chiral biaryl derivatives as chiral auxiliaries and/or ligands for asymmetric synthesis, as well as their structural role in bioactive natural products, continuous efforts have been undertaken to propose efficient methods for their atropo-selective synthesis. As a consequence, proposing robust and reliable analytical tools that are able to discriminate the spectral signal of atropisomeric enantiomers becomes crucial to evaluate the enantiomeric excesses of mixtures. In this work, we show how several multi-nuclear 1D/2D-NMR techniques using homopolypeptide chiral liquid crystals as aligning solvents can provide a panel of analytical possibilities (through differences of chemical shift anisotropies, dipolar and quadrupolar residual couplings) to spectrally discriminate enantiomers of a large collection of trisubstituted axially chiral biphenyls. Approaches involving 31P, 13C and 2H 1D- or 2D-NMR experiments at natural abundance levels are explored. Among noteworthy results, the first examples of spectral enantioseparations using 31P nuclei as nuclear probe are reported. Finally, the roles of electronic factors and shape anisotropy in the efficiency of chiral discrimination mechanisms are examined and discussed. Molecular modeling calculations were carried out to establish the electronic profile of these analytes in order to understand and rationalize the 13C–{1H} NMR results.
Introduction
Axially chiral biaryl derivatives possess a peculiar stereochemical motif able to generate a couple of stereoisomers. The stereogenic structural motif is present in various potentially bioactive natural compounds and exhibits a wide range of biological properties.1 For instance, one can mention the well-known vancomycin (a clinically used antibiotic glycopeptide)2 or steganacin (a cytotoxic tubulin-binding dibenzocyclooctadiene lignan).3 In fact, the biaryl scaffold is a privileged structure for pharmaceutical research as its incorporation assures frequently high entry rates.4 In addition, the stereogenic axes provide rigid molecular frameworks for highly efficient tools in asymmetric synthesis.5 Concomitantly, atropisomeric C1-symmetric biaryls play an important and effective role as chiral auxiliaries and/or ligands for asymmetric synthesis. Consequently, continuous efforts have been undertaken by organic chemists to develop efficient methods for the atropo-selective synthesis of ligands based on the biphenyl, binaphthyl, or other biaryl backbones.6 The conformational stability of bridged biaryls can be strongly increased by incorporation of ortho-substituents, the associated rotational energy barrier primary depending on their number and their bulkiness.7 Generally, ortho-trisubstituted biphenyls show no stereo-labile properties, and hence no rapid enantiomerization at room temperature is expected (see Fig. 1a).8 | ||
| Fig. 1 (a) General structure of enantiomeric couples of ortho-trisubstituted biphenyls investigated here, along with the systematic atomic numbering used. The aromatic cycle B contains the Z substituent and quaternary carbon atoms are displayed in green. (b) Structures of chiral compounds 1 to 17 defined by series: (I): dibromo derivatives (1 to 6); (II): chloro–bromo derivatives (7 to 11); (III): monophosphorous derivatives (12 to 15); (IV): miscellaneous derivatives (16 and 17). The stereodescriptors reported in the four series correspond to structures drawn. As short notation associated to atropisomers, we will use the stereodescriptors (S/R) instead of (aS/aR). | ||
So far, both chiral supercritical fluid chromatography and chiral gas chromatography have been the main methods used to separate the enantiomers of such peculiar chiral compounds.8,9 Although adequate in numerous cases, chromatographic approaches present some well-known specific drawbacks for systematic implementation (price of chiral columns, for instance). Furthermore, the determination of experimental conditions leading to enantiomeric resolution is sometimes highly time-consuming. As a consequence, proposing (simple) analytical alternatives involving other techniques, such as NMR spectroscopy, to chemists is a valuable task.
In the past, liquid-state NMR methods involving mainly chiral derivatizing agents (MPTA or Mosher's acid) in combination with or without lanthanide shift reagents have been proposed to discriminate enantiomers of (bridged or not) chiral biaryl atropisomers.10 Although successful, these approaches require the presence of accessible reactive groups (–COOH, –OH, –NH2,…) to generate (in situ or not) diastereoisomers.
This prerequisite can be overcome when using NMR in lyotropic chiral liquid crystals (CLC). This breakthrough approach was revealed to be a powerful and flexible tool, providing a robust and general alternative to the multitude of isotropic NMR methods for the differentiation of enantiomers of chiral molecules,11 but also enantiotopic elements in prochiral molecules.12–14 Compared to classical NMR approaches using chiral derivatizing or solvating chiral agents, NMR in CLC requests no specific functional groups inside the analyte,15 while all magnetically active nuclei (even at very low natural abundance levels) can provide effective probes.
Using a chiral aligned environment, the intermolecular interactions between each enantiomer (or each enantiotopic direction) and the CLC differ, and hence the internuclear vectors, i–j, are not oriented the same on average, SSi–j ≠ SRi–j (or Spro-Si–j ≠ Spro-Ri–j).11–13 A doubling of the spectral information/patterns for a given nuclear site indicates therefore that the enantiorecognition phenomenon occurs and is revealed on the NMR spectrum by a difference of residual chemical shift anisotropies (CSA), dipolar couplings (D) or quadrupolar couplings (ΔνQ) (for spin I > 1/2) (see Fig. S1, ESI†).16,17 As a first example, deuterium NMR in CLC was used to analyze the intramolecular dynamic processes of chiral and prochiral deuterated ortho-disubstituted biaryls (derivatives of 1-(4-methylphenyl)naphthalene).14
In this work, we show how multi-nuclear 1D/2D-NMR using chiral anisotropic solvents (homopolypeptide CLC) can provide various analytical possibilities to spectrally separate enantiomers of a large collection of ortho-trisubstituted axially chiral biphenyls (see Fig. 1). From an analytical viewpoint, anisotropic NMR results will be discussed in terms of spectral enantiodiscrimination efficiency and an attempted rationalization of the results is proposed. For this purpose, the seventeen analytes investigated have been classified into four series of structurally related molecules (I to IV) depending on the similarity of their substitution patterns, as displayed in Fig. 1b.
Experimental section
Synthesis
The synthesis of these atropisomeric biaryls was recently reported using an original, modular approach (see Fig. S2, ESI†).8 Briefly speaking, it is based on (a) the preparation of ortho,ortho′-dibromobiphenyls bearing an additional substituent in the 6-position via a transition metal-free aryl–aryl coupling (the ‘ARYNE coupling’),18 (b) the regioselective introduction of an enantiopure p-tolylsulfinyl group as a traceless chiral auxiliary allowing the separation of atropo-diastereoisomers by simple crystallization, (c) the chemoselective functionalization of this auxiliary and (d) subsequent regioselective functionalization of the remaining bromine atoms. During all these chemical transformations, the configuration of the biaryl axis is maintained, and hence no racemization was found to occur.8 The diphosphine 16 was obtained by means of catalytic C–P coupling.19Material for oriented NMR samples
In this study, homopolypeptide CLC samples were composed of poly-γ-benzyl-L-glutamate (PBLG), purchased from Sigma and dissolved in chloroform.11,20 The degree of polymerization of PBLG is equal to 743 (MW = 162 900 g mol−1). The mass of solute in the samples varies from 19 to 100 mg, while the molar variation ranking from 2.1 × 10−6 (sample 14) to 1.8 × 10−4 mol per enantiomer (sample 17). Table S1 (ESI†) lists the exact composition (samples 1 to 17) and if the chloroform was protonated or deuterated. The preparation of (sealed) anisotropic NMR tubes and practical aspects have been reported in previous papers (also see ESI†).10,12,13NMR spectroscopy
13C, 13C–{1H} and 31P–{1H} 1D/2D-NMR spectra were recorded on routine 9.4 T Bruker (Avance I) NMR spectrometers equipped with either a 5 mm BBO, TBI or QXO probe. Unless otherwise specified, the sample temperature was set to 298 K. NAD–{1H} 2D NMR spectra were performed on 14.1 T Bruker (Avance II) spectrometer equipped with a 5 mm 2H selective cryoprobe,17,21 and the WALTZ-16 CPD sequence was used to decouple proton (0.5 W).11 Specific experimental details are given in the figure captions.Molecular modeling and DFT calculations
Geometry optimizations and electronic structure determinations were carried out using the Gaussian 09 program running on the “IDA” cluster of the University of Paris-Sud.22 Density functional theory (DFT) with self-consistent reaction field (SCRF) Tomasi's polarized continuum model (PCM) for solvation23 was used in all calculations to describe implicitly the solvent (chloroform) for energy minimizations and for description of orbitals. All computations were performed with the hybrid method B3LYP, whereas electronic correlation and exchange were respectively described by the use of the Becke24 and Lee–Yang–Parr25 functionals. Relativistic effective core potentials (ECP) were used to describe electrons of heavy atoms (Br and Cl) with the valence double z quality basis sets Lanl2dz.26 The standard 6-311G(d,p) basis sets were used for the rest of the atomic orbitals' descriptions of H, C, O and P atoms. Local minima conditions per molecule were confirmed with the calculation of harmonic vibrational frequencies of all structures. None of the predicted vibrational spectra has any imaginary frequency (data not shown), implying that the optimized geometry of each of the molecules under study lies at a local point on the potential energy surface. The electronic properties such as Molecular Electrostatic Potential (MEP), frontier molecular HOMO–LUMO orbital energies and Mulliken atomic charges have been obtained with the same level of theory as previously described.Results and discussion
For a global view of the results, Table 1 (see also Table S2, ESI†) summarizes the essential data for the analytes and the sets of experimental results. As all results were obtained with very similar experimental conditions (T ≈ 298 K, W/W of PBLG of 14%), we will then follow with their interpretation in terms of chiral discrimination mechanisms (noted in short as CDMs). In particular, for the 13C NMR results, attempts to correlate the number of 13C discriminated sites (noted in short as NDS(13C)) and the possible solute–PBLG electrostatic interactions in combination with molecular shape recognition effects will be proposed and discussed.| Series | Solute | Mass (mg) | 10−5 × nR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) orS/mole orS/mole |
μ mol (scalar)a/D | Hydrogen bond | Oxygen atom | 13C–{1H}b NMR | 2H–{1H}b NMR | 31P–{1H}b NMR |
|---|---|---|---|---|---|---|---|---|---|
| a The molecular electric dipole moment, μmol, is calculated for the lowest-energy conformer. b Number of sites (13C, 2H, 31P) spectrally discriminated. c /: spectrum not recorded. d US: unexpoitable NAD spectrum. e For one site 2H, two interpretations of results are possible. f 13C signals are also decoupled from 31P signals. | |||||||||
| I (Br/Br) | 1 (I) | 19.9 | 2.27 | 2.132 | N | N | 1/12 | /c | — |
| 2 (COOH) | 54.5 | 7.65 | 2.529 | Y | Y | 12/13 | USd | — | |
| 3 (CHO) | 61.0 | 9.00 | 2.933 | N | Y | 9/13 | 7/8e | — | |
| 4 (OMe) | 100.0 | 14.60 | 4.213 | N | Y | 10/13 | 7/8e | — | |
| 5 (Me) | 21.2 | 3.25 | 3.104 | N | N | 6/13 | NS | — | |
| 6 (COOMe) | 72.2 | 9.75 | 2.963 | N | Y | 8/13 | 6/8 | — | |
| II (Cl/Br) | 7 (I) | 21.1 | 2.68 | 2.213 | N | N | 3/12 | / | — |
| 8 (COOH) | 59.5 | 9.55 | 2.357 | Y | Y | 11/13 | US | — | |
| 9 (CHO) | 62.6 | 10.60 | 2.737 | N | Y | 11/13 | 6/8e | — | |
| 10 (Me) | 19.0 | 3.38 | 3.179 | N | Y | 6/13 | NS | — | |
| 11 (COOMe) | 70.1 | 10.75 | 3.006 | N | Y | 8/13 | 6/8 | — | |
| III (Cl/Br) | 12 (PPh2) | 26.0 | 2.88 | 4.166 | N | N | 4/24f | / | 0/1 |
| 13 (POPh2) | 96.2 | 10.26 | 4.622 | N | Y | 12/24f | / | 0/1 | |
| 14 (PCy2) | 25.3 | 2.73 | 3.048 | N | N | 3/24f | / | 1/1 | |
| 15 (POCy2) | 20.1 | 2.09 | 5.146 | N | Y | 2/24f | / | 0/1 | |
| IV | 16 (P2)e | 30.4 | 2.73 | 5.290 | N | N | 12/36f | / | 2/2 |
| 17 (Me/OMe) | 100.6 | 18.20 | 2.377 | N | N | 6/14 | 7/9e | — | |
1H 1D-NMR spectroscopy
Even for small-size molecules, the number and the magnitude of (short and long-range) 1H–1H residual dipolar couplings significantly increase the linewidth to obtain rather low-resolution 1H spectra where no fine structures clearly emerge. Some exceptions can be found with molecules possessing isolated methyl groups, for instance. Compounds 4 to 6, 10, 11 and 17 are typical examples. Contrarily to isotropic 1H NMR, an uncoupled methyl group exhibits a triplet structure in a LC (instead of a single resonance) due to the intramethyl 1H–1H dipolar couplings (see Fig. S3, ESI†). In a CLC, two triplets with different splittings (|3DSHH| ≠ |3DRHH|) centered on very close 1H chemical shifts (due to a small difference of 1H CSA) are generally detected.For analytes, 6 and 11, a single triplet (with |3DHH| = 58 and 52 Hz, respectively) is observed, thus revealing no resolved discrimination through a difference of DHH. Two reasons may explain this absence of enantiodiscriminations: (i) the rather low sensitivity of D(1H–1H) to a difference of orientational ordering (compared to 2H quadrupolar interaction, for instance); (ii) the complex conformational dynamics of ligand Y (here, up to three rotors), which generally leads to averaging down the order parameters of each internuclear vector along the chain, and subsequently reduces spectral enantiodifferences. For solutes 5, 10 and 17, a symmetric 1H spectral pattern of six lines is observed for methyl bound to the ring (see Fig. S3, ESI†). This structure can be analyzed as follows: (i) a dedoubled triplet if the methyl group is dipolarly coupled with one of the aromatic protons; (ii) two triplets centered on two 1H chemical shifts due to a surprisingly large 1H CSA. Two approaches involving the components of mixture can be proposed to assess the origin of the spectral pattern. They consist of either recording the 1H spectrum in ALC made of a racemic mixture of PBLG and PBDG (PBLG's enantiomer)27 or using the enantiopure compound (when available) in the CLC. Irrespective of the method used, it appears here that the doubling of triplets has its origin from the dipolar coupling with the ortho-position aromatic proton as simply exemplified in the case of 10 (see Fig. S1, ESI†). Spin-manipulation based alternatives for simplifying the coupling pattern of 1H signals should be possible but they are beyond the scope of this paper and have not been explored.
1H-(de)coupled 31P NMR spectroscopy
Compounds 12 to 16 possess 31P nuclei, which are both sensitive and 100% abundant, and hence easy to detect. Spectral enantiodiscriminations can be observed through the difference of 31P CSA, leading to two independent resonances, one for each enantiomer, or a difference of 31P–1H RDCs. For the series of monophosphorous derivatives, no exploitable spectral separations based on 31P–1H RDC differences were obtained on the 31P 1D-NMR spectra, mainly due to the presence of various long-range 31P–1H RDCs, which obscure spectra. When protons are decoupled, spectra are significantly simplified. Contrarily to 12, 13 and 15, the presence of two 31P resonances for solute 14 indicates that enantiomers are discriminated on the basis of 31P CSA differences (|Δσ| = 14 Hz, Δν1/2 = 3.5 Hz) (see Fig. 2a). Surprisingly, monophosphine oxide biaryl moieties 13 and 15 are not spectrally discriminated despite the expected increase of the electronic shielding anisotropy of the phosphorous nucleus due to the presence of the oxygen atom, which produces bigger 31P CSA susceptible to lead (a priori) to larger enantiodiscriminations. Variable temperature 31P NMR experiments (range of 30 K) did not permit enantiodiscriminations neither for 13 nor for 15. The last strongly suggests that the energy gap of the interconversion barrier between enantiomers is importantly increased by the fact that aryls' free-rotation is sterically hindered by the presence of the oxide and thus only one enantiomer is favored.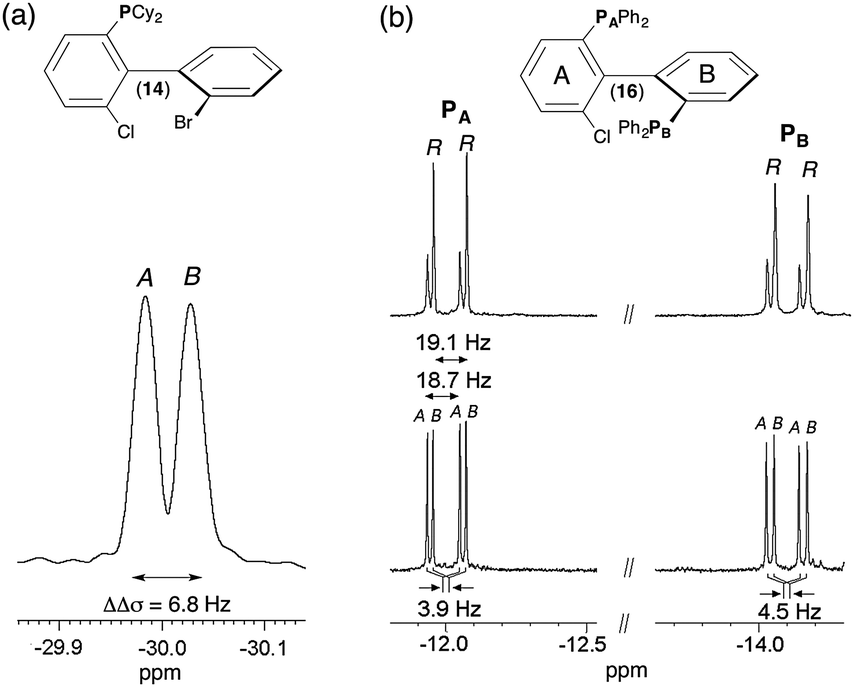 | ||
| Fig. 2 161.9 MHz 31P–{1H} 1D-NMR spectrum of (a) (R/S)-14 in PBLG/CHCl3 at 335 K and (b) of 16 in racemic series (bottom) and enantioenriched series (R isomer) (top) in PBLG/CDCl3 at 298 K (ref. δ(80% of H3PO4) = 0 ppm). 1000 scans (a) and 5000 scans (b) are added and soft line-sharpened Gaussian filtering is applied. Note the 31P CSA of 3.9 Hz and 4.5 Hz for PA and PB of 16. The assignment of PA and PB atoms is given in ESI† (see Fig. S14 to S18 in Section SI–SIII). The italicized notations “A/B” stand for the chiral stereodescriptors of enantiomers A and B. | ||
Finally, the case of chiral biaryl-based diphosphane 16 is rather peculiar. Indeed in the liquid state, this molecule possesses two anisochronous 31P atoms (at room temperature) resonating at two distinct chemical shifts (δ(31PA) = −12.0 ppm and δ(31PB) = −14.1 ppm) and mutually coupled as first evidenced in 2011.28 This spin–spin coupling finds its origin via a “through-space” scalar coupling (noted J(31PA–31PB) = 22.7 Hz) and not via the intramolecular five-bond connectivity, which should lead to a small scalar 5J(31PA–31PB) coupling. The spectral assignments of PA and PB atoms derive from the analysis of 1H–31P 2D HMBC, 31P 2D J-resolved and 1H–1H 2D COSY experiments shown in ESI† (Section SIII). If two doublets (AX spin system) are observed on the isotropic 31P–{1H} spectrum, four resonances (Δν1/2 < 0.8 Hz) are detected at each 31P site in a racemic series (see Fig. S2a and S4, ESI†). This doubling of lines indicates enantiodiscrimination. Three spectral situations corresponding to a difference of either 31P CSA or 31P–31P RDC or from both contributions can explain the presence of two pairs of doublets for each 31P site (see Fig. S5, ESI†). The assignment of 31P resonances was assessed by comparing the 1D spectrum of 16 to the one recorded with an enantioenriched mixture (enriched in R isomer, ee = 51.3%) using similar experimental conditions (sample composition and temperature). As seen in Fig. 2b, the peak intensity difference between enantiomers allows their associated signals to be undoubtedly assigned. Various homonuclear 2D experiments confirm qualitatively the presence of each enantiomer, regardless of the absolute configuration of NMR signals (vide infra and ESI†). According to the enantio-assignment made, the analysis of 31P–{1H} 1D spectrum of 16 indicates that total couplings between the two 31P nuclei for each isomer, |TA(31P–31P)| and |TB(31P–31P)| with T = J + 2D, are very close and equal to 18.7 and 19.1 Hz, respectively while each doublet is shifted by 3.9 Hz (31PA) and 4.5 Hz (31PB). Assuming a negative value for TA or B, the magnitude of D(31P–31P) is equal to −20.3 and −20.5 Hz for R and S, respectively. Conversely, if T(31P–31P)A or B is positive, D(31P–31P)A or B becomes equal to −2 or −1.8 Hz, respectively. Among the two anisotropic contributions (D and CSA), only the 31P CSA (ΔΔσ = 3.9 Hz and 4.5 Hz) is the relevant NMR interaction here that can be in fine efficiently exploited to evaluate the ee. As CSA is directly proportional to the magnetic field strength, operating with higher magnetic field spectrometers should guarantee larger discriminations. Interestingly, the results obtained here are the two first examples of enantiodifferentiation using 31P–{1H} NMR, so far. Previous studies using 31P–{1H} or 31P NMR as the analytical technique had failed in discriminating enantiomers of phosphorous compounds.29
1H-decoupled 13C 1D-NMR
Although less sensitive compared to 1H or 31P NMR, anisotropic 13C–{1H} NMR at natural abundance is an excellent and competitive method to discriminate enantiomers, in particular when sp2 hybridized carbon atoms are present in the analyte. The gain in sensitivity of commercially available cryogenic probes with respect to conventional ones (up to a factor of 4 to 5) enables the experimental acquisition times of qualitative 13C–{1H} NMR to be reduced to less than 1–2 hour(s),30 even working with small amounts of solutes or with analytes of high MW. However, when a sufficient amount of solute is available (10–30 mg), depending on the enantiomeric mole number (from 2.27 × 10−5 to 18.20 × 10−5) and the S/N ratio (SNR) desired, all 13C–{1H} 1D spectra were recorded with a moderate magnetic field (9.4 T) using acquisition times in the range of 3–10 hours.All experimental data related to the number of discriminated 13C sites in PBLG are presented in Table 1, while Table S3 (ESI†) lists the value of all δ(13C) and the differences of CSA (ΔΔσ) for each of them.
For derivatives of series I and II, the 1D analysis of 13C–{1H} NMR spectra in CLC can be easily performed by counting the number of 13C lines and then comparing with the isotropic 13C–{1H} NMR spectra where no discrimination occurs (see Fig. S6, ESI†). For all of them, several spectral enantiodiscriminations occur both in the CH and quaternary aromatic carbon atoms, but the number of differentiated 13C sites varies from one (solute 1) to twelve sites (solute 2), with spectral difference from 1 Hz (limit of discrimination) to 14 Hz. Note that carbon atoms belonging to the Y substituent (see Fig. 1) can provide further potential 13C sites for discrimination like in the case of 4, 9 and 10, for instance (see Table S3, ESI†).
Two illustrative examples of 13C–{1H} 1D spectra (solutes 2 and 8) are given in Fig. 3. The variations of δ(13C) between 2 and 8 (and between each solute) result in the well-known electronic effects (+I, −I and +M, −M) of various substituents on the rings (see also Fig. S4, ESI†). For 2 and 8, about 90% of 13C sites are discriminated, thus affording a multiple choice for measuring enantiomeric excess (ee) of mixture. However, from a quantitative viewpoint, the choice of the best sites is clearly governed by three parameters (i) the spectral frequency differences between enantiomer signals; (ii) SNR; and (iii) signal overlapping of analytes with solvent signals. Typically, for 2 and 8, the C-4 atom (SNR ≈ 80–100) provides the best site for quantitative purposes when the PBLG/CHCl3 chiral system is used. Carbon atom C-9 could also provide a discrimination site but the signal overlay with very broad resonances of PBLG aromatic signals complicates the evaluation of enantiomeric purity. Finally, the quaternary C-6 and C-8 (for 2) or C-6 (for 8) carbons also provide large separations (≈11 Hz), but their low SNR (≈30–35) excludes them from an accurate determination of large ee's. The overall interspectral analysis of all analytes indicates that biaryls with an aldehyde (3 and 9), ether (4) and ester (6 and 11) substituent possess numerous enantiodiscriminated sites (60 to 80%), but these sites show smaller spectral differences (1 to 3.5 Hz).
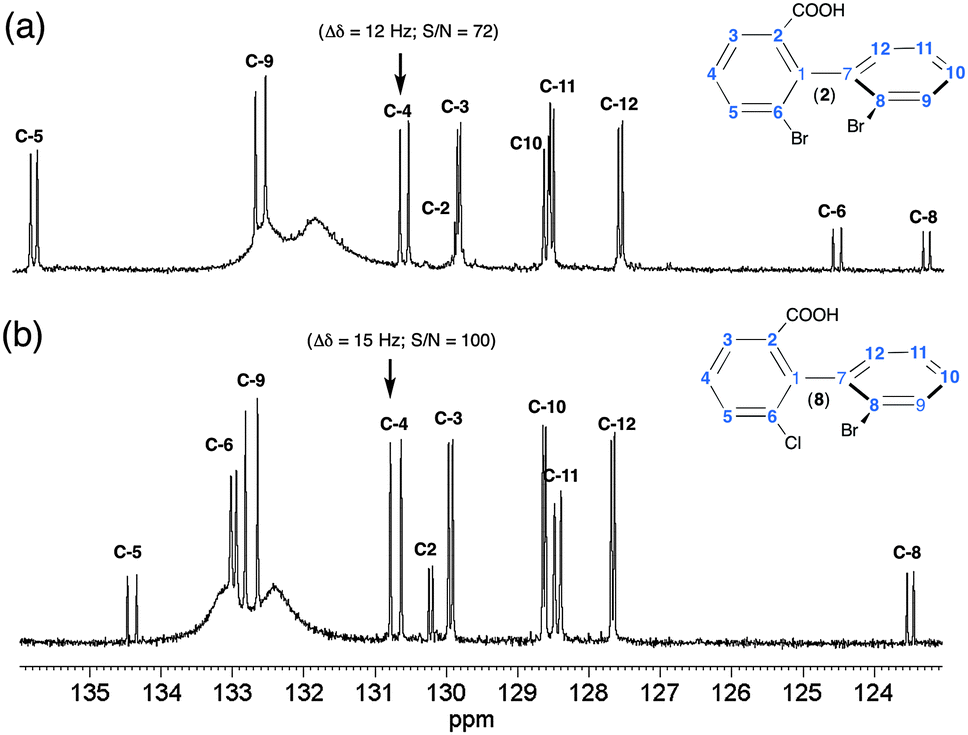 | ||
| Fig. 3 100.4 MHz 13C–{1H} 1D spectra (BBO probe) of (a) (R/S)-2 and (b) (R/S)-8 recorded in PBLG/CHCl3. Only the region ranging from 123 to 136 ppm is displayed (carbon atoms C-1/C-7 and from carboxyl group not shown). Note the doubling of numerous 13C signals (compared to isotropic spectra) associated to the spectral discrimination of R/S isomers. The very broad resonances observed around 132–134 ppm originate from ortho/meta aromatic 13C signals of the benzyl group of the PBLG side chain. | ||
For methylated biaryls (5 and 10), the ratio of enantiodiscriminated sites does not exceed 50% while the lowest number of sites (<25%) is obtained for the iodo derivatives (1 and 7). 13C–{1H} 1D-NMR spectra of 7 and 9 are given in the ESI.† Similar experimental conditions (co-solvent, w/w of polypeptide and temperature) for all solutes allow 13C spectral comparisons. Furthermore, the 13C CSA is weakly sensitive to small variations of T or sample concentration.
Due to the diversity of contributing factors/effects to the CDMs (molecular shape, electronic properties and/or conformational dynamics), the attempt to establish qualitative correlations between the enantiodiscrimination efficiency and molecular properties is far from being trivial. Nevertheless, it is important to investigate them in order to rank their role and evaluate their respective predominance contributing to the CDMs. This is a prerequisite step toward a global insight of the phenomenon, and subsequently the possibility to predict spectral results for any given analyte.
Considering the rather high degree of structural homology (ortho-three-substituted biaryl) of analytes in series I and II, we have first attempted to simply correlate (and explain) the 13C NDS and the range of spectral separations (ΔΔσ(13C)) (weak (1–3 Hz), medium (4–8 Hz) or large (> 9 Hz)) to the magnitude of the global dipolar moment of the molecule, μmol, calculated by molecular modeling. For this purpose, the dipole moments (in CHCl3) for all biaryls in their lowest-energy conformation have been calculated using solvent-dependent density functional theory (SCRF-DFT) Mulliken charge distribution analysis (see Experimental section for details). The scalar value is listed in Table 1, while the value on the three-axis components (x, y, z) and their vectorial representation are reported in Table S4 (ESI†). A first inspection of the molecular modelling results shows that: (i) the variation of μmol does not exceed 7% when Br is replaced by the chlorine atom; and (ii) the nature of substituent Y modifies significantly μmol, from 2.1 (1) to 4.2 D (4).
The comparison of NMR results between series I and II shows rather similar results (NDS(13C) and ΔΔσ(13C)), thus indicating that the replacement of Br by Cl in position 6 (ring A) does not change strongly the global molecular properties (dipole moment and shape) of the solute towards the efficiency of CDMs. In contrast, in each series, the difference of properties of ligand Y has a larger impact on both criteria. Except for solutes 1 and 7, which both exhibit the smallest values of μmol and NDS (with small ΔΔσ), the analysis of results for other analytes indicates that there is no simple dependency (i.e. a monotonous variation) between both spectral criteria and the magnitude of global dipole moment. In clear, the larger NDS does not occur for the biggest μmol. This absence of direct correlation suggests that the nature of substituent Y and the associated specific electronic properties (presence of labile hydrogens able to form intermolecular hydrogen bondings (HB) or the presence of electronegative oxygen in carbon–oxygen double bond, for instance) play a crucial role in the efficiency of CDM for this series of biaryls, independently to the global molecular dipole moment. Thus, the best results obtained with acid derivatives 2 and 8, and not for the methyl ester analogues (6 and 11), point out that the possibility of forming HB between the substituent Y and the oxygen of the carboxylate group of the PBLG side chain is of primary importance in the CDM. Schematically, the role of HB can be understood as follows. Contrarily to ordering mechanisms (mainly due to the coupling between the solute quadrupole moment and the electric field gradient of the solvent), the CDMs involve short-range intermolecular interactions that derive from the repulsive forces correlated with the size and shape (and the shape-anisotropy) of the solute.31 Hence, the CDM efficiency is strongly dependent on the average distance between the solute and the PBLG chiral helix. In this context, irrespective of the magnitude of the global dipole moment, HB can be seen as a crucial local electronic interaction susceptible to bring the solute nearest the fibers (at small distances), in turn promoting better enantiodiscriminations, this effect being particularly strong when the labile hydrogen is highly topologically accessible, as in the case of 2 and 8.
When no HB are possible, other attractive specific electronic interactions can play important roles in the CDM, in particular to reduce the average solute–PBLG distance. Although probably less efficient than HB, these (secondary) interactions then become key parameters governing the efficiency of discrimination. Clearly the presence of an electronegative oxygen atom with accessible lone pairs (aldehydic or ester groups for instance) appears as an important electronic parameter favoring fiber-solute electrostatic interactions (van der Waals type). In contrast (vide infra), it is a priori expected that CDM are much less efficient for biaryls devoid of any groups susceptible to promote any attractive intramolecular interactions (case 5 and 10). This simply explains why better results are obtained for methoxy, carbonyl or ester groups (NDS varying from 8/13 to 10/13), whereas the situation is much less favorable for methyl group (NDS = 6/13). From the spectral enantiodiscrimination viewpoint, the case of methoxy or aldehydic derivatives (3, 4, 9, 17) could be qualified as an “intermediate” situation for which only enantiodiscriminations with moderate spectral differences are expected and occur.
As an illustrative example of the experimental quantification of ee's of enantioenriched chiral biaryl mixtures, Fig. 4 compares the signals of four 13C sites of 10 in racemic (top) and enantioenriched mixtures (R) (bottom). Thus, in the enantioenriched series, a single resonance is observed at quaternary carbons (C-1 and C-7) for which the SNR is smaller (137 and 114) compared to para methine sites (C-4 and C-10) (SNR = 317 and 375). In contrast, for the latter case, a very weak signal can be found at the foot of the most intense 13C signal, indicating that the mixture is not enantiopure but only enantioenriched (see Fig. 4b, right panel). Evaluation of peak areas by deconvolution indicates an ee of about 95% (in good agreement with the chromatographic results),8 while the absence of signals for the minor enantiomer at quaternary carbon atoms (C-1 and C-7) could suggest an ee of 100%. This example points out the importance of sites selected for quantitative measurement, in particular when a reliable evaluation of ee is necessary. The analysis of 13C–{1H} spectra of molecules of series IV is analytically much more challenging for two reasons: (i) the important number of sp2 carbon atoms (up to 24 sites) in the aromatic region ranging from 123 to 145 ppm (12 and 13); and (ii) the weak distribution of chemical shifts of sp3 carbon atoms in the aliphatic region (from 25 to 34 ppm) of cyclohexyl rings (14 and 15). For all phosphorous analytes, the determination of δ(13C) listed here was achieved from the analysis and the intercomparison of 13C–{1H} and 13C–{1H, 31P} 1D-NMR spectra and 13C–{1H} and 13C–{1H, 31P} J-modulated 1D-NMR spectra. All δ(13C) measured (and the associated |ΔΔσ|) at 9.4 T are reported in Table S3 (ESI†). Globally, the results from the 13C enantiodiscrimination side are rather mediocre and disappointing because only very few carbon sites (with tiny spectral differences) show useful separation both in phenyl and cyclohexyl rings. Actually, two reasons may explain the absence of numerous and large differentiations. The first one is related to the presence of sp3 carbon atoms in cyclohexyl groups (14 and 15), which are less susceptible to discrimination on the basis of 13C chemical shift anisotropy (spherical electronic screening). The second one could originate from the global shape of the molecule. Indeed, the presence of a biphenylphosphine (oxide or not) moiety significantly increases the size of the structure (four cycles), and concomitantly leads to a more globular molecular topology (regardless of the conformational dynamics). In a simple (static) schematic view, increasing of the number of (aromatic or aliphatic) cycles reduces the geometrical shape anisotropy (compared to molecules of series I and II), and in turn the efficiency of shape recognition mechanisms, which are also another key parameter in the CDM.17b To illustrate this idea, Fig. 5 shows the electronic topologies of 3 and 12 displayed in terms of the DFT-computed electrostatic potential contour plots associated to the optimized electronic structures. As seen, the surfaces for these two examples significantly differ from each other. Thus, solute 3 shows a rather cylindrical topology (with a diameter (D) of about 8 Å and a length (L) of about 11 Å, leading to a D/L ratio of 0.72) whereas 12 has a roughly spherical topology (with D ≈ 15 Å). Actually, the former is representative of the topology (rod-like type) adopted by biaryls of series I and II (including also 17 of series IV), whose D/L ratios differ accordingly with the nature of the X and Z substituents. In contrast, the latter (a rather spherical shape) is representative of series III (including also analyte 16 of series IV), whose D varies between 15 and 18 Å (see Table S4, ESI†). Reasonably, the shape recognition mechanisms where the topological anisotropy plays a substantial role are expected to be less efficient in the second case.
 | ||
| Fig. 4 Comparison of 13C–{1H} 1D signals of carbons C-1, C-4, C-7 and C-10 of 10 in (a) racemic and (b) enantiopure series (R) recorded with 4k and 8k scans added, respectively. For those four sites, enantiodiscrimination is observed. The ee measured is about 95%. | ||
 | ||
| Fig. 5 Two examples of electrostatic potential (ESP) surfaces (by plotting only their positive contribution and restraining the contour level with isovalues of 0.005 a.u. for both cases) associated to (a) 3 and (b) 12. | ||
1H-coupled 13C 1D-NMR
At a first glance, and compared to 13C–{1H} NMR in CLC, 13C NMR might be seen to be of limited practical interest for two reasons: (i) the complexity of spectral pattern due to the presence of both short range (1DCH) and long range (nDCH, n = 2, 3) RDCs; and (ii) the distribution of 13C signals on numerous lines, thus reducing the SNR, and hence the accuracy of the ee. However, this approach must be kept in the panel of tools because it can also provide a solution to reveal chiral discriminations. In particular, it can be a useful alternative to 13C–{1H} NMR when molecules possess few or no sp, sp2 carbon atoms. Besides, using simple heteronuclear 2D-NMR experiments (see below), it becomes possible to simplify the spectral analysis of fine 13C–1H structures, while the measurement of peak volumes on 2D maps allows the determination of the ee when the signal of each enantiomer has been identified.Another potential source of useful information can be found from the analysis of 13C signals of substituent Y (see Fig. 1), in particular when small (isolated) groups such as methyl or methoxy are present. Indeed such moieties can lead to very simple spectral patterns to analyze since they are primarily governed by direct 1DCH, which in turn gives rise to quadruplet structures (of relative intensities 1:3:3:1). Such a relevant spectral situation exists for 4, for which two slightly shifted quadruplets (ΔΔσ = 2 Hz) with two different total couplings (|1TACH| = 156 Hz and |1TBCH| = 149 Hz) appear on the 13C spectrum. Assuming that T is positive, we obtain 1DACH = −3.5 Hz and 1DBCH = −7 Hz (1JCH = + 163 Hz). The presence of further doubling is due to one remote dipolar coupling, whose magnitudes differ for each enantiomer. The differences of fine structures for the four components of the quadruplet originate from the various combinations of lines, which also depend on the difference of 13C CSA (2 Hz) and the short- and long-range 13C–1H RDCs as well. The assignment of 13C resonances shown in Fig. 6b has also been supported by the analysis of the associated 13C–1H heteronuclear T-resolved 2D map. In this example, the shielded component of the quadruplet provides the best site for a quantitative measurement of ee (see Fig. 6).
 | ||
| Fig. 6 Comparison of 13C signals of the methyl group of (R/S)-4 dissolved in PBLG/CHCl3 recorded with (a) and without (b) 1H decoupling (4k scans added). Inset (c): zoom on resonances. Inset (d): zoom on the deshielded components of two quadruplets. Integrations of lines (or groups of lines) associated to each enantiomer and showing that ee = 0%. | ||
 | ||
| Fig. 7 (a) Expanded region (centered on C-10 and C-11 sites) of 13C–1H T-resolved 2D spectra of (R/S)-2 in PBLG/CHCl3 (see Fig. 2a). (b) Two vertical slices extracted from the T-resolved map project two dedoubled triplets, one per enantiomer. Each of the (1:2:1) intensity patterns in (b) is highlighted with filled and unfilled circles for visualization purposes. The T values (in Hz) given in (b) are half of the true values. | ||
Heteronuclear correlation 2D-NMR approaches
Except for particular cases (as discussed previously), the analysis of proton-coupled 13C 1D-NMR in CLC cannot be simply performed, and hence heteronuclear correlation 2D-NMR experiments are needed to tentatively extract the useful spectral information for quantitative purposes. During the last decade, a large panel of heteronuclear 2D experiments (involving HSQC scheme) has been explored to extract one bond 1H–13C dipolar couplings.32 However, the control of 1H–13C polarization transfer efficiency (for quantitative measurement of ee dissolved in CLC) can be subtle and time-consuming for routine NMR users. Considering the framework of this study, we only focused our purpose on 2D experiments based on the well-known heteronuclear “J-resolved” schemes.33 As in CLC, the T couplings replace the J couplings, so the experiments were renamed “T-resolved” 2D experiments, but the pulse scheme remains identical. As expected, the 13C chemical shifts are refocused during t1 while the T(13C–1H) couplings are removed during acquisition by 1H decoupling. As the 13C–1H T couplings evolve only during half of the t1 evolution period (gated-decoupled method), the T values are scaled down by a factor of 2. Modified sequences of the basic “T-resolved” experiment might be proposed. For instance, with the view of simplifying the coupling structures in F1, a BIRD cluster can be incorporated to differentiate long-range from direct couplings.34 Additionally, the sensitivity could be improved by incorporating INEPT or DEPT pulse trains as an initial transfer step.35 Nevertheless, one can be faced with either distorted lines or important differences with the transfer efficiency, leading to less accurate ee measurements.Fig. 7 displays the region of the T-resolved 2D spectrum where 1H–13C coupling patterns and 13C chemical shifts associated to C-10 and C-11 atoms of 2 appear in F1 and F2 dimensions, respectively. The analysis of the map allows the relevant information for each carbon site and each enantiomer (noted A and B) to be separated. For each of them, the spectral pattern is dominated by the direct 1TCH coupling, which is different for each enantiomer (|1TA/BCH(C-10)| = 459/528 Hz and |1TA/BCH(C-11)| = 106/82 Hz), as seen on the map. Interestingly, we can measure a large difference of 1TCH (Δ1TCH) of about 70 Hz at site C-10. As 1TA/BCH = 1JA/BCH + 21DA/BCH, the magnitude of 1TA/BCH(C-11) suggests that the sign of 1DA/BCH is negative (compared to 1JA/BCH, which is always positive and ranging from 150–160 Hz for aromatic carbons).36 Note that a similar spectral situation was also observed for the C-11 site of 3 (see Fig. S5, ESI†). Indeed, here again the 1TA/BCH(C-11) value (66/74 Hz) is smaller than 1JCH(C-11), thus indicating that 1DA/BCH<0. The further splittings (at C-10) and triplets (at C-11) observed on the map originate from the nTCH long-range couplings. For both sites, the separation of coupling patterns in F1 is facilitated by the 13C chemical shifts difference between enantiomers. The presence of two triplets (for each enantiomer) is quite unusual but can be explained if C-11 is coupled identically with two inequivalent aromatic protons in the vicinity of C-11 (62 and 64 Hz for A and B, respectively).
The large magnitude of 1DA/BCH (148 and 170 Hz) for C-10 whereas 1JCH = +164 Hz indicates that the C10–H direction is strongly ordered. Actually, the analysis of other carbon sites of 2 (and 8, also) confirms a rather strong degree of molecular alignment compared to other solutes, leading to large (and unusual) 1DCH values. This result suggests the presence of HB in the orientation mechanisms of 2 (and 8) leading to a decrease of the average distance between the analyte and the fiber, and hence an increase of the average degree of alignment of the solute. Locally, it is expected that the ordering of each C–H vector (SCH), and in turn the associated RDC value, increase.
In a framework of a crude two-site interaction model, we can simply write that the Sij order parameter is a weighted sum of two situations corresponding to the case where the solute is either close to the polypeptide fibers (bonded) or at remote location (free):
| SObsij = Pbonded(Sbondedij) + Pfree(Sfreeij) | (1) |
| Obsi = Pbonded(Obsbondedi) + Pfree(Obsfreei) | (2) |
31P–31P correlation 2D-NMR experiments
As explained previously, the comparison of the 31P–{1H} 1D-NMR spectrum of diphosphino biphenyl 16 recorded in racemic and enantioenriched series leads to a rapid assignment of the absolute configuration of lines in the spectrum of the racemic mixture (vide supra). However, this is only possible when an enantioenriched mixture or an enantiopure compound is available and the absolute configuration of the major isomer is known. When the racemic mixture is only available, the assignment of various 31P peaks of (±)-16 is obviously not straightforward. Indeed the positions of 31P resonances can be explained by a difference of 31P CSA or 31P RDC, or both (see Fig. 2). To clear up this ambiguity, various homonuclear 2D-NMR approaches were tested to correlate the 31P resonances to each enantiomer of the mixture: the 31P–31P COSY, T-resolved and INADEQUATE 2D11,37 experiments, no knowledge of 31P–31P RDC being requested for the first two. Experimental maps and comments on the experimental results are proposed in ESI.†The NAD 2D-NMR approach
The main feature of NAD NMR is mainly its low sensitivity due to the very weak natural abundance of deuterium nuclei (1.5 × 10−2%), namely 100 fold less than 13C nuclei. Besides using CLC, the intensity of NAD signals for a given 2H site is reduced by a factor four when spectral discrimination occurs. Indeed, the single 2H peak observed in achiral liquids (see Fig. S2, ESI†) is now split into four resonances (two quadrupolar doublets), hence reducing the SNR, and subsequently the error in the ee value, in particular when the ee's are large. Technically, this situation can be partly overcome using high-field NMR spectrometers, equipped with cryogenic probes when possible. However, the efficiency/interest of this tool will depend primarily on the available amount of analyte, concomitantly to its MW. In this study, the MW of the solutes ranges from 282 to 557 g mol−1, while the available amounts vary from 20 to 100 mg (half of these masses for each enantiomer). Under these conditions, NAD 2D-NMR experiments in CLC have been only recorded for solutes 2 to 4, 6, 9, 11 and 17, for which a sufficient mass of solute was available (see Table 1); this corresponds to a mole number varying from 7.65 to 18.2 × 10−5 mol, namely a mole number of monodeuterated isotopomers [2H] varying from 11.8 to 28.2 × 10−9 mol. The number of discriminated 2H sites for each analyte is reported in Table 1 (see also Table S2, ESI†).The analytical interest of NAD 2D-NMR is the possibility to separate the useful information on two spectral dimensions (see Fig. S12 to S14, ESI†). Thus on the tilted NAD Q-COSY Fz map of 17 (Fig. S12, ESI†), we can easily assess that seven 2H sites (over nine) show spectral discrimination (77%). The presence of three DQ (instead of 4) associated to sites 10 and 12 resonating at the same δ(2H) can lead to two possible interpretations from the discrimination viewpoint: (i) two quadrupolar doublets (QD) for site 12 and one for site 10; and (ii) two QD for sites 10 and 12, considering that both internal doublets for each site possess the same splitting. Actually, the second analysis is less probable for three reasons: (i) the differences of RQCs (ΔΔνQ = |ΔνRQ − ΔνSQ|) for each aromatic site are quite similar (ranging from 210 to 355 Hz for the outer one and from 167 to 278 for the inner one); (ii) in the structure, the C–2H10 and C–2H4 bonds are collinear (para position), and hence the order parameters (and their associated ΔνQ's) are expected to be similar for both C–2H vectors (124 and 140 Hz respectively); and (iii) the absence of discrimination at both sites. The analysis of the aliphatic region indicates that both methyl and methoxy groups are discriminated (until the baseline) with RQCs differences of 24.0 and 23.4 Hz, respectively, namely 12 Hz between R and S components of doublets. Due to the free rotation of methyl deuterons around the C–C bond (1 rotor) or C–O–C bonds (2 rotors), the RQC values are averaged down compared to the RQCs of aromatic 2H sites (from 3–10 fold less). Interestingly, the contribution of three deuterons to the NAD signals increases the SNR (162 and 146) compared to the aromatic sites, thus providing the two best sites to accurately determine the enantiomeric excesses.
Except for analytes 2 and 8 (see below), the distribution in magnitude of the RQCs of the aromatic 2H QDs observed on the NAD 2D map is globally quite similar for the various analytes (see Fig. S12 and S13, ESI,† for instance). In contrast, the RQCs measured for the 2H sites of the flexible Y substituent show large variations in magnitude. In order to illustrate this, Fig. 8 compares the NAD 1D signals of the aldehyde groups of 3 and 9 and methyl groups of 4/17 and 11/17 (see Fig. S15, ESI† for the full 2D map of 6 and 11). For each case, the spectral enantiodiscrimination on Y occurs with differences of RQCs, |ΔΔνQ|, varying from 5 Hz (11) to 35 Hz (4), while the average of the corresponding RQCs varies from 129 Hz to 28.5 Hz. The large variation of SNR between the methine and methyl group originates from the number of 2H nuclei contributing to signals (1 to 3) but also the mass (60 mg to 100 mg) and the MW (277 to 370) of each sample, and possibly some variations of isotopic ratio from one site to another (but not between enantiomers).
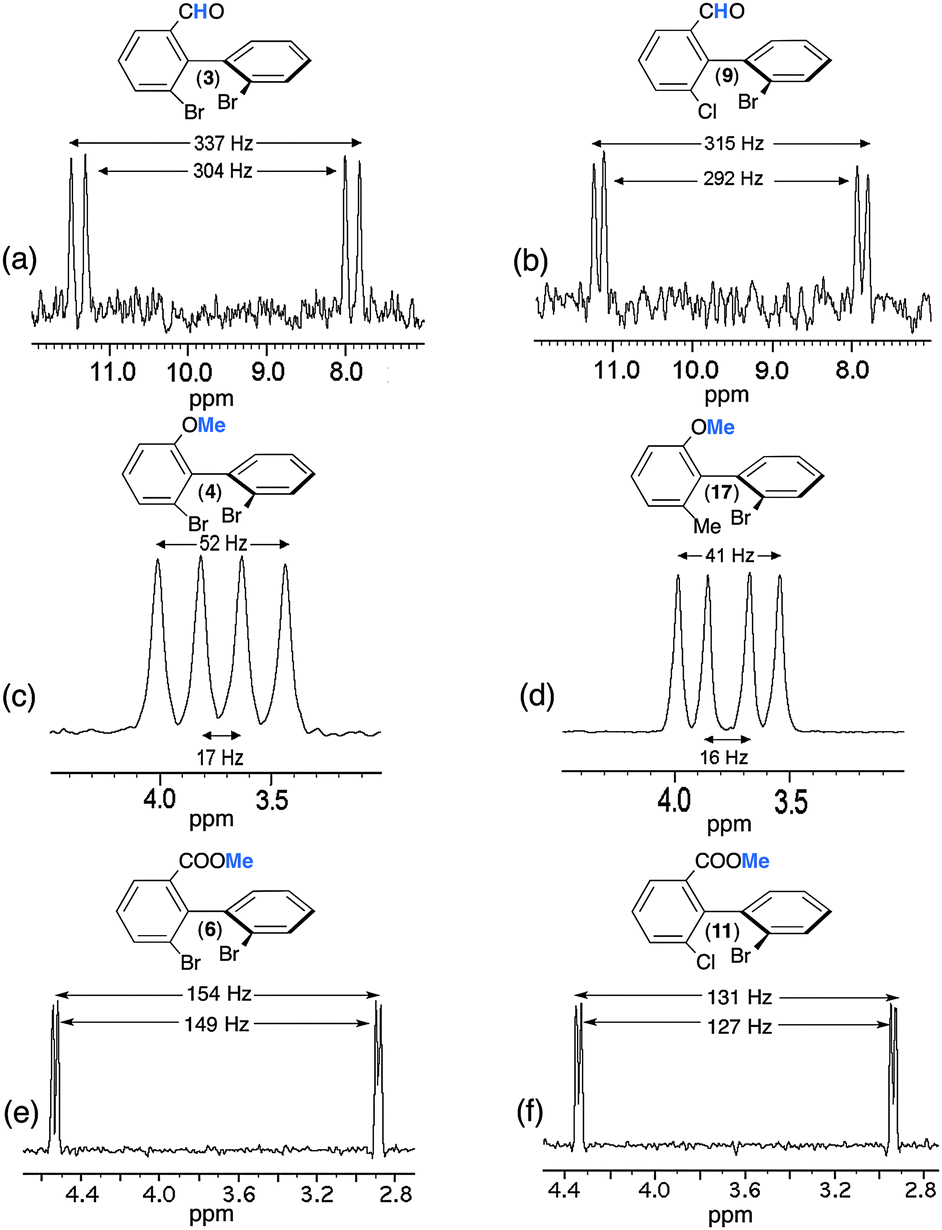 | ||
| Fig. 8 92.1 MHz proton-decoupled NAD 1D-NMR signals of (a and b) the methine of the aldehyde group of (R/S)-3 and (R/S)-9, of (c and d) the methyl of the methoxy group of (R/S)-4 and (R/S)-17 and of (e and f) the methyl of the ester group of (R/S)-6 and (R/S)-11, all of them dissolved in PBLG/CHCl3 at 295 K. All patterns are extracted from their tilted NAD Q-COSY Fz map. Except for e and f (see Fig. S10, ESI†), an exponential filtering (LB = 2 Hz) is applied on both dimensions. | ||
Various comments can be made on this series of results, in particular on the variation of RQCs and the magnitude of enantiodiscriminations for the different 2H sites. Indeed, the comparison of results indicates that there is not a simple correlation between the magnitude of RQCs, |ΔνQ|, the RQC's difference between enantiomers, |ΔΔνQ|, and the number of rotors in the ligand Y (one for 3 and 9), two for 4 and 17, and three for 6 and 11. Actually, various (sometimes contradictory) effects, such as the difference of electronic properties of each ligand (aldehyde, ether and ester group) and the position (number of rotors) of the 2H site in the ligand can be evoked to explain the results. Thus, the large magnitude of RQCs for 3 and 9 associated to a strong degree of alignment of the C–H direction could suggest a strong site-specific interaction (charge-transfer interactions) between the carbonyl polar group and the PBLG fiber, despite the free rotation around the C–C bond. The large difference of |ΔνQ's| (from 34 to 150) and |ΔΔνQ's| (from 35 to 5) observed for the methyl groups in 4 (17) and 6 (11) is much more subtle to explain/understand. They both involve significant differences of averaged orientation of the C–D directions, the number of rotors (2 and 3) between the ring and the 2H site, and the electronic properties of the ligand. On the basis of the rotor number, it could be expected that the |ΔνQ's| for 6 and 11 would be larger than 4 and 17. This trend is not experimentally observed whereas larger |ΔΔνQ's| are measured for those latter compared to 6 and 11. This illustrates the difference of electronic interaction between ether or carboxylate groups and the PBLG side chain (which can promote a more or less stronger alignment of the Y ligand), but also the ability of the 2H site to sense the chirality of the biaryl skeleton versus its distance and the number of rotors in the flexible part. Results obtained for the methyl group (6 and 11) suggest that a physical interaction between PBLG and the carboxylate group might generate a higher degree of alignment for the COOMe moiety, and hence for the terminal methyl group, but in this case with a weaker enantiodiscrimination efficiency.
Finally, it must be noticed that the NAD 2D-NMR spectra of the carboxylic acids (2 and 8) were not analytically exploitable whereas 13C–{1H} NMR had provided well-resolved spectra with excellent results in terms of spectral quality and enantiodiscriminations (see Table 1, Table S3, ESI†). Indeed, 2D maps of 2 and 8 are made of weakly resolved NAD QDs of low intensity, not distinctly emerging from noise (even with strong exponential apodisation), and showing splittings ranging from 1500 to 2000 Hz (see Fig. S14a/b, ESI†). These magnitudes of RQCs are unusually large for small solutes oriented in weakly aligning media as those prepared with the PBLG polymer, whereas the symmetrical shape, the linewidth (3 Hz) and the splitting (around 500 Hz) of chloroform both indicate a homogeneous and uniform mesophase that complies with standards when w/w (PBLG) = 14%. No significant enhancement was obtained by several rehomogenizations of the sample (new cycles of centrifugations) or by a sample temperature variation.
Although a priori unexpected, the low quality of NAD NMR spectra for 6 and 8 (due to unusually large RQCs) can be explained by the presence of HB, and again understood in the frame of the simple model proposed above. Derived from eqn (2), we can write that:
| ΔνObsQ(2H) = Pbonded(ΔνbondedQ(2H)) + Pfree(ΔνfreeQ(2H)) | (3) |
All these results and arguments point out openly the important role of HB in the ordering mechanisms of the solute (in particular the degree of molecular orientation) and the related analytical consequences according to the NMR properties of the observed nucleus. Thus, the presence of HB involving COOH groups was a major advantage in terms of 13C enantiodiscrimination (on the basis of 13C CSAs) since 85% of 13C sites showed discriminations, but can lead to undesirable effects visible on the NAD spectra (on basis of 2H RQCs). This beneficial difference results from a more complex and “dilute” dependence of the 13C CSA through the electronic screen tensor (for each 13C site) toward the molecular ordering of a solute compared to ΔνQ. This rather contradictory situation associated to 13C and 2H NMR illustrates nicely some versatile aspects of anisotropic NMR as well as the subtle balance between the orientation and the chiral discrimination.
Importance and role of factors involved in the CDM
In the frame of the understanding and the phenomenological description of CDMs in polypeptide CLC (and in particular in PBLG), the analysis of the various factors (and their respective role) governing or involving the efficiency of CDM is a necessary step before proposing models describing the phenomenon or a starting point for any computational modeling of the system. A priori, it is difficult to dissociate the topological properties (shape anisotropy) and the electronic profile of a solute due to their strong inherent intrication. However, for a qualitative description of contributive factors, we may propose this artificial separation.From the analysis of 13C–{1H} NMR results of this study (data from seventeen solutes) related to previous studies, we evidenced that the topological factors are of primary importance.11,16,17b,39 According to the degree of shape anisotropy of solutes (for instance, spheroid, cylinder, spiral,…), the efficiency of global shape recognition mechanisms (which are closely related to steric exclusion effects) is basically different, the best situation being met with a spiral topology rather than a spheroid one.
As shape recognition mechanisms are short-range interactions that are highly active when the solute is in the closest vicinity of the polypeptide, they are strongly dependent on possible local electronic (electrostatic) interactions between the solute and the chiral fiber, and in particular the flexible side chain of the polymer. Hence, the electronic local properties related to the nature of substituents (presence of HB, strength of C–O dipole moment, steric hindrance) in combination with the global properties of the analyte (the global dipole moment) play a key role in the solute–PBLG interactions and their capability to “maintain” or not the solute close to the chiral fiber, namely when the CDM are the most efficient.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” bonds in 2, 3, 4 and 6 has been computed by DFT method, and an attempt to correlate NDS (and their magnitude) was performed. The correlation curve (μC–Oversus% of discriminated 13C sites) is plotted in Fig. 9. Regardless of the electro-donor or electro-attractor character of each substituent within the biaryls, the analysis of μC–O values is informative. Thus, we can notice that the variation of μC–O trends is rather linear for 2, 3 and 4 for series I whereas electro-donor (ED)/electro-attractor (EA) properties towards biaryl electronic density differ from each other (EA for 2 and 3, and ED for 4). The divergence observed for 6 (compared to the linearity observed for 2, 3 and 4) is rather surprising because μC–O(6) is very similar to μC–O(2). Indeed, for an isovalue of μC–O, we might expect similar NDS. This situation suggests that HB is the primary driving force of 2 to interact with PBLG. Once no labile proton is available, solute 6 only has the C–O dipole as driving force, such as 3 and 4 in order to interact with PBLG by means of a positive dipole within the mobile arm. This occurrence perfectly illustrates the multivariable dependency of NDS.
O” bonds in 2, 3, 4 and 6 has been computed by DFT method, and an attempt to correlate NDS (and their magnitude) was performed. The correlation curve (μC–Oversus% of discriminated 13C sites) is plotted in Fig. 9. Regardless of the electro-donor or electro-attractor character of each substituent within the biaryls, the analysis of μC–O values is informative. Thus, we can notice that the variation of μC–O trends is rather linear for 2, 3 and 4 for series I whereas electro-donor (ED)/electro-attractor (EA) properties towards biaryl electronic density differ from each other (EA for 2 and 3, and ED for 4). The divergence observed for 6 (compared to the linearity observed for 2, 3 and 4) is rather surprising because μC–O(6) is very similar to μC–O(2). Indeed, for an isovalue of μC–O, we might expect similar NDS. This situation suggests that HB is the primary driving force of 2 to interact with PBLG. Once no labile proton is available, solute 6 only has the C–O dipole as driving force, such as 3 and 4 in order to interact with PBLG by means of a positive dipole within the mobile arm. This occurrence perfectly illustrates the multivariable dependency of NDS.
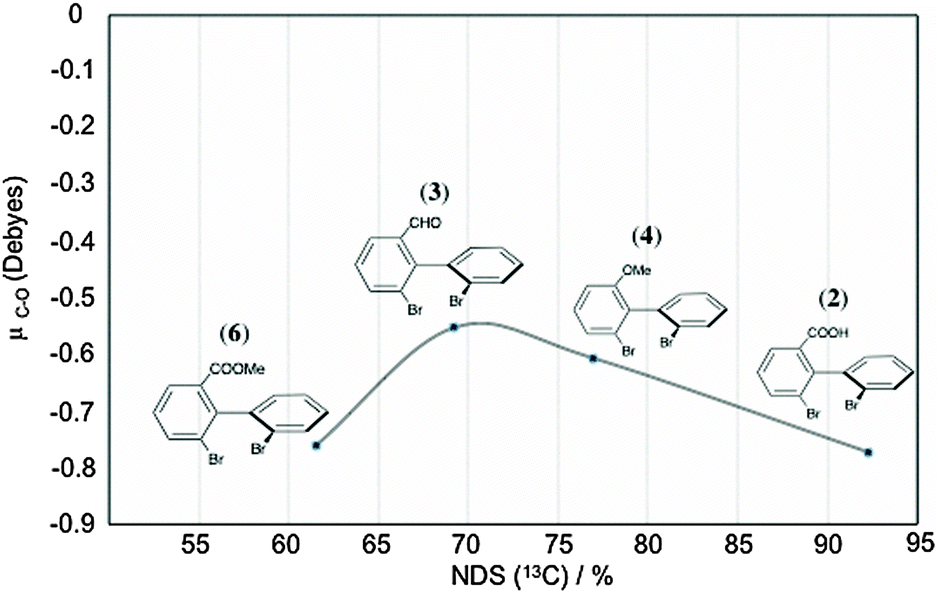 | ||
| Fig. 9 Variation of the predicted C–O dipole moment (obtained by Mulliken charge distribution analysis, see details in the Experimental Section) as a function of the percentage of NDS(13C) for biaryl solutes (2, 3, 4 and 6) of series I (see Table 1). | ||
From the chemical reactivity viewpoint, it is known that we can relate the Lewis bases (HOMOs with rich electronic density) with the Lewis acids (LUMOs with deficiency in electronic density).40 In our case, the π-stacking interaction (which can be though of as being a specific kind of dipole–dipole interaction)41 could also be explained in terms of frontier molecular orbital approach, and hence regarded as a Lewis base (HOMO) interacting with an acidic moiety (LUMO). In this context, dipole–dipole π-stacking interactions between biaryls (HOMO) and PBLG benzylic rings (LUMO) can be reasonably proposed.
To understand our purpose, we have determined the location and the electronic density charge of HOMOs for six model compounds of the series I (1, 2, 6) and II (7, 8, 11), and then correlated both pieces of information with the NDS revealed by 13C NMR. To illustrate our purpose, Fig. 10 proposes such a graphical correlation. As seen in the figure, the resulting effect due to the various activation/deactivation contributions of the three substituents leads to the HOMOs of biaryls being located either on ring A (1, 7) or on ring B (2, 6, 8, 11). Note however that the replacement of Br by Cl atom on ring A (series I and II) only slightly modifies the electronic density charge but not the position of the HOMOs on the rings.
 | ||
| Fig. 10 Representation of the highest occupied molecular orbital electronic densities of some biaryls of series I (red zone) and series II (grey zone). HOMOs were plotted with the same contour levels and their relative charges were normalized for all species at the same conditions. | ||
Excluding all electronic effects discussed previously and just limiting the discussion to the π-stacking interaction, a correlation between the NDS(13C) and the location of HOMOs, their electronic density charge, and also the steric hindrance on each ring (number and size of substituents) can be proposed. Thus, biaryl species (1, 7) with a Y substituent (I atom) that localizes the electronic density of HOMOs within the di-substituted ring A (1 and 7) present the lowest number of discriminated 13C sites. In contrast, once the Y group relocalizes the electronic density charge of HOMOs at ring B (mono-substituted), the NDS is considerably enhanced. Besides, Y groups like COOH not only re-localize the electronic density charge on ring B, but also increase the electronegativity of HOMOs (observed by negative red HOMO lobes for COOH-dibromide-biaryl solute 2) due to the COOH's deactivating nature on ring A. The last could be conceived as if COOH increases the Lewis basic nature of biaryls and thus increases the “reactivity” towards a Lewis acid (PBLG benzyl ring). Finally, the presence of a lower steric hindrance in ring B (a single mono-atomic ortho-substituent) with respect to ring A (two ortho-substituents with complex dynamics for Y) reinforces the idea that HOMOs at ring B are more susceptible to establishing π-stacking interactions with the LUMOs of PBLG rings (compared to HOMOs at site A). In other words, ring B is more sterically free to form dipole–dipole π-stack interactions with PBLG.
Actually, for these six solutes, the variation of NDS can be globally understood as follows. For solutes 2 and 8, three highly favorable electronic factors enhance the efficiency of CDM (thus leading to a maximal NDS), the presence of HB, the strength of C–O dipole and the location of HOMOs at ring B. For solutes 6 and 11, HB is impossible, and only two favorable electronic factors exist (the strength of C–O dipole and the location of HOMOs at ring B), thus reducing the efficiency of CDM (and the NDS). For solutes 1 and 7, the absence of important elements (HB and C–O dipole) on the Y substituent leads to the less favorable situation with respect to CDM, thus leading to smaller NDS in the series.
Conclusions
New emerging pure enantiomeric structures need robust analytical tools to determine the enantiopurity of a given class of chiral compounds, in particular when routine methods failed or requested instrumentation specific accessories.The aim of this work was: (i) to propose a panel of simple and robust 1D/2D-NMR experiments in CLCs to investigate the chiral biaryls, without any need for NMR expertise, (ii) to present the relevant analytical subtleties to understand all NMR observables in CLCs and their consequences on spectra; and (iii) to analyze and correlate all data to provide new insights into the factors playing a role within the CDMs of polypeptide CLC, thus leading to a better understanding of all interaction mechanisms and enantiorecognition phenomena.
Among the appealing results of the present study, the analysis of NDS and the magnitude of enantiodiscriminations in the 13C NMR spectra revealed the importance of the global molecular shape anisotropy of analytes, the role of (local) electronic properties of substituents (compared to global properties) to maintain the solute in the vicinity of the PBLG fibers and, last but not least, the subtle balance between the electronic effects favoring interaction with PBLG and the steric repulsion associated with the size of substituents.
From the results reported in this work, it appears that NMR in polypeptide CLC should be considered as a valuable tool to analyze the enantiopurity of molecular structures belonging to the fascinating family of (bridged and non-bridged) biaryl atropisomers.
Acknowledgements
This research work was supported by the CNRS and the MENR. P.L. and F.L. are very much grateful to the CNRS for the continuous financial support of fundamental research. J.-E.H.-P. thanks Labex CHARM3AT and V.J. is very grateful to the IFCPAR/CEFIPRA for their postdoctoral fellowship.References
- G. Bringmann, G. Günther, M. Ochse, O. Schupp and S. Tasler, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk, G. W. Kirby, R. E. Moore and C. Tamm, Springer, New York, 2001, vol. 82, p. 1 Search PubMed.
- (a) A. V. R. Rao, M. K. Gurjar, K. L. Reddy and A. S. Rao, Chem. Rev., 1995, 95, 2135 CrossRef CAS; (b) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096 CrossRef; (c) O. Baudoin and F. Guéritte, in Studies in Natural Product Chemistry, ed. Atta-Ur-Rahman, Elsevier, 2003, vol. 29, p. 355 Search PubMed; (d) T. Leermann, P. E. Broutin, F. R. Leroux and F. Colobert, Org. Biomol. Chem., 2012, 10, 4095 RSC.
- (a) S. M. Kupchan, R. W. Britton, M. F. Ziegler, C. J. Gilmore, R. J. Restivo and R. F. Bryan, J. Am. Chem. Soc., 1973, 95, 1335 CrossRef CAS PubMed; (b) R. W.-J. Wang, L. I. Rebhun and S. M. Kupchan, Cancer Res., 1977, 37, 3071 CAS; (c) F. Zavala, D. Guenard, J.-P. Robin and E. Brown, J. Med. Chem., 1980, 23, 546 CrossRef CAS PubMed; (d) R. Dhal, E. Brown and J.-P. Robin, Tetrahedron, 1983, 39, 2787 CrossRef CAS; (e) K. Tomioka, T. Ishiguro, H. Mizuguchi, N. Komeshima, K. Koga, S. Tsukagoshi, T. Tsuruo, T. Tashiro, S. Tanida and T. Kishi, J. Med. Chem., 1991, 34, 54 CrossRef CAS PubMed; (f) D. B. M. Wickramaratne, T. Pengsuparp, W. Mar, H.-B. Chai, T. E. Chagwedera, C. W. W. Beecher, N. R. Farnsworth, A. D. Kinghorn, J. M. Pezzuto and G. A. Cordell, J. Nat. Prod., 1993, 56, 2083 CrossRef CAS PubMed; (g) D. L. Sackett, Pharmacol. Ther., 1993, 59, 163 CrossRef CAS PubMed; (h) B. Yalcouye, S. Choppin, A. Panossian, F. R. Leroux and F. Colobert, Eur. J. Org. Chem., 2014, 6285 CrossRef CAS.
- (a) A. A. Patchett and R. P. Nargund, in Annu. Rep. Med. Chem., ed. G. L. Trainor, Academic Press, New York, 2000, section IV, vol. 35, p. 289 Search PubMed; (b) F. Leroux, Curr. Med. Chem., 2005, 12, 1623 CrossRef CAS PubMed; (c) F. Leroux, ChemBioChem, 2004, 5, 644 CrossRef CAS PubMed; (d) F. Leroux, T. U. Hutschenreuter, C. Charriere, R. Scopelliti and R. W. Hartmann, Helv. Chim. Acta, 2003, 86, 2671 CrossRef CAS; (e) E. Baston and F. R. Leroux, Recent Pat. Anti-Cancer Drug Discovery, 2007, 2, 31 CrossRef CAS PubMed.
- (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed; (b) M. McCarthy and P. J. Guiry, Tetrahedron, 2001, 57, 3809 CrossRef CAS; (c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS; (d) T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS PubMed; (e) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed; (f) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (g) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed; (h) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC.
- (a) G. Bringmann, R. Walter and R. Weirich, Angew. Chem., Int. Ed., 1990, 29, 977 CrossRef; (b) M. Putala, Enantiomer, 1999, 4, 243 CAS; (c) P. Lloyd-Williams and E. Giralt, Chem. Soc. Rev., 2001, 30, 145 RSC; (d) C. Bolm, J. P. Hildebrand, K. Muñiz and N. Hermanns, Angew. Chem., 2001, 113, 3382 CrossRef; (e) Angew. Chem., Int. Ed., 2001, 40, 3284 Search PubMed; (f) J. Hassan, M. Sévignon, C. Gozzi, E. Schultz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- A. Ahmed, R. A. Bragg, J. Clayden, L. W. Lai, C. McCarthy, J. H. Pink, N. Westlund and S. A. Yasin, Tetrahedron, 1998, 54, 13277 CrossRef CAS.
- F. R. Leroux, A. Berthelot, L. Bonnafoux, A. Panossian and F. Colobert, Chem. – Eur. J., 2012, 18, 14232 CrossRef CAS PubMed.
- (a) J. Graff, T. Debande, J. Praz, L. Guenee and A. Alexakis, Org. Lett., 2013, 15, 4270 CrossRef CAS PubMed; (b) Q. Perron and A. Alexakis, Adv. Synth. Catal., 2010, 352, 2611 CrossRef CAS.
- (a) K. Kabuto, F. Yasuhara and S. Yamaguchi, Tetrahedron Lett., 1980, 21, 307 CrossRef CAS; (b) K. Kabuto, F. Yasuhara and S. Yamaguchi, Tetrahedron Lett., 1981, 22, 659 CrossRef CAS; (c) A. Alexakis, J. C. Frutos and P. Mangeney, Tetrahedron Lett., 1994, 35, 5125 CrossRef CAS.
- M. Sarfati, P. Lesot, D. Merlet and J. Courtieu, Chem. Commun., 2000, 2069 RSC.
- (a) C. Aroulanda, D. Merlet, J. Courtieu and P. Lesot, J. Am. Chem. Soc., 2001, 123, 12059 CrossRef CAS PubMed; (b) P. Lesot, Z. Luz, C. Aroulanda and H. Zimmermann, Magn. Reson. Chem., 2014, 52, 581 CrossRef CAS PubMed.
- P. Lesot, C. Aroulanda, H. Zimmerman and Z. Luz, Chem. Soc. Rev., 2015, 44, 2330 RSC.
- (a) P. Lesot, O. Lafon, H. B. Kagan and P. Fan, Chem. Commun., 2006, 389 RSC; (b) O. Lafon, P. Lesot, C. A. Fan and H. B. Kagan, Chem. – Eur. J., 2007, 13, 3772 CrossRef CAS PubMed.
- (a) T. Wenzel, Discrimination of Chiral Compounds using NMR Spectroscopy, John Wiley & Sons, INC publication, Wiley-Interscience, 2007 Search PubMed; (b) T. J. Wenzel and C. D. Chisholm, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 1 CrossRef CAS PubMed; (c) T. J. Wenzel and C. D. Chisholm, Chirality, 2011, 23, 190 CrossRef CAS PubMed; (d) S. R. Chaudhari and N. Suryaprakash, J. Indian Inst. Sci., 2014, 94(4), 485 Search PubMed.
- (a) A. Meddour, P. Berdagué, A. Hedli, J. Courtieu and P. Lesot, J. Am. Chem. Soc., 1997, 119, 4502 CrossRef CAS; (b) P. Lesot, O. Lafon, J. Courtieu and P. Berdagué, Chem. – Eur. J., 2004, 10, 3741 CrossRef CAS PubMed; (c) P. Tzvetkova, B. Luy and S. Simova, in Topics in Chemistry and Material Science 5, Current Issues in Organic Chemistry, ed. R. D. Nikolova, S. Simova, P. Denkova and G. N. Vayssilov, Heron Press Ltd, Birmingham, 2011, pp. 70–77 Search PubMed.
- (a) P. Lesot, D. Merlet, A. Loewenstein and J. Courtieu, Tetrahedron: Asymmetry, 1998, 9, 1871 CrossRef CAS; (b) P. Lesot, M. Sarfati and J. Courtieu, Chem. – Eur. J., 2003, 9, 1724 CrossRef CAS PubMed; (c) P. Lesot and J. Courtieu, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 128 CrossRef CAS; (d) P. Lesot, In Deuterium NMR of Liquid-Crystalline Samples at Natural Abundance, eMagRes, 2013, 2(3), 315, DOI:10.1002/9780470034590.Emrstm1318.
- L. Bonnafoux, F. R. Leroux and F. Colobert, Beilstein J. Org. Chem., 2011, 7, 1278 CrossRef CAS PubMed.
- L. Bonnafoux, R. Gramage-Doria, F. Colobert and F. R. Leroux, Chem. – Eur. J., 2011, 17, 11008 CrossRef CAS PubMed.
- (a) C. Aroulanda, M. Sarfati, J. Courtieu and P. Lesot, Enantiomer, 2001, 6, 281 CAS; (b) C. M. Thiele and S. Berger, Org. Lett., 2003, 5, 705 CrossRef CAS PubMed; (c) P. Lesot, O. Lafon, C. Aroulanda and R. Dong, Chem. – Eur. J., 2008, 14, 4082 CrossRef CAS PubMed.
- H. Kovacs, D. Moskau and M. Spraul, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 131 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) J. Tomasi, R. Cammi, B. Mennucci, C. Cappelli and S. Corni, Phys. Chem. Chem. Phys., 2002, 4, 5697 RSC; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- C. Canlet, D. Merlet, P. Lesot, A. Meddour, A. Loewenstein and J. Courtieu, Tetrahedron: Asymmetry, 2000, 11, 1911 CrossRef CAS.
- L. Bonnafoux, L. Ernst, F. R. Leroux and F. Colobert, Eur. J. Inorg. Chem., 2011, 3387 CrossRef CAS.
- A. Meddour, J. Uziel, J. Courtieu and S. Jugé, Tetrahedron: Asymmetry, 2006, 17, 1424 CrossRef CAS.
- A. M. Marathias, P. A. Tate, N. Papaioannou and W. Massefski, Chirality, 2010, 22, 838 CrossRef PubMed.
- (a) E. E. Burnell and C. A. De Lange, Chem. Rev., 1998, 98, 2359 CrossRef CAS PubMed; (b) E. E. Burnell and C. A. De Lange, NMR of Ordered Liquids, Kluwer, Academic, Dordrecht, 2003, ch. 10 to 14 included Search PubMed.
- (a) A. Enthart, J. C. Freudenberger, J. Furrer, H. Kessler and B. Luy, J. Magn. Reson., 2008, 192, 314 CrossRef CAS PubMed; (b) T. Reinsperger and B. Luy, J. Magn. Reson., 2014, 239, 110 CrossRef CAS PubMed.
- G. A. Morris, in Two-Dimensional J-Resolved 2D Spectroscopy, Encyclopedia of Magnetic Resonance, ed. R. K. Harris and R. E. Wasylishen, Wiley, Chichester, 2009, http://DOI: 10.1002/9780470034590.emrstm0579.pub2 Search PubMed.
- (a) J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504 CrossRef CAS; (b) A. Bax, J. Magn. Reson., 1983, 53, 517 CAS; (c) A. Lupulescu, G. L Olsen and L. Frydman, J. Magn. Reson., 2013, 218, 141 CrossRef PubMed.
- (a) G. A. Morris and R. Freeman, J. Am. Chem. Soc., 1979, 101, 760 CrossRef CAS; (b) D. M. Doddrell, D. T. Pegg and M. R. Bendall, J. Magn. Reson., 1982, 48, 323 CAS; (c) G. A. Morris, J. Am. Chem. Soc., 1980, 102, 428 CrossRef CAS.
- H. O. Kalinowski, S. Berger and S. Braun, Carbon-13 NMR Spectroscopy, J. Wiley and Sons, Chichester, 1984 Search PubMed.
- (a) A. Bax, R. Freeman and S. P. Kempsell, J. Am. Chem. Soc., 1980, 102, 4849 CrossRef CAS; (b) A. Bax, R. Freeman, T. A. Frenkiel and M. H. Levitt, J. Magn. Reson., 1981, 43, 478 CAS; (c) P. Lesot, J. W. Emsley and J. Courtieu, Liq. Cryst., 1998, 25, 123 CrossRef CAS.
- J. W. Emsley, P. Lesot and D. Merlet, Phys. Chem. Chem. Phys., 2004, 6, 522 RSC.
- P. Lesot, Z. Serhan, C. Aroulanda and I. Billault, Magn. Reson. Chem., 2012, 50, S2 CrossRef CAS PubMed.
- K. Fukui, T. Yonezawa and H. Shingu, J. Chem. Phys., 1952, 20, 722 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A description of experimental details and DFT calculation, analytical details, chemical pathway, background of NMR in CLC, further 1D/2D NMR spectra, and NMR assignment data of compound 16 are given. Composition of oriented samples, details on anisotropic NAD NMR, further anisotropic NMR spectra and extra discussion for some compounds. See DOI: 10.1039/c5nj01434d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |