
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding in the crystal structures of 1,2-diiodo alkenes†
Christina
Hettstedt
,
Peter
Mayer
and
Konstantin
Karaghiosoff
*
Department of Chemistry, Ludwig-Maximilians University (LMU), Butenandtstr. 5–13 (Haus D), 81377 Munich, Germany. E-mail: klk@cup.uni-muenchen.de; Fax: +49-89-2180-77492; Tel: +49-89-2180-77426
First published on 20th August 2015
Abstract
The crystal structures of 1,2-diiodoolefins are governed by extensive halogen bonding involving I⋯I, I⋯O and I⋯C interactions. These interactions are discussed considering nine new crystal structures determined in our laboratory and several additional crystal structures from the literature that complement our data.
Introduction
Halogen bonding is currently a widely discussed topic in the literature. In the last 15 years an increasing number of reviews (approximately 50) have been published on this subject. This type of intermolecular interaction was discovered about two centuries ago1 but was investigated in detail only in the last few years (for detailed mechanism and the theory of halogen bonding see ref. 2–6). Halogen bonds are relevant in the field of crystal engineering3 as well as in synthetic chemistry and material science.4 Many theoretical investigations5 have been published and halogen bonding has proved to be important in medicinal chemistry as well as in chemical biology.6 In 2013, the official IUPAC definition was released which states amongst others that typical halogen bonds R–X⋯Y–Z are formed between a halogen bond donor (R–X, X = electrophilic halogen atom, e.g. I2, CH3Br, halonium ion) and a halogen bond acceptor Y (Y = lone pair possessing atom, a π-system or an anion).7 Additionally, the contact distance of X⋯Y has to be shorter than the sum of the van der Waals radii, the R–X bond length should be elongated and the contact angle R–X⋯Y should be near to 180°.7Generally, iodine containing molecules form stronger halogen bonds than corresponding bromine, chlorine or fluorine containing derivatives.3c In most cases reported in the literature structures showing halogen bonding were obtained by forming co-crystals between two different compounds, one of which acts as the halogen bond donor and the other as the acceptor. A typical example is provided by the co-crystals of phenazine and 1,2,3,4-tetrafluoro-5,6-diiodo benzene8 or 4,4′-bipyridine and tetraiodoethene.9 The other possible case – halogen bonds in crystals where the halogen bond donor and acceptor are included in the same molecule – is much less discussed. This includes structures of compounds containing e.g. iodine as halogen bond donor and nitrogen, oxygen, a halogen or a π-system as acceptor (I⋯I, I⋯O, I⋯N, I⋯C(π)).
Diiodoolefines having additional halogen bond acceptor functionalities in the organic framework provide good examples for such structures. In these compounds halogen bonding should play a key role in determining the features of the crystal structure. In order to put light on the importance of halogen bonding as directing interaction in the formation of the structure in the solid state we have determined and investigated the crystal structures of a series of 1,2-diiodoolefines (Fig. 1). In this paper our aim is to investigate weak halogen bonds of iodine with the less strong halogen bond acceptors oxygen, iodine itself and aromatic, aliphatic or acetylenic π-systems. The systems we include in this study consist of the new crystal structures of nine 1,2-diiodoolefines determined in our laboratory and the crystal structures of several 1,2-diiodolefines from the literature that complement our data.
 | ||
| Fig. 1 Additional (E)-iodoolefines that have been synthesized from the corresponding alkyne. | ||
Particular attention is directed towards how the molecules are arranged in the crystal with respect to the optimal space filling and at the same time optimal formation of weak interactions. This is of course dependent on the substituents and their acceptor abilities, which are connected to the double bond. We would like to find a kind of “ranking” of the interactions in terms of which interaction is responsible for the structural arrangement in these weak systems and which interactions are subordinate. What is stronger in this case: hydrogen bonds (HBs) or halogen bonds (XBs). From this systematic consideration we try to find general trends that could be suitable for crystal engineering of pure compounds/crystals concerning future applications.
Results and discussion
Synthesis
1,2-Diiodoolefines 1–8 (Fig. 1) were all synthesized by 1,2-addition of I2 to the corresponding acetylenes according to literature procedures.10 The unsymmetrical diiodoolefine esters 5 and 6 are new and were prepared from the corresponding alkynes via the CuI catalyzed procedure published by Duan et al. (Fig. 2).11 | ||
| Fig. 2 Synthetic pathway of the new 1,2-diiodoolefinic esters 5 and 6. | ||
In all cases the trans isomers were obtained except for the acetal 7, where a mixture of the cis (7a) and trans (7b) derivatives was formed. From this mixture the two isomers could be separated by fractional crystallization and were structurally investigated via single crystal X-ray diffraction.
General remarks concerning the crystal structures
Single crystals, suitable for X-ray diffraction, of all compounds were obtained by recrystallization from EtOAc. Fig. 6 shows their molecular structures. A feature of most structures is a disorder of the diiodoalkene molecules over two positions. Only the structures of compounds 7a, 7b and 5 show no disorder. Here we discuss the higher occupied positions of the disordered atoms. For the diphenyldiiodide 1 and the alcohol derivative 4 the disorder concerns over 80% of the molecule. Hence the values of the atom distances and halogen bonds of these molecules have to be treated carefully in discussion and comparison with other data. Nevertheless, we include these data in the comparisons here, because the detected values of compounds 1 and 4 lie in the medium range of all here discussed compounds. Figures showing the disorder of all compounds are contained in the ESI.† Of the literature compounds from CSD there is only one disordered (RIDTOO12). The structures of all literature compounds discussed here and some additional ones for the statistics with their names from CSD are depicted in Fig. 3 and 4. | ||
| Fig. 3 (Z)- and cyclic diiodo compounds from the literature with their names from CSD. | ||
 | ||
| Fig. 4 (E)-Diiodo olefines from the literature with their names from CSD. | ||
The azide WIFVAJ13 depicted in the bottom of Fig. 4 would fit perfectly in the topic of this comparison but the quality of the structural data is inadequate. That's why we exclude this data from this discussion. We also found the crystal structures of C2H4I214 and C2H2I214 in CSD which would also fit very well in the discussion here. Unfortunately the structures are from the year 193514 and thus the cif files contain not enough data for showing the structures with modern methods.
Crystal structures
The crystal structure of 8 contains the second shortest C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (1.186(4) Å, Fig. 5) and the shortest C–I (trans) bond (2.109(4) Å) of our structures which is located under the shorter C–I bonds in this discussion (Fig. 8). The carboxyl group is strongly twisted out of the double bond plane (82.7(4)° for OMe and −104.7(5)° for CO) compared to dimethyl fumarate, which is completely planar.15
O bond (1.186(4) Å, Fig. 5) and the shortest C–I (trans) bond (2.109(4) Å) of our structures which is located under the shorter C–I bonds in this discussion (Fig. 8). The carboxyl group is strongly twisted out of the double bond plane (82.7(4)° for OMe and −104.7(5)° for CO) compared to dimethyl fumarate, which is completely planar.15
 | ||
| Fig. 5 Statistical distribution of the CO atom distances of all diiodides discussed here, separated in cis/trans and literature compounds and ours. The trend goes to longer bonds compared to the literature value of 1.19 Å.22 The reference of Allen et al.21 (1.199 Å) is more appropriate. | ||
 | ||
| Fig. 6 Overview over the asymmetric units or molecular structures of our compounds 1–8 with the specific atom numbering. Symmetry codes: 1 [(a) (1 − x, 1 − y, 2 − z)]; 3 [(a) (1 − x, y, 1.5 − z)]; 8 [(a) (2 − x, −y, 1 − z)]. DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
The crystal structure is governed by the shortest I⋯O halogen bonds (3.005(3) Å) occurring in this work (Fig. 9) with an almost linear C–I⋯O angle (173.1(1)°) and a CO⋯I angle (117.4(2)°) that indicates an interaction between one of the lone pairs of the oxygen as halogen bond acceptor and the iodine as donor. This interaction is generated by both iodine atoms, and both carbonyl groups function as halogen bond acceptors, forcing the molecule to form four halogen bonds. Due to this, the molecules form layers parallel to the bc-plane that are stacked along the a-axis (Fig. 7).
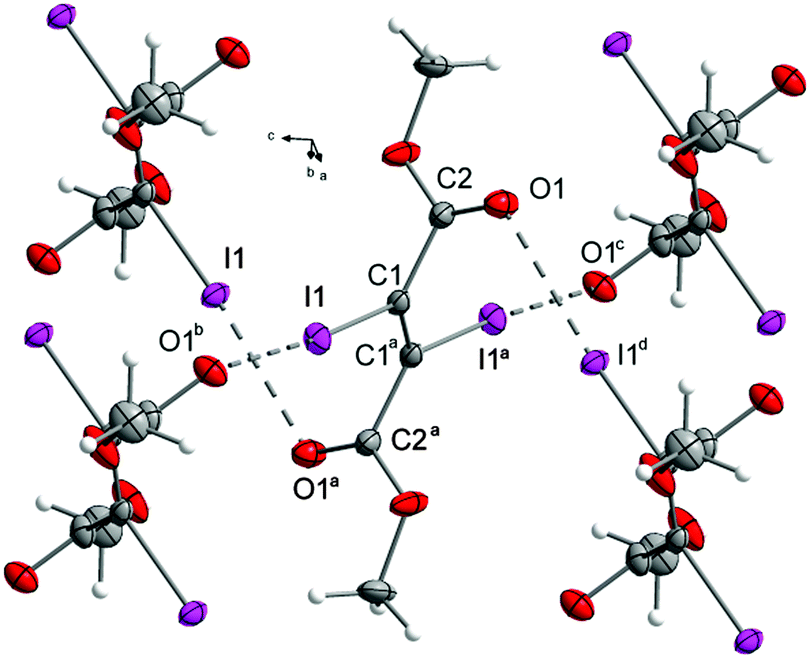 | ||
| Fig. 7 Network in the crystal structure of the diester 8. Symmetry codes: (a) (2 − x, −y, 1 − z), (b) (x, 0.5 − y, 0.5 + z), (c) (2 − x, −0.5 + y, 0.5 − z), (d) (x, 0.5 − y, −0.5 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
 | ||
| Fig. 8 Statistical distribution of the C–I atom distances of all diiodides discussed here, separated in cis/trans and literature compounds and ours. C2I218 was excluded because of the very short C–I distance (1.987(3) Å), F3CI19 and C2I420 are included in the literature trans category. cis Diido compounds, which are mostly cyclic, have shorter C–I atom distances than linear ones and those with trans configuration. They are even shorter than the literature value given for the CAr–I distance (2.095 Å).21 | ||
Looking at the smaller, oxygen containing molecules from the literature (RIDTOO,12 GIWTEM,16 GIWTIQ,16 NIMGOF17), where no large substituent or phenyl ring is attached to the double bond, some common features and also differences in the crystal structures compared to 8 can be detected.
RIDTOO12 is the only carboxylic acid containing molecule, that does not form I⋯O halogen bonds (the less occupied disordered part seems to form an I⋯O halogen bond but its arrangement fits perfectly in the network built up by the I⋯I interactions). The structure is dominated by hydrogen bonds that cause the molecules to form dimers. These dimers are arranged in a way that a complex network is formed under assistance of I⋯I halogen bonds that are the shortest here observed (3.801(1) Å) intermolecular I⋯I atom distances (Fig. 10). Additionally, the molecular structure is almost planar (10.2(3)°) which most probably indicates that the twisting of all other carboxyl groups is due to halogen bonding.
In GIWTIQ16 the hydrogen at the double bond is substituted by a methyl group. This steric increase of the substituent leads to a completely different crystal structure. The molecules form also dimers interconnected via hydrogen bonds. The carboxyl group is twisted out of the double bond plane by 81.2(9)° and participates in I⋯O halogen bonding (3.089(6) Å). The I⋯O halogen bond ranges among the shorter ones of the here detected distances (Fig. 9). Additionally, the molecules form also long I⋯I halogen bonds (3.938(9) Å, Fig. 10). All intermolecular interactions lead to the formation of a complex network.
 | ||
| Fig. 9 Statistical distribution of the intermolecular I⋯O halogen bond lengths of all diiodides discussed here, separated in cis/trans and literature compounds and ours. The maximum distance is the sum of the van der Waals radii of the involved atoms (3.50 Å). | ||
 | ||
| Fig. 10 Statistical distribution of the intermolecular I⋯I halogen bond lengths of all diiodides discussed here, separated in cis/trans and literature compounds and ours. The maximum distance is the sum of the van der Waals radii of the involved atoms (3.96 Å). | ||
The bulkiness of the CH2OMe group included in the molecules of GIWTEM16 instead of H or Me seems to be high enough to prevent the molecules from I⋯I halogen bond formation. The molecules form chains via hydrogen bonds parallel to the bc-plane. Again the carboxyl group is twisted out of the double bond plane by 80.3(9)° and the CO bond forms I⋯O halogen bonds (3.308(8) Å) which ranges amongst the longer here detected intermolecular I⋯O distances (Fig. 9). The steric increase of the CH2OMe substituent leads to an elongation of the I⋯O halogen bond compared to the value found in the crystal structure of GIWTOQ.16 The molecules form also chains interconnected via the I⋯O halogen bonds which results in the formation of layers.
If both substituents at the double bond of GIWTEM16 are replaced by CH2OH groups as it is the case for NIMGOF17 the molecules form hydrogen- and I⋯O halogen bonds (3.429(1) Å) as already described in the original literature.17 The molecules do not form dimers via hydrogen bonds and due to the high flexibility of the molecule there again is the possibility to form I⋯I halogen bonds (3.875(3) Å) which range like the I⋯O intermolecular distances amongst the longer ones (Fig. 10). All intermolecular interactions form a complex network.
Substitution of one CH2OH group by a phenyl ring leads to compound 4. As observed for the carboxyl groups in the previously discussed crystal structures, here the phenyl ring is twisted out of the double bond plane (−80.9(7)°). The hydroxyl groups make hydrogen bonds (Fig. 11) forming right twisted helical chains along the c-axis with four molecules forming a turn (Fig. 12).
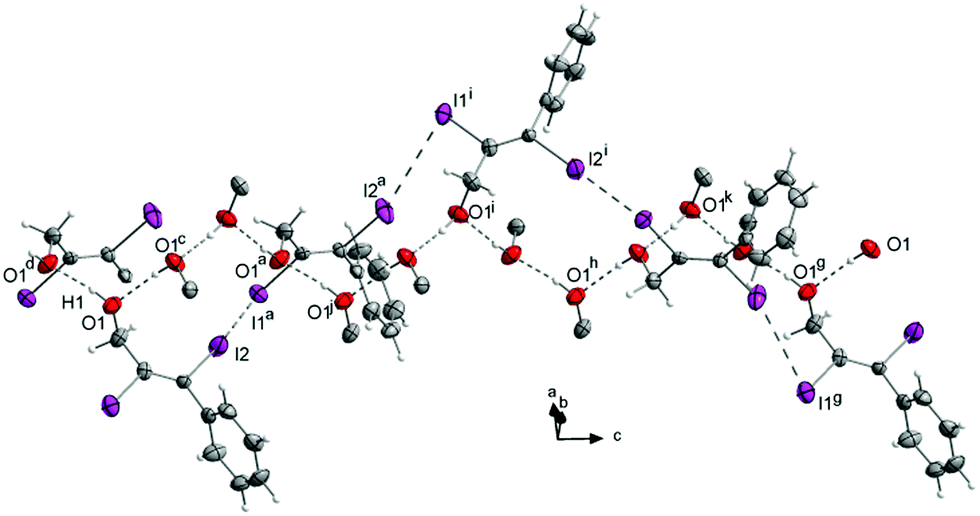 | ||
| Fig. 11 Crystal structure of 4. One turn of the helix formed by the I⋯I halogen bonds (I2 to I1g) includes three turns of the helix formed via hydrogen bonds (O1 to O1j; O1j to O1h; O1h to O1g) d(O–H) = 0.84(2) Å, d(O1⋯O1d) = 2.693(3) Å, ∠ O1–H1⋯O1d = 169.6(2)°. Some (I)CC(I)Ph residues are omitted for clarity. Symmetry codes: (a) (−0.25 + x, 1.25 − y, 0.75 + z), (c) (1.25 − x, 0.25 + y, 0.25 + z), (g) (x, y, 3 + z), (h) (x, y, 2 + z), (i) (1.5 − x, 1 − y, 1.5 + z), (j) (x, y, 1 + z), (k) (1.5 − x, 1 − y, 2.5 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
 | ||
| Fig. 12 Helix along the c-axis in the crystal structure of 4. All C–H hydrogen atoms are omitted for clarity. Symmetry codes: (a) (−0.25 + x, 1.25 − y, 0.75 + z), (b) (1.5 − x, 1 − y, −0.5 + z), (c) (1.25 − x, 0.25 + y, 0.25 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
The distance between the turns d(O1⋯O1f) is 7.532(3) Å. Starting from the asymmetric unit the molecules in the crystal also form a helix interconnected by I1⋯I2e halogen bonds (3.927(3) Å) with I2 as the halogen bond donor and I1 as the corresponding acceptor. The turns of this second helix are three times as large as the helix formed by the hydrogen bonds (d(O1⋯O1g) = 22.597(2) Å) (Fig. 12). The molecules within the helix form chains along the c-axis via I1⋯C7f contacts (3.563(1) Å).
In the crystal structure there are also I⋯O1a (3.420(3) Å) contacts (Fig. 13). The I1⋯I2e interaction is one of the longest, here observed I⋯I halogen bonds (3.927(3) Å, Fig. 10). The I⋯C(π) interaction is the atom distance concerning in the medium range (Fig. 14). Although the direction of the C–I bond and the orientation of the phenyl ring indicate almost no interaction. The I⋯O halogen bond ranges among the longest here detected intermolecular I⋯O interactions (Fig. 9). Consequently the main interactions that are responsible for this arrangement of the molecules in the crystal are the hydrogen bonds. However, despite the quite long I⋯I distances, I⋯I halogen bonds are also of importance in stabilizing the structure, as indicated by the corresponding bond angles (Tables in ESI†).
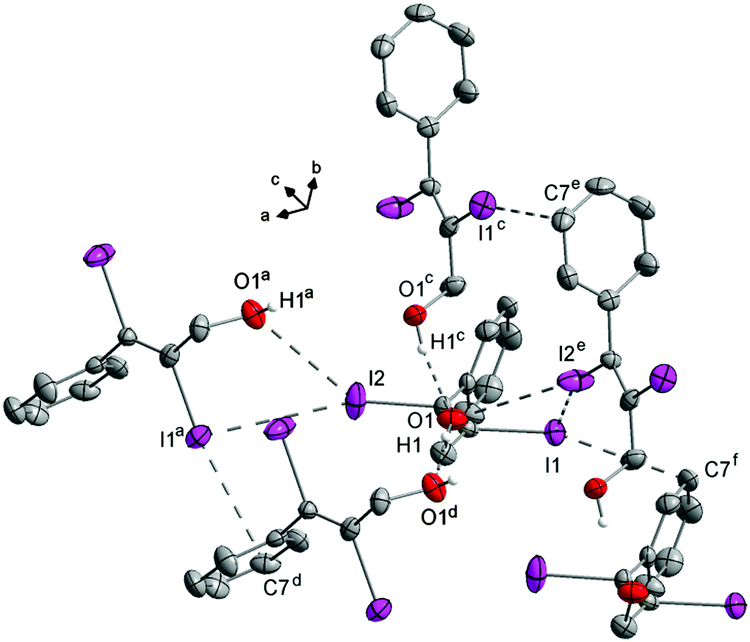 | ||
| Fig. 13 All intermolecular interactions emanating from the asymmetric unit. All C–H hydrogen atoms are omitted for clarity. Symmetry codes: (a) (−0.25 + x, 1.25 − y, 0.75 + z), (c) (1.25 − x, 0.25 + y, 0.25 + z), (d) (−0.25 + x, 1.25 − y, −0.25 + z), (e) (1.25 − x, 0.25 + y, −0.75 + z), (f) (x, y, −1 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
Replacing CH2OH by a methyl ester group as in the case of compound 5 the structural dominating interactions change completely. In the molecular structure of 5 the C1–I1 (2.118(1) Å) atom distance is a little elongated compared to the C2–I2 (2.109(1) Å) bond length, which compares well to the value of 8. The CO atom distance (1.197(1) Å) of 5 is the longest CO bond compared to our other structures (6, 8) but in the medium range of all structures (Fig. 5) and longer than the literature value.21 In this crystal structure the phenyl ring is also twisted out of the double bond plane (91.0(5)°), same observations can be made for the carbonyl group (51.0(6)°), but this angle is smaller than expected and observed for the above discussed structures.
The crystal structure of 5 is governed by I⋯O (3.143(1) Å) and I⋯C(π) (3.372(1) Å) halogen bonds (Fig. 15). The intermolecular I⋯O distance ranges among the shorter ones compared to all structures here and is the second shortest interaction of this kind observed for our substances (Fig. 9).
The intermolecular I2⋯C7(π) distance is the shortest of all substances in this comparison and highly directional (Fig. 14). The C2–I2 bond points directly towards the C7–C8 bond of the phenyl ring of the adjacent molecule. The two kinds of halogen bonds make the molecules form chains along the b-axis in the crystal structure of 5 (Fig. 15) and in fact there is no interaction between the chains other than van der Waals interactions.
 | ||
| Fig. 14 Statistical distribution of the intermolecular I⋯C(π) halogen bond lengths of all diiodides discussed here, separated in cis/trans and literature compounds and ours. The maximum distance is the sum of the van der Waals radii of the involved atoms (3.65 Å). | ||
 | ||
| Fig. 15 Chains along the b-axis in the crystal structure of the methylester 5. Symmetry codes: (a) (x, 1 + y, z), (b) (x, −1 + y, z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
Changing the ester from methyl to ethyl as in the case of 6 there again can be observed a different arrangement of the molecules in the crystal (Fig. 16).
 | ||
| Fig. 16 Chains along the b-axis in the crystal structure of the ethylester 6. Symmetry codes: (a) (x, −1 + y, z), (b) (x, 1 + y, z), (c) (1 − x, 2 − y, 1 − z), (d) (1 − x, 1 − y, 1 − z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
The C–I bonds of 6 are much more similar than those of the methylester 5. The C1–I1 bond (2.116(5) Å) that is part of I⋯C(π) interactions (3.530(3) Å) is the longer one. The phenyl substituent and the ester carbonyl group are twisted out of the double bond plane about 87.2(5)° and 104.0(5)°, respectively. The latter value is comparable with the value that can be found in the crystal structure of the diester 8. The I⋯C(π) halogen bond length lies in the medium range of all halogen bonds of this sort here detected (Fig. 14) and is responsible for the formation of chains that are dominating the crystal structure of 6. The steric increase caused by the insertion of the CH2 group pushes the molecules away from each other and the intermolecular interactions become weaker. This results also in longer I⋯O interactions (3.326(2) Å, 3.498(2) Å) compared to the values that can be detected in the crystal structure of 5. Compared to all values for the I⋯O interactions in this comparison, the data of 6 belong to the longer ones (Fig. 9).
Changing the substituent from an ester to an ethyl acetal, there are two different isomers. For the (Z) isomer (7a) the two C–I distances are clearly different; the longer C2–I2 distance (2.115(3) Å) is associated with the phenyl bonded carbon atom and I2 does not form any halogen bonds. I1 with the shorter C1–I1 distance (2.091(3) Å) forms I1⋯O2b halogen bonds (3.330(2) Å) that range between the longer I⋯O halogen bonds here observed (Fig. 9). This observation is unusual, because for halogen bonding a slight elongation of the C–I bond (typically 2.1 Å)22 would be expected.2,7 The phenyl ring is clearly twisted out of the double bond plane (65.2(4)°) but not that much like in the structures discussed before. The molecules form zig-zag chains along the b-axis interconnected via the I⋯O contacts. There are no further interactions between the chains, which are packed in the crystal to give an optimal space filling (Fig. 17). Here we have the border line between steric hindrance and the ability to form halogen bonds, where also is the need for a little geometrical flexibility. Same observations can also be made in the crystal structure of MIQKUU,23 the only acyclic cis compound from the literature. Here no halogen bonds can be observed and the molecule is quite rigid.
 | ||
| Fig. 17 Zigzag chains governed by short intermolecular I⋯O contacts in b-direction, in the crystal structure of compound 7a. Symmetry codes: (a) (0.5 − x, 0.5 + y, 1.5 − z), (b) (0.5 − x, −0.5 + y, 1.5 − z), (c) (x, −1 + y, z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
The arrangement of the (E) isomer 7b in the crystal is completely different compared to the (Z) isomer 7a. In contrast to the (Z) isomer 7a the C–I atom distances in the (E) isomer 7b are with 2.124(5) Å (C1–I1) and 2.131(5) Å (C2–I2) longer. The corresponding dihedral angle of the phenyl ring (73.4(6)°) is larger but despite its inclusion in I⋯C(π) halogen bonds (3.441(5) Å) the twisting angle is smaller than in the structures discussed before. This I2⋯C7(π) interaction is the second shortest observed in this comparison (Fig. 14) and the corresponding C2–I2 bond is elongated compared to the other one. These interactions result in the formation of chains along the c-axis (Fig. 18) as could also be observed in the crystal structure of 5. No interactions between iodine and oxygen are observed.
 | ||
| Fig. 18 Crystal structure of compound 7b. View of the chains along the c-axis resulting from intermolecular I⋯C(π) interactions. Symmetry codes: (a) (x, 0.5 − y, −0.5 + z), (b) (x, 0.5 − y, 0.5 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
Keeping the phenyl substituent as a constant and changing the oxygen containing substituent against a H leads us to compound 3. In this crystal structure the C–I bond lengths are different and the phenyl ring is twisted out of the double bond plane (80.9(2)°). Here also I⋯C(π) halogen bonds (3.573(1) Å) are the dominant intermolecular interactions which ranges under the longer ones in this discussion (Fig. 14). The molecules of 3 form chains along the a-axis head to tail connected via the I⋯C(π) halogen bonds (Fig. 19). Here we have an example that shows, that a decrease in steric hindrance leads to a maximum of intermolecular interaction.
 | ||
| Fig. 19 Crystal structure of compound 3. View of the chains along the a-axis resulting from intermolecular I⋯C(π) interactions. Symmetry codes: (a) (1 − x, y, 1.5 − z), (b) (0.5 − x, 0.5 − y, 2 − z), (c) (0.5 + x, 0.5 − y, −0.5 + z), (d) (1.5 − x, 0.5 − y, 1 − z), (e) (1 + x, y, −1 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
The largest change in crystal packing is caused by the substitution of the H to methyl. The asymmetric unit of compound 2 contains three independent molecules. Molecule 1 (contains I1) forms I⋯C(π) halogen bonds (3.543(5) Å) whose lengths lie in the medium range of all here detected I⋯C(π) interactions (Fig. 14). The phenyl ring of M1 is twisted out of the double bond plane about −79.0(1)°. These molecules form chains along the b-axis via the I⋯C(π) interactions (Fig. 20).
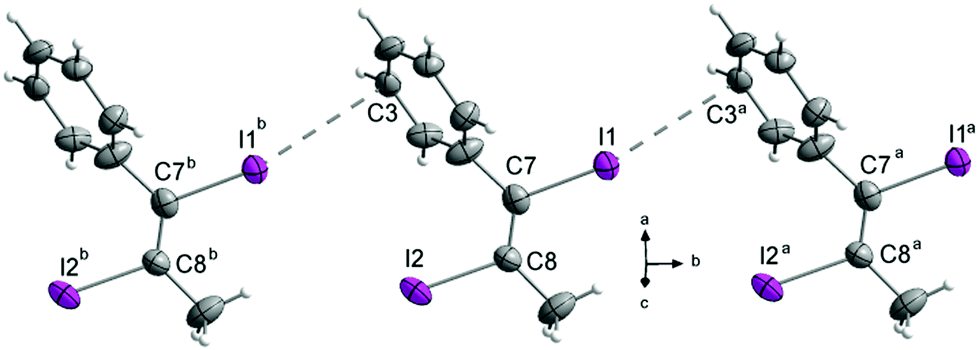 | ||
| Fig. 20 I⋯C(π) Halogen bonds in the crystal structure of 4 forming chains in b-direction. Symmetry codes: (a) (x, 1 + y, z), (b) (x, −1 + y, z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
In molecules 2 (containing I3) and 3 (containing I5) the phenyl rings are also twisted out of the double bond plane (−86.6(8)° for M2 and 72.1(8)° for M3). Each molecule forms a helical chain that can be converted into each other by symmetry. In both helices the alternating molecules M1 and M2 are interconnected via I⋯I halogen bonds (I3⋯I6a 3.884(6) Å, I4⋯I5c 3.885(5) Å, Fig. 21) that range in the middle of all here detected I⋯I interactions (Fig. 10). The helices itself are not connected with each other. Looking at the angles of the halogen bonds, there are two different tendencies. Two of the four angles are near to linear (I3⋯I6–C25 159.0(2)°, I5⋯I4–C16 173.2(2)°) and the other two are near to rectangular (I6⋯I3–C17 91.5(2)°, I4⋯I5–C26 97.7(2)°). These values indicate that I4 and I6 act as halogen bond donors and I3 and I5 act as halogen bond acceptors. Remarkable is that the corresponding C–I bonds of the donor atoms (I4/I6) are shorter compared to the C–I bond lengths of the acceptor atoms (I3/I5) because the opposite was expected, considering the XB definition of the IUPAC2. The helices are arranged parallel to the cell edges of the unit cell and the chains interconnected via I⋯C(π) interactions are arranged between two helix strands (Fig. 22). The independent occurrence of I⋯C and I⋯I halogen bonds in the same crystal structure lets us conclude, that in this case the energy of both interactions is very similar. This left twisted helical chain arrangement is very similar to the crystal structure of 4.
 | ||
| Fig. 21 Helical double strand chains along the b-axis in the crystal structure of 2, formed by I⋯I halogen bonds. Symmetry codes: (a) (x, 1 + y, z), (b) (x, −1 + y, z), (c) (−x, −0.5 + y, 0.5 − z), (d) (−x, 0.5 + y, 0.5 − z), (e) (1 + x, y, −1 + z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
 | ||
| Fig. 22 The helices form quad shaped subunits which form strands along the c-axis in the crystal structure of 2. The molecules that form I⋯C(π) halogen bonds are between the strands of quad shaped units. DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
Changing methyl to phenyl, as it is the case for compound 1 again I⋯C(π) halogen bonds (3.543(3) Å) are the dominant interaction. This intermolecular distance is in the medium range of all interactions of this type here observed (Fig. 14). Also the overlap of the C1–I1 line with the adjacent phenyl substituent is warranted. The latter again is twisted out of the double bond plane (−80.5(8)°) as observed for the phenyl substituents of all other compounds discussed here. Via this intermolecular interaction the molecules form chains (Fig. 23).
 | ||
| Fig. 23 I⋯C(π) Halogen bonds in the crystal structure of 1 forming chains. Symmetry codes: (a) (1 − x, 1 − y, 2 − z), (b) (−x, 1 − y, 1 − z), (c) (−1 + x, y, −1 + z), (d) (1 + x, y, 1 + z), (e) (2 − x, 1 − y, 3 − z). DIAMOND representation, thermal ellipsoids are drawn at 50% probability level. | ||
In the crystal structure of EMUZIW24 where one phenyl substituent is replaced by a bicyclic lactone there are two molecules in the asymmetric unit. Both substituents of both molecules are twisted largely out of the double bond plane although no I⋯C(π) halogen bonds occur in this crystal structure due to the steric hindrance of the large substituents. One molecule of the asymmetric unit forms I⋯I halogen bonds (3.835(4) Å) and chains interconnected via those. The other molecule forms also chains but those are interconnected by I⋯O halogen bonds (3.347(5) Å).
In the crystal structure of ULEFUN25 the molecule contains two terminal diiodo olefinic moieties and is generally quite flexible. The molecules are arranged in a way that leads to maximum halogen bond formation, four out of four iodine atoms form halogen bonds, including I⋯I, I⋯O and I⋯C(π) interactions.
In the molecular structure of RUWRUX26 there are two diiodo olefinic moieties nearby and twisted in a way that makes conjugation of the double bond unlikely. The molecule itself is quite flexible and is twisted in a way that a maximum halogen bond formation is possible. The asymmetric unit persists of two molecules with all in all eight iodine atoms. Seven of them form halogen bonds.
Discussion and conclusion
Looking at the similarities that can be found in the crystal structures of the compounds discussed above, a competition between the formation of hydrogen bonds and the formation of halogen bonds can be observed when both is possible. Also a competition between the different halogen bonding acceptors is present. All acids and alcohols form hydrogen bonds which always seem to be the dominant interaction and all others subordinate themselves. When halogen bonds occur in the crystal structure then the participating halogen bond acceptor like CO2R or Phenyl is twisted out of the double bond plane to ensure optimal contact conditions. This happens in a range that is clearly larger than single steric effects would induce. The reverse case is not always warranted. This observation lets us conclude that the energy of the halogen bond is higher than the energy the molecule wins via the conjugation.Under exclusion of the very small molecule RIDTOO12 where mostly hydrogen bonds and I⋯I halogen bonds are relevant for the crystal structure formation, a general statement concerning the I⋯O halogen bond formation can be made. Is an oxygen containing functional group included in the molecule that could act as halogen bond acceptor, then it functions as such. In the ester or acid groups occurring here, the included carbonyl group always forms the halogen bond. Those CO functions seems to be the stronger halogen bond acceptor than the OH or OR groups that are also present. Exceptions are small molecules like RIDTOO12 where stronger interactions like hydrogen bonds are possible. Also exceptions are rigid molecules with stericly demanding substituents in the cis configuration like 7a and MIQKUU,23 where very weak or even no halogen bonds under participation of the oxygen occur.
As one can clearly see for the crystal structures that are built up by the molecules arranged in chains (7b, 1, 2, 3, 5) mostly I⋯C(π) interactions are responsible for the chains. In the crystal structure of 5 the chains are built up by I⋯O interactions supported by the I⋯C(π) halogen bonds, resulting in the strongest I⋯C(π) and the second strongest I⋯O interactions.
When there are only iodine and a π-system present in a molecule to act as halogen bond acceptor as is the case for 1, 2, 3, C2I2 and C2I4 then, except for C2I4 and parts of the crystal structure of 2, the heteroatomic I⋯C(π) interaction is always preferred in the crystal structure instead of the homoatomic I⋯I halogen bond. In this study no iodine interactions with olefinic double bonds could be observed. The main interactions occur between an iodine atom and the π-system of a phenyl ring. In the crystal structure of C2I2 the halogen bond acceptor is the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond. In the crystal structure of RETRIR27 where a phenyl ring and a triple bond are included in the same molecule, the triple bond is the favored halogen bond acceptor.
C triple bond. In the crystal structure of RETRIR27 where a phenyl ring and a triple bond are included in the same molecule, the triple bond is the favored halogen bond acceptor.
For the I⋯I halogen bond formation it is noticeable that they occur either in crystal structures consisting of smaller molecules like RIDTOO,12 GIWTIQ,16 NIMGOF,172 and 4. The steric demand of the substituents of those molecules is either small or the substituents cannot act as halogen bond acceptor or both. That gives the iodine atoms the possibility to converge and interact. Or they occur in the crystal structures of larger flexible molecules with higher iodine content. There the molecules are able to twist themselves in a way of maximum halogen bond formation where also I⋯I halogen bonds occur. Stating which intermolecular interaction is dominant in such molecules is hardly possible.
With this background the similarity (despite the opposite twisting: 2 left, 4 right) of the structural arrangement in the crystal structures of compounds 2 and 4 is remarkable, considering the diversity of interactions (O⋯H, I⋯O, I⋯I, I⋯C(π)) occurring in the crystal structure of 4. In the relevant part of the crystal structure of 2 there are solely I⋯I halogen bonds. Nevertheless both crystal structures are dominated by helical chains with four molecules in a turn but the turns in the crystal structure of 4 are smaller because there the molecules are connected by hydrogen bonds. The corresponding helix built up by the I⋯I halogen bonds is three times as large. In the crystal structure of 2 the asymmetric unit contains two independent molecules that form helices that are symmetry generated and also twisted into each other without connection. For the steric demand methyl and CH2OH seems to be equivalent in this case.
Discussing the question, whether the twisting of the phenyl ring at the CC double bond is correlated due to the formation of halogen bonding to iodine the two isomers KAFBOJ28 and KAFBID28 can be considered (Fig. 24). In the case of KAFBOJ28 clear halogen bonding of iodine to the π-system can be observed in the crystal. On the other hand, in the case of KAFBID28 only hydrogen bonding and no halogen bonding is found. Looking at the torsion angles, regarding the phenyl substituents the twisting of the phenyl ring of KAFBOJ is with 74.4(7)° much larger than the twisting in KAFBID (37.6(6)°). The larger twisting in the case of KAFBOJ is most probably caused by the effect of halogen bonding, which fits well to the observations made in the related diiodoolefins 1–7.
 | ||
| Fig. 24 Structures of KAFBOJ and KAFBID from the literature. | ||
Experimental section
Materials and methods
All chemicals were commercially available and were used as received. NMR spectra were recorded with a JEOL EX 400 Eclipse instrument operating at 400.128 MHz (1H) and 100.626 MHz (13C). Chemical shifts are referred to Me4Si (1H, 13C) as external standards. All spectra were measured, if not mentioned otherwise, at 25 °C. The assignment of the signals in the 1H and 13C NMR spectra is based on 2D (1H,1H-COSY45, 1H,13C-HMQC and 1H,13C-HMBC) experiments. Mass spectrometric data were obtained with a JEOL Mstation JMS 700 spectrometer using the direct EI mode. The molecular structures in the crystalline state were determined by single crystal X-ray diffraction. For data collection an Xcalibur3 diffractometer equipped with a Spellman generator (voltage 50 kV, current 40 mA) and a Kappa CCD detector with an X-ray radiation wavelength of 0.71073 Å was used. The data collection was performed with the CrysAlis CCD software29 and the data reduction with the CrysAlis RED software.30 The structures were solved with SIR-92 or with SIR-2004, refined with SHELXL-97 and finally checked using PLATON.31 The absorptions were corrected by SCALE3 ABSPACK multiscan method.32 All relevant data and parameters of the X-ray measurements and refinements are given in Table 1. CCDC 1037179 (1), 1037183 (2), 1037178 (3), 1037181 (4), 1037184 (5), 1037182 (6), 988932 (7a), 988931 (7b) and 1037180 (8).| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7a | 7b | 8 |
|---|---|---|---|---|---|---|---|---|---|
| Formula | C14H10I2 | C9H8I2 | C8H6I2 | C9H8I2O | C10H8I2O2 | C11H10I2O2 | C13H16I2O2 | C13H16I2O2 | C6H6I2O4 |
| M | 432.02 | 369.95 | 355.93 | 385.95 | 413.96 | 427.99 | 458.06 | 458.06 | 395.91 |
| T, [K] | 173(2) | 200(2) | 100(2) | 173(2) | 100(2) | 173(2) | 100(2) | 100(2) | 100(2) |
| Color, habit | Colorless plate | Colorless block | Colorless block | Colorless plate | Colorless block | Colorless block | Colorless plate | Colorless plate | Colorless block |
| Cryst. size, [mm] | 0.40 × 0.25 × 0.08 | 0.30 × 0.15 × 0.10 | 0.22 × 0.12 × 0.05 | 0.35 × 0.10 × 0.05 | 0.4 × 0.15 × 0.05 | 0.20 × 0.15 × 0.10 | 0.417 × 0.176 × 0.053 | 0.323 × 0.181 × 0.021 | 0.20 × 0.20 × 0.10 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Tetragonal | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c | C2/c | I41/a | C2/c | P21/n | P21/n | P21/c | P21/c |
| a, [Å] | 5.7078(3) | 18.9368(9) | 10.1365(4) | 23.9673(5) | 20.5998(15) | 12.8366(5) | 11.3978(4) | 10.7818(7) | 8.6795(4) |
| b, [Å] | 17.4128(8) | 7.6070(3) | 15.9425(6) | 23.9673(5) | 7.1799(2) | 7.0467(2) | 7.2145(2) | 10.2566(6) | 7.5937(2) |
| c, [Å] | 7.0077(4) | 22.9348(12) | 5.7295(2) | 7.5322(3) | 18.253(2) | 14.2790(6) | 18.1044(6) | 14.5072(9) | 8.5520(4) |
| α, [°] | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β, [°] | 110.984(6) | 109.349(6) | 94.279(4) | 90 | 120.835(11) | 96.053(4) | 98.253(3) | 109.917(7) | 108.798(5) |
| γ, [°] | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| V, [Å3] | 650.30(6) | 3117.2(3) | 923.31(6) | 4326.7(2) | 2318.1(4) | 1284.41(9) | 1473.30(8) | 1508.32(16) | 533.59(4) |
| Z | 2 | 12 | 4 | 16 | 8 | 4 | 4 | 4 | 2 |
| ρ calcd, [g cm−3] | 2.206 | 2.365 | 2.560 | 2.370 | 2.372 | 2.213 | 2.065 | 2.017 | 2.464 |
| μ, [mm−1] | 4.806 | 5.993 | 6.739 | 5.769 | 5.399 | 4.876 | 4.258 | 4.159 | 5.871 |
| Irradiation, [Å] | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 | 0.71069 |
| F(000) | 400 | 2016 | 640 | 2816 | 1520 | 792 | 864 | 864 | 360 |
| Index ranges | −7 ≤ h ≤ 7 | −23 ≤ h ≤ 22 | −14 ≤ h ≤ 14 | −28 ≤ h ≤ 29 | −28 ≤ h ≤ 28 | −17 ≤ h ≤ 18 | −15 ≤ h ≤ 15 | −14 ≤ h ≤ 14 | −10 ≤ h ≤ 10 |
| −21 ≤ k ≤ 21 | −9 ≤ k ≤ 9 | −22 ≤ k ≤ 22 | −20 ≤ k ≤ 29 | −5 ≤ k ≤ 5 | −9 ≤ k ≤ 9 | −9 ≤ k ≤ 9 | −13 ≤ k ≤ 13 | −9 ≤ k ≤ 9 | |
| −8 ≤ l ≤ 8 | −19 ≤ l ≤ 28 | −8 ≤ l ≤ 8 | −7 ≤ l ≤ 9 | −31 ≤ l ≤ 31 | −20 ≤ l ≤ 20 | −24 ≤ l ≤ 14 | −19 ≤ l ≤ 19 | −10 ≤ l ≤ 10 | |
| Reflns collected | 8934 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 609 609 |
9343 | 10866 |
13308 |
16765 |
24970 |
13331 |
5167 |
| Reflns unique | 1266 | 6098 | 1361 | 2116 | 2876 | 3760 | 3647 | 3719 | 1052 |
| Reflns obsd | 1099 | 4352 | 1126 | 1501 | 2264 | 2747 | 3215 | 2919 | 923 |
| R int | 0.0303 | 0.0363 | 0.0311 | 0.0326 | 0.0596 | 0.0364 | 0.042 | 0.049 | 0.0298 |
| Params refined | 67 | 319 | 75 | 161 | 133 | 156 | 166 | 166 | 60 |
| θ range, [°] | 4.14–25.98 | 4.17–26.0 | 4.17–30.09 | 4.32–33.58 | 4.18–32.50 | 4.26–32.35 | 4.2–28.3 | 4.3–28.3 | 4.77–32.46 |
| R 1, wR2 [I > 2σ(I)] | 0.0162, 0.0368 | 0.0282, 0.0525 | 0.0176, 0.0404 | 0.0225, 0.0412 | 0.0250, 0.0531 | 0.0232, 0.0424 | 0.0231, 0.0566 | 0.0307, 0.0656 | 0.0174, 0.0420 |
|
R
1, wR2(alldata) |
0.0210, 0.0374 | 0.0457, 0.0552 | 0.0245, 0.0415 | 0.0386, 0.0432 | 0.0367, 0.0547 | 0.0416, 0.0449 | 0.0294, 0.0531 | 0.0472, 0.0593 | 0.0211, 0.0427 |
| GooF | 1.058 | 0.857 | 1.027 | 0.890 | 0.940 | 0.917 | 1.098 | 1.029 | 0.969 |
| δpmax, δpmin, [e nm−3] | −0.462, 0.339 | −0.822, 1.536 | −0.626, 0.592 | −0.443, 0.304 | −0.758, 1.395 | −0.957, 0.878 | −0.68, 1.06 | −0.682, 1.285 | −0.499, 0.671 |
Syntheses
7a: δH (270.17 MHz, CDCl3) 7.38–7.26 (5H, m, HAr), 3.92 (1H, s, CH), 3.42, 3.36 (4H, AB-system, 2JHH = 9.4 Hz, CH2), 1.17 (6H, t, 3JHH = 7.1 Hz, CH3).
δ
C (67.93 MHz, CDCl3) 143.8 (Ci), 128.9 (Cp), 128.5 (Cm), 127.8 (Co), 122.4 (CI), 115.7 (Ph–CI), 99.1 (CH), 62.6 (CH2), 15.1 (CH3).
7b: δH (270.17 MHz, CDCl3) 7.38–7.19 (5H, m, HAr), 8.83 (1H, s, CH), 3.77, 3.66 (4H, AB-system, 2JHH = 9.5 Hz, CH2), 1.33 (6H, t, 3JHH = 7.1 Hz, CH3).
δ
C (67.93 MHz, CDCl3) 147.2 (Ci), 128.6 (Cm), 128.5 (Cp), 128.3 (Co), 107.2 (CH), 106.5 (CI), 96.8 (Ph–CI), 63.0 (CH2), 15.3 (CH3).
δ H (270.17 MHz, CDCl3) 7.49–7.28 (5H, m, HAr), 3.93 (3H, s, CH3).
δ
C (67.93 MHz, CDCl3) 166.9 (CO), 145.1 (Ci), 129.3 (CAr), 128.7 (CAr), 128.0 (CAr), 98.6 (C–I), 85.7 (C–I), 53.7 (CH3).
δ C (270.17 MHz, CDCl3) 7.62–7.15 (5H, m, HAr), 4.40 (2H, q, J = 7.2 Hz, CH2), 1.42 (3H, t, J = 7.2 Hz, CH3).
δ
C (67.93 MHz, CDCl3) 166.5 (CO), 145.1 (Ci), 129.1 (CAr), 128.3 (CAr), 128.0 (CAr), 97.9 (C–I), 86.2 (C–I), 63.0 (CH2), 14.0 (CH3).
Acknowledgements
Financial support from the Department of Chemistry, Ludwig-Maximilian University of Munich is gratefully acknowledged. The authors are thankful to Prof. T. M. Klapötke for the generous allocation of diffractometer time and his continuous support.Notes and references
- (a) M. Colin, Ann. Chim., 1814, 91, 252 Search PubMed; (b) F. Guthrie, J. Chem. Soc., 1863, 16, 239 RSC.
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7 CrossRef CAS PubMed.
- (a) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed; (b) A.-L. Barres, M. Allain, P. Frère and P. Batail, Isr. J. Chem., 2014, 54(5–6), 689–698 CrossRef CAS PubMed; (c) X. Ding, M. Tuikka and M. Haukka, Recent Adv. Crystallogr., 2012, 143–168 Search PubMed; (d) P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12(12), 5835–5838 CrossRef CAS; (e) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC; (f) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC; (g) P. Metrangolo, T. Pilati, G. Terraneo, S. Biella and G. Resnati, CrystEngComm, 2009, 11, 1187–1196 RSC; (h) K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC; (i) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed; (j) D. M. P Mingos, P. Metrangolo and G. Resnati, Structure and Bonding-Halogen Bonding Fundamentals and Applications, Springer-Verlag, Berlin Heidelberg, 2008 DOI:10.1007/978-3-540-74330-9; (k) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed; (l) M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed; (m) P. Metrangolo, T. Pilati, G. Resnati and A. Stevenazzi, Curr. Opin. Colloid Interface Sci., 2003, 8, 215–222 CrossRef CAS; (n) P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7(12), 2511–2519 CrossRef CAS.
- (a) F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC; (b) R. W. Troff, T. Mäkelä, F. Topic, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617–1637 CrossRef CAS PubMed; (c) T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; (d) S. H. Jungbauer, Synlett, 2013, 2624–2628 CAS; (e) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46(11), 2686–2695 CrossRef CAS PubMed; (f) R. Bertania, P. Sgarbossa, A. Venzo, F. Lelj, M. Amatic, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef PubMed; (g) M. Formigué, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36–45 CrossRef PubMed; (h) P. Metrangolo, G. Resnati, T. Pilati, R. Liantonio and F. Meyer, J. Polym. Sci., Polym. Chem. Ed., 2007, 45, 1–15 CrossRef CAS PubMed.
- (a) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC; (b) P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed; (c) O. I. Titov, D. A. Shulga, V. A. Palyulin and N. S. Zefirov, Dokl. Chem., 2013, 450(1), 139–143 CrossRef CAS; (d) J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63(12), 1598–1607 CrossRef CAS; (e) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC; (f) A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC; (g) P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13(2), 305–311 CrossRef CAS PubMed; (h) A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- (a) M. O. Zimmermann, A. Lange, R. Wilcken, M. B. Cieslik, T. E. Exner, A. C. Joerger, P. Koch and F. M. Boeckler, Future Med. Chem., 2014, 6(6), 617–639 CrossRef CAS PubMed; (b) M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed; (c) R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed; (d) P. Metrangolo and G. Resnati, Nat. Chem., 2012, 4(6), 437–438 CrossRef CAS PubMed; (e) Y. Lu, Y. Liu, Z. Xu, H. Li, H. Liu and W. Zhu, Expert Opin. Drug Discovery, 2012, 7(5), 375–383 CrossRef CAS PubMed; (f) S. K. Grant and E. A. Lunney, Chem. Biol., 2011, 18(1), 3–4 CrossRef CAS PubMed; (g) E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC; (h) P. Metrangolo and G. Resnati, Science, 2008, 321, 918–919 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. A. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85(8), 1711–1713 CrossRef CAS.
- D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13(9), 3224–3231 RSC.
- R. B. Walsh, C. W. Padgett, P. Metrangolo, G. Resnati, T. W. Hanks and W. T. Pennington, Cryst. Growth Des., 2001, 1, 165–175 CAS.
- (a) L. Su, C.-Y. Lei, W.-Y. Fan and L.-X. Liu, Synth. Commun., 2011, 41, 1200–1207 CrossRef CAS PubMed; (b) R. A. Hollins and M. P. A. Campos, J. Org. Chem., 1979, 44, 3931–3934 CrossRef CAS; (c) V. L. Heasley, D. F. Shellhamer, L. E. Heasley, D. B. Yaeger and G. E. Heasley, J. Org. Chem., 1980, 45, 4649–4652 CrossRef CAS.
- J. Duan, W. R. Dolbier and Q.-Y. Chen, J. Org. Chem., 1998, 63, 9486–9489 CrossRef CAS.
- J. P. Donahue, Synth. Commun., 2006, 36, 3461–3471 CrossRef CAS PubMed.
- D. Fischer, H. Tomeba, N. K. Pahadi, N. T. Patil and Y. Yamamoto, Angew. Chem., Int. Ed., 2007, 46, 4764–4766 CrossRef CAS PubMed.
- H. P. Klug, Z. Kristallogr., 1935, 90, 495–507 CAS.
- H. Kooijman, J. W. Sprengers, M. J. Agerbeek, C. J. Elsevier and A. L. Spek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o917–o918 CAS.
- S. Langle, S. I. Ngi, E. Anselmi, M. Abarbri, J. Thibonnet and A. Duchene, Synthesis, 2007, 1724–1728 CAS.
- M. S. Loiten, B. Dalhus, B. Flaertoft and J. Klaveness, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 555–557 Search PubMed.
- J. D. Dunitz, H. Gehrer and D. Britton, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1989–1994 CrossRef CAS.
- S. J. Clarke, J. K. Cockcroft and A. N. Fitch, Z. Kristallogr., 1993, 206, 87–95 CrossRef CAS.
- R. D. Bailey, L. L. Hook, R. P. Watson, T. W. Hanks and W. T. Pennington, Cryst. Eng., 2000, 3, 155–171 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- A. F. Holleman and N. Wiberg, Lehrbuch der anorganischen Chemie, de Gruyter & Co, Berlin, 1995, Anhang, IV Search PubMed.
- R. Singha, S. Dhara and J. K. Ray, RSC Adv., 2013, 3, 23989–23992 RSC.
- G. Raffa, S. Belot, G. Balme and N. Monteiro, Org. Biomol. Chem., 2011, 9, 1474–1478 CAS.
- K. Shah, M. R. Shaha and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o568 CAS.
- F. Piron, N. Vanthuyne, B. Joulin, J.-V. Naubron, C. Cismas, A. Terec, R. A. Varga, C. Roussel, J. Roncali and I. Grosu, J. Org. Chem., 2009, 74, 9062–9070 CrossRef CAS PubMed.
- J. Barluenga, I. Llorente, L. J. Alvarez-Garcia, J. M. Gonzalez, P. J. Campos, M. R. Diaz and S. Garcia-Granda, J. Am. Chem. Soc., 1997, 119, 6933–6934 CrossRef CAS.
- W. Xu and Q.-Y. Chen, J. Org. Chem., 2002, 67, 9421–9427 CrossRef CAS PubMed.
- CrysAlis CCD, version 1.171.27p5 beta (release 01–04–2005 CrysAlis171.NET; compiled Apr 1 2005,17:53:34), Oxford Diffraction Ltd., Oxfordshire, UK Search PubMed.
- CrysAlis RED, version 1.171.27p5 beta (release 01–04–2005 CrysAlis171.NET; compiled Apr 1 2005, 17:53:34), Oxford Diffraction Ltd., Oxfordshire, UK Search PubMed.
- (a) SIR-92, A Program for Crystal Structure Solution: A. Altomare, G. L. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef; (b) A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS; (c) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS; (d) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (e) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1999 Search PubMed; (f) L. A. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1999 Search PubMed.
- SCALE3 ABSPACK – An Oxford Diffraction program (1.0.4, gui:1.0.3), Oxford Diffraction Ltd., Oxfordshire, UK., 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing additional analytical data, figures of the disorder and data for the crystal structures. CCDC 988931, 988932 and 1037178–1037184. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj00821b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |