
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One ligand fits all: lanthanide and actinide sandwich complexes comprising the 1,4-bis(trimethylsilyl)cyclooctatetraenyl (=COT′′) ligand†‡
Janek
Rausch
a,
Christos
Apostolidis
*b,
Olaf
Walter
b,
Volker
Lorenz
a,
Cristian G.
Hrib
a,
Liane
Hilfert
a,
Marcel
Kühling
a,
Sabine
Busse
a and
Frank T.
Edelmann
*a
aChemisches Institut der Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, 39106 Magdeburg, Germany. E-mail: frank.edelmann@ovgu.de; Fax: +49 391 6712933; Tel: +49 391 6758327
bEuropean Commission, Joint Research Centre, Institute for Transuranium Elements, P.O. Box 2340, 76125 Karlsruhe, Germany. E-mail: christos.apostolidis@ec.europa.eu
First published on 4th June 2015
Abstract
The series of anionic lanthanide(III) sandwich complexes of the type [Ln(COT′′)2]− (COT′′ = 1,4-bis(trimethylsilyl)cyclooctatetraenyl dianion) has been largely extended by the synthesis of eight new derivatives ranging from lanthanum to lutetium. The new compounds [Li(DME)3][Ln(COT′′)2] (Ln = Y (1), La (2), Pr (3), Gd (4), Tm (6), Lu (8)) and [Li(THF)4][Ln(COT′′)2] (Ln = Ho (5), Tm (7)) were prepared in good yields following a straightforward synthetic protocol which involves the treatment of LnCl3 with 2 equiv. of in situ-prepared Li2COT′′ in either DME (=1,2-dimethoxyethane) or THF. The neutral actinide sandwich complexes An(COT′′)2 (An = Th (9), U (10)) and An(COT′′′)2 (COT′′′ = 1,3,6-tris(trimethylsilyl)cyclooctatetraenyl dianion; An = Th (11), U (12)) were synthesized in a similar manner, starting from ThCl4 or UCl4, respectively. The COT′′ ligand imparts excellent solubility even in low-polar solvents as well as excellent crystallinity to all new compounds studied. All twelve new f-element sandwich complexes have been structurally authenticated by single-crystal X-ray diffraction. All are nearly perfect sandwich complexes with little deviation from the coplanar arrangement of the substituted COT′′ rings. Surprisingly, all six [Li(DME)3][Ln(COT′′)2] complexes covering the entire range of Ln3+ ionic radii from La3+ to Lu3+ are isostructural (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). Compound 10 is the first uranocene derivative for which 13C NMR data are reported.
). Compound 10 is the first uranocene derivative for which 13C NMR data are reported.
1. Introduction
Second only to the omnipresent cyclopentadienyl complexes, the dianionic 10π-cyclooctatetraenyl ligand C8H82−, commonly abbreviated as COT, plays an important role in the organometallic chemistry of lanthanides and actinides for almost 50 years. There is a general understanding that the large, flat C8H82− ring is ideally suited for overlapping with the f-orbitals of the large lanthanide and actinide ions.1 Early work in this area was mainly focused on complexes bearing unsubstituted COT ligands.2Scheme 1 shows some prototypical lanthanide COT complexes which are considered milestones in the development of organolanthanide chemistry using COT ligands. Notable are the anionic sandwich complexes K[Ln(COT)2] (A),3 the dimeric half-sandwich mono(cyclooctatetraenyl)lanthanide chlorides [(COT)Ln(μ-Cl)(THF)2]2 (B),4 the mixed-sandwich complexes (COT)LnCp (C),5 and the so-called cerocene, Ce(COT)2 (D).6 The chemistry of such lanthanide COT complexes has already been summarized in several comprehensive review articles.2,7 | ||
| Scheme 1 Prototypical lanthanide COT sandwich and half-sandwich complexes. | ||
In the case of actinides, the discovery of uranocene, U(COT)2 (Scheme 2, An = U), by Streitwieser and Müller-Westerhoff in 1968 had a considerable impact on the development of organoactinide chemistry.11,12 Following the preparation of uranocene, other tetravalent actinidocenes An(C8H8)2 (Scheme 2; An = Th, Pa, Np, Pu) have also been reported.12 The bonding in uranocene is considered to be less ionic than in the lanthanide sandwich complexes K[Ln(COT)2] (A) and Ce(COT)2 (D). Uranocene is also significantly more stable than cerocene and the thorium analogue Th(COT)2, which can be explained by a higher degree of covalency due to a stronger participation of the 5f and 6d orbitals in the uranium–cyclooctatetraenyl bonding. Recent work by Ephritikhine and co-workers has demonstrated that the chemistry of actinidocenes continues to produce very interesting results.13
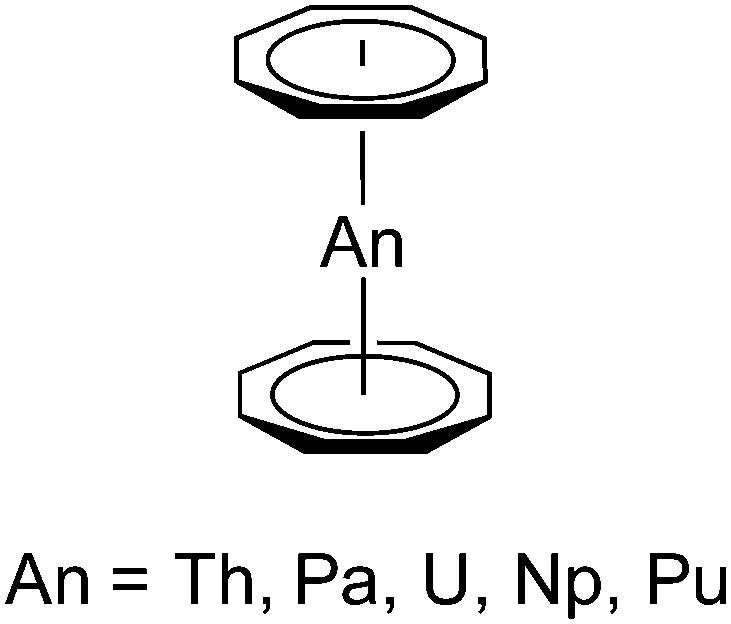 | ||
| Scheme 2 Schematic representation of the neutral actinidocenes An(C8H8)2 (An = Th, Pa, U, Np, Pu). | ||
In general, the use of the unsubstituted COT ligand in organolanthanide and -actinide chemistry has several disadvantages in terms of low solubility and/or poor crystallinity. For example, the most important series of precursors in lanthanide COT chemistry, the chloro-bridged mono(COT) dimers [(COT)Ln(μ-Cl)(THF)2]2 (Scheme 1, B),4 lack good solubility even in THF. Moreover, commercially available cyclooctatetraene is very expensive. As a consequence, more soluble alternative starting materials such as (COT)LnI(THF)n (Ln = Tm, n = 2; Ln = La, Ce, Pr, Nd, Sm, n = 3)2,8 and [(COT)Ln(μ-O3SCF3)(THF)]2 (Ln = Ce, Pr, Nd, Sm)9 have been reported in the literature, but their use as precursors in organolanthanide chemistry still remains limited.2 More recently, lanthanide COT chemistry received fundamental new impulses through the use of bulky silyl-substituted cyclooctatetraenyl ligands. The initial idea originated from the pioneering work of Cloke et al., who first employed the 1,4-bis(trimethylsilyl)cyclooctatetraenyl dianion (=COT′′) in organo-f-element chemistry.10 In many cases, using the bulky COT′′ ligand did in fact improve the solubility of the products, but occasionally also led to novel molecular structures and coordination geometries.7g,10,14 Typical examples include the unprecedented cluster-centered Pr/Li multidecker sandwich complex of the composition [Pr(COT′′)]2[Pr2(COT′′)2]2Li2(THF)2Cl815 as well as the first linear rare-earth metal triple-decker complexes Ln2(COT′′)3 (Ln = Nd, Gd, Dy, Ho, Er).14,16–18 Previously reported anionic lanthanide sandwich complexes comprising [Ln(COT′′)2]− anions include the THF solvates [Li(THF)4][Ln(COT′′)2] with Ln = Y,19 Ce,19 Pr,19 Nd,14,19 Sm,19 Gd17 and Dy17 as well as the DME adducts [Li(DME)3][Ln(COT′′)2] (Ln = Ce,14,20 Dy,17 Er21), Li(DME)Tb(COT′′)222 and Li(THF)(DME)Dy(COT′′)2.23 A notable neutral lanthanide sandwich complex containing COT′′ is the cerocene derivative Ce(COT′′)2.24 Sterically even more demanding is the 1,3,6-tris(trimethylsilyl)cyclooctatetraenyl dianion ligand (=COT′′′), which has also been successfully employed in organolanthanide24–26 and -actinide chemistry.24,27
Recent findings by Murugesu and co-workers revealed that some of the anionic [Ln(COT′′)2]− sandwich complexes behave as organometallic single-molecule magnets.17,20,21,23 Due to the renewed interest in this class of compounds, we carried out a broader investigation on lanthanide and actinide COT′′ sandwich complexes. In this contribution we report the synthesis and structural characterization of the new anionic lanthanide sandwich complexes [Li(DME)3][Ln(COT′′)2] (Ln = Y (1), La (2), Gd (4), Tm (6), Lu (8)) and [Li(THF)4][Ln(COT′′)2] (Ln = Pr (3), Ho (5), Tm (7)) as well as the neutral actinide sandwich complexes An(COT′′)2 (An = Th (9), U (10)) and An(COT′′′)2 (An = Th (11), U (12)). Most recently, after this work had been completed, Murugesu et al. reported the synthesis, structure, and magnetic properties of the closely related uranium(III) sandwich complex [Li(DME)3][U(COT′′)2] and the isostructural and isoelectronic lanthanide analogue [Li(DME)3][Nd(COT′′)2]. This work also included the synthesis and structural characterization of the neutral uranocene derivative U(COT′′)2 (10), although its preparation involved a different synthetic route (vide infra).27
2. Results and discussion
The general synthetic protocol for preparing the anionic lanthanide sandwich complexes is outlined in Scheme 3. The synthesis starts with the well-established preparation of 1,4-bis(trimethylsilyl)cycloocta-2,5,7-triene from 1,5-cyclooctadiene according to Cloke et al.10 The dilithium reagent Li2COT′′ can be conveniently prepared by metalation of 1,4-bis(trimethylsilyl)cycloocta-2,5,7-triene with n-butyllithium,10 and the resulting solutions can be used in situ for further reactions. However, it is also possible to isolate crystalline adducts of Li2COT′′, e.g. [Li(TMEDA)]2(COT′′) (TMEDA = N,N,N′,N′-tetramethylethylenediamine),10c [Li(DME)]2(COT′′),28a and [Li(THF)2]2[Li2(COT′′)2].28b The latter two adducts have been structurally characterized through X-ray diffraction. [Li(TMEDA)]2(COT′′) was shown to be an inverse sandwich complex with the two Li+ ions coordinated to the bridging 1,4-bis(trimethylsilyl)cyclooctatetraene dianion ring in an η3-allyl-like fashion.28a [Li(THF)2]2[Li2(COT′′)2] contains two Li+ ions sandwiched between two COT′′ rings and two Li(THF)2+ units attached to the outside of the COT′′ rings.28b In the present study, however, it was found to be more convenient to use in situ-prepared THF solutions of Li2COT′′ rather than isolated samples. Accordingly, the anionic lanthanide sandwich complexes 1–8 were prepared by treatment of selected anhydrous lanthanide trichlorides, LnCl3, with 2 equiv. of Li2COT′′ in THF solution as outlined in Scheme 3. In the case of the THF adducts [Li(THF)4][Ln(COT′′)2] (Ln = Ho (5), Tm (7)), purification was achieved by recrystallization of the crude products from toluene. The DME adducts [Li(DME)3][Ln(COT′′)2] (Ln = Y (1), La (2), Pr (3), Gd (4), Tm (6), Lu (8)) were obtained by extraction of the reaction products with toluene followed by recrystallization from DME after addition of n-pentane. The products were isolated in moderate to good yields (57–75%) in the form of yellow or yellow-green (Tm: red), highly air-sensitive crystalline solids. It has been noted earlier that DME is the solvent of choice for crystallizing these anionic lanthanide sandwich complexes.14,20 The DME solvates are readily crystallized and the resulting crystals do not lose DME even under vacuum or upon prolonged storage in the dry-box. In contrast, crystals of the THF adducts are less stable with respect to loss of solvent and become opaque upon storing in the dry-box. | ||
| Scheme 3 Synthetic route to the anionic lanthanide sandwich complexes 1–8. | ||
Meaningful NMR spectra could be obtained only for the diamagnetic products [Li(DME)3][Y(COT′′)2] (1), [Li(DME)3][La(COT′′)2] (2), and [Li(DME)3][Lu(COT′′)2] (8) as well as for the paramagnetic praseodymium derivative 3. In all four cases the 1H and 13C NMR data were in good agreement with the formation of the expected anionic sandwich complexes. The observation of only one signal in the 29Si NMR spectra (1: 0.7 ppm, 2: 0.5 ppm, 3: −46 ppm, 8: 0.8 ppm) indicated high purity of the materials. Moreover, the IR spectra of the DME adducts on one hand and the THF adducts on the other hand were found to be almost superimposable. All the new complexes were structurally characterized through single-crystal X-ray crystallography. Crystallographic data for 1–8 are summarized in Tables 1 and 2. The most significant bond lengths and angles are listed in Table 3.
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Empirical formula | C40H78LiO6Si4Y | C40H78LaLiO6Si4 | C40H78LiO6PrSi4 | C40H78GdLiO6Si4 | C44H80HoLiO4Si4 | C40H78LiO6Si4Tm |
| a (Å) | 11.445(2) | 11.557(2) | 11.532(2) | 11.428(2) | 11.358(2) | 11.410(2) |
| b (Å) | 12.219(2) | 12.262(3) | 12.286(3) | 12.216(2) | 11.657(2) | 12.239(2) |
| c (Å) | 18.477(4) | 18.340(4) | 18.388(4) | 18.406(4) | 19.603(4) | 18.503(4) |
| α (°) | 99.13(3) | 98.83(3) | 98.82(3) | 98.84(3) | 87.12(3) | 99.12(3) |
| β (°) | 102.20(3) | 102.56(3) | 102.49(3) | 102.36(3) | 82.38(3) | 102.17(3) |
| γ (°) | 99.68(3) | 98.57(3) | 99.04(3) | 99.44(3) | 77.39(3) | 100.11(3) |
| V (Å3) | 2438.7(8) | 2461.6(9) | 2464.1(9) | 2428.0(8) | 2509.9(9) | 2434.9(8) |
| Z | 2 | 2 | 2 | 2 | 2 | 2 |
| Formula weight | 863.23 | 913.23 | 915.23 | 931.57 | 957.31 | 943.25 |
| Space group |
P |
P |
P |
P |
P |
P |
| T (K) | 153(2) | 133(2) | 153(2) | 133(2) | 153(2) | 153(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calcd (g cm−3) | 1.176 | 1.232 | 1.234 | 1.274 | 1.267 | 1.287 |
| μ (mm−1) | 1.332 | 1.003 | 1.124 | 1.503 | 1.707 | 1.959 |
| R(Fo or Fo2) | 0.0467 | 0.0381 | 0.0267 | 0.0333 | 0.0491 | 0.0319 |
| R w(Fo or Fo2) | 0.1018 | 0.0944 | 0.0662 | 0.0883 | 0.0859 | 0.0771 |
| 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|
| Empirical formula | C44H80LiO4Si4Tm | C40H78LiLuO6Si4 | C28H48Si4Th | C28H48Si4U | C34H64Si6Th | C34H64Si6U |
| a (Å) | 11.358(2) | 11.471(2) | 9.830(3) | 12.6598(12) | 20.9990(12) | 23.381(3) |
| b (Å) | 11.657(2) | 12.051(2) | 9.898(3) | 20.3542(18) | 25.1674(15) | 21.170(3) |
| c (Å) | 19.603(4) | 18.459(4) | 17.038(5) | 24.645(2) | 17.8458(10) | 18.125(2) |
| α (°) | 87.12(3) | 99.79(3) | 79.262(4) | 90 | 90 | 90 |
| β (°) | 82.38(3) | 101.72(3) | 80.692(4) | 90 | 114.3160(10) | 107.923(2) |
| γ (°) | 77.39(3) | 100.05(3) | 80.891(3) | 90 | 90 | 90 |
| V (Å3) | 2509.9(9) | 2403.6(8) | 1.5932(8) | 6350.6(10) | 8594.7(9) | 8536.3(18) |
| Z | 2 | 2 | 2 | 8 | 8 | 8 |
| Formula weight | 961.31 | 949.29 | 729.06 | 735.05 | 873.43 | 879.42 |
| Space group |
P |
P |
P |
Pbca | C2/c | P21/c |
| T (K) | 153(2) | 143(2) | 100(2) | 173(2) | 200(2) | 200(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calcd (g cm−3) | 1.272 | 1.312 | 1.520 | 1.538 | 1.350 | 1.369 |
| μ (mm−1) | 1.899 | 2.192 | 4.845 | 5.277 | 3.657 | 3.991 |
| R(Fo or Fo2) | 0.0390 | 0.0557 | 0.0472 | 0.0238 | 0.0301 | 0.0421 |
| R w(Fo or Fo2) | 0.1015 | 0.1473 | 0.1084 | 0.0838 | 0.0675 | 0.1127 |
| Y (1) | La (2) | Pr (3) | Gd (4) | Ho (5) | Tm (6) | Tm (7) | Lu (8) | |
|---|---|---|---|---|---|---|---|---|
| Ln–C | 2.649 | 2.785 | 2.740 | 2.680 | 2.607 | 2.624 | 2.615 | 2.609 |
| C–C | 1.413 | 1.414 | 1.414 | 1.416 | 1.414 | 1.417 | 1.414 | 1.412 |
| Si–C | 1.865 | 1.867 | 1.866 | 1.867 | 1.868 | 1.867 | 1.868 | 1.864 |
| Li–O | 2.135 | 2.133 | 2.134 | 2.134 | 1.921 | 2.135 | 1.919 | 2.123 |
| Ln–Ctr | 1.900 | 2.084 | 2.020 | 1.940 | 1.873 | 1.861 | 1.850 | 1.845 |
| Ctr–Ln–Ctr | 176.19 | 175.59 | 175.80 | 175.74 | 177.6 | 176.6 | 177.8 | 176.86 |
All eight lanthanide complexes were found to form large crystals quite readily. X-ray quality single-crystals of the DME solvates [Li(DME)3][Ln{C8H6(SiMe3)2}2] Ln = Y (1), La (2), Pr (3), Gd (4), Tm (6), Lu (8) were obtained by recrystallization from solvent mixtures of DME and n-pentane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature. Single-crystals of the THF adducts [Li(THF)4][Ln(COT′′)2] (Ln = Ho (5), Tm (7)) were grown from concentrated solutions in THF at r.t. The molecular structure of the THF adduct [Li(THF)4][Ho(COT′′)2] (5) is shown in Fig. 1, and Fig. 2 shows the molecular structure of [Li(DME)3][Lu(COT′′)2] (8) as a representative DME adduct.
:1) at room temperature. Single-crystals of the THF adducts [Li(THF)4][Ln(COT′′)2] (Ln = Ho (5), Tm (7)) were grown from concentrated solutions in THF at r.t. The molecular structure of the THF adduct [Li(THF)4][Ho(COT′′)2] (5) is shown in Fig. 1, and Fig. 2 shows the molecular structure of [Li(DME)3][Lu(COT′′)2] (8) as a representative DME adduct.
 | ||
| Fig. 1 Molecular structure of [Li(THF)4][Ho(COT′′)2] (5). | ||
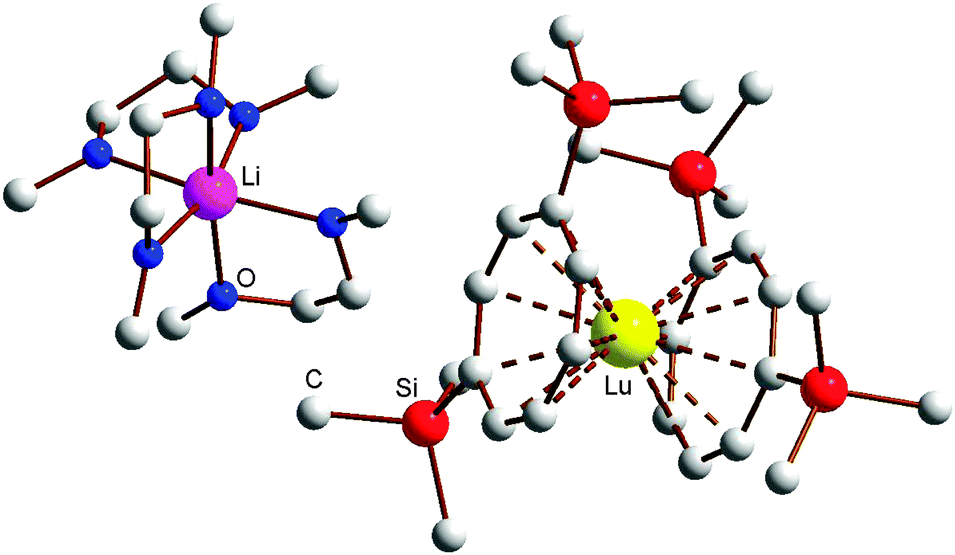 | ||
| Fig. 2 Molecular structure of [Li(DME)3][Lu(COT′′)2] (8). | ||
As can be seen from Table 3, the average Ln–C bond lengths vary between 2.785 Å in the lanthanum complex 2 and 2.609 Å in the lutetium derivative 8. The difference of 0.176 Å can be attributed to the lanthanide contraction. Certainly the most significant result is the finding that all known [Li(DME)3][Ln(COT′′)2] complexes (Ln = Y (1), La (2), Ce,14,20 Pr (3), Nd,27 Gd (4), Dy,17 Er,21 Tm (6), Lu (8)) and also the recently reported [Li(DME)3][U(COT′′)2]27 crystallize in the triclinic space group P and are isostructural. The same can be said about the series of known [Li(THF)4][Ln(COT′′)2] complexes,14,17,19 including the new derivatives [Li(THF)4][Ho(COT′′)2] (5) and (b) [Li(THF)4][Tm(COT′′)2] (7). The Ctr–Ln–Ctr angles (Ctr = ring centroid) are found to be in the very narrow range between 177.8° for [Li(THF)4][Tm(COT′′)2] (7) and 175.59° for [Li(DME)3][Ln(COT′′)2] (2). Thus there is only little deviation from the ideal linear sandwich arrangement. Clearly, the bulky cyclooctatetraenyl ligand COT′′ is ideally suited for studying homologous series of lanthanide and actinide sandwich complexes comprising the full range of ionic radii possible. Table 4 provides an overview of all anionic lanthanide sandwich complexes of the types [Li(THF)4][Ln(COT′′)2] and [Li(DME)3][Ln(COT′′)2] reported thus far in order to show which gaps have been filled by the present study.
In a similar manner, the closely related neutral actinidocenes An(COT′′)2 (An = Th (9), U (10)) have also been prepared. As outlined in Scheme 4, these sandwich complexes were prepared in a straightforward manner by reaction of anhydrous ThCl4 or UCl4 with 2 equiv. of in situ-prepared Li2COT′′. Due to the high solubility of all the reactants in THF, the reactions were finished after 2 h stirring at r.t. In contrast, reactions of AnCl4 with the unsubstituted K2COT normally take days.11,12 Bright yellow Th(COT′′)2 (9) and dark green (dichroitic red/green) U(COT′′)2 (10) were both isolated in high yields of ca. 80%. Purification could be achieved either by high-vacuum sublimation at 240 °C or by slow crystallization from the oily crude products. In this context it is interesting to note that Murugesu et al. very recently prepared compound 10via a two-step synthesis where UIIII3(1,4-dioxane)1.5 and [Li(THF)2]2[Li2(COT′′)2]28b were first combined in THF to afford the anionic uranium(III) sandwich complex [Li(DME)3][U(COT′′)2] which was then oxidized to the uranium(IV) sandwich 10 using FeCl2.27
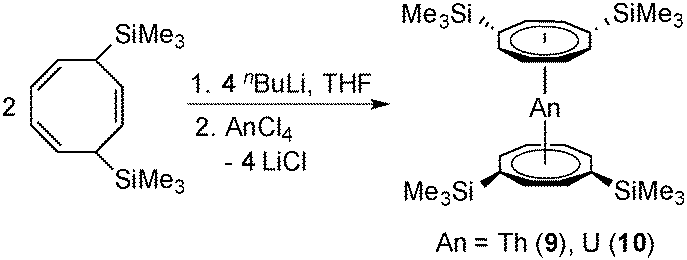 | ||
| Scheme 4 Synthetic route to the neutral actinidocenes An(COT′′)2 (An = Th (9), U (10)). | ||
For comparison, two neutral actinide sandwich complexes comprising the bulky 1,3,6-tris(trimethylsilyl)cyclooctatetraenyl ligand (COT′′′) have also been prepared. These compounds have earlier been mentioned in two communications, but structural characterization through X-ray diffraction was lacking.24 Both compounds were prepared according to the straightforward synthetic protocol illustrated in Scheme 5. In this case, the use of the potassium precursor K2COT′′′ provided products 11 and 12 in yields of around 80% after crystallization from concentrated solutions in n-pentane. Like their tetrasubstituted congeners 9 and 10, thorium compound 11 forms bright yellow crystals, while crystals of 12 appear dichroitic red/green. Both complexes are highly soluble in common organic solvents, including hydrocarbons.
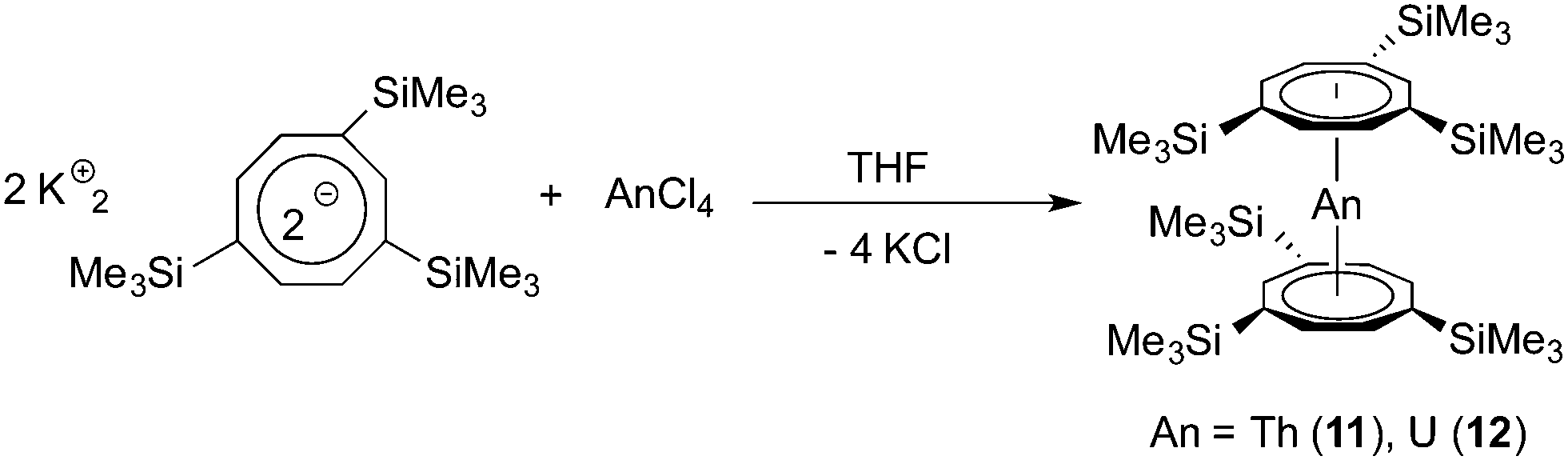 | ||
| Scheme 5 Synthesis of the neutral actinidocenes An(COT′′′)2 (An = Th (11), U (12)). | ||
All four silyl-substituted actinidocenes 9–12 have been structurally characterized through single-crystal X-ray diffraction. Crystallographic data for 9–12 are summarized in Table 2; selected bond lengths and angles are listed in Table 4. The molecular structures are depicted in Fig. 3 and 4. As can be seen from the structural data listed in Table 4, the overall structural features of all four actinidocene derivatives studied here are very similar. According to the unsymmetrical substitution pattern on the cyclooctatetraenyl rings leading to steric interactions, all complexes show a slight distorsion from the ideal linear arrangement with Ctr–M–Ctr angles of about 174°. As expected, evidence for actinide contraction is found which is reflected in ∼5 pm shorter M–C as well as in ∼7 pm shorter M–Ctr distances in the uranium complexes as compared to the thorium species (Table 5).
 | ||
| Fig. 3 Molecular structures of Th(COT′′)2 (9) and U(COT′′)2 (10). | ||
 | ||
| Fig. 4 Molecular structures of Th(COT′′′)2 (11) and U(COT′′′)2 (12). | ||
| Th (9) | U (10) | Th (11) | U (12) | |
|---|---|---|---|---|
| M–C |
2.696–2.745
(2.718) |
2.642–269.0
(2.663) |
2.709–2.763
2.705–2.762 (2.735/2.732) |
2.643–2.727
2.647–2.727 (2.679/2.681) |
| M–Ctr |
1.987
1.999 (1.993) |
1.921/1.913
(1.917) |
2.012
2.010 (2.011) |
1.942, 1.944
1.938, 1.945 (1.942) |
| Ctr–M–Ctr | 172.9 | 173.0 | 174.3, 175.3 | 174.3, 174.9 |
| Pl/Pl | 7.1 | 7.4 | 6.3 | 5.7/6.0 |
In the following, the structural and spectroscopic characterization of 10 as a typical example will be discussed in more detail. The molecular structure of 10 can be clearly described as being of the well-known uranocene type (Fig. 5). Accordingly, in the molecular structure the central uranium atom is placed between the two cyclooctatetraenyl rings with U–Ctr distances of 1.913 or 1.921 Å, comparable to previously reported uranocene derivatives29 (Table 4). However, the trimethylsilyl substituents in 1,4-positions of the cyclooctatetraenyl ring lead to an arrangement in the solid state where on one side of the molecule a stronger steric interaction between the two cyclooctatetraenyl rings results. Si1 and Si4 are found to be in closer steric environment than Si2 and Si3, giving rise to a significant repulsion on this side of the rings. This has an influence on the bond lengths and angles in that the two cyclooctatetraenyl rings do not bind symmetrically to the central uranium atom. The U–C bond distances cover a range between 2.642 and 2.690(4) Å with the longer bond lengths found on the side with the stronger steric interactions, whereas the shortest U–C bond length is observed for U1–C22 with 2.642(4) Å. Accordingly, the two cyclooctatetraenyl rings are not coordinated coplanar with respect to the uranium center. This results in a Ctr–U–Ctr angle of 7.0° and a tilt angle between the two ring planes of 7.4° with the opening to the side of Si1/Si4 (Table 4 and Fig. 5). This is further reflected in the corresponding distances between opposing carbon atoms of the two COT′′ rings in the staggered structure. With 4.047 and 4.070 Å the distances C1–C17 and C2–C18 are remarkably longer than those between C5 and C21 or C6 and C22, which are with 3.627 or 3.614 Å significantly shorter. These structural findings clearly show that compound 10 shows typical uranocene structural features29a but with a significant distorsion caused by steric effects due to the trimethylsilyl substituents at the COT rings. A significantly stronger tilting of the two cyclooctatetraenyl rings has been observed in the 1,4-bis(triphenylsilyl)-substituted system where ring-to-ring C–C distances between 3.468 and 4.247 Å and a tilt angle of 11.4° have been found.30
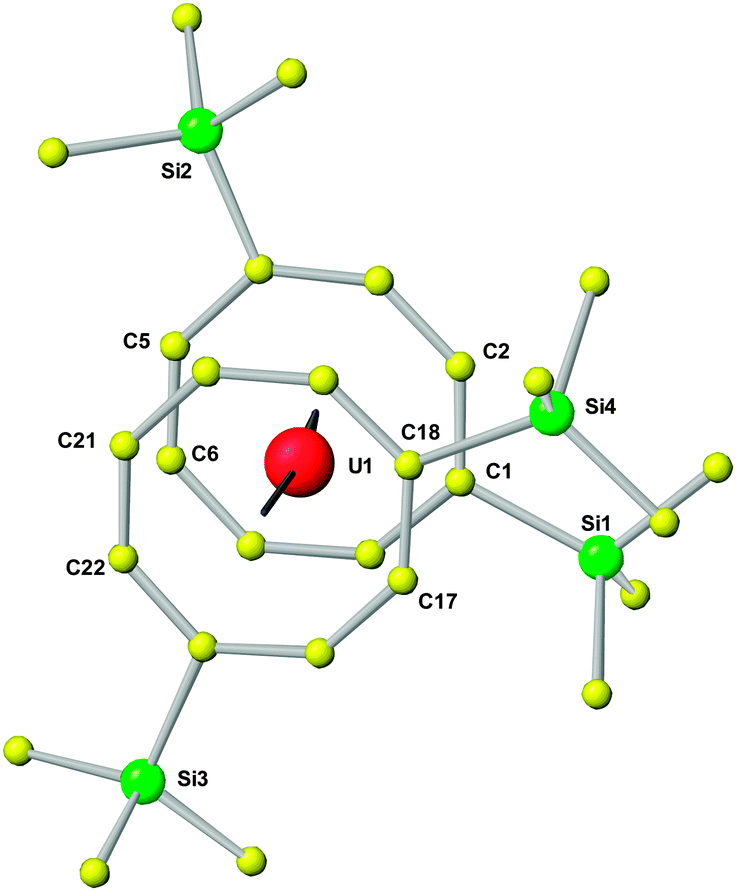 | ||
| Fig. 5 Top view of the molecular structure of U(COT′′)2 (10). | ||
The spectroscopic data of the complexes 9–12 are in good agreement with their structural features. As expected, the IR spectra of 9–12 are all very similar, showing the comparable molecular constitution of these actinidocenes. Frequencies arising from the COT′′ ligand increase slightly upon complexation as compared to K2COT′′. However, the spectra are more complicated than those of the unsubstituted actinidocenes as the SiMe3-substituents give rise to strong absorptions themselves and cause a distorsion from the ideal D8h-symmetry observed in the actinidocenes, leading to a higher number of observed frequencies.31 However, the general congruency of the IR and FIR spectra clearly shows the similarity in the structural features of the complexes 9–12. The other spectroscopic data will be highlighted taking again compound 10 as example. In contrast to the corresponding Th-complex 9, the uranocene derivative 10 exhibits a 5f2-electron configuration causing paramagnetism and an intensely red color in transmission. These findings are confirmed by the UV-vis data (Fig. 6), showing that below 450 nm the absorption of the complex is strongly increasing. The absorptions at 592, 618, 635 nm are caused by strong charge transfer transitions typical for actinocene complexes, however being more intense in symmetry-distorted systems.32 In the range of 800 to 2000 nm, the UV-vis spectrum does not show any significant differences between the solid state and the solution, indicating that the solid state structure is retained in solution and no adduct formation takes place. Accordingly, the absorptions at 980, 1322, 1486, 1710, 1755, 1793, 1865 nm are caused by f–f transitions, which are characteristic of U(IV)-organometallics.33 The f–f transitions are in this case of higher intensity than for the unsubstituted uranocene due to the observed distortion of the complex symmetry by the SiMe3 substituents, which causes an increase in the intensity for the symmetry-forbidden f–f transitions. These are, however, between 10 to 100 times less intense than the charge transfer absorptions.
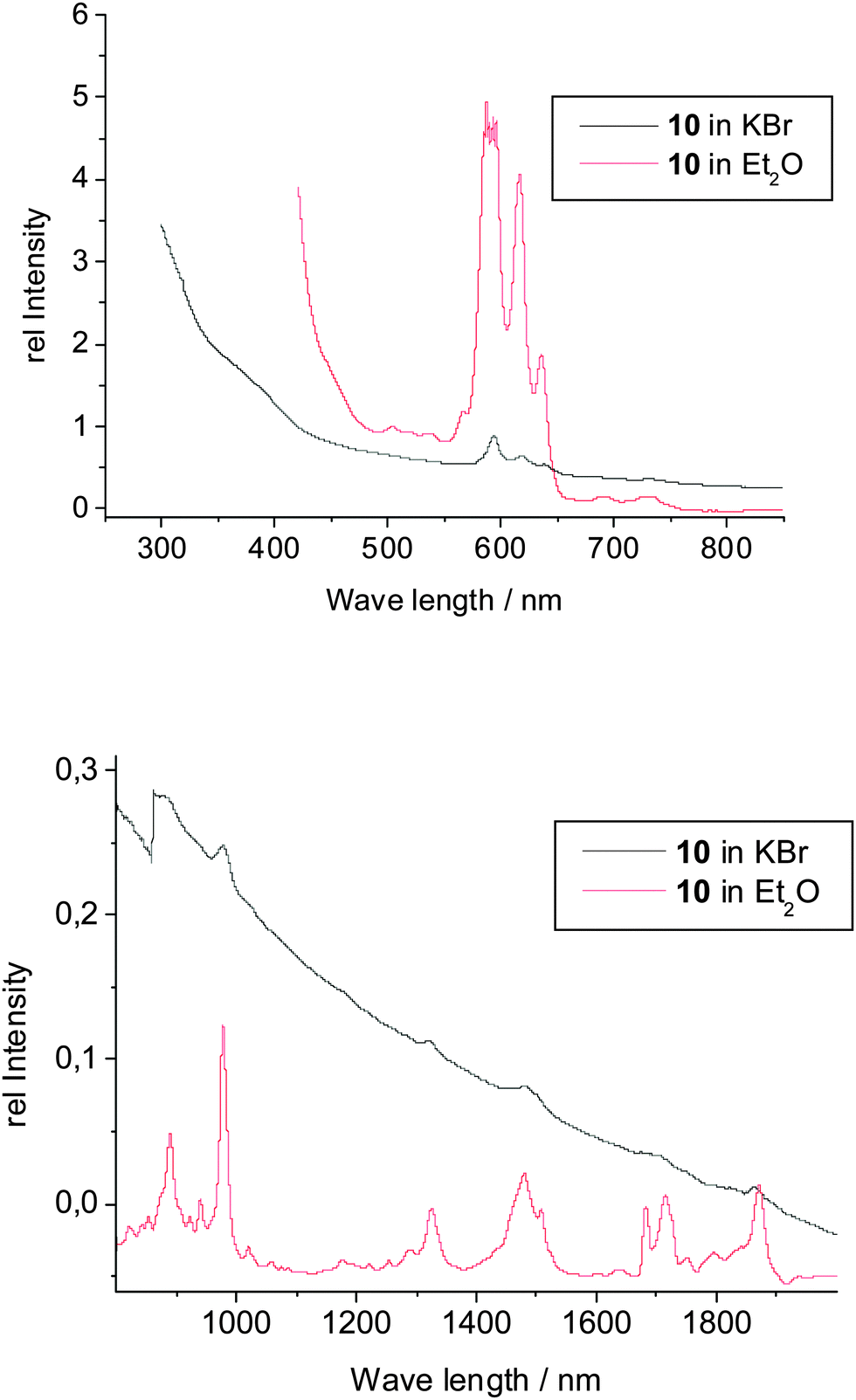 | ||
| Fig. 6 UV-vis spectra of U(COT′′)2 (10). | ||
The paramagnetism of 10 is also clearly seen from its NMR data (Fig. 7–9), where for all signals a typical upfield shift is observed.7a,34 In good agreement with the solid state structure, the 1H NMR spectrum of 10 (Fig. 7) shows four well-separated singlets at −9.99, −25.20, −39.63, and −45.62 ppm. The latter three each correspond to four ring protons, whereas the first resonance can be clearly assigned to the protons of the SiMe3 substituents. In the two-dimensional HH-correlated spectrum the resonances at −39.63 and −45.62 ppm (β-position to the SiMe3-substituents) are assigned to the protons in the (CH)4-chain of the COT′′ ring, whereas the resonance at −25.20 ppm corresponds to the ring protons positioned between the two trimethylsilyl substituents in 1,4-positions (Fig. 8).
 | ||
| Fig. 7 1H NMR spectrum of U(COT′′)2 (10). | ||
 | ||
| Fig. 8 HH correlated NMR spectrum of U(COT′′)2 (10). | ||
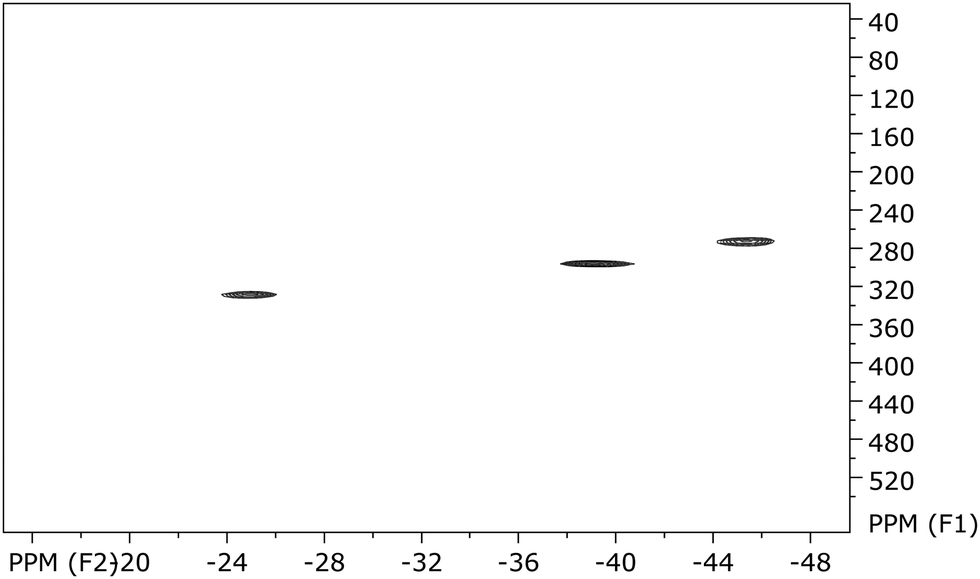 | ||
| Fig. 9 CH correlated NMR spectrum of U(COT′′)2 (10). | ||
This assignment is in good agreement with the published data where a strong influence of the paramagnetism on the chemical shifts in uranocene derivatives is described.34 However, in this paper, for the first time, the 13C chemical shifts of a uranocene complex are reported. The carbon resonance of the SiMe3 groups was localized at −3.5 ppm. The proton resonance at −25.20 ppm exhibits a cross peak at 325.9 ppm in the 13C frequency, whereas the two coupling H-atoms of the aromatic ring at −39.63 ppm and −45.62 ppm give rise to carbon resonances as well at low field shifts with 293.8 and 270.3 ppm, respectively (Fig. 9). The observation of carbon frequencies at these low fields is in agreement with theoretical predictions.35
3. Conclusions
In summarizing the results reported here, the series of anionic lanthanide(III) sandwich complexes of the type [Ln(COT′′)2]− (COT′′ = 1,4-bis(trimethylsilyl)cyclooctatetraenyl dianion) has been largely extended through the synthesis of eight new derivatives ranging from lanthanum to lutetium. Surprisingly, neither the ionic radius nor the oxidation state of the f-element ion (Ln3+/An4+) have a pronounced influence on the structural features of the compounds [Li(DME)3][Ln(COT′′)2] (1–8; Ln = Y, La, Pr, Gd, Tm, Lu), [Li(THF)4][Ln(COT′′)2] (5, 7; Ln = Ho, Tm), An(COT′′)2 (9, 10; An = Th, U) and An(COT′′′)2 (11, 12; An = Th, U). In all cases the slight deviation from the ideal sandwich structure is in the same range. Through this comparative study anionic sandwich complexes containing the [Ln(COT′′)2]− anions have now become available for the entire series of rare-earth metals. This should allow for more detailed investigations e.g. of the magnetic properties in the course of future studies.4. Experimental section
4.1 General procedures
All operations were performed with rigorous exclusion of air and water in oven-dried or flame-dried glassware under an inert atmosphere of dry argon, employing standard Schlenk, high-vacuum and glovebox techniques (MBraun MBLab; <1 ppm O2, <1 ppm H2O). THF, DME, toluene, and cyclopentane were dried over sodium/benzophenone and freshly distilled under a nitrogen atmosphere prior to use. All glassware was oven-dried at 120 °C for at least 24 h, assembled while hot, and cooled under high vacuum, prior to use. The starting materials, anhydrous LnCl3 (Ln = Ce, Nd),36 ThCl4,37 UCl4,38 C8H8(SiMe3)2,10 Li2(COT′′),10 and K2COT′′′24,25 were prepared according to the published procedures. The NMR spectra were recorded in C6D6 or d8-THF solutions on a Bruker AVANCE 600 (1H: 600.1 MHz; 13C: 150.9 MHz) or a Bruker AVANCE 400 (5 mm BB, 1H: 400.1 MHz; 13C: 100.6 MHz) (Ln compounds), or a Bruker-AVANCE 250 (5 mm TBI, 1H: 250.1 MHz; 13C: 62.5 MHz) (An compounds). 1H and 13C shifts are referenced to internal solvent resonances and reported in parts per million relative to TMS. IR (KBr) spectra were recorded using a Perkin-Elmer FT-IR 2000 spectrometer. UV-Vis spectra were registered on a Perkin-Elmer Lambda 2 spectrometer. Mass spectra (EI, 70 eV) were run on a MAT 95 apparatus. Microanalyses of the compounds were performed using a Leco CHNS 932 apparatus. Metal analyses were performed via ICP AES.4.2 Preparation of the anionic lanthanide sandwich complexes 1–8 (general synthetic protocols)
4.3 Preparation of the actinide sandwich complexes An(COT′′)2 (An = Th (9), U (10)) (general synthetic protocol)
∼200 mg of anhydrous AnCl4 (An = Th, U; ∼0.5 mmol) were treated with 2.05 equiv. of freshly prepared Li2(COT′′) in 25 mL of THF. Due to the high solubility of all reactants the reactions were finished after 2 h stirring at r.t. The solvent was evaporated and n-pentane (20 mL) was added, yielding an intense yellow solution (Th) and a red-green solution for the U complex 10. Filtration and removal of the solvents afforded the crude complexes in ∼80% yield. From the waxy yellow thorium complex 9 (m.p. 135 °C) single-crystals were obtained by recrystallization in a closed ampule at 240 °C under high vacuum. From the oily, crude uranium complex 10, single-crystals grew in the refrigerator during storage at 4 °C for several months. Sublimation under identical conditions as described for Th led to the formation of red crystals in a green oil.4.4 Preparation of the actinide sandwich complexes An(COT′′′)2 (An = Th (11), U (12))
The two polysilylated actinidocenes 11 and 12 were prepared by treatment of AnCl4 (An = Th, U) with 2 equiv. of K2(COT′′′), following the procedure reported by Edelmann and Kanellakopulos et al. (cf.Scheme 5).24b Bright yellow 11 and dichroitic red/green 12 were isolated in high yields of around 80% after recrystallization from n-pentane.4.5 Crystal structure determination
The intensity data of the lanthanide sandwich complexes 1–8 were collected on a Stoe IPDS 2T diffractometer with MoKα radiation. The data were collected with the Stoe XAREA program using ω-scans.39 The space groups were determined with the XRED32 program. The structures were solved by direct methods (SHELXS-97) and refined by full matrix least-squares methods on F2 using SHELXL-97.40 Data collection parameters are summarized in Tables 1 and 2. Single-crystal X-ray analyses of the actinide complexes 9–12 were performed on a Bruker Apex II Quazar diffractometer at given temperature, collecting two or four spheres of data with an irradiation time of 10 to 40 s per frame, applying a combination of ω- and φ-scans. Maximum θ-values were in the range of 28°. Completeness of data to θ ≤ 25° was higher than 99%. For more information refer to Table 2. Integration of the data proceeded with SAINT,41 the data were corrected for Lorentz- and polarisation effects, and an experimental absorption correction with SADABS41 was performed. For searches relating to single-crystal X-ray diffraction data, the Cambridge Structural Database was used. The structures have been solved by direct methods and refined to a minimum R-value with SHELXL-201342via full-matrix least-squares on F2. In the case of compound 9, a second type of crystals could be isolated with a different elementary cell showing a strong disorder. The data have been deposited at the CCDC with the CCDC 1049928 but will not be discussed here in detail as due to the disorder the overall standard deviations for all values are significantly higher.Acknowledgements
Financial support by the Otto-von-Guericke-Universität Magdeburg is gratefully acknowledged. Special thanks are due to Ms Desirée Schneider for preparing the ChemDraw Schemes.References
- Review: F. T. Edelmann, Complexes of Scandium, Yttrium and Lanthanide Elements, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2006, p. 1 Search PubMed.
- C. Meermann, K. Ohno, K. W. Törnroos, K. Mashima and R. Anwander, Eur. J. Inorg. Chem., 2009, 76 CrossRef CAS PubMed , and references cited therein.
- (a) F. Mares, K. O. Hodgson and A. Streitwieser, J. Organomet. Chem., 1970, 24, C68 CrossRef CAS; (b) K. O. Hodgson, F. Mares, D. Starks and A. Streitwieser, J. Am. Chem. Soc., 1973, 85, 8650 CrossRef; (c) S. A. Kinsley, A. Streitwieser and A. Zalkin, Organometallics, 1985, 4, 52 CrossRef CAS; (d) A. L. Wayda, Organometallics, 1983, 2, 565 CrossRef CAS; (e) J. Jin, Z. Jin, G. Wei, W. Chen and Y. Zhang, Chin. J. Inorg. Chem., 1993, 9, 326 CAS; (f) U. Kilimann, M. Schäfer, R. Herbst-Irmer and F. T. Edelmann, J. Organomet. Chem., 1994, 469, C15 CrossRef CAS; (g) S. Anfang, G. Seybert, K. Harms, G. Geiseler, W. Massa and K. Dehnicke, Z. Anorg. Allg. Chem., 1998, 624, 1187 CrossRef CAS; (h) G. W. Rabe, M. Zhang-Presse, J. A. Golen and A. L. Rheingold, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m255 CAS.
- (a) F. Mares, K. O. Hodgson and A. Streitwieser, J. Organomet. Chem., 1971, 28, C24 CrossRef CAS; (b) K. O. Hodgson and K. N. Raymond, Inorg. Chem., 1972, 11, 171 CrossRef CAS; (c) K. O. Hodgson and K. N. Raymond, Inorg. Chem., 1972, 11, 3030 CrossRef CAS; (d) A. L. Wayda, Organometallics, 1983, 2, 565 CrossRef CAS; (e) J. Xia, Z. Jin, G. Wei and W. Chen, Jiegou Huaxue, 1992, 11, 113 CAS.
- (a) J. D. Jamerson, A. P. Masino and J. Takats, J. Organomet. Chem., 1974, 65, C33 CrossRef CAS; (b) A. Westerhoff and H. J. De LiefdeMeijer, J. Organomet. Chem., 1976, 116, 319 CrossRef; (c) K. Wen, Z. Jin and W. Chen, J. Chem. Soc., Chem. Commun., 1991, 680 CAS.
- A. Greco, S. Cesca and G. Bertolini, J. Organomet. Chem., 1976, 113, 321 CrossRef CAS.
- (a) A. Streitwieser and S. A. Kinsley, in Fundamental and Technological Aspects of Organo-f-Element Chemistry, ed. T. J. Marks and I. L. Fragalà, NATO ASI Series, D. Reidel, Boston, 1985, vol. 155, p. 77 Search PubMed; (b) A. Streitwieser and T. R. Boussie, Eur. J. Solid State Inorg. Chem., 1991, 28, 399 CAS; (c) F. T. Edelmann, New J. Chem., 1995, 19, 535 CAS; (d) F. T. Edelmann, Angew. Chem., Int. Ed., 1995, 34, 2466 CrossRef CAS PubMed; (e) F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851 CrossRef CAS PubMed; (f) H.-D. Amberger, F. T. Edelmann, J. Gottfriedsen, R. Herbst-Irmer, S. Jank, U. Kilimann, M. Noltemeyer, H. Reddmann and M. Schäfer, Inorg. Chem., 2009, 48, 760 CrossRef CAS PubMed; (g) F. T. Edelmann, New J. Chem., 2011, 35, 517 RSC.
- K. Mashima and H. Takaya, Tetrahedron Lett., 1989, 30, 3697 CrossRef CAS.
- (a) U. Kilimann, M. Schäfer, R. Herbst-Irmer and F. T. Edelmann, J. Organomet. Chem., 1994, 469, C10 CrossRef CAS; (b) H. Schumann, J. Winterfeld, H. Hemling, F. E. Hahn, P. Reich, K.-W. Brzezinka, F. T. Edelmann, U. Kilimann, M. Schäfer and R. Herbst-Irmer, Chem. Ber., 1995, 128, 395 CrossRef CAS PubMed; (c) U. Reißmann, P. Poremba, M. Noltemeyer, H.-G. Schmidt and F. T. Edelmann, Inorg. Chim. Acta, 2000, 303, 156 CrossRef.
- (a) J. M. Bellama and J. B. Davidson, J. Organomet. Chem., 1975, 86, 69 CrossRef CAS; (b) N. C. Burton, F. G. N. Cloke, P. B. Hitchcock, H. C. de Lemos and A. A. Sameh, J. Chem. Soc., Chem. Commun., 1989, 1462 RSC; (c) N. C. Burton, F. G. N. Cloke, S. C. P. Joseph, H. Karamallakis and A. A. Sameh, J. Organomet. Chem., 1993, 462, 39 CrossRef CAS.
- A. Streitwieser and U. Müller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef CAS.
- Review: D. Seyferth, Organometallics, 2004, 23, 3562 CrossRef CAS.
- (a) A. Hervé, N. Garin, P. Thuéry, M. Ephritikhine and J.-C. Berthet, Chem. Commun., 2013, 49, 6304 RSC; (b) J.-C. Berthet, P. Thuéry, N. Garin, J.-P. Dognon, T. Cantat and M. Ephritikhine, J. Am. Chem. Soc., 2013, 135, 10003 CrossRef CAS PubMed.
- A. Edelmann, V. Lorenz, C. G. Hrib, L. Hilfert, S. Blaurock and F. T. Edelmann, Organometallics, 2013, 32, 1435 CrossRef CAS.
- V. Lorenz, A. Edelmann, S. Blaurock, F. Freise and F. T. Edelmann, Organometallics, 2007, 26, 4708 CrossRef CAS.
- V. Lorenz, S. Blaurock, C. G. Hrib and F. T. Edelmann, Organometallics, 2010, 29, 4787 CrossRef CAS.
- J. J. Le Roy, M. Jeletic, S. I. Gorelski, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502 CrossRef CAS PubMed.
- J. J. Le Roy, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2014, 136, 8003 CrossRef CAS PubMed.
- P. Poremba, U. Reißmann, M. Noltemeyer, H.-G. Schmidt, W. Brüser and F. T. Edelmann, J. Organomet. Chem., 1997, 544, 1 CrossRef.
- J. J. Le Roy, I. Korobkov, J. E. Kim, E. J. Schelter and M. Murugesu, Dalton Trans., 2014, 43, 2737 RSC.
- J. J. Le Roy, I. Korobkov and M. Murugesu, Chem. Commun., 2014, 50, 1602 RSC.
- V. Lorenz, A. Edelmann, S. Blaurock, F. Freise and F. T. Edelmann, Organometallics, 2007, 26, 6681 CrossRef CAS.
- M. Jeletic, P.-H. Lin, J. J. Le Roy, I. Korobkov, S. I. Gorelsky and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 19286 CrossRef CAS PubMed.
- (a) U. Kilimann, R. Herbst-Irmer, D. Stalke and F. T. Edelmann, Angew. Chem., Int. Ed., 1994, 33, 1618 CrossRef PubMed; (b) C. Apostolidis, F. T. Edelmann, B. Kanellakopulos and U. Reißmann, Z. Naturforsch., 1999, 54b, 960 Search PubMed.
- A. Edelmann, S. Blaurock, V. Lorenz, L. Hilfert and F. T. Edelmann, Angew. Chem., Int. Ed., 2007, 46, 6732 CrossRef CAS PubMed.
- A. Edelmann, C. G. Hrib, S. Blaurock and F. T. Edelmann, J. Organomet. Chem., 2010, 695, 2732 CrossRef CAS PubMed.
- J. J. Le Roy, S. I. Gorelsky, I. Korobkov and M. Murugesu, Organometallics, 2015, 34, 1415 CrossRef CAS.
- (a) P. Poremba, H. G. Schmidt, M. Noltemeyer and F. T. Edelmann, Organometallics, 1998, 17, 986 CrossRef CAS; (b) M. Jeletic, F. A. Perras, S. I. Gorelsky, J. L. Le Roy, I. Korobkov, D. L. Bryce and M. Murugesu, Dalton Trans., 2012, 41, 8060 RSC.
- (a) A. Avdeef, K. N. Raymond, K. O. Hodgson and A. Zalkin, Inorg. Chem., 1972, 11, 1083 CrossRef CAS; (b) K. O. Hodgson and K. N. Raymond, Inorg. Chem., 1973, 12, 458 CrossRef CAS; (c) L. K. Templeton, D. H. Templeton and R. Walker, Inorg. Chem., 1976, 15, 3000 CrossRef CAS; (d) A. Zalkin, D. H. Templeton, S. R. Berryhill and W. D. Luke, Inorg. Chem., 1979, 18, 2287 CrossRef CAS; (e) A. Zalkin, D. H. Templeton, W. D. Luke and A. Streitwieser Jr., Organometallics, 1982, 1, 618 CrossRef CAS; (f) A. Zalkin, D. H. Templeton, R. Kluttz and A. Streitwieser Jr., Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 327 CrossRef.
- V. Lorenz, B. M. Schmiege, C. G. Hrib, J. W. Ziller, A. Edelmann, S. Blaurock, W. J. Evans and F. T. Edelmann, J. Am. Chem. Soc., 2011, 133, 1257 CrossRef CAS PubMed.
- (a) H. P. Fritz, Adv. Organomet. Chem., 1964, 1, 239 CrossRef CAS; (b) L. Hocks, J. Goffart, G. Duyckaert and P. Teyssie, Spectrochim. Acta, Part A, 1974, 30, 907 CrossRef; (c) V. T. Aleksanyan, I. A. Garbusova, T. M. Chernyshova, Z. V. Todres, M. R. Leonov and N. I. Gramateeva, J. Organomet. Chem., 1981, 217, 169 CrossRef CAS.
- (a) A. Streitwieser and U. Müller-Westerhoff, J. Am. Chem. Soc., 1973, 95, 8644 CrossRef CAS; (b) R. Bohlander, PhD thesis, University of Karlsruhe, 1986; (c) N. Magnani, C. Apostolidis, A. Morgenstern, E. Colineau, J.-C. Griveau, H. Bolvin, O. Walter and R. Caciuffo, Angew. Chem., Int. Ed., 2011, 50, 1696 CrossRef CAS PubMed.
- C. R. Graves, A. E. Vaughn, E. J. Schelter, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2008, 47, 11879 CrossRef CAS PubMed , and references cited therein.
- (a) A. Streitwieser, Jr., R. Q. Kluttz, K. A. Smith and W. D. Luke, Organometallics, 1983, 2, 1873 CrossRef; (b) W. Jahn, K. Yünlü, W. Oroschin, H.-D. Amberger and R. D. Fischer, Inorg. Chim. Acta, 1984, 95, 85 CrossRef CAS; (c) A. Streitwieser Jr., M. H. Lyttle, H.-K. Wang, T. Boussie, A. Weinländer and J. P. Solar, J. Organomet. Chem., 1995, 501, 245 CrossRef.
- (a) A. W. Spiegl, Doctoral dissertation, University of Erlangen-Nürnberg, 1978; (b) R. D. Fischer, NMR-spectroscopy of Organo-f-Element and Pre-Lanthanoid Complexes: Some Current Trends, in Fundamental and Technological Aspects of Organo-f-Element Chemistry, ed. T. J. Marks and I. L. Fragala and D. Reidel Publishing Company, 1985, pp. 277–326 Search PubMed.
- J. H. Freeman and M. L. Smith, J. Inorg. Nucl. Chem., 1958, 7, 224 CrossRef CAS.
- D. C. Bradley, M. A. Saad and W. wardlaw, J. Chem. Soc., 1954, 2002 RSC.
- J. A. Hermann, J. F. Suttle and H. R. Hoekstra, Inorg. Synth., 1957, 5, 143 CAS.
- Stoe, XAREA, Program for X-ray Crystal Data Collection, (XRED32 included in XAREA) Stoe, 2002 Search PubMed.
- (a) G. M. Sheldrick, SHELXL-97 Program for Crystal Structure Refinement, Universität Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Solution, Universität Göttingen, Germany, 1997 Search PubMed.
- Bruker APEX, SAINT, SADABS, programs for X-ray Crystal Data Collection, integration and absorption correction, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Basil Kanellakopulos on the occasion of his 80th birthday. |
| ‡ CCDC 1052366–1052372 (1–8) and 1049924–1049927 (9–12). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00991j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |