
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraction and complexation of alkali and alkaline earth metal cations by lower-rim calix[4]arene diethylene glycol amide derivatives†
Igor
Sviben
ab,
Nives
Galić
a,
Vladislav
Tomišić
*a and
Leo
Frkanec
*b
aDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: vtomisic@chem.pmf.hr; Fax: +385 1 46 06 131; Tel: +385 1 46 06 136
bLaboratory of Supramolecular and Nucleoside Chemistry, Department of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, Bijenička 54, 10000 Zagreb, Croatia. E-mail: frkanec@irb.hr; Fax: +385 1 46 80 195; Tel: +385 1 46 80 217
First published on 26th May 2015
Abstract
Novel calix[4]arene derivatives, 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetrakis(N-(2-(2-methoxyethoxy)ethyl)carbamoyl methoxy)calix[4]arene (1) and 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetra(N,N-bis(2-(2-methoxyethoxy)ethyl)carbamoyl methoxy)calix[4]arene (2), were prepared by introducing diethylene glycol subunits at the lower calixarene rim. The complexation affinities of these compounds towards alkali and alkaline earth metal cations were studied at 25 °C in acetonitrile and methanol by means of spectrophotometric, conductometric, and potentiometric titrations. The stability constants of the corresponding complexes with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (cation:ligand) stoichiometry were determined (in some cases only estimated), and their values obtained by different methods were in good agreement. The complexes with secondary-amide derivative (1) were observed to have much lower stabilities than those with tertiary-amide derivative (2). This was presumably mostly caused by the presence of intramolecular NH⋯O
:1 (cation:ligand) stoichiometry were determined (in some cases only estimated), and their values obtained by different methods were in good agreement. The complexes with secondary-amide derivative (1) were observed to have much lower stabilities than those with tertiary-amide derivative (2). This was presumably mostly caused by the presence of intramolecular NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds in the former case. It was found that solvent significantly affected the stability of the complexes; the prepared calixarenes showed considerably higher affinities for cations in acetonitrile than in methanol. Generally, the ligands studied showed better binding abilities for alkaline earth cations than for alkali metal cations. The extraction of metal picrates from water to dichloromethane by the complexation of metal ions with ligands 1 and 2 was also investigated. In accordance with the complex stabilities, all cations were extracted fairly well (in some cases even excellently) with the tertiary-amide derivative, whereas this was not the case in the extraction experiments with the other ligand studied.
C hydrogen bonds in the former case. It was found that solvent significantly affected the stability of the complexes; the prepared calixarenes showed considerably higher affinities for cations in acetonitrile than in methanol. Generally, the ligands studied showed better binding abilities for alkaline earth cations than for alkali metal cations. The extraction of metal picrates from water to dichloromethane by the complexation of metal ions with ligands 1 and 2 was also investigated. In accordance with the complex stabilities, all cations were extracted fairly well (in some cases even excellently) with the tertiary-amide derivative, whereas this was not the case in the extraction experiments with the other ligand studied.
Introduction
The complexation properties of calixarene derivatives have been extensively studied due to their ability to selectively bind a wide variety of guest species.1–3 Numerous macrocyclic receptors have been prepared by the functionalization of calixarenes at the lower and/or upper rim. By choosing an adequate number of phenolic units and appropriate substituents, it has been possible to prepare selective and efficient calixarene hosts for cations,4,5 anions,4,6,7 and neutral species.4,8–10 The p-tert-butylcalix[4]arene derivatives containing carbonyl groups at the lower rim (ketones, esters, oramides) were reported to possess excellent properties for binding alkali, alkaline earth, and transition metal cations.5,11 Secondary-11–13 and tertiary-amide14–16 calixarene derivatives were shown to have particularly high affinities towards alkali and alkaline earth metal cations. The formation of intramolecular NH⋯OC hydrogen bonds in tetrasubstituted secondary-amide derivatives was proven to have a strong influence on the binding properties of such ligands.12,13,17–24 In addition to the abovementioned calixarene size and the nature of the binding groups, the affinity of such calixarenes towards metal cations depends strongly on the reaction medium, i.e. on the solvent used.11–14,17,25–31
Several ethylene glycol-based calixarene derivatives have been reported previously.1,32–39 The first oxyalkylated p-tert-butylphenol-formaldehyde oligomers (both linear and cyclic) were developed as demulsifiers for the petroleum industry.1 Calixarene crown ethers with ethylene glycol substituents at the lower rim were shown to selectively bind sodium, potassium, and cesium cations.32,33 Another interesting group of compounds are bis-calixarenes, also known as calix-tubes, which possess two calixarene skeletons bridged by ethylene glycol or various polyethylene glycol (PGE) chains.34 Calixarene derivatives with a large number of attached (poly)ethylene glycol groups have increased hydrophilic character, and have found application as agents for the extraction of metal cations from aqueous to organic phases.35–37 In order to optimize the structure of calix[4]arene derivatives for metal-ion biphasic extraction and phase-transition catalysis, Shinkai et al.35 prepared several amphiphilic calix[4]arene derivatives by introducing hydrophobic groups at the upper calixarene rim and oligo(ethylene glycol) chains at the lower rim. In the course of the extraction process, the latter hydrophilic groups penetrate to the aqueous layer, whereas the hydrophobic calixarene basket remains in a non-polar organic layer. In this way, such compounds can efficiently bind cations in the aqueous phase and transfer them to the organic phase. Interestingly, the length of the hydrophilic chains does not significantly affect the ligand extraction and phase-transfer catalysis properties. On the other hand, the receptor can be improved in these respects by increasing its overall lipophilicity, which can be accomplished by appending large lipophilic functionalities to the upper calixarene rim.34 Roundhill et al.36 synthesized a number of polyethylene glycol functionalized calix[4]arenes by introducing PEG groups at both calixarene rims, and the obtained compounds were envisaged as potential efficient metal-ion host molecules and extracting agents. A larger ethylene glycol-based calix[6]arene derivative37 was investigated as a catalyst and extraction agent for alkali metal cations, whereby the solvent effect on the ligand abilities was proven to be of great importance. Shi and Zhang38 reported a water-soluble p-tert-butycalix[8]arene bearing PEG chains which was capable of efficiently binding organic molecules and ions in its hydrophobic cavity. Most recently, pegylated octopus-shaped calix[4]arenes with different degrees of polyoxyethylation of their lower-rim substituents were described as promising supramolecular drug delivery platforms.39,40 To the best of our knowledge, there is only one calix[4]arene amide derivative bearing diethylene glycol chains bound to the amide nitrogen atoms reported in the literature,41 which was studied as an anion41 and amino acid42–44 receptor.
In this work, we present the syntheses of calix[4]arene amide derivatives with diethylene glycol functionalities appended to amide groups at the lower rim, and the study of their complexation affinities towards alkali and alkaline earth metal cations in two solvents with different solvation and hydrogen-bonding abilities (methanol, MeOH, and acetonitrile, MeCN). In addition, the efficiencies of the extraction of cations from water to chloroform with both ligands have been investigated and discussed.
Results and discussion
Syntheses
The syntheses of diethylene glycol calixarenes 1 and 2 were performed in several reaction steps, as shown in Scheme 1. Compound 1 was prepared via aminolysis (i) of the p-tert-butylcalix[4]arene tetraethyl ester with 2-(2-methoxyethoxy)ethanamine with 90% yield, as described in the Experimental section. Compound 2 was synthesized by the modified method described previously, using tetra acid chloride p-tert-butylcalix[4]arene,45 starting from the corresponding tetraester (cone conformation) by (ii) hydrolysis to the tetraacid, (iii) activation to the acid chloride, and (iv) amide bond formation (65% yield). For both compounds, an additional purification step was required to ensure that all of the ions were removed, and this was done in a similar manner to that described in ref. 46. Details of the purification procedure are described in the Experimental section.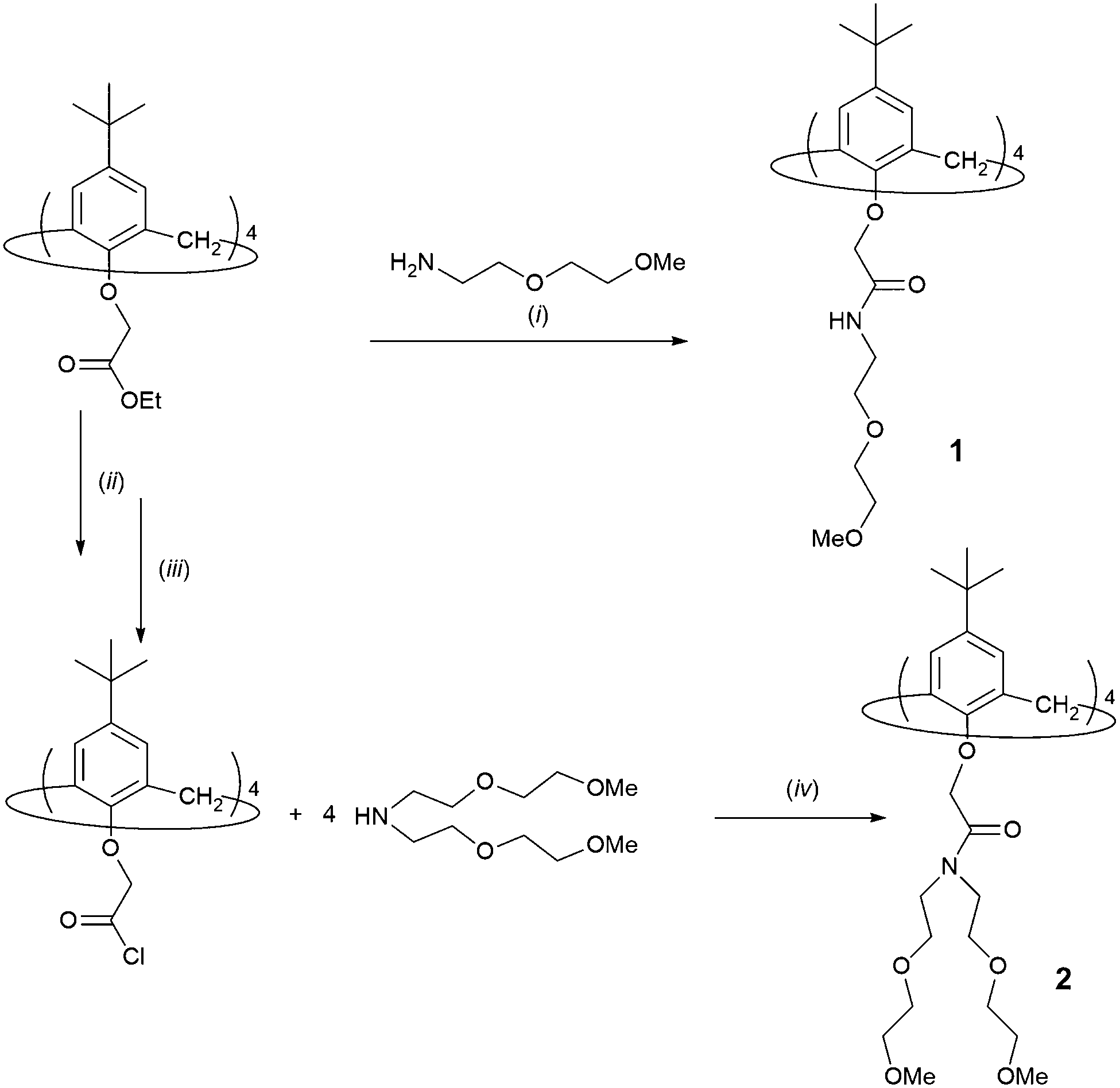 | ||
| Scheme 1 Syntheses of compounds 1 and 2. Reagents and conditions: (i) tert-butanol, r.t.; (ii), (a) NaOH, EtOH/H2O, Δ; (b) HCl (iii) SOCl2 Δ; (iv) Et3N, CH2Cl2 (Ar), 0 °C → r.t. | ||
Calixarene derivatives 1 and 2 were characterized by spectroscopic methods and mass spectrometry. The 1H NMR spectra of compounds 1 and 2 (CDCl3) showed the pattern characteristic of p-tert-butyl-calix[4]arene in a cone conformation and approximately C4 symmetry of tetrasubstituted calix[4]arene.19 Two singlets appeared, one corresponding to the tert-butyl groups (1.10 ppm, 1.09 ppm) and another due to the calixarene aromatic protons (6.79 ppm, 6.77 ppm). In addition, two doublets assigned to the equatorial (3.16 ppm, 3.26 ppm) and axial (4.65 ppm, 5.07 ppm) bridging methylene protons could be found. In the spectrum of compound 1, a rather high chemical shift of the amide protons (7.94 ppm) indicated the presence of intramolecular NH⋯OC hydrogen bonds between the amide groups of the lower-rim substituents.19,20 The FTIR data were fully in agreement with the NMR results. In the spectrum of 1, the NH stretching band at 3373 cm−1 corresponded to intramolecular hydrogen-bonds between the amide groups. Positive ESI mass spectra of compounds 1 and 2 were acquired in acetonitrile. The signals of singly-[M + H]+ and doubly-[M + 2H]2+ protonated ions, as well as those of adducts with alkali metal cations [M + Na]+, [M + H + Na]2+, and [M + H + K]2+ were observed (see ESI,† Fig. S1 and S2). The [M + H]+ ions of the ligands were isolated and MS/MS experiments were performed at different collision energies. The product ion spectra are shown in Fig. S3 and S4 (ESI†). The fragmentation of both derivatives was very similar; the loss of substituent(s) on the amide nitrogen atom was observed as well as the cleavage of C–O bonds, including phenolic and ether oxygen atoms. The formation of a carbocation was noticed in the case of 2, and the signal at m/z 262 was assigned to the fragment ion C12H24O3N+. The proposed fragmentation pathways of 1 and 2 are shown in Schemes S1 and S2 (ESI†).
Cation complexation studies
A hypochromic effect on the UV spectra of the acetonitrile solutions of 1 and 2 was observed upon the stepwise addition of LiClO4, NaClO4, KClO4, RbNO3, CsNO3, Mg(ClO4)2, Ca(ClO4)2, Sr(ClO4)2, and Ba(ClO4)2 (Fig. S5–S18, ESI†) solutions. In addition, an isosbestic point at 256 nm appeared in the case of titrations of ligand 1 with Mg(ClO4)2 (Fig. S12, ESI†) and Ba(ClO4)2 (Fig. 1). Isosbestic points were also observed in titrations of ligand 2 with Rb+ (251 nm), Cs+ (254 nm), and Ba2+ (255 nm) (Fig. S10, S11, and S18, ESI†).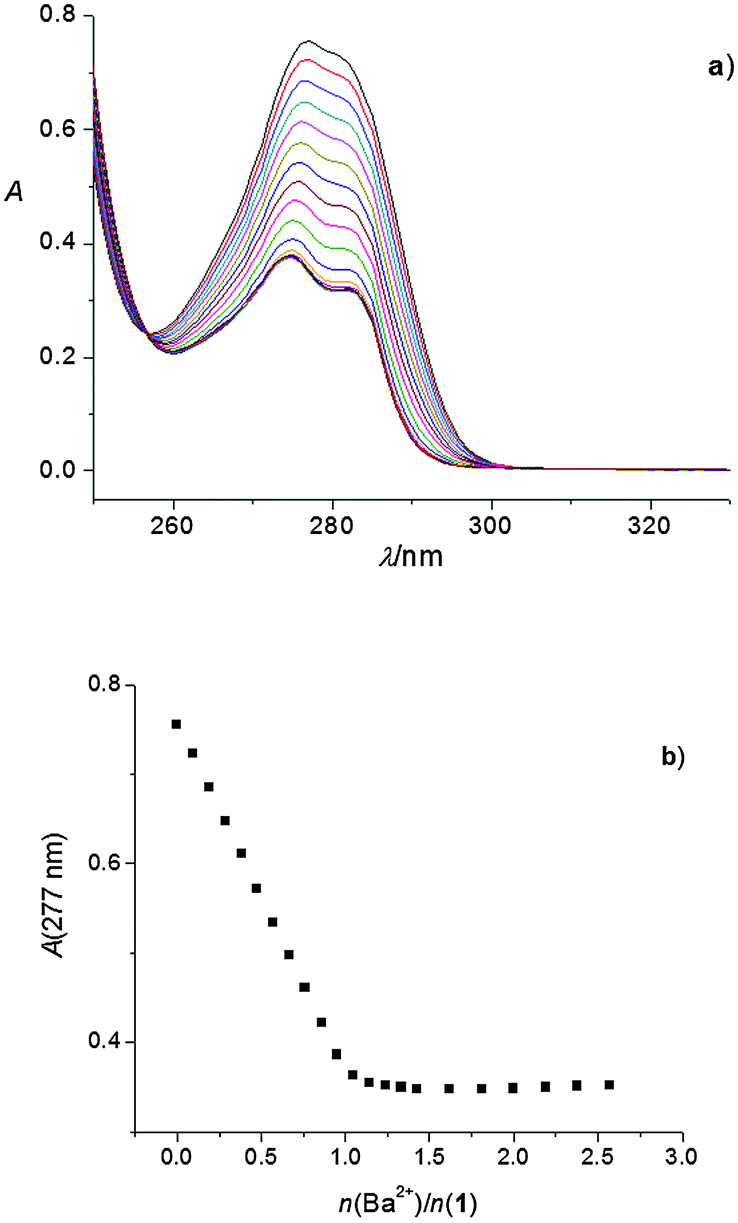 | ||
| Fig. 1 (a) Spectrophotometric titration of 1 (c = 1.89 × 10−4 mol dm−3) with Ba(ClO4)2 (c = 1.80 × 10−3 mol dm−3) in acetonitrile. l = 1 cm; t = (25.0 ± 0.1) °C; n(Ba2+)/n(1) = 0 (top curve)–2.57 (bottom curve); the spectra are corrected for dilution. (b) Dependence of absorbance at 277 nm on the n(Ba2+)/n(1) ratio. | ||
The linear dependence of absorbance vs. the amount of cation added up to the ratio n(cation)/n(ligand) ≈ 1, followed by a break in the titration curve, indicated a strong complexation and the formation of 1:1 complexes (Fig. 1; the corresponding stability constants could only be estimated, Table 1). This was observed in all the abovementioned titrations, except in that of 1 with KClO4 and RbNO3, and those of both 1 and 2 with CsNO3. In the case of complexes K1+ and Cs2+, their stability constants could be determined spectrophotometrically (Fig. 2, Fig. S11, ESI,†Table 1). Addition of RbNO3 and CsNO3to the calixarene derivative 1 acetonitrile solution had no significant effect on its UV spectrum, indicating that under the conditions used no observable complexation took place.
| Cation | logK |
|||
|---|---|---|---|---|
| MeCN | MeOH | |||
| 1 | 2 | 1 | 2 | |
| a Spectrophotometric determinations. b Conductometric determinations. c Potentiometric determination (Ic = 0.01 mol dm−3 (Et4NClO4)). d No complexation was observed. | ||||
| Li+ | >5a,b | >5a,b | —d | 4.69(2)b |
| Na+ | 7.09(3)c | >8c | 3.50(5)a | 7.45(1)c |
| 3.84(5)b | ||||
| K+ | 3.59(5)a | >5a,b | —d | >5a,b |
| 3.57(7)b | ||||
| Rb+ | —d | >5a,b | —d | 4.16(7)a |
| 4.02(1)b | ||||
| Cs+ | —d | 3.9(1)a | —d | 2.10(1)a |
| 4.06(1)b | ||||
| Mg2+ | >5a,b | >5a,b | 2.9(2)b | —d |
| Ca2+ | >5a,b | >5a,b | >5a,b | >5a,b |
| Sr2+ | >5a,b | >5a,b | 4.44(3)a | >5a,b |
| 4.27(3)b | ||||
| Ba2+ | >5a,b | >5a,b | 3.29(3)a | >5a,b |
| 3.0(1)b | ||||
 | ||
| Fig. 2 (a) Spectrophotometric titration of 1 (c = 2.30 × 10−4 mol dm−3) with KClO4 (c = 2.24 × 10−3 mol dm−3) in acetonitrile. l = 1 cm; t = (25.0 ± 0.1) °C; n(K+)/n(1) = 0 (top curve)–5.93 (bottom curve); the spectra are corrected for dilution. (b) Dependence of absorbance at 277 nm on n(K+)/n(1) ratio. ■ experimental; – calculated. | ||
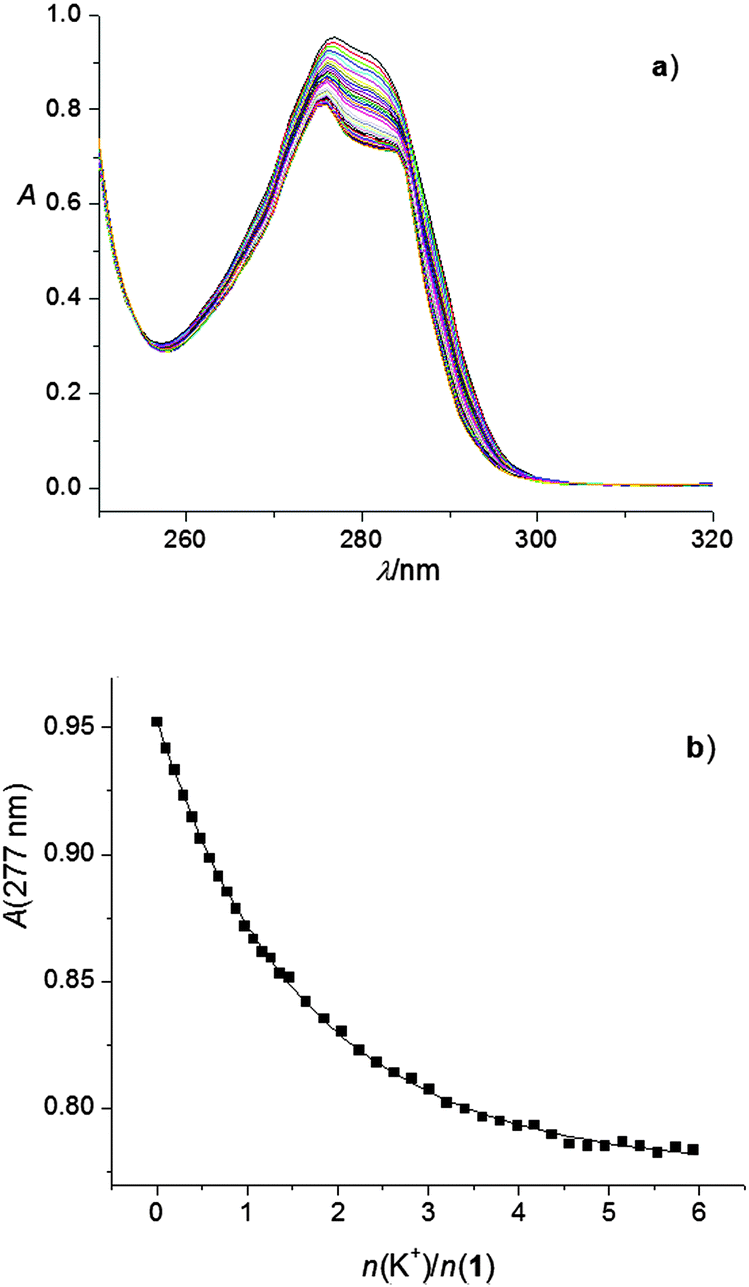
To corroborate the findings obtained by spectrophotometry, conductometric titrations of acetonitrile solutions of alkali and alkaline earth cation salts with calixarene derivatives 1 and 2 were carried out (Fig. S19–S33, ESI†). In most cases, a linear decrease in the molar conductivities with the addition of calixarene solutions was recorded up to a break in the titration curve at the molar ratio n(ligand)/n(cation) ≈ 1, indicating, as with the spectrophotometric titrations, strong complexation and the formation of 1:1 complexes (an example can be seen in Fig. 3). The exceptions were titrations of KClO4 with 1 (Fig. 4) and CsNO3 with 2 (Fig. S25, ESI†). By processing the data of these titrations, the stability constants of K1+ and Cs2+ were determined. In all the above cases, the decrease in the molar conductivity was due to the lower electric mobility of the larger complexes compared to the free metal cations. As with the spectrophotometric experiments, during the conductometric titrations of RbNO3 and CsNO3 acetonitrile solutions with ligand 1, no complexation was observed under the experimental titration conditions.
 | ||
| Fig. 3 Conductometric titration of Mg(ClO4)2 (c = 1.81 × 10−4 mol dm−3) with 1 (c = 1.90 × 10−3 mol dm−3) in acetonitrile; t = (25.0 ± 0.1) °C. | ||
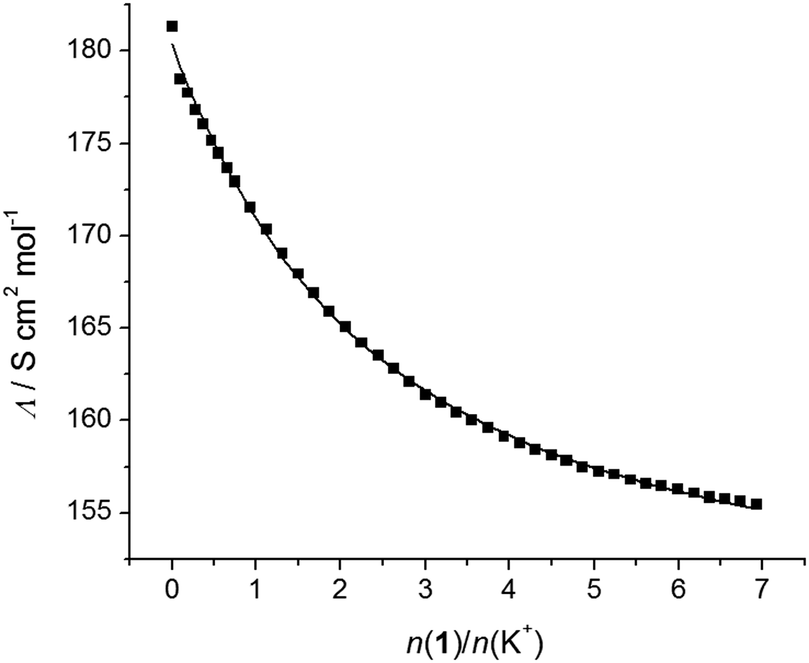 | ||
| Fig. 4 Conductometric titration of KClO4 (c = 1.09 × 10−4 mol dm−3) with 1 (c = 1.90 × 10−3 mol dm−3) in acetonitrile; t = (25.0 ± 0.1) °C. ■ experimental; – calculated. | ||
The stability constants of the Na1+ and Na2+ complexes in acetonitrile were too high for spectrophotometric or conductometric determination. For that reason, direct potentiometry using a sodium-selective glass electrode was applied. The potentiometric titration curves for both ligands showed a steep p[Na] jump at the 1:1 n(ligand)/n(cation) ratio, which is in accordance with the results of the previously mentioned methods. However, the p[Na] jump was steeper in the case of titration with tertiary-amide derivative 2, indicating the higher stability of the corresponding complex. The jump was even too steep (Fig. S34, ESI†) to allow accurate calculation of the Na2+ complex stability constant; therefore, it could only be estimated. On the other hand, the stability constant of the Na1+ complex was determined by processing the corresponding potentiometric titration data (Fig. 5 and Table 1).
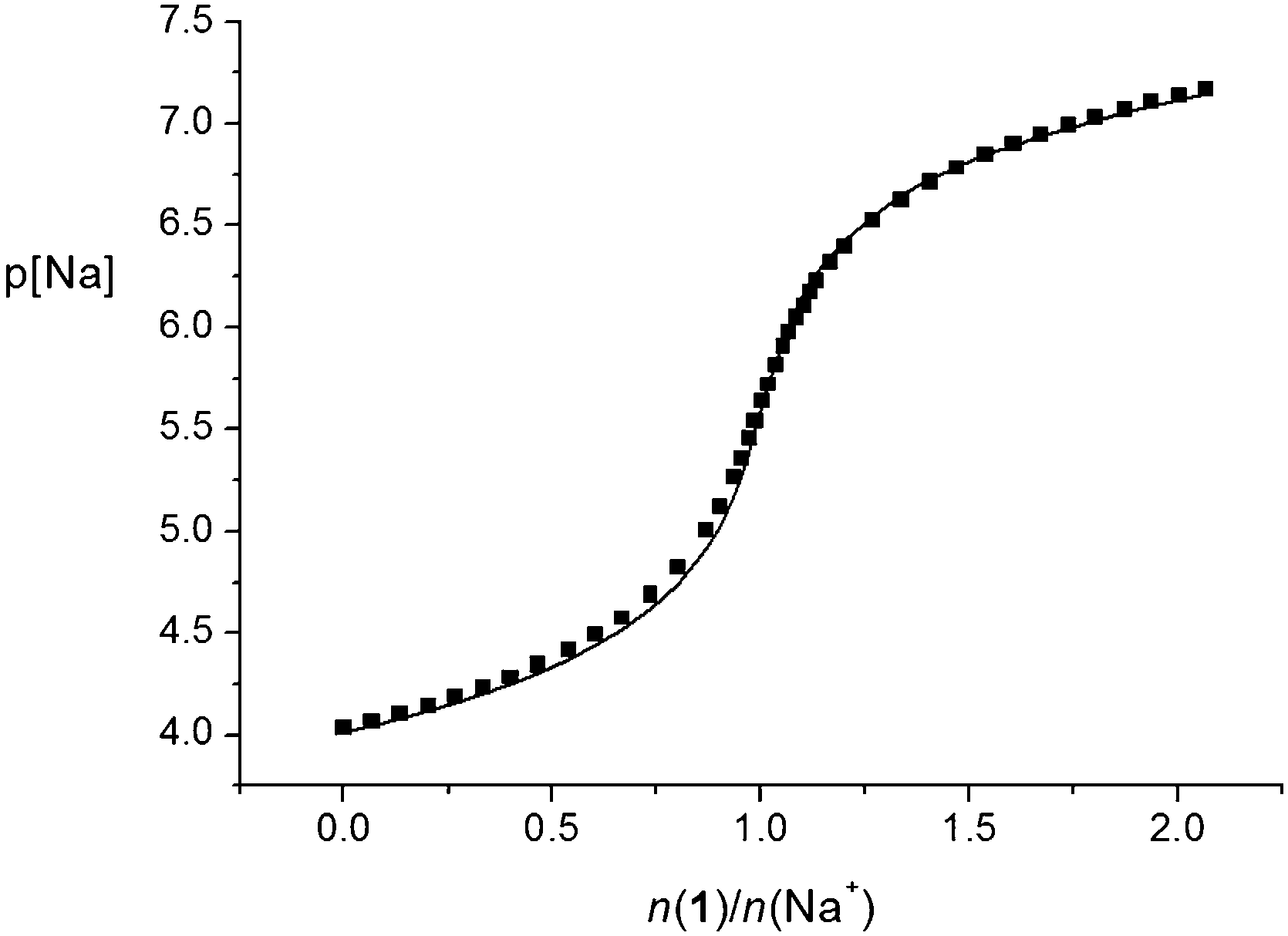 | ||
| Fig. 5 Potentiometric titration of NaClO4 (c = 9.78 × 10−5 mol dm−3) with 1 (c = 9.97 × 10−4 mol dm−3) in acetonitrile. V0(NaClO4) = 30.3 cm3Ic = 0.01 mol dm−3 ((C2H5)4NClO4); t = (25.0 ± 0.1) °C. ■ experimental; –calculated. | ||
The spectral changes observed upon the addition of salt solutions to ligand solution in methanol were basically similar to those described above (Fig. S35–S45, ESI†), i.e. the spectra exhibited hypochromic effects and in some cases isosbestic points were observed. For secondary-amide derivative 1, linear dependence of the absorbance was observed only upon the addition of Ca(ClO4)2 solution (Fig. S41, ESI†), whereas during titration of tertiary-amide derivative 2, this dependence was observed in titrations with all salt solutions except those of RbNO3 and CsNO3 (Fig. S39 and S40, ESI†).
Apart from the high-stability complex Ca12+, spectrophotometric measurements also showed the formation of Na1+, Ba12+ (Fig. S36 and S44, ESI†) and Sr12+ complexes (Fig. 6). The stability constants of these complexes are given in Table 1. As can be concluded from the described experimental results, calixarene derivative 2 in methanol forms high-stability complexes with most of the investigated cations (the exceptions are Rb+ and Cs+). The determined stability constant of the Rb2+ complex is given in Table 1, whereas no observable complexation of cesium cation with ligand 2 took place.
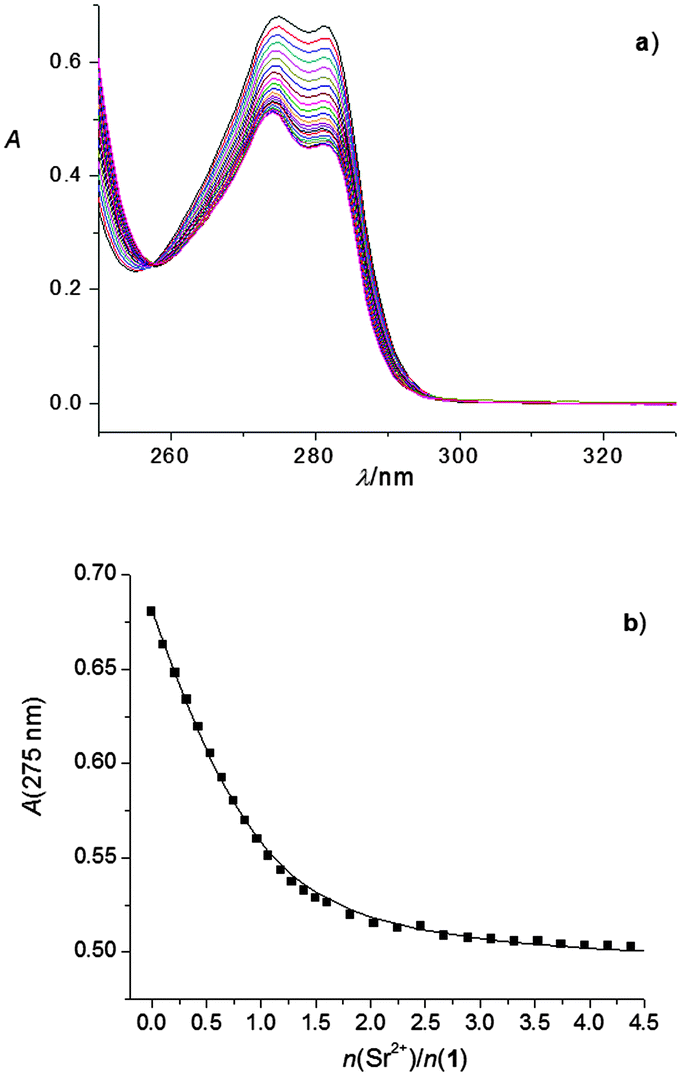 | ||
| Fig. 6 (a) Spectrophotometric titration of 1 (c = 1.95 × 10−4 mol dm−3) with Sr(ClO4)2 (c = 2.20 × 10−3 mol dm−3) in methanol. l = 1 cm; t = (25.0 ± 0.1) °C; n(Sr2+)/n(1) = 0 (top curve)–4.38 (bottom curve); the spectra are corrected for dilution. (b) Dependence of the absorbance at 275 nm on the n(Sr2+)/n(1) ratio. ■ experimental; – calculated. | ||
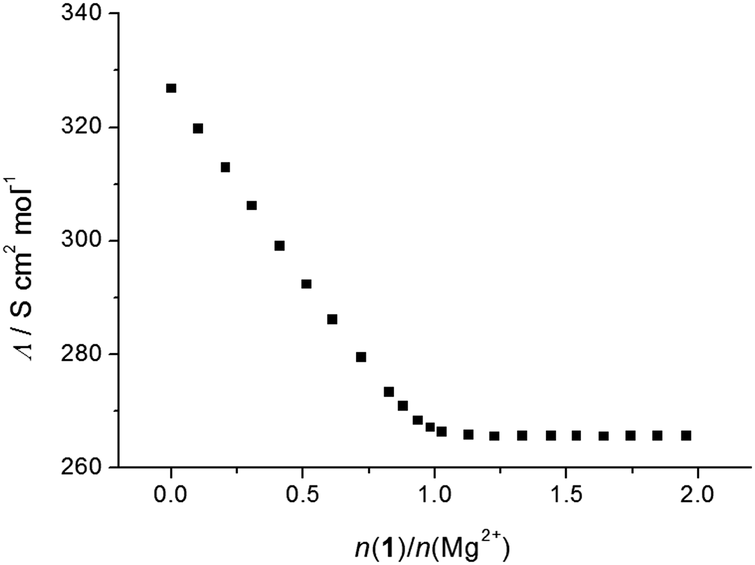
The above results were confirmed by conductometric titrations. By processing of the corresponding data, the stability constants of the Na1+, Mg12+, Sr12+, Ba12+, Li2+, and Rb2+ complexes given in Table 1 were determined (the corresponding titrations are shown in ESI,† Fig. S46–S51). On the other hand, because of their high values, the equilibrium constants for the reactions of formation of the Ca12+, Na2+, K2+, Ca22+, Sr22+, and Ba22+ species were only estimated (Table 1; Fig. S52–S57, ESI†). During the conductometric titrations of LiClO4, NaClO4, and RbNO3 with 1 and that of CsNO3 with both ligands in methanol, negligible and irregular changes in molar conductivity were observed, indicating that no complexation took place. It should be noted that the formation of Mg12+ complex was observed conductometrically, although no complexation was detected using spectrophotometry. The stability constant of the Na2+ complex in methanol was, as in acetonitrile, too high for spectrophotometric and conductometric determinations. For that reason, it was determined by means of direct potentiometric titration (Fig. 7 and Table 1). As the stability of the sodium complex with 1 was much lower (Table 1), considerably higher concentrations of both ligand and sodium perchlorate were needed to determine its stability constant potentiometrically. Because of the limited solubility of calixarene 1 in methanol, these measurements were not conducted.
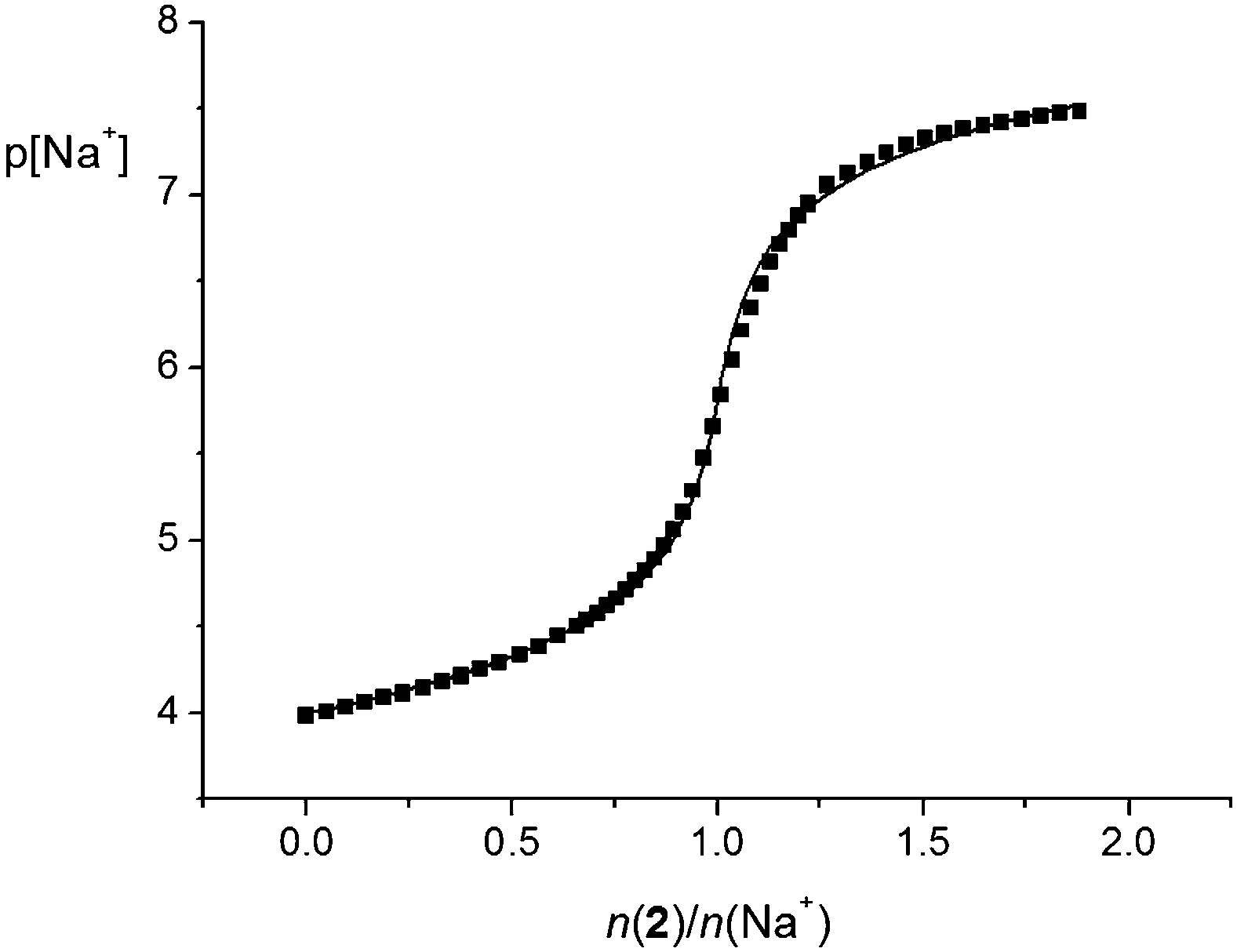 | ||
| Fig. 7 Potentiometric titration of NaClO4 (c = 1.00 × 10−4 mol dm−3) with 2 (c = 7.13 × 10−4 mol dm−3) in methanol. V0(NaClO4) = 30.3 cm3Ic = 0.01 mol dm−3 ((C2H5)4NClO4); t = (25.0 ± 0.1) °C. ■ experimental; – calculated. | ||
Given the data listed in Table 1, it is evident that the affinity of ligand 2 for alkali and alkaline earth metal cations is much higher than that of ligand 1. The main reason for the difference can be readily explained by taking into account the presence of N–H⋯OC intramolecular hydrogen bonds in the latter case. Namely, in order for a cation to form a complex with 1, these bonds must be disrupted to allow reorganization of the amide groups. Amide hydrogen atoms are in fact competing with the cation for the carbonyl oxygen. Tertiary amide derivatives, such as 2, do not have the ability to form the aforementioned hydrogen bonds; therefore, no unfavorable competition between the cation and –NH group can take place. Secondary-amide derivative 1 binds smaller Li+ and Na+ cations very well. Its affinity for the larger K+ is lower, whereas Rb+ and Cs+ are too large to fit into its hydrophilic cavity. On the other hand, this compound binds all alkaline earth metal cations quite strongly in acetonitrile, as expected due to their size and charge. Tertiary-amide derivative 2 forms highly stable complexes with all cations, except with the largest Cs+ (even in that case, the complex stability is moderate). This size- and charge-dependent selectivity is even more pronounced in methanol. As in MeCN, in MeOH both ligands 1 and 2 showed higher affinity towards alkaline earth compared to alkali metal cations (the only exception is complexation with Ba2+ because of its relatively large size). However, the complex stabilities in methanol are considerably lower (Table 1). This strong solvent influence can be explained by different solvation of all species taking part in the complexation reaction, i.e. free cation, free ligand, and the complex. The influence of cation solvation is most pronounced in the case of the smallest cations, such as Li+ and Mg2+. Although they form highly stable complexes with 1 in acetonitrile, the presence of these complexes was hardly, or not at all, observed in methanol solution. Methanol, as a hydrogen bonding solvent, strongly solvates small cations,47 thus making the substitution of its molecules by ligand binding sites unfavorable. The cation solvation effect is of course present in the complexation of other metal cations, but it is not as dominant as in the cases of Li+ and Mg2+. The ligand solvation effect on the studied equilibria should be presumably more pronounced in reactions with 1 than in those with 2. In both cases, methanol molecules compete with the cations for binding sites by forming hydrogen bonds with amide carbonyl oxygen atoms. However, contrary to 2, in the case of 1, which is a secondary amide derivative, MeCN molecules can (as a proton acceptor) form H-bonds with the amide NH groups of the receptor. This in turn leads to the disruption of the intramolecular N–H⋯OC hydrogen bonds and to an orientation of the carbonyl groups that is favorable for the complexation of metal ions. Obviously, the solvation of the complex also plays an important role in determining its thermodynamic stability. However, on the basis of the data presented in this paper, we cannot say much about this effect.
It should be also mentioned that the process of solvent molecule inclusion into the hydrophobic cavities of calixarenes 1 and 2 and their complexes could occur. This phenomenon is more pronounced in acetonitrile than in methanol, and could be quite important in determining the equilibria of the complexation reactions.12,13,28,48–50
Extraction properties
The abilities of compounds 1 and 2 to extract alkali and alkaline earth metal cations from water to dichloromethane were explored according to the procedure described in detail in the Experimental section. As can be seen by inspecting the data in Table 2, secondary-amide derivative 1 moderately extracted sodium, potassium, and calcium picrates from the aqueous to the organic phase. Other examined picrate salts were extracted poorly (or not at all) by this compound. On the other hand, tertiary-amide derivative 2 was shown to be a much better extractant of alkali and alkaline earth metal cations. Most of these cations were extracted very well (95–100%) by 2, the exceptions being rubidium, cesium, and magnesium ions, which were not extracted as efficiently. It is interesting to note that the extraction efficiency was 100% in the case of sodium cation, which was not observed earlier with the other calix[4]arene tertiary-amide derivatives.16,45 The results described are obviously directly correlated with the stabilities of the corresponding complexes, i.e. by the structural features of the compounds studied and their amphiphilic character.| Li+ | Na+ | K+ | Rb+ | Cs+ | Mg2+ | Ca2+ | Sr2+ | Ba2+ | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.35(2) | 10.9(2) | 7.8(1) | 3.0(1) | 3.69(6) | 1.7(1) | 6.55(9) | 2.59(6) | 2.3(1) |
| 2 | 96.0(1) | 100.0(1) | 96.96(8) | 75.6(1) | 58.31(8) | 68.4(1) | 99.6(2) | 98.5(2) | 93.3(1) |
Conclusion
Diethylene glycol calix[4]arene derivatives 1 and 2 were designed and synthesised as compounds comprising hydrophobic and relatively hydrophilic parts as well as binding sites, which ensure the strong complexation of alkali and alkaline earth metal cations. Hence, due to these properties, strong cation binding associated with high extraction abilities was expected for these compounds. The complexation of alkali and alkaline earth cations by these ligands was studied by several experimental methods. The solvent effect on the complexation reactions was also examined using two solvents with different solvation and hydrogen-bonding abilities, namely acetonitrile and methanol. In both the solvents, the affinity for binding alkali and alkaline earth metal cations was found to be much higher in the case of tertiary-amide derivative 2 compared to compound 1, which comprises secondary-amide subunits. This can be explained mostly by taking into account the presence of intramolecular NH⋯OC hydrogen bonds in 1, which cannot be formed in 2. In the process of cation complexation, these bonds must be disrupted, which is energetically quite demanding and therefore this significantly reduces the stability of the complex.
The role of hydrogen-bonding is also important for the explanation of the solvent effect on the equilibria of the complexation reactions. Namely, the carbonyl-oxygen binding sites of compound 1 are most probably “blocked” by the formation of H-bonds with methanol molecules as proton donors, whereas such bonds cannot be established with acetonitrile. When the difference in cation solvation in the two solvents is also considered, the reason for the much higher complex stabilities in MeCN in comparison to MeOH becomes obvious.
In accordance with the abovementioned results, the structural differences between compounds 1 and 2 were shown to be of utmost importance in determining their abilities for the extraction of metal cations from water to dichloromethane. Thus, contrary to 1, calix[4]arene derivative 2 was found to be a very good, even excellent extractant of alkali and alkaline earth metal picrates, with 100% efficiency in the case of sodium cation. Therefore, it can be concluded that due to its amphiphilic character and strong cation-binding ability, compound 2 can be considered to be a promising reagent for the extraction of metal ions from aqueous to organic phases.
Experimental
General
All reagents used in the syntheses were of the best grade commercially available and were not further purified. Solvents were purified by standard procedures.51 Analytical TLC was performed on silica gel plates (SiO2, Merck 60 F254). Melting points were determined on a Kofler hot-bench apparatus and were not corrected. 1H NMR and 13C NMR spectra were recorded with a Bruker Avance 300 or 600 MHz spectrometer (d in ppm relative to (CH3)4Si as an internal standard, J values are in Hz). IR spectra were recorded by means of an ABB Bomem MB102 FTIR spectrometer. MS measurements were conducted on an Agilent 6410 Triple Quadrupole mass spectrometer. The salts used for the investigation of the calixarene complexation reactions were LiClO4 (Fluka, p.a. and Sigma Aldrich 99.99%), NaClO4·H2O (Fluka p.a.), NaClO4 (Sigma Aldrich 98+%), KClO4 (Merck, p.a.), RbNO3 (Sigma, 99.7%), CsNO3 (Sigma, 99.5%), CsCl (Merck, p.a.), Mg(ClO4)2 (Aldrich, 99%), Ca(ClO4)2 (Aldrich, 99%), Sr(ClO4)2 (Aldrich, p.a.), and Ba(ClO4)2 (Fluka, p.a.). The solvents, acetonitrile (Merck, Uvasol and Baker, HPLC Grade) and methanol (Baker, HPLC Grade and Sigma-Aldrich, 99.9%, spectrophotometric grade) were used without further purification. In all potentiometric titrations the ionic strength was maintained by the addition of Et4NClO4 (Fluka, p.a.). The compounds p-tert-calix[4]arene ethyl ester,52 the tetra acid chloride of p-tert-calix[4]arene,15 2-(2-methoxyethoxy)ethylamine,53 and bis(2-(2-methoxyethoxy)ethyl)amine54 used in the synthesis of 1 and 2 (Scheme 1) were prepared according to procedures described in the literature.Synthesis of 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetra(N-(2-(2-methoxyethoxy)ethyl)carbamoylmethoxy)calix[4]arene (1)
Preparation of compound 1 by aminolysis: p-tert-calix[4]arene ethyl ester52 (1.93 g, 1.5 mmol) and 2-(2-methoxyethoxy)ethylamine53 (2.63 g, 25 mmol) were added to 10 ml of i-butanol. The mixture was maintained for several weeks at room temperature. After determining that no calixarene ethyl ester was present in the solution by means of 1H NMR spectroscopy, the solvent was evaporated and the precipitate was dissolved in CH2Cl2. The solution was extracted first with HCl (1 mol dm−3) and then with water (the mixing of water and dichloromethane solution resulted in an emulsion which separated after 1 hour). The solvent was removed under vacuum, the solid residue was dissolved in methanol, water was added to the solution, and compound 1 was precipitated. The precipitate was separated by suction filtration. After drying, compound 1 was obtained as a white powder. Rf = 0.64 (10% CH3OH/CH2Cl2); tt = 173–174 °C; yield 90%. An additional purification step was required to ensure that all of the ions were removed, and that was done in a similar manner to that described in ref. 40. Compound 1 was dissolved in methanol, followed by the addition of deionised water. Subsequently, the solution was heated to evaporate the methanol, leaving a precipitate in the aqueous solution. The precipitated ligand 1 was collected by suction filtration, washed with deionised water, and dried for 12 hours at 110 °C prior to use in physico-chemical experiments.1H NMR spectra (600 MHz, CDCl3): δH/ppm = 1.10 (36H, s, C(CH3)3), 3.26 (4H, d, J = 13.3, ArCH2eqAr), 3.35 (12H, s, OCH3), 3.52–3.62 (32H, m, OCH2, NCH2), 4.62 (8H, s, OCH2CO), 4.65 (4H, d, J = 13.3, ArCH2axAr), 6.79 (8H, s, ArH), 7.94 (4H, t, J = 6.3, NH). 13CNMR spectra (75 MHz, CDCl3): δC/ppm = 31.33 (C(CH3)3), 31.69 (Ar–CH2–Ar) 33.83, (C(CH3)3), 39.12 (NHCH2CH2), 58.69 (OCH3), 69.74 (OCH2CO), 70.01 (OCH2CH2O), 71.88 (NCH2CH2O), 74.38 (OCH2CH2O), 125.76 (o-Ar–C), 132.74 (m-Ar–C), 145.53 (Ar–C–t-Bu), 153.32 (p-Ar–C), 170.03 (CONH). IR (KBr) νmax/cm−1 = 3373 (s, NH), 2960 (s, CH), 1674 (s, CO amidI), 1542 (m, NH, amidII), 1479 (m, CH2), 1198 (s, COC), 1126 (s, COC). HRMS: m/z [M + H]+ exact mass, C72H108N4O16: 1285.7839; found: 1285.7852.
Synthesis of 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetra(N,N-bis(2-(2-methoxyethoxy)ethyl)carbamoylmethoxy)calix[4]arene (2)
Bis(2-(2-methoxyethoxy)ethyl)amine54 (1.4 g, 6.3 mmol) and triethylamine (1.0 mL, 7.2 mmol) were added to the cooled (0 °C) dry CH2Cl2 (20 mL) under argon atmosphere. Then, the acid chloride of calix[4]arene tetraacetic acid (the cone conformer)15 (1.24 g, 1.3 mmol) in dry CH2Cl2 (10 mL) was added at once with a syringe. The reaction mixture was allowed to warm to room temperature and stirred for 24 h. After filtration of the solid, the filtrate was extracted with HCl (1.0 mol dm−3) and water. Dichloromethane was removed under vacuum and the product 2 was obtained as a pale yellow wax. Ligand 2 was additionally purified by continuous liquid–liquid extraction. A dichloromethane solution of 2 was placed in a continuous extractor and extracted with deionised water for 36 hours (the water phase was replaced 3 times) in order to remove ion traces from the product. The resulting CH2Cl2 solution was again extracted several times with deionized water. The presence of charged species was checked conductometrically. After evaporating CH2Cl2 under vacuum, the compound 2 was obtained as a waxy solid with a yield of 59%, Rf = 0.75 (6% CH3OH/CH2Cl2). Prior to use, ligand 2 was dried for approximately 12 hours at 110 °C.1H NMR (600 MHz, CDCl3): δH (ppm) = 1.09 (36H, s, C(CH3)3), 3.16 (4H, d, J = 12.9, ArCH2eqAr), 3.34 (12H, s, OCH3), 3.38 (12H, s, OCH3), 3.43–3.66 (64H, m, OCH2, NCH2), 5.02 (8H, s, OCH2CO), 5.07 (4H, d, J = 12.8, ArCH2axAr), 6.77 (8H, s, ArH). 13C NMR (75 MHz, CDCl3): δC (ppm) = 31.42 (C(CH3)3), 32.18 (Ar–CH2–Ar), 33.73 (C(CH3)3), 46.60 (NCH2CH2), 47.82 (NCH2CH2), 58.96 (OCH3), 69.74 (OCH2CO), 69.33 (NCH2CH2O), 70.07 (NCH2CH2O), 70.23 (OCH2CH2O), 70.54 (OCH2CH2O), 71.38 (OCH2CH2O), 71.91 (OCH2CH2O), 125.21 (o-Ar–C), 133.58 (m-Ar–C), 144.32 (Ar–C–t-Bu), 153.70 (p-Ar–C), 170.19 (CONH). IR (KBr) νmax (cm−1) = 2953 (s, CH), 2871 (s, CH), 1653 (s, CO), 1482 (s, CH2), 1196 (s, COC), 1120 (s, COC). HRMS: m/z [M + Na]+ exact mass, C92H148N4O24: 1716.0381; found: 1716.0386.
Spectrophotometry
UV titrations were performed at (25.0 ± 0.1) °C by means of a Varian Cary 5 double-beam spectrophotometer equipped with a thermostating device. The spectral changes of ligand solution (V0 = 2.0 cm3, c0 = 1.5 × 10−4 to 3.0 × 10−4 mol dm−3) were recorded upon stepwise addition of an alkali metal salt solution (c = 2.0 × 10−3 to 3.5 × 10−3 mol dm−3) directly into a measuring cell with a 1 cm path length (Hellma, Suprasil QX). Absorbances were sampled at 1 nm intervals, with an integration time of 0.2 s. Titrations for each system were performed in triplicate. The obtained spectrophotometric data were processed using the SPECFIT program.55,56Conductometry
Conductometric titrations were carried out at (25.0 ± 0.1) °C by means of a Metrohm 712 conductometer. The cell constant ((0.861 ± 0.001) cm−1) was determined before each experiment using 0.01 mol dm−3 aqueous KCl solution. The alkali metal salt solution (V0 = 20.0 to 22.0 cm3, c0 = 8.0 × 10−5 to 2.0 × 10−4 mol dm−3) was titrated with a ligand solution (c = 1.0 × 10−4 to 2.5 × 10−3 mol dm−3) in a thermostated vessel. The measured conductivities were corrected for the conductivity of the solvent. Titrations for each system were done in triplicate. The obtained data were processed by the OriginPro 7.5 program.Potentiometry
The stability constants of Na1+ and Na2+ complexes were determined by potentiometric titrations of 30.3 cm3 NaClO4 solution (c0 = 1.0 × 10−4 mol dm−3) with solutions of ligands 1 and 2 (c = 1.0 × 10−3 mol dm−3) in a thermostated titration vessel (25.0 ± 0.1) × C. The ionic strength of the solutions was set to 0.01 mol dm−3 using Et4NClO4. The indicator electrode was a sodium-selective glass electrode (Metrohm, 6.0501.100) with an Ag/AgCl reference electrode (Metrohm, 6.0733.100) filled with an acetonitrile or methanol solution of Et4NCl (c = 0.01 mol dm−3). The working and reference half-cells were connected with a salt bridge containing 0.01 mol dm−3 Et4NClO4. A Metrohm 827 pH Lab was used for electromotivity measurements. The cell was calibrated by the incremental addition of NaClO4 solution (c = 1.0 × 10−3 mol dm−3) to 30.0 cm3 solution of Et4NClO4 (c = 0.01 mol dm−3). In each calibration experiment a Nernst-like behaviour was observed, with the slope of the E vs. p[Na] plot being about −58 mV. Each titration was repeated three times, and the obtained potentiometric data were analysed with the HYPERQUAD program.57Picrate extractions
Aqueous solutions of metal picrates (c0 = 1.5 × 10−4 mol dm−3) were prepared in situ by dissolving the metal hydroxide (0.001 mol) in picric acid. (c = 1.5 × 10−4 mol dm−3, 25 mL). Solutions (c0 = 2.0 × 10−4 mol dm−3) of the calixarene derivatives were prepared in dichloromethane. Equal volumes (5 mL) of the two solutions were shaken vigorously for 20 min in a 100 mL flask. The solutions were left to stand in a thermostated bath at (25.0 ± 0.1) °C for 24 h until phase separation was complete. The concentration of picrate ion in the organic phase was then determined spectrophotometrically as described by Pedersen.58 Control blank experiments showed that no picrate extraction occurred in the absence of a calixarene. The extraction percentage (%E) has been calculated as %E = [(A0 − A)/A0] × 100, where A0 and A are the initial and final absorbances of the metal picrate at 356 nm before and after the extraction.Acknowledgements
This work was partially supported by the Ministry of Science, Education and Sports of the Republic of Croatia and by the Croatian Science Foundation (projects 7387 and IP-2014-09-7309). The authors thank Mrs Biserka Kolar for the technical assistance.Notes and references
- C. D. Gutsche, Calixarenes: An Introduction, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008 Search PubMed.
- Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001 Search PubMed.
- V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713 CrossRef PubMed.
- B. Mokhtari, K. Pourabdollah and N. Dalali, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 1 CrossRef CAS.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893 CrossRef CAS PubMed.
- S. E. Matthews and P. D. Beer, Supramol. Chem., 2005, 17, 411 CrossRef CAS PubMed.
- P. Lhoták, Top. Curr. Chem., 2005, 255, 65 Search PubMed.
- P. Cuřínová, M. Pojarová, J. Budka, K. Lang, I. Stibor and P. Lhoták, Tetrahedron, 2010, 66, 8047 CrossRef PubMed.
- S. Smirnov, V. Sidorov, V. Pinkhassik, J. Havlicek and I. Stibor, Supramol. Chem., 1997, 8, 187 CrossRef CAS PubMed.
- G. Arena, A. Contino, E. Longo, G. Spoto, A. Arduini, A. Pochini, A. Secchi, C. Massera and F. Ugozzolli, New J. Chem., 2004, 28, 56 RSC.
- F. Arnaud-Neu, M. A. McKervey and M.-J. Schwing-Weill, in Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001, ch. 21, pp. 385–406 Search PubMed.
- G. Horvat, V. Stilinović, T. Hrenar, B. Kaitner, L. Frkanec and V. Tomišić, Inorg. Chem., 2012, 51, 6264 CrossRef CAS PubMed.
- G. Horvat, V. Stilinović, B. Kaitner, L. Frkanec and V. Tomišić, Inorg. Chem., 2013, 52, 12702 CrossRef CAS PubMed.
- A. F. Danil de Namor, T. T. Matsufuji-Yasuda, K. Zegarra-Fernandez, O. A. Webb and A. El Gamouz, Croat. Chem. Acta, 2013, 86, 1 CrossRef CAS.
- F. Arnaud-Neu, G. Barrett, S. Cremin, M. Deasy, G. Ferguson, S. J. Harris, A. J. Lough, L. Guerra, M. A. McKervey, M. J. Schwing-Weill and P. Schwinte, J. Chem. Soc., Perkin Trans. 2, 1992, 1119 RSC.
- F. Arnaud-Neu, S. Barboso, F. Berny, A. Casnati, N. Muzet, A. Pinalli, R. Ungaro, M.-J. Schwing-Weill and G. Wipff, J. Chem. Soc., Perkin Trans. 2, 1999, 1727 RSC.
- V. Tomišić, N. Galić, B. Bertoša, L. Frkanec, V. Simeon and M. Žinić, J. Inclusion Phenom. Macrocyclic Chem., 2005, 53, 263 CrossRef PubMed.
- J. C. Mareque Rivas, C. H. Schwalbe and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9478 CrossRef CAS PubMed.
- L. Frkanec, A. Višnjevac, B. Kojić-Prodić and M. Žinic, Chem. – Eur. J., 2000, 6, 442 CrossRef CAS.
- E. Nomura, M. Takagaki, C. Nakaoka, M. Uchida and H. Taniguchi, J. Org. Chem., 1999, 64, 3151 CrossRef CAS PubMed.
- N. Galić, N. Burić, R. Tomaš, L. Frkanec and V. Tomišić, Supramol. Chem., 2011, 23, 389 CrossRef PubMed.
- Y. L. Cho, D. M. Rudkevich, A. Shivanyuk, K. Rissanen and J. Rebek Jr., Chem. – Eur. J., 2000, 6, 3788 CrossRef CAS.
- I. Stibor, M. Ružičkova, R. Krátký, M. Vindyš, J. Havlíček, E. Pinkhassik, P. Lhoták, A. R Mustafina, Y. E. Morozova, E. Kh. Kazakova and V. P. Gubskaya, Collect. Czech. Chem. Commun., 2001, 66, 641 CrossRef CAS.
- L. Baldini, F. Sansone, F. Scaravelli, A. Casnati and R. Ungaro, Supramol. Chem., 2010, 22, 776 CrossRef CAS PubMed.
- A. F. Danil de Namor, R. G. Hutcherson, F. J. Sueros Velarde, M. L. Zapata-Ormachea, L. E. Pulcha Salazar, I. Al Jammaz and N. Al Rawi, Pure Appl. Chem., 1998, 70, 769 CAS.
- A. F. Danil de Namor, R. M. Cleverley and M. L. Zapata-Ormachea, Chem. Rev., 1998, 98, 2495 CrossRef PubMed.
- A. F. Danil de Namor, in Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001, ch. 19, pp. 346–364 Search PubMed.
- A. F. Danil de Namor, D. Kowalska, E. E. Castellano, O. E. Piro, F. J. Sueros Velarde and J. Villanueva Salasa, Phys. Chem. Chem. Phys., 2001, 3, 4010 RSC.
- J. Požar, T. Preočanin, L. Frkanec and V. Tomišić, J. Solution Chem., 2010, 39, 835 CrossRef.
- J. Požar, G. Horvat, M. Čalogović, N. Galić, L. Frkanec and V. Tomišić, Croat. Chem. Acta, 2012, 85, 541 CrossRef.
- M. Tranfić Bakić, D. Jadreško, T. Hrenar, G. Horvat, J. Požar, N. Galić, V. Sokol, R. Tomaš, S. Alihodžić, M. Žinić, L. Frkanec and V. Tomišić, RSC Adv., 2015, 5, 23900 RSC.
- L. Baldini, F. Sansone, A. Casnati and R. Ungaro, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. G. A. Philip and J. W. Steed, John Wiley & Sons, Chichester, 2012, pp. 863–895 Search PubMed.
- A. Casnati and R. Ungaro, in Calixarene in Action, ed. L. Madolini and R. Ungaro, Imperial College Press, London, 2000, pp. 62–84 Search PubMed.
- P. Schmitt, D. P. Beer, M. G. B. Drew and P. D. Sheen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1840 CrossRef CAS PubMed.
- K. Araki, A. Yanagi and S. Shinkai, Tetrahedron, 1993, 49, 6763 CrossRef CAS.
- J. Shen, H. F. Koch and M. Roundhill, J. Inclusion Phenom. Macrocyclic Chem., 2000, 38, 57 CrossRef CAS.
- E. Nomura, H. Taniguchi, K. Kawaguchi and Y. Otsujit, J. Org. Chem., 1993, 58, 4709 CrossRef CAS.
- Y. Shi and Z. Zhang, J. Chem. Soc., Chem. Commun., 1994, 375 RSC.
- C. Tu, L. Zhu, P. Li, Y. Chen, Y. Su, D. Yan, X. Zhu and G. Zhou, Chem. Commun., 2011, 47, 6063 RSC.
- D. Momekova, D. Budurova, E. Drakalska, S. Shenkov, G. Momekov, B. Trzebicka, N. Lambov, E. Tashev and S. Rangelov, Int. J. Pharm., 2012, 410, 436 Search PubMed.
- A. Hamdi, R. Abidi and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 193 CrossRef CAS.
- A. Medvedovici, F. Albu, A. Hamdi, R. Souane, L. Kim, L. Mutihac and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 545 CrossRef CAS.
- L. Kim, A. Hamdi, A. D. Stancu, R. Souane, L. Mutihac and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 55 CrossRef CAS.
- A. Hamdi, R. Souane, L. Kim, R. Abidi, L. Mutihac and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2009, 64, 95 CrossRef CAS.
- F. Arnaud-Neu, G. Barrett, S. Fanni, D. Marrs, W. McGregor, M. A. McKervey, M.-J. Schwing-Weill, V. Vetrogon and S. Wechsler, J. Chem. Soc., Perkin Trans. 2, 1995, 453 RSC.
- A. Moser, G. P. A. Yap and D. Detellier, J. Chem. Soc., Dalton Trans., 2002, 428 RSC.
- A. D'Aprano, M. Salomon and V. Mauro, J. Solution Chem., 1995, 24, 685 CrossRef.
- A. F. Danil de Namor, N. A. de Sueros, M. A. McKervey, G. Barrett, F. A. Neu and M. J. Schwing-Weill, J. Chem. Soc., Chem. Commun., 1991, 1546 RSC.
- A. F. Danil de Namor, S. Chahine, E. E. Castellano and O. E. Piro, J. Phys. Chem. A, 2005, 109, 6743 CrossRef CAS PubMed.
- A. F. Danil de Namor, S. Chahine, D. Kowalska, E. E. Castellano and O. E. Piro, J. Am. Chem. Soc., 2002, 124, 12824 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann (Elsevier), Oxford, UK, 6th edn, 2009 Search PubMed.
- F. Arnaud-Neu, E. M. Collins, M. Deasy, G. Ferguson, S. J. Harris, B. Kaitner, A. J. Lough, M. A. McKervey, E. Marques, B. L. Ruhl, M. J. Schwing-Weill and E. M. Seward, J. Am. Chem. Soc., 1989, 111, 8681 CrossRef CAS.
- G. Madhusudhan, G. Ravibabu and G. Reddy, Indian J. Chem., 2010, 49B, 96 CAS.
- A. Jain, S. G. Huang and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 5057 CrossRef CAS.
- H. Gamp, M. Maeder, C. J. Meyer and A. D. Zuberbuhler, Talanta, 1985, 32, 95 CrossRef.
- H. Gamp, M. Maeder, C. J. Meyer and A. D. Zuberbuhler, Talanta, 1985, 32, 257 CrossRef.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- C. J. Pedersen, Fed. Proc., 1968, 27, 1305 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI mass spectra of compounds 1 and 2 and the results of MS/MS experiments. Additional spectrophotometric, conductometric, and potentiometric titrations. See DOI: 10.1039/c5nj00805k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |