
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of substitution pattern and enhanced π-conjugation on a family of thiophene functionalized 1,5-dithia-2,4,6,8-tetrazocines†
François
Magnan
ab,
Ilia
Korobkov
a and
Jaclyn
Brusso
*ab
aDepartment of Chemistry, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada. E-mail: jbrusso@uottawa.ca; Fax: +1 6135625170; Tel: +1 6135625800 ext. 7091
bCentre for Catalysis Research and Innovation, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
First published on 23rd July 2015
Abstract
A versatile, one-pot synthetic route to prepare a series of 1,5,2,4,6,8-dithiatetrazocines (DTTA) functionalized with 4-hexylthiophen-2-yl, 5-(thiophen-2′-yl)thiophen-2-yl and 5-(5′-hexylthiophen-2′-yl)thiophen-2-yl is described. The latter two compounds mark the first reported examples of π-extended DTTA systems. Comparative spectroscopic, computational and structural studies were carried out on this family of heterocycles to investigate the influence of the substitution pattern, in addition to enhanced conjugation, on the optoelectronic and solid-state properties. These studies revealed a slight increase in the optical energy gap upon displacement of the alkyl chain from the 5- to the 4-position in hexylthienyl functionalized DTTA, whereas extension of the conjugation diminishes the HOMO–LUMO gap. In the solid-state, variation in alkyl substituent plays an important role, contrary to what was previously observed for aryl functionalized DTTA derivatives.
Introduction
Electroactive organic molecules have seen significant development over the past two decades owing to their potential use in next-generation electronic devices.1–3 Low-cost, miniaturization, tunable optoelectronic properties, mechanical flexibility and processability are a few of the key advantages organic based materials offer compared to their more traditional inorganic counterparts.4–6 In regards to tuning the optoelectronic properties in organic semiconductors (OSC), this may be achieved via modulating the substituents or by introduction of heteroatoms into the conjugated core. To that end, sulphur and nitrogen containing heterocycles are commonly observed motifs in OSCs as incorporating these atoms into planar π-conjugated frameworks provides several benefits over their all carbon variants (e.g., acenes). For example, S–N heterocycles have been shown to demonstrate: (i) improved stability towards air oxidation due to the presence of the electronegative atoms, which lowers the energy of the highest occupied molecular orbital (HOMO); (ii) the potential for structural control through S⋯N and N⋯HC interactions; and (iii) the possibility of strong orbital overlap between the larger π-orbitals of sulphur leading to enhanced electronic communication between molecules.7–14 With respect to the rational design of electroactive materials, organization in the solid-state is of utmost importance as their electronic properties are critically dependent on the extent of molecular order. Thus the packing arrangements afforded by planar π-conjugated molecules, in addition to the intermolecular interactions exhibited by sulphur and nitrogen moieties, are key to the implementation of S–N heterocycles into molecular electronics. As such, ideal systems should exhibit π–π stacking in the solid-state, strong intermolecular communication, as well as high thermal and ambient stability.In that regard, 1,5,2,4,6,8-dithiatetrazocine (DTTA) is an attractive building block for electroactive substrates as not only does it meet these criteria, but it can also be modulated by virtue of the 3,7-susbtituents (Chart 1). First discovered in 1981,15 the molecular structure of DTTA has been shown to be highly dependent on the nature of the substituents. For example, the C2N4S2 ring can exist in either a planar, 10π electron aromatic structure or in a bent conformation where the ring is folded about the axis of the two sulphur atoms with a transannular S⋯S contact, as shown in Chart 1. Although this structural dichotomy is substituent dependent, the planar structure is most commonly observed except when strongly electron-donating substituents are employed (e.g., –NMe2).16 In the solid-state, planar DTTAs commonly form π-stacked structures with close intermolecular contacts and tend to be incredibly robust with high tolerance to acidic, basic, oxidizing and thermal conditions.15,17 Thus, incorporation of the DTTA framework in the design of OSCs, either polymeric or molecular in nature, offers many potential avenues to explore.
 | ||
| Chart 1 Planar and bent configurations of dithiatetrazocine and examples of thienyl (1) and bithienyl (2) functionalized derivatives. | ||
Remarkably, the majority of research reported on DTTA focuses on the synthesis of phenyl- and amino-functionalized derivatives, with very little emphasis on their electronic properties or potential as electroactive substrates.17–21 In regards to the latter, we recently reported the synthesis and detailed study of the optoelectronic and solid-state properties of thienyl-functionalized DTTA (1a) and its hexylated analogue 1b. While thienyl groups are stronger π-donors than phenyl substituents, 1a and 1b were isolated as planar, π-conjugated aromatic molecules.22 To investigate the role of the substitution pattern on the optoelectronic and solid-state properties, we herein report the synthesis and characterization of 3,7-bis(4-hexylthiophen-2-yl)-1,5,2,4,6,8-dithiatetrazocine (1c). As well, in order to enhance the intermolecular interactions, we sought to increase the degree of conjugation. As such, the first examples of bithienyl-substituted DTTAs (3,7-bis([5-(thiophen-2′-yl)thiophen-2-yl])-1,5,2,4,6,8-dithiatetrazocine (2a) and 3,7-bis([5-(5′-hexylthiophen-2′-yl)thiophen-2-yl])-1,5,2,4,6,8-dithiatetrazocine (2b)) are also presented. These studies demonstrate the versatility of the synthetic procedure in the development of DTTAs functionalized with thienyl moieties. As well, comparative optical, computational and single crystal X-ray diffraction studies were performed on this family of thienyl (1a–c) and bithienyl (2a–b) functionalized DTTAs to probe the influence that both the substitution pattern and extension of the conjugation have on the optoelectronic and solid-state properties.
Results and discussion
Synthesis
While a number of amino and phenyl functionalized DTTAs have been reported, examples of thienyl-substituted derivatives remain scarce,17,18,22 with the first reported synthesis involving the reaction of 2-thienylamidine with sulphur dichloride affording 1,5-bisthienyl-2,4,6,8-dithiatetrazocine (1a) in 7% yield.17 Although 1a can be prepared via this route, a higher yielding approach based on methodology reported by Boeré et al.19 involves the formation of the dithiadiazolium cation (5a) from the corresponding persilylated amidine, followed by reduction with triphenylantimony (SbPh3) in the presence of oxygen (Route A, Scheme 1).22 Following the Boeré protocol, crude 4-hexylthienyl functionalized DTTA (1c) can be isolated as a dark yellow solid in two steps from 2-cyano-4-hexylthiophene (3c) with an overall yield of 35%. Recrystallization from hot chlorobenzene afforded 1c as bright yellow needles. | ||
| Scheme 1 Synthesis of thienyl-functionalized dithiatetrazocines 1a–c and 2a–b ((a) R1 = H, R2 = H; (b) R1 = C6H13, R2 = H; (c) R1 = H, R2 = C6H13). Reagents and conditions: (i) LiN(TMS)2·Et2O/TMSCl/toluene; (ii) S2Cl2/CH3CN; (iii) LiN(TMS)2·Et2O/S2Cl2/Et2O; (iv) Ph3Sb/O2/CH3CN. | ||
While route A is more efficient than the originally reported synthetic procedure, it requires isolation and purification of 4-hexylthiophene-2-N,N,N′-tris(trimethylsilyl)carboximidamide via vacuum distillation. This can unfortunately lead to varying yields due to the instability of the silylated amidine, especially with respect to moisture. In order to alleviate these issues, a slightly different procedure was explored (Route B), inspired by the work of Rawson and coworkers.23 As outlined in Scheme 1, following treatment of 3c with LiN(TMS)2·Et2O the reaction was quenched with sulphur monochloride to afford the dithiadiazolium salt directly without isolation of the silylated amidine intermediate. Subsequent treatment of the dithiadiazolium cation 5c with SbPh3 in the presence of oxygen afforded 1c as a dark yellow solid in 15–20% yield. Although the yields for the one-pot synthesis of 1c are admittedly lower than that of Route A, the latter route carries the advantage of higher throughput as it eliminates the need of isolating the persilylated amidine intermediate, where purification tends to be difficult. Thus following Route B outlined in Scheme 1, compounds 1c, 2a and 2b can be prepared from the corresponding nitrile (i.e., 3c, 4a and 4b). Compared to 1c, the π-extended systems 2a and 2b are copper-coloured and red, respectively, indicative of the expected bathochromic shift associated with extension of the conjugation. While 2a was recrystallized as copper-like flakes from hot toluene, recrystallization from chloroform afforded 2b as red shards.
Spectroscopic and computational studies
In order to probe the effect of increased conjugation, as well as to explore the substituent effect (e.g., 5-hexyl vs. 4-hexylthiophene) on the optoelectronic properties of DTTAs, spectroscopic and computational studies were performed on 1c, 2a and 2b and compared to the previously reported derivatives 1a and 1b;22 the results of which are presented in Fig. 1 and summarized in Table 1 (for more information see ESI,† Tables S1–4). The photophysical properties were investigated via absorption and photoluminescence studies on dichloromethane (DCM) solutions, in addition to solid-state diffuse reflectance studies. The UV-Vis spectrum of 1c is comprised of two regions; one consisting of intense bands absorbing at 300–380 nm, while the other contains a weaker broad band centred at 450 nm. Such an absorption profile is consistent with a planar, aromatic structure, as the bent configuration leads to absorption restricted to the UV region of the electromagnetic spectrum (i.e., below 270 nm).15 Compared to the previously reported 5-hexylthienyl derivative 1b,22 the main absorption band of 1c is shifted hypsochromically by 11 nm (505 cm−1), from 472 to 461 nm, which is consistent with the 5 nm blue shift predicted by TD DFT calculations (vide infra). The absorption profiles of the π-extended complexes 2a and 2b are similar to 1c; however, a much less defined vibronic structure is observed. This may be attributed to a less rigid structure resulting from flexibility of the peripheral thienyl substituents.24 Furthermore, the spectrum of 2a and 2b are bathochromically shifted with respect to 1a and 1b by 58 and 55 nm (4357 and 3900 cm−1), respectively, as is expected due to the increased conjugation afforded by the additional thiophene moieties.25–27 Based on the onset of UV-Vis absorption, extension of conjugation from 1a to 2a and 1b to 2b leads to a decrease in the optical energy gap of 0.26 eV and 0.21 eV, respectively. DFT calculations show concordant results, with a decrease in the HOMO–LUMO energy gap of 0.37 and 0.33 eV with increased conjugation (i.e., for 1a → 2a and 1b → 2b, respectively). The diffuse reflectance measurements (ESI,† Fig. S1) are in good agreement with the solution based absorption profiles; that is, a continuous bathochromic shift from 1c to 2b is also observed in the solid-state. | ||
| Fig. 1 Experimental absorption spectra of 1c (black), 2a (red) and 2b (blue) in DCM solutions (top). TD DFT calculated absorption spectra of 1c (black), 2a (red) and 2b (blue). Insets: zoom-in on the low energy absorptions between 400–700 nm. | ||
| 1c | 2a | 2b | |
|---|---|---|---|
| a Solution measurements performed in HQ–GC grade DCM. b Solid measurements performed with an Agilent Praying Mantis module. c DFT/B3LYP/6-311+G(d,p) calculations. d Molar extinction coefficient for λabsmax. e Calculated from corresponding λedge. f From TD DFT calculations performed on optimized geometries. g Oscillator strength of the reported transition. | |||
| λ absmax (nm) | 343 | 395 | 404 |
| ε (M−1 cm−1) | 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
59400 |
66200 |
| λ absedge (nm) | 504 | 547 | 564 |
| E optgap (eV) | 2.45 | 2.27 | 2.19 |
| λ DRmax (nm) | 450 | 487 | 507 |
| λ DRedge (nm) | 505 | 568 | 605 |
| E DRgap , (nm) | 2.44 | 2.17 | 2.04 |
|
λ
calcdmaxf (nm) |
345 | 415 | 426 |
| f , | 1.0581 | 1.5882 | 1.6323 |
|
λ
calcdedgef (nm) |
565 | 644 | 676 |
|
E
calcdgapf,e (eV) |
2.19 | 1.92 | 1.84 |
DFT calculations, performed at the B3LYP/6-311+G(d,p) level of theory, reveal that the LUMO in 1c, 2a and 2b (where R = Me for 1c and 2b) consists of a nodal plane that bisects the DTTA ring through the two carbon atoms and, as a result, no electron density resides on the thienyl groups (see ESI,† Fig. S2). Consequently, substituent modification should have little effect on the energy level of this orbital. By contrast, electron density in the HOMO is spread over the entire tricyclic (1c) or pentacyclic (2a and 2b) framework. Thus any variation in the substituents (e.g., alkylation) should influence these orbital energies, and is therefore responsible for the observed shifts in the absorption profiles within this family of compounds (see ESI,† Fig. S3). Interestingly, geometry optimization for the pentacyclic derivatives, 2a and 2b, reveals a degree of rotation about the peripheral thienyl moieties away from planarity, contrary to the tricyclic derivatives (1a–c), which are calculated to be planar.
TD DFT calculations were performed on optimized geometries of 1c, 2a and 2b (where R = Me for 1c and 2b) in order to probe the optical transitions; the results are presented in Fig. 1 and the twenty vertical excitation energies calculated for each compound are listed in the ESI,† Table S3. Overall, the calculated absorption profiles are consistent with those experimentally obtained; the calculated maximum absorption wavelengths and bathochromic shifts associated with extension of the conjugation match the experimental behaviour well. Energy gaps obtained from these calculations are shifted by 0.25–0.52 eV compared to those determined from optical measurements, but the trends are consistent such that Egap is largest for 1c and smallest for 2b. The highest wavelength absorption, which is associated with the HOMO to LUMO transition, has relatively low oscillator strength (f ∼ 0.02). The band with the largest oscillator strength (f > 1) corresponds to excitation of the HOMO to LUMO+1 for each compound and, in the case of 2a, also includes excitation from the HOMO−2 to LUMO. Furthermore, the oscillator strength predicted by computational methods for each derivative correlates well with the measured molar extinction coefficients (see ESI,† Table S2).
Interestingly, in this study 1c, 2a and 2b were found to be very weakly fluorescent, unlike what was previously observed for 1a and 1b.22 As such, the photoluminescence was probed on DCM solutions, demonstrating very weak emission bands whose overall shape was independent of excitation wavelength (ESI,† Fig. S4). To preclude the possibility that the observed photoluminescence was simply Raman scattering of the solvent, blank fluorescence spectrums were also acquired under similar conditions. These showed an even weaker photoluminescence band, distinct from what was observed in the presence of the DTTA and whose spectral features were dependent on excitation wavelength. Thus this study demonstrates that 1c, 2a and 2b weakly emit in the violet (1c) and blue-green (2a and 2b) regions of the visible spectrum.
Structural analysis
Since long-range molecular order and π-orbital overlap are crucial to semiconductor efficiency as they provide a pathway for electronic communication, structures consisting of π-stacked planar molecules are often considered ideal. To confirm the planarity of the DTTA ring and to investigate the effect of the substitution pattern on intermolecular interactions, the solid-state structure of 1c, 2a and 2b were studied by single crystal X-ray diffraction. Pertinent crystallographic data for the three derivatives is given in Table 2. In each compound, the planarity of the eight-membered heterocyclic ring coupled with the symmetrical bond lengths, which correlate well to previously reported DTTAs,18,19,28 is indicative of an aromatic system. Thus, as expected, thienyl-functionalization affords essentially planar tricyclic (1c) and pentacyclic (2a and 2b) frameworks, which is consistent with the photophysical properties.| 1c | 2a | 2b | |
|---|---|---|---|
| a δ is the mean interplanar separation between molecular planes along the π stack. b τ is the tilt angle between the mean molecular plane and the stacking axis. | |||
| Formula | C22H30N4S4 | C17H10N4S6 | C30H34N4S6 |
| fw | 478.74 | 474.66 | 642.96 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 11.837(4) | 5.6479(6) | 6.6627(4) |
| b (Å) | 5.482(2) | 28.280(3) | 9.3450(6) |
| c (Å) | 18.312(6) | 6.4275(7) | 13.5348(7) |
| α (°) | 90 | 90 | 98.497(3) |
| β (°) | 92.953(12) | 110.342(6) | 102.934(4) |
| γ (°) | 90 | 90 | 107.792(3) |
| V (Å3) | 1186.8(7) | 962.58(18) | 760.54(8) |
| Z | 2 | 2 | 2 |
| D calc (Mg m−3) | 1.340 | 1.638 | 1.404 |
| T (K) | 200 | 200 | 200 |
| μ (mm−1) | 0.418 | 0.724 | 0.478 |
| 2Θmax (°) | 56.728 | 56.662 | 56.688 |
| No. of total reflections | 13302 |
11549 |
7798 |
| No. of unique reflections | 2886 | 2354 | 3723 |
| R int | 0.1728 | 0.0722 | 0.0199 |
| R 1, wR2 (on F2) | 0.0746, 0.1580 | 0.1155, 0.1987 | 0.0424, 0.0853 |
| δ (Å) | 3.46 | 3.53 | 3.50 |
| τ (°) | 39.3 | 38.7 | 21.7 |
Crystals of the tricyclic derivative 1c belong to the monoclinic space group P21/n and consist of slipped π-stacks that align in a herringbone arrangement (Fig. 2). The crystal structure reveals that the tricyclic framework is essentially planar, with a torsion angle of 5.9° between the central DTTA ring and the thienyl moieties. Furthermore, while the planarity of the hexyl groups with respect to the π-conjugated core enables tight π-stacking, the resulting alkyl–alkyl interactions limit inter-stack communication. Consequently, the only close contacts observed in the structure of 1c are those within the π-stacks (e.g., S1⋯S2′ = 3.613(2) Å; C3⋯S1′ = 3.482(4) Å; C1⋯C1′ = 3.429(6) Å). This type of effect has been noted in other alkyl-substituted π-conjugated systems, in which the long hydrocarbon chains act as buffers that effectively isolate the π-stacked molecules from one another.29–31 Comparison to the previously reported 5-hexylthienyl functionalized DTTA (1b),22 which crystallizes in the triclinic space group P, reveals that switching the hexyl chain from the 5- to the 4-position of the thienyl substituent leads to enhanced symmetry. Nonetheless, a number of similarities exist between the structures of 1b and 1c, especially when looking down the perpendicular direction to the π-conjugated core. For example, in both structures a thiophene moiety on one molecule overlaps with the DTTA ring of a neighbouring molecule below (see ESI,† Fig. S6). This degree of slippage within the π-stacked structure, which can be quantified in terms of the slippage angle τ, is nearly identical (cf. 37.6° vs. 39.3° for 1b and 1c, respectively). This leads to a relatively consistent interplanar separation between molecules (3.44 Å for 1b; 3.46 Å for 1c). While it has been observed in other π-conjugated molecules32,33 and heterocycles34 that minor changes in substituents result in dramatic changes in slippage angle, here it appears the interaction between the donor thienyl groups with the acceptor DTTA core dominates the packing arrangement.
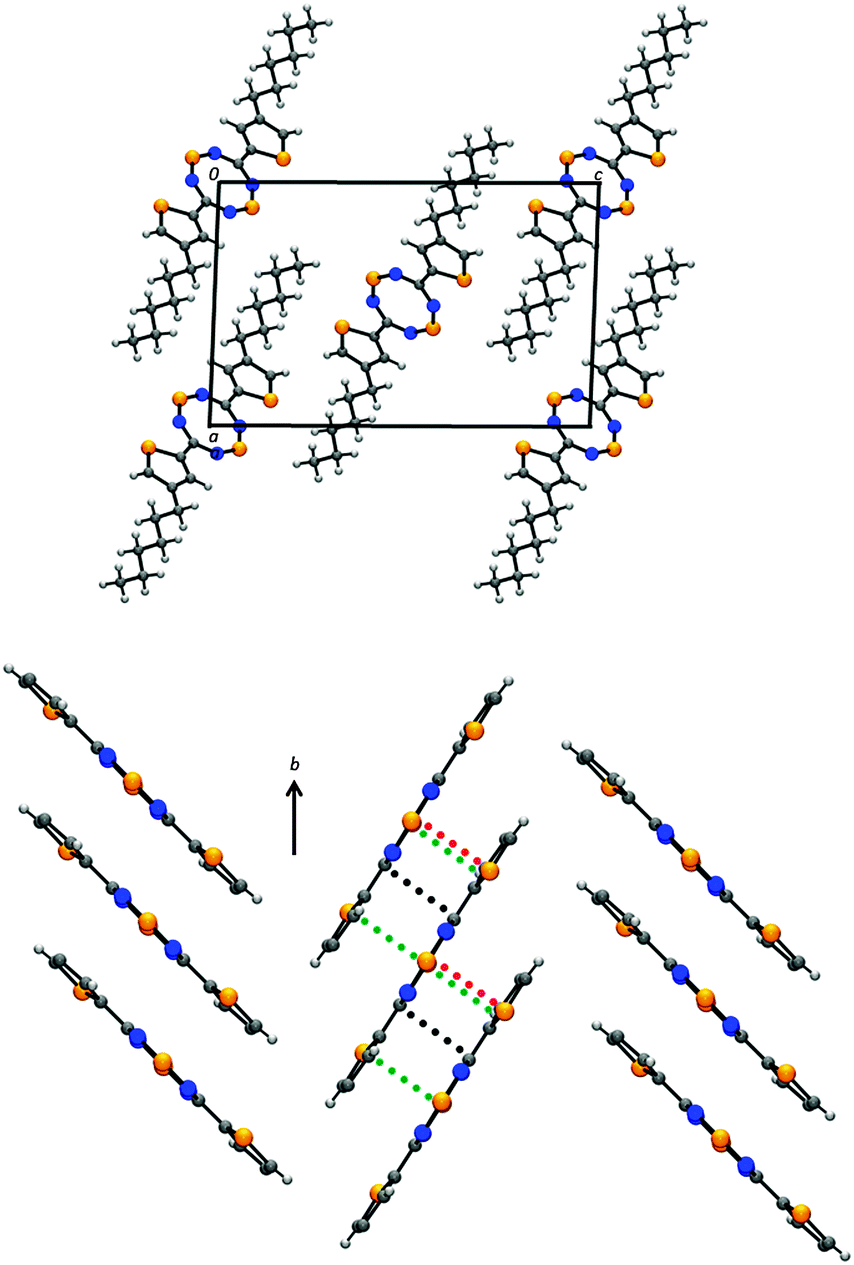 | ||
| Fig. 2 Unit cell (top) and herringbone array of slipped π-stacks (bottom) of 1c. Hexyl groups have been removed in the slipped π-stacked structure for clarity. Close intermolecular contacts are shown; C⋯C′ in black; S⋯S′ in red; S⋯C′ in green. | ||
The crystal structure of the bithienyl derivative 2a also belongs to the monoclinic space group P21/n and consists of herringbone arrays of slipped π-stacks (Fig. 3). Within the molecular framework, there is disorder about the outer thienyl substituents, similar to that of 1a and 1b in addition to other thiophene-substituted heterocycles.9,18,22,35 This disorder involves a 180° rotation about the C5–C6 bond that affects the positions of the C7 and S3 atoms and was modelled as 0.60 and 0.40 occupancy of the two positions. The major contributor in 2a, presented in Fig. 3, is comprised of a cis-bithiophene in a trans-conformation about the DTTA ring. As well, while the central tricyclic core is essentially planar, with a torsion angle of only 5.25° between the DTTA ring and the inner thienyl moieties, the outer thienyl groups twist at a torsion angle of 29.55° reducing the planarity of the pentacyclic framework. Nonetheless, numerous close π–π interactions exist within the π-stacked structure (e.g., C7⋯S2′ = 3.44(2) Å; C3⋯S1′ = 3.424(6) Å; S1⋯S1′ = 3.463(2) Å). This increase in the number of close contacts, compared to 1a, is likely due to the enhanced π-conjugation while maintaining a relatively close interplanar separation (3.53 Å for 2avs. 3.55 Å for 1a).
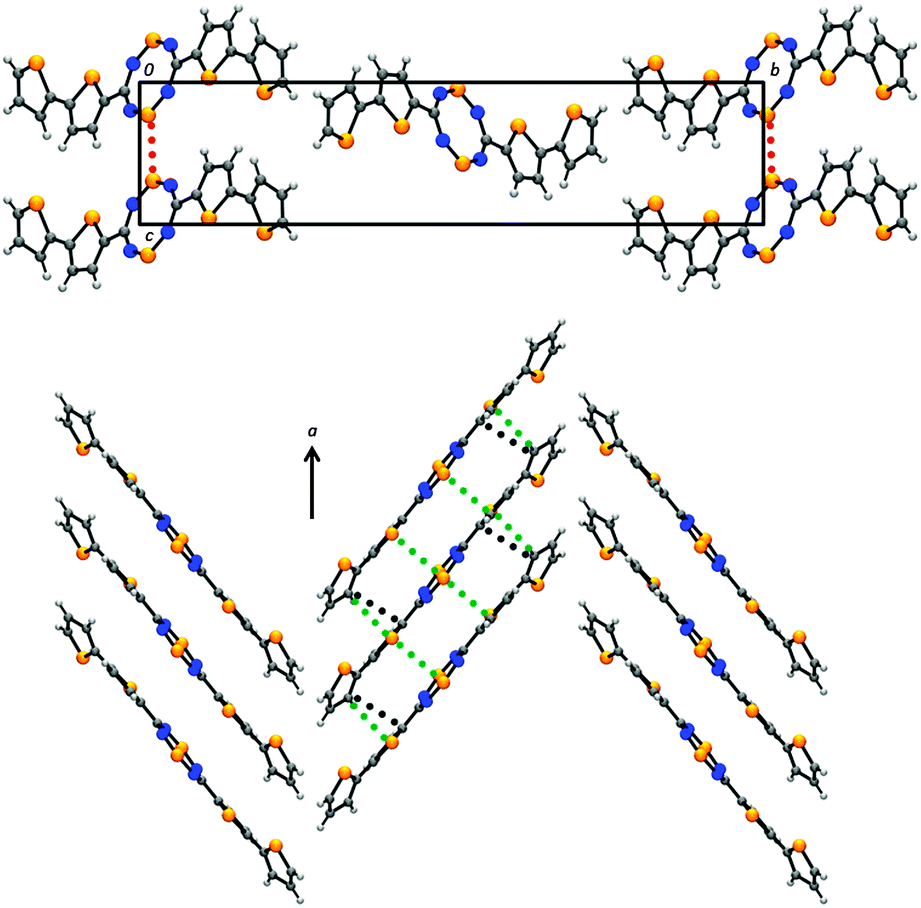 | ||
| Fig. 3 Unit cell (top) and herringbone array of slipped π-stacks (bottom) of 2a. Close intermolecular contacts are shown; C⋯C′ in black; S⋯S′ in red; S⋯C′ in green. | ||
Similar to the 5-hexylthienyl derivative 1b, crystals of its π-extended analogue 2b belong to the triclinic space group P and consist of slipped π-stacks (Fig. 4). However, contrary to the former, in which numerous close π–π interactions exist within the slipped π-stack, the structure of 2b reveals only two close intra-stack contacts (S2⋯S2′ = 3.5686(6) Å; C1⋯C7′ = 3.415(2) Å). Interestingly, these interactions occur between neighbouring thienyl moieties, which is inconsistent with the other thienyl substituted DTTAs where the structure is dominated by the donor–acceptor interaction between the DTTA ring on one molecule and the thienyl groups of another. This dichotomy is exemplified by considering the slippage angle τ, which is significantly altered in 2b (21.7°) compared to the other thienyl functionalized DTTAs (τ = 36–39°). This may be attributed to alkyl–alkyl interactions dominating the packing arrangement in 2b, compared to the other thienyl substituted DTTA derivatives (i.e., 1a–c and 2a) the π–π interactions between the donor thienyl groups and central DTTA acceptor appear to dictate the solid-state structure. Overall, the pentacyclic framework in 2b is more planar than in the case of 2a, with torsion angles of only 1.8° and 8.7° for the inner and outer thienyl moieties, respectively. As well, while there are limited contacts within the slipped π-stacks, additional inter-stack contacts exist between carbon atoms of the thienyl substituents on neighbouring stacks (C7⋯C3′ = 3.568(6) Å).
 | ||
| Fig. 4 Slipped π-stacked structure (top) and looking down the stacking direction (bottom) of 2b. Close intermolecular contacts are shown; C⋯C′ in black; S⋯S′ in red. Hexyl groups have been removed in the bottom figure for clarity. | ||
Conclusions
To summarize, a family of dithiatetrazocines functionalized with 4-hexylthiophen-2yl (1c), 5-(thiophen-2′-yl)thiophen-2yl (2a) and 5-(5′-hexylthiophen-2′-yl)thiophen-2yl (2b) substituents were prepared in a one pot synthesis from the corresponding nitrile, demonstrating the versatility of the synthetic procedure reported here. The latter two compounds (i.e., 2a and 2b) mark the first reported examples of π-extended DTTA systems. Comparative optical, computational and X-ray diffraction studies were carried out on this family of heterocycles to investigate the influence of the substitution pattern, in addition to enhanced conjugation, on the optoelectronic and solid-state properties. More specifically, displacement of the alkyl chain from the 5- to the 4-position in hexylthienyl functionalized DTTA (i.e., 1bvs.1c) results in a relatively small hypsochromic shift in the absorption profile, indicative of a slight increase in the optical energy gap. At the same time, such variation in the placement of the alkyl substituent on the π-conjugated core leads to enhanced symmetry in the solid-state affording a herringbone arrangement of slipped π-stacks. Extension of the conjugation via additional thienyl moieties on either side of the central DTTA ring leads to a decrease of the HOMO–LUMO gap, as witnessed by the bathochromic shifts in the absorption profiles for 2a and 2b with respect to their mono-thienyl analogues (i.e., 1a and 1b, respectively). As is expected, enhanced conjugation afforded by the presence of additional thienyl groups in 2a also results in an increase in the number of intermolecular contacts in the solid-state, which bodes well for its implementation into optoelectronic devices. On the other hand, comparison of the structures between 1b and 2b reveal a decrease in the number of π–π interactions with increasing conjugation. This may be attributed to alkyl–alkyl interactions dominating the packing arrangement in 2b, whereas in all the other thienyl substituted DTTA derivatives (i.e., 1a–c and 2a) the π–π interactions between the donor thienyl groups and central DTTA acceptor appear to dictate the solid-state structure.Experimental section
General procedures
Hydroxylamine hydrochloride, N-methylpyrrolidone (NMP), triphenylantimony (SbPh3) and N-bromosuccinimide (NBS) were obtained commercially from VWR and used as received. Sulphur monochloride (S2Cl2) was obtained commercially from Sigma Aldrich and used as received. LiN(TMS)2·Et2O,36 2-formyl-4-hexylthiophene (made with nBuli in-lieu of tBuLi),37 5-formyl-2,2′-bithiophene38 and 5-formyl-5′-hexyl-2,2′-bithiophene39 were prepared as previously reported. All solvents were ACS grade; dry solvents were obtained by passing them through activated alumina on a J.C. Meyer solvent purification system. Melting points were taken using a Mel-Temp apparatus and are uncorrected. NMR spectra were run in CDCl3 solutions at room temperature on a Bruker Avance 400 MHz spectrometer. All spectra are referenced to the deuterated solvent peak at 7.26 ppm. IR spectra were recorded on an Agilent Technologies Cary 630 FT-IR spectrometer. UV-visible spectra were measured with a Varian Cary Series 6000 UV-Vis-NIR spectrophotometer and the fluorescence spectra were obtained using a Varian Cary Eclipse fluorescence spectrometer. UV-Visible and fluorescence spectra were measured on HQGC-grade dichloromethane solutions with 1 cm precision quartz cuvettes. Diffuse reflectance measurements were performed with an Agilent Praying Mantis module. Elemental analyses were performed by G.G. Hatch Stable Isotope Laboratory, Ottawa, ON, K1N 6N5.Crystal growth
Bright yellow needles of 1c suitable for X-ray analysis were grown by slowly cooling a saturated chlorobenzene solution. Copper-coloured plates of 2a suitable for X-ray analysis were grown by slowly cooling a saturated toluene solution. Red plates of 2b suitable for X-ray analysis were grown by slowly cooling a saturated chloroform solution.X-ray measurements
Data collection results for compounds 1c, 2a and 2b represent the best data sets obtained in several trials for each sample. The crystals were mounted on thin glass fibers using paraffin oil. Prior to data collection crystals were cooled to 200.15° K. Data were collected on a Bruker AXS KAPPA single crystal diffractometer equipped with a sealed Mo tube source (wavelength 0.71073 Å) APEX II CCD detector. Raw data collection and processing were performed with APEX II software package from BRUKER AXS.40 Diffraction data for 1c and 2a samples were collected as a sequence of 0.3° ω scans at 0, 120, and 240° in ϕ. Due to lower unit cell symmetry, diffraction data for 2b were collected as a sequence of 0.3° ω scans at 0, 90, 180 and 270° in ϕ, in order to ensure adequate data redundancy. Initial unit cell parameters were determined from 60 data frames with 0.3° ω scan each collected at the different sections of the Ewald sphere. Semi-empirical absorption corrections based on equivalent reflections were applied.41 Systematic absences in the diffraction data-set and unit-cell parameters were consistent with monoclinic P21/c (No. 14) for compound 1c, monoclinic P21/n (No. 14, alternative settings) for compound 2a and triclinic P (No. 2) for compound 2b. Solutions in the centrosymmetric space groups for all three compounds yielded chemically reasonable and computationally stable refinement results. The structures were solved by direct methods, completed with difference Fourier synthesis, and refined with full-matrix least-squares procedures based on F2.
Diffraction data for the crystal of the complex 1c was collected to 0.75 Å resolution, however due to small crystal size and weak diffraction it was discovered that both R(int) and R(sigma) exceeded 35% for the data below 0.95 Å resolution. Based on R(sigma), value data were truncated to 0.90 Å resolution for refinement. Asymmetric unit for crystallographic model of 1c consists of one target molecule located in the inversion center of the space group.
Structural model of 2a consists of one molecule of compound located in the inversion center of the space group. On the final refinement stages, unusual values of thermal parameters for sulphur atoms as well as several carbon atoms in the model indicated the presence of disorder in the orientation of 5-membered ring S(3)–C(6) to C(8). Such disorder unrelated to symmetry was interpreted as an 180° rotation of the fragment around the C(5)–C(6) bond. Initial value of the occupancies for disordered fragment was refined. However, at the final refinement cycles occupancies of the fragment were fixed at a 60:40 ratio. Several sets of geometry constraints (SADI) and thermal motion parameters constraints (SIMU, RIGU) were introduced to ensure reasonable molecular geometry and acceptable thermal motion values for these fragments.
Solution of structure for 2b revealed that one molecule of target compound was located in the inversion center of the space group.
For all three compounds all non-hydrogen atoms were refined in anisotropic thermal motion approximation. All hydrogen atomic positions were calculated based on the geometry of related non-hydrogen atoms. All hydrogen atoms were treated as idealized contributions during the refinement. All scattering factors are contained in several versions of the SHELXTL program library, with the latest version used being v.6.12.42 Crystallographic data and selected data collection parameters are reported in Table 1.
Computational chemistry
All calculations were carried out using the Gaussian09 program package.43 The geometries of the studied compounds were investigated using the hybrid density functional B3LYP with the 6-311+G(d,p) basis set. Optimized structures were used to examine the orbital energies and HOMO–LUMO gaps. The calculated UV-vis spectra and excitation energies were determined using TD DFT which employed the optimized B3LYP ground states. The solvent model and solvent was dichloromethane, and twenty excited states were considered.:1 DCM/Hexanes) yielded the title product as a red oil which crystallizes upon standing (6.06 g, 73% yield). The product was identified by comparison to the 1H NMR spectrum reported in literature.451H NMR (δ, CDCl3, RT, 400 MHz): 7.47 (d, J = 3.95 Hz, 1H), 7.08 (d, J = 3.65 Hz, 1H), 7.02 (d, J = 3.95 Hz, 1H), 6.71 (dt, J1 = 3.65 Hz, J2 = 0.95 Hz, 1H), 2.79 (t, J = 7.70 Hz, 2H), 1.66 (m, 2H), 1.32 (m, 6H), 0.88 (t, J = 6.80 Hz, 3H).
Acknowledgements
This work was supported by the University of Ottawa, the Canadian Foundation for Innovation, the National Sciences and Engineering Council of Canada and the Ontario Research Fund. J.L.B. is grateful to the Ontario Ministry of Research and Innovation for an Early Researcher Award.Notes and references
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- C. Zhang, P. Chen and W. Hu, Chem. Soc. Rev., 2015, 44, 2087 RSC.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef CAS PubMed.
- P. Tyagi, J. Mater. Chem., 2011, 21, 4733 RSC.
- K. Takemoto and M. Kimura, RSC Adv., 2014, 4, 64589 RSC.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- S. Steinberger, A. Mishra, E. Reinold, J. Levichkov, C. Uhrich, M. Pfeiffer and P. Bauerle, Chem. Commun., 2011, 47, 1982 RSC.
- T. Kono, D. Kumaki, J. Nishida, S. Tokito and Y. Yamashita, Chem. Commun., 2010, 46, 3265 RSC.
- M. Karikomi, C. Kitamura, S. Tanaka and Y. Yamashita, J. Am. Chem. Soc., 1995, 117, 6791 CrossRef CAS.
- M. P. Cava, M. V. Lakshmikantham, R. Hoffmann and R. M. Williams, Tetrahedron, 2011, 67, 6771 CrossRef CAS PubMed.
- A. Mishra, C.-Q. Ma and P. Baeuerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS PubMed.
- T. Torroba, J. Prakt. Chem., 1999, 341, 99 CrossRef CAS.
- R. Tang, F. Zhang, Y. Fu, Q. Xu, X. Wang, X. Zhuang, D. Wu, A. Giannakopoulos, D. Beljonne and X. Feng, Org. Lett., 2014, 16, 4726 CrossRef CAS PubMed.
- G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670 CrossRef CAS PubMed.
- I. Ernest, W. Holick, G. Rihs, D. Schomburg, G. Shoham, D. Wenkert and R. B. Woodward, J. Am. Chem. Soc., 1981, 103, 1540 CrossRef CAS.
- R. Gleiter, R. Bartetzko and D. Cremer, J. Am. Chem. Soc., 1984, 106, 3437 CrossRef CAS.
- M. Amin and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1989, 2495 RSC.
- T. M. Barclay, A. W. Cordes, R. T. Oakley and K. E. Preuss, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 3250 CrossRef.
- R. T. Boeré, K. H. Moock, S. Derrick, W. Hoogerdijk, K. Preuss, J. Yip and M. Parvez, Can. J. Chem., 1993, 71, 473 CrossRef.
- A. D. Bond, D. A. Haynes and J. M. Rawson, Can. J. Chem., 2002, 80, 1507 CrossRef CAS.
- R. A. Pascal and R. P. Lesperance, J. Am. Chem. Soc., 1994, 116, 5167 CrossRef CAS.
- S. S. Afjeh, A. A. Leitch, I. Korobkov and J. L. Brusso, RSC Adv., 2013, 3, 23438 RSC.
- C. P. Constantinides, D. J. Eisler, A. Alberola, E. Carter, D. M. Murphy and J. M. Rawson, CrystEngComm, 2014, 16, 7298 RSC.
- L. Zoephel, V. Enkelmann, R. Rieger and K. Muellen, Org. Lett., 2011, 13, 4506 CrossRef CAS PubMed.
- A. A. Leitch, A. Mansour, K. A. Stobo, I. Korobkov and J. L. Brusso, Cryst. Growth Des., 2012, 12, 1416 CAS.
- A. A. Leitch, K. A. Stobo, B. Hussain, M. Ghoussoub, S. Ebrahimi-Takalloo, P. Servati, I. Korobkov and J. L. Brusso, Eur. J. Org. Chem., 2013, 5854 CrossRef CAS PubMed.
- S. F. Robertson, A. A. Leitch, I. Korobkov, D. V. Soldatov and J. L. Brusso, Can. J. Chem., 2014, 92, 1106 CrossRef CAS.
- M. Parvez, R. T. Boeré, S. Derrick and K. H. Moock, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 2116 CrossRef.
- M. Q. He and F. X. Zhang, J. Org. Chem., 2007, 72, 442 CrossRef CAS PubMed.
- X. Guo, S. H. Wang, V. Enkelmann, M. Baumgarten and K. Mullen, Org. Lett., 2011, 13, 6062 CrossRef CAS PubMed.
- H. T. Black, S. B. Liu and V. S. Ashby, Org. Lett., 2011, 13, 6492 CrossRef CAS PubMed.
- A. A. Leitch, A. Mansour, K. A. Stobo, I. Korobkov and J. L. Brusso, Cryst. Growth Des., 2012, 12, 1416 CAS.
- C. Kitamura, Chem. Rec., 2012, 12, 506 CrossRef CAS PubMed.
- C. M. Robertson, A. A. Leitch, K. Cvrkalj, D. J. T. Myles, R. W. Reed, P. A. Dube and R. T. Oakley, J. Am. Chem. Soc., 2008, 130, 14791 CrossRef CAS PubMed.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS.
- R. T. Boeré, R. T. Oakley and R. W. Reed, J. Organomet. Chem., 1987, 331, 161 CrossRef.
- A. Dessi, G. B. Consiglio, M. Calamante, G. Reginato, A. Mordini, M. Peruzzini, M. Taddei, A. Sinicropi, M. L. Parisi, F. F. de Biani, R. Basosi, R. Mori, M. Spatola, M. Bruzzi and L. Zani, Eur. J. Org. Chem., 2013, 1916 CrossRef CAS PubMed.
- H. Wang, J. L. Wang, S. C. Yuan, H. Pei and W. W. Pei, Tetrahedron, 2005, 61, 8465 CrossRef CAS PubMed.
- P. Frere, J. M. Raimundo, P. Blanchard, J. Delaunay, P. Richomme, J. L. Sauvajol, J. Orduna, J. Garin and J. Roncali, J. Org. Chem., 2003, 68, 7254 CrossRef CAS PubMed.
- APEX Software Suite, W. Bruker AXS, Madison, 2005 Search PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. G. Fox, Inc., Wallingford CT, 2009.
- Z. Li, J. Ding, N. Song, J. Lu and Y. Tao, J. Am. Chem. Soc., 2010, 132, 13160 CrossRef CAS PubMed.
- E. Kurach, D. Djurado, J. Rimarcik, A. Kornet, M. Wlostowski, V. Lukes, J. Pecaut, M. Zagorska and A. Pron, Phys. Chem. Chem. Phys., 2011, 13, 2690 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optical characterization, computational data and crystallography. CCDC 1403503–1403505. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01345c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |